

Abstract
The urgency to find complementary therapies to current SARS-CoV-2 vaccines, whose effectiveness is preserved over time and not compromised by the emergence of new and emerging variants, has become a critical health challenge. We investigate the possibility of jamming the opening of the Receptor Binding Domain (RBD) of the spike protein of SARS-CoV-2 with small compounds. Through in silico screening, we identified two potential candidates that would lock the Receptor Binding Domain (RBD) in a closed configuration, preventing the virus from infecting the host cells. We show that two drugs already approved by the FDA, mithramycin and dihydroergotamine, can block infection using concentrations in the μM range in cell-based assays. Further STD-NMR experiments support dihydroergotamine’s direct interaction with the spike protein. Overall, our results indicate that repurposing of these compounds might lead to potential clinical drug candidates for the treatment of SARS-CoV-2 infection.
Introduction
Since the onset of the COVID-19 pandemic, there has been a significant improvement in the way the disease is managed, largely due to the development of various therapeutic agents, vaccines, and monoclonal antibodies.1
One of the most significant impacts of the COVID-19 pandemic is the emergence of long-term side effects of the infection, also known as long-covid.2−11 It is currently unclear if there is a correlation between the severity of the infection and the likelihood of developing long-covid symptoms. However, it is known that not all current vaccines prevent the onset of infection due to emerging variants and the subsequent development of long-covid.12 Some treatment options have been shown to be effective in specific patient groups,13,14 but monoclonal antibodies have reduced neutralizing efficacy against new variants,15,16 and vaccines are limited by variable efficacy, the emergence of vaccine-resistant viral variants, cost, and availability.17,18 Currently, the only prophylactic agents for immunocompromised patients are monoclonal antibodies (e.g., tixagevimab/cilgavimab), from which the viral spike can escape by rapidly evolving new variants.15 There are no other approved agents for pharmacological prophylaxis against COVID-19 (https://www.who.int/publications/i/item/WHO-2019-nCoV-prophylaxes-2021-1).
Therefore, it is essential to develop a therapy that can be administered at the first sign of an infection. Ideally, this treatment should be made available to the public at the earliest and at a low cost to ensure accessibility to as many people as possible.
An effective way to identify new treatments meeting the aforementioned criteria is to search for FDA-approved drugs that target crucial physiological processes of the virus.19−28 Currently, there are several studies that look for potential binding to either the SARS-CoV-2 main protease (MPro) (https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html) or for an overall screening to the proteins that allow the virus to replicate (Exscalate4CoV project (https://www.cineca.it/news/exscalate4cov-progetto-di-riferimento-europa-il-coronavirus)). Another important target is the Receptor Binding Domain (RBD) of the spike glycoprotein on the surface of SARS-CoV-2.20,21 This protein is the primary means by which the virus infects human cells. The ability of SARS-CoV-2 to infect human cells requires the exposure of the RBD in the open state of the spike protein.29−31 Once the RBD is exposed, it can bind to the ACE2 human receptor, the primary entry point for the SARS-CoV-2 virus, and initiate the process leading to viral entry into the cell and ultimately infection.
Several studies have identified FDA approved drugs that bind to the spike protein, but none have comprehensively studied their ability to block viral entry into cells.20,32 Recently, Brevini et al.33 published the first study evaluating the use of a repurposed drug for the potential treatment of COVID-19 by inhibiting viral entry into cells that express the ACE2 receptor. The study included laboratory testing to assess the effectiveness of the drug in downregulating ACE2 expression. This research demonstrated the potential of drug repurposing as a method for finding effective treatments. However, downregulating receptors on healthy cells throughout the body carries the risk of potential side effects. As an alternative approach, we decided to focus on directly targeting the opening of the RBD in the viral spike protein.27−29 Through in silico screening of over 6,000 compounds we identified two already FDA-approved drugs, mithramycin and dihydroergotamine, as substances that could lock the RBD in a closed configuration thus preventing the virus from infecting cells.29−31 These two drugs emerged as promising candidates by in vitro testing for their cytotoxicity and their ability to halt entry of pseudo SARS-CoV-2 viruses into cells expressing the ACE2 receptor. We found that at 3.12 and 1.56 μM concentrations dihydroergotamine and mithramycin, respectively, were able to reduce 6-fold the entry of pseudoviruses (PsV). In addition, by saturation transfer difference (STD) Nuclear Magnetic Resonance (NMR) spectroscopy, we showed that soluble dihydroergotamine binds to the spike protein. Our results suggest that mithramycin and dihydroergotamine are suitable candidate compounds for drug repurposing against COVID-19.
Results
Identification of FDA-Approved Mithramycin and Dihydroergotamine
We used cryo-EM derived atomic models of the spike protein (PDB ID 6VYB and 6VXX) and Autodock Vina molecular docking software (see Methods) to screen a total of 5940 compounds, including both FDA-approved and non-FDA-approved drugs. The docking search space encompasses the entire spike protein structure, and hence it includes but is not limited to the region already considered in the study of the D. E. Shaw research lab (“Molecular Dynamics Simulations Related to SARS-CoV-2”, D. E. Shaw Research Technical Data, 2020. http://www.deshawresearch.com/resources_sarscov2.html) (Figure S2a, S3a). We screened 1466 FDA-approved and 4274 non-FDA-approved drugs (see Supporting Information (SI) for Vina scores). Out of the FDA group we selected the top 10 with the highest binding affinity in the apical region (lowest Vina score, see Figure 1a).
Figure 1.

VINA docking: (a) Ranking of the FDA-approved drugs from the VINA docking calculations on the Spike glycoprotein of the first variant of SARS-CoV-2 virus. (b) Average opening of the RBD as a function of the interaction strength between the drug and the Spike protein. Four molecules showed the best ability to block the opening at the lowest strengths.
While the docking analysis can provide useful information about potential binding between molecules and the receptor binding domain (RBD) of the spike protein, it cannot accurately predict which molecules will mechanically block the opening of the RBD. This is because the ability of a molecule to block the opening of the RBD depends on the specific residues involved in the binding, which cannot be reliably predicted by the docking analysis alone.
To address this issue, it was necessary to perform further computational analysis using a different approach. For example, using a coarse-grained representation of the spike protein can be an effective way to sample the configurations near the native state, which can provide more accurate predictions about the ability of molecules to block the opening of the RBD. This approach is also more efficient in terms of computational resources, as coarse-grained models typically require less computational power to run than more detailed models.
To this end we modeled the spike protein using a coarse-grained representation that we previously found to be extremely efficient in sampling the configuration close to the native state (Methods).34 In our model the interactions between the residues are defined as those present in the native structure taken from the closed conformation of the spike (PDB ID 6VXX), the dihedral angles are also taken from the atomic model and guarantee a fast-folding dynamics for the entire complex.35 The second step consisted of letting the RBD domain free to move from the closed to the open state; for this we removed the interdomain interactions between the RBD and the rest of spike. By not including any interactions between the RBD and the spike protein, we are considering the worst-case scenario where the ligand alone will have to do all the work of locking down the RBD.
We ran a coarse-grained simulation of the Spike protein alone until we observed the full opening of the RBD domain (106 Steps). The opening is monitored by measuring the smallest distance between the atoms of the RBD domain and the rest of the spike protein. We then performed 4 simulations for each the 10 best FDA approved drugs and measured the motion of the RBD domain compared to the closed state, for a total of 40 simulations. From the AutoDock calculations we selected the first in the list which corresponds to the poses with the highest binding affinity. Interestingly the poses we found are visually different from the ones identified by D. E. Shaw research lab (“Molecular Dynamics Simulations Related to SARS-CoV-2”, D. E. Shaw Research Technical Data, 2020. http://www.deshawresearch.com/resources_sarscov2.html) even if the search regions are similar. From the selected poses, we identified the contacts between the drug atoms and the Spike ones in the pose conformation. Each of the 4 simulations is performed at a different value of the ligand-spike interaction energy EI [0.1,1,5,10] in reduced units. With these simulations, we are testing the quality of the mechanical coupling between the ligand and the spike protein. In other words, we are checking which of the ligands is capable of blocking the opening already at the weakest binding strengths. The best ligands will be those that efficiently block the opening of the RBD at the lowest interaction strength. Hence our scoring function will be the min of EI that blocks the opening of the RBD.
The results of the simulations show quite a difference in the ability of the ligands to hamper the RBD opening (Figure 1b). Strikingly, Conivaptan (DB00872), which according to Vina docking simulations displays the highest binding affinity (−9.9 kcal/mol Vina Score), cannot block the opening of the RDB at any strength EI (0.1–10 internal units). Similarly, lomitapide (DB08827) works well only at the highest binding strength EI = 10. Differently, we found linaclotide, actinomycin, dihydroergotamine, and mithramycin (see chemical structure in Figure 2) to be the most promising candidates for in vitro testing as they were the compounds that blocked the opening even at the lowest binding strength EI = 0.1 (Figure 1b). Linaclotide is a peptide with sequence CCEYCCNPACTGCY. It is used to alleviate the symptoms of irritable bowel syndrome. Actinomycin is a chemotherapy medication used to treat a number of cancer types. This includes Wilms tumor, rhabdomyosarcoma, Ewing’s sarcoma, trophoblastic neoplasm, testicular cancer, and certain types of ovarian cancer.36 mithramycin and dihydroergotamine have been also computationally studied by other groups in the search of antivirals against SARS-CoV-2,18,24,25 but none of them has been tested in vitro or in vivo.19,25,26 Mithramycin is an antibiotic that inhibits the synthesis of the bacterial RNA (https://www.fermentek.com/product/mithramycin-plicamycin) and is not currently on the market because it is not approved for cancer therapy. Dihydroergotamine is a widely used antimigraine drug approved in 1946, still on the market also as a nasal spray formulation (ideal for the intended use against respiratory disease) with well-known record of dosage and side effects on patients.
Figure 2.

Molecular structure of the drugs isolated through our computational screening process: (a) Actinomycin D (C62H86N12O16 1255.438 g·mol–1). (b) Mithramycin (also known as Plicamycin) (C52H76O24 1085.156 g·mol–1). (c) Linaclotide (C59H79N15O21S6 1526.73 g·mol–1). (d) Dihydroergotamine Mesylate (C33H37N5O5 583.689 g·mol–1).
Biocompatibility and Neutralization Assays
Prior to neutralization assays we performed a biocompatibility test with the four selected drugs.37 The results showed that actinomycin is toxic at all evaluated concentrations, such a result is not surprising considering that actinomycin is an oncological drug. Dihydroergotamine and mithramycin are harmless to the treated cells up to a concentration <25.0 μM. Linaclotide, on the other hand, is nontoxic at all concentrations (Figure 3).
Figure 3.

Cytotoxicity of drugs: (a) HEK293T and (b) HEKT293T-ACE2 cells after 3 days of treatment with DMSO, mithramycin (M), dihydroergotamine (D), linaclotide (L), actinomycin (AC) drugs. All data are shown as mean ± SD. For more details about p-values, see Table S1 in the Supporting Information.
In addition to the cytotoxicity assays, cell entry neutralization assays for SARS-CoV-2 pseudoviruses (PsV) were performed. PsV-SARS-CoV-2 incorporation was significantly reduced in actinomycin-treated cells at all concentrations used (Figure 4a). The same effect was observed with dihydroergotamine and mithramycin at concentrations of 3.12 and 1.56 μM, respectively (Figure 4b,c). Linaclotide did not demonstrate the ability to block viral entry at any of the concentrations tested (Figure 4d). However, the observed toxicity of actinomycin on the treated cells (Figure 3) could be biasing the neutralization effect observed in Figure 4, leading to the conclusion that the concentrations of actinomycin studied are not optimal for viral neutralization treatment.
Figure 4.

Neutralization assays: Neutralizing effect on PsV infection by different drugs (a) Linaclotide, (b) Actinomycin, (c) Dihydroergotamine, and (d) Mithramycin at different concentrations (1.56, 3.1, 6.2, 12.5, 25, 50 uM). The 293T and 293T-ACE2 cells were used as a negative control without drug (Control and ACE2) and with drug (XControl, XACE2), respectively. The infection of PsV were evaluated in 293T cells (PsV Control) as a nonincorporation control and in 293T-ACE2 cells as a positive control (PsVACE2). The luminescence exposition was measured after 72 h of treatment. All data are shown as mean ± SD. For more details about p-values see Table S1 in the Supporting Information. The values in the table indicate p < 0.001 statistically significant data analyzed by two-way ANOVA.
Thus, dihydroergotamine and mithramycin emerge as the most promising drug candidates for further preclinical and clinical study.
Saturation Transfer Difference Nuclear Magnetic Resonance Spectroscopy
To assess the interaction between mithramycin and dihydroergotamine and the spike protein, we used saturation transfer difference Nuclear Magnetic Resonance spectroscopy. In the case of mithramycin the limited water solubility hampered the NMR binding studies as no signal could be detected in the 1H NMR spectrum. On the contrary, weak but clear signals were observed for dihydroergotamine when used at low concentrations (Figure 5a top and Figure S1). Thus, a 1H-STD-NMR experiment was performed by employing a 10 μM spike protein ectodomain and 400 μM dihydroergotamine. Aliphatic irradiation (d 1H −1 ppm) produced clear STD signals for most aromatic protons of dihydroergotamine (Figure 5c), confirming its binding to the spike protein, which involves both aromatic moieties of the ligand molecule. The very weak signals for the aliphatic protons, already in the 1H NMR spectrum, precluded further analysis. A control experiment was performed by acquiring the same 1H-STD-NMR experiment in the absence of the protein, which produced no STD signals (Figure 5d). Binding was further confirmed by comparing the 1H NMR spectra of dihydroergotamine in the absence (free) and presence of spike protein (bound) (Figure 5a), which showed a clear line broadening effect in the presence of the protein, indicative of binding. These results provide an experimental proof of the direct interactions of dihydroergotamine to the spike protein as predicted by the docking simulations in silico (Figure S2–S4). Unfortunately, the weak NMR signals did not allow confirmation or dismissal of the binding mode of the drug predicted in silico, only that the aromatic residues are involved. It is important to emphasize that our focus is on targeting the virus’s ability to bind to the exterior of human cells, rather than the drug’s ability to enter the cell and remain active. As a result, our findings should be valid regardless of the type of cell line used to perform the assays.
Figure 5.
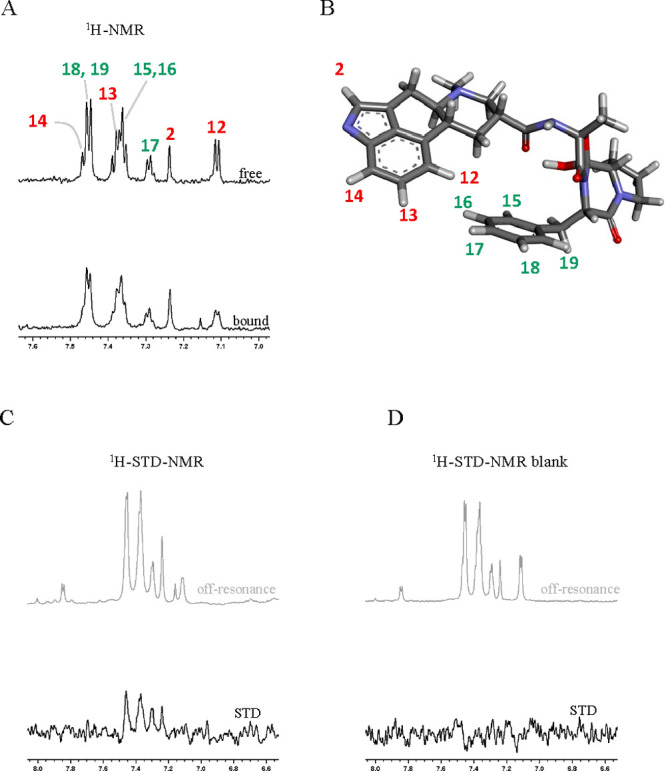
STD-NMR experiments. (a) 1H NMR spectra of dihydroergotamine zoomed at the aromatic region, free (above) and in the presence of Spike protein (below). Annotations indicate the signal assignment according to the numbering shown in panel B. Both spectra were acquired exactly under the same experimental conditions and processed with the same parameters (lb = 2 Hz). (b) 3D structure of dihydroergotamine as obtained by X-ray crystallography (PDB ID 4IAQ) and proton numbering used for signal assignment. (c) 1H-STD-NMR experiment (lb = 6 Hz) of dihydroergotamine in the presence of Spike protein: STD spectrum below and corresponding off-resonance52 spectrum on top. (d). 1H-STD-NMR experiment (lb = 6 Hz) of dihydroergotamine in the absence of Spike protein (control): STD spectrum below and corresponding off-resonance52 spectrum on top.
Discussion
In the search for compounds to combat recurrent outbreaks of the SARS-CoV-2 pandemic, our study identified mithramycin and dihydroergotamine as potential ligands that block the opening of the receptor binding domain (RBD) of the SARS-CoV-2 spike protein.
This study focuses on directly targeting the opening of the RBD in the viral spike protein of SARS-CoV-2, rather than downregulating receptors on healthy cells throughout the body, which carries the risk of potential side effects.32 We propose mithramycin as a substance that could lock the RBD in a closed configuration. Neither mithramycin nor dihydroergotamine were tested before in an in vitro assay proving their ability to block the viral entry. Additionally, it is the first study evaluating the use of a repurposed drug for the potential treatment of COVID-19 by inhibiting viral entry into cells that express the ACE2 receptor.30,38 Administration at nontoxic concentrations of these drugs has proven to be an effective treatment that significantly reduced viral infection. However, only the binding between dihydroergotamine and the spike protein could be experimentally confirmed.
Compared to the other proposed drugs,13−18 nasal administration of dihydroergotamine is an attractive alternative route. According to our assay dihydroergotamine is active starting from concentrations of 3.1 μM. Considering that the molecular weight of dihydroergotamine is 673.679 Da, our results suggest that a concentration of 2 mg/L would block the infection, a number which is compatible with the prescribed dosage of the nasal spray formulation of dihydroergotamine mesylate (1–2 mg/L).39
Due to the physicochemical characteristics that favor cell interaction and a pharmacological effect at low doses of the drug, the method of administration (which is harmless and highly efficient for patients), its availability on the market, and its biocompatibility, dihydroergotamine is a potential candidate for use in clinical studies. Analysis of the docking site of dihydroergotamine on the spike protein (see Figures S3–S5) reveals that it does not share any residues in common with the top 10 mutations registered in the literature.40 This means that the drug is likely to maintain its efficacy against the virus, even in the presence of these mutations. Moreover, the large list of potential binding partner residues of dihydroergotamine makes its efficacy very robust against point mutations. This means that even if there are additional mutations in the virus beyond the top 10, dihydroergotamine is still likely to be effective in binding to the spike protein and preventing viral entry into human cells.
This research demonstrates the potential of drug repurposing as a method for finding effective treatments for COVID-19 and offers a ranking of their potential efficacy in blocking the opening of the RDB domain.
Drug repurposing has been gaining popularity as a way to find new treatments for various diseases including COVID-19. This is mainly because it can save time and resources by repurposing drugs that have already been developed and tested for safety and efficacy. However, traditional drug repurposing strategies typically involve screening large libraries of existing drugs to identify potential candidates based on their ability to interact with a specific target. This approach can be time-consuming and often leads to a large number of false positives, making it difficult to identify the most promising candidates.
Our study introduces a nuanced, multiscale strategy as a fresh paradigm for pinpointing drugs that inhibit key allosteric transitions in proteins, specifically targeting the receptor binding domain (RBD) in the SARS-CoV-2 spike protein. Unlike prior research primarily reliant on high-throughput screenings, we harmonize computational in silico models with subsequent experimental validation. Our computational engine leverages coarse-grained simulations aimed at locking down the RBD’s allosteric activity—a focus notably absent in much of the existing literature. The distinct advantage of using coarse-grained simulations for studying the RBD opening process in the spike protein, as compared to traditional full atomistic approaches, lies in their ability to explore larger time scales. This facilitated the rapid screening of a diverse range of drugs and their binding poses. Additionally, the reduced resolution of our simulations contributes to the robustness of the results, making them less susceptible to model-specific biases. This computational rigor enabled us to identify mithramycin and dihydroergotamine as promising candidates, whose efficacy was later confirmed through both cell-based assays and STD-NMR experiments. This holistic validation is particularly noteworthy given the technical intricacy and specialized skill set required for spike-protein assays. Our research breaks new ground by offering a cohesive methodology with proven potential in identifying FDA-approved drugs for repurposing, including dihydroergotamine, for which we provide compelling experimental evidence. Furthermore, our approach facilitates a more precise evaluation of drug efficacy in hindering RBD transitions, making it a potentially game-changing framework applicable to a broad spectrum of infectious diseases.
In the long term, our multiscale approach could be a valuable framework for identifying drugs that effectively block allosteric transitions in viral or bacterial proteins at the initial stages of infection. Our methodology allows for the development of new treatments against viral or bacterial infections that complement traditional competitive binding strategies. It is well-known that achieving competitive binding can be challenging, as pathogens have evolved to have extremely high binding affinity for their target receptors. On the other hand, targeting proteins from pathogens directly reduces the risk of potential side effects associated with interference with the physiological activities of human cells.
In conclusion, our research introduces a method for discovering novel therapies by inhibiting allosteric transitions in proteins that are crucial for viral infection. By applying this approach to target the RBD opening in the SARS-CoV-2 spike protein, we identified mithramycin and dihydroergotamine as potential candidates for further clinical trials.
Methods
AutoDock Vina Protocol
The crystallographic structure of the SARS-CoV-2 spike glycoprotein in the closed state was recently deposited in the Protein Data Bank (PDB ID: 6VXX). The protein was prepared computing gestgeiser charges and autodock atom type by the means of prepare_receptor4.py, a script included in MGLTools 1.5.6.
Due to the focus on drug repurposing, the list and the structures of the ligands used in this work are drawn from FDA and world approved drug catalogues of ZINC15. These catalogues include drugs approved by the FDA and drugs approved by other major jurisdictions, including tautomer and different protonation states for a total of 5753 compound (FDA = 1466, other jurisdictions = 4287). The compounds were prepared for docking using prepare_ligand4.py.
The structure-based virtual screening was carried out using AutoDock Vina.41 The grid parameters are reported in Table 1.
Table 1.
| 6VXX Apicala | 6VXX Lateralb | ||
|---|---|---|---|
| Center | Center | ||
| X | 210.11 Å | X | 224.74 Å |
| Y | 210.92 Å | Y | 185.22 Å |
| Z | 269.26 Å | Z | 242.51 Å |
| Dimension | Dimension | ||
|---|---|---|---|
| X | 46.88 Å | X | 52.50 Å |
| Y | 46.88 Å | Y | 44.25 Å |
| Z | 46.88 Å | Z | 38.25 Å |
The list of selected residues from the Apical Grid is 338–339, 341–356, 363–378, 380, 396–411, 414–424, 432–444, 447–454, 465–468, 489, 491–501, 503–513, 1307.
The list of selected residues from the Lateral Grid is 2, 37–42, 53, 87–88, 117, 130, 165–169, 193, 195, 199–204, 223–232, 234, 236–237, 271–272, 323–335, 336–344, 346–352, 354–365, 366–377, 378–387, 388–405, 406–419, 420–428, 429–437, 452–454, 462–468, 489–493, 495, 504, 508–525, 526–534, 539–548, 552, 560–565, 567, 576–583, 585, 746–749, 751–752, 755, 969–986, 987–995, 1306–1307.
Coarse-Grained Protein Simulations
In our study, we used coarse-grained molecular dynamics simulations to investigate the structural changes in the SARS-CoV-2 spike protein in the presence of two repurposed drugs: mithramycin and dihydroergotamine. We identified the most promising drugs and their bound poses using AutoDock and then simulated the bound state using the LAMMPS package testing for their ability to lock the RBD domain.42,43
To accurately model the spike protein, we used a computationally efficient description of the native state, known as a GO-type protein model.44 This type of model is based on the idea that the folding landscape of a protein follows the principle of minimum frustration, and therefore, the configurational space close to the native state can be modeled accurately.45
All attractive interactions between the spike protein and the drugs were represented using a sigmoidal-type potential called UATT, which we extensively used in coarse-grained protein models to represent residue–residue interactions.34,46,47
where rc= 4.0 Å is the hard-core radius of each residue; the solvent is implicit. While all excluded volume interactions are represented with a Weeks–Chandler–Andersen potential, a Lennard-Jones potential shifted to zero from the distance corresponding to the minimum to infinite.48
Additionally, we removed all interdomain interactions between the RBD and the rest of the spike protein to model the worst-case scenario where the drug has to do all the work to keep the spike protein closed.
The drugs were represented as rigid bodies with all of their atoms, and the interactions between the drug atoms and the spike protein were attractive only according to the UATT potential for the bonds identified during the docking simulations. We relied on the AutoDock ranking and assumed it to be accurate. We then used the coarse-grained simulations to test if the drugs were capable of impeding the opening of the spike protein. For each drug pose we performed 10 coarse-grained simulations averaging the minima atomic distance between the RBD and rest of Spike protein atoms to quantify the opening of the RBD.
Experimental Section
Plasmids for Spike Protein Expression
The sequences of all plasmids used in this study are described elsewhere,49 and they are available at Genbank (File S1) and also at GitHub (https://github.com/jbloomlab/SARS-CoV-2_lentiviral_pseudotype/tree/master/plasmid_maps). The plasmids were the following: HDM-tat1b (NR-52518), pRC-CMV-Rev1b (NR-52519), pHAGE-CMV-Luc2-IRES-ZsGreen-W (NR-52516), HDM-Hgpm2 (NR-52517), pHAGE2-CMV-ZsGreen-W (NR-52520), HDM-nCoV-Spike-IDTopt-ALAYT (NR-52515), and HDM-IDTSpike-fixK (NR-52514). All plasmids have ampicillin resistance.
HEK-293T ACE2 Cells and Pseudotyped Lentiviral Particles Production
The methodology used to create the 293T cell constitutively expressing the human ACE2 cellular receptor followed the protocol previously reported.49 Briefly, to produce pseudotyped lentiviral particles (PsV) we seeded HEK293T cells in DMEM at 10% FBS. After 16–24 h (50–70% cellular confluent), the cells were transfected with the plasmids required for lentiviral production. The transfection cocktail, 1000 μL of DMEM with no FBS, contained 15 μg of Luciferase-IRES-ZsGreen, 3.3 μg each of plasmids HDM-Hgpm2, pRC-CMV-Rev1b, and HDM-tat1b and 5.1 μg SARS-CoV-2 spike, and then 60 μL of BioT reagent was added in mixing gently and incubating at RT for 5 min. The cocktail was added to the cells growing in a 175T flask, top-up with additional 9 mL of DMEM with no FBS. The cells were incubated during 2 h at 37 °C with gentle shaking every 15 min; after 2 h, further 15 mL of DMEM with 5% FBS was added to the flask. At 60 h post transfection, the supernatant containing secreted pseudotyped-SARS-CoV-2 particles was harvested and filtered through a 0.45 μm filter. PsV was either stored at 4 °C for immediate use or frozen at −80 °C for use at later stages.
Titration of SARS-CoV-2 S-Pseudotyped Viruses
To assess the titer of the pseudotyped particles a luciferase assay was performed as previously described.49 In short, a 96-well cell-culture plate was seeded with 1.25 × 104 293T-ACE2 cells per well in 100 μL of growth media. Serial dilution of the virus sample to be titrated were prepared in a final volume of 160 μL growth media. After 12 and 24 h postseeding, the serial dilutions were added into the different wells; a final concentration of 5 μg/mL Polybrene (Sigma-Aldrich, TR-1003-G) was also added to each well to promote infection. The plate was left incubating during 2 h with gentle shaking each 30 min, then further 100 μL of DMEM at 5% FBS were added (final volume 200 μL of DMEM 2.5% FBS per well). At 48–60 h postinfection, the cells were collected for luciferase analysis. 90 μL of media were removed from each well leaving ∼50 μL due to evaporation; to this 50 μL of luciferase reagent Bright-Glo Luciferase Assay System (Promega, E2610) were added and incubated for 2 min. After mixing well, 90 μL solution from each well was transferred to a black 96-well-plate and read in a luminometer (VICTOR Nivo Multimode Microplate Reader).
Biocompatibility Assays
To determine the biocompatibility of the drugs and the concentration range to use, 293T and 293T-ACE2 cells were seeded with 1.25 × 104 cells per well using a 96-well culture plate and maintained in a cell incubator at 37 °C, 5% CO2, and 80% humidity. After 24 h post cell seeding, the medium was removed and replaced with fresh media and the following conditions explored: (i) DMSO at 0.015, 0.031, 0.062, 0.12, 0.25, 0.5, 1% v/v as a control of drug vehicle; (ii) only medium as a cellular control; and (iii) drugs at different concentrations 0.78 μM, 1.56 μM, 3.12 μM, 6.25 μM, 12.5 μM, 25.0 μM as a treatment during 72 h. A MTT assay for different drug in 293T-ACE2 and 293T-ACE2 cells lines was performed to compare toxicity response in control and treated cells.50 Absorbance of samples at 540 nm was measured, and the percent of cell survival was calculated.
Neutralization Assay of SARS-CoV-2 S-Pseudotyped Viruses
To determinate the neutralization of PsV entry we used published protocols with minor modifications.49 For the neutralization assay, 293T and 293T-ACE2 cells were seeded at 1.25 × 104 cells per well, respectively, in 100 μL of growth media using 96-well culture plate, which were maintained in an incubator at 37 °C, 5% CO2, and 80% humidity. After 24 h post cell seeding, the medium was removed, and the cells washed with PBS twice. 75 μL of fresh media was added to each well. Then the cells were differentially treated using DMEM supplemented with Polybrene (0.75 ug per well) and additional components as follows: (i) only medium as a cellular control; (ii) with control vehicle DMSO at 0.015, 0.031, 0.062, 0.12, 0.25, 0.5, 1% v/v; (iii) with PsV (60 μL of the PsV dilution select into each well) as positive control; (iv) with PsV and different drugs at increasing concentrations 0.78, 1.56, 3.12, 6.25, 12.5, 25.0 μM. The plates were then incubated for 2 h at 37 °C and with gentle shaking every 15 min. Afterwards, 75 μL of DMEM with 5% FBS was added to each well so that the final FBS concentration was at 2.5% v/v. Plates were incubated for 60 h at 37 °C, and then the cellular viability was assayed via MTT as mentioned above.
Spike Protein Production and Purification
The ectodomain of the S spike protein (BEI construct NR-52394) was expressed by transient transfection of HEK293F suspension cells and purified from clarified cell supernatants 7 days post-transfection using a nickel affinity column and size-exclusion chromatography as previously described.51 Then, the spike protein and previously purified ligand according to Barnes et al. (2020) were mixed and incubated 2 h at 4 °C.51
Drug Purity
Actinomycin and mithramycin, both from Sigma, have HPLC purity >95% and ≥90%, respectively. Dihydroergotamine from LGC Labor GmbH and Linaclotide from Toronto Research Chemicals both have an HPLC purity of ≥95%. Ultrapure quality DMSO, which was purchased from ThermoFisher, was used to solubilize these drugs and make the stock solution.
Dyhydroergotamine/Spike Protein Complexation in Solution
A stock solution of 2.5 × 104 μM dihydroergotamine in deuterated DMSO was prepared. 8 μL of it was diluted in ultrapure water (1:100), frozen in liquid nitrogen, and lyophilized for 3 days. After the solvent had been completely removed, the solid drug was then resuspended in 492 μL of a 10 μM solution of spike protein in phosphate saline (PBS) buffer pH 7.4 in D2O, resulting in a sample of 10 μM spike protein and 400 μM dihydroergotamine (1:40 ratio).
For the control sample, the same procedure was performed, but the solid dihydroergotamine was resuspended in phosphate saline (PBS) buffer, pH 7.4, in D2O.
Detection Complexation in Solution of Spike Protein/Dihydroergotamine by Saturation Transfer Difference (STD-NMR)
All the NMR spectra were acquired on an 800 MHz BRUKER AVANCE III spectrometer equipped with a TCI cryoprobe with z-gradient coil, and TopSpin 3.2.7 (BRUKER) software was employed for data acquisition and processing. 5 mm Shigemi NMR tubes were used. All spectra were acquired at 310 K. Two samples containing 400 μM of dihydroergotamine were prepared in the absence and presence of the spike protein as described above. H-STD-NMR52 experiments were performed by acquiring 3942 scans, employing a Gauss-shaped pulse of 50 ms and 50 dB, with −1 and 100 ppm for the on and off resonance irradiation, respectively.
Statistical Analysis and 2D LigPlot+ Diagrams
Statistical assays were carried out on STATISTICA (Statasoft, Inc. 2007, version 7, Microsoft). One-way study of variance (ANOVA) was carried out for data related to the cytotoxicity of drug and PsV neutralization. All errors reported are the standard deviation of the mean. Schematic 2D diagrams of protein–ligand complexes from AutoDock and simulated file input were generated by LigPlot+ v.2.2.5.
Acknowledgments
We would like to thank Prof H.W. Frijlink for fruitful discussions about the hypothetical dosage for a treatment based on dihydroergotamine. We thank the support of the computing infrastructure of the i2BASQUE academic network and the ATLAS machine at the DIPC. This research was supported by the Spanish Ministerio de Economía y Competitividad (MINECO) (FIS2017-89471-R to I.C.). This research was supported by Programa Red Guipuzcoana de Ciencia, Tecnología e Información (2019-CIEN-000051-01 to I.C.). I.C. acknowledges support from try BIKAINTEK program (grant No. 008-B1/2020). I.C. acknowledges support from the COST Action CA17139 EUTOPIA. This work was supported by grants of the Agencia Estatal de Investigacion (Spain) for projects RTI2018-095700–B-I00 and PID2021-126130OB-I00 (NGAA), project PDI2021-1237810B–C21 (JJB & AA), and CIBERES and CIBERehd, initiatives of Instituto de Salud Carlos III (ISCIII). We thank the Basque Government for COVID-19 CO-2020/00001 grant to NGAA and JJ-B. SS is supported by the ISCII-Health programme. This research was also funded by the Italian Ministry of University and Research (MIUR), grants “Dipartimenti di Eccellenza” (Legge 232/2016, Articolo 1, Comma 314–337) and PRIN (Grant No. 2017483NH8) to F.P., B.C and P.C.
Glossary
Abbreviations
- CCR2
CC chemokine receptor 2
- CCL2
CC chemokine ligand 2
- CCR5
CC chemokine receptor 5
- TLC
thin layer chromatography.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c02921.
Extra NMR data and the models obtained with the docking simulations (PDF)
Author Contributions
IC designed the study. IC and GM (with the supervision of FP) conducted the molecular docking studies. SS performed biochemical and cellular experiments and statistical analysis with the supervisions of NGAA. AV and JE-O helped with protein production and inhibition viral experiments. AA carried out STD experiments and analyzed NMR data with JJ-B. IC, GM, BC and SS wrote the manuscript. F.P. and NGAA reviewed and edited the manuscripts. All authors approved the findings and conclusions of the final manuscript.
Spanish Ministerio de Economía y Competitividad (MINECO) (FIS2017–89471-R) Programa Red Guipuzcoana de Ciencia, Tecnología e Información (2019-CIEN-000051–01). BIKAINTEK program (grant No. 008-B1/2020). COST Action CA17139 EUTOPIA. Agencia Estatal de Investigacion (Spain) for projects RTI2018–095700–B-I00, PID2021–126130OB-I00, PDI2021–1237810B–C21 and CEX2021-001136-S. Basque Government COVID-19 CO-2020/00001. Italian Ministry of University and Research (MIUR), grants “Dipartimenti di Eccellenza” (Legge 232/2016, Articolo 1, Comma 314–337) and PRIN (Grant No. 2017483NH8).
The authors declare no competing financial interest.
Supplementary Material
References
- Bartoszko J. J.; Siemieniuk R. A. C.; Kum E.; Qasim A.; Zeraatkar D.; Ge L.; Han M. A.; Sadeghirad B.; Agarwal A.; Agoritsas T.; Chu D. K.; Couban R.; Darzi A. J.; Devji T.; Ghadimi M.; Honarmand K.; Izcovich A.; Khamis A.; Lamontagne F.; Loeb M.; Marcucci M.; McLeod S. L.; Motaghi S.; Murthy S.; Mustafa R. A.; Neary J. D.; Pardo-Hernandez H.; Rada G.; Rochwerg B.; Switzer C.; Tendal B.; Thabane L.; Vandvik P. O.; Vernooij R. W. M.; Viteri-García A.; Wang Y.; Yao L.; Ye Z.; Guyatt G. H.; Brignardello-Petersen R. Prophylaxis against Covid-19: Living Systematic Review and Network Meta-Analysis. BMJ 2021, 373, n949. 10.1136/bmj.n949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveendran A. V.; Jayadevan R.; Sashidharan S. Long COVID: An Overview. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2021, 15 (3), 869–875. 10.1016/j.dsx.2021.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hakeim H. K.; Al-Rubaye H. T.; Al-Hadrawi D. S.; Almulla A. F.; Maes M. Long-COVID Post-Viral Chronic Fatigue and Affective Symptoms Are Associated with Oxidative Damage, Lowered Antioxidant Defenses and Inflammation: A Proof of Concept and Mechanism Study. Mol. Psychiatry 2023, 28, 564. 10.1038/s41380-022-01836-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Borst B.; Peters J. B.; Brink M.; Schoon Y.; Bleeker-Rovers C. P.; Schers H.; van Hees H. W. H.; van Helvoort H.; van den Boogaard M.; van der Hoeven H. Comprehensive Health Assessment 3 Months after Recovery from Acute Coronavirus Disease 2019 (COVID-19). Clinical Infectious Diseases 2021, 73 (5), e1089–e1098. 10.1093/cid/ciaa1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahase E. Covid-19: What Do We Know about “Long Covid”?. BMJ 2020, 370, m2815. 10.1136/bmj.m2815. [DOI] [PubMed] [Google Scholar]
- Miyazato Y.; Morioka S.; Tsuzuki S.; Akashi M.; Osanai Y.; Tanaka K.; Terada M.; Suzuki M.; Kutsuna S.; Saito S. Prolonged and Late-Onset Symptoms of Coronavirus Disease 2019. Open forum infectious diseases 2020, 7, ofaa507. 10.1093/ofid/ofaa507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.; Li X.; Geng D.; Mei N.; Wu P.-Y.; Huang C.-C.; Jia T.; Zhao Y.; Wang D.; Xiao A. Cerebral Micro-Structural Changes in COVID-19 Patients–an MRI-Based 3-Month Follow-up Study. EClinicalMedicine 2020, 25, 100484. 10.1016/j.eclinm.2020.100484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis A.; Wamil M.; Alberts J.; Oben J.; Cuthbertson D. J.; Wootton D.; Crooks M.; Gabbay M.; Brady M.; Hishmeh L. Multiorgan Impairment in Low-Risk Individuals with Post-COVID-19 Syndrome: A Prospective. Community-Based Study. BMJ. Open 2021, 11 (3), e048391 10.1136/bmjopen-2020-048391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong S. J. Long COVID or Post-COVID-19 Syndrome: Putative Pathophysiology, Risk Factors, and Treatments. Infect Dis 2021, 53 (10), 737–754. 10.1080/23744235.2021.1924397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah W.; Hillman T.; Playford E. D.; Hishmeh L. Managing the Long Term Effects of Covid-19: Summary of NICE, SIGN, and RCGP Rapid Guideline. bmj 2021, 372, n136. 10.1136/bmj.n136. [DOI] [PubMed] [Google Scholar]
- Townsend L.; Dowds J.; O’Brien K.; Sheill G.; Dyer A. H.; O’Kelly B.; Hynes J. P.; Mooney A.; Dunne J.; Ni Cheallaigh C. Persistent Poor Health after COVID-19 Is Not Associated with Respiratory Complications or Initial Disease Severity. Ann. Am. Thorac Soc. 2021, 18 (6), 997–1003. 10.1513/AnnalsATS.202009-1175OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik J. A.; Ahmed S.; Mir A.; Shinde M.; Bender O.; Alshammari F.; Ansari M.; Anwar S. The SARS-CoV-2 Mutations versus Vaccine Effectiveness: New Opportunities to New Challenges. J. Infect Public Health 2022, 15 (2), 228–240. 10.1016/j.jiph.2021.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigel J. H.; Tomashek K. M.; Dodd L. E.; Mehta A. K.; Zingman B. S.; Kalil A. C.; Hohmann E.; Chu H. Y.; Luetkemeyer A.; Kline S.; et al. Remdesivir for the Treatment of Covid-19. New England Journal of Medicine 2020, 383 (19), 1813–1826. 10.1056/NEJMoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colchicine in Patients Admitted to Hospital with COVID-19 (RECOVERY): A Randomised, Controlled, Open-Label, Platform Trial. Lancet Respir Med. 2021, 9 (12), 1419–1426. 10.1016/S2213-2600(21)00435-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y.; Wang J.; Jian F.; Xiao T.; Song W.; Yisimayi A.; Huang W.; Li Q.; Wang P.; An R.; et al. Omicron Escapes the Majority of Existing SARS-CoV-2 Neutralizing Antibodies. Nature 2022, 602 (7898), 657–663. 10.1038/s41586-021-04385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier D. A.; De Marco A.; Ferreira I. A. T. M.; Meng B.; Datir R. P.; Walls A. C.; Kemp S. A.; Bassi J.; Pinto D.; Silacci-Fregni C.; et al. Sensitivity of SARS-CoV-2 B. 1.1. 7 to MRNA Vaccine-Elicited Antibodies. Nature 2021, 593 (7857), 136–141. 10.1038/s41586-021-03412-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer O. Covid-19: Countries Are Learning What Others Paid for Vaccines. BMJ: British Medical Journal (Online) 2021, 372, n281. 10.1136/bmj.n281. [DOI] [PubMed] [Google Scholar]
- Callaway E. The Unequal Scramble for Coronavirus Vaccines--by the Numbers. Nature 2020, 584 (7822), 506–508. 10.1038/d41586-020-02450-x. [DOI] [PubMed] [Google Scholar]
- Kouznetsova V. L.; Huang D.; Tsigelny I. F. Potential COVID-19 Protease Inhibitors: Repurposing FDA- Approved Drugs. Phys. Biol. 2021, 18, 025001. 10.1088/1478-3975/abcb66. [DOI] [PubMed] [Google Scholar]
- Murugan N. A.; Kumar S.; Jeyakanthan J.; Srivastava V. Searching for Target-Specific and Multi-Targeting Organics for Covid-19 in the Drugbank Database with a Double Scoring Approach. Sci. Rep 2020, 10 (1), 1–16. 10.1038/s41598-020-75762-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vita S.; Chini M. G.; Lauro G.; Bifulco G. Accelerating the Repurposing of FDA-Approved Drugs against Coronavirus Disease-19 (COVID-19). RSC Adv. 2020, 10 (67), 40867–40875. 10.1039/D0RA09010G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trezza A.; Iovinelli D.; Santucci A.; Prischi F.; Spiga O. An Integrated Drug Repurposing Strategy for the Rapid Identification of Potential SARS-CoV-2 Viral Inhibitors. Sci. Rep 2020, 10 (1), 1–8. 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon S.; Ko M.; Lee J.; Choi I.; Byun S. Y.; Park S.; Shum D.; Kim S. Identification of Antiviral Drug Candidates against SARS-CoV-2 from FDA-Approved Drugs. Antimicrob Agents Chemother. 2020, 64, 1. 10.1128/AAC.00819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M.; Chang S. Y.; Byun S. Y.; Ianevski A.; Choi I.; Pham Hung d’Alexandry d’Orengiani A.-L.; Ravlo E.; Wang W.; Bjøras M.; Kainov D. E.; Shum D.; Min J.-Y.; Windisch M. P. Screening of FDA-Approved Drugs Using a MERS-CoV Clinical Isolate from South Korea Identifies Potential Therapeutic Options for COVID-19. Viruses 2021, 13, 651. 10.3390/v13040651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O’Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; Tummino T. A.; Huettenhain R.; Kaake R. M.; Richards A. L.; Tutuncuoglu B.; Foussard H.; Batra J.; Haas K.; Modak M.; Kim M.; Haas P.; Polacco B. J.; Braberg H.; Fabius J. M.; Eckhardt M.; Soucheray M.; Bennett M. J.; Cakir M.; McGregor M. J.; Li Q.; Meyer B.; Roesch F.; Vallet T.; Mac Kain A.; Miorin L.; Moreno E.; Naing Z. Z. C.; Zhou Y.; Peng S.; Shi Y.; Zhang Z.; Shen W.; Kirby I. T.; Melnyk J. E.; Chorba J. S.; Lou K.; Dai S. A.; Barrio-Hernandez I.; Memon D.; Hernandez-Armenta C.; Lyu J.; Mathy C. J. P.; Perica T.; Pilla K. B.; Ganesan S. J.; Saltzberg D. J.; Rakesh R.; Liu X.; Rosenthal S. B.; Calviello L.; Venkataramanan S.; Liboy-Lugo J.; Lin Y.; Huang X.-P.; Liu Y.; Wankowicz S. A.; Bohn M.; Safari M.; Ugur F. S.; Koh C.; Savar N. S.; Tran Q. D.; Shengjuler D.; Fletcher S. J.; O’Neal M. C.; Cai Y.; Chang J. C. J.; Broadhurst D. J.; Klippsten S.; Sharp P. P.; Wenzell N. A.; Kuzuoglu D.; Wang H.-Y.; Trenker R.; Young J. M.; Cavero D. A.; Hiatt J.; Roth T. L.; Rathore U.; Subramanian A.; Noack J.; Hubert M.; Stroud R. M.; Frankel A. D.; Rosenberg O. S.; Verba K. A.; Agard D. A.; Ott M.; Emerman M.; Jura N.; von Zastrow M.; Verdin E.; Ashworth A.; Schwartz O.; D’Enfert C.; Mukherjee S.; Jacobson M.; Malik H. S.; Fujimori D. G.; Ideker T.; Craik C. S.; Floor S. N.; Fraser J. S.; Gross J. D.; Sali A.; Roth B. L.; Ruggero D.; Taunton J.; Kortemme T.; Beltrao P.; Vignuzzi M.; García-Sastre A.; Shokat K. M.; Shoichet B. K.; Krogan N. J. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molavi Z.; Razi S.; Mirmotalebisohi S. A.; Adibi A.; Sameni M.; Karami F.; Niazi V.; Niknam Z.; Aliashrafi M.; Taheri M.; Ghafouri-Fard S.; Jeibouei S.; Mahdian S.; Zali H.; Ranjbar M. M.; Yazdani M. Identification of FDA Approved Drugs against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) and 3-Chymotrypsin-like Protease (3CLpro), Drug Repurposing Approach. Biomedicine and Pharmacotherapy 2021, 138, 111544 10.1016/j.biopha.2021.111544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti R.; Shukla S. K. Molecules against COVID-19: An in Silico Approach for Drug Development. Journal of Electronic Science and Technology 2021, 19 (1), 100095. 10.1016/j.jnlest.2021.100095. [DOI] [Google Scholar]
- Milani M.; Donalisio M.; Bonotto R. M.; Schneider E.; Arduino I.; Boni F.; Lembo D.; Marcello A.; Mastrangelo E. Combined in Silico and in Vitro Approaches Identified the Antipsychotic Drug Lurasidone and the Antiviral Drug Elbasvir as SARS-CoV2 and HCoV-OC43 Inhibitors. Antiviral Res. 2021, 189, 105055. 10.1016/j.antiviral.2021.105055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A. C.; Park Y.-J.; Tortorici M. A.; Wall A.; McGuire A. T.; Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281. 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Zhang Y.; Li Y.; Xia L.; Guo Y.; Zhou Q. Structural Basis for the Recognition of the SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444. 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D.; Wang N.; Corbett K. S.; Goldsmith J. A.; Hsieh C.-L.; Abiona O.; Graham B. S.; McLellan J. S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molecular Dynamics Simulations Related to SARS-CoV-2. D. E. Shaw Research Technical Data. [Google Scholar]
- Brevini T.; Maes M.; Webb G. J.; John B. V.; Fuchs C. D.; Buescher G.; Wang L.; Griffiths C.; Brown M. L.; Scott W. E.; Pereyra-Gerber P.; Gelson W. T. H.; Brown S.; Dillon S.; Muraro D.; Sharp J.; Neary M.; Box H.; Tatham L.; Stewart J.; Curley P.; Pertinez H.; Forrest S.; Mlcochova P.; Varankar S. S.; Darvish-Damavandi M.; Mulcahy V. L.; Kuc R. E.; Williams T. L.; Heslop J. A.; Rossetti D.; Tysoe O. C.; Galanakis V.; Vila-Gonzalez M.; Crozier T. W. M.; Bargehr J.; Sinha S.; Upponi S. S.; Fear C.; Swift L.; Saeb-Parsy K.; Davies S. E.; Wester A.; Hagström H.; Melum E.; Clements D.; Humphreys P.; Herriott J.; Kijak E.; Cox H.; Bramwell C.; Valentijn A.; Illingworth C. J. R.; Dahman B.; Bastaich D. R.; Ferreira R. D.; Marjot T.; Barnes E.; Moon A. M.; Barritt A. S.; Gupta R. K.; Baker S.; Davenport A. P.; Corbett G.; Gorgoulis V. G.; Buczacki S. J. A.; Lee J.-H.; Matheson N. J.; Trauner M.; Fisher A. J.; Gibbs P.; Butler A. J.; Watson C. J. E.; Mells G. F.; Dougan G.; Owen A.; Lohse A. W.; Vallier L.; Sampaziotis F. FXR Inhibition May Protect from SARS-CoV-2 Infection by Reducing ACE2. Nature 2023, 615, 134. 10.1038/s41586-022-05594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meesaragandla B.; García I.; Biedenweg D.; Toro-Mendoza J.; Coluzza I.; Liz-Marzán L. M.; Delcea M. H-Bonding-Mediated Binding and Charge Reorganization of Proteins on Gold Nanoparticles. Phys. Chem. Chem. Phys. 2020, 22 (8), 4490–4500. 10.1039/C9CP06371D. [DOI] [PubMed] [Google Scholar]
- Whitford P. C.; Noel J. K.; Gosavi S.; Schug A.; Sanbonmatsu K. Y.; Onuchic J. N. An All-Atom Structure-Based Potential for Proteins: Bridging Minimal Models with All-Atom Empirical Forcefields. Proteins 2009, 75 (2), 430–441. 10.1002/prot.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linaclotide Monograph for Professionals - Drugs.com. https://www.drugs.com/monograph/linaclotide.html?references=1 (accessed 2023–01–20).
- Stagnoli S.; Luna M. A.; Villa C. C.; Alustiza F.; Niebylski A.; Moyano F.; Correa N. M.; Falcone R. D. Unique Catanionic Vesicles as a Potential “Nano-Taxi” for Drug Delivery Systems. In Vitro and in Vivo Biocompatibility Evaluation. RSC Adv. 2017, 7 (9), 5372–5380. 10.1039/C6RA27020D. [DOI] [Google Scholar]
- Li W.; Moore M. J.; Vasilieva N.; Sui J.; Wong S. K.; Berne M. A.; Somasundaran M.; Sullivan J. L.; Luzuriaga K.; Greenough T. C.; Choe H.; Farzan M. Angiotensin-Converting Enzyme 2 Is a Functional Receptor for the SARS Coronavirus. Nature 2003, 426 (6965), 450–454. 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin V.; Hoekman J.; Aurora S. K.; Shrewsbury S. B. Nasal Delivery of Acute Medications for Migraine: The Upper Versus Lower Nasal Space. Journal of Clinical Medicine 2021, 10 (11), 2468. 10.3390/jcm10112468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guruprasad L. Human SARS CoV-2 Spike Protein Mutations. Proteins 2021, 89 (5), 569. 10.1002/prot.26042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimpton S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117 (1), 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- LAMMPS.
- Baumketner A.; Jewett A.; Shea J. E. E. Effects of Confinement in Chaperonin Assisted Protein Folding: Rate Enhancement by Decreasing the Roughness of the Folding Energy Landscape. J. Mol. Biol. 2003, 332 (3), 701–713. 10.1016/S0022-2836(03)00929-X. [DOI] [PubMed] [Google Scholar]
- Onuchic J. N.; Wolynes P. G. Theory of Protein Folding. Curr. Opin Struct Biol. 2004, 14 (1), 70–75. 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Toro-Mendoza J.; Maio L.; Gallego M.; Otto F.; Schulz F.; Parak W. J.; Sanchez-Cano C.; Coluzza I. Bioinspired Polyethylene Glycol Coatings for Reduced Nanoparticle–Protein Interactions. ACS Nano 2023, 17, 955. 10.1021/acsnano.2c05682. [DOI] [PubMed] [Google Scholar]
- Magi Meconi G.; Sasselli I. R.; Bianco V.; Onuchic J. N.; Coluzza I. Key Aspects of the Past 30 Years of Protein Design. Rep. Prog. Phys. 2022, 85 (8), 086601 10.1088/1361-6633/ac78ef. [DOI] [PubMed] [Google Scholar]
- Weeks J. D.; Chandler D.; Andersen H. C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54 (12), 5237–5247. 10.1063/1.1674820. [DOI] [Google Scholar]
- Crawford K. H. D.; Eguia R.; Dingens A. S.; Loes A. N.; Malone K. D.; Wolf C. R.; Chu H. Y.; Tortorici M. A.; Veesler D.; Murphy M.; Pettie D.; King N. P.; Balazs A. B.; Bloom J. D. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses 2020, 12, 513. 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyermann J.; Lochmann D.; Zimmer A. A Practical Note on the Use of Cytotoxicity Assays. Int. J. Pharm. 2005, 288 (2), 369–376. 10.1016/j.ijpharm.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Barnes C. O.; West A. P.; Huey-tubman K. E.; Robbiani D. F.; Nussenzweig M. C.; Bjorkman P. J.; Barnes C. O.; West A. P.; Huey-tubman K. E.; Hoffmann M. A. G.; Sharaf N. G. Structures of Human Antibodies Bound to SARS- CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies Ll Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 828–842. 10.1016/j.cell.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viegas A.; Manso J.; Nobrega F. L.; Cabrita E. J. Saturation-Transfer Difference (STD) NMR: A Simple and Fast Method for Ligand Screening and Characterization of Protein Binding. J. Chem. Educ. 2011, 88 (7), 990–994. 10.1021/ed101169t. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.