

Abstract
Anaplastic lymphoma kinase (ALK) rearrangements occur in about 5% of nonsmall cell lung cancer (NSCLC) patients. Despite being first recognized as EML4-ALK, fusions with several additional genes have been identified, all of which cause constitutive activation of the ALK kinase and subsequently lead to tumor development. ALK inhibitors first-line crizotinib, second-line ceritinib, and alectinib are effective against NSCLC patients with these rearrangements. Patients progressing on crizotinib had various mutations in the ALK kinase domain. ALK fusion proteins are activated by oligomerization through the fusion partner, which leads to the autophosphorylation of the kinase’s domain and consequent downstream activation. The proposed computational study focuses on understanding the activation mechanism of ALK and ATP binding of wild-type (WT) and I1171N/S/T mutations. We analyzed the conformational change of ALK I1171N/S/T mutations and ATP binding using molecular docking and molecular dynamics simulation approaches. According to principal component analysis and free energy landscape, it is clear that I1171N/S/T mutations in Apo and ATP showed different energy minima/unstable structures compared to WT-Apo. The results revealed that I1171N/S/T mutations and ATP binding significantly supported a change toward an active-state conformation, whereas WT-Apo remained inactive. We demonstrated that I1171N/S/T mutations are persistent in an active state and independent of ATP. The I1171S/T mutations showed greater intermolecular H-bonds with ATP than WT-ATP. The molecular mechanics Poisson–Boltzmann surface area analysis revealed that the I1171N/S/T mutation binding energy was similar to that of WT-ATP. This study shows that I1171N/S/T can form stable bonds with ATP and may contribute to a constitutively active kinase. Based on the Y1278–C1097 H-bond and E1167–K1150 salt bridge interaction, I1171N strongly promotes the constitutively active kinase independent of ATP. This structural mechanism study will aid in understanding the oncogenic activity of ALK and the basis for improving the ALK inhibitors.
Introduction
Anaplastic lymphoma kinase (ALK) rearrangements lead to constitutive oncogenic activation.1 ALK belongs to the insulin receptor kinase superfamily, which plays a significant role in brain development.2,3 Chromosomal rearrangements are the most prevalent of ALK alterations. ALK-tyrosine kinase alterations are constituted by ALK fusion genes that induce cell proliferation, resulting in anaplastic large-cell lymphoma (ALCL) and nonsmall-cell lung cancer (NSCLC).4 ALK mutations are found in about 3–7% of all lung cancers, and patients younger than average are likely to have these mutations.5 ALK mutations were more common in adenocarcinoma patients with acinar histology or who had never smoked.6,7 ALK mutations do not coincide with other NSCLC-related oncogenic mutations like EGFR or RAS.8
The ALK protein comprises a signal peptide, a long extracellular domain, a transmembrane segment, and a short intracellular kinase domain. The ALK kinase domain consists of two lobes: a large carboxy-terminal lobe (C-lobe), a small amino-terminal lobe (N-lobe), and a hinge region.3 These lobes incorporate highly conserved α-helices and β strands. The protein kinase domain ranges from 1116 to 1392 amino acid residues; these K/D/D motifs specifying Lys, Asp, and Asp amino acids illustrate the catalytic activity of ALK.9 The ATP binding site is located in the interlobe cleft, also known as the ALK active site; it allows the catalytic kinase reaction to proceed. The N-lobe is dynamically inflexible and unable to bind to ATP in the inactive state, but it migrates toward the C-lobe when it is active.10 The extracellular domain interacts with the ligand, inducing tyrosine kinase domain dimerization through transphosphorylation. The dimerization of the tyrosine kinase domain induces ALK gene expression. It promotes the Ras/Raf/MEK/ERK1/2 pathway, JAK-STAT (Janus activated kinase/signal transducer and activator of transcription) pathway, PI3K (phosphatidylinositol-3 kinase)/Akt pathway, and PLC (phospholipase-C)-γ pathway essential for cell proliferation and cell survival.3 The hydrophobic regulatory spine (R-spine) comprises residues, namely, I1171, C1182, H1247, F1271, and D1311, which are vital in determining the active and inactive conformation of ALK.9,11,12
Mutations in ALK that resulted in the missense substitution of a single amino acid in the tyrosine kinase domain may facilitate the constitutive and ligand-independent kinase activity.13,14 The I1171 amino acid residue position in the ALK kinase domain acquires I1171 T/N/S mutations.15,16 The prevalence rates of ALK mutations at position I1171 N/S/T are 5, 31, and 7.1%.17 These mutations result in resistance to ALK drugs. Currently, I1171N is known to confer resistance to crizotinib, a first-generation ALK drug in NSCLC patients.15 Recent reports suggest superior resistance of I1171T mutation against alectinib, a second-generation ALK drug in neuroblastoma patients.18 The major lobes of the ALK kinase domain are connected by the amino acid residue at position I1171 (found in the N-lobe) in the R-spine required to maintain active conformation.15 The ALK rearrangement with an echinoderm microtubule-associated protein like-4 (EML-4) or nucleophosmin contributes to the I1171 mutation.19 Several experimental studies focused on the drug response of the acquired ALK I1171 mutation to identify the ALK inhibitors and overcome the drug resistance.15,20−24 In addition, few computational studies have highlighted the significance of ALK mutation at I1171 and drug resistance using molecular docking and molecular dynamics approaches.25−28 Karabencheva et al. (2014) exposed significant conformational effects on crucial interactions necessary for the activation of ALK enzyme by atomistic molecular dynamics simulations (MDSs) on wild-type (WT) and nine clinically significant mutants, including the I1171N mutation.25 He et al. (2018) revealed the mechanisms of alectinib resistance induced by the I1171N, V1180L, and L1198F mutations via molecular dynamics.26 Salifu et al. (2023) elucidated that the compound mutations I1171N + F1174I and I1171N + L1198H distorted the activation loop and affected the phosphorylation.27 Another study by Liang et al. (2021) demonstrated that gilteritinib overcame lorlatinib-resistant ALK double mutation (I1171N/F1174I).28
The proposed computational study investigated the putative activation mechanism of different missense mutations at position I1171 in the ALK protein of NSCLC in Apo and ATP-bound states. We performed MDS for 200 ns using Gromacs (2021.2 version) to elucidate the structural transitions of the interaction between the ATP and ALK WT and I1171N/S/T mutations.29 We evaluated the free energy landscape (FEL) and dynamic cross-correlation matrix (DCCM) to determine the dynamic effects of Apo and ATP-bound I1171 mutations to understand the conformational changes of essential structural elements and atomistic insights involved in ALK activation. These insights into the atomic level activation mechanism of the ALK I1171 mutation can help us understand the structural characteristics and conformational changes of drug-resistant ALK mutations.
Methodology
Structure Preparation
We retrieved the ALK structure PDB ID 3LCT (1093–1410 residues) from the protein data bank (PDB).12 To provide the initial ALK WT structure with Mg, the ALK PDB ID-3LCT was superimposed with the insulin receptor kinase (IRK) PDB ID-3BU5 (structure with Mg), and Mg was placed in the optimum position of the ALK 3LCT structure.30 We utilized Discovery Studio to fill the missing residues and induce the I1171N/S/T mutations in the WT structure with Mg. Energy minimization was performed for ALK WT and I1171N/S/T structures coupled with Mg using a Swiss-PDB-viewer.31 Similarly, we downloaded the ATP structure from PubChem in the structure data file (SDF).32 Subsequently, we converted the ATP from SDF to PDB format with an Open Babel Web server.33 We initially performed 10 ns MDS with the CHARMM36 force field using GROMACS 2021.2 version software to optimize all the four Apo 3D structures of ALK WT and I1171N/S/T before docking.34,35
Molecular Docking Using AutoDock Vina
The optimized four Apo 3D structures of ALK WT and I1171N/S/T were then docked with ATP in triplicates using AutoDock Vina software.36 The ATP binding residues (His1124, Lys1150, Asp1270, Glu1197, Leu1198, and Met1199) of the ALK human protein were obtained from the UniProt entry Q9UM73.37 First, all the four proteins (WT and I1171N/S/T mutant) and ATP structures were prepared using default parameters in AutoDockTools 1.5.7 and saved in PDBQT formats.38 The grid box size for the ATP binding sites of each protein structure was obtained using AutoDockTools 1.5.7.38 Later, molecular docking was carried out for all the four proteins with ATP in triplicate and obtained nine different conformations for each docked structure using AutoDock Vina 4.2 software. Before proceeding to MDS, the best-docked conformations were selected based on the binding energies, lowest rmsds, number of H-bonds, and least distance between Mg and ATP.
Interaction Analysis
The Discovery Studio visualizer analyzed the ATP-docked ALK WT and I1171N/S/T 3D structures.39 Using this software, we visualized a better 3D surface of the complexes. We also elucidated the complex interactions and contact residues with ATP through 2D structural interactions.
MDS and Analysis
The MDS was initiated for the ATP-docked conformation of ALK WT and I1171N/S/T (ATP-bound) and Apo ALK WT and I1171N/S/T (ATP-unbound) structures to evaluate the binding mechanism of ATP and conformational changes upon mutations. CHARMM-GUI generated topology files for eight different systems, namely, WT-Apo, WT-ATP, I1171N-Apo, I1171N-ATP, I1171S-Apo, I1171S-ATP, I1171T-Apo, and I1171T-ATP, with the CHARMM36 force field.40 Using those topology files, the MDS was performed for 200 ns in GROMACS 2021.2 software with default parameters.35 Furthermore, MDS analyses such as the root-mean-square deviation (rmsd), root-mean-square fluctuation (RMSF), radius of gyration (Rg), solvent-accessible surface area (SASA), number of intermolecular H-bonds, intramolecular H-bonds, and molecular mechanics Poisson–Boltzmann surface area (MM/PBSA) analyses were evaluated throughout the run time. The DCCM and rmsd vs Rg correlation plots were also assessed to investigate the proteins’ correlated collective motions, compactness, and stability.
Principal Component Analysis, Essential Dynamics, and MM/PBSA Analysis
A widely used statistical strategy to reduce data dimensionality is principal component analysis (PCA). The minimization is accomplished by classifying the directions in which the data variance is greatest, also known as PCs. Typically, a small number of components through several variables represent each sample. Additionally, PCA may be utilized to find large-amplitude differential movements in MD trajectories. We used PCA and FEL analysis to examine the stability, conformational sampling, and atomic movements of the MD trajectories of Apo and ATP-bound ALK WT and I1171 mutations.41 To calculate the protein–ligand interactions, the MM/PBSA method has been widely used as a reliable and effective free energy calculation method in MD trajectories.42 Using the MM/PBSA calculation, we calculated the total binding energy of governing residues around the ATP active site pockets.
Results
Molecular Docking
Molecular docking enabled us to check the binding patterns of ATP with ALK WT and I1171N/S/T mutations; this aids in determining the binding energy and locating the interaction regions. The molecular docking performed in AutoDock Vina resulted in nine different conformations of ATP binding toward each protein. We merged ALK-Mg along with different conformations of ATP and saved them in PDB format. We further calculated the distance between Mg and ATP for each protein complex using the Discovery Studio visualizer. After the docking process, the obtained binding energy and distance between Mg and ATP for the nine different conformations with triplicates for each protein are shown in Supporting Information Table S1. We selected the best conformations based on the criteria and displayed their average binding energy and distance between Mg and ATP of the triplicates in Table 1. We took the single best structure from these triplicates and utilized them for further analysis.
Table 1. Binding Energy, Selected Conformation Number, and Distance between Mg and ATP of the ATP-Bound ALK WT and I1171N/S/T Mutations in Triplicatesa.
| ALK | conformation | distance (Å) Mg – ATP | binding energy (kcal/mol) | average binding energy (kcal/mol) |
|---|---|---|---|---|
| ALK WT-ATP1-T1 | 1 | 5.551 | –5.9 | –6.1 |
| ALK WT-ATP1-T2* | 1 | 5.536 | –6 | |
| ALK-WT ATP1-T3 | 6 | 5.881 | –6.4 | |
| ALK-I1171T-ATP1-T1 | 2 | 3.179 | –6.5 | –6.533333333 |
| ALK-I1171T-ATP1-T2* | 3 | 2.239 | –6.5 | |
| ALK-I1171T-ATP1-T3 | 2 | 5.065 | –6.6 | |
| ALK-I1171S-ATP1-T1* | 1 | 5.061 | –6.1 | –6.2 |
| ALK-I1171S-ATP1-T2 | 3 | 5.107 | –6.3 | |
| ALK-I1171S-ATP1-T3 | 2 | 5.065 | –6.2 | |
| ALK-I1171N-ATP1-T1 | 4 | 6.098 | –5.8 | –6.2 |
| ALK-I1171N-ATP1-T2* | 1 | 4.821 | –6.9 | |
| ALK-I1171N-ATP1-T3 | 2 | 5.013 | –5.9 |
T1, T2, and T3 represent triplicate 1, triplicate 2, and triplicate 3, respectively. “*” denoted that the structures were further taken for MDS studies.
Interaction Analysis
The interaction analysis of the selected structures was performed using the Discovery Studio visualizer, and Figure 1 illustrates the 3D cartoon structure (Figure 1A,D,G,J), 3D atomic interactions (Figure 1B,E,H,K), and their 2D interactions (Figure 1C,F,I,L) for ATP-bound ALK WT and I1171N/S/T mutations. ALK WT binds to ATP and establishes the binding affinity of −6 kcal/mol with two conventional H-bonds (Arg1253 and Met1199) and other contact residues (Arg1253, Arg1254, Gly1269, Leu1122, Gly1202, Glu1197, Leu1198, Ala1148, Leu1196, Val1130, Gly1123, Asp1203, Leu1256, and Asp1270; Figure 1B,C). ALK I1171N binds to ATP and establishes a binding affinity of −6.9 kcal/mol with five conventional H-bonds (Asp1270, Glu1197, Met1199, Lys1150, and Gly1269) and other contact residues (Asp1270, Glu1167, Arg1253, and Gly1202) (Figure 1E,F). Similarly, ALK I1171S binds to ATP and establishes the binding affinity of −6.1 kcal/mol with three conventional H-bonds (Leu1122, Met1199, and Asp1270) and other contact residues (Ala1148, Leu1198, Lys1150, Glu1197, Leu1196, Leu1180, Leu1256, Leu1122, Gly1202, Arg1253, Asp1203, Asn1254, Gly1269, Val1130, and Asp1270) (Figure 1H,I). ALK I1171T binds to ATP and establishes a binding affinity of −6.5 kcal/mol with seven conventional H-bonds (Lys1150, Asn1254, Arg1275, and Asp1270) and other contact residues (Leu1150, Glu1167, Arg1275, Asn1254, Asp1249, and Asp1270) (Figure 1K,L). The ATP binding residues for WT and I1171N/S/T mutations varied, but we observed one constant conventional H-bond formation with residue Asp1270 in ALK I1171N/S/T mutations.
Figure 1.

Molecular docking results of best-docked structures of ALK WT and I1171N/S/T with ATP from the triplicates were picturized using the Discovery Studio visualizer. We demonstrated the docked 3D cartoon structures of ALK WT-ATP (A), I1171N-ATP (D), I1171S-ATP (G), and I117T-ATP (J), where the ALK structures are represented in the purple cartoon, Zn represented in green CPK, and ATP represented in blue stick with orange color surface mesh wire. The 2D atomic structure illustrated the ATP interaction residues in the WT (B), I1171N (E), I117S (H), and I1171T (K) structures. The 2D structure showed the type of ATP interaction with WT (C), I1171N (D), I117S (E), and I1171T (F). The range of bright green color to fade green color interaction represents the conventional hydrogen bond, van der Waals, and carbon–hydrogen bond. The purple interaction represents π-sigma, whereas the pink interaction represents π-alkyl. The orange color interaction represents attractive charge, π-cation, and π-anion.
MDS and Analysis
MDS was performed for the Apo and ATP-docked structures of ALK WT and I1171N/S/T mutations to evaluate the stability, structural aspect, and dynamic behavior throughout the 200 ns run time. We also investigated the structural changes of ALK mutations caused by different amino acid substitutions at I1171 found in NSCLC. The eight-topology files were generated in the CHARMM36 force field utilizing the CHARMM-GUI input generator and performed in GROMACS 2021.2. The system was energy-minimized and equilibrated using NVT and NPT ensembles, followed by MDS for 200 ns. The protein’s structural conformation throughout the simulation is revealed by rmsd analysis, indicating the protein’s stability. The backbone rmsds of all of the proteins in the simulation converged after 40 ns. Henceforth, all the analyses were performed in the 40–200 ns of the stable structures (Figure 2A,C,E,G). The moving average of rmsds was evaluated and displayed in Figure 2A,C,E,G, which shows a similar stable conformation. We further determined the stability through statistical validation. We have calculated the probability distribution of backbone rmsds from 40 to 200 ns for all of the simulations, which shows no statistical difference in all systems, stating that the simulations were stable from 40 to 200 ns (Figure 2B,D,F,H). The average rmsd values of stable WT-Apo, WT-ATP, I1171N-Apo, I1171N-ATP, I1171S-Apo, I1171S-ATP, I1171T-Apo, and I1171T-ATP are 0.25 ± 0.03, 0.29 ± 0.04, 0.27 ± 0.04, 0.31 ± 0.04, 0.25 ± 0.03, 0.25 ± 0.04, 0.27 ± 0.02, and 0.23 ± 0.04 nm, respectively (Figure 2). The above observation premises that all the simulations reached equilibrium and that the proteins produced stable structures. The simulation time was sufficient and attained an equilibrium structure after 40 ns of MDS, suggesting that the simulation was appropriate for further comprehensive analysis.
Figure 2.

rmsd analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states over 200 ns MDS. The X-axis denotes the time in ns, and the Y-axis displays the rmsd values in nm. (A) The rmsd analysis of WT-Apo and WT-ATP is represented in black and navy blue, respectively. The rmsd moving averages (Mov-Avg) of WT-Apo and WT-ATP are represented in yellow and purple, respectively. (B) The rmsd probability distributions of WT-Apo and WT-ATP are represented in black and navy blue, respectively. (C) The rmsd analyses of I1171N-Apo and I1171N-ATP are represented in green and pear color, respectively. The rmsd moving averages (Mov-Avg) of WT-Apo and WT-ATP are represented in yellow and orange, respectively. (D) The rmsd probability distributions of I1171N-Apo and I1171N-ATP are represented in green and pear color, respectively. (E) rmsd analysis of I1171S-Apo and I1171S-ATP, where I1171S-Apo and I1171S-ATP are represented in mustard and cyan color, respectively. The rmsd moving averages (Mov-Avg) of WT-Apo and WT-ATP are represented in red and yellow. (F) The rmsd probability distributions of I1171S-Apo and I1171S-ATP are represented in mustard and cyan, respectively. (G) rmsd analysis of I1171T-Apo and I1171T-ATP, where I1171T-Apo and I1171T-ATP are represented in red and orange, respectively. The rmsd moving average (Mov-Avg) of WT-Apo and WT-ATP are represented in blue and yellow, respectively. (H) The rmsd probability distributions of I1171T-Apo and I1171T-ATP are represented in red and orange, respectively.
The RMSF was generated for each residue throughout the MDS, where the peaks signify regional fluctuations of the protein chain. According to the RMSF graph, all the residues of WT-Apo have fluctuated from 0.1 to 0.6 nm, whereas WT-ATP fluctuated from 0.1 to 0.8 nm. We observed higher fluctuation up to 0.6 nm at residue 1140 in WT-ATP compared to Apo ALK. The average RMSF values for WT-Apo and WT-ATP are 0.14 and 0.16 nm, respectively (Figure 3A). We observed a higher RMSF value of 0.4 nm for I1171N-ATP around residue 1220 compared to I1171N-Apo. The average RMSF values for I1171N-Apo and I1171N-ATP are 0.16 and 0.15 nm, respectively (Figure 3B). The RMSF value for I1171S-ATP was higher by 0.5 nm around residue 1290 than that of I1171S-Apo. The average RMSF values for I1171S-Apo and I1171S-ATP are 0.11 and 0.12 nm, respectively (Figure 3C). We observed higher RMSF values for I1171T-ATP around the residues, such as 1140, 1160, 1190, 1310, and 1340. The average RMSF values for I1171T-Apo and I1171T-ATP are 0.12 and 0.13 nm (Figure 3D). A higher RMSF value signifies greater mobility, flexibility, and unstable protein structure. The RMSF analysis revealed that ATP-WT, I1171S-ATP, and I1171T-ATP showed higher RMSF averages, indicating a more flexible and unstable structure than their respective Apo (ATP unbound) state.
Figure 3.

RMSF analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states throughout 200 ns MDS. The X-axis denotes the residue number, and the Y-axis displays the RMSF values in nanometer. (A) RMSF analysis of WT-Apo and WT-ATP, where WT-Apo and WT-ATP are represented in black and navy blue color, respectively. (B) RMSF analysis of I1171N-Apo and I1171N-ATP, where I1171N-Apo and I1171N-ATP are represented in green and pear color, respectively. (C) RMSF analysis of I1171S-Apo and I1171S-ATP, where I1171S-Apo and I1171S-ATP are represented in mustard and cyan color, respectively. (D) RMSF analysis of I1171T-Apo and I1171T-ATP, where I1171T-Apo and I1171T-ATP are represented in red and orange, respectively.
Rg represents the overall dimension and compactness of the protein structure. It also demonstrates how the protein 3D structure is densely packed with secondary structures. The unfolded proteins fluctuated over time. In contrast, stable folded proteins were expected to have a relatively constant Rg value. The average Rg values of stable WT-Apo, WT-ATP, I1171N-Apo, I1171N-ATP, I1171S-Apo, I1171S-ATP, I1171T-Apo, and I1171T-ATP are 2.09 ± 0.02, 2.09 ± 0.03, 2.10 ± 0.02, 2.14 ± 0.02, 2.06 ± 0.01, 2.08 ± 0.01, 2.07 ± 0.01, and 2.08 ± 0.01 nm, respectively (Figure 4). We noticed that the Apo WT and I1171N/S/T mutations exhibited minimum Rg compared to the respective ATP-bound state, which signified that a tight package of proteins can be seen in the Apo state. We also observed that the I1171N mutations had a Rg value significantly higher than those of the others, increased volume, and reduced compactness caused by the I1171N mutation in both Apo and ATP-bound states. The rmsd vs Rg correlation plots for the Apo and ATP-bound states of ALK WT and I1171N/S/T mutations throughout the simulation time are presented in Figure 5. In WT-Apo, the distribution of the analyzed conformers was positioned at the rmsd value ranging from ∼0.15 to ∼0.32 nm, corresponding to the Rg value between ∼2.05 and ∼2.13 nm (Figure 5A). In contrast, the WT-ATP distribution showed different rmsd values ranging from ∼0.18 to ∼0.38 nm and Rg value from ∼2.04 to ∼2.15 nm (Figure 5B). In I1171N-Apo, the distribution of the analyzed conformers was positioned at the rmsd value ranging from 0.12 to 0.38 nm, corresponding to the Rg value between ∼2.04 and ∼2.16 nm (Figure 5C). In contrast, the I1171N-ATP corresponding distribution showed slightly different rmsd values ranging from 0.18 to 0.4 nm and Rg value from ∼2.10 to ∼2.19 nm (Figure 5D). In I1171S-Apo, the distribution of the analyzed conformers was positioned at the rmsd value ranging from 0.15 to 0.32 nm, corresponding to the Rg value between ∼2.04 nm and ∼2.10 nm (Figure 5E). In contrast, the I1171S-ATP corresponding distribution showed a slightly different rmsd value ranging from 0.12 to 0.36 nm and Rg value from ∼2.06 to ∼2.11 nm (Figure 5F). In I1171T-Apo, the distribution of the analyzed conformers was positioned at the rmsd value ranging from 0.12 to 0.36 nm, corresponding to the Rg value between 2.04 and 2.12 nm (Figure 5G). In contrast, the I1171T-ATP corresponding distribution showed different rmsd values ranging from 0.14 to 0.32 nm and Rg value from 2.04 to 2.11 nm (Figure 5H). The correlation plots revealed that the dynamics of WT-Apo is distinct from that of I1171N/S/T and WT-ATP mutations.
Figure 4.

2D (A,C,E,G) and 3D (B,D,F,H) representation of Rg analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states during 200 ns MDS. 2D (A) and 3D (B) Rg analysis of WT-Apo and WT-ATP, where WT-Apo and WT-ATP are represented in black and navy blue, respectively. 2D (C) and 3D (D) Rg analysis of I1171N-Apo and I1171N-ATP, where I1171N-Apo and I1171N-ATP are represented in green and pear, respectively. 2D (E) and 3D (F) Rg analyses of I1171S-Apo and I1171S-ATP, where I1171S-Apo and I1171S-ATP are represented in mustard and cyan, respectively. 2D (G) and 3D (H) Rg analysis of I1171T-Apo and I1171T-ATP, where I1171T-Apo and I1171T-ATP are represented in red and orange, respectively.
Figure 5.

Correlation plot of rmsd vs Rg for backbone atoms of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states throughout 200 ns MDS. The X-axis denotes rmsd in nanometer, and the Y-axis displays Rg in nanometer.
Structural Property Analysis
SASA indicates the affinity between polar solvents and proteins. SASA was estimated from the 200 ns simulation for all the eight systems to understand the stability and conformation changes upon Apo and ATP-bound WT and I1171 mutations (Figure 6). The average SASA values of stable WT-Apo, WT-ATP, I1171N-Apo, I1171N-ATP, I1171S-Apo, I1171S-ATP, I1171T-Apo, and I1171T-ATP are 164.01 ± 3.19, 167.33 ± 4.20, 169.05 ± 5.25, 169.79 ± 3.55, 154.47 ± 2.46, 163.26 ± 2.37, 157.62 ± 2.91, and 159.53 ± 3.24 nm2, respectively (Figure 6). The ATP-bound WT and I1171 mutations showed higher SASA values compared to Apo. A higher SASA value for ATP-bound structures indicates a stronger affinity for solvents compared to Apo.
Figure 6.

SASA analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states throughout 200 ns MDS. The X-axis denotes the time in nanosecond, and the Y-axis displays the area in square nanometer. (A) SASA analysis of WT-Apo and WT-ATP, where WT-Apo and WT-ATP are represented in black and navy blue, respectively. (B) SASA analysis of I1171N-Apo and I1171N-ATP, where I1171N-Apo and I1171N-ATP are represented in green and pear, respectively. (C) SASA analysis of I1171S-Apo and I1171S-ATP, where I1171S-Apo and I1171S-ATP are represented in mustard and cyan, respectively. (D) SASA analysis of I1171T-Apo and I1171T-ATP, where I1171T-Apo and I1171T-ATP are represented in red and orange, respectively.
Protein’s hydrogen bonds (H-bonds) are a crucial noncovalent structural factor and a key determinant of protein stability. The intramolecular H-bond formation in Apo and ATP-bound ALK WT was analyzed throughout the simulation (Supporting Information Figure 1). The average numbers of intramolecular H-bonds of stable WT-Apo, WT-ATP, I1171N-Apo, I1171N-ATP, I1171S-Apo, I1171S-ATP, I1171T-Apo, and I1171T-ATP are 194.05 ± 7.67, 191.53 ± 7.04, 197.33 ± 7.49, 190.49 ± 7.98, 196.35 ± 6.95, 193.79 ± 7.23, 199.42 ± 7.09, and 201.17 ± 7.67, respectively (Supporting Information Figure 1). The intramolecular H-bonds disrupt ATP binding in WT and I1171N/S/T. Compared to WT, mutations showed a higher number of H-bonds, which makes the structure more rigid and restricts the movement/motion of the proteins. The intermolecular H-bonds in the ATP-bound ALK WT and I1171N, I1171S, and I1171T mutations were analyzed throughout the simulation (Figure 7). The WT-ATP-bound stable structure formed a maximum of eight H-bonds with an average of 3.04 ± 0.80 (Figure 7A), whereas the I1171N-ATP-bound stable structure formed a maximum of six H-bonds with an average of 2.89 ± 0.65 (Figure 7B), the I1171S-ATP-bound stable structure formed a maximum of eight H-bonds with an average of 3.77 ± 0.78 (Figure 7C), and the I1171T-ATP-bound stable structure formed a maximum of nine H-bonds with an average of 4.11 ± 1.48 (Figure 7D). The I1171S/T mutations showed greater H-bonds with ATP than WT, which resembles the stable bond formation with ATP. Likewise, WT and I1171N showed a similar number of H-bonds formed with ATP. The Apo I1171N/S/T mutations showed higher intramolecular H-bonds than WT, which makes the structure more rigid.
Figure 7.

Intermolecular hydrogen bond analysis of ALK WT and I1171N/S/T mutations with ATP over the 200 ns MDS. The X-axis denotes the time in nanoseconds, and the Y-axis displays H-bonds in number. The intermolecular hydrogen bond analysis for (A) WT-ATP showed navy blue, (B) I1171N-ATP in pear, (C) I1171S-ATP in cyan, and (D) I1171T-ATP in orange.
PCA and Essential Dynamics
PCA, which produces eigenvectors, was used to examine the effects of the overall motion of the protein caused by binding of ATP in WT and I1171 mutations. This technique allows the use of the covariance matrix computation and diagonalization. WT-Apo occupied the conformational space of PC1 (−6 to 6 nm) and PC2 (−5 to 6 nm), but upon ATP binding, a change in overall motion and expansion of conformational space of PC1 (−8 to 5 nm) and PC2 (−5 to 5 nm) in WT-ATP is observed (Figure 8A). Similarly, the diagonalized trace covariance matrix values for WT-Apo and WT-ATP are 24.127 and 30.669, respectively. I1171N-Apo occupied the conformational space of PC1 (−6 to 8 nm) and PC2 (−7 to 6 nm); then, upon ATP binding of I1171N, a change in the overall motion is observed, but a similar conformational space of PC1 (−6 to 8 nm) and PC2 (−5 to 4 nm) in I1171N-ATP is shown (Figure 8B). This analysis confirms that the 11171N mutation makes the protein active since it resembles WT-ATP. Similarly, the diagonalized trace covariance matrix values for I1171N-Apo and I1171N-ATP are 30.677 and 26.227, respectively. The I1171S-Apo mutation occupied the conformational space of PC1 (−5 to 5 nm) and PC2 (−4.5 to 5 nm), whereas I1171S-ATP occupied the conformational space of PC1 (−6 to 6 nm) and PC2 (−4 to 3.5 nm) (Figure 8C). Similarly, the diagonalized trace covariance matrix values for I1171S-Apo and I1171S-ATP are 17.624 and 19.351, respectively. The I1171T mutation occupied the conformational space of PC1 (−5 to 5 nm) and PC2 (−7 to 5 nm), while I1171T-ATP occupied the conformational space of PC1 (−5.8 to 6 nm) and PC2 (−5 to 4.7 nm) (Figure 8D). Similarly, the diagonalized trace covariance matrix values for I1171T-Apo and I1171T-ATP are 21.241 and 22.730, respectively. In a 2D projection of PCA, if the protein/complex takes up less phase space, it is considered stable, whereas if the protein/complex takes up more space, it is considered less stable. We observed larger phase space in ATP-bound WT, indicating that these are less stable than the Apo state. Similarly, we noticed that the WT occupied less phase space than I1171N/S/T mutations in both Apo and ATP-bound states, suggesting that the WT was more stable. Moreover, the trace covariance matrix values confirm that ATP-bound WT and I1171S/T mutations occupied significantly higher values than the Apo state. It demonstrated increased flexibility and showed a different manner from the Apo state. WT-ATP and I1171N-Apo revealed identical values higher than the WT, suggesting that I1171N mutation causes ALK to remain constitutively active and flexible. Similarly, I1171S/T mutations showed lower values compared to WT, indicating that these mutations cause ALK to be more rigid.
Figure 8.

PCA analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states during 200 ns MDS. The X-axis denotes PC 1 in nanometer, and the Y-axis displays PC 2 in nanometer. (A) PCA analysis of WT-Apo and WT-ATP, where WT-Apo and WT-ATP are represented in black and navy blue, respectively. (B) PCA analysis of I1171N-Apo and I1171N-ATP, where I1171N-Apo and I1171N-ATP are denoted in green and pear, respectively. (C) PCA analysis of I1171S-Apo and I1171S-ATP, where I1171S-Apo and I1171S-ATP are denoted in mustard and cyan, respectively. (D) PCA analysis of I1171T-Apo and I1171T-ATP, where I1171T-Apo and I1171T-ATP are denoted in red and orange, respectively.
To further examine the impact of ATP binding on the conformational dynamics of ALK WT and I1171N/S/T, the correlative movements of ATP for interacting with ALK WT and I1171N/S/T mutations must be detected. The findings of the DCCM analysis, as shown in Figure 9, revealed that the global dynamics of all the systems were strikingly similar. Besides that, ATP binding had little effect on the original secondary structures of ALK. However, a thorough inspection of DCCM reveals the differences between the apo and ATP-bound WT ALK protein (Figure 9A,B). The significant differences were discovered in several positions: the ALK WT-Apo protein’s few positions had blue, representing significant anticorrelated motions. The anticorrelated motions were significantly reduced in ALK WT-ATP around the ATP binding sites 1197–1199 and 1293 positions. Similarly, DCCM analysis showed that I1171N-Apo (Figure 9C), I1171N-ATP (Figure 9D), I1171S-ATP (Figure 9F), I1171T-Apo (Figure 9G), and I1171T-ATP (Figure 9H) had a greater reduction of anticorrelated motions (reduced blue color), whereas I1171NS-Apo (Figure 9E) had a minor reduction of anticorrelated motions. These results indicate that the correlative motions increased in the WT-Apo compared to WT-ATP. Similarly, we noted that the Apo and ATP-bound state of I1171N/S/T mutations had higher correlative collective motions than WT-Apo. The DCCM analysis of Apo and ATP-bound ALK WT and I1171N/S/T mutations was consistent with the former analysis outcomes.
Figure 9.

DCCM of the backbone atoms around their mean positions of ALK (A) WT-Apo, (B) WT-ATP, (C) ALK I1171N-Apo, (D) I1171N-ATP, (E) ALK I1171S-Apo, (F) I1171S-ATP, (G) I1171T-Apo, and (H) I1171T-ATP. The color bar varies from red to blue (+1 to −1), which represents the level of correlation and anticorrelation movements. The significant change in DCCM is highlighted with arrows.
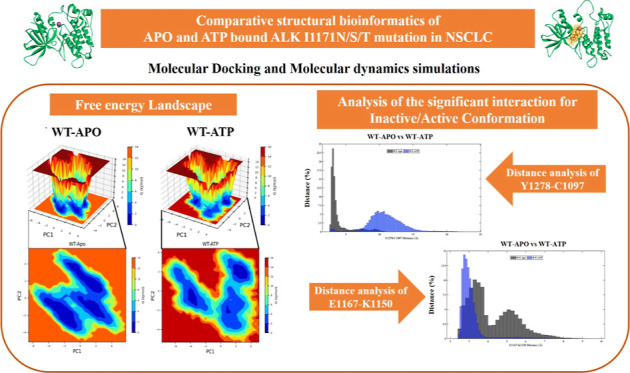
Additionally, we analyzed FEL to check whether ATP binding affects the conformational stability of the ALK WT and I1171N/S/T protein conformational stability. FEL can offer incredible insights into the many conformational states of which the protein is capable of in the simulation. The 2D and 3D FEL contour maps were plotted using the first two PCs (PC1 and PC2) and obtained ΔG values for each protein simulation. The deeper blue color indicates conformational states with minimal energy. The ΔG values range from 0 to 13.6 and 14.4 kJ/mol for WT and WT-ATP (Figure 10A,B). WT-Apo obtained three larger minimum energies, which indicated a more stable structure. In WT-ATP, we observed different conformational motions and a lower minimum energy than the Apo state. The ΔG values range from 0 to 14.2 and 14.6 kJ/mol for I1171N and I1171N-ATP (Figure 10C,D). I1171N showed a single stable global minimum energy, whereas the I1171N-ATP-bound state showed dispersed three minimum global energies with an obvious conformational change upon ATP binding in I1171N. The ΔG values range from 0 to 15 and 14.7 kJ/mol for I1171S and I1171S-ATP (Figure 10E,F). I1171S-ATP showed less or similar minima energy with conformational shift throughout the transition state than the I1171S-Apo state. The ΔG values range from 0 to 15 and 15.2 kJ/mol for I1171S and I1171S-ATP (Figure 10G,H). I1171T showed a wide global minima energy, but I1171T-ATP showed a single and reduced minima energy. The WT displayed three precise global minima energies of the different conformational states. Observing several energy minima in the conformational space attained by I1171 mutations of the Apo and ATP-bound states indicates that the protein has been severely destabilized.
Figure 10.

2D contour and 3D free energy landscape maps of ALK (A) WT-Apo, (B) WT-ATP, (C) ALK I1171N-Apo, (D) I1171N-ATP, (E) ALK I1171S-Apo, (F) I1171S-ATP, (G) I1171T-Apo, and (H) I1171T-ATP over 200 ns MDS. The free energy is expressed in kJ/mol and shown visually in color.
MM/PBSA Analysis
The average structures were extracted from the stable trajectories of the four systems: ATP-bounded ALK WT and I1171N/S/T mutation complexes. Using the MM/PBSA calculation, the binding energy values of governing residues within 5 Å at the ATP active site pockets of ATP-bounded ALK WT and I1171N/S/T mutations were taken from the res.dat file. From these values, we obtained the binding energy at the ATP active site for the ATP-bound WT and I1171 mutations using governing residues such as L1122, G1125, V1130, A1148, L1198, G1202, and L1256. The binding energy based on the governing residues’ contribution for WT-ATP, I1171N-ATP, I1171S-ATP, and I1171T-ATP is illustrated in Figure 11. The sum of the governing residue binding energies for WT-ATP, I1171N-ATP, I1171S-ATP, and I1171T-ATP was −30.064, −31.743, −27.591, and −24.919 kJ/mol, respectively. The binding energy of I1171N showed a higher binding energy with ATP than WT. At the same time, I1171S/T was relatively similar to the WT. The binding energy obtained from molecular docking also demonstrated an almost similar value for the I1171N/S/T and WT mutations.
Figure 11.

Binding energy of governing residues within 5 Å at the active site pockets of ATP-bound ALK WT and I1171N/S/T mutations estimated using their stable trajectories.
Analysis of a Significant Interaction Involved in ALK Activation
We analyzed the significant interaction involved in ALK inactive and active conformations by measuring their distance and hydrogen bond interaction in all conformations of WT-ALK and I1171N/S/T mutations with Apo/ATP-bound states of MD trajectories using VMD. The significant interaction keeps the C-helix in an inactive conformation by forming a hydrogen bond of the A-loop helix Tyr1278 (Y1278) residue, the first tyrosine residue that facilitates the autophosphorylation process, with N-terminal-turn Cys1097 (C1097). The distance between Y1278–C1097 was measured throughout the MD trajectories and is provided in Supporting Information Figure 2. The distance distribution of the hydroxyl group Y1278 with backbone nitrogen C1097 in all of the simulations is plotted in Figure 12A–D. The distance distribution of Y1278–C1097 for WT-Apo was within 5 Å, whereas WT-ATP showed more than 5 up to 15 Å (Figure 12A). In the I1171N mutations in both Apo and ATP-bound states, the distance distribution of Y1278–C1097 was almost similar; it ranges from 5 to 15 Å (Figure 12B). In the I1171S mutation in both Apo and ATP-bound states, the distance distribution of Y1278–C1097 was within 5 Å (Figure 12C). In I1171T-Apo, we noticed that the distance distribution of Y1278–C1097 was within 5 Å but deviated in I1171T-ATP; it ranges from 3 to 9 Å (Figure 12D). The hydrogen bond formed between residues Y1278 and C1097 was measured throughout the MD trajectories (Supporting Information Figure 3). The average number of H-bonds between Y1278 and C1097 of all of the trajectories is mentioned in Table 2. A stable H-bond formed in WT-Apo, but no H-bond can be seen in WT-ATP after 10 ns of simulation (Supporting Information Figure 3). The average H-bond for I1171N/T mutations in Apo and ATP-bound states was less than that in WT-Apo.
Figure 12.

Distance frequency of Y1278–C1097 (A–D) and E1167–K1150 (E–H) throughout the MDSs of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states.
Table 2. Average Number of H-Bonds between the Y1278–C1097 and E1167–K1150 Interactions over the MDSs of ALK WT and I1171N/S/T Mutations in Apo and ATP-Bound States.
| average H-bonds between
interaction |
||
|---|---|---|
| MD-trajectories | Y1278–C1097 | E1167–K1150 |
| WT-Apo | 0.088346 | 0.239138 |
| I1171N-Apo | 0.017149 | 0.502625 |
| I1171S-Apo | 0.100095 | 0.39898 |
| I1171T-Apo | 0.033648 | 0.438528 |
| WT-ATP | 0.0013 | 0.318734 |
| I1171N-ATP | 0.010049 | 0.532423 |
| I1171S-ATP | 0.095545 | 0.40978 |
| I1171T-ATP | 0.034898 | 0.109695 |
In contrast, I1171S showed a stronger H-bond interaction in Apo and ATP-bound states than WT-Apo (Table 2). The C-helix enables the correct placement of Glu1167 (E1167) and Lys1150 (K1150) for salt bridge building, which is also a characteristic of the active conformation. First, we measured the distance between E1167 and K1150 throughout MD trajectories (Supporting Information Figure 4). The distance distribution of residue E1167 with K1150 in the overall simulations was represented in percentage (Figure 12E–H). We noticed that the distance between E1167 and K1150 was far in WT-Apo compared to WT-ATP. In WT-Apo, the E1167–K1150 distance ranges from 3 to 8 Å, whereas WT-ATP showed active conformation within 4 Å (Figure 12E). The E1167–K1150 distance for I1171N-Apo and ATP-bound states is within 4 Å, the same as that for WT-ATP (Figure 12F). The E1167–K1150 distance for I1171S-Apo and ATP-bound states is within 5 Å (Figure 12G). The E1167−K1150 distance for I1171T-Apo ranges from 2.5 to 6 Å, whereas F1174T−ATP showed 2.5 to 10.5 Å (Figure 12H). The hydrogen bond formed between residues E1167–K1150 was measured throughout the MD trajectories (Supporting Information Figure 5). The average number of H-bonds between E1167 and K1150 of all the trajectories is mentioned in Table 2. The average number of H-bonds in the E1167–K1150 salt bridge was higher in WT-ATP than in WT-Apo. We noticed that the average number of H-bonds in the E1167–K1150 salt bridge was higher in I1171N/S/T mutations in the Apo state than in WT-ATP. Similarly, the average number of H-bonds in E1167–K1150 were higher in I1171N/S mutations in the ATP-bound state, but I1171T-ATP showed the least interaction compared to WT-Apo/ATP.
Discussion
In a subgroup of NSCLC, Soda et al. identified the EML4-ALK fusion gene in 2007.43 It is the most common fusion compared with other fusion partners of ALK.44 Exclusive of EGFR and KRAS mutations, the ALK rearrangement in NSCLC is responsible for 3–7%.43,45 Several tyrosine kinase inhibitors have been upended against ALK mutational rearrangements. Crizotinib, the first-generation TKI targeting ALK, was approved by the FDA in 2011,8 although the crizotinib tolerance level is elevated in patients, resulting in treatment-related adverse events (TRAEs).46 ALK I1171, a missense mutation, acquired resistance against crizotinib and alectinib TKIs. Alectinib, a second-generation ALK tyrosine kinase inhibitor, binds to the ATP binding site on ALK.47 The presence of the αC-helix I1171N mutation reportedly triggered resistance to alectinib.15 Alectinib resistance by I1171 mutations is rendered due to increased steric hindrance and disruption of the hydrogen bonding between the mutations and the drug that alters the ALK’s ATP-binding pocket.8,47 I1171N stabilizes the activated conformation of tyrosine kinase via destabilization of the ALK inhibitor.8 Secondary treatments against I1171 mutations are provided through ceritinib, a second-generation drug.15 Ceritinib interacts with the hinge region of the protein kinase domain, proving to be efficient against I1171. The postcrizotinib treatments are essential for normal CNS functioning; ceritinib is known to cross the blood–brain barrier. Brigatinib, a second-generation TKI, is a ceritinib analogue; studies have shown that brigatinib affects the activating mutations in the ALK protein.44 A recent report showed that only ceritinib is sensitive to the I1171 mutational variants. The I1171 resistance mechanism is independent of the ALK-fusion partner, as mutations are observed in both EML4-ALK and HIP1-ALK.15 Recent MD simulations on I1171 suggest that the mutation results in conformational dynamics of αC-helices interfering with the ATP-binding pocket, affecting the binding of alectinib.48 The missense mutation I1171 induces conformational changes such that the affinity between alectinib and ALK mutations is diminished. Numerous allosteric effects of ATP binding on kinase activation have been identified.49−51 Many clinical studies are being conducted to see if any ALK-TKIs can replace crizotinib as the first-line treatment for ALK-positive NSCLC.52−54 Similarly, several computational studies have identified the first- and second-line inhibitors’ drug resistance mechanisms caused by ALK mutations at the atomic level.55−57 Hu et al. (2020) demonstrated that the acquired mechanisms of lorlatinib resistance are caused by several mutations in ALK, including I1171N, L1196M, and G1202R.58 The mechanism of drug resistance brought by ALK I1171N/F1174I that is resistant to lorlatinib therapy in NSCLC was explained mechanistically by Liang et al.28 The harmful consequences of the compound ALK mutations, I1171N/F1174I and I1171N/L1178H, and their role in the etiology of NSCLC were studied.27 Even though there has been extensive research on ALK drug identification and the resistance mechanism of inhibitors, the activation mechanism of ALK is currently unclear. Understanding the mechanism of ALK activation will lead to possible ALK inhibitors. Hence, we performed molecular docking for ALK WT and I1171N/S/T mutations with ATP to elucidate the binding affinity and structural conformation of ATP. We subjected the ALK WT and I1171N/S/T mutations in Apo and ATP-bound states to MD simulation for 200 ns and studied the structural stability, conformational changes, and mechanism of ATP.
In the current study, we found that ALK WT docked with ATP has a binding affinity of −6 kcal/mol, whereas the mutations I1171N, I1171S, and I1171T have binding affinity values of −6.9, −6.1, and −6.5 kcal/mol, respectively (Figure 1). The I1171N/T mutations showed a higher binding affinity for ATP than the WT. The I1171S mutation exhibited almost similar binding affinity similar to WT. This analysis confirms that the I1171N/S/T mutations have a robust binding affinity toward ATP compared to WT. The ALK protein develops resistance to TKIs due to the significant interaction of ATP in the I1171N/S/T mutations. Somatic mutations in the genes encoding RTKs commonly accumulate at evolutionarily conserved residues, such as the DFG motif in the kinase activation loop and close to the nucleotide-binding pocket. D, F, and G, three conserved residues, are crucial for ATP binding and catalytic activity; it is present in the residue D1270, F1271, and G1272 of the C-lobe in the ALK structure.9,59−61 ATP occurs in various molecular recognition and catalytic activities across biochemistry; its magnesium chelate, ATP-Mg, is the most prevalent bioactive form.62 From Figure 1, it can be seen that molecular docking of ALK WT and I1171N/S/T with ATP in the presence of Mg showed that all the structures formed H-bonds with D1270, further confirming the significant interaction of ATP in the DFG region and its responsible for catalytic activity. The ALK kinase domain, like other kinases, consists of a conserved N-lobe and a C-lobe linked with a hinge region to form a cleft that offers an ATP binding pocket, enabling the catalytic kinase reaction to take place.12 Most ALK TKI resistance mutations discovered so far cluster near the ATP/TKI binding site in the kinase domain, causing a steric hindrance for inhibitor binding that ALK fusion kinase activity to continue even in the presence of TKI.63 These resistance mutations might result after ALK TKI therapy in NSCLC patients, which include I1171, F1174, F1245, and L1196 gatekeeper mutations, provide constitutive activity of ALK, and are capable of inducing oncogenic transformation.9 To overcome the acquired drug resistance in ALK-targeted therapies, knowing the activation mechanism and structural changes of the I1171N, S, and T mutations will enable us to find better drug targets. Furthermore, MD analysis for 200 ns was executed for the complex structures obtained from docking along with the Apo ALK WT and I1171N/S/T mutations to check the structural variation, stability upon ATP binding, and structural characteristics of different amino acid substitutions in I1171.
To determine whether all the simulations were stable, we examined the rmsds and the probability distribution of the backbone rmsds from 40 to 200 ns.64 After the MD simulation, the rmsd analyses of WT-Apo and WT-ATP both were stable after 40 ns, suggesting that both structures reached equilibrium (Figure 2A). We observed a greater RMSF value in WT-ATP than Apo, indicating the flexibility at the ATP-bound state (Figure 3A). Based on the Rg analysis, the WT-Apo structure is more compact than the WT-ATP structure (Figure 4A). SASA analysis revealed that WT-ATP has a stronger affinity for solvents (Figure 6A). The intramolecular hydrogen bond of WT-ATP was lesser than the Apo state, suggesting more flexibility; this corresponds to RMSF (Supporting Information Figure 1). Furthermore, the diagonalized trace covariance matrix stated that WT-ATP was more flexible than Apo. PCA and FEL demonstrate that ATP-bound structures are unstable/conformational motion change with lower minima energy. He et al.’s (2018) findings revealed that mutations and ATP supported a change toward an active-like conformation, whereas WT-Apo ALK is mostly preferred in an inactive conformation.65 Our study also demonstrated the active-state conformation in the WT-ATP structure.
In I1171N-Apo vs I1171N-ATP, we observed stable structures after 40 ns of the simulation time (Figure 2C). There was no significant change in the RMSF value for the I1171N mutations in either state (Figure 3B). According to Rg (Figure 4C) and SASA (Figure 6B) analyses, the I1171N-ATP structure is less compact and has a greater affinity for solvents than the I1171N-Apo structure. A recent study by Salifu et al. (2023) revealed that ALK compound mutations such as I1171N + F1174I and I1171N + L1198H had lower values for RMSF, Rg, and SASA, which implies that the mutations are smaller and more compact than the WT. Furthermore, they exposed that these mutations also altered residues that make up the ALK structure’s activation loop, indicating that they may be phosphorylation disruptors.27 Intramolecular H-bonds are disturbed when ATP binds to I1171N, reducing the number of H-bonds (Supporting Information Figure 1). PCA and FEL demonstrate that the I1171N mutation in Apo and ATP states showed different energy minima and unstable structures than WT. In a previous investigation, tumor samples from an NSCLC patient who took alectinib were shown to have αC helix I1171N mutation.15 The I1171N mutation impacts the structural dynamics of the αC helix in the ALK–alectinib complex, as evidenced by the noticeable large fluctuations in the αC helix and nearby A-loop.48 Here, we obtained significantly higher RMSF for I1171N than for WT-Apo, equivalent to ALK active conformation (WT-ATP). These analyses prove that the I1171N mutation in ALK resulted in an active-like conformation.
In I1171S-Apo vs I1171S-ATP, both the structures converged after 40 ns and produced stable structures for the analysis (Figure 2E). The flexibility of I1171S increased with ATP-binding since the RMSF value was marginally higher in the bound state than in the Apo state (Figure 3C). In Rg (Figure 4E) and SASA (Figure 6C) analyses, we observed that the I1171S-ATP structure was less compact and had a stronger affinity. After ATP bound to I1171S, the number of intramolecular hydrogen bonds was decreased. The intramolecular H-bonds were reduced when ATP bound to the I1171S mutation (Supporting Information Figure 1). These reduced H-bonds make I1171S-ATP more flexible. Takahashi et al. recommend that the I1171S single mutation was initially acquired following administration of crizotinib, and consecutive ALK therapy reduced the I1171S single mutation and development of new G1269A mutation after lorlatinib-generated I1171S + G1269A mutation. By overcoming these resistant mutations, they found that brigatinib and ceritinib could decrease ALK phosphorylation at lower dosages.16
In I1171T-Apo vs I1171T-ATP, the rmsd analysis revealed that both structures attained equilibrium after 40 ns (Figure 2G). The RMSF value was higher in the ATP-bound state than that in Apo, indicating increased flexibility (Figure 3D). The Rg (Figure 4G) and SASA (Figure 6D) analyses led us to conclude that the I1171T-ATP structure was less compact and had a higher affinity for solvents. Intramolecular H-bonds of I1171T remained the same even after ATP interaction (Supporting Information Figure 1). Apo and ATP I1171T were shown to have an unstable structure and several minima regions compared to WT. A previous study reported that the ALK-I1171T mutation results in a gain of function. Based on the pharmacological inhibition profile of ALK-I1171T in response to several ALK TKIs, ceritinib inhibits ALK-I1171 11-fold better than crizotinib.66 The rmsd vs Rg correlation plot revealed that WT-Apo showed different compactness and stability from the WT-ATP and I1171N/S/T mutations’ Apo and ATP-bound states. PCA and FEL demonstrate that I1171S and I1171T mutations in Apo and ATP-bound states showed different energy minima and unstable structures than WT. Similar to PCA and FEL analysis, the DCCM plot also showed reduced anticorrelation motions in WT-ATP, I1171N/T Apo, and I1171N/S/T ATP than WT-Apo, which correspond to an active state of the structures. The significant changes in DCCM are demonstrated with arrows in Figure 9.
According to intermolecular H-bonds, WT and I1171N showed an equivalent interaction with ATP throughout the 200 ns run. In contrast, I1171N/S showed greater interaction with ATP. This reveals that all the mutations may have a potent transition into an active conformation state (Figure 7). The I1171N/S/T mutations showed higher intramolecular H-bonds than the WT, which makes the structure more rigid (Supporting Information Figure 1). The earlier study noticed that the R1275Q mutation stabilizes the active conformation by rigidifying the αC in a conformation. They also noticed that the Y1278S mutation increased the activation at the loss of the π-stacking hydrophobic group, which plays a crucial role in maintaining the inactive conformation.65 The amino acid isoleucine at the 1171 position maintains the hydrophobicity; mutations that change the amino acid to asparagine, serine, and threonine will change the properties from nonpolar to polar, reducing the hydrophobicity, and the addition of H-bond interactions, which transforms ALK to an active state.
In the ALK kinase domain, the initial residue that is to be phosphorylated is Y1278. The small proximal A-loop helix has a special inhibitory structural characteristic that aligns it against the long C-helix in the ALK kinase domain. It has C-1097 containing a turn motif that blocks the substrate binding. Y1278 is not permitted for phosphorylation in this configuration because it is bound to C1097 in the N- terminal β-sheet.11,12 From the average H-bond interaction between Y1278–C1097, it is clear that I1171N/T mutations are active conformation since a weaker H-bond interaction can be seen in both Apo and ATP-bound states. The I1171S mutation in both Apo and ATP states showed a stronger H-bond interaction of Y1278–C1097 than WT-Apo; it resembles the characteristics of an inactive conformation. This finding implies that altering the orientation of the N-turn and C-helix may be involved in the activation effect. MDS over 100 ns of I1171N mutation observed different frequencies of stable hydrogen between Y1278 and C1097 in the I1171N and WT mutations, which resembles inactive conformation.25 However, our study showed that the distance between Y1278 and C1097 was increased and that there was reduction of the hydrogen bond after 100 ns of simulations, suggesting active conformation of the mutation. The E1167–K1150 salt bridge also characterizes the active conformation of ALK.25 The average number of H-bonds in the E1167–K1150 salt bridge was higher in I1171N/S/T mutations in the Apo state than in WT-ATP, similar to that in the active state devoid of ATP.
We further took snapshots of the structures at 100 ns intervals for each of the eight systems to examine the structural changes caused by mutation and ATP binding. The A-loop is a common site for mutations in the kinase domain that induces constitutive activity. To check the structural changes at the A-loop helix alters the Y1278 residue interaction (Supporting Information Figure 6). After 200 ns simulation, the WT secondary structure comprises an A-loop helix from residue 1272–1277, whereas upon I1171N/S mutations, a reduced A-loop helix was noticed (from residue 1272–1275); these structural modifications may allow phosphorylation to take place in the kinase domain. Similarly, upon obtaining I1171T mutation, the addition of helix (from residue 1272–1280) is carried out. ALK activation is promoted by ATP, which produces a more compact active site. F1174 and R1275 preserve the domain in autoinhibited conformation.9 In the WT-ATP structure, the A-loop helix was observed from residues 1274–1278; this transformation is due to the binding of ATP in the ALK domain. After the ATP binding upon I1171N/S/T after 200 ns simulation, we observed helix extension compared to WT-ATP. These minor structural changes may be associated with constitutive activity upon mutations.
We used secondary structure analysis to compare the secondary structural changes of WT and I1171N/S/T mutations in Apo and ATP-bound states in all conformations (Supporting Information Figure 7). This analysis shows that structural changes are minor, but the orientation affects the phosphorylation in I1171N mutations by resembling the WT-ATP characteristics of the active conformation. Thus, it is concluded that I1171N mutation orientation changes will modify the behavior of ALK and transform it into an active state. Similarly, MM/PBSA analysis revealed that I1171N had a higher binding energy, possibly due to structural changes in the A-loop. I1171S/T also showed a similar WT binding affinity toward ATP. Furthermore, the WT and I1171N/S/T mutations have an equal binding affinity toward ATP and may contribute to kinase activity. Here, we demonstrated the structural transition in the A-loop upon I1171N mutation and ATP binding, resulting in kinase activity.
Conclusions
Molecular docking enabled ATP binding affinity and patterns with WT and I1171N/S/T mutations. The MDS demonstrated the conformational transfiguration triggered by I1171N/S/T mutations that activate the ALK kinase activity. We further revealed atomistic insights on ALK WT and I1171N/S/T mutations in Apo and ATP-bound states. The I1171N/S/T mutations had a reduced low energy minimum that resembled the unstable structure, whereas WT-apo had a large low energy minimum that resembled a stable structure. WT-ATP and I1171N/S/T mutations showed similar reduced low energy minima resembling the active state conformation. The I1171N mutation and I1171N-ATP binding strongly promote constitutively more active kinase than I117S/T; this mutation, independent of ATP, remains active. The explored structural mechanism may be useful for understanding the oncogenic activity of ALK and providing a basis for developing potent inhibitors for ALK.
Acknowledgments
Ambritha Balasundaram gratefully acknowledges the Indian Council of Medical Research (ICMR), India, for providing a Senior Research Fellowship [BMI/11(05)/2022]. The authors would like to take this opportunity to thank the management of Vellore Institute of Technology (VIT), Vellore, India, for providing the necessary facilities and encouragement to carry out this work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c05785.
Binding energy and distance between Mg and ATP of the ATP-bounded ALK WT and I1171N/S/T mutations in all conformations in triplicates; intramolecular hydrogen bond analysis of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states; distance and number of H-bonds between Y1278 and C1097 of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states throughout the MD trajectories; salt bridge distance and number of H-bonds between E1167–K1150 of ALK WT and I1171N/S/T mutations in Apo and ATP-bound states throughout the MD trajectories; snapshots of the ALK WT and I1171N/S/T mutations in Apo and ATP-bound states over the entire simulations; and overall percentage of secondary structures for ALK WT and I1171N/S/T mutations in Apo and ATP-bound states (PDF)
Author Contributions
A.B. and C.G.P.D. were involved in the design of the study. A.B. was involved in the data collection and experimentation. A.B. was involved in acquiring, analyzing, and interpreting the results. A.B. drafted the manuscript. C.G.P.D. supervised the entire study and was involved in study design, acquiring, analyzing, and understanding the data and critically reviewing the manuscript. All authors edited and approved the submitted version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Javanmardi N.; Fransson S.; Djos A.; Sjöberg R. M.; Nilsson S.; Truvé K.; Kogner P.; Martinsson T. Low Frequency ALK Hotspots Mutations In Neuroblastoma Tumours Detected By Ultra-Deep Sequencing: Implications For ALK Inhibitor Treatment. Sci. Rep. 2019, 9 (1), 2199. 10.1038/s41598-018-37240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holla V. R.; Elamin Y. Y.; Bailey A. M.; Johnson A. M.; Litzenburger B. C.; Khotskaya Y. B.; Sanchez N. S.; Zeng J.; Shufean M. A.; Shaw K. R.; Mendelsohn J.; Mills G. B.; Meric-Bernstam F.; Simon G. R. ALK: A Tyrosine Kinase Target for Cancer Therapy. Cold Spring Harbor Mol. Case Stud. 2017, 3 (1), a001115. 10.1101/mcs.a001115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Anaplastic Lymphoma Kinase (ALK): Structure, Oncogenic Activation, and Pharmacological Inhibition. Pharmacol. Res. 2013, 68 (1), 68–94. 10.1016/j.phrs.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Morris S. W.; Kirstein M. N.; Valentine M. B.; Dittmer K. G.; Shapiro D. N.; Saltman D. L.; Look A. T. Fusion of a Kinase Gene, ALK, to a Nucleolar Protein Gene, NPM, in Non-Hodgkin’s Lymphoma. Science 1994, 263 (5151), 1281–1284. 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- Zappa C.; Mousa S. A. Non-Small Cell Lung Cancer: Current Treatment and Future Advances. Transl. Lung Cancer Res. 2016, 5 (3), 288–300. 10.21037/tlcr.2016.06.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura K.; Takeuchi K.; Togashi Y.; Nomura K.; Ninomiya H.; Okui M.; Satoh Y.; Okumura S.; Nakagawa K.; Soda M.; Lim Choi Y.; Niki T.; Mano H.; Ishikawa Y. EML4-ALK Fusion Is Linked to Histological Characteristics in a Subset of Lung Cancers. J. Thorac. Oncol. 2008, 3 (1), 13–17. 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- Inamura K.; Takeuchi K.; Togashi Y.; Hatano S.; Ninomiya H.; Motoi N.; Mun M.; Sakao Y.; Okumura S.; Nakagawa K.; Soda M.; Lim Choi Y.; Mano H.; Ishikawa Y. EML4-ALK Lung Cancers Are Characterized by Rare Other Mutations, a TTF-1 Cell Lineage, an Acinar Histology, and Young Onset. Mod. Pathol. 2009, 22 (4), 508–515. 10.1038/modpathol.2009.2. [DOI] [PubMed] [Google Scholar]
- Kwak E. L.; Shaw A. T.; Ou S.-H. I.; Varella-Garcia M.; Stubbs H.; Gandhi L.; Ratain M. J.; Wilner K.; Iafrate A. J. Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2010, 363, 1693. 10.1056/nejmoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallberg B.; Palmer R. H. The Role of the ALK Receptor in Cancer Biology. Ann. Oncol. 2016, 27, iii4–iii15. 10.1093/annonc/mdw301. [DOI] [PubMed] [Google Scholar]
- Chen H.; Ma J.; Li W.; Eliseenkova A. V.; Xu C.; Neubert T. A.; Miller W. T.; Mohammadi M. A Molecular Brake in the Kinase Hinge Region Regulates the Activity of Receptor Tyrosine Kinases. Mol. Cell 2007, 27 (5), 717–730. 10.1016/j.molcel.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi R. T.; Saccardo M. B.; Ardini E.; Menichincheri M.; Rusconi L.; Magnaghi P.; Orsini P.; Avanzi N.; Borgia A. L.; Nesi M.; Bandiera T.; Fogliatto G.; Bertrand J. A. Crystal Structures of Anaplastic Lymphoma Kinase in Complex with ATP Competitive Inhibitors. Biochemistry 2010, 49 (32), 6813–6825. 10.1021/bi1005514. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Jia Y.; Li N.; Sun X.; Ng K.; Ambing E.; Gao M.-Y.; Hua S.; Chen C.; Kim S.; Michellys P.-Y.; Lesley S. A.; Harris J. L.; Spraggon G. Crystal Structure of the ALK (Anaplastic Lymphoma Kinase) Catalytic Domain. Biochem. J. 2010, 430 (3), 425–437. 10.1042/BJ20100609. [DOI] [PubMed] [Google Scholar]
- Mesaros E. F.; Ott G. R.; Dorsey B. D. Anaplastic Lymphoma Kinase Inhibitors as Anticancer Therapeutics: A Patent Review. Expert Opin. Ther. Pat. 2014, 24 (4), 417–442. 10.1517/13543776.2014.877890. [DOI] [PubMed] [Google Scholar]
- Palmer R. H.; Vernersson E.; Grabbe C.; Hallberg B. Anaplastic Lymphoma Kinase: Signalling in Development and Disease. Biochem. J. 2009, 420 (3), 345–361. 10.1042/BJ20090387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou S.-H. I.; Greenbowe J.; Khan Z. U.; Azada M. C.; Ross J. S.; Stevens P. J.; Ali S. M.; Miller V. A.; Gitlitz B. I1171 Missense Mutation (Particularly I1171N) Is a Common Resistance Mutation in ALK-Positive NSCLC Patients Who Have Progressive Disease While on Alectinib and Is Sensitive to Ceritinib. Lung Cancer 2015, 88 (2), 231–234. 10.1016/j.lungcan.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Takahashi K.; Seto Y.; Okada K.; Uematsu S.; Uchibori K.; Tsukahara M.; Oh hara T.; Fujita N.; Yanagitani N.; Nishio M.; Okubo K.; Katayama R. Overcoming Resistance by ALK Compound Mutation (I1171S + G1269A) after Sequential Treatment of Multiple ALK Inhibitors in Non small Cell Lung Cancer. Thorac. Cancer 2020, 11 (3), 581–587. 10.1111/1759-7714.13299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman B.; Groen H. J. M.; Schuuring E.; Hiltermann T. J. N.; Timens W.; den Dunnen W. F. A.; van den Berg A.; ter Elst A.; van Kruchten M.; Kluiver J. L.; Hiddinga B. I.; Hijmering-Kappelle L. B. M.; Groves M. R.; Vilacha J. F.; van Kempen L. C.; van der Wekken A. J. Actionability of On-Target ALK Resistance Mutations in Patients With Non-Small Cell Lung Cancer: Local Experience and Review of the Literature. Clin. Lung Cancer 2022, 23 (2), e104–e115. 10.1016/j.cllc.2021.06.011. [DOI] [PubMed] [Google Scholar]
- Umapathy G.Oncogenic ALK Signaling in Neuroblastoma; University of Gothenburg, 2017. [Google Scholar]
- Toyokawa G.; Hirai F.; Inamasu E.; Yoshida T.; Nosaki K.; Takenaka T.; Yamaguchi M.; Seto T.; Takenoyama M.; Ichinose Y. Secondary Mutations at I1171 in the ALK Gene Confer Resistance to Both Crizotinib and Alectinib. J. Thorac. Oncol. 2014, 9 (12), e86–e87. 10.1097/JTO.0000000000000358. [DOI] [PubMed] [Google Scholar]
- Shiba-Ishii A.; Johnson T. W.; Dagogo-Jack I.; Mino-Kenudson M.; Johnson T. R.; Wei P.; Weinrich S. L.; McTigue M. A.; Walcott M. A.; Nguyen-Phuong L.; Dionne K.; Acker A.; Kiedrowski L. A.; Do A.; Peterson J. L.; Barth J. L.; Yeap B. Y.; Gainor J. F.; Lin J. J.; Yoda S.; Hata A. N. Analysis of Lorlatinib Analogs Reveals a Roadmap for Targeting Diverse Compound Resistance Mutations in ALK-Positive Lung Cancer. Nat. Cancer 2022, 3 (6), 710–722. 10.1038/s43018-022-00399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda S.; Lin J. J.; Lawrence M. S.; Burke B. J.; Friboulet L.; Langenbucher A.; Dardaei L.; Prutisto-Chang K.; Dagogo-Jack I.; Timofeevski S.; Hubbeling H.; Gainor J. F.; Ferris L. A.; Riley A. K.; Kattermann K. E.; Timonina D.; Heist R. S.; Iafrate A. J.; Benes C. H.; Lennerz J. K.; Mino-Kenudson M.; Engelman J. A.; Johnson T. W.; Hata A. N.; Shaw A. T. Sequential ALK Inhibitors Can Select for Lorlatinib-Resistant Compound ALK Mutations in ALK-Positive Lung Cancer. Cancer Discov. 2018, 8 (6), 714–729. 10.1158/2159-8290.CD-17-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyokawa G.; Inamasu E.; Shimamatsu S.; Yoshida T.; Nosaki K.; Hirai F.; Yamaguchi M.; Seto T.; Takenoyama M.; Ichinose Y. Identification of a Novel ALK G1123S Mutation in a Patient with ALK-Rearranged Non-Small-Cell Lung Cancer Exhibiting Resistance to Ceritinib. J. Thorac. Oncol. 2015, 10 (7), e55–e57. 10.1097/JTO.0000000000000509. [DOI] [PubMed] [Google Scholar]
- Pan Y.; Deng C.; Qiu Z.; Cao C.; Wu F. The Resistance Mechanisms and Treatment Strategies for ALK-Rearranged Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 713530. 10.3389/fonc.2021.713530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal K.; Peters M. L. B.; VanderLaan P. A.; Rangachari D.; Kobayashi S. S.; Costa D. B. Activity of Brigatinib in the Setting of Alectinib Resistance Mediated by ALK I1171S in ALK-Rearranged Lung Cancer. J. Thorac. Oncol. 2019, 14 (1), e1–e3. 10.1016/j.jtho.2018.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabencheva T. G.; Lee C. C.; Black G. W.; Donev R.; Christov C. Z. How Does Conformational Flexibility Influence Key Structural Features Involved in Activation of Anaplastic Lymphoma Kinase?. Mol. BioSyst. 2014, 10 (6), 1490–1495. 10.1039/C4MB00141A. [DOI] [PubMed] [Google Scholar]
- He M.; Li W.; Zheng Q.; Zhang H. A Molecular Dynamics Investigation into the Mechanisms of Alectinib Resistance of Three ALK Mutants. J. Cell. Biochem. 2018, 119 (7), 5332–5342. 10.1002/jcb.26666. [DOI] [PubMed] [Google Scholar]
- Salifu E. Y.; Rashid I. A.; Soliman M. E. S. Impact of Compound Mutations I1171N + F1174I and I1171N + L1198H on the Structure of ALK in NSCLC Pathogenesis: Atomistic Insights. J. Biomol. Struct. Dyn. 2023, 41 (10), 4735–4743. 10.1080/07391102.2022.2072390. [DOI] [PubMed] [Google Scholar]
- Liang S.; Wang Q.; Qi X.; Liu Y.; Li G.; Lu S.; Mou L.; Chen X. Deciphering the Mechanism of Gilteritinib Overcoming Lorlatinib Resistance to the Double Mutant I1171N/F1174I in Anaplastic Lymphoma Kinase. Front. Cell Dev. Biol. 2021, 9, 808864. 10.3389/fcell.2021.808864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl; Abraham; Hess; Van Der Spoel. GROMACS 2021.2 Source Code; Zenodo; 2021.
- Wu J.; Tseng Y. D.; Xu C.-F.; Neubert T. A.; White M. F.; Hubbard S. R. Structural and Biochemical Characterization of the KRLB Region in Insulin Receptor Substrate-2. Nat. Struct. Mol. Biol. 2008, 15 (3), 251–258. 10.1038/nsmb.1388. [DOI] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18 (15), 2714–2723. 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Kim S.; Thiessen P. A.; Bolton E. E.; Chen J.; Fu G.; Gindulyte A.; Han L.; He J.; He S.; Shoemaker B. A.; Wang J.; Yu B.; Zhang J.; Bryant S. H. PubChem Substance and Compound Databases. Nucleic Acids Res. 2016, 44 (D1), D1202–D1213. 10.1093/nar/gkv951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croitoru A.; Park S.-J.; Kumar A.; Lee J.; Im W.; MacKerell A. D.; Aleksandrov A. Additive CHARMM36 Force Field for Nonstandard Amino Acids. J. Chem. Theory Comput. 2021, 17 (6), 3554–3570. 10.1021/acs.jctc.1c00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A.; Martin M. J.; Orchard S.; Magrane M.; Agivetova R.; Ahmad S.; Alpi E.; Bowler-Barnett E. H.; Britto R.; Bursteinas B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. 10.1093/nar/gkaa1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30 (16), 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawar S. S.; Rohane S. H. Review on Discovery Studio: An Important Tool for Molecular Docking. Asian J. Res. Chem. 2021, 14 (1), 1–3. 10.5958/0974-4150.2021.00014.6. [DOI] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29 (11), 1859–1865. 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- Altis A.; Otten M.; Nguyen P. H.; Hegger R.; Stock G. Construction of the Free Energy Landscape of Biomolecules via Dihedral Angle Principal Component Analysis. J. Chem. Phys. 2008, 128 (24), 245102. 10.1063/1.2945165. [DOI] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Moreno E. Gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17 (10), 6281–6291. 10.1021/acs.jctc.1c00645. [DOI] [PubMed] [Google Scholar]
- Soda M.; Choi Y. L.; Enomoto M.; Takada S.; Yamashita Y.; Ishikawa S.; Fujiwara S.; Watanabe H.; Kurashina K.; Hatanaka H.; Bando M.; Ohno S.; Ishikawa Y.; Aburatani H.; Niki T.; Sohara Y.; Sugiyama Y.; Mano H. Identification of the Transforming EML4-ALK Fusion Gene in Non-Small-Cell Lung Cancer. Nature 2007, 448 (7153), 561–566. 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- Brenner A. K.; Gunnes M. W. Therapeutic Targeting of the Anaplastic Lymphoma Kinase (ALK) in Neuroblastoma—A Comprehensive Update. Pharmaceutics 2021, 13 (9), 1427. 10.3390/pharmaceutics13091427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K.; Choi Y. L.; Soda M.; Inamura K.; Togashi Y.; Hatano S.; Enomoto M.; Takada S.; Yamashita Y.; Satoh Y.; Okumura S.; Nakagawa K.; Ishikawa Y.; Mano H. Multiplex Reverse Transcription-PCR Screening for EML4-ALK Fusion Transcripts. Clin. Cancer Res. 2008, 14 (20), 6618–6624. 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- Sharma G.; Mota I.; Mologni L.; Patrucco E.; Gambacorti-Passerini C.; Chiarle R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers 2018, 10 (3), 62. 10.3390/cancers10030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam M. W.; Borenäs M.; Lind D. E.; Cervantes-Madrid D.; Umapathy G.; Palmer R. H.; Hallberg B. Alectinib, an Anaplastic Lymphoma Kinase Inhibitor, Abolishes ALK Activity and Growth in ALK-Positive Neuroblastoma Cells. Front. Oncol. 2019, 9, 579. 10.3389/fonc.2019.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.; Li W.; Zheng Q.; Zhang H. A Molecular Dynamics Investigation into the Mechanisms of Alectinib Resistance of Three ALK Mutants. J. Cell. Biochem. 2018, 119 (7), 5332–5342. 10.1002/jcb.26666. [DOI] [PubMed] [Google Scholar]
- Chataigneau T.; Lemoine D.; Grutter T. Exploring the ATP-Binding Site of P2X Receptors. Front. Cell. Neurosci. 2013, 7, 273. 10.3389/fncel.2013.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda Z. H.; Shan Y.; Kim E. T.; Shaw D. E.; Seeliger M. A. A Dynamically Coupled Allosteric Network Underlies Binding Cooperativity in Src Kinase. Nat. Commun. 2015, 6 (1), 5939. 10.1038/ncomms6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Deng R.; Jiang H.; Song H.; Li S.; Shen Q.; Huang W.; Nussinov R.; Yu J.; Zhang J. The Mechanism of ATP-Dependent Allosteric Protection of Akt Kinase Phosphorylation. Structure 2015, 23 (9), 1725–1734. 10.1016/j.str.2015.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hida T.; Nokihara H.; Kondo M.; Kim Y. H.; Azuma K.; Seto T.; Takiguchi Y.; Nishio M.; Yoshioka H.; Imamura F.; Hotta K.; Watanabe S.; Goto K.; Satouchi M.; Kozuki T.; Shukuya T.; Nakagawa K.; Mitsudomi T.; Yamamoto N.; Asakawa T.; Asabe R.; Tanaka T.; Tamura T. Alectinib versus Crizotinib in Patients with ALK-Positive Non-Small-Cell Lung Cancer (J-ALEX): An Open-Label, Randomised Phase 3 Trial. Lancet 2017, 390 (10089), 29–39. 10.1016/S0140-6736(17)30565-2. [DOI] [PubMed] [Google Scholar]
- Mizuta H.; Okada K.; Araki M.; Adachi J.; Takemoto A.; Kutkowska J.; Maruyama K.; Yanagitani N.; Oh-hara T.; Watanabe K.; Tamai K.; Friboulet L.; Katayama K.; Ma B.; Sasakura Y.; Sagae Y.; Kukimoto-Niino M.; Shirouzu M.; Takagi S.; Simizu S.; Nishio M.; Okuno Y.; Fujita N.; Katayama R. Gilteritinib Overcomes Lorlatinib Resistance in ALK-Rearranged Cancer. Nat. Commun. 2021, 12 (1), 1261. 10.1038/s41467-021-21396-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters S.; Camidge D. R.; Shaw A. T.; Gadgeel S.; Ahn J. S.; Kim D.-W.; Ou S.-H. I.; Pérol M.; Dziadziuszko R.; Rosell R.; Zeaiter A.; Mitry E.; Golding S.; Balas B.; Noe J.; Morcos P. N.; Mok T. Alectinib versus Crizotinib in Untreated ALK-Positive Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377 (9), 829–838. 10.1056/NEJMoa1704795. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wang J.; Zhu W. Mutation L1196M-Induced Conformational Changes and the Drug Resistant Mechanism of Anaplastic Lymphoma Kinase Studied by Free Energy Perturbation and Umbrella Sampling. Phys. Chem. Chem. Phys. 2017, 19 (44), 30239–30248. 10.1039/C7CP05418A. [DOI] [PubMed] [Google Scholar]
- McCoy M. D.; Madhavan S. A Computational Approach for Prioritizing Selection of Therapies Targeting Drug Resistant Variation in Anaplastic Lymphoma Kinase. AMIA Summits Transl. Sci. Proc. 2018, 2018, 160–167. [PMC free article] [PubMed] [Google Scholar]
- Ni Z.; Zhang T.-C. Computationally Unraveling How Ceritinib Overcomes Drug-Resistance Mutations in ALK-Rearranged Lung Cancer. J. Mol. Model. 2015, 21 (7), 175. 10.1007/s00894-015-2716-z. [DOI] [PubMed] [Google Scholar]
- Hu J.; Zhang B.; Yao F.; Fu Y.; Chen D.; Li D.; Du N.; Lizaso A.; Song J.; Zhang L.; Li X. Acquired Multiple Mutations ALK I1171N, L1196M and G1202R Mediate Lorlatinib Resistance in EML4-ALK-Rearranged Malignant Pleural Mesothelioma: A Case Report. Ther. Adv. Respir. Dis. 2020, 14, 175346662093577. 10.1177/1753466620935770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Lovly C. M. Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol. Cancer 2018, 17 (1), 58. 10.1186/s12943-018-0782-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P.; Torkamani A.; Schork N. J.; Hegele R. A. Kinase Mutations in Human Disease: Interpreting Genotype-Phenotype Relationships. Nat. Rev. Genet. 2010, 11 (1), 60–74. 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- Medves S.; Demoulin J.-B. Tyrosine Kinase Gene Fusions in Cancer: Translating Mechanisms into Targeted Therapies. J. Cell. Mol. Med. 2012, 16 (2), 237–248. 10.1111/j.1582-4934.2011.01415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelens F. P.; Leonov H.; de Groot B. L.; Grubmüller H. ATP-Magnesium Coordination: Protein Structure-Based Force Field Evaluation and Corrections. J. Chem. Theory Comput. 2021, 17 (3), 1922–1930. 10.1021/acs.jctc.0c01205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainor J. F.; Shaw A. T. Emerging Paradigms in the Development of Resistance to Tyrosine Kinase Inhibitors in Lung Cancer. J. Clin. Oncol. 2013, 31 (31), 3987–3996. 10.1200/JCO.2012.45.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y.; Lu Z.; Li C.; Qi R.; Chang H.; Han L.; Han W. Molecular Dockings and Molecular Dynamics Simulations Reveal the Potency of Different Inhibitors against Xanthine Oxidase. ACS Omega 2021, 6 (17), 11639–11649. 10.1021/acsomega.1c00968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M.-Y.; Li W.-K.; Zheng Q.-C.; Zhang H.-X. Conformational Transition of Key Structural Features Involved in Activation of ALK Induced by Two Neuroblastoma Mutations and ATP Binding: Insight from Accelerated Molecular Dynamics Simulations. ACS Chem. Neurosci. 2018, 9 (7), 1783–1792. 10.1021/acschemneuro.8b00105. [DOI] [PubMed] [Google Scholar]
- Guan J.; Fransson S.; Siaw J. T.; Treis D.; Van den Eynden J.; Chand D.; Umapathy G.; Ruuth K.; Svenberg P.; Wessman S.; Shamikh A.; Jacobsson H.; Gordon L.; Stenman J.; Svensson P.-J.; Hansson M.; Larsson E.; Martinsson T.; Palmer R. H.; Kogner P.; Hallberg B. Clinical Response of the Novel Activating ALK-I1171T Mutation in Neuroblastoma to the ALK Inhibitor Ceritinib. Cold Spring Harbor Mol. Case Stud. 2018, 4 (4), a002550. 10.1101/mcs.a002550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.