

Abstract
Background:
The lack of efficient preventive interventions against Alzheimer’s disease (AD) calls for identifying efficient modifiable risk factors for AD. As diabetes shares many pathological processes with AD, including accumulation of amyloid plaques and neurofibrillary tangles, insulin resistance, and impaired glucose metabolism, diabetes is thought to be a potentially modifiable risk factor for AD. Mounting evidence suggests that links between AD and diabetes may be more complex than previously believed.
Objective:
To examine the pleiotropic architecture of AD and diabetes mellitus (DM).
Methods:
Univariate and pleiotropic analyses were performed following the discovery-replication strategy using individual-level data from 10 large-scale studies.
Results:
We report a potentially novel pleiotropic NOTCH2 gene, with a minor allele of rs5025718 associated with increased risks of both AD and DM. We confirm previously identified antagonistic associations of the same variants with the risks of AD and DM in the HLA and APOE gene clusters. We show multiple antagonistic associations of the same variants with AD and DM in the HLA cluster, which were not explained by the lead SNP in this cluster. Although the ε2 and ε4 alleles played a major role in the antagonistic associations with AD and DM in the APOE cluster, we identified non-overlapping SNPs in this cluster, which were adversely and beneficially associated with AD and DM independently of the ε2 and ε4 alleles.
Conclusions:
This study emphasizes differences and similarities in the heterogeneous genetic architectures of AD and DM, which may differentiate the pathogenic mechanisms of these diseases.
Keywords: Alzheimer’s disease, diabetes, pleiotropy, apolipoprotein E gene, major histocompatibility complex
INTRODUCTION
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder with limited interventions available to partly ameliorate its symptoms. Alzheimer’s Association (AA) alarms about sharp increases in AD with age and over time that increase AD-related emotional, physical, and economic burdens on people, their families, societies, and health systems [1]. Prior studies argue that AD is closely related to diabetes [2, 3] implicating AD even as a type 3 diabetes [4, 5].
Diabetes mellitus (DM)—an endocrine/metabolic disorder in aging—occurs when the body becomes resistant to insulin or does not make enough insulin and it can be considered a modifiable risk factor for AD. Insulin plays a key role in the brain including food intake control and regulation of cognitive function. Insulin/insulin-like-growth-factor (IGF) signaling is involved in synaptic formation; neuronal plasticity; learning; memory; neuronal stem cell activation; neurite growth and repair [6]. Insulin resistance induces hyperinsulinemia that leads to inhibition of insulin-degrading enzyme—a regulator of amyloid beta (Aβ) concentrations in neuronal and microglial cells—and decreasing Aβ clearance [7]. Dysfunction of the insulin/PI3K/Akt signaling pathway leads to hyperphosphorylation of the microtubule-associated protein tau in the brain of AD patients through the GSK-3β kinase and the formation of neurofibrillary tangles [8]. Several cardiovascular risk factors can increase the severity of DM and AD including obesity, high blood pressure, high cholesterol, and triglycerides [1]. These and other evidence support insulin deficiency, insulin resistance, and, consequently, impaired glucose metabolism as potential mechanisms linking DM and AD [9–16].
The analyses of summary statistics from genome-wide association studies of AD and DM have been launched to gain insights into potential commonalities and differences in the genetic architectures of these traits. They provided mixed evidence suggesting that some genetic variants could be involved in the regulation of both AD and DM in a concordat manner, while the other variants can confer their risks in an antagonistic manner when the same variant increases the risk of one trait but decreases it for the other trait [17–22]. A growing literature suggests that the links between AD and DM might be more complex than previously believed [22–25].
In this study, we leveraged individual-level data from 10 large-scale datasets, rather than summary statistics from published studies, to identify loci harboring pleiotropic associations of single nucleotide polymorphisms (SNPs) with AD and DM. Having individual-level data, we examined whether the same variants in each locus conferred risks of AD and DM in a consistent or antagonistic manner.
METHODS
Study cohorts
We used data on populations of European ancestry from three Alzheimer’s Disease Centers (ADCs), which are a part of the National Institute on Aging (NIA) Alzheimer’s Disease Genetics Consortium (ADGC) initiative [26], the NIA collaborative study from the Alzheimer’s Disease Sequencing Project (ADSP) [27], the Atherosclerosis Risk in Communities (ARIC) study [28], the Cardiovascular Health Study (CHS) [29], the Framingham Heart Study (FHS) parental and offspring cohorts [30], the Multi-Site Collaborative Study for Genotype-Phenotype Associations in Alzheimer’s disease (GenADA) [31, 32], the NIA Late-Onset Alzheimer Disease Family-Based Study (LOADFBS) [33], the Multi-Ethnic Study of Atherosclerosis (MESA) [34], the Religious Orders Study and Memory and Aging Project (ROSMAP) data [35], and the UK Biobank (UKB) [36]. The basic characteristics of the available samples are given in Table 1.
Table 1.
Basic characteristics of genotyped participants of European ancestry in the selected studies.
| Sample | All | AD cases | DM cases | ||||||
|---|---|---|---|---|---|---|---|---|---|
| N | Men (%) | Age, mean (SD), years | N | Men (%) | Age, mean (SD), years | N | Men (%) | Age, mean (SD), years | |
| ADGC | 5349 | 44.59 | 78.53 (8.5) | 3585 | 46.97 | 79.72 (7.71) | |||
| ADSP | 1568 | 60.52 | 76.86 (10.92) | 998 | 61.72 | 74.20 (12.02) | |||
| ARIC | 9631 | 47.00 | 75.34 (6.85) | 2044 | 53.91 | 75.23 (7.03) | |||
| CHS | 3262 | 39.79 | 83.63 (5.16) | 201 | 34.83 | 85.35 (4.86) | 440 | 50.68 | 83.08 (5.11) |
| FHS | 3761 | 43.84 | 79.23 (10.13) | 361 | 31.02 | 89.41 (6.94) | 660 | 56.52 | 79.48 (9.06) |
| GenADA | 1588 | 38.98 | 75.76 (8.59) | 806 | 42.18 | 78.05 (8.60) | 143 | 44.76 | 77.23 (7.37) |
| LOADFBS | 3299 | 38.83 | 75.62 (11.61) | 1507 | 35.70 | 85.50 (8.45) | 309 | 52.10 | 77.99 (10.01) |
| MESA | 2526 | 47.74 | 72.69 (9.92) | 382 | 53.14 | 73.10 (9.77) | |||
| ROSMAP | 871 | 32.95 | 86.28 (4.65) | 486 | 30.25 | 87.31 (3.91) | |||
| UKB, 60+ | 333208 | 45.99 | 69.32 (5.10) | 913 | 51.04 | 72.67 (4.63) | 24723 | 61.22 | 68.78 (6.85) |
AD denotes Alzheimer’s disease; DM denotes diabetes mellitus; SD denotes standard deviation.
ADGC is the National Institute on Aging (NIA) AD Genetics Consortium (ADGC) dataset, which includes data from three AD Centers (ADC1, ADC2, and ADC3); ADSP is the NIA AD Sequencing Project; ARIC is the Atherosclerosis Risk in Communities study; CHS is the Cardiovascular Health Study; FHS is the Framingham Heart Study parental and offspring cohorts; GenADA is the Multi-Site Collaborative Study for Genotype-Phenotype Associations in AD; LOADFBS is the NIA Late-Onset Alzheimer Disease Family-Based Study; MESA is the Multi-Ethnic Study of Atherosclerosis; ROSMAP is the Religious Orders Study and Memory and Aging Project; and UKB is the UK Biobank dataset.
Age was defined as the age of death or censoring for ADGC, ARIC, CHS, FHS, MESA, ROSMAP, and UKB. For LOADFBS and GenADA, age was provided at the launch of the study. ADSP defined age as the age at the onset of AD or the age at the last follow-up.
The UKB sample was limited to subjects who were 60 years and older.
Blank cells indicate not available information.
AD phenotype
We used AD affection status defined by investigators from ADGC, ADSP, FHS, GenADA, LOADFBS, and ROSMAP primarily based on the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association criteria [37, 38]. In CHS, AD affection status was determined using the International Classification of Disease codes, ninth revision (ICD-9, 331.0). In UKB, AD definition was done using ICD-10 (G30, F00) codes.
DM phenotype
Diabetes mellitus (DM) phenotype was defined based on having fasting blood glucose levels of 126 mg/dl or larger or using glucose-lowering medications in MESA (2003 American Diabetes Association fasting criteria algorithm) [39]. FHS and ARIC defined DM as having fasting blood glucose ≥126 mg/dl, non-fasting blood glucose ≥200 mg/dl, or taking glucose-lowering medications. In CHS and UKB, DM was defined based on ICD-9 (250) and ICD-10 (E11) codes. In LOADFBS and GenADA, DM was self/proxy reported. We did not separate type 1 and type 2 diabetes because the vast majority of DM cases were of type 2 and the largest sample of DM in UKB (Table 1) was defined based on ICD-10 E11 (type 2 diabetes mellitus) code.
Genotypes
To facilitate cross-platform comparison, we imputed SNPs for all studies except UKB using the HRC reference panel at the Michigan Imputation Server (MIS) [40]. The following genotyping arrays were used for imputation: Affymetrix 6.0 (~1M SNPs) chip in ARIC and MESA; Illumina HumanCNV370v1 (~370K SNPs) in the CHS; Affymetrix 500K (~500K SNPs) in FHS and GenADA; Illumina Human 610Quadv1B (~610K SNPs) in the LOADFBS; Illumina Human 660WQuadv1A (~600K SNPs) and Illumina HumanOmniExpress-12v1C (~700K SNPs) in the ADGC; the quality-controlled whole-genome sequencing (WGS) GATK pipeline in ADSP; WGS, Illumina HumanOmniExpress, and Affymetrix GeneChip 6.0 (from Synapse, https://www.synapse.org/#!Synapse:syn17008936) in the ROSMAP cohort. SNPs, which were submitted to the MIS, were selected by using Rayner’s quality control tools with the HRC reference panel (https://www.well.ox.ac.uk/~wrayner/tools/).
For the UKB cohort, the imputation was performed by the UK Biobank team using HRC and UK10K panel (https://biobank.ndph.ox.ac.uk/ukb/label.cgi?id=100319).
The imputation process resulted in about 6742K overlapping SNPs in most cohorts, which were used in the analyses.
Statistical Analyses
We followed the discovery-replication strategy and performed univariate (i.e., phenotype-specific) and pleiotropic analyses as detailed below. The discovery analysis used studies, which included both AD and DM phenotypes, i.e., CHS, FHS, GenADA, LOADFBS, and UKB (Table 1 and Figure 1). For replication analysis, we used independent datasets assessing AD in each of three ADC samples (ADC1, ADC2, and ADC3), ADSP, ROSMAP, and DM from ARIC and MESA.
Figure 1. A chart representing the flow of the analyses.
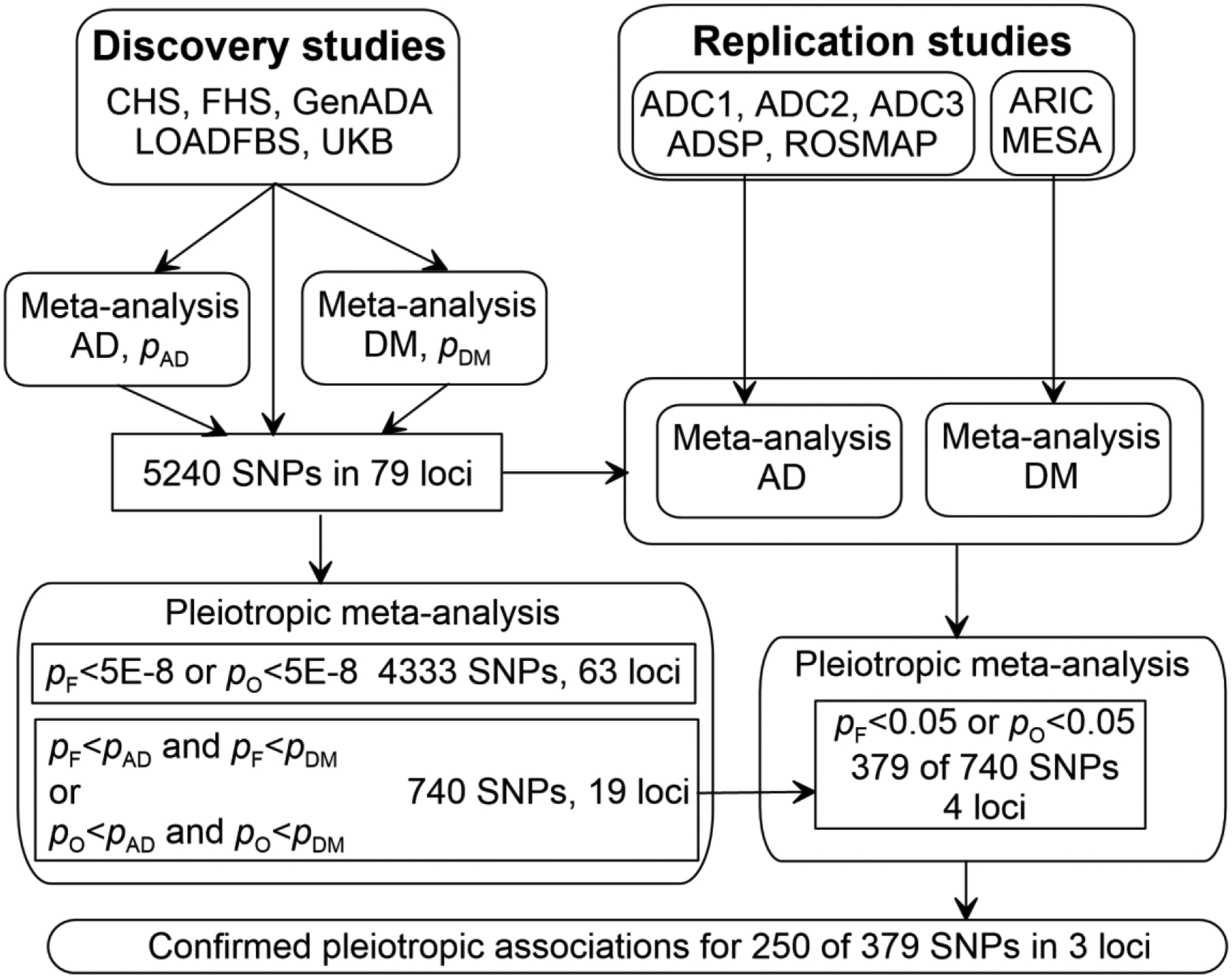
Symbols pAD, pDM, pF, and pO denote values from the univariate meta-analyses of Alzheimer’s disease (AD) and diabetes mellitus (DM) and pleiotropic meta-analysis using Fisher’s method and the omnibus test, respectively. Study abbreviations are given in Table 1.
Our univariate analyses focused on AD and DM separately. We conducted a genome-wide scan at the discovery stage and examined associations for the selected SNPs (selection is detailed in the Results section) at the replication stage in each study (except FHS and LOADFBS) separately using Plink [41]. The logistic regression model was adjusted by basic covariates, i.e., sex, age, and first five principal components (PCs) (all studies); year of birth (ARIC, CHS); field center (ARIC, CHS, MESA); an indicator of ROS or MAP sample in ROSMAP. Because FHS and LOADFBS included participants from families, we used models from the GCTA package to adjust for familial correlation [42]. Other adjustments in FHS and LOADFBS included sex, age, first five PCs, and the cohort indicator in FHS. Mediation effects of the lead SNPs in the selected loci were evaluated using the same models with additional adjustment by the lead SNP in each locus separately.
The results of the univariate analyses were aggregated using fixed-effects meta-regression with inverse-variance weighting implemented in METAL software [43]. This test provided summary statistics (effect sizes, standard errors, and p-values) for AD and DM separately and for the discovery and replication samples separately.
Pleiotropic meta-analysis was implemented using Fisher’s method and an omnibus test to aggregate the summary statistics for AD and DM. Fisher’s method [44] combines p-values from the meta-analysis of AD and DM assuming that p-values are from independent tests. The omnibus test [45–47] adjusts the analyses for correlation between phenotypes and takes into account the direction of the effect. In the case of two phenotypes, it takes the following form:
Here is a chi-squared distribution with K=2 degrees of freedom equal to the number of phenotypes, from which the p-value is calculated. Parameter is a z-score of the associations of SNPs with an ith phenotype (i=1,2), is an estimated effect size and is its standard error. The is the 2×2 correlation matrix of the z-scores. We evaluated the correlation between AD and DM in each study, in which both phenotypes were available, and constructed one correlation coefficient as an average weighted by the study sample size, r=0.02. The same correlation coefficient was used at the discovery and replications stages. Fisher’s method and omnibus test adjust for tests with multiple phenotypes by increasing the degree of freedom.
In the case if the signs of the correlation between AD and DM () and the product of z-scores are the same, the p-value can be larger in the omnibus test than in Fisher’s method. This situation indicates mediation pleiotropy. The opposite signs indicate the antagonistic genetic heterogeneity when the p-value can be smaller in the omnibus test than in Fisher’s method [48–50].
Pleiotropic associations
Pleiotropic effects can be naturally defined using the inherent property of Fisher’s method and the omnibus test, which defines the probability of events. That is, assuming no correlation between phenotypes, a smaller p-value in Fisher’s method compared to those in the meta-analyses of AD and DM implies a larger probability of associations with both these phenotypes than the probabilities of the associations with each of these phenotypes separately. The same logic holds for the omnibus test, except that it corrects for correlation between phenotypes.
RESULTS
Univariate and pleiotropic analyses of AD and DM identified 250 SNPs in three loci
Univariate analysis at the discovery stage identified 5186 SNPs with high imputation quality (r2≥0.75), which attained genome-wide (GW) significance (p<5E-8) in at least one individual study or a univariate meta-analysis across five studies for AD or DM. In addition, we included 54 SNPs for which the significance of the associations was below the GW threshold after removing the estimates from studies with lower imputation quality. These 5240 SNPs were mapped to 79 loci on all chromosomes, except chromosome 21 (Figure 1, Supplementary Table 1).
Pleiotropic meta-analysis at the discovery stage identified that 4333 SNPs in 63 loci attained the GW significance either in Fisher’s method or the omnibus test. We further excluded SNPs by requiring that either Fisher’s or omnibus p-value was less than p-values in the meta-analysis of AD and DM (see “Pleiotropic associations” section in Methods) and SNPs, which were not available or excluded in the replication studies. This selection resulted in 740 SNPs with pleiotropic associations in 19 loci.
Replication analysis of independent datasets identified a subset of 379 of 740 SNPs in four loci with pleiotropic associations having the same effect directions in the meta-analyses of each disease separately (AD or DM) at the discovery and replication stages and attaining p<0.05 either in Fisher’s method or the omnibus test.
Univariate and pleiotropic meta-analyses of the results from the discovery and replication stages confirmed pleiotropic associations for 250 of 379 SNPs by showing smaller p-values in these analyses compared to those at the discovery stage. This set included seven SNPs,—the APOE ε4 encoding rs429358 SNP and six more SNPs in the APOE cluster,—for which Fisher’s, omnibus, and AD p-values were nearly zero, p<10−302. We considered them as pleiotropic because they attained either GW (four SNPs) or suggestive-effect (three SNPs with p~1E-7) significance for DM. These 250 SNPs with pleiotropic effects were on chromosomes 1p12 (rs5025718 mapped to NOTCH2), 6p21.32, and 6p21.33 (174 SNPs mapped to an HLA gene cluster), and 19q13.32 (75 SNPs mapped to an APOE gene cluster) (Tables 2 and 3, and Supplementary Table 1).
Table 2.
The results of the univariate and pleiotropic meta-analyses of the associations of the lead SNPs in three clusters with Alzheimer’s disease (AD) and diabetes mellitus (DM) from the discovery and replication samples.
| SNP ID | Position | Cytoband | Cluster | Alleles | Sex | Pleiotropy | Univariate AD | Univariate DM | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P Fisher | P omnibus | β AD | SEAD | P AD | β DM | SEDM | P DM | ||||||
| rs5025718 | 119935662 | 1p12 | NOTCH2 | C/T | M&W | 3.88E-10 | 6.37E-10 | 0.111 | 0.038 | 3.48E-03 | 0.076 | 0.013 | 4.30E-09 |
| W | 1.21E-04 | 1.02E-04 | 0.058 | 0.060 | 3.36E-01 | 0.071 | 0.017 | 2.88E-05 | |||||
| M | 1.93E-06 | 3.10E-06 | 0.150 | 0.052 | 4.06E-03 | 0.084 | 0.020 | 2.80E-05 | |||||
| rs9275476 | 32704847 | 6p21.32 | HLA | T/C | M&W | 3.38E-25 | 1.62E-25 | −0.241 | 0.042 | 1.16E-08 | 0.125 | 0.014 | 4.75E-19 |
| W | 4.43E-13 | 3.22E-13 | −0.216 | 0.064 | 7.42E-04 | 0.122 | 0.018 | 1.81E-11 | |||||
| M | 1.13E-12 | 9.82E-13 | −0.261 | 0.059 | 1.03E-05 | 0.129 | 0.022 | 3.44E-09 | |||||
| rs9275599 | 32714652 | 6p21.32 | HLA | C/T | M&W | 6.73E-24 | 3.40E-24 | −0.205 | 0.042 | 1.20E-06 | 0.130 | 0.014 | 9.63E-20 |
| W | 4.28E-14 | 2.66E-14 | −0.205 | 0.065 | 1.46E-03 | 0.133 | 0.019 | 8.31E-13 | |||||
| M | 4.13E-10 | 3.31E-10 | −0.192 | 0.059 | 1.08E-03 | 0.126 | 0.022 | 1.48E-08 | |||||
| rs429358 | 44908684 | 19q13.32 | APOE | T/C | M&W | <1E-302 | <1E-302 | 1.305 | 0.031 | <1E-302 | −0.080 | 0.013 | 1.91E-10 |
| W | 1.42E-136 | 4.84E-138 | 1.179 | 0.048 | 5.57E-133 | −0.080 | 0.016 | 7.98E-07 | |||||
| M | 1.89E-210 | 2.58E-212 | 1.371 | 0.044 | 6.58E-209 | −0.079 | 0.020 | 5.86E-05 | |||||
Column Position is a chromosome position in base pairs given in Genome Reference Consortium Human Build 38 (GRCh38).
Column Alleles shows major/minor alleles. A minor allele was used as an effect allele.
Column Sex indicates samples of men (M) and women (W) combined (M&W) and separately.
Column Pleiotropy shows p-values from the pleiotropic meta-analysis using Fisher’s method (PFisher) and the omnibus test (Pomnibus).
Columns Univariate AD and Univariate DM show effect sizes (β), standard errors (SE), and p-values (P) from the univariate meta-analysis of AD and DM, respectively.
Table 3.
Summary information on 249 SNPs showing pleiotropic associations with Alzheimer’s disease (AD) and diabetes mellitus (DM) in the APOE and HLA gene clusters.
| Disease | Chr | Cluster | Lead-SNP-unadjusted models | Lead-SNP-adjusted models | ||
|---|---|---|---|---|---|---|
| NSNPs | Significance | NSNPs | Significance | |||
| AD | 6 | HLA | 174 | 1.16E-8≤p≤3.29E-2 | 91 | 3.68E-3≤p≤4.69E-2 |
| DM | 6 | HLA | 174 | 9.63E-20≤p≤3.79E-7 | 105 | 7.47E-12≤p≤3.90E-2 |
| AD | 19 | APOE | 75 | p≤1.70E-14 | 36 | 1.81E-11≤p≤4.74E-2 |
| DM | 19 | APOE | 75 | 1.91E-10≤p≤4.66E-2 | 4 | 1.76E-2≤p≤4.27E-2 |
Chr denotes chromosome
Columns Lead-SNP-unadjusted models and Lead-SNP-adjusted models show the number of AD- or DM-associated SNPs (NSNPs) and the range of p-values in the univariate meta-analyses of the discovery and replication samples in HLA and APOE gene clusters. Detailed information on the univariate and pleiotropic associations is given in Supplementary Table 1 for the lead-SNP-unadjusted models and Supplementary Tables 2 and 3 for the lead-SNP-adjusted models in the APOE and HLA gene clusters, respectively.
Pleiotropic association in the NOTCH2 1p12 region
The minor allele of the rs5025718 NOTCH2 SNP was adversely associated with DM at the GW level (Table 2) showing consistent effect directions in all studies, except LOADFBS (Supplementary Table 1). The same allele was also adversely associated with AD in all studies except GenADA and ADC3.
Antagonistic pleiotropic associations in the APOE and HLA gene clusters
All 75 pleiotropic SNPs in the APOE locus showed antagonistic associations with AD and DM, i.e., if a minor allele conferred a risk of AD then the same allele was also associated with decreased risk of DM (Table 2 and Supplementary Table 1). As expected, the APOE ε4 allele showed the strongest adverse association with AD, β=1.305, p<1E-302. This allele also showed the most significant favorable association with DM, β=−0.080, p=1.91E-10. Likewise, all 174 SNPs mapped to an HLA gene cluster showed antagonistic associations with AD and DM (Table 2 and Supplementary Table 1). Although the most significant associations with AD (rs9275476) and DM (rs9275599) were observed for different SNPs in this cluster, these SNPs were in nearly perfect linkage disequilibrium (LD) with r2=87%.
The role of sex in the associations of the lead SNPs in the NOTCH2, APOE, and HLA gene clusters with AD and DM.
Consistent with previous studies [51, 52], our analysis fitting the models for each sex separately showed a somewhat smaller (but significantly, p=3.32E-3, chi-square test [53]) risk of AD for rs429358 in men than women (Table 2). No other significant differences in the risks of AD or DM between men and women for the same genetic variants were identified.
Mediation of pleiotropic associations by the lead SNPs in the APOE and HLA gene clusters
Next, we examined whether pleiotropic associations of the identified SNPs with AD and DM in the APOE and HLA gene clusters could be mediated by the lead SNPs. As lead SNPs, we selected ε4-encoding rs429358 in the APOE cluster and rs9275476 in the HLA cluster.
All 75 pleiotropic SNPs in the APOE cluster were associated with AD at the GW level (Supplementary Table 1). After adjustment of the model by rs429358, the significance of all associations decreased, i.e., p-values increased (Supplementary Table 2). Still, 36 SNPs were associated with AD at p<0.05, including three AD-associated SNPs at the GW level (Table 3 and Supplementary Table 2). For 19 of these 36 SNPs, the effect directions in the rs429358 adjusted model were the same as in the unadjusted model. For the remaining 17 SNPs, including the three AD-associated SNPs at the GW level, the effects changed directions from positive (adverse) association with AD in the unadjusted model to negative (beneficial) association with AD in the rs429358 adjusted model.
These 75 pleiotropic SNPs were associated with DM at 1.91E-10≤p≤0.0466 in the unadjusted model (Table 3 and Supplementary Table 1). Only four of them were associated with DM at p<0.05 after adjustment by rs429358. The effect directions for them were the same in the adjusted and unadjusted models. Minor alleles of two SNPs—rs440277 and rs73052307—were associated with both AD and DM oppositely at p<0.05 in the rs429358 adjusted models.
Of 174 pleiotropic SNPs associated with AD in the HLA cluster, 91 SNPs remained significant at p<0.05 after adjustment by rs9275476 (Table 3 and Supplementary Table 3). For DM, 105 SNPs were significant at p<0.05 after adjustment by rs9275476, including 26 DM-associated SNPs at the GW level. None of the associations with AD or DM changed the directions in the rs9275476 adjusted models. All 91 AD-associated SNPs were also oppositely associated with DM.
Mediation of the associations with AD and DM by the ε4 and ε2 alleles in the APOE cluster
Change of the effect direction for the same allele in the model adjusted for the ε4-encoding rs429358 SNP compared to the unadjusted model suggests a potential role of the ε2 allele (encoded by a minor allele of rs7412). We selected 36 SNPs associated with AD at p<0.05 (Table 3 and Supplementary Table 2) and identified 11 clumps (plink 1.9) with LD between SNPs from different clumps less than r2=0.7 (Supplementary Table 4). Then, we evaluated associations of 11 SNPs representing these clumps with AD using a model adjusted for rs429358 and rs7412. Favorable GW significant associations with AD—represented by APOC1 rs438811 SNP (β=−0.322, p=1.81E-11)—were explained by the protective association of the ε2 allele with AD, i.e., highly significant strong protective effect for rs438811 became weak and non-significant (Table 4). For the other six SNPs favorably associated with AD in the rs429358 adjusted models, adjustment by rs7412 showed no significant associations (p>0.05) for three SNPs (rs111371860, rs11668327, rs10119) and significant protective associations for the remaining three SNPs (rs440277, rs118170342, rs1081105). For two (rs10414043 and rs79701229) of the four SNPs adversely associated with AD in the rs429358 adjusted models, adjustment by rs7412 had little effect and resulted in non-significant associations for the remaining two SNPs, rs12972156 and rs2927481 (Table 4).
Table 4.
The results of the meta-analysis of the associations of SNPs representing 11 clumps in the APOE gene cluster on chromosome 19 with Alzheimer’s disease after adjustment for the ε4- and ε2-encoding SNPs.
| SNP ID | Position | Alleles | Gene | Gene distance | β | SE | P-value | Direction | I2 | P-het |
|---|---|---|---|---|---|---|---|---|---|---|
| rs2927481 | 44833992 | C/A | NECTIN2 | 12305 | 0.039 | 0.029 | 1.72E-01 | ++-?+++-+- | 0.3 | 4.31E-01 |
| rs111371860 | 44842530 | A/T | NECTIN2 | 3767 | −0.109 | 0.066 | 9.75E-02 | +---+-?-+- | 65.9 | 2.81E-03 |
| rs440277 | 44857967 | G/A | NECTIN2 | 0 | −0.058 | 0.029 | 4.03E-02 | --+----+-- | 0 | 5.72E-01 |
| rs79701229 | 44881674 | G/A | NECTIN2 | 0 | 0.209 | 0.091 | 2.15E-02 | +++---+-++ | 0 | 5.45E-01 |
| rs12972156 | 44884202 | C/G | NECTIN2 | 0 | 0.081 | 0.046 | 7.64E-02 | +++-++?-++ | 49.8 | 4.33E-02 |
| rs11668327 | 44895376 | G/C | TOMM40 | 0 | −0.036 | 0.041 | 3.86E-01 | --+--+?++- | 3.3 | 4.07E-01 |
| rs118170342 | 44898611 | T/C | TOMM40 | 0 | −0.123 | 0.062 | 4.76E-02 | ------?++- | 0 | 6.38E-01 |
| rs10119 | 44903416 | G/A | TOMM40 | 0 | −0.041 | 0.041 | 3.18E-01 | +-+--+?-+- | 46.6 | 5.97E-02 |
| rs1081105 | 44909698 | A/C | APOE | 305 | −0.174 | 0.067 | 9.90E-03 | ------?+-- | 41.1 | 9.34E-02 |
| rs10414043 | 44912456 | G/A | APOC1 | 1869 | 0.136 | 0.056 | 1.55E-02 | +++-++?-++ | 39.9 | 1.01E-01 |
| rs438811 | 44913484 | C/T | APOC1 | 841 | −0.057 | 0.070 | 4.21E-01 | +?+---?-++ | 81.5 | 3.24E-06 |
Column Position is a chromosome position in base pairs given in Genome Reference Consortium Human Build 38 (GRCh38).
Column Alleles shows major/minor alleles. A minor allele was used as an effect allele.
SE denotes a standard error.
Column Direction shows signs of the effects in individual cohorts, ordered as ADC1, ADC2, ADC3, ADSP, CHS, FHS, GENADA, LOADFS, ROSMAP, and UKB (see Table 1 footnotes for abbreviations). Positive (+) and negative (−) signs indicate positive and negative values of beta. Question mark (?) refers to not available estimates.
I2 is a heterogeneity coefficient across cohorts; P-het is the heterogeneity p-value.
Of four SNPs associated with DM in the rs429358 adjusted models (Supplementary Table 2, rs57537848, rs11666329, rs440277, rs73052307), we selected three SNPs, as rs57537848 and rs11666329 were in perfect LD (r2=100%). Adjustment of the rs429358-adjusted models for DM by rs7412 had a trivial effect on the estimates for these three SNPs (Table 5 and Supplementary Table 2). SNPs associated with AD and DM at p<0.05 did not overlap (Tables 4 and 5).
Table 5.
The results of the meta-analysis of the associations of SNPs with diabetes mellitus in the APOE gene cluster on chromosome 19 adjusted for the ε4- and ε2-encoding SNPs.
| SNP ID | Position | Alleles | β | SE | P-value | Direction | I2 | P-het |
|---|---|---|---|---|---|---|---|---|
| rs11666329 | 44851039 | G/A | −0.022 | 0.009 | 1.55E-02 | --+---- | 26.6 | 2.26E-01 |
| rs440277 | 44857967 | G/A | 0.019 | 0.010 | 4.29E-02 | -++++++ | 0 | 6.55E-01 |
| rs73052307 | 44881148 | T/C | 0.030 | 0.013 | 2.66E-02 | --++++- | 12.8 | 3.32E-01 |
Column Position is a chromosome position in base pairs given in Genome Reference Consortium Human Build 38 (GRCh38). All SNPs are mapped to the NECTIN2 gene.
Column Alleles shows major/minor alleles. A minor allele was used as an effect allele.
SE denotes a standard error.
Column Direction shows signs of the effects in individual cohorts, ordered as ARIC, CHS, FHS, GENADA, LOADFS, MESA, and UKB (see Table 1 footnotes for abbreviations). Positive (+) and negative (−) signs indicate positive and negative values of beta. Question mark (?) refers to not available estimates.
I2 is a heterogeneity coefficient across cohorts; P-het is the heterogeneity p-value.
DISCUSSION
Our univariate and pleiotropic analyses following the discovery-replication strategy identified three clusters on chromosomes 1p12 (rs5025718 SNP mapped to NOTCH2 gene), 6p21.32–33 (174 SNPs mapped to the HLA gene cluster), and 19q13.32 (75 SNPs mapped to the APOE gene cluster). The results for the HLA and APOE gene clusters support previous findings, including antagonistic relationships when the same allele conferred a risk of one trait but was beneficially associated with the other trait [17, 22]. Our sex-stratified analysis showed consistent associations of the lead SNPs in these clusters with AD and DM in men and women with the only significant difference indicating a smaller AD risk for rs429358 in men than women [51, 52].
The NOTCH2 gene appears to be a plausible candidate for pleiotropic predisposition to AD and DM, which was not reported in recent pleiotropic analyses powered by summary statistics from large-scale GWAS of AD and diabetes [17, 54]. Unlike the HLA and APOE gene clusters, NOTCH2 was characterized by concordant directions of the associations with AD and DM. The Notch signaling pathway was extensively studied for its links with neurodegeneration, AD, and cardiovascular conditions including diabetes [55–59]. In addition, rs10923931 mapped to the NOTCH2 gene was reported as a diabetes-associated variant [60]. However, unlike potentially pleiotropic rs5025718 NOTCH2 SNP (Table 2), rs10923931 was associated with DM in our study (β=0.084, p=1.44E-9), but not with AD (β=0.060, p=0.124).
Our analysis showed that the majority of SNPs in the HLA cluster were still associated with AD (91 of 174 SNPs) and DM (105 of 174 SNPs) after adjustment for the potential mediation effect of the lead SNP. We also observed that all 91 AD-associated SNPs were associated with DM in the same antagonistic manner as the lead SNP (Table 2 and Supplementary Table 3). The presence of many disease-associated SNPs in the lead-SNP adjusted analysis and their antagonistic associations with AD and DM is in line with the complex structure of the HLA cluster, which includes 224 genes. The critical role of this cluster in the immune system and the dense coverage of the short chromosome region by many genes suggest a complex interplay of different genes in the immune defense that is likely the result of evolutionary adaptation to various conditions in the past [61]. The evolutionary-driven complexity of this cluster makes it difficult to identify a specific pathogenic mechanism of age-related diseases such as AD and DM [62]. This complexity also suggests that disease risks can be attributed not only to individual variants but also to their combinations [61].
In the APOE gene cluster, the major contribution to the AD and DM risks was from the ε4 allele, as evidenced by the fact that adjustment of the model by the ε4-encoding rs429358 SNP resulted in weaker associations for the other SNPs, many of which became non-significant. However, our analysis also showed GW’s significant favorable associations with AD (Table 3) even though the same variants were adversely associated with AD in the ε4-unadjusted model at the GW level. These GW significant favorable associations were explained by the contribution of the ε2 allele as they became non-significant after adjustment by rs7412 (Table 4, rs438811). Nevertheless, our analysis conditional on rs429358 and rs7412 SNPs identified the ε4- and ε2- independent adverse and beneficial associations with AD and DM. Unlike the HLA gene cluster, SNPs associated with AD and DM in the APOE gene cluster did not overlap (Tables 4 and 5). Antagonistic associations of the ε4 allele with AD and DM in the APOE cluster, complemented by the non-overlapping AD- and DM-associated SNPs, support different genetic mechanisms for AD and DM in this cluster.
Prior epidemiological studies suggested links between DM and AD (see Introduction). Mounting evidence indicates, however, that DM may be tighter linked to non-AD neuropathology, such as cerebrovascular pathology and vascular dementia, rather than AD pathology [23–25]. These insights from neuropathological studies are complemented by the results of correlation analyses, which suggest a weak genetic and phenotypic correlation between AD and DM, e.g., r=0.09 [63], and phenotypic, e.g., r=0.02 (in this study and in [48]). These results are further complemented by the increasing body of studies, including the current work, reporting antagonistic associations of the same genetic variants with AD and DM [17, 22]. Mechanisms driving divergence between the AD and DM pathologies are currently unclear; this divergence may impact, however, potential interventional strategies aimed to manage DM-related conditions to affect the incidence of AD [22].
Deeper analyses of the genetic architectures of AD and DM may help in identifying the overlap and divergence of pathogenic mechanisms of these highly heterogeneous diseases. Possible explanations of the divergence and overlap could be related to more pronounced roles of combinations of genetic variants in the form of haplotypes or combinations of genotypes, than previously believed, in susceptibility to AD and DM. Indeed, prior studies showed the substantial impact of such combinations on the AD risk [64–68], including strong modulation of the association of the ε4 allele with AD by other variants in the APOE gene cluster [69]. The effects of combinations of genetic variants can be driven by evolutionary adaptation and survival selection during the life course. These genetic mechanisms can be affected by environmental factors contributing to AD and DM heterogeneity. Discovering such genetic mechanisms will contribute to identifying more homogeneous pathogenic mechanisms of AD and DM.
Concluding, our study leveraging individual-level data reports a potentially novel pleiotropic NOTCH2 gene, on chromosome 1p12, with a minor allele of rs5025718 concordantly conferring risks of both AD and DM. Our study also supports antagonistic associations of the same variants with the risks of AD and DM in the HLA and APOE gene clusters previously reported by studies utilizing summary statistics from large-scale meta-analyses. Access to the individual-level data allowed us to identify the effects of SNPs, which were independent of the lead SNPs in the HLA and APOE gene clusters. We report antagonistic associations of the same variants with the risks of both AD and DM in the HLA gene clusters, independently of the lead SNP. We also found non-overlapping SNPs in the APOE gene cluster, which effects were not explained by the ε2 and ε4 alleles. This study supports the view that pathogenic mechanisms of highly heterogeneous conditions such as AD and DM may have different origins. Dissecting overlaps and differences of these mechanisms may be facilitated by the analyses of the roles of combinations of genetic variants in the form of haplotypes or combinations of genotypes.
Supplementary Material
Acknowledgments:
This article was prepared using data obtained through dbGaP (accession numbers phs000372.v1 [ADGC], phs000572, v.8 [ADSP], phs000280, v.5 [ARIC], phs000007.v32 [FHS], phs000287.v7 [CHS], phs000219, v.1 [GenADA], phs000168.v2 [LOADFBS], phs000209, v.13 [MESA]), Synapse (https://www.synapse.org/#!Synapse:syn3219045) (ROSMAP), and UK biobank repository (UKB). See the extended acknowledgment in the Supplemental Acknowledgement file.
Funding:
This research was supported by Grants R01 AG047310, R01 AG061853, R01 AG065477, and R01 AG070488 from the NIA. The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript. The content is solely the authors’ responsibility and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: none
Supplementary material includes: Supporting Acknowledgement and Supplementary Tables 1–4 in Excel format.
Data Availability Statement:
The data were obtained through designated repositories with controlled access. Data users cannot share this data.
References
- [1].Report AsA (2022) 2022 Alzheimer’s disease facts and figures. Alzheimers Dement 18, 700–789. [DOI] [PubMed] [Google Scholar]
- [2].Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R (2001) Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol 154, 635–641. [DOI] [PubMed] [Google Scholar]
- [3].Irie F, Fitzpatrick AL, Lopez OL, Kuller LH, Peila R, Newman AB, Launer LJ (2008) Enhanced risk for Alzheimer disease in persons with type 2 diabetes and APOE epsilon4: the Cardiovascular Health Study Cognition Study. Arch Neurol 65, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis 7, 63–80. [DOI] [PubMed] [Google Scholar]
- [5].de la Monte SM, Wands JR (2008) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tumminia A, Vinciguerra F, Parisi M, Frittitta L (2018) Type 2 Diabetes Mellitus and Alzheimer’s Disease: Role of Insulin Signalling and Therapeutic Implications. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100, 4162–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang Y, Huang NQ, Yan F, Jin H, Zhou SY, Shi JS, Jin F (2018) Diabetes mellitus and Alzheimer’s disease: GSK-3beta as a potential link. Behav Brain Res 339, 57–65. [DOI] [PubMed] [Google Scholar]
- [9].Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Suzuki SO, Kanba S, Kiyohara Y, Iwaki T (2010) Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology 75, 764–770. [DOI] [PubMed] [Google Scholar]
- [10].Cholerton B, Baker LD, Craft S (2013) Insulin, cognition, and dementia. Eur J Pharmacol 719, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ebrahimpour S, Zakeri M, Esmaeili A (2020) Crosstalk between obesity, diabetes, and alzheimer’s disease: Introducing quercetin as an effective triple herbal medicine. Ageing Res Rev 62, 101095. [DOI] [PubMed] [Google Scholar]
- [12].Xu W, Qiu C, Gatz M, Pedersen NL, Johansson B, Fratiglioni L (2009) Mid- and late-life diabetes in relation to the risk of dementia: a population-based twin study. Diabetes 58, 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Jash K, Gondaliya P, Kirave P, Kulkarni B, Sunkaria A, Kalia K (2020) Cognitive dysfunction: A growing link between diabetes and Alzheimer’s disease. Drug Dev Res 81, 144–164. [DOI] [PubMed] [Google Scholar]
- [14].Zhang X, Tong T, Chang A, Ang TFA, Tao Q, Auerbach S, Devine S, Qiu WQ, Mez J, Massaro J, Lunetta KL, Au R, Farrer LA (2023) Midlife lipid and glucose levels are associated with Alzheimer’s disease. Alzheimers Dement 19, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Luth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J, Arendt T, Munch G (2005) Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb Cortex 15, 211–220. [DOI] [PubMed] [Google Scholar]
- [16].Chen J, Mooldijk SS, Licher S, Waqas K, Ikram MK, Uitterlinden AG, Zillikens MC, Ikram MA (2021) Assessment of Advanced Glycation End Products and Receptors and the Risk of Dementia. JAMA Netw Open 4, e2033012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hao K, Di Narzo AF, Ho L, Luo W, Li S, Chen R, Li T, Dubner L, Pasinetti GM (2015) Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol Aspects Med 43–44, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang XF, Lin X, Li DY, Zhou R, Greenbaum J, Chen YC, Zeng CP, Peng LP, Wu KH, Ao ZX, Lu JM, Guo YF, Shen J, Deng HW (2017) Linking Alzheimer’s disease and type 2 diabetes: Novel shared susceptibility genes detected by cFDR approach. J Neurol Sci 380, 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Meng L, Wang Z, Ji HF, Shen L (2022) Causal association evaluation of diabetes with Alzheimer’s disease and genetic analysis of antidiabetic drugs against Alzheimer’s disease. Cell Biosci 12, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ghiam S, Eslahchi C, Shahpasand K, Habibi-Rezaei M, Gharaghani S (2022) Exploring the role of non-coding RNAs as potential candidate biomarkers in the cross-talk between diabetes mellitus and Alzheimer’s disease. Front Aging Neurosci 14, 955461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gao L, Cui Z, Shen L, Ji HF (2016) Shared Genetic Etiology between Type 2 Diabetes and Alzheimer’s Disease Identified by Bioinformatics Analysis. J Alzheimers Dis 50, 13–17. [DOI] [PubMed] [Google Scholar]
- [22].Hardy J, de Strooper B, Escott-Price V (2022) Diabetes and Alzheimer’s disease: shared genetic susceptibility? Lancet Neurol 21, 962–964. [DOI] [PubMed] [Google Scholar]
- [23].Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, Bennett DA (2006) Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67, 1960–1965. [DOI] [PubMed] [Google Scholar]
- [24].Abner EL, Nelson PT, Kryscio RJ, Schmitt FA, Fardo DW, Woltjer RL, Cairns NJ, Yu L, Dodge HH, Xiong C, Masaki K, Tyas SL, Bennett DA, Schneider JA, Arvanitakis Z (2016) Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12, 882–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dos Santos Matioli MNP, Suemoto CK, Rodriguez RD, Farias DS, da Silva MM, Leite REP, Ferretti-Rebustini REL, Farfel JM, Pasqualucci CA, Jacob Filho W, Arvanitakis Z, Naslavsky MS, Zatz M, Grinberg LT, Nitrini R (2017) Diabetes is Not Associated with Alzheimer’s Disease Neuropathology. J Alzheimers Dis 60, 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beecham GW, Bis JC, Martin ER, Choi SH, DeStefano AL, van Duijn CM, Fornage M, Gabriel SB, Koboldt DC, Larson DE, Naj AC, Psaty BM, Salerno W, Bush WS, Foroud TM, Wijsman E, Farrer LA, Goate A, Haines JL, Pericak-Vance MA, Boerwinkle E, Mayeux R, Seshadri S, Schellenberg G (2017) The Alzheimer’s Disease Sequencing Project: Study design and sample selection. Neurol Genet 3, e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sharrett AR (1992) The Atherosclerosis Risk in Communities (ARIC) Study. Introduction and objectives of the hemostasis component. Ann Epidemiol 2, 467–469. [DOI] [PubMed] [Google Scholar]
- [29].Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, Kuller LH, Manolio TA, Mittelmark MB, Newman A, et al. (1991) The Cardiovascular Health Study: design and rationale. Ann Epidemiol 1, 263–276. [DOI] [PubMed] [Google Scholar]
- [30].Cupples LA, Heard-Costa N, Lee M, Atwood LD (2009) Genetics Analysis Workshop 16 Problem 2: the Framingham Heart Study data. BMC Proc 3 Suppl 7, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L, Hosford D, Barnes MR, Briley JD, Borrie M, Coletta N, Delisle R, Dhalla D, Ehm MG, Feldman HH, Fornazzari L, Gauthier S, Goodgame N, Guzman D, Hammond S, Hollingworth P, Hsiung GY, Johnson J, Kelly DD, Keren R, Kertesz A, King KS, Lovestone S, Loy-English I, Matthews PM, Owen MJ, Plumpton M, Pryse-Phillips W, Prinjha RK, Richardson JC, Saunders A, Slater AJ, St George-Hyslop PH, Stinnett SW, Swartz JE, Taylor RL, Wherrett J, Williams J, Yarnall DP, Gibson RA, Irizarry MC, Middleton LT, Roses AD (2008) Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 65, 45–53. [DOI] [PubMed] [Google Scholar]
- [32].Filippini N, Rao A, Wetten S, Gibson RA, Borrie M, Guzman D, Kertesz A, Loy-English I, Williams J, Nichols T, Whitcher B, Matthews PM (2009) Anatomically-distinct genetic associations of APOE epsilon4 allele load with regional cortical atrophy in Alzheimer’s disease. Neuroimage 44, 724–728. [DOI] [PubMed] [Google Scholar]
- [33].Lee JH, Cheng R, Graff-Radford N, Foroud T, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study G (2008) Analyses of the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study: implication of additional loci. Arch Neurol 65, 1518–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr., Kronmal R, Liu K, Nelson JC, O’Leary D, Saad MF, Shea S, Szklo M, Tracy RP (2002) Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol 156, 871–881. [DOI] [PubMed] [Google Scholar]
- [35].Negash S, Bennett DA, Wilson RS, Schneider JA, Arnold SE (2011) Cognition and neuropathology in aging: multidimensional perspectives from the Rush Religious Orders Study and Rush Memory And Aging Project. Curr Alzheimer Res 8, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, Liu B, Matthews P, Ong G, Pell J, Silman A, Young A, Sprosen T, Peakman T, Collins R (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12, e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944. [DOI] [PubMed] [Google Scholar]
- [38].McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011) The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Genuth S, Alberti KG, Bennett P, Buse J, Defronzo R, Kahn R, Kitzmiller J, Knowler WC, Lebovitz H, Lernmark A, Nathan D, Palmer J, Rizza R, Saudek C, Shaw J, Steffes M, Stern M, Tuomilehto J, Zimmet P, Expert Committee on the D, Classification of Diabetes M (2003) Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 26, 3160–3167. [DOI] [PubMed] [Google Scholar]
- [40].Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C (2016) Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang J, Lee SH, Goddard ME, Visscher PM (2011) GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Willer CJ, Li Y, Abecasis GR (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fisher RAS (1970) Statistical methods for research workers, Oliver and Boyd, Edinburgh. [Google Scholar]
- [45].Xu X, Tian L, Wei LJ (2003) Combining dependent tests for linkage or association across multiple phenotypic traits. Biostatistics 4, 223–229. [DOI] [PubMed] [Google Scholar]
- [46].Bolormaa S, Pryce JE, Reverter A, Zhang Y, Barendse W, Kemper K, Tier B, Savin K, Hayes BJ, Goddard ME (2014) A multi-trait, meta-analysis for detecting pleiotropic polymorphisms for stature, fatness and reproduction in beef cattle. PLoS Genet 10, e1004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, Smith JA, Yanek LR, Sun YV, Edwards TL, Chen W, Nalls M, Fox E, Sale M, Bottinger E, Rotimi C, Consortium CB, Liu Y, McKnight B, Liu K, Arnett DK, Chakravati A, Cooper RS, Redline S (2015) Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet 96, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kulminski AM, Loika Y, Huang J, Arbeev KG, Bagley O, Ukraintseva S, Yashin AI, Culminskaya I (2019) Pleiotropic Meta-Analysis of Age-Related Phenotypes Addressing Evolutionary Uncertainty in Their Molecular Mechanisms. Front Genet 10, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kulminski AM, Loika Y, Nazarian A, Culminskaya I (2020) Quantitative and Qualitative Role of Antagonistic Heterogeneity in Genetics of Blood Lipids. J Gerontol A Biol Sci Med Sci 75, 1811–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kulminski AM, Huang J, Loika Y, Arbeev KG, Bagley O, Yashkin A, Duan M, Culminskaya I (2018) Strong impact of natural-selection-free heterogeneity in genetics of age-related phenotypes. Aging (Albany NY) 10, 492–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Snyder HM, Asthana S, Bain L, Brinton R, Craft S, Dubal DB, Espeland MA, Gatz M, Mielke MM, Raber J, Rapp PR, Yaffe K, Carrillo MC (2016) Sex biology contributions to vulnerability to Alzheimer’s disease: A think tank convened by the Women’s Alzheimer’s Research Initiative. Alzheimers Dement 12, 1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C, Pasquier F, Dubois B, Tognoni G, Fievet N, Brouwers N, Bettens K, Arosio B, Coto E, Del Zompo M, Mateo I, Epelbaum J, Frank-Garcia A, Helisalmi S, Porcellini E, Pilotto A, Forti P, Ferri R, Scarpini E, Siciliano G, Solfrizzi V, Sorbi S, Spalletta G, Valdivieso F, Vepsalainen S, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Hanon O, Piccardi P, Annoni G, Seripa D, Galimberti D, Licastro F, Soininen H, Dartigues JF, Kamboh MI, Van Broeckhoven C, Lambert JC, Amouyel P, Campion D (2011) APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry 16, 903–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Allison PD (1999) Comparing logit and probit coefficients across groups. Sociological Methods & Research 28, 186–208. [Google Scholar]
- [54].Bone WP, Siewert KM, Jha A, Klarin D, Damrauer SM, Program VAMV, Chang KM, Tsao PS, Assimes TL, Ritchie MD, Voight BF (2021) Multi-trait association studies discover pleiotropic loci between Alzheimer’s disease and cardiometabolic traits. Alzheimers Res Ther 13, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kapoor A, Nation DA (2021) Role of Notch signaling in neurovascular aging and Alzheimer’s disease. Semin Cell Dev Biol 116, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Polychronidou E, Vlachakis D, Vlamos P, Baumann M, Kossida S (2015) Notch signaling and ageing. Adv Exp Med Biol 822, 25–36. [DOI] [PubMed] [Google Scholar]
- [57].Alberi L, Hoey SE, Brai E, Scotti AL, Marathe S (2013) Notch signaling in the brain: in good and bad times. Ageing Res Rev 12, 801–814. [DOI] [PubMed] [Google Scholar]
- [58].Mathieu P, Adami PV, Morelli L (2013) Notch signaling in the pathologic adult brain. Biomol Concepts 4, 465–476. [DOI] [PubMed] [Google Scholar]
- [59].Mirtschink P, Chavakis T (2018) The Missed Notch to Bring Down Diabetes. Trends Endocrinol Metab 29, 448–450. [DOI] [PubMed] [Google Scholar]
- [60].Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jorgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjogren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Wellcome Trust Case Control C, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40, 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Arnaiz-Villena A, Suarez-Trujillo F, Juarez I, Rodriguez-Sainz C, Palacio-Gruber J, Vaquero-Yuste C, Molina-Alejandre M, Fernandez-Cruz E, Martin-Villa JM (2022) Evolution and molecular interactions of major histocompatibility complex (MHC)-G, -E and -F genes. Cell Mol Life Sci 79, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Arnaiz-Villena A, Juarez I, Suarez-Trujillo F, Lopez-Nares A, Vaquero C, Palacio-Gruber J, Martin-Villa JM (2021) HLA-G: Function, polymorphisms and pathology. Int J Immunogenet 48, 172–192. [DOI] [PubMed] [Google Scholar]
- [63].Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier JG, Harold D, Fitzpatrick AL, Valladares O, Moutet ML, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Del Zompo M, Fox NC, Choi SH, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujic-Comic H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernandez I, Kamboh MI, Brundin R, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT Jr., Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu CK, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo A, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frolich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kolsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hull M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin LW, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jockel KH, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann HE, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nothen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossu P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues JF, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz J, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, M COD, DeStefano AL, Jones L, Haines JL, Deleuze JF, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang LS, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert JC, Pericak-Vance MA, Alzheimer Disease Genetics C, European Alzheimer’s Disease I, Cohorts for H, Aging Research in Genomic Epidemiology C, Genetic, Environmental Risk in Ad/Defining Genetic P, Environmental Risk for Alzheimer’s Disease C (2019) Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zhou X, Chen Y, Mok KY, Kwok TCY, Mok VCT, Guo Q, Ip FC, Chen Y, Mullapudi N, Alzheimer’s Disease Neuroimaging I, Giusti-Rodriguez P, Sullivan PF, Hardy J, Fu AKY, Li Y, Ip NY (2019) Non-coding variability at the APOE locus contributes to the Alzheimer’s risk. Nat Commun 10, 3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lescai F, Chiamenti AM, Codemo A, Pirazzini C, D’Agostino G, Ruaro C, Ghidoni R, Benussi L, Galimberti D, Esposito F, Marchegiani F, Cardelli M, Olivieri F, Nacmias B, Sorbi S, Tagliavini F, Albani D, Martinelli Boneschi F, Binetti G, Santoro A, Forloni G, Scarpini E, Crepaldi G, Gabelli C, Franceschi C (2011) An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer’s disease. J Alzheimers Dis 24, 235–245. [DOI] [PubMed] [Google Scholar]
- [66].Linnertz C, Anderson L, Gottschalk W, Crenshaw D, Lutz MW, Allen J, Saith S, Mihovilovic M, Burke JR, Welsh-Bohmer KA, Roses AD, Chiba-Falek O (2014) The cis-regulatory effect of an Alzheimer’s disease-associated poly-T locus on expression of TOMM40 and apolipoprotein E genes. Alzheimers Dement 10, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nazarian A, Loika Y, He L, Culminskaya I, Kulminski AM (2022) Genome-wide analysis identified abundant genetic modulators of contributions of the apolipoprotein E alleles to Alzheimer’s disease risk. Alzheimers Dement 18, 2067–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kulminski AM, Philipp I, Loika Y, He L, Culminskaya I (2021) Protective association of the epsilon2/epsilon3 heterozygote with Alzheimer’s disease is strengthened by TOMM40-APOE variants in men. Alzheimers Dement 17, 1779–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kulminski AM, Philipp I, Shu L, Culminskaya I (2022) Definitive roles of TOMM40-APOE-APOC1 variants in the Alzheimer’s risk. Neurobiol Aging 110, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data were obtained through designated repositories with controlled access. Data users cannot share this data.