

Abstract
Quantitative proteomics in large cohorts is highly valuable for clinical/pharmaceutical investigations but often suffers from severely compromised reliability, accuracy, and reproducibility. Here, we describe an ultra-high-resolution IonStar method achieving reproducible protein measurement in large cohorts while minimizing the ratio compression problem, by taking advantage of the exceptional selectivity of ultra-high-resolution (UHR)-MS1 detection (240k_FWHM@m/z = 200). Using mixed-proteome benchmark sets reflecting large-cohort analysis with technical or biological replicates (N = 56), we comprehensively compared the quantitative performances of UHR-IonStar vs a state-of-the-art SWATH-MS method, each with their own optimal analytical platforms. We confirmed a cutting-edge micro-liquid chromatography (LC)/Triple-TOF with Spectronaut outperforms nano-LC/Orbitrap for SWATH-MS, which was then meticulously developed/optimized to maximize sensitivity, reproducibility, and proteome coverage. While the two methods with distinct principles (i.e., MS1- vs MS2-based) showed similar depth-of-analysis (∼6700–7000 missing-data-free proteins quantified, 1% protein-false discovery rate (FDR) for entire set, 2 unique peptides/protein) and good accuracy/precision in quantifying high-abundance proteins, UHR-IonStar achieved substantially superior quantitative accuracy, precision, and reproducibility for lower-abundance proteins (a category that includes most regulatory proteins), as well as much-improved sensitivity/selectivity for discovering significantly altered proteins. Furthermore, compared to SWATH-MS, UHR-IonStar showed markedly higher accuracy for a single analysis of each sample across a large set, which is an inadequately investigated albeit critical parameter for large-cohort analysis. Finally, we compared UHR-IonStar vs SWATH-MS in measuring the time courses of altered proteins in paclitaxel-treated cells (N = 36), where dysregulated biological pathways have been very well established. UHR-IonStar discovered substantially more well-recognized biological processes/pathways induced by paclitaxel. Additionally, UHR-IonStar showed markedly superior ability than SWATH-MS in accurately depicting the time courses of well known to be paclitaxel-induced biomarkers. In summary, UHR-IonStar represents a reliable, robust, and cost-effective solution for large-cohort proteomic quantification with excellent accuracy and precision.
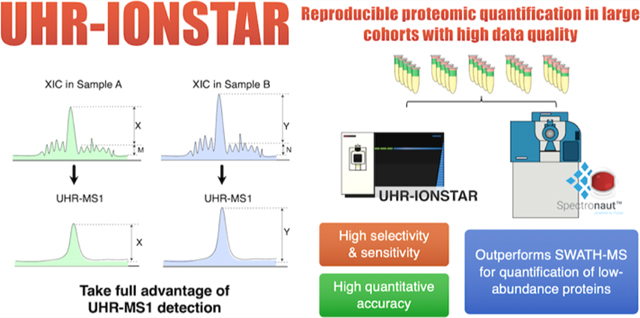
Graphical Abstract
1. INTRODUCTION
For quantitative proteomics in pharmaceutical and clinical investigations, it is often necessary to analyze large numbers of biological samples (N > 30) to achieve sufficient power of statistics and to reduce false-positive discoveries arising from interindividual variability.1 In theory, label-free quantification methods have unlimited replicate capacity and therefore the potential to quantify many samples in the same batch, reducing the intractable problem of batch effect.1−3 Consequently, these methods are often preferred over isotope-labeling approaches for large-cohort quantification.1 Nonetheless, despite strenuous efforts, label-free quantification of large cohort remains challenging. The most prevalent data-dependent acquisition (DDA) methods are susceptible to undersampling problem and therefore suboptimal quantification of low-abundance proteins with high missing data;4,5 moreover, the missing-data problem substantially deteriorates when the sample number increases.6 To enable more reproducible protein measurement across large sample sets, MS2 data-independent acquisition (MS2-DIA) methods were developed, which could reduce missing data down to ∼10% of quantified proteins in sizable cohorts, by triggering MS2 scans sequentially in preset m/z windows.7 MS2-DIA methods, most prominently the SWATH,8 represent a tremendous advance in large-cohort analysis, and a large body of literature has shown its excellent capacity in reproducible protein measurement across relatively large sample sets.7 Yet MS2-DIA often suffers from problems such as difficulty in interpreting MS2 spectra from multiple cofragmented precursors9 and high false positives as well as compromised quantitative accuracy for low-abundance proteins.10
Another promising strategy for large-cohort analysis is MS1 ion current-based. Since the peptide precursor MS1 ion currents are extracted as the sole quantitative feature while the accompanying DDA-MS2 spectra are utilized merely to assign peptide ID to quantitative features, the quantification is completely independent of the stochastic MS2 acquisition and thereby greatly reducing missing data compared to DDA-MS2 methods.1,11 While this strategy opens the possibility for reproducible protein measurement in large biological cohorts, nonetheless, this potential had not been fully explored, largely owing to the lack of an optimal strategy taking full advantage of the high selectivity carried by high-resolution MS1.1 To address this need, we developed an IonStar pipeline enabling accurate, precise quantification of large cohorts with very low levels of missing data and false-positive discovery.2 The method demonstrated the quantification of >7000 unique proteins in a batch of 100 tissue samples with high precision, and 99.8% of these proteins were quantified free of missing data.2 While the method provided reliable quantification of large cohorts as demonstrated in many projects,1 like the majority of quantitative proteomic methods, IonStar too suffers from the ratio compression problem, especially when quantifying low-abundance proteins. Ratio compression roots from interfering signals in complex biological matrices, leading to underestimation of the differences between samples12,13 (Figure 1a). Furthermore, because running multiple technical replicates is not feasible for large sample sets,1 the ability of reliable protein quantification in each sample by a single analysis is essential to attain reliable large-cohort quantification. However, this critical parameter has rarely been assessed or optimized for quantitative proteomics. Finally, it would be highly valuable to perform an extensive, comparative study of various state-of-the-art methods capable of large-cohort proteomic quantification. For example, currently the most advanced Triple-TOF instruments combining with Spectronaut data processing pipeline represent a high-performance SWATH-MS procedure, which have been demonstrated to achieve reproducible, in-depth quantification in relatively large biological sample sets.8 It would be valuable and interesting as well to comparatively investigate the quantitative performances by SWATH-MS vs IonStar; from a technical perspective, these two methods are, respectively, MS2- and MS1-based; though MS2-based methods have generally been considered more sensitive/selective than MS1-based methods owing to the high specificity by tandem mass spectrometry (MS) analysis,5 the advent of high-resolution MS1 measurement is likely a gamechanger: high-resolution MS1 detection enables the extraction of precursor ion currents within an extremely narrow m/z window, resulting in excellent selectivity and sensitivity.1 Moreover, the MS1 signal intensity of a peptide is substantially higher (often by >10-fold) than its MS2 product ions, enabling sensitive MS1-based quantification if the chemical noise could be effectively reduced by high-resolution detection.1 Consequently, an updated comparison of the performances of MS2- vs MS1-based quantification in terms of sensitivity, selectivity, and quantitative accuracy/precision is timely and imperative. From the perspective of the application, such a comparative investigation will provide valuable information to facilitate fit-for-purpose protocol development for large-cohort analysis.
Figure 1.

Development and characterization of ultra-high-resolution (UHR)-IonStar strategy. (a) Rationale of reducing ratio compression by UHR-MS1 measurement. Mixing matrix noises (M, N) and true MS responses (X, Y) for quantification of a low-abundance peptide causes a negative bias in the observed ratio (sample B/A). UHR-MS1 detection, with higher selectivity, would substantially lower matrix noise and alleviate the ratio compression problem. (b) Investigation of MS1-peak-width vs m/z, which indicated that ±5 and ±10 ppm m/z extraction windows are optimal, respectively, for 240 and 120k settings (FWHM@m/z = 200); (c) in an 11-week evaluation, UHR-IonStar achieved very low deviations in m/z measurement (note: not m/z error) and peak areas of peptides, as exemplified by representative peptides; this enables reliable large-cohort quantification; and (d) representative chromatograms showing 240k MS1-resolution improved sensitivity/selectivity for the quantification of low-abundance species.
To address the above issues, here we developed an ultra-high-resolution (UHR, FWHM = 240k@m/z = 200) IonStar strategy, which substantially improves quantitative selectivity and alleviates the ratio compression problem. By combining UHR-MS1 precursor measurement along with an optimized narrow-window extraction of precursor ion currents, this approach represents a significant advance over the previous version of IonStar, which drastically reduces interfering signals and permits accurate protein measurement with exceptional quantitative performances, especially for low-abundance species.1 Furthermore, a graphical user interface (GUI) was implemented. The performances of UHR-IonStar were extensively compared with a state-of-the-art SWATH-MS method, in terms of quantitative accuracy, precision, false-positive discovery of altered proteins, and the performance for a single analysis of each biological sample, using carefully designed benchmark proteomic sample sets reflecting proteomic quantification of large biological cohorts. Consistent with literature,14 a cutting-edge micro-liquid chromatography (LC)/Triple-TOF system was found to markedly outperform the LC/Orbitrap system for SWATH-MS, and the former was therefore selected for SWATH-MS. The procedure was rigorously developed and optimized, including the use of the most advanced micro-LC/Triple-TOF 6600 system, twodimensional (2D) fractionation (strong anion exchange and then high-pH reversed-phase LC (RPLC)) to build a comprehensive, project-specific spectral library, an optimized, robust capillary-LC separation to ensure reproducible quantification, and data analysis by the most advanced Spectronaut Pulsar X software package. Finally, to evaluate the performance of the two methods in the analysis of a biological project, we conducted a time-course quantification of the protein changes in paclitaxel-treated pancreatic cancer cells (N = 36), where the molecular-level biological cascades induced by the drug have been very well characterized by numerous previous studies.
2. EXPERIMENTAL SECTION
2.1. Protein Sample Preparation.
All samples including spike-in benchmark sets and biological samples were subjected to an exhaustive, reproducible extraction with a surfactant cocktail buffer.15 The extracts were then processed with a surfactant-aided-precipitation/on-pellet trypsin-based digestion procedure for robust, efficient, and reproducible digestion among large cohorts.15 Details are given in the Supporting Information (SI).
2.2. UHR-IonStar Quantitative Pipeline.
2.2.1. LC-MS Data Acquisition.
A trapping-nano-RPLC system consisted of a microtrap and a nanocolumn was employed for a 2 h separation of peptides; a selective trapping/delivery strategy was used to ensure continuous, robust analysis of hundreds of samples.11 An Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific), with MS1 survey scan at a resolution of 240 000 (FWHM@m/z = 200), was employed for peptide identification and quantification, under conditions optimized in this study. Details are given in the Supporting Information.
2.2.2. Data Processing and UHR-IonStar v1.4 (Released with This Work).
Protein identifications were based on stringent false discovery rate (FDR) control on the entire data set level and requirement of ≥2 unique peptides/protein (details in the Supporting Information). The UHR-IonStar data processing pipeline was developed based on our previously described IonStar data processing method,2,11 to take full advantage of the 240k-resolution MS1 measurement. The package can be downloaded at https://github.com/JunQu-Lab/UHR-IonStar/releases. The procedure involves using the SIEVE algorithm (Thermo Fisher Scientific) for chromatographic alignment and frame generation, followed by data processing modules using a GUI interface in UHR-IonStar v1.4 released with this publication. The chromatographic alignment was performed with a ChromAlign algorithm for inter-run retention time correction. The quantitative MS1 features were generated in a data-independent manner using the direct ion current extraction strategy, and the defined m/z-RT extraction window was optimized for ultra-high-resolution MS1 measurement via a narrow m/z window width (±5 ppm m/z) coupled with a customized retention time width (e.g., 1.0 min). One sample run in the middle of the batch is typically selected for the reference of peak alignment. Main functions of UHR-IonStar v1.4 include: (i) extraction of quantitative features using narrow, precisely defined windows; (ii) generation of quantitative frame report and quantitative analysis on framethen peptide-level; (iii) dataset-wide normalization and quality control using multivariate outlier removal; and (iv) aggregation of qualified peptides. For data set containing mixed species, a “deconvoluted proteomic quantification” module is implemented for unambiguous quantification of species-specific proteins. A variety of postquantification analysis and visualization features are implemented as well (details in the Supporting Information).
2.3. SWATH-MS Quantitative Pipeline.
To maximize the performance of SWATH-MS, the platform was meticulously optimized and developed. First, a comprehensive spectral library was built via extensive peptide fractionation by strong anion exchange (SAX) followed by high-pH RPLC prior to LC-MS analysis. Second, a unique and highly efficient microflow-LC/Triple-TOF 6600 system (Sciex) with high loading capacity and exceptional reproducibility was developed for sensitive, in-depth, and robust protein quantification in large sample cohorts, with rapid scan speed enabling sequential narrow, variable m/z window acquisition. Third, the data processing pipeline based on Spectronaut Pulsar X (version 12.0.20491.0.14754, Biognosys) was optimized. Details of optimized conditions are given in the Supporting Information.
2.4. Comparison of the SWATH-MS and UHR-IonStar.
We employed two types of benchmark samples to extensively evaluate the performance of SWATH-MS vs UHR-IonStar: (1) Two hybrid proteome benchmark sample sets, both prepared by spiking small, variable amounts of Escherichia coli (E. coli) and yeast proteins (mimicking, low-abundance, altered proteins) into a large, constant background of human cell proteins representing unchanged proteins. The first set has five groups of technical replicates (N = 5/group), while the second set mimics large-cohort analysis of biological replicates, which includes 56 samples, each with a distinct, strategically designed protein composition to reflect individual variation. The detailed design is given in Table S1. Samples were analyzed by LC-MS in a randomized sequence to avoid bias. To determine the significantly different proteins, we employed a cutoff threshold of 30% changes (e.g., 3 times of the median intragroup coefficient of variability (CV)% for technical replicates) and p < 0.05 by Student t-test. The false-positive discovery was rigorously monitored. (2) a very-well-characterized biological system, paclitaxel-treated human cancer cells, where dysregulated biological processes have been very well characterized by numerous publications in decades. A time-course study is performed to comprehensively capture drug-induced biological cascades and to describe the time courses of key dysregulated proteins. The cutoff for the discovery of significantly altered proteins was determined by an Experimental Null method described previously.24 More details are given in the Supporting Information.
2.5. Functional Analysis.
Gene ontology terms of biological process and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were analyzed with functional annotation tool embedded in Database for Annotation, Visualization and Integrated Discovery (DAVID), Bioinformatics Resources v6.8 (https://david.ncifcrf.gov/).
3. RESULTS AND DISCUSSION
3.1. UHR-IonStar Greatly Improved the Performance of MS1-Based Quantification with Drastically Reduced Ratio Compression for Low-Abundance Proteins.
Achieving highly selective peptide detection with minimized interference is a key prerequisite for reliable proteomic quantification but represents a common challenge especially for lower-abundance species. Selectivity-related issues may severely compromise quantification and cause problems such as ratio compression.12 Ratio compression, a prevalent issue for both isotope-labeling and label-free quantification, is caused by the inclusion of signals from contaminant ions with close m/z,10,16 leading to underestimation of quantitative ratios between groups. As illustrated in Figure 1a, this problem “compresses” the calculated intergroup abundance ratio of a changed protein toward unity because the interfering signals are mostly from unchanged matrix proteins. Apparently, achieving high selectivity is the key to minimize interfering signals and thus improving sensitivity and accuracy for low-abundance protein measurement, and alleviating the ratio compression problem. For MS1-based methods, we previously showed that improved selectivity can be realized by higher-resolution peptide precursor measurement, which effectively reduces the inclusion of interfering signals in quantification.1,11,17 Here, we hypothesized that ultra-high-resolution MS1 detection coupled with an optimized quantitative feature extraction would substantially improve the selectivity/sensitivity for quantification of low-abundance proteins as well as enabling more precise peptide matching among samples, which significantly enhances quantitative selectivity, accuracy/precision, and thereby ameliorating ratio compression problem (Figure 1a). To examine this, a UHR-IonStar procedure was developed (cf. Section 2) by employing a 240k resolution (FWHM@m/z = 200, on an ultra-high-field (UHF) Orbitrap) MS1 measurement with an optimized data processing method that effectively extracts MS1 ion currents with a precisely defined, narrow (±5 ppm) m/z window. As shown in Figure 1b, this window width permits selective extraction of MS1 peaks without losing sensitivity for up to m/z = 1500, at the 240k-resolution setting. One important prerequisite for UHR-IonStar is reproducible m/z measurement (e.g., with m/z correction, deviation from the mean of entire set <2.5 ppm is desired), which is essential to ensure reliable, quantitative procurement of peak areas across a large cohort. It was found that m/z deviations of >95% peptides in benchmark samples were within 1.5 ppm during a 11-week evaluation period, as exemplified in Figure 1c. This extraction approach, coupled with the reproducible IonStar trapping-LC/MS method,11 resulted in consistent peptide peak areas procured by UHR-IonStar, with >95% peptides showed deviation % <20% as exemplified in Figure 1c over the 11-week period, which laid a solid foundation for reliable large-cohort quantification. Quantitative performance of the method was evaluated with a mixed-proteome benchmark sample set (i.e., the technical replicate set in Table S1a), where the human protein content remains constant (70%), and E. coli and yeast protein levels are low and vary among the five groups of samples (A−G). The low-abundance yeast and E. coli proteins simulate the quantitative behaviors of regulatory proteins, which are mostly of lower abundance in a typical biological system. Each group contains five technical replicates (N = 35, Table S1a).
We first compared the quantitative performance of UHR-IonStar (240k resolution and UHR-IonStar v1.4 with ±5 ppm m/z extraction window) against the previous version of IonStar (120k resolution with ±10 ppm m/z window by previous IonStar package2,11). Similar numbers of missing-data-free proteins (≥2 unique peptides/protein, 1% protein identification FDR for the entire set, same criteria below), 6657 for UHR-IonStar and 6606 for IonStar, were, respectively, quantified across 35 runs, suggesting that UHR-IonStar did not result in a significant decrease of quantifiable proteins despite slightly lower scan numbers per sample (<5%). This is likely owing to the fact that quantification by UHR-IonStar is based on MS1 signal and independent of MS2 spectra. UHR-IonStar substantially decreased chemical interference and enhanced sensitivity and selectivity for low-abundance peptides, as exemplified in Figures 1d and S1. Apparently, the UHR-IonStar provided a more accurate quantification of both E. coli and yeast proteins in each comparison (Figures 2a and S2a). Moreover, the error of median ratio by UHR-IonStar was lower in all comparisons; for example, IonStar showed significant ratio compression (error % = −27.24%, measured-median-ratio = 5.09 vs the true-value = 7.0) when comparing E. coli proteins in group G vs A, while UHR-IonStar yielded a much more accurate ratio of 6.80 with error %= −2.96% (Figure 2a). The comparison of quantifying yeast proteins showed a similar trend (Figure S2b, SI). These results indicated that UHR-IonStar effectively alleviates the ratio compression problem. The run-to-run reproducibility of quantified proteins was assessed by correlating quantitative values of proteins from two replicate analyses as defined previously.2,11 Reproducibility was excellent for both methods (R2 ∼ 0.99), while UHR-IonStar showed further improved reproducibility for lower-abundance proteins (Figure S2c, SI). The median intragroup CV for protein quantification in technical replicates by the UHR-IonStar was 7%, lower than that by IonStar (12%, Figure 2b), suggesting that the enhanced selectivity by UHR-IonStar improves quantitative precision. Furthermore, we evaluated the capacity of discovering the significantly altered proteins by the two methods. Clearly, UHR-IonStar discovered more true positives (i.e., E. coli proteins and yeast proteins) and fewer false positives (i.e., human proteins) as significantly different across the six comparisons (Figure 2c). Finally, we used p-values to calculate true-positive rate (TPR) and false-positive rate (FPR) of the quantified proteins on a receiver operating characteristic (ROC) curve (Figure 2d). UHR-IonStar achieved higher areas under curve (AUC) in each comparison, indicating a better sensitivity/selectivity. Overall, our studies showed UHR-IonStar provided much ameliorated ratio compression, as well as markedly improved quantitative performance in terms of sensitivity, selectivity, and quantitative accuracy/precision.
Figure 2.

Quantitative performance of IonStar (120k-MS1) vs UHR-IonStar (240k-MS1). Evaluation was performed using a mixed-proteome benchmark sample set (Table S1). (a) log2 ratios of quantified E. coli proteins of each comparison (horizontal solid line/theoretical ratio), (b) coefficient variations of quantified proteins, (c) true and false positives of significantly altered proteins, (d) ROCs of quantified proteins by the two methods. One-sided Student t-test and one-sided DeLong’s test were employed to determine the statistical significance of comparing the two methods on the CV and ROC plots, respectively. ***, p < 0.001.
3.2. Comparative Evaluations of Quantitative Performances by UHR-IonStar and a State-of-the-Art SWATH-MS Method.
As discussed above, a comparative investigation of MS1-based vs MS2-DIA methods, both capable of reproducible large-cohort analysis, has yet been conducted. Here, we extensively and unbiasedly compared the UHR-IonStar with a state-of-the-art SWATH-MS method, on which a large body of literature has showed much-improved quantitative quality over MS2-DDA-based methods.8,18 For SWATH-MS, analytical platforms based on either Orbitrap or Triple-TOF are previously reported.7,19 Using a cutting-edge Spectronaut package for data analysis, we extensively compared the performances of the two platforms after individualized optimization: the same nano-LC/UHF-Orbitrap system used for UHR-IonStar vs a cutting-edge micro-LC/Triple-TOF 6600 system. With the benchmark sets in Figure 3, it was found that the micro-LC/Triple-TOF 6600 platform clearly outperformed the nano-LC/UHF-Orbitrap system for SWATH-MS, in that the former quantified >30% more missing-data-free proteins, while the commonly quantified proteins by the two platforms showed comparable accuracy, precision, and false-positive discoveries. This result agrees with most recent studies showing that an advanced Triple-TOF is ideal for SWATH-MS owing to its excellent absolute sensitivity and fast scan speed, which permits selective acquisition of quantitative features via narrow, variable acquisition windows and more data points per LC peak.14 As the comparison of the LC-MS platforms for SWATH-MS is out-of-scope of this study, detailed results and discussion are not shown.
Figure 3.

Comparison of quantitative performances of UHR-IonStar vs SWATH-MS in analyzing technical (a–d) and biological (e–g) replicates. (a) Tri-proteome (E. coli, yeast, human) technical replicate benchmark set, with low, variable amount of E. coli proteins (more details are in Table S1a, groups A–E); (b) Reproducibility examined by correlating protein quantitative values from replicate LC-MS runs of the same pooled sample. The R2 was shown for proteins in the highest 75% and lowest 25% in abundance. (c) Log2 ratio of quantified low-abundance E. coli proteins (lowest 25% in abundance) in technical replicates by UHR-IonStar and SWATH-MS. (d) Intragroup coefficient variations of low-abundance proteins quantified by UHR-IonStar and SWATH-MS in technical replicates, ***One-sided Student t-test p < 0.001. (e) Simulated (i.e., mimicking) biological replicate set consisting of seven groups A′–G′, eight samples/group, each with different E. coli protein contents (N = 56 distinct samples in total, Table S1b). (f) Median ratios (against the mean of group A′) of low-abundance E. coli proteins in each individual sample by a single analysis with UHR-IonStar or SWATH-MS. Black lines: theoretical ratios in individual samples. Quantitative errors are shown in Figure S4. (g) ROCs of low-abundance proteins are quantified by UHR-IonStar and SWATH-MS for comparison of each simulated biological replicate groups. (***, One-sided DeLong’s test p < 0.001 comparing the two methods).
3.2.1. Development and Optimization of a State-of-the-Art SWATH-MS Strategy to Achieve Maximal Proteomic Coverage and Quantitative Accuracy/Precision.
Three measures were taken: (1) a highly comprehensive, project-specific spectral library was established with an extensive 2D high-performance liquid chromatography (HPLC) fractionation (strong anion exchange followed by high-pH-reversed-phase separation into 72 concatenated fractions) prior to LC-MS identification (cf. Supporting Information). Under stringent cutoff criteria, this high-quality spectral library contains 10 249 protein groups, a >50% increase over a previously published work on a very similar mixed-proteome sample set.20 Such a high-quality library markedly improves depth, sensitivity, and reliability of proteomic quantification; (2) with the emphasis on dynamic precursor isolation windows, critical SWATH-MS parameters were meticulously optimized. The ultrafast scan speed on the state-of-the-art Triple-TOF 6600 instrument enabled narrow, variable window widths, which markedly enhance spectral matching performance of SWATH-MS. (3) A capillary-LC separation system was vigorously optimized, which greatly facilitated sensitive, reproducible, and robust SWATH-MS analysis of large cohorts. Compared to a nano-flow LC, this capillary-LC approach achieved similar sensitivity by taking advantage of its high loading capacity using a strategy we described recently,21 with much-improved robustness and exceptional separation performances, which are important for large-cohort analysis. Moreover, significantly improved quantitative performances were observed using capillary-LC over the nano-LC on the Triple-TOF platform. More details are given in the Supporting Information section.
3.2.2. Comparing Quantitative Performances by UHR-IonStar vs SWATH-MS Using Mixed-Proteome Sets Mimicking Large-Cohort Quantification Using Technical or Biological Replicates.
First, the comparison was conducted by analyzing technical replicates (N = 5/group) of the mixed-proteome set described in Figure 3a and Table S1a (group A−E). Totally 6802 proteins and 6657 proteins were quantified with stringent false-positive control, ≥2 unique peptides per protein and without missing data across all runs by SWATH-MS and UHR-IonStar, respectively. The SWATH-MS method optimized here achieved much-improved depth than these previously reported on similar benchmark samples.20 To ensure an unbiased comparison, we evaluated the quantitative performance of the 5504 proteins commonly quantified by both strategies. While the run-to-run reproducibility for higher-abundance proteins (i.e., the highest 75% proteins in abundance) is good for both methods, UHR-IonStar showed much-improved reproducibility for lower-abundance proteins. For proteins of the lowest 25% in abundance, R2 = 0.92−0.94 by UHR-IonStar vs R2 = 0.3−0.4 by SWATH-MS, as exemplified in Figure 3b. As lower-abundance proteins encompass the majority of key regulators/markers,22,23 we then focused the comparison on the lowest 25% proteins (i.e., the lower quartile) in abundance. The higher reproducibility by UHR-IonStar resulted in improved quantitative precision, with a median CV of 10.7% for low-abundance proteins vs 17% by SWATH-MS (p < 0.001, Figure 3d). For low-abundance E. coli and yeast proteins, UHR-IonStar achieved better quantitative accuracy (<5% median error) compared to SWATH-MS (up to 15%) as shown in Figure 3c. Meanwhile, both methods performed well for higher-abundance proteins (Figure S3a–c). Additionally, the ability of UHR-IonStar to discover significantly different proteins was superior, as shown in Figure S3d.
While benchmarking using technical replicates is valuable, nonetheless, running technical replicates is not feasible in a typical large-cohort analysis, where each biological sample is only measured once owing to the limitation of throughput. Therefore, it is critical to evaluate the quantitative performance for a single analysis of each biological sample across a large cohort. Here, we devised a set mimicking the analysis of a large cohort of biological samples, containing 56 distinct mixed-proteome samples (seven groups A′–G′, N = 8/group, Figure 3e), each sample with strategically varied levels of E. coli and yeast proteins (Table S1b, SI). With the emphasis on lower-abundance proteins, we compared the accuracy of quantifying protein ratios in individual samples, as exemplified in Figure 3f. In all samples, UHR-IonStar achieved substantially more accurate one-time-measurement of low-abundance E. coli proteins than SWATH-MS, while the latter showed an apparent ratio compression problem in most samples. As shown in Figure S4a, UHR-IonStar showed markedly lower quantitative error % (<13% across individual samples), compared to SWATH-MS (up to 48% error) across the set.
In this set, all E. coli proteins are true positives because the levels of E. coli proteins in groups B′–G′ are designed to be significantly higher (p-value of one-sided Student t-test <0.05) than group A′. We evaluated the performance of discovering lower abundance, significantly different proteins by UHR-IonStar vs SWATH-MS via ROC curves, as shown in Figure 3g. Cutoffs were set to p-value <0.05 and ≥30% changes, as determined by an Experimental Null method we described previously.24 Again, for the analysis of this simulated biological set, UHR-IonStar showed superior performance in the sensitive and selective discovery of differential proteins (as evident by higher AUC of UHR-IonStar, p < 0.001 by one-sided DeLong’s test), especially for comparison with ≤2.5-fold differences. Moreover, UHR-IonStar discovered more true positives than SWATH-MS for lower-abundance proteins, as shown in Figure S4b.
For the quantification of higher-abundance proteins (i.e., the highest 75% proteins in abundance) in technical and simulated biological replicates, both UHR-IonStar and SWATH-MS achieved accurate quantification (data not shown).
3.2.3. Comparison of UHR-IonStar and SWATH-MS in Quantification of a Well-Characterized Biological Sample Set: A Time-Course Study of Drug-Responsive Proteins Induced by Paclitaxel in Cancer Cells.
Apparently, the best approach to compare quantitative proteomic methods is by the analysis of real biological sample sets, but is difficult because the relative expression levels of proteins in such sets are unknown. Here, we circumvent this issue using a very-well-characterized pharmaceutical system for benchmark: cancer cells treated by paclitaxel, an extremely well-researched antimitotic agent that inhibits cell replications by disrupting mitotic spindle formation and arresting cell cycle/growth with subsequent apoptosis. Since the year of 2000, >30 000 papers on its biological effects have been published, rendering it one of the most-studied drugs.25,26 The main paclitaxel-induced biological cascades have been extensively characterized.27 Here, we employed paclitaxel-treated pancreatic cancer cells to compare UHR-IonStar and SWATH-MS. To comprehensively capture the drug-induced biological cascades that tend to occur at various time points, we designed a time-course study consisted of nine time groups (treatment for 1–72 h and vehicle control) with four biological replicates per point (N = 36 in total, shown in Figure 4a).
Figure 4.

Comparison of UHR-IonStar and SWATH-MS in paclitaxel-treated pancreatic cancer cells. (a) Very-well-characterized system for the benchmark of methods: time-course study in paclitaxel-treated cancer cells, with nine time points and four biological replicates per point (N = 36 in total), more details in the Experimental Procedure section, SI. (b) Discovery of 12 biological processes and 8 signaling pathways that are well recognized as altered by paclitaxel, respectively, by SWATH-MS and UHR-IonStar. Significance scores are shown in color. (c) Quantification of the time courses of tubulin isoforms by SWATH and UHR-IonStar. The tubulins are well known to be consistently induced by paclitaxel, and that tubulin α and β form 1:1 dimer, where the depolymerization is inhibited by paclitaxel. Therefore, a smooth, consistently up induction, with overlapped trends of tubulin α and β isoforms, should be observed.
UHR-IonStar and SWATH-MS, respectively, quantified 6021 and 5355 missing-data-free proteins (≥2 unique peptides/protein), as shown in Tables S2 and S3. UHR-IonStar showed much lower intragroup CV% than SWATH (p < 0.001 by one-sided t-test, Figure S5). While the altered proteins of high abundance are commonly discovered by both methods with excellent ratio correlation (e.g., R2 = 0.8–0.9 shown in Figure S6a), the discovery of significantly changed, lower-abundance proteins (i.e., most regulatory proteins) is quite different by the two methods, showing poor correlation of intergroup ratios (Figure S6b). To objectively compare the capacity to accurately reflect drug-induced protein changes, two aspects are assessed: First, we evaluated the two methods’ ability to discover protein changes implicated in the widely recognized biological cascades induced by paclitaxel. From many research articles on the mechanism of actions of paclitaxel, 12 most-recognized biological processes associated with paclitaxel-related toxicity were summarized in three categories: microtubule/cell cycle regulation, DNA regulation, cell proliferation/apoptosis,28 as well as eight well-known signaling pathways that are profoundly dysregulated by the drug. Based on the analysis of biological process and KEGG pathways with a functional annotation tool embedded in DAVID, both methods correctly identified dysregulated cell proliferation and apoptosis processes (more details described in Section 2.5). Nevertheless, while UHR-IonStar discovered all three dysregulated processes in paclitaxel-induced DNA regulations,29 SWATH-MS discovered only one with a suboptimal p-value as calculated by DAVID (Figure 4b). Furthermore, UHR-IonStar was able to discover all six biological processes as dysregulated in the “cell cycle regulation” category, which has been broadly recognized following paclitaxel treatment,30 whereas SWATH-MS identified only three processes. Most importantly, microtubule depolymerization, the key mechanism of action by paclitaxel,31 was discovered by UHR-IonStar with an excellent EASE score (equivalent to p-value <0.05), but surprisingly, not by SWATH-MS (Figure 4b). Among the eight KEGG pathways, UHR-IonStar discovered seven as dysregulated in paclitaxel-treated cells with high confidence, while SWATH-MS discovered only one (Figure 4b).
Second, we compared the capacity of the two methods to accurately/quantitatively describe the expression–time profiles of well-known dysregulated proteins. The consistency of the time profiles of significantly altered proteins, where each time point group is independently quantified, can faithfully reflect the quantitative accuracy/precision of methods.32 It has been well known that paclitaxel inhibits microtubule depolymerization and triggers the synthesis of both tubulin α and β, an important negative feedback loop of drug-induced microtubule damage.33 As the induction is constant during paclitaxel treatment,33 a smooth, continuous increase of tubulins over time is warranted. Moreover, because tubulin α and β present at 1:1 ratio in a heterodimer and that the drug effectively inhibits decoupling of this dimer, the relative expression vs time profiles of both tubulin α and β, including their isoforms, should mostly overlap. Though both methods indicated roughly ∼60% elevation of tubulins after 72 h of incubation, nonetheless, the measured temporary profiles were quite different (Figure 4c). The time courses depicted by SWATH-MS were quite bumpy with many fluctuations, and the induction–time profiles of various tubulin isoforms did not overlap well. By comparison, the time courses of tubulins measured by UHR-IonStar showed well-defined trends of upregulation, which were much smoother than these by SWATH-MS (Figure 4c), agreeing well with the expected continuous synthesis and accumulation of tubulins after paclitaxel-induced microtubule stabilization.33 Furthermore, all of the α and β isoforms of tubulin showed largely superimposing time profiles of consistent induction, which again is in line with the drug action.
4. CONCLUSIONS
For pharmaceutical/clinical proteomic investigation, the capacity for reliable large-cohort analysis is highly desirable but difficult to achieve owing to daunting technical challenges.1 Recently, MS1-based quantitative methods showed high promise for reproducible/accurate protein measurement of a large number of samples within one analytical batch, and thereby eliminating the intractable problems associated with batch effects.1−3 Here, we described a new MS1-based strategy, UHR-IonStar, which takes advantage of the extraordinary selectivity of ultra-high-resolution MS1 measurement, achieving high run-to-run reproducibility, low interference, and excellent quantitative precision and accuracy, and alleviating the ratio compression problem that is prevalent in quantitative proteomics. Comparing to the previous version of IonStar, the UHR-IonStar substantially improved quantitative performances with only a very minor (<2%) decrease of quantified proteins. It is important to note that a UHF-Orbitrap instrument is necessary to achieve optimal selectivity for UHR-IonStar.
We then compared UHR-IonStar with a state-of-the-art SWATH-MS method. While both techniques can be adapted to large-cohort analysis in one batch, their principles for quantification are quite distinct: MS1- vs MS2-based. On one hand, MS2-based methods carry the high selectivity/sensitivity intrinsic to tandem-MS analysis; on the other hand, the MS1 signal intensity of a peptide precursor is >10-fold higher than its product ions,1 potentially enabling more sensitive quantification if high specificity can be achieved by a high-resolution MS. Given the recent advances in ultra-high-resolution MS, a comparative study of the MS1- vs MS2 methods would be of immense interest and urgently needed as well. Here, the two methods were comprehensively compared; it is important to note that this study is not aimed at comparing only the data processing pipelines of UHR-IonStar vs SWATH-MS using the same LC-MS platform, but rather comparing the two quantitative strategies under their own optimal LC-MS platforms. In this study, it was found for SWATH-MS, an optimized, advanced micro-LC/Triple-TOF 6600 platform outperforms the nano-LC/Orbitrap system, which also corroborates with recent reports.14
The comparison was conducted in three steps: first, the basic quantitative parameters of the two methods were benchmarked by measuring mixed-proteome samples in technical replicates; second, with a strategically designed large sample cohort consisting of 56 distinct mixed-proteome samples in seven groups, we assessed the accuracy, precision, and ability to discover changed proteins across the groups, as well as the accuracy in a single analysis of each individual sample, a particularly important parameter for large-cohort quantification yet inadequately investigated.
Both methods performed well for quantification of higher-abundance proteins; nonetheless, for lower-abundance proteins that encompass the majority of key regulatory proteins and biomarkers,22,23 UHR-IonStar showed substantially better quantitative reproducibility, accuracy, and precision, as well as superior performance in protein measurement by a single analysis of an individual biological sample. Moreover, UHR-IonStar demonstrated markedly better sensitivity/selectivity in discovering altered proteins among groups, which is more pronounced for protein changes ≤2.5-fold, a range where the majority of discovered protein changes in biological systems appear to fall into, as evident in our extensive literature research (examples in Figure S7). Again, the superior quantitative performance of UHR-IonStar roots from effective utilization of the high sensitivity/selectivity by ultra-high-resolution measurement.
Third, the two methods were further compared in a sizable time-course investigation of drug-responsive proteins in cancer cells after paclitaxel treatment (N = 36). Though the exact folds of changes of altered proteins are unknown, the biological processes, signaling pathways, and key proteins dysregulated by paclitaxel have been extremely well characterized by decades of extensive research, rendering it an excellent benchmark system for evaluating quantitative proteomic methods. Despite UHR-IonStar and SWATH-MS commonly discovered a number of higher-abundance proteins as significantly altered with good correlations, the discovery of lower-abundance, altered proteins by the two methods are quite different. Comparing to SWATH-MS, UHR-IonStar was able to discover substantially more biological processes and signaling pathways, which are well recognized to be induced by paclitaxel. Moreover, UHR-IonStar showed much better precision and accuracy in the quantitative depiction of the time courses of tubulin α/β isoforms, which are well known to be constantly upregulated by paclitaxel.
In summary, UHR-IonStar represents a practical and reliable tool for large-cohort proteomic quantification with high accuracy, precision, and excellent sensitivity/selectivity for the discovery of significantly altered proteins. Comparing to SWATH-MS, UHR-IonStar showed better performance in quantification of lower-abundance proteins and the capacity to discover relatively moderate changes, without the need of building a library. Potential advantages of SWATH-MS include: first, once the library is built for SWATH-MS, data analysis is less computation-intensive; by comparison, a computer with 128 GB memory is recommended when performing large-cohort analysis with UHR-IonStar; running time is ∼2 h/sample for a typical desktop personal computer (PC). Second, the SWATH-MS method appeared to be more tolerant to low-quality, poorly characterized samples and large variation and/or errors in sample preparation, as suggested by our pilot study (data not shown). The results in this study are important to inform and rationalize the planning of large-cohort quantification. Finally, procedures described here, such as using simulated biological replicate sets and well-characterized biological systems to benchmark quantitative methods, are valuable for the technical development/evaluation of quantitative proteomic techniques.
Supplementary Material
ACKNOWLEDGMENTS
This work is partially supported by NIH grants DK124020 (J.Q.) and HL103411 (J.Q.), a DOD grant W81XWH1910805 (J.Q.) and a CPT consortium grant. AbbVie partially sponsored and funded the study; contributed to the design; participated in the collection, analysis, and interpretation of data, and in writing, reviewing, and approval of the final publication.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c05002.
Experimental procedure; results and discussions; improvement of sensitivity/selectivity for quantification of low-abundance peptides by ultra-high-resolution (UHR, 240k@m/z = 200) MS1 measurement (Figure S1); evaluation of quantitative data quality by IonStar (120k) and UHR-IonStar (240k) (Figure S2); evaluation of quantitative performances of UHR-IonStar and SWATH-MS in technical replicates (Figure S3); comparison of UHR-IonStar and SWATH-MS in quantifying the simulated biological sample set (N = 56) (Figure S4); intragroup CVs of quantified proteins by UHR-IonStar and SWATH-MS in each treatment group, as shown with box plots (Figure S5); correlations of the ratios of significantly altered proteins (discovered by at least one method) in paclitaxel-treated sample set, as measured by UHR-IonStar vs SWATH-MS (Figure S6); percentage of discovered significantly changed proteins with ≤2.5-fold changes in proteomic literature (different methods summarized from randomly selected papers) (Figure S7); designs of mix-proteome benchmark sets reflecting quantification with technical replicates and biological replicates (Table S1) (PDF)
Quantitative proteins_UHR-IonStar_Paclitaxel-treated cells (Table S2) (XLSX)
Quantitative proteins_SWATH-MS_Paclitaxel-treated cells (Table S3) (XLSX)
Quantitative proteins_120K_IonStar (Table S4) (XLSX)
Quantitative proteins_UHR-Ionstar_technical replicates (Table S5) (XLSX)
Quantitative proteins_SWATH-MS_technical replicates (Table S6) (XLSX)
Quantitative proteins_UHR-Ionstar_biological replicates (Table S7) (XLSX)
Quantitative proteins_SWATH-MS_biological replicates (Table S8) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c05002
The authors declare no competing financial interest.
Contributor Information
Xue Wang, Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States; AbbVie Bioresearch Center, Worcester, Massachusetts 01605, United States.
Liang Jin, AbbVie Bioresearch Center, Worcester, Massachusetts 01605, United States.
Chenqi Hu, AbbVie Bioresearch Center, Worcester, Massachusetts 01605, United States.
Shichen Shen, Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, New York 14214, United States.
Shuo Qian, Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States.
Min Ma, Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States.
Xiaoyu Zhu, Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, New York 14214, United States.
Fengzhi Li, Department of Pharmacology and Therapeutics, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States.
Jianmin Wang, Department of Biostatistics and Bioinformatics, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States.
Yu Tian, AbbVie Bioresearch Center, Worcester, Massachusetts 01605, United States.
Jun Qu, Department of Pharmaceutical Sciences, University at Buffalo, SUNY, Buffalo, New York 14214, United States; Department of Cell Stress Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York 14203, United States.
REFERENCES
- (1).Wang X; Shen S; Rasam SS; et al. Mass Spectrom. Rev. 2019, 38, 461–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Shen X; Shen S; Li J; et al. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E4767–E4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brenes A; Hukelmann E; Bensaddek D; et al. Mol. Cell. Proteomics 2019, 18, 1967–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Geib T; Sleno L; Hall RA; et al. J. Am. Soc. Mass Spectrom. 2016, 27, 1404–1410. [DOI] [PubMed] [Google Scholar]
- (5).Tu C; Li J; Sheng Q; et al. J. Proteome Res. 2014, 13, 2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bruderer R; Bernhardt OM; Gandhi T; et al. Mol. Cell. Proteomics 2015, 14, 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Collins BC; Hunter CL; Liu Y; et al. Nat. Commun. 2017, 8, No. 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ludwig C; Gillet L; Rosenberger G; et al. Mol. Syst. Biol. 2018, 14, No. e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gillet LC; Navarro P; Tate S; et al. Mol. Cell. Proteomics 2012, 11, No. O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wu JX; Song XM; Pascovici D; et al. Mol. Cell. Proteomics 2016, 15, 2501–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shen X; Shen S; Li J; et al. J. Proteome Res. 2017, 16, 2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ting L; Rad R; Gygi SP; et al. Nat. Methods 2011, 8, 937–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Karp NA; Huber W; Sadowski PG; et al. Mol. Cell. Proteomics 2010, 9, 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rajski Ł; Beraza I; Ramos MJG; et al. Anal. Methods 2020, 12, 2027–2038. [Google Scholar]
- (15).Shen SC; An B; Wang X; et al. Anal. Chem. 2018, 90, 10350–10359. [DOI] [PubMed] [Google Scholar]
- (16).Fuller HR; Morris GE Quantitative Proteomics Using iTRAQ Labeling and Mass Spectrometry. In Integrative Proteomics; Leung HC; Man TK; Flores R, Eds.; IntechOpen: London, U.K., 2012; pp 347–362. [Google Scholar]
- (17).Tu CJ; Sheng QH; Li J; et al. J. Proteome Res. 2014, 13, 5888–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Krasny L; Bland P; Kogata N; et al. J. Proteomics 2018, 189, 11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Amon S; Meier-Abt F; Gillet LC; et al. Mol. Cell. Proteomics 2019, 18, 1454–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Navarro P; Kuharev J; Gillet LC; et al. Nat. Biotechnol. 2016, 34, 1130–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhang M; An B; Qu Y; et al. Anal. Chem. 2018, 90, 1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Misra S; Kumar A; Kumar P; et al. Proteomics Clin. Appl. 2017, 11, No. 1700007. [DOI] [PubMed] [Google Scholar]
- (23).Zhang C; Shi Z; Han Y; et al. J. Proteome Res. 2019, 18, 461–468. [DOI] [PubMed] [Google Scholar]
- (24).Shen X; Hu Q; Li J; et al. J. Proteome Res. 2015, 14, 4147–4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kampan NC; Madondo MT; McNally OM; et al. BioMed Res. Int. 2015, 2015, No. 413076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Gan L; Chen S; Wang YW; et al. Cancer Res. 2009, 69, 8386–8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Weaver BA Mol. Biol. Cell 2014, 25, 2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ren XL; Zhao BB; Chang HJ; et al. Mol. Med. Rep. 2018, 17, 8289–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Branham MT; Nadin SB; Vargas-Roig LM; et al. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2004, 560, 11–17. [DOI] [PubMed] [Google Scholar]
- (30).Choi YH; Yoo YH Oncol. Rep. 2012, 28, 2163–2169. [DOI] [PubMed] [Google Scholar]
- (31).Xiao H; Verdier-Pinard P; Fernandez-Fuentes N; et al. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 10166–10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Nouri-Nigjeh E; Sukumaran S; Tu C; et al. Anal. Chem. 2014, 86, 8149–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Gasic I; Boswell SA; Mitchison TJ PLoS Biol. 2019, 17, No. e3000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.