

Abstract
The central dogma of molecular biology dictates the general flow of molecular information from DNA that leads to a functional cellular outcome. In skeletal muscle fibers, the extent to which global myonuclear transcriptional alterations, accounting for epigenetic and post-transcriptional influences, contribute to an adaptive stress response is not clearly defined. In this investigation, we leveraged an integrated analysis of the myonucleus-specific DNA methylome and transcriptome, as well as myonuclear small RNA profiling to molecularly define the early phase of skeletal muscle fiber hypertrophy. The analysis of myonucleus-specific mature microRNA and other small RNA species provides new directions for exploring muscle adaptation and complemented the methylation and transcriptional information. Our integrated multi-omics interrogation revealed a coordinated myonuclear molecular landscape during muscle loading that coincides with an acute and rapid reduction of oxidative metabolism. This response may favor a biosynthesis-oriented metabolic program that supports rapid hypertrophic growth.
Keywords: RNA sequencing, small RNA sequencing, RRBS, mitochondrial respiration, oxidative metabolism, epigenetics
Graphical Abstract
Graphical Abstract.

Introduction
Skeletal muscle cells (myofibers) are syncytia containing hundreds of nuclei (myonuclei) in an expansive cytoplasmic volume.1,2 A unique feature of myofibers is the remarkable capacity for growth in response to stimuli such as mechanical overload (MOV).3 Several mechanisms underlie the cellular and molecular responses to MOV that contribute to skeletal muscle hypertrophy, including increases in mammalian target of rapamycin complex 1 (mTORC1) signaling, ribosome biogenesis, and protein synthesis rates.3 In addition to these well-characterized processes, emerging evidence suggests a role for novel mechanisms, including epigenetic changes and alterations to microRNA (miRNA) expression.3
The role of miRNAs in gene repression through the RNA-induced silencing complex (RISC)-mediated targeting of transcripts in the cytoplasm is well established. However, mature miRNAs are also localized to the nucleus. Emerging work shows miRNA post-transcriptional regulatory functions within the nucleus.4–6 Several studies identified a cytoplasmic-nuclear transport mechanism for mature miRNAs7,8 and direct miRNA targeting of nuclear-retained noncoding transcripts.9–11 Moreover, transcriptional gene silencing has also been reported, whereby miRNAs guide the RNA-induced transcriptional silencing (RITS) complex to promoter-associated transcripts, altering DNA methylation and chromatin structure.12–15 Specific miRNAs have since been found to play a role in the formation of epigenetic silencing complexes to induce hypermethylation-mediated transcriptional gene regulation in various cell types.16–18 However, the role of myonuclear miRNAs in skeletal muscle adaptation is not understood.
We previously investigated the myonuclear DNA methylome and transcriptome, separately, in response to an acute hypertrophic stimulus in the plantaris muscles of mice.19,20 Specifically, myonuclei were labeled with green fluorescent protein (GFP) before MOV and purified following 72 h of MOV by fluorescent-activated nuclei sorting (FANS). We then isolated DNA and RNA for myonuclear reduced representation bisulfite sequencing (RRBS) or myonuclear RNA sequencing (RNA-seq), respectively.19,20 Due to the incompletely defined role of DNA methylation, it can be difficult to assign individual CpG sites to specific genes to infer transcriptional regulation. Also, simply comparing differentially expressed (DE) genes with hypo- and hypermethylated promoter regions typically underestimates the degree of overlap. In the current investigation, we integrated epigenetic alterations with myonuclear gene expression changes by using a novel multi-omics integration method adapted from chromatin immunoprecipitation sequencing (ChIP-seq) and RNA-seq analyses. The binding and expression target analysis (BETA) integration approach lends a holistic understanding of the CpG sites that are significantly contributing to gene expression changes.21 Furthermore, we used our conditional myonuclear labeling approach to perform small RNA-seq on purified myonuclei. Mature miRNAs were then computationally combined with the myonuclear mRNA transcriptome. Collectively, we link myofiber-specific epigenetic and post-transcriptional factors to reduced metabolism-related gene expression following 72 h of MOV; this included hypermethylation of downregulated mitochondrial respiratory chain complex genes. The downregulation of oxidative phosphorylation (OXPHOS) after acute MOV may be due to pyruvate shunting from the mitochondria in favor of aerobic glycolysis and biosynthetic precursor production for growth.22
Materials and Methods
Animals
All animal studies were performed in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of the University of Kentucky. Mice were housed in a temperature- and humidity-controlled room, maintained on a 14:10-h light-dark cycle, and food and water were provided ad libitum throughout the entire study. We used the myofiber-specific Tet-On system to label myonuclei as previously described for the HSA-rtTA+/−;TRE-H2B-GFP+/− (HSA-GFP) mice.23 Mice (female, ∼3 mo of age) were given doxycycline (0.5 mg/mL, 2% sucrose) for 5 d followed by a minimum 1-wk washout period prior to MOV (3 wk maximum). This strategy leads to the labeling of approximately 90%-95% of myonuclei, with negligible off-target labeling of nonmyonuclei.23
Mechanical Overload
Following doxycycline treatment and a washout period, mice underwent sham surgery or synergist ablation, as previously described.19,20,24 Sham surgery involved the same steps of synergist ablation without removal of muscle. Following surgeries, mice resumed ambulatory cage behavior for 72 h. At this time, mice were euthanized in the morning by intraperitoneal injection of sodium pentobarbital, followed by cervical dislocation. Plantaris muscles were Dounce homogenized and subjected to FANS for purification of myonuclei according to our published protocols.19,20,25,26 DNA and RNA from myonuclei were isolated as previously reported.19,20
DNA Methylation Data Processing and Statistical Analysis
We used our previously published myonuclear RRBS dataset for DNA methylation analysis.19 Quality control and adapter sequence trimming were performed using FastQC and Cutadapt, respectively, as parts of the Trim Galore wrapper. Low-quality base calls (Phred score <20) were removed prior to trimming adapter sequences. Bismark aligner was used to align the sequence reads to the bisulfite-converted GRCm39 genome prior to data processing. Coverage (.cov) files produced from Bismark aligner were used for data analysis in the methylKit R package.27 MethylKit was used to pool samples into their respective groups to maximize read coverage across the genome using a minimum read cutoff of 10 reads per base, and minimum base coverage of 1 sample per group. Differentially methylated regions (DMRs) were determined by genomic ranges for every gene promoter as defined by the mm39.bed file obtained from National Center for Biotechnology Information. Fisher’s exact test with sliding linear model correction for false discovery28 was used to qualify both differentially methylated sites and differentially methylated promoters within the dataset. Percent methylation and percent differential methylation were then obtained from methylKit following analysis.
Myonuclear mRNA Sequencing Data Processing and Statistical Analysis
We used our previously published myonuclear RNA-seq dataset for myonuclear transcriptome analysis.20 Quality control was performed using FastQC and Cutadapt was used to trim adapter sequences. STAR aligner was used to align sequenced reads to the GRCm39 genome. Binary alignment map (.bam) files resulting from STAR aligner were further processed using SAMtools. Bam files were indexed and unmapped reads and reads with low coverage were filtered prior to generating gene read counts. Limma29 was used to determine differential gene expression between sham and MOV plantaris myonuclear mRNA and raw P-values were adjusted for false discovery using the Benjamini–Hochberg false discovery rate (FDR) correction.
Binding and Expression Target Analysis Integration for Combining Gene Expression With DNA Methylation
BETA is a software that provides an integrated analysis of transcription factor binding to genomic DNA and transcript abundance using ChIP-seq and transcriptomics (RNA-seq) datasets.21 BETA takes into consideration the distance of the regulatory element relative to the transcription start site (TSS) by modeling the effect of regulation using a natural log function termed the regulatory potential (eqn 1), as described previously by Tang and colleagues.30 In this study, we adapted this software to integrate CpG methylomics with RNA-seq data instead. To do so, CpG islands (whether hypo- or hyper-methylated) were converted into “methylation peaks” similar to transcription factor binding peaks, which are built using the GRCm39 CpG island bedfile downloaded from the UCSC Genome Browser. Only genes differentially expressed with adjusted P < .05 from RNA sequencing analysis were included as input for gene expression. The BETA basic command was run with the following parameters: “-c 0.05—df 0.05—da 500.”
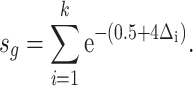 |
(1) |
In our study, a CpG island (k) within 100 kb of TSS of gene (g) is included in the calculation for the regulatory potential score (s). The distance between the CpG island and the TSS is expressed as a ratio relative to 100 kb (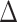 ). The scoring is weighted based on the distance from the TSS (higher for smaller distances, lower for larger distances).
). The scoring is weighted based on the distance from the TSS (higher for smaller distances, lower for larger distances).
BETA Integration Pathway Analysis
Pathway analysis was performed using the enrichR R package. Up- and downtarget genes resulting from BETA integration of DNA methylation and RNA-seq datasets were included in independent up and down gene sets. The 2023 Gene Ontology (GO) database including biological processes, molecular functions, and cellular components was used to annotate up and down gene sets and determine enriched ontologies for each gene set. GOplot R package was used to combine log2 fold change for each gene with their respective enriched pathways.
Myonuclear Small RNA-seq
The myonuclear RNA isolated from MOV plantaris muscles of mice that underwent synergist ablation (n = 4) or control plantaris muscles of mice that underwent sham surgery (n = 3) was analyzed by small RNA-seq. Library preparation was performed by Norgen Biotek (ON, Canada) using a Norgen Biotek Small RNA Library Prep Kit (Cat. 63600), and small RNA-seq was performed by Norgen Biotek using an Illumina NextSeq 500 platform. Data analysis was performed using the exceRpt small RNA-seq Pipeline (v4.6.2.) (Genboree Bioinformatics Research Laboratory). The databases miRbase version 22 for miRNAs, gtRNAdb for tRNAs, RNAdb, piRNAbank, & RNAcentral for piRNAs, and the reference genome GENCODE version 22 (mm10) were used for analyses.
miRNA Target Enrichment and Network-Based Analysis
Targets of differentially expressed (DE) miRNAs (adj. P < .05) were identified using miRTarBase Release 9.0.31 Differentially expressed genes from the myonuclear mRNA-seq analyses were compared to genes identified as targets of DE miRNAs, and pathway analysis was performed on overlapping genes using the EnrichR web-based tool.32–34
To identify potential miRNA targets and pathways that may have been missed by the miRTarBase database, an in-silico prediction of the miRNA target genes was performed. Target genes were detected using both miRanda and RNAhybrid tools. We used a custom python script to select shared miRNA: target gene interactions between the two software programs. Minimal free energy was set as ΔG°= −18 kJ. Each algorithm compares the 3’UTR gene region to the miRNA seed sequence and assesses for complementarity that suggests miRNA binding at the gene target.
Gene ontology (sources; Reactome, Wikipathways, BioCarta) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were carried out in cluster profiler on the predicted target genes.35 The GO terms and KEGG pathways with adj. P < .05 were considered to be significantly enriched.
To further assess miRNA-target regulatory network interactions that are not evident through pairwise analyses, we used the web tool MIENTURNET, which utilizes network-based visualization and analysis of miRNA-target interactions.36 The list of all DE myonuclear miRNAs was provided as input, which produced a network of miRNA-target interactions along with topological properties of all nodes.
High-Resolution Respirometry
For mitochondrial respiration experiments, 6–8-mo-old female C57BL/6 J mice were randomized to synergist ablation (n = 7) or sham surgery (n = 7). After 72 h, mice were euthanized via CO2 inhalation followed by cervical dislocation. Plantaris muscles were removed, cut lengthwise into small samples, and immediately placed in ice-cold biopsy preservation solution (BIOPS) relaxing and preservation solution (10 m m Ca-EGTA buffer, 0.1 µm free calcium, 20 m m imidazole, 20 m m taurine, 50 m m K-MES, 0.5 m m DTT, 6.56 m m MgCl2, 5.77 m m adenosine triphosphate (ATP), 15 m m phosphocreatine, pH 7.1). Fiber bundles were mechanically separated using forceps in a petri dish on ice, followed by transfer intro BIOPS containing saponin at a final concentration of 50 μg/mL. Samples were agitated on ice for 30 min, then transferred to MiR05 mitochondrial respiration medium (Oroboros Instruments, Innsbruck, Austria) for a 10 min wash. High-resolution oxygen consumption measurements were conducted at 37°C in MiR05 supplemented with 20 m m creatine monohydrate using an Oroboros O2k Oxygraph (Oroboros Instruments). Fiber bundles of ∼2 mg wet mass were added to O2k chambers, and a substrate inhibitor titration (SUIT) protocol was performed as follows: 10 m m pyruvate, 2 m m malate, and 10 m m glutamate (complex I leak); 4 m m ADP (to initiate OXPHOS); 10 µm cytochrome c (to test integrity of the mitochondrial membrane); 10 m m succinate (to initiate electron flow through complex II); 250 n m titrations (up to 1.5 μm) of carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (maximum respiration/ET capacity); 10 µm rotenone (to inhibit complex I); and 5 µm antimycin A (to inhibit complex III for background correction). All samples had a cytochrome c response < 15%. Oxygen flux (JO2), expressed as pmol/s was normalized to sample wet weight, and differences between sham and MOV samples were assessed using an unpaired t-test, with significance set at P < .05.
Results
MOV Robustly Alters the Contribution of the Myonuclear DNA Methylome to Activate and Repress Gene Expression
Mechanical overload induced hypermethylation of at least 1 CpG in 3988 promoters, 5092 exons, 7343 introns, and 3242 intergenic CpG sites and hypomethylation of 17 812 promoters, 13 722 exons, 12 508 introns, and 16 895 intergenic CpG sites across the genome (Figure 1A and B). Binding and expression target analysis revealed 116 upregulated and 203 downregulated gene targets associated with significant differentially methylated sites (Figure 1C). Gene set enrichment of upregulated and downregulated targets from BETA indicated biases toward upregulation of growth-associated gene ontologies and downregulation of mitochondrial and metabolic gene ontologies (Figure 1D). Specifically, downregulated targets included genes encoding mitochondrial respiratory chain complex subunits (ie, NADH: ubiquinone oxidoreductase subunits A9, A10, B9, Ndufa9, Ndufa10, Ndufb9 and cytochrome c oxidase subunits 4I1 and 5B, Cox4i1, Cox5b). Upregulated targets included growth factor genes, including nerve growth factor (Ngf), heparin-binding EGF-like growth factor (Hbegf), and fibroblast growth factor receptor 4 (Fgfr4). Examples of RNA-seq and RRBS integration are provided for a metabolism-related gene (6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, Pfkfb3, Figure S1A) and a cytoskeleton-related gene (obscurin like cytoskeletal adaptor 1, Obsl1, Figure S1B). Both RNA-seq read coverage data (top) and RRBS % methylation per CpG site (bottom) are shown.
Figure 1.

Myonuclear DNA methylation directly downregulates expression of genes encoding mitochondrial respiratory chain proteins. Integrative analysis combining reduced representation bisulfite sequencing (RRBS) and RNA sequencing (RNA-seq) data provides insight into the genes directly regulated by DNA methylation following 72 h of mechanical overload (MOV)-induced muscle hypertrophy. (A) Overview of myonuclear DNA methylome showing the number of significantly differentially methylated CpG sites falling within the promoter (2000 bp upstream of transcription start site), exonic, and intronic regions as well as CpG islands; MOV (n = 3) versus sham (n = 2). (B) Schematic showing the limitations of methylation analysis where significantly differentially methylated CpG sites are assigned to a specific gene because of promoter region. CpG sites may fall outside of the promoters and still participate in the regulation of genes in its vicinity. (C) BETA integration analysis of myonuclear RRBS and RNA-seq data comparing up- and downregulated genes relative to background (significance indicated by P-values in parentheses). (D) Chord plot showing up- and downregulated genes identified by BETA integration corresponding to significantly enriched gene ontologies; legend represent log2 fold difference shown in red/blue heatmap for RNA-seq data [MOV (n = 4) versus sham (n = 3)].
Small RNA-seq Reveals a Modest Increase in the Proportion of Myonuclear miRNAs After 72 h of MOV
We investigated the relative biotype distribution of small RNAs in myonuclei using small RNA-seq (Figure 2A and B). Although miRNA abundance dominated proportions of small RNAs present in myonuclei, small RNA biotypes other than miRNAs were also detected. The fraction of transcriptome-aligned reads mapping to different biotypes of RNAs in sham and MOV myonuclei varied significantly with respect to miRNAs, protein coding RNAs (mRNAs), and circular RNAs (Figure 2C). MOV myonuclei had a higher proportion of miRNAs compared to sham (44.74% versus 39.69%, P = .018), while protein-coding RNA abundance was lower in MOV compared to sham myonuclei (11.17% versus 24.38%, P = .016). Detection of mRNA species is consistent with other studies showing uniquely mapped protein-coding RNAs by small RNA-seq.37,38
Figure 2.

Differentially expressed (DE) miRNAs and prediction of biological processes targeted by DE miRNAs. The fraction of transcriptome-aligned reads mapping to different biotypes of RNAs in (A) sham (n = 3) and (B) MOV (n = 4) myonuclei. Small RNA biotypes other than miRNAs are detected. (C) Differences in relative biotype distribution between sham and overload tested using an independent t-test. snoRNA: small nucleolar RNA, tRNA: transfer RNA, piRNA: Piwi-interacting RNA, snRNA: small nuclear RNA, and lincRNA: long intergenic noncoding RNAs. *P < .05. (D) Volcano plot indicating mean expression level for each miRNA, red dots represent significant DE miRNAs (adj. P < .05). (E) Network-based pathway enrichment analysis of genes identified in silico as myonuclear-miRNA targets of myonuclear DE genes. Arrows represent relationships and color represents the adj. P-value.
DE Myonuclear miRNAs After 72 h of MOV Target Genes Associated With Mitochondrial Function and Metabolism
A total of 48 miRNAs were differentially expressed (21 upregulated, 27 downregulated) in myonuclei from MOV muscle compared to sham (Figure 2D). Of the top 10 miRNAs by expression level in myonuclei, 6 were differentially expressed with MOV relative to sham (Table S1). Targets of DE miRNAs identified using miRTarBase that corresponded with DE myonuclear mRNAs were found to be significantly enriched for several metabolism-related pathways, including TCA cycle and respiratory electron transport, pyruvate metabolism, and ATP synthesis ( Figure S2A).
Using the in-silico target prediction tools, miRanda and RNAhybrid, a total of 13,651 genes were identified as potential targets of all the DE myonuclear miRNAs. Given that a single miRNA is predicted to have many target genes, there was significant overlap between DE miRNA target genes. Of this list, 1618 genes were identified as DE following 72 h of MOV (Table S2). Gene ontology analysis of these genes indicated enrichment of pathways for generation of precursor metabolites and energy positive regulation of the cell, adhesion, and energy derivation by oxidation of organic compounds (Table S3). Further analysis of these pathways showed abundant activity in the central metabolic pathways, including cellular respiration and mitochondrial ATP synthesis coupled with electron transport (Figure 2E).
Network-Based Analysis Reveals miRNA-Based Regulation of Metabolic and Ubiquitin Ligase Genes
The MIENTURNET web tool36 was used to perform regulatory network analysis of DE myonuclear miRNAs. This tool identified 73 genes that were predicted to be significant miRNA network-based targets (Table S4). Of these genes, 12 were DE in our myonuclear mRNA-seq, including peroxisome proliferator-activated receptor alpha (Ppara) and solute carrier family 16 member 13 (Slc16a1), which are both involved in lipid metabolism39,40 (Figure 3A). These genes, which were significantly downregulated in the myonuclear mRNA-seq analysis, were identified as predicted targets of several DE myonuclear miRNAs. Interestingly, the miRNAs targeting these downregulated genes were also significantly lower in MOV compared to sham (Figure S2B and C), which may represent regulatory functions of nuclear miRNAs beyond post-transcriptional gene silencing. Other significant miRNA network-based DE targets in our myonuclear mRNA-seq included ubiquitin protein ligase E3 component n-recognin 3 (Ubr3) and WW domain containing E3 ubiquitin protein ligase 1 (Wwp1), both of which encode E3 ubiquitin-ligase molecules (Figure 3A).41,42 Both genes were significantly downregulated in MOV compared to sham in the myonuclear mRNA-seq analysis. The miRNAs targeting these genes were significantly upregulated in myonuclei in MOV relative to sham (Figure S2D).
Figure 3.

Downregulation of oxidative metabolism genes following mechanical overload accompanied by reduced mitochondrial respiration of permeabilized plantaris muscle fiber bundles. (A) Interaction graph of selected miRNAs (blue circles) and their target genes (yellow circles) built in MIENTURNET. (B) Overview of substrate inhibitor titration (SUIT) protocol showing mean ± SD of oxygen flux (JO2) for each measurement state in sham (n = 7) and MOV (n = 7) samples [complex I leak; complex I oxidative phosphorylation (OXPHOS); complex II OXPHOS; combined complex I + II OXPHOS; ET Capacity: electron transfer capacity (maximum, noncoupled state)]. Data in (B) shown in (C-G) with individual data points and statistical analysis (unpaired t-test) between groups. Ns: not significant, *P < .05, **P < .01.
Reduced Mitochondrial Respiration Following 72 h of MOV
To assess whether the signature of downregulated myonuclear metabolic pathways following acute MOV coincides with an effect on mitochondrial function, we assessed mitochondrial respiration in permeabilized plantaris muscle fibers following 72 h of sham or MOV (Figure 3B). Oxygen flux (JO2), a direct measure of oxygen consumption rate, was significantly lower in MOV compared to sham during several measurement states (complex I leak: P = .011; combined complex I + II-linked OXPHOS: P = .009; and ET capacity: P = .027) (Figure 3C–E). Although OXPHOS was lower in MOV compared to sham during complex I and complex II, differences were not statistically significant (P = .060 and .108, respectively) (Figure 3F and G).
Discussion
Skeletal muscle myofibers form a multinucleated syncytium with the ability to quickly enhance cellular size and DNA content in response to an acute MOV stimulus.43,44 Resident myonuclei dramatically upregulate transcriptional output during early stages of MOV-induced hypertrophy (72 h).45 We previously interrogated myonuclear DNA methylation via RRBS and found evidence of growth-related regulation by myonuclei following 72 h of MOV. This signature was characterized by hypomethylation of mTOR signaling genes as well as Myc, a master regulator of ribosome biogenesis.19 Furthermore, we performed myonuclear RNA-seq, which identified enrichment of cell cycle regulators including Myc, and elevations in extracellular matrix (ECM) and RHO-GTPase genes in response to 72 h of MOV.20 In this work, we significantly expand our prior work to reveal the coordinated methylation and transcriptional regulation of metabolic genes with MOV that corresponds to lower mitochondrial respiration, and is complemented by the myonuclear miRNA landscape. Although there is not always a direct relationship between mRNA levels, protein levels, and ultimately phenotype and function in skeletal muscle,46 we show here that early molecular responses during rapid hypertrophy correspond well with the measurable cellular outcome of mitochondrial respriation.
In general, significantly hypermethylated CpG sites that fall within the promoter region tend to repress a target gene and vice versa for the hypomethylated promoter CpG sites. Oftentimes, CpG sites fall into regions of the genome that may be the promoter of one gene but in close proximity of another gene upstream. In such cases, the role of the CpG sites in the promoter of one gene may be similar or different compared to its role downstream of another gene. Also, the overall number of differentially methylated sites within a region may impact the magnitude of gene expression regulation as well, even if that function is not yet mechanistically defined for a given gene. As such, using an integrated analysis by combining the methylomics data with the transcriptomics data facilitates a more comprehensive understanding of epigenomic regulation. One approach that allows for mismatched sample sizes across analyses is BETA, which was first developed for integrating ChIP-seq and RNA-seq datasets. In the current investigation, after integration of the RRBS data with the myonuclear RNA-seq, our data suggest that epigenetic modifications in myonuclear DNA may drive the observed reductions in oxidative metabolism gene expression. Integrative analysis specifically points to downregulated genes induced by DNA hypermethylation, and these genes are enriched for pathways representing mitochondrial respiratory chain complexes I, III, and IV and mitochondrial organization. Furthermore, upregulated GOs included growth factor activity and ECM organization, suggesting that growth-associated alterations in myonuclear gene expression may be linked to changes in DNA methylation as well. Together, upregulated and downregulated targets from BETA analysis reveal that epigenetic modifications to the DNA methylome contribute to modulation of the myonuclear transcriptome that aligns with altered muscle metabolism.
A novel finding of the present work is the identification of mature miRNAs within myonuclei and our observation that the relative distribution of myonuclear miRNAs is increased after 72 h of MOV. Notably, miRNAs are transcribed into primary transcripts (pri-miRNAs) and processed into precursor miRNAs (pre-miRNAs) in the nucleus by Drosha, followed by export to the cytoplasm, where they are processed into mature miRNAs by Dicer.47 Traditionally, microRNA function has been thought to be restricted to the cytoplasm; however, deep sequencing of nuclear and cytoplasmic pools independently showed that a majority of miRNAs were imported into the nucleus,48 and nuclear-cytoplasmic shuttling machinery has also been identified.7,49 In the current study, we identified several DE miRNAs by small RNA-seq of isolated myonuclei in MOV compared to sham. Target prediction using either a standard miRNA-target interaction database or in silico prediction demonstrated that mRNA targets of DE myonuclear miRNAs were enriched for oxidative metabolism-related pathways. The miRNA-mRNA interaction network also included key metabolic regulatory genes as well as ubiquitin ligase E3 components. While some miRNAs that were elevated after MOV were predicted to target downregulated genes, we also observed examples of downregulated miRNAs predicted to target downregulated genes. This could potentially be explained by the fact that both RNA activation and transcriptional gene silencing have been demonstrated in nuclei via recruitment of chromatin modifying proteins to chromosomal DNA, altering DNA methylation.12,15,50 Taken together, these data suggest that in addition to classical post-transcriptional silencing by 3′UTR targeting, myonuclear miRNA-mediated adaptation to MOV may also involve epigenetic mechanisms that contribute to the transcriptional control of myonuclear genes implicated in skeletal muscle metabolism as well as protein degradation. Our data provide further support for the contribution of miRNA-mediated mechanisms to the molecular responses to exercise.46 It is important to note that while cytoplasmic and nonmyonuclear miRNAs may also play a role in the hypertrophic response to MOV,51 our current investigation focused on myonuclei, which represent only ∼50%-60% of the total nuclei in murine skeletal muscle.25 Interestingly, miRNA alterations in myonuclei may not coincide with changes in their miRNA distribution. For example, we found that levels of miR-1, which decrease with MOV in whole muscle,52,53 were not significantly different in myonuclei between MOV and sham. This suggests that while miR-1 transcription may not change in myonuclei in response to MOV, the decrease in miR-1 in whole muscle may reflect export via exosomes, as previously reported.52
We found that downregulated oxidative metabolism signatures after 72 h of MOV were accompanied by functional reductions in mitochondrial respiration. In addition to reduced maximal respiration (ET capacity) in the noncoupled state, respiratory capacity of mitochondria was also significantly reduced in MOV muscle both with saturating levels of ADP (OXPHOS) and during respiration supported by endogenous substrates (leak). Although this observation may appear intriguing considering skeletal muscle hypertrophy and associated increases in protein synthesis are bioenergetically costly processes,54 others report similar reductions in oxidative metabolism-related gene expression55 as well as reductions in oxidative phosphorylation proteins and peroxisome proliferator acivated receptor gamme (PPARG) coactivator 1 alpha (Pgc-1α) levels in response to MOV.56 These data, along with evidence for increased glucose uptake and utilization in growing muscle cells,57,58 have led to the hypothesis that hypertrophying muscle fibers reprogram their metabolism in a manner characterized by “aerobic glycolysis,” commonly known as the “Warburg effect.”22 Increased glucose utilization may allow for increased nucleotide and rRNA synthesis, which may facilitate synergist ablation-induced muscle growth by increasing translational capacity.59 Further support for this metabolic reprogramming comes from reports indicating that glucose uptake, lactate secretion, and the pentose phosphate pathway increased in mice after MOV induced by synergist ablation.60–62 In C2C12 myotubes, glucose incorporates into muscle protein and RNA, which is enhanced during hypertrophy and controlled by phosphorylation of AKT and 3-phosphoglycerate dehydrogenase (PHGDH),63 a serine-synthesizing enzyme. In our data, Phgdh mRNA is upregulated in myonuclei during MOV (adj. P = .02) and has previously been linked to rapid developmental muscle hypertrophy in vivo,64 suggesting a high rate of glucose usage and biosynthetic metabolism during MOV. Thus, reduced mitochondrial respiration following acute MOV may be a consequence of inhibited pyruvate entry into mitochondria rather than dysfunctional mitochondria.65 Interestingly, 72 h of MOV reduces myonuclear Myh2 mRNA content (oxidative fast-twitch myosin 2A), whereas prolonged MOV (8 wk) ultimately induces an aerobic fiber-type shift toward myosin 2A fibers alongside 2A hypertrophy.66 Despite these observations, the mechanisms regulating energy metabolism reprogramming in rapidly hypertrophying skeletal myofibers remained unclear. The present work furthers our understanding of metabolic gene regulation in response to MOV.
Our study had limitations that are worth addressing. First, our sample size for the integrative analysis using BETA was limited and exclusive to females, which likely reduced the power to identify significant genes and subsequently the significant pathways associated with MOV. Still, analytical integration of multi-omics can enhance statistical power and synergistic insights compared to single-omics approaches.67–70 Another limitation of the study is the supraphysiological nature of the synergist ablation model, which allows for very rapid hypertrophy, limiting the generalizability of the current findings to humans where muscle hypertrophy is typically slower. Worth noting, however, is that functional hypertrophy of type 2 muscle fibers in humans can be as high as 8%-14% per week during short-term tapering following heavy training.71–74 Synergist ablation induces inflammation associated with the surgical procedure.75,76 Since inflammatory stress and signaling can result in reduced mitochondrial respiration,77 it is possible that this contributes to the synergist ablation-induced effects on mitochondrial metabolism. On balance, the epigenetic and transcriptional data presented here suggest that the reduction in mitochondrial metabolism is a “programmed” response, and not necessarily due to acute mitochondrial damage by inflammation.
By integration of DNA methylation and myonuclear RNA-seq data, we demonstrate that epigenetic modifications in myonuclear DNA may play a role in the downregulation of nuclear oxidative metabolism gene expression with MOV. Furthermore, small RNA-seq profiling revealed a hitherto undescribed involvement of myonuclear miRNAs in the regulation of myonuclear oxidative metabolism-related gene expression. Our data provide new avenues for exploration of the molecular mechanisms driving epigenetic modifications in myonuclei.
Supplementary Material
Acknowledgments
The authors thank Charlotte A. Peterson, Ph.D., for discussions and useful insights on the manuscript. The graphical abstract was created with BioRender.com.
Contributor Information
Ahmed Ismaeel, Department of Physiology, College of Medicine, University of Kentucky, Lexington, KY 40508, USA; Center for Muscle Biology, University of Kentucky, Lexington, KY 40536, USA.
Nicholas T Thomas, Center for Muscle Biology, University of Kentucky, Lexington, KY 40536, USA; Department of Athletic Training and Clinical Nutrition, College of Health Sciences, University of Kentucky, Lexington, KY 40536, USA.
Mariah McCashland, Department of Nutrition and Health Sciences, University of Nebraska–Lincoln, Lincoln, NE 68583, USA.
Ivan J Vechetti, Department of Nutrition and Health Sciences, University of Nebraska–Lincoln, Lincoln, NE 68583, USA.
Sebastian Edman, Department of Women’s and Children’s Health, Karolinska Institutet, Solna 17177, Sweden.
Johanna T Lanner, Department of Physiology and Pharmacology, Karolinska Institutet, Solna 17177, Sweden.
Vandré C Figueiredo, Department of Biological Sciences, Oakland University, Rochester, MI 48309, USA.
Christopher S Fry, Center for Muscle Biology, University of Kentucky, Lexington, KY 40536, USA; Department of Athletic Training and Clinical Nutrition, College of Health Sciences, University of Kentucky, Lexington, KY 40536, USA.
John J McCarthy, Department of Physiology, College of Medicine, University of Kentucky, Lexington, KY 40508, USA; Center for Muscle Biology, University of Kentucky, Lexington, KY 40536, USA.
Yuan Wen, Department of Physiology, College of Medicine, University of Kentucky, Lexington, KY 40508, USA; Center for Muscle Biology, University of Kentucky, Lexington, KY 40536, USA; Division of Biomedical Informatics, Department of Internal Medicine, College of Medicine, University of Kentucky, Lexington, KY 40536, USA.
Kevin A Murach, Department of Health, Human Performance, and Recreation, Exercise Science Research Center, University of Arkansas, Fayetteville, AR 72701, USA.
Ferdinand von Walden, Department of Women’s and Children’s Health, Karolinska Institutet, Solna 17177, Sweden.
Funding
Y.W. was supported by the National Institutes of Health (K99 AR081367). K.A.M. was supported by the National Institutes of Health (R00 AG063994 and R01 AG080047). F.v.W. was supported by the Swedish Research Council (2022-01392), AFM-Telethon (23137), SMDF, Åke Wiberg, Swedish Medical Association, and the Swedish Research Council for Sport Science (2022/10). I.J.V. was supported by the National Institutes of Health (P20GM104320-07).
Conflict of Interest
None declared.
Data Availability
The new data generated underlying this article are available in Gene Expression Omnibus and can be accessed with GSE245715.
References
- 1. Kim M, Franke V, Brandt B et al. Single-nucleus transcriptomics reveals functional compartmentalization in syncytial skeletal muscle cells. Nat Commun. 2020;11(1):6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bruusgaard JC, Liestol K, Ekmark M, Kollstad K, Gundersen K. Number and spatial distribution of nuclei in the muscle fibres of normal mice studied in vivo. J Physiol. 2003;551(2):467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roberts MD, McCarthy JJ, Hornberger TA et al. Mechanisms of mechanical overload-induced skeletal muscle hypertrophy: current understanding and future directions. Physiol Rev. 2023;103(4):2679–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roberts TC. The microRNA biology of the mammalian nucleus. Mol Ther Nucleic Acids. 2014;3(8):e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stavast CJ, Erkeland SJ. The non-canonical aspects of microRNAs: many roads to gene regulation. Cells. 2019;8(11):1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu H, Lei C, He Q, Pan Z, Xiao D, Tao Y. Nuclear functions of mammalian microRNAs in gene regulation, immunity and cancer. Mol Cancer. 2018;17(1):64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castanotto D, Lingeman R, Riggs AD, Rossi JJ. CRM1 mediates nuclear-cytoplasmic shuttling of mature microRNAs. Proc Natl Acad Sci USA. 2009;106(51):21655–21659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishi K, Nishi A, Nagasawa T, Ui-Tei K. Human TNRC6A is an Argonaute-navigator protein for microRNA-mediated gene silencing in the nucleus. RNA. 2013;19(1):17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fasanaro P, Greco S, Lorenzi M et al. An integrated approach for experimental target identification of hypoxia-induced miR-210. J Biol Chem. 2009;284(50):35134–35143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leucci E, Patella F, Waage J et al. microRNA-9 targets the long non-coding RNA MALAT1 for degradation in the nucleus. Sci Rep. 2013;3(1):2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hansen TB, Wiklund ED, Bramsen JB et al. miRNA-dependent gene silencing involving Ago2-mediated cleavage of a circular antisense RNA. EMBO J. 2011;30(21):4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim DH, Saetrom P, Snove O Jr., Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci USA. 2008;105(42):16230–16235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan Y, Zhang B, Wu T et al. Transcriptional inhibiton of Hoxd4 expression by miRNA-10a in human breast cancer cells. BMC Mol Biol. 2009;10(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gonzalez S, Pisano DG, Serrano M. Mechanistic principles of chromatin remodeling guided by siRNAs and miRNAs. Cell Cycle. 2008;7(16):2601–2608. [DOI] [PubMed] [Google Scholar]
- 15. Younger ST, Corey DR. Transcriptional regulation by miRNA mimics that target sequences downstream of gene termini. Mol Biosyst. 2011;7(8):2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adilakshmi T, Sudol I, Tapinos N. Combinatorial action of miRNAs regulates transcriptional and post-transcriptional gene silencing following in vivo PNS injury. PLoS One. 2012;7(7):e39674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benhamed M, Herbig U, Ye T, Dejean A, Bischof O. Senescence is an endogenous trigger for microRNA-directed transcriptional gene silencing in human cells. Nat Cell Biol. 2012;14(3):266–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zardo G, Ciolfi A, Vian L et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood. 2012;119(17):4034–4046. [DOI] [PubMed] [Google Scholar]
- 19. Von Walden F, Rea M, Mobley CB et al. The myonuclear DNA methylome in response to an acute hypertrophic stimulus. Epigenetics. 2020;15(11):1151–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murach KA, Liu Z, Jude B et al. Multi-transcriptome analysis following an acute skeletal muscle growth stimulus yields tools for discerning global and MYC regulatory networks. J Biol Chem. 2022;298(11):102515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang S, Sun H, Ma J et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat Protoc. 2013;8(12):2502–2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wackerhage H, Vechetti IJ, Baumert P et al. Does a hypertrophying muscle fibre reprogramme its metabolism similar to a cancer cell?. Sports Med. 2022;52(11):2569–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Iwata M, Englund DA, Wen Y et al. A novel tetracycline-responsive transgenic mouse strain for skeletal muscle-specific gene expression. Skelet Muscle. 2018;8(1):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirby TJ, McCarthy JJ, Peterson CA, Fry CS. Synergist ablation as a rodent model to study satellite cell dynamics in adult skeletal muscle. Methods Mol Biol. 2016;1460:43–52. [DOI] [PubMed] [Google Scholar]
- 25. Wen Y, Englund DA, Peck BD, Murach KA, McCarthy JJ, Peterson CA. Myonuclear transcriptional dynamics in response to exercise following satellite cell depletion. iScience. iScience. 2021;24(8):102838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wen Y, Dungan CM, Mobley CB, Valentino T, von Walden F, Murach KA. Nucleus type-specific DNA methylomics reveals epigenetic “memory” of prior adaptation in skeletal muscle. Function( Oxf). 2021;2(5):zqab038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akalin A, Kormaksson M, Li S et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13(10):R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang H-Q, Tuominen LK, Tsai C-J. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics. 2011;27(2):225–231. [DOI] [PubMed] [Google Scholar]
- 29. Ritchie ME, Phipson B, Wu D et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tang Q, Chen Y, Meyer C et al. A comprehensive view of nuclear receptor cancer cistromes. Cancer Res. 2011;71(22):6940–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang HY, Lin YC, Cui S et al. miRTarBase update 2022: an informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022;50(D1):D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen EY, Tan CM, Kou Y et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuleshov MV, Jones MR, Rouillard AD et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xie Z, Bailey A, Kuleshov MV et al. Gene set knowledge discovery with Enrichr. Curr Protoc. 2021;1(3):e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Licursi V, Conte F, Fiscon G, Paci P. MIENTURNET: an interactive web tool for microRNA-target enrichment and network-based analysis. BMC Bioinf. 2019;20(1):545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Godoy PM, Bhakta NR, Barczak AJ et al. Large differences in small RNA composition between human biofluids. Cell Rep. 2018;25(5):1346–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chettimada S, Lorenz DR, Misra V, Wolinsky SM, Gabuzda D. Small RNA sequencing of extracellular vesicles identifies circulating miRNAs related to inflammation and oxidative stress in HIV patients. BMC Immunol. 2020;21:(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Biswas D, Ghosh M, Kumar S, Chakrabarti P. PPARalpha-ATGL pathway improves muscle mitochondrial metabolism: implication in aging. FASEB J. 2016;30(11):3822–3834. [DOI] [PubMed] [Google Scholar]
- 40. Perry MC, Dufour CR, Tam IS, B'Chir W, Giguere V. Estrogen-related receptor-alpha coordinates transcriptional programs essential for exercise tolerance and muscle fitness. Mol Endocrinol. 2014;28(12):2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hirata Y, Nomura K, Senga Y et al. Hyperglycemia induces skeletal muscle atrophy via a WWP1/KLF15 axis. JCI Insight. 2019;4(4):e124952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li T, Giagtzoglou N, Eberl DF et al. The E3 ligase Ubr3 regulates Usher syndrome and MYH9 disorder proteins in the auditory organs of Drosophila and mammals. eLife. 2016;5:e15258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCarthy JJ, Mula J, Miyazaki M et al. Effective fiber hypertrophy in satellite cell-depleted skeletal muscle. Development. 2011;138(17):3657–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Koopmans PJ, Zwetsloot KA, Murach KA. Going nuclear: molecular adaptations to exercise mediated by myonuclei. Sports Med Health Sci. 2023;5(1):2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kirby TJ, Patel RM, McClintock TS, Dupont-Versteegden EE, Peterson CA, McCarthy JJ. Myonuclear transcription is responsive to mechanical load and DNA content but uncoupled from cell size during hypertrophy. MBoC. 2016;27(5):788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bishop DJ, Hoffman NJ, Taylor DF, Saner NJ, Lee MJC, Hawley JA. Discordant skeletal muscle gene and protein responses to exercise. Trends Biochem Sci. 2023;48(11):927–936. [DOI] [PubMed] [Google Scholar]
- 47. Huang V, Li LC. miRNA goes nuclear. RNA Biol. 2012;9(3):269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liao JY, Ma LM, Guo YH et al. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3' trailers. PLoS One. 2010;5(5):e10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weinmann L, Hock J, Ivacevic T et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136(3):496–507. [DOI] [PubMed] [Google Scholar]
- 50. Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105(5):1608–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hitachi K, Tsuchida K. Role of microRNAs in skeletal muscle hypertrophy. Front Physiol. 2013;4(eCollection 2013):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vechetti IJ Jr., Peck BD, Wen Y et al. Mechanical overload-induced muscle-derived extracellular vesicles promote adipose tissue lipolysis. FASEB J. 2021;35(6):e21644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 2007;102(1):306–313. [DOI] [PubMed] [Google Scholar]
- 54. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun. 2021;12(1):330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chaillou T, Lee JD, England JH, Esser KA, McCarthy JJ. Time course of gene expression during mouse skeletal muscle hypertrophy. J Appl Physiol. 2013;115(7):1065–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Uemichi K, Shirai T, Hanakita H, Takemasa T. Effect of mechanistic/mammalian target of rapamycin complex 1 on mitochondrial dynamics during skeletal muscle hypertrophy. Physiol Rep. 2021;9(5):e14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hosios AM, Hecht VC, Danai LV et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev Cell. 2016;36(5):540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Semsarian C, Sutrave P, Richmond DR, Graham RM. Insulin-like growth factor (IGF-I) induces myotube hypertrophy associated with an increase in anaerobic glycolysis in a clonal skeletal-muscle cell model. Biochem J. 1999;339(2):443–451. [PMC free article] [PubMed] [Google Scholar]
- 59. Mesquita PHC, Vann CG, Phillips SM et al. Skeletal muscle ribosome and mitochondrial biogenesis in response to different exercise training modalities. Front Physiol. 2021;12(eCollection 2021):725866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Augert G, Van de Werve G, Le Marchand-Brustel Y. Effect of work-induced hypertrophy on muscle glucose metabolism in lean and obese mice. Diabetologia. 1985;28(5):295–301. [DOI] [PubMed] [Google Scholar]
- 61. Weyrauch LA, McMillin SL, Witczak CA. Insulin resistance does not impair mechanical overload-stimulated glucose uptake, but does alter the metabolic fate of glucose in mouse muscle. Int J Mol Sci. 2020;21(13):4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Valentino T, Figueiredo VC, Mobley CB, McCarthy JJ, Vechetti IJ Jr. Evidence of myomiR regulation of the pentose phosphate pathway during mechanical load-induced hypertrophy. Physiol Rep. 2021;9(23):e15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stadhouders LEM, Smith JAB, Gabriel BM et al. Myotube growth is associated with cancer-like metabolic reprogramming and is limited by phosphoglycerate dehydrogenase. Exp Cell Res. 2023;433(2):113820. [DOI] [PubMed] [Google Scholar]
- 64. Brown DM, Williams H, Ryan KJ et al. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) and serine biosynthetic pathway genes are co-ordinately increased during anabolic agent-induced skeletal muscle growth. Sci Rep. 2016;6(1):28693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. 2021;599(6):1745–1757. [DOI] [PubMed] [Google Scholar]
- 66. Fry CS, Lee JD, Jackson JR et al. Regulation of the muscle fiber microenvironment by activated satellite cells during hypertrophy. FASEB J. 2014;28(4):1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sathyanarayanan A, Gupta R, Thompson EW, Nyholt DR, Bauer DC, Nagaraj SH. A comparative study of multi-omics integration tools for cancer driver gene identification and tumour subtyping. Brief Bioinform. 2020;21(6):1920–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Graw S, Chappell K, Washam CL et al. Multi-omics data integration considerations and study design for biological systems and disease. Mol Omics. 2021;17(2):170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Planell N, Lagani V, Sebastian-Leon P et al. STATegra: multi-omics data integration—a conceptual scheme with a bioinformatics pipeline. Front Genet. 2021;12(eCollection 2021):620453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Vahabi N, Michailidis G. Unsupervised multi-omics data integration methods: a comprehensive review. Front Genet. 2022;13(eCollection 2022):854752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Luden N, Hayes E, Galpin A et al. Myocellular basis for tapering in competitive distance runners. J Appl Physiol. 2010;108(6):1501–1509. [DOI] [PubMed] [Google Scholar]
- 72. Murach K, Raue U, Wilkerson B et al. Single muscle fiber gene expression with run taper. PLoS One. 2014;9(9):e108547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Trappe S, Costill D, Thomas R Effect of swim taper on whole muscle and single muscle fiber contractile properties. Med Sci Sports Exerc. 2001;33(1):48–56. [PubMed] [Google Scholar]
- 74. Neary JP, Martin TP, Quinney HA. Effects of taper on endurance cycling capacity and single muscle fiber properties. Med Sci Sports Exerc. 2003;35(11):1875–1881. [DOI] [PubMed] [Google Scholar]
- 75. Terena SM, Fernandes KP, Bussadori SK, Deana AM, Mesquita-Ferrari RA. Systematic review of the synergist muscle ablation model for compensatory hypertrophy. Rev Assoc Med Bras. 2017;63(2):164–172. [DOI] [PubMed] [Google Scholar]
- 76. Novak ML, Billich W, Smith SM et al. COX-2 inhibitor reduces skeletal muscle hypertrophy in mice. Am J Physiol-Regul, Integr Comp Physiol. 2009;296(4):R1132–R1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cherry AD, Piantadosi CA. Regulation of mitochondrial biogenesis and its intersection with inflammatory responses. Antioxid Redox Signal. 2015;22(12):965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The new data generated underlying this article are available in Gene Expression Omnibus and can be accessed with GSE245715.