

Dear Sir,
Neurodegeneration with brain iron accumulation (NBIA) are the inherited neurodegenerative diseases associated with iron deposition predominantly in the basal ganglia. They are characterized mainly by dystonia, parkinsonism, spasticity, neuropsychiatric abnormalities, cognitive impairment, and optic atrophy/pigmentary retinopathy. Till date, around 10 genetically distinct subtypes of NBIA have been recognized.[1,2] COASY protein-associated neurodegeneration or CoPAN is characterized by oromandibular dystonia, dysarthria, spastic–dystonic paraparesis, parkinsonism, and cognitive impairment. There are a limited number of cases of CoPAN reported worldwide.[3] Hereby, we report a 21-year-old male with spastic paraparesis, dysarthria, dysphagia, and parkinsonism. Whole exome sequencing showed nonsense variant in exon 8 of COASY gene suggestive of CoPAN.
A 21-year-old male patient born out of non-consanguineous parentage with uneventful perinatal history, motor, and language milestone presented with gait difficulty due to stiffness in legs with falls since the age of 8 years. From the age of 12 years, patient developed speech disturbance in the form of slurring of words with nasal twang which was slowly progressive, and by age of 14 years, his speech was not comprehensible. He developed dysphagia to liquids followed by solids since the age of 14 years. He had urgency with urge continence since age of 19 years. Patient has reduced interest in daily activities, decreased social interaction, forgetfulness for recent events, and decline in learning of 1 year duration. He had developed tremulousness of upper limbs while holding objects and inability to write. There were no visual, hearing disturbances. There were no seizures, myoclonus, and tics. He did not have history of jaundice, diurnal fluctuations. There was no family history of similar complaints in parents or siblings. Systemic examination was unremarkable. Neurological examination showed bilateral pes cavus. Cognitive testing showed impairment in attention and memory domains. Speech was slurred, strained quality with merging of syllables. Fundus examination was normal. Saccades and pursuits were normal. Hypomimic facies was present. Motor examination showed spasticity in both lower limbs (grade 3 according to modified Ashworth scale) and grade 1 spasticity tone both upper limbs. The upper limb power was 5/5 and 4/5 in lower limbs (assessment hindered by spasticity). Deep tendon reflexes were grade 2 + in upper limbs and grade 4 + in lower limbs with ill-sustained ankle clonus with absent jaw jerk. Sensory examination was normal. Plantar responses were extensor. Facial dystonia in the form of frontalis contraction and perioral contraction was present. Lingual dystonia was present. There was bilateral finger dystonic posturing. He had bilateral moderate severity of bradykinesia in both upper limbs. There were mild rest tremors in right upper limb (grade 1) with bilateral postural and simple kinetic tremors of upper limbs. Grade 2 postural instability was present. Cerebellar examination was hindered by spasticity of lower limbs. Patient had spastic quadriparesis, symmetrical parkinsonism with dystonia, spastic dysarthria, and cognitive impairment. A clinical diagnosis of complicated hereditary spastic paraplegia versus NBIA spectrum disorder was considered. Complete hemogram, renal, hepatic, and thyroid functions were normal. Serum copper, ceruloplasmin, ferritin, and 24 hrs. of urinary copper levels were normal. Peripheral smear for the presence of acanthocytes was negative. Brain and whole spine magnetic resonance imaging (MRI) showed mild cerebral atrophy, mineralization in bilateral globus pallidus (GP) with no “eye-of-tiger” sign [Figure 1], and mild cord atrophy.
Figure 1.
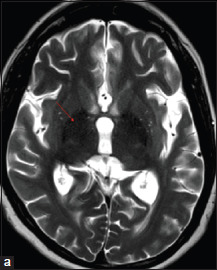
Brain MRI axial T2-weighted image (a) showing hypointensity in globus pallidus (red arrow)
Somatosensory evoked potentials of median nerve and tibial nerves showed normal cortical potentials. Nerve conduction studies were normal. Whole exome sequencing showed a homozygous nonsense variation in exon 8 of the COASY gene (chr17:g. 40717299C > T; Depth: 97x; ENST00000590958.1) that resulted in a stop codon and premature truncation of the protein at codon 482 (p.Arg482Ter). The variant was classified as likely pathogenic. A final diagnosis of NBIA spectrum disorder, CoPAN, was made. He was treated with oral baclofen (30mg/day) and levodopa/carbidopa (300mg/day) but showed no significant improvement.
The most common NBIA subtype in pantothenate kinase-associated neurodegeneration (PKAN). One of the less common NBIA is the CoPAN. CoA synthase is a bifunctional enzyme located in the mitochondrial matrix catalyzing the last two steps in the CoA biosynthetic pathway.[4] The mutation of the gene COASY causing NBIA was first identified in 2014 by Dusi et al.[5] One proband had symptoms of gait difficulty from age of 2 years, poor academic ability, oromandibular dystonia, dysarthria, and spastic dystonic paraparesis, and at age of 25 years, the proband had spastic bradykinetic-rigid syndrome associated with mild dystonia and with distal areflexia in the lower limbs. Brain MRI showed bilateral hypointensity in the GP associated with a central region of hyperintensity in the anteromedial portion. A homozygous missense mutation, a c. 1495C > T transition causing an amino acid change p.Arg499Cys, was noted. Another proband from another family had spastic tetraparesis with moderate mental and language impairment, lost independent ambulation at age 15. Neurological examination at age 17 years showed oromandibular dystonia with dysarthria, spastic–dystonic tetraparesis, and parkinsonian features (rigidity and abnormal postural reflexes). Brain MRI showed hypointensity in the GP. A compound heterozygote for the same mutation, c. 1495C > T (p.Arg499Cys) and for a c. 175C > T transition, resulting in a premature p.Gln59* stop codon was identified.[5] Annesi et al. (2016) from Italy reported same homozygous missense mutation (p Arg499Cys). The proband had gait difficulty at the age of 4 years followed by inattention, hyperactivity, dysarthria, oromandibular dystonia, and dystonic–bradykinetic–rigid syndrome at 17 years of age. Brain MRI showed bilaterally symmetrical, hyperintense signal changes in the anterior medial globus pallidus with surrounding hypointensity on T2-weighted images.[3] Evers et al. (2017) from Germany reported two siblings with CoPAN. They had early age at onset of symptoms of intellectual disability, microcephaly, short stature, ataxia, spastic paraparesis, pyramidal signs, and behavioral abnormalities. Brain MRI showed bilateral T2 hyperintensity and swelling of caudate nucleus, putamen, and thalamus without “eye-of-tiger” sign. They had novel variant c. 641C > T; p.(Ala214Val) and already reported variant c. 1495C > T; p.(Arg499Cys) found in COASY.[6] Our patient had clinical phenotype of pes cavus, spastic tetraparesis, spastic dysarthria, cranial and limb dystonia, parkinsonism, and cognitive impairment similar to reported CoPAN phenotype. Brain MRI showed bilateral T2 hypointensity in GP without “eye-of-tiger” sign similar to other with a novel homozygous nonsense mutation (p.Arg482Ter).
CoPAN is a distinctive NBIA subtype similar to the common subtype PKAN but without pigmentary retinopathy. There are very few reports of CoPAN worldwide, and this case adds to the list. CoPAN should be considered in patients with early-onset spastic tetraparesis with dystonia–parkinsonism, GP hypointensity with or without “eye-of-tiger” sign.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
Authors thank MedGenome Labs Ltd, for Genetic analysis.
REFERENCES
- 1.Hayflick SJ. A Brief history of NBIA gene discovery. J Mov Disord 2023. doi:10.14802/jmd. 23014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Svetel M, Dragašević N, Petrović I, Novaković I, Tomić A, Kresojević N, et al. . NBIA syndromes: A step forward from the previous knowledge. Neurol India 2021;69:1380–8. [DOI] [PubMed] [Google Scholar]
- 3.Annesi G, Gagliardi M, Iannello G, Quattrone A. Mutational analysis of COASY in an Italian patient with NBIA. Parkinsonism Relat Disord 2016;28:150–1. [DOI] [PubMed] [Google Scholar]
- 4.Gregory A, Hayflick SJ. Genetics of neurodegeneration with brain iron accumulation. Curr Neurol Neurosci Rep 2011;11:254–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dusi S, Valletta L, Haack TB, Tsuchiya Y, Venco P, Pasqualato S, et al. . Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am J Hum Genet 2014;94:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evers C, Seitz A, Assmann B, Opladen T, Karch S, Hinderhofer K, et al. . Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger’s eye. Am J Med Genet A 2017;173:1878–86. [DOI] [PubMed] [Google Scholar]