

Abstract
Metabolic dysfunction-associated steatotic liver disease (MASLD) is the commonest cause of chronic liver disease worldwide and represents an unmet precision medicine challenge. We established a retrospective national cohort of 940 histologically defined patients (55.4% men, 44.6% women; median body mass index 31.3; 32% with type 2 diabetes) covering the complete MASLD severity spectrum, and created a secure, searchable, open resource (SteatoSITE). In 668 cases and 39 controls, we generated hepatic bulk RNA sequencing data and performed differential gene expression and pathway analysis, including exploration of gender-specific differences. A web-based gene browser was also developed. We integrated histopathological assessments, transcriptomic data and 5.67 million days of time-stamped longitudinal electronic health record data to define disease-stage-specific gene expression signatures, pathogenic hepatic cell subpopulations and master regulator networks associated with adverse outcomes in MASLD. We constructed a 15-gene transcriptional risk score to predict future hepatic decompensation events (area under the receiver operating characteristic curve 0.86, 0.81 and 0.83 for 1-, 3- and 5-year risk, respectively). Additionally, thyroid hormone receptor beta regulon activity was identified as a critical suppressor of disease progression. SteatoSITE supports rational biomarker and drug development and facilitates precision medicine approaches for patients with MASLD.
Subject terms: Translational research, Data integration, Genetic databases
SteatoSITE is an open resource that integrates histopathologic assessments, transcriptomic data and longitudinal electronic health records for a cohort of 940 patients with metabolic dysfunction-associated steatotic liver disease.
Main
Metabolic dysfunction-associated steatotic liver disease (MASLD), previously termed non-alcoholic fatty liver disease (NAFLD)1, is a growing and underappreciated global public health concern affecting more than one in four adults (over two billion people) and imposing a substantial burden of ill health and socioeconomic problems2,3. It is characterized by the presence of cardiometabolic risk factors such as obesity, type 2 diabetes, hypertension and dyslipidemia and is associated with increased morbidity and mortality from cardiovascular disease, chronic kidney disease and many cancers. The histopathological spectrum of MASLD includes isolated fatty liver (simple steatosis) and metabolic dysfunction-associated steatohepatitis (MASH), previously termed non-alcoholic steatohepatitis (NASH), the inflammatory subphenotype of MASLD that can lead to progressive liver fibrosis, cirrhosis and hepatocellular carcinoma (HCC). Notably, over the past ten years, there has been a tenfold increase in the number of patients with MASLD-related cirrhosis who require a liver transplant4, and Bayesian modeling forecasts a staggering global prevalence rate of 55.7% by 2040 (ref. 5).
The pathogenesis of MASLD is complex and multifactorial6,7. The primary insult of excess lipid accumulation (‘substrate overload’) is followed by variable contributions from pathogenic drivers including lipotoxicity and immune-mediated inflammation, and disease progression is further enhanced or suppressed by modifiers such as genetic variants8 and gut microbiota dysbiosis9,10. Crucially, not all people with a fatty liver go on to develop adverse liver-related events or die11, but there is a critical lack of prognostic biomarkers to enable innovations in clinical care pathways and personalized approaches in MASLD. The importance of histological fibrosis in predicting outcomes in MASLD has been highlighted by several studies11–13, with discrete fibrosis stages dramatically influencing the risk of all-cause mortality and liver-related morbidity and mortality. Yet fibrosis progression rates and outcome prediction in patients remain uncertain, and clinical trials that have been used post hoc to understand the natural history of MASLD are limited by selection bias and rigid entry criteria14.
There are no licensed pharmacotherapies for MASLD. Numerous drugs with plausible biological targets have failed to show robust efficacy in late-phase interventional trials, especially in patients with cirrhosis15. Consequently, the field is shifting focus to combination drug regimens, and, increasingly, researchers are leveraging patient-centered big data to bolster drug discovery efforts16. However, the most efficacious drugs and therapeutic combinations to mitigate adverse clinical outcomes in MASLD remain undefined.
To address these issues and provide a resource suitable for the development of precision medicine strategies in MASLD, we established SteatoSITE: to our knowledge, the world’s first data commons17 for MASLD research. SteatoSITE integrates multimodal multiscale retrospective data at a national level from 940 cases across Scotland, consisting of quantitative histopathological assessment of archival liver tissues, hepatic bulk RNA sequencing (RNA-seq) and routine clinical data extracted from electronic health records (EHRs) and other administrative sources. Distinctively, SteatoSITE is outcome-rich, with data curated in a longitudinal time-stamped format comprising 5.67 million days of clinical follow-up, enabling translational analyses to elucidate the pathogenesis of MASLD and accelerating the discovery of potential prognostic biomarkers and tractable therapeutic targets. Here, we describe and use this unique resource, providing a comprehensive analysis of hepatic gene expression and cell-type composition across the complete MASLD spectrum by identifying transcription-factor-regulated gene networks (regulons) associated with disease progression and developing a novel transcriptome-based risk-prediction model for hepatic decompensation.
Results
Cohort composition
SteatoSITE is a secondary-care, outcome-enriched cohort of 940 cases from the three participating NHS Scotland Biorepositories (Lothian, Greater Glasgow, and Clyde and Grampian) (Fig. 1a), representing the full histological spectrum from normal liver tissue to MASLD-related cirrhosis. Demographic and phenotypic characteristics of the cohort are shown in Table 1. Cases with a liver tissue sample acquired between January 2000 and October 2019 were selected. All patients were ≥18 years of age at the tissue sampling date. Data from EHRs and national datasets were retrieved, where available, from a period between ten years before the tissue sampling date until May 2020, comprising more than 5.67 million days (or roughly 15,547 years) of comprehensive routine clinical data, and annotated patient timelines were created (Fig. 1b and Supplementary Table 1). Further detail of the temporal coverage of blood test results is shown in Supplementary Table 2. Indications for biopsy or resection on the basis of a review of the information submitted with each specimen as part of routine clinical care are shown in Supplementary Table 3. The post-tissue sampling periods for biopsies, resections and explants are shown in Extended Data Fig. 1a.
Fig. 1. Clinicopathological correlations.

a, SteatoSITE Data Commons overview. b, Schematic diagram in which horizontal lines are individual patient timelines decorated with a variable amount of multimodal data preceding or following the date of liver tissue sampling (time zero, represented by the vertical yellow line). c, Alluvial diagram illustrating the relationship between the scored histopathological features of liver samples. d,e, Kaplan–Meier time-to-event analysis with log-rank test P value for all-cause mortality (P < 0.0001) (d) and hepatic decompensation events (P < 0.0001) (e) in patients with biopsies showing early disease (fibrosis stages F0 to F2), bridging fibrosis (stage F3) and cirrhosis (stage F4). A&E, accident and emergency; SFTP, Secure File Transfer Protocol.
Table 1.
Demographics and clinical characteristics of SteatoSITE cohort
| NASH-CRN fibrosis stage | |||||
|---|---|---|---|---|---|
| Variable | 0 | 1 | 2 | 3 | 4 |
| Age, mean (s.d.) | |||||
| Years | 49.8 (13.9) | 53 (14.5) | 56.8 (13.6) | 59.1 (11.5) | 59.9 (9.8) |
| Gender, number (%) | |||||
| Men | 137 (55.5) | 124 (59.6) | 92 (60.5) | 85 (50.3) | 83 (50.6) |
| Women | 110 (44.5) | 84 (40.4) | 60 (39.5) | 84 (49.7) | 81 (49.4) |
| Ethnicity, number (%) | |||||
| White | 153 (16.3) | 137 (14.6) | 90 (9.6) | 106 (11.3) | 106 (11.3) |
| Asian | 8 (0.9) | 6 (0.6) | 3 (0.3) | 3 (0.3) | 2 (0.2) |
| African | 1 (0.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Unknown | 85 (9) | 65 (6.9) | 59 (6.3) | 60 (6.4) | 56 (6) |
| BMI | |||||
| Median (IQR) | 30 (7.3) | 32.2 (10.3) | 31 (5.2) | 33.1 (7) | 31.7 (7.4) |
| Unknown BMI, number | 175 | 146 | 116 | 120 | 105 |
| Diabetes, number (%) | |||||
| Yes | 33 (13.4) | 48 (23.1) | 42 (27.6) | 79 (46.7) | 105 (64) |
| No | 214 (86.6) | 160 (76.9) | 110 (72.4) | 90 (53.3) | 59 (36) |
| SIMD quintile, number (%) | |||||
| 1 | 21 (2.2) | 27 (2.9) | 23 (2.4) | 29 (3.1) | 25 (2.7) |
| 2 | 19 (2) | 30 (3.2) | 14 (1.5) | 16 (1.7) | 26 (2.8) |
| 3 | 28 (3) | 19 (2) | 17 (1.8) | 21 (2.2) | 24 (2.6) |
| 4 | 23 (2.4) | 18 (1.9) | 10 (1.1) | 14 (1.5) | 23 (2.4) |
| 5 | 43 (4.6) | 37 (3.9) | 32 (3.4) | 36 (3.8) | 26 (2.8) |
| Unknown SIMD | 113 (12) | 77 (8.2) | 56 (6) | 53 (5.6) | 40 (4.3) |
| Liver enzymes, median (IQR) | |||||
| ALT (U per l) | 66 (78) | 90 (106.5) | 85 (109.5) | 77.5 (98.75) | 53 (110.25) |
| AST (U per l) | 38 (29) | 49 (42) | 48 (36.25) | 53 (46) | 44.5 (37.5) |
| ALP (U per l) | 88 (50.5) | 82.5 (43.75) | 81 (47) | 93.5 (53.5) | 107 (69) |
| Bilirubin (µmol per l) | 11 (9) | 11 (8.25) | 12 (10) | 14 (12) | 19 (37.5) |
| Other laboratory results, median (IQR) | |||||
| Prothrombin time (s) | 12 (2) | 12 (2) | 12 (2) | 13 (1.25) | 14 (7) |
| INR | 1 (0.1) | 1 (0.1) | 1 (0.1) | 1.1 (0.2) | 1.2 (0.4) |
| Albumin (g per l) | 40 (10) | 39 (8) | 38 (12) | 37 (9) | 33.5 (16) |
| Platelets (×109 per l) | 237 (100) | 230 (94) | 213 (96.5) | 198 (104.75) | 130.5 (103) |
| Creatinine (µmol per l) | 73 (17) | 72 (21) | 75 (18.25) | 70 (22) | 71 (26) |
| Non-invasive tests, median (IQR) | |||||
| FIB-4 | 1 (1) | 1.1 (1.2) | 1.4 (1.2) | 1.8 (1.8) | 2.4 (1.9) |
| Transient elastography (kPa) | 8.2 (5.7) | 10.9 (5.5) | 12.3 (6.9) | 14.4 (9.4) | 27.3 (34.3) |
| Prescription, number (%) | |||||
| Statin | 19 (7.7) | 32 (15.4) | 18 (11.8) | 27 (16) | 32 (19.5) |
| Non-selective beta blocker | 1 (0.4) | 1 (0.5) | 5 (3.3) | 4 (2.4) | 7 (4.3) |
| Specimen type, number (%) | |||||
| Biopsy | 198 (80.2) | 150 (72.1) | 100 (65.8) | 115 (68) | 96 (58.5) |
| Explant | 0 (0) | 0 (0) | 0 (0) | 2 (1.2) | 54 (32.9) |
| Resection | 49 (19.8) | 58 (27.9) | 52 (34.2) | 52 (30.8) | 14 (8.5) |
| Total case number | 247 | 208 | 152 | 169 | 164 |
ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate transaminase; FIB-4, fibrosis 4; INR, international normalized ratio; IQR, interquartile range; s.d., standard deviation; U per l, units per liter; µmol per l, mircomoles per liter.
Extended Data Fig. 1. Additional histopathological analyses.

a, Clinical follow-up depth (in days) according to specimen type. b, Clinical assessment of the presence or absence of a histological diagnosis of steatohepatitis versus NASH-CRN stage. c, Alluvial diagram illustrating the relationship between the scored SAF and modified Ishak stage features. d, A representative whole-slide image of a PSR-stained section with the associated tissue mask and applied classifier showing PSR-positive (red) and fat droplets (purple) from the QuPath quantitative pathology pipeline; only pixels present within the tissue mask are used for PSR and fat quantitation. Pixel classification using the same classifier always returns the same values so was only undertaken once. Scale bar, 2 mm. e, The amount of steatosis from image quantification versus NAS steatosis assigned scores from the 922 cases with quantifiable whole-slide images. f, The PSR percentage from image quantification versus assigned NASH-CRN scores from the 922 cases with quantifiable whole-slide images. g, The PSR percentage from image quantification versus assigned modified Ishak scores from the 922 cases with quantifiable whole-slide images. All boxplots show median (centre line), first and third quartiles (lower and upper box limits), 1.5× interquartile range (whiskers). Computationally-derived scar proportion can be used to stratify the risk of all-cause mortality (h) and hepatic decompensation (i); log–rank test P-values < 0.0001. j, Count of presence or absence of hepatocellular carcinoma (HCC) in resection cases versus assigned NASH-CRN stage. k, Kaplan–Meier time-to-event analysis for HCC in patients with biopsies showing early disease (fibrosis stages F0 to F2), bridging fibrosis (stage F3) and cirrhosis (stage F4); log–rank test P-value = 0.015.
Histopathological characterization
Scans of hematoxylin and eosin (H&E)- and picrosirius red (PSR)-stained sections from each case were assessed by one of three consultant pathologists with a specialist interest in liver pathology. From the H&E-stained sections, NAFLD activity scores (NASs), components of the Steatosis, Activity and Fibrosis (SAF) scoring system and a score-independent clinical histological diagnosis of NASH were given. From the PSR-stained sections, a NASH-Clinical Research Network (NASH-CRN) and modified Ishak score for staging of fibrosis were given. The inter-rater agreement levels of the scoring pathologists (Supplementary Table 4), assessed on a set of 20 cases after the scoring-harmonization exercise, were at the upper end of the expected ranges18,19. Spearman’s rho correlation coefficient was used to assess the relationship between modified Ishak fibrosis scores and NASH-CRN fibrosis scores, demonstrating the expected strong positive correlation (rs = 0.98, P < 2.2 × 10−16).
Of the 940 cases, 659 were needle biopsies, 225 were from hepatic resections and 56 were explants for clinically end-stage NAFLD cirrhosis. A clinical histological diagnosis of NASH was given in 455 (48.4%) cases (Extended Data Fig. 1b). Roughly equal numbers of cases were scored at each point of the NASH-CRN fibrosis scale. The relationship of the NAS components and the NASH-CRN fibrosis score are shown in Fig. 1c, and those of the SAF system with modified Ishak fibrosis stage are shown in Extended Data Fig. 1c.
A pixel classifier was trained to identify fat and PSR-positive tissue in scans of PSR-stained sections (Extended Data Fig. 1d). All classified images were manually quality-controlled, and any images containing large fragments of liver capsule, large portal tracts or hilar tissue or with an artifact that was easily ignored during subjective assessment but erroneously computationally classified were removed. The derived fat percentage of the tissue correlated with the assigned steatosis score of the NAS (and SAF) systems, as expected (rs = 0.53, P < 2.2 × 10−16; Extended Data Fig. 1e). The derived PSR-positive percentage of the tissue also correlated with both NASH-CRN (rs = 0.61, P < 2.2 × 10−16; Extended Data Fig. 1f) and modified Ishak (rs = 0.61, P < 2.2 × 10−16; Extended Data Fig. 1g) assigned scores.
Association of histological features with clinical outcomes
The extensive annotation of individual case timelines with clinical data, anchored by the specimen date and retrieved from the EHRs and national datasets, allowed the relationship between data derived from the histological sections and patient outcomes to be examined. Using the 659 needle biopsy cases only, the clear and expected relationship between assigned NASH-CRN fibrosis stage and all-cause mortality was observed (Fig. 1d). Stepwise increases in mortality were evident through progression from stage F0 to F4. An unbiased algorithmic approach to cluster survival curves20 created two clusters (F0,1,2) and (F3,4), supporting the hypothesis that fibrous bridging of vascular structures is a critical pathophysiological event with prognostic importance in progressive fibrogenesis.
Standard Cox regression modeling of all-cause mortality using age at biopsy, gender and NASH-CRN fibrosis stage as covariates showed that age and NASH-CRN fibrosis stage F4 were independently associated with higher all-cause mortality (Supplementary Table 5). Median intervals to death and censoring of included cases are shown in Supplementary Table 6. A ‘pathology scoring only’ model was also examined; only fibrosis stage was predictive of survival on multivariate analysis (Supplementary Table 7).
The predictive value of the computationally derived PSR-positive proportion in the biopsy cases where a value was available was also separately examined. Increased PSR content in a biopsy was associated with increased risk of death, hazard ratio 1.06 (95% confidence interval 1.045–1.075, P < 2 × 10−16). To illustrate the potential value of computational pathology in providing prognostic information about overall survival, maximally selected rank statistics were used to determine the optimal PSR percentage cutpoint that divided samples into high- and low-risk groups; the Kaplan–Meier estimator curves are shown in Extended Data Fig. 1h.
The annotated patient timeline of 183 of the 659 biopsy cases contained an outcome coding for at least one of the events defined by expert consensus to represent decompensation in cirrhotic patients21 or, using UK Operations/Procedure coding data, identifying activity relating to cirrhosis-related hospital admissions22. This high number of events contrasts with a published prospective observational study11. In SteatoSITE, using only the 106 cases for which an event coding related to decompensation was not present on the patient timeline before the biopsy, F3 and F4 NASH-CRN fibrosis stage was predictive of subsequent decompensation (Fig. 1e). Kaplan–Meier estimator curves for liver-related events (excluding death) associated with F0, F1 and F2 were placed in the same cluster by an unbiased clustering approach, but those associated with F3 and F4 were distinct.
Cox regression modeling of hepatic decompensation events on the cause-specific hazard, with death as a competing risk, using age at biopsy, gender and NASH-CRN fibrosis stage, showed that NASH-CRN fibrosis stages F3 and F4 were associated with increased decompensation events (Supplementary Table 8); Fine–Gray regression for the proportional-hazards modeling of the subdistribution hazard is also reported. Median intervals to decompensation or censoring (death or end of follow-up) of included cases are shown in Supplementary Table 9. A ‘pathology scoring only’ model for hepatic decompensation (with death as a competing risk) was also examined; only fibrosis stage was predictive of hepatic decompensation on multivariate analysis (Supplementary Table 10).
The value of the computationally derived PSR-positive proportion in the biopsy cases where a value was available was also separately examined for hepatic decompensation, with death as a competing risk. Increased PSR content in a biopsy was associated with increased risk of hepatic decompensation, hazard ratio 1.07 (95% confidence interval 1.05–1.08, P < 2 × 10−16). To illustrate the potential value of computational pathology in providing prognostic information about hepatic decompensation, maximally selected rank statistics were used to determine the optimal PSR percentage cutpoint that divided samples into high- and low-risk groups; the Kaplan–Meier estimator curves are shown in Extended Data Fig. 1i.
Finally, the development of HCC is a low-frequency outcome in MASLD. The complete SteatoSITE cohort includes 80 patients with a coding of HCC at any point in their annotated timeline. The increased risk of HCC in patients with non-cirrhotic MASLD is also well-recognized23,24. The SteatoSITE cohort includes 227 resection specimens, with HCC being present in the annotated patient timeline in 44 of these. Notably, 36 of these cases did not have histopathological evidence of cirrhosis in the background liver (Extended Data Fig. 1j). Focusing only on the liver biopsy cases in which no event coding for HCC was present before the biopsy, ten patients received a subsequent coding of HCC in the follow-up period included in the data commons, and assigned NASH-CRN F4 fibrosis stage at biopsy was predictive of the subsequent development of HCC (Extended Data Fig. 1k).
Transcriptomic profiling
The SteatoSITE cohort includes high-quality hepatic RNA-seq data in 668 out of the 940 total cases (comprising 538 biopsies, 39 explants and 130 resections), after applying prespecified quality-control criteria appropriate for archival formalin-fixed paraffin-embedded (FFPE) samples, including the percentage of RNA fragments > 200 nucleotides (DV200 ) > 30% (ref. 25). Overall, larger liver-resection tissues yielded poorer RNA quality compared with much smaller needle biopsy specimens, likely related to sample fixation.
Normal liver controls (n = 39) were also retrieved from the biorepositories for comparative RNA-seq analysis. To confirm the suitability of these control samples, we showed that their transcriptional profile strongly correlated with normal liver samples from independent hepatic RNA-seq datasets26,27 (average rs > 0.9, adjusted P < 2 × 10−16; Extended Data Fig. 2). After applying quality-control checks, 34 control samples were used for further analyses.
Extended Data Fig. 2. Validation of normal liver transcriptome.

Correlation (corrplot) of transcriptomic profiles of normal SteatoSITE samples (n = 39) with published normal liver reference data (n = 57). The colour scale corresponds to the strength and direction of correlations.
We performed RNA-seq variant calling to detect the most relevant and replicated single-nucleotide polymorphisms (SNPs) identified as risk modifiers of MASLD progression (rs738409, rs72613567, rs58542926 and rs641738 in the genes patatin-like phospholipase domain containing 3 (PNPLA3), hydroxysteroid 17-beta dehydrogenase 13 (HSD17B13), transmembrane 6 superfamily member 2 (TM6SF2) and membrane-bound O-acyltransferase domain containing 7 (MBOAT7), respectively). The SNP frequency across histological fibrosis stages is shown in Supplementary Table 11. Sequencing coverage of these genes, and therefore the ability to call variants, was variable. However, the rs738409 C>G p.I148M variant in PNPLA3 (the strongest genetic risk factor for MASLD and its severity) was successfully called in 612 of 668 MASLD samples with RNA-seq available. The prevalence of GG, GC and CC genotypes among cases where a call was possible was 16.8%, 27.8% and 54.7%, respectively. After controlling for fibrosis stage, genotype status for predefined variants had no substantial effect on outcomes (data not shown), consistent with findings from a previous long-term follow-up study of 901 individuals with MASLD in which overall mortality was not affected by any genetic variant28.
For differential gene expression analysis, samples were grouped according to NAS and different fibrosis stages (F0/F1, F2, F3 and F4) and compared with normal liver control samples. The data structure was examined by principal component analysis (PCA). As shown in Fig. 2a, the first two principal components (PCs) explained 8.86% and 7.94% of the observed variation in gene expression. There was modest segregation according to the fibrosis stage.
Fig. 2. Hepatic transcriptomic analysis and summary bioinformatics.

a, PCA plot for 616 MASLD samples and 34 normal liver control samples after including batch effects as covariates in the linear model. The first PC is displayed on the x axis and the second PC on the y axis, with the corresponding percentage of total variance explained by each PC. Dots represent individual samples colored according to NASH-CRN fibrosis stage. b, UpSet plot of DEGs showing the unique DEGs belonging to individual sets (fibrosis stages F0/1, F2, F3, F4) and the intersection of DEGs across all fibrosis stages. Set sizes are presented as bars, and their composition is described by the bottom panel. c,d, Dot plots showing the downregulated (c) and upregulated (d) Kyoto Encyclopedia of Genes and Genomes pathways (Benjamini and Hochberg false discovery rate q < 0.05 and fold-change of ≥1) obtained from one-sided gene-set-enrichment analysis for different fibrosis stages (F0/1, F2, F3, F4). The size of the dot is on the basis of gene count enriched in the pathway, and the color of the dot shows the pathway enrichment significance. e, FC of the top 20 differentially expressed genes in each fibrotic stage for both men and women. Exemplar dysregulated genes between men and women are highlighted for illustration. The gray color indicates that the genes were not statistically significantly dysregulated for that specific stage. f, Dot plot highlighting the differences between the Reactome pathways (q < 0.05 and FC ≥1) obtained by one-sided GSEA in men and women in stage F4 compared with controls. setSIZE indicates the total number of genes present in each specific gene set. P.adjust indicates the Benjamini and Hochberg adjusted P values. GeneRatio is k/n, where k is the overlap between the genes of interest and the gene set and n is the number of all unique genes of interest. CAM, cell adhesion molecule; ECM, extracellular matrix; HCM, hypertrophic cardiomyopathy; JAK, Janus kinase; MAPK, mitogen-activated protein kinase; STAT, signal transducer and activator of transcription.
When samples were classified according to fibrosis stage, we found 218, 478, 1,114 and 2,800 differentially expressed protein-coding genes when comparing F0/F1, F2, F3 and F4 livers with normal liver controls, respectively, and 99 genes differentially expressed in common across stages versus controls (Fig. 2b).
Kyoto Encyclopedia of Genes and Genomes pathways linked to extracellular matrix were enriched from stage F2 onwards, as expected. Notably, other upregulated pathways were those linked to cardiomyopathies (q < 0.05; Fig. 2c). In contrast, downregulated genes were enriched for pathways associated with signaling, predominantly in early disease (F0/1), and fatty acid metabolism and peroxisome pathways were enriched from F2 to F4 (q < 0.05; Fig. 2d).
When the samples were grouped according to NAS, we identified 203 differentially expressed genes when comparing NAS ≤ 2 (‘low NAS’) with controls, 621 in the NAS 3–4 group (‘borderline NAS’) and 793 in the NAS > 4 group (‘high NAS’); 182 genes were shared across all NAS groups (Extended Data Fig. 3a). Gene set enrichment analysis (GSEA) revealed, among others, significantly enriched gene ontology terms that participate in ‘translation’ and the ‘immune system’ (q < 0.05; Extended Data Fig. 3b–d).
Extended Data Fig. 3. SteatoSITE RNA-seq according to NAS scores.

a, UpSet plot of differentially assessed genes (DEGs), showing DEGs belonging to individual sets (according to NAFLD activity score (NAS) ≤ 2, NAS 3/4 and NAS ≥ 5) and the intersection of DEGs across all NAS sets. Set sizes are presented as bars and their composition is described by the bottom panel. b, Top 10 molecular functions (gene ontology (GO) terms) identified with one-sided gene set enrichment analysis based on the differentially expressed genes of NAS ≤ 2; c, NAS 3/4; d, NAS ≥ 5 (Benjamin and Hochberg false discovery rate q < 0.05 and fold-change of ≥1).
As previous studies have highlighted the sexually dimorphic nature of MASLD29, we also determined gender-specific differences in gene expression and biological pathway enrichment across the MASLD histological spectrum. After stratifying patients by gender and fibrosis stage, we performed differential gene expression analysis to discern shared and gender-specific molecular profiles. In men, we identified 156, 253, 1,167 and 2,767 differentially expressed genes in F0/F1, F2, F3 and F4, respectively, when compared with controls. In women, we identified 383, 768, 1,285 and 2,997 genes in each fibrosis stage, respectively. This indicates that there are more dysregulated genes in women than men. The top 20 differentially expressed genes in each fibrosis stage for both genders are presented in Fig. 2e. Additionally, we performed enrichment analysis between men and women at each stage of fibrosis and observed gender-specific differences in biological pathways. To illustrate, in Fig. 2f, we show that for stage F4, most of the identified pathways are common to both genders, apart from ‘bile acid and bile salt metabolism’ and ‘phase I-functionalization of compounds’, which are only present in men. The enriched pathways obtained in other fibrosis stages are shown in Extended Data Fig. 4.
Extended Data Fig. 4. SteatoSITE RNA-seq gender-focused gene enrichment analysis.

Reactome pathways obtained from one-sided gene set enrichment analysis in both men and women for a, F0/1 vs control; b, F2 vs controls; c, F3 vs controls (Benjamin and Hochberg false discovery rate q < 0.05 and fold-change ≥ 1).
Hence, SteatoSITE enables a comparison of whole-liver gene expression profiles according to histological disease stage. To maximize the accessibility and utility of this resource, we developed an open-access gene browser (https://shiny.igc.ed.ac.uk/SteatoSITE_gene_explorer/), allowing high-level assessment and visualizations of user-selected gene expression according to fibrosis stage.
Cell-type characterization by single-cell deconvolution
Using single-cell RNA-seq (scRNA-seq), we have previously identified distinct populations of scar-associated monocyte-derived macrophages, mesenchymal cells and endothelial cells that populate the fibrotic niche in patients with advanced cirrhosis and interact to regulate liver fibrogenesis30. However, the presence of specific pathogenic cellular subpopulations across the full MASLD disease spectrum and how these cells relate to clinical outcomes remain uncertain. To address this, we performed deconvolution of the SteatoSITE bulk RNA-seq data using a publicly available fully annotated scRNA-seq reference dataset compiled from healthy and cirrhotic patients30 to estimate the proportions of specific hepatic cell types in each SteatoSITE sample and correlate them with histopathological features and patient outcomes.
This analysis demonstrated that the proportion of hepatic scar-associated macrophages (SAMacs), a key regulator of liver fibrosis30, correlated positively with liver fibrosis stage and steatohepatitis activity across the full MASLD spectrum (Fig. 3a). In contrast, tissue-resident macrophage (Kupffer cell) proportions declined substantially with more advanced fibrosis (Fig. 3a). We validated these transcriptomic findings at the protein level using a bespoke MultiOmyx liver multiplex immunofluorescence (mIF) assay in an independent MASLD histological dataset (n = 43), confirming a statistically significant positive correlation of hepatic HLA-DR+CD9+CD14+ SAMac numbers with liver fibrosis stage (Fig. 3b). Positive correlations between mIF SAMac numbers and steatosis, lobular inflammation and ballooning scores were less pronounced than fibrosis stage (Extended Data Fig. 5a–c), mirroring our findings from the deconvolution analysis (Fig. 3a).
Fig. 3. Hepatic bulk RNA-seq data with clinical annotation.

a, Corrplots of macrophage, mesenchyme, endothelia and B cell subpopulation proportion correlations with NAS components and NASH-CRN fibrosis stage. *P < 0.05, **P < 0.01, ***P < 1 × 10−6. Color and size of circles indicate the Pearson correlation coefficients. b, Representative H&E-stained sections and multiplex immunofluorescent staining to identify subpopulations of SAMacs, Kupffer cells and tissue monocytes. Selected markers shown. Arrowheads indicate stated cell type. Correlation of number of stated cell type per mm2 tissue with fibrosis stage (n = 43; Spearman’s rho and P value indicated). Scale bars, 100 µm. c,d, Corrplots of macrophage, mesenchyme, endothelia and B cell subpopulation proportion correlations with 1-, 3- and 5-year all-cause mortality (c) and decompensation risk (d). *P < 0.05, **P < 0.01. Color and size of circles indicate the Pearson correlation coefficients.
Extended Data Fig. 5. Correlation of scar-associated macrophage numbers with NAFLD activity score components.

Scar-associated macrophage numbers determined by multiplex immunofluorescence correlated positively with assigned components of the NAFLD activity score (n = 43). Spearman’s rho and P-value indicated for ballooning (a), steatosis (b), and inflammation (c).
Deconvolution analysis also revealed statistically significant positive correlations between fibrosis and the proportion of scar-associated mesenchymal cells, hepatic arterial endothelial cells and lymphatic endothelial cells (Fig. 3a), in keeping with the key roles for mesenchymal cell activation and progressive arterialization of the hepatic microcirculation with loss of normal specialized liver sinusoidal endothelial cell phenotype (‘capillarization’) in the development of liver fibrosis across the full MASLD spectrum. Interestingly, expansion of plasma cells was also associated with more advanced hepatic fibrosis (Fig. 3a).
Finally, the proportions of cell types, derived by single-cell deconvolution analysis of SteatoSITE bulk RNA-seq data, were examined for their value in predicting adverse clinical outcomes. Strikingly, increased hepatic proportions of SAMacs, scar-associated mesenchymal cells, hepatic arterial endothelial cells, lymphatic endothelial cells and plasma cells were predictive of higher future all-cause mortality (Fig. 3c) and hepatic decompensation events (Fig. 3d). In contrast, higher proportions of more homeostatic liver resident cell types such as vascular smooth muscle cells and liver sinusoidal endothelial cells were protective against future mortality or hepatic decompensation (Fig. 3c,d). Hence, in addition to histology and bulk transcriptomics, changes in the cellular composition of the liver may offer key prognostic information in patients with MASLD.
Transcriptional risk prediction of hepatic decompensation
The annotated patient timelines in SteatoSITE allow predictive tools to be developed. To demonstrate, we used the SteatoSITE RNA-seq data and associated clinical outcomes to develop a novel transcriptome-based risk-prediction model for hepatic decompensation. Such transcriptional risk scores (TRSs) on the basis of transcript abundance are physiologically closer to the phenotype of interest, require smaller training samples and offer greater portability across diverse ancestry groups than polygenic risk scores using genomic variants (SNPs)31,32.
As the initial feature set, we used the 1,127 protein-encoding genes that were differentially expressed in advanced (F3/F4) compared with early (F0/F1) stage disease (P < 0.05, log fold change (FC) ≥1). Univariate Cox regression identified 955 differentially assessed genes (DEGs) as significantly related to decompensation events (P < 0.01). To develop a sparse feature set, ten runs of a tenfold lasso-regularized Cox regression were performed, and the coefficients for selected genes were derived (Extended Data Fig. 6a,b). The final TRS predicting hepatic decompensation was composed of the expression of 15 genes: metallothionein 1F (MT1F), potassium voltage-gated channel subfamily H member 7 (KCNH7), collagen type XXV alpha 1 chain (COL25A1), RASD family member 2 (RASD2), connective tissue growth factor (CTGF), stanniocalcin 1 (STC1), glial-cell-derived neurotrophic factor (GDNF), paired related homeobox 1 (PRRX1), fibroblast growth factor 7 (FGF7), lipocalin-like 1 (LCNL1), dipeptidase 1 (DPEP1), chordin-like 2 (CHRDL2), LIM homeobox 6 (LHX6), POU class 4 homeobox 1 (POU4F1) and cadherin 16 (CDH16). (Expression across fibrosis stages as plotted by the SteatoSITE gene browser is shown in Extended Data Fig. 7.)
Extended Data Fig. 6. Regularised Cox regression model.

a, Plot of the optimal value of logarithmic lambda (n = 100) and the cross-validation error. Data presented as mean value +/− SD. b, Coefficient values across the logarithmic lambda. c, Kaplan-Meier plot demonstrating no separation of the event curves of patients with biopsies showing F2 fibrosis using the transcriptional risk score; log rank test P-value = 0.82.
Extended Data Fig. 7. Gene expression of the prognostic signature across the fibrotic stages.

Plots of the normalised expression of the named 15 gene constituents of the transcriptional risk score by NASH-CRN fibrosis stage (n = 368), as visualised by the Shiny transcriptome browser app. All boxplots show median (centre line), first and third quartiles (lower and upper box limits), 1.5× interquartile range (whiskers). P-values of one-way ANOVA with post-hoc Tukey test.
The risk scores were calculated using the formula indicated in the Methods. According to the median risk score, patients were split into high- and low-risk groups (Fig. 4a). Interestingly, samples with a higher risk score had not only a higher fibrosis stage but also a higher hepatocyte ballooning score (Fig. 4b). Time-dependent receiver operating characteristic (ROC) curves were used to assess the predictive ability of the model at specific times after biopsy; the areas under the ROC curves (AUCs) were 86.24% (standard error (SE) 5.11), 80.97% (SE 4.62) and 83.26% (SE 3.86) for 1-, 3- and 5-year risk of decompensation events, respectively (Fig. 4c).
Fig. 4. Prediction of risk of future decompensation events in advanced fibrosis using hepatic bulk RNA-seq data.

a, Correlation between the risk scores and the time of the decompensation events. b, Heatmap of the gene expression profile of the prognostic signature. c, Time-dependent ROC curves and AUC analyses by the expression of the 15 genes. d, Kaplan–Meier curves (with 95% confidence intervals) showing high- and low-risk groups for all biopsy cases; log-rank test P value < 0.0001. e, Kaplan–Meier curves demonstrating separation of patients into high- and low-risk groups in both mild (F0/1) and severe (F3/4) fibrosis stages; log-rank test P value.
All the biopsy samples could be stratified using the TRS into high- and low-risk groups with substantially different decompensation trajectories. Over postbiopsy follow-up of up to 20 years, those with a high-risk TRS had a cumulative decompensation event probability of more than 0.65 compared with a cumulative probability of 0.11 in those with a low-risk TRS (Fig. 4d). Next, to derive further prognostic information beyond routine fibrosis staging, samples with mild (F0/F1) or advanced (F3/F4) scarring were stratified using the TRS into groups at high or low risk of future decompensation (Fig. 4e). Although there were insufficient F3/F4 patients with a low TRS to enable further categorization of these patients, application of the TRS augmented risk stratification in MASH patients with early-stage fibrosis. Stratification of biopsies with F2 fibrosis is shown in Extended Data Fig. 6c.
Master regulator analysis of disease progression
We sought to further exploit the rich RNA-seq dataset using the high-risk genes identified as prognostically important components of the TRS to derive further biological understanding of the related transcriptional regulatory network (TRN) in MASLD. The TRN, consisting of transcription factors and regulated target genes, was inferred from the whole-gene-expression dataset. Regulons, a set of genes regulated by a specific transcription factor, were constructed for all transcription factors cataloged by ref. 33. A regulon activity score for each sample was estimated using a two-tailed GSEA approach. Regulons cluster on the basis of activity into two broad groups: those with high activity in advanced fibrosis and low activity in early stages and those showing the opposite pattern (Fig. 5a).
Fig. 5. The relationship of THRB with disease progression.

a, Heatmap of estimated transcriptional network (regulon) activity across the SteatoSITE dataset. b, Patterns of activity and cluster membership of the three regulons (THRB, AEBP2, BNC2). c, Relationships of THRB, AEBP1, BNC2 regulatory networks with significant numbers of component genes from the TRS as gene target members in disease progression. Red edges, positive regulation of gene target; blue edges, negative regulation of gene target; blue nodes, net negative gene regulation; red nodes, net positive gene regulation; gray nodes, net neutral gene regulation; green nodes, TRS gene member. d, Ranked THRB regulon differential activity plot confirms the negative relationship between THRB regulon activity and disease stage (left and center), and the Kaplan–Meier plot indicates that lower THRB regulon activity is predictive of hepatic decompensation (right); log-rank test P value. e, In histologically identical high-risk fibrosis stages (F3 and F4), low THRB activity identifies patients at high risk of hepatic decompensation event; log-rank test P value < 0.0001. f, Example individual two-tailed gene set enrichment plots of target genes in the THRB regulon (left, two-tailed gene set enrichment testing with Benjamini and Hochberg (false discovery rate) adjusted P values < 0.001) from two patients with identical F4 stage (cirrhosis) on PSR-stained sections (right), where the patient with low THRB differential regulon activity (top, blue) progressed to a hepatic decompensation event 224 days after biopsy in contrast to a patient with high THRB regulon activity who did not experience a decompensation event during the 4,500 days until censoring (bottom, red). Scale bars, 2 mm. dES, differential enrichment score.
To identify which transcriptional networks lie upstream of the expression of high-risk genes representing the TRS, master regulator analysis34 was undertaken to identify the statistical significance of the overlap between the regulon (gene targets) of each transcription factor and the 15 genes of the TRS, corrected for multiple hypothesis testing. Three regulons (gene networks regulated by AE binding protein 1 (AEBP1), thyroid hormone receptor beta (THRB) and zinc finger protein basonuclin zinc finger protein 2 (BNC2)) contained substantially greater numbers of the TRS genes than expected by chance, indicating that these three networks may be critical to MASLD disease progression leading to decompensation events.
To examine the patterns of activity of these regulons during progression of disease stage, the mean activity scores for each regulon from each NASH-CRN fibrosis stage were used as pseudo-timeseries data for unsupervised soft clustering. AE binding protein 2 (AEBP2) regulon activity is in a cluster of regulons whose activity is low in F0–2 but increased in F3 and F4, whereas BNC2 regulon activity increases more uniformly from F0 to F4. In contrast, THRB regulon activity is high in F0 and F1 but decreases as scarring progresses from F2 to F4 (Fig. 5b).
The relationship between the inferred AEBP1, THRB and BNC2 regulons is shown in Fig. 5c, annotated with the direction of regulation of each gene target. Most gene targets for AEBP1 and BNC2 are positively regulated, whereas THRB exerts largely negative regulation of gene targets shared with both AEBP1 and BNC2.
Given the potential suppressive role for THRB over disease-promoting core gene targets, and because THRB agonists are in current clinical trials for MASLD, we examined THRB regulon activity in more detail. In biopsy cases with no hepatic decompensation before biopsy, differential regulon activity was estimated and the predictive value of THRB regulon activity for decompensation events examined (Fig. 5d). A ranked THRB regulon differential activity plot confirms the negative relationship between THRB regulon activity and disease stage in the biopsy-restricted subset, and the Kaplan–Meier plot indicates that lower THRB regulon activity is predictive of hepatic decompensation.
However, the predictive value of THRB regulon activity may only reflect association with disease stage. To determine whether THRB regulon activity was predictive of hepatic decompensation in samples of matched fibrosis stages, maximally selected rank statistics were used to determine the optimal THRB regulon activity cutpoint that divided samples into high- and low-risk groups. Using this cutpoint on histologically low-risk samples with F0 and F1 fibrosis, THRB regulon activity identified a subset of MASLD patients with high risk of hepatic decompensation (Extended Data Fig. 8). Deriving a new cutpoint in only the histologically advanced stages F3 and F4 allowed stratification into two distinct groups with either rapid or slower progression to a decompensation event (Fig. 5e).
Extended Data Fig. 8. Additional predictive and personalisation value of estimated THRB regulon activity in F0 and F1 stage disease.
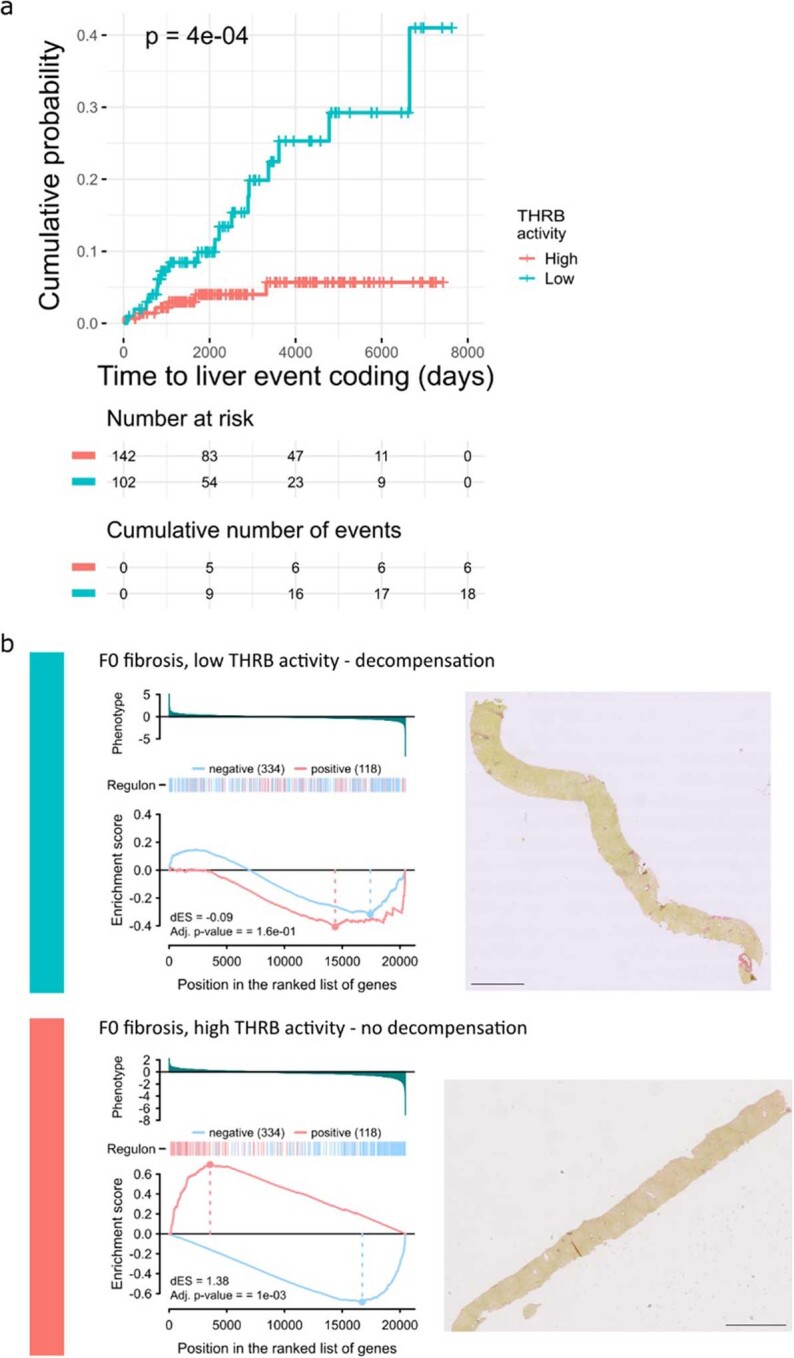
a, In histologically identical low-risk fibrosis stages (F0 and F1 biopsy cases), low THRB activity identifies patients at high risk of progression to a hepatic decompensation event; log rank test P-value. b, Example individual 2-tailed gene set enrichment plots of target genes in the THRB regulon (left, 2-tailed gene set enrichment testing with Benjamini and Hochberg (false discovery rate) adjusted P-values) from two patients with identical F0 stage (no scarring) on PSR stained sections (right), where the patient with low THRB differential regulon activity (top, blue) progressed to a hepatic decompensation event 3000 days after biopsy in contrast to a patient with high THRB regulon activity who did not experience a decompensation event during the 4400 days until censoring (bottom, red). Scale bars, 2 mm.
The potential for a personalized approach, for clinical trials or routine practice, using estimated THRB regulon activity is illustrated by case studies from the SteatoSITE dataset. Two cases scored F4 (cirrhosis) on PSR-stained sections with high and lower estimated THRB activity are shown in Fig. 5f; the patient with estimated THRB regulon activity below the derived threshold (top) had a coded decompensation event 224 days after biopsy, but the patient with THRB activity above the cutpoint (bottom) did not experience a coded decompensation event in more than 4,500 days of follow-up. At the other end of the disease stage spectrum, two cases that scored F0 on the PSR-stained sections are shown in Extended Data Fig. 8; the case with high estimated THRB regulon activity derived from RNA-seq (bottom) was not associated with hepatic decompensation, whereas the case with low estimated THRB regulon activity (top) was.
Discussion
We have established what is to the best of our knowledge a unique resource comprising extensive histopathological assessments, transcriptome analysis and rich clinical data across the full MASLD severity spectrum. SteatoSITE is a dataset of broad utility that will enable discovery science, digital pathology research, artificial intelligence approaches and translational studies in MASLD (a subset of the umbrella grouping of steatotic liver disease (SLD)) in the future. Our approach is complementary to continuing global initiatives in MASLD such as biomarker-focused research being conducted by the Liver Investigation: Testing Marker Utility in Steatohepatitis and Non-Invasive BioMarkers of MetaBolic Liver DiseasE35 consortia, as well as ‘omics repositories such as http://liverproteome.org/ and https://www.livercellatlas.org/index.php that contain MASLD and human subsections, respectively. However, crucially, gene signatures and histopathological metrics in SteatoSITE are linked to clinical outcomes to enable rational diagnostic and treatment approaches for patients with different stages and activity of disease to be determined. Indeed, an authoritative expert review stated that “for more refined treatment of NASH, orthogonal approaches that integrate genetic, clinical and pathological datasets may yield treatments for specific subphenotypes of the disease”36. We believe that SteatoSITE provides the relevant multiscale and multimodal data required to meet this challenge.
Consistent with previously published observational data37, we began by showing the strong positive association between histological fibrosis stage and future clinical outcomes including all-cause mortality, HCC and a composite outcome of hepatic decompensation events. These findings validated the broad content and data integration of the SteatoSITE cohort. We went on to perform a comprehensive bioinformatic analysis of the hepatic bulk RNA-seq data, identifying key genes and enriched gene functions/pathways that characterize discrete histological subtypes and generating disease stage-specific transcriptional profiles to inform drug-target discovery and the development of clinically relevant biomarkers in MASLD. In addition, we provide an open-access web-based gene browser so the scientific community can interactively explore and visualize the SteatoSITE RNA-seq data, thus increasing the accessibility and effect of our work.
As studies have highlighted the sexually dimorphic nature of MASLD38, we also performed gender-specific analyses to explore differences in gene expression and molecular pathways. Across the MASLD disease spectrum, we identified transcriptional differences and gender-biased enrichment of molecular pathways, such as bile acid and bile salt metabolism and O-linked glycosylation in stage F4 livers. Further work is needed to determine the effect of such differences on disease progression in MASLD, as well as their relevance to biomarkers, therapeutic targets and treatment responses in men and women.
The advent of single-cell transcriptomic technologies is transforming our understanding of the pathobiology of liver diseases including MASLD, identifying key pathogenic cell types and candidate therapeutic targets39–41. However, the study of these cell populations across the full disease spectrum remains limited, with minimal data defining which specific cell states are associated with adverse clinical outcomes. Here, we used a single-cell deconvolution approach to interrogate changes in cellular composition in the SteatoSITE cohort, confirming expansion of SAMacs, scar-associated mesenchymal cells, plasma cells, hepatic arterial endothelial cells and lymphatic endothelial cells during MASLD progression. Crucially, expansion of these populations also correlates with poor long-term patient prognosis, highlighting their potential role as therapeutic targets in MASLD and informing novel immunohistochemical approaches to better characterize liver biopsy tissue and identify high-risk patient subphenotypes. Indeed, multiplex immunohistochemistry is already being used to improve stratification in patients with a range of cancers42–44, so future refinement of our liver macrophage-focused high-plex MultiOmyx assay to include specific markers for pathogenic mesenchymal, endothelial and plasma cells will enable a comprehensive assessment of the liver fibrotic niche from a single slide and may facilitate improved identification of high-risk MASLD patients compared to existing histological approaches.
As the individual patient-level datapoints in SteatoSITE are temporally defined, researchers can perform time-to-event predictions using survival-analysis methods. Here, we illustrated this by showing that hepatic gene expression data in our cohort had a high predictive value for risk of future hepatic decompensation events, over and above standard fibrosis staging. The transition from compensated cirrhosis to decompensated cirrhosis occurs at a rate of about 5% to 7% per year45. Decompensation represents a key prognostic inflection point in the natural history of chronic liver disease, as the median survival drops from more than 12 years for compensated cirrhosis to about 2 years for decompensated cirrhosis45. However, in patients with advanced fibrosis or cirrhosis (F3/F4), individual risk of decompensation is variable and hard to predict, and preventative therapies are lacking. On the basis of automated variable selection methods to reduce overfitting, we identified a 15-gene panel and derived a risk score that could classify stage-defined MASLD patients into high- or low-risk groups for decompensation46,47. To the best of our knowledge, there are no TRSs reported in MASLD; but notably, a TRS approach outperformed static genotypic risk assessment in distinguishing Crohn’s disease from healthy samples and predicting complications48. Genes represented in our TRS include five members predicted to be secreted proteins, according to The Human Protein Atlas (https://www.proteinatlas.org), which could be explored further in suitably well-annotated clinical samples. Although not previously reported in MASLD, CHRDL2 is upregulated in a range of tumor tissues including HCC49, while STC1, CTGF50, GDNF and FGF7 are all linked to hepatic fibrogenesis51,52. Additionally, STC1 encodes a secreted glycoprotein reported as a potential serum biomarker for hepatitis B virus-associated liver fibrosis51, and secreted FGF7 protein53 was identified as a prognostic marker in cholangiocarcinoma. The predictive model requires external validation, ideally in prospective longitudinal studies. However, to our knowledge, there are at present no available MASLD datasets with integrated pathology, transcriptomics and clinical outcome data to enable this.
Finally, to search for new and highly effective upstream therapeutic targets, we used high-risk genes from the TRS to uncover core gene networks and master regulators likely to exert influence over disease progression and patient outcomes. Intriguingly, one regulon—THRB—was suppressive of the other two identified (AEBP1 and BCN2, which are linked to hepatic fibrogenesis and MASLD progression54,55). Moreover, THRB regulon activity not only decreased with advancing fibrosis stage but also predicted future hepatic decompensation (beyond standard fibrosis scoring). Liver-targeted thyromimetics selectively activating THRB, such as resmetirom (MGL-3196) and VK2809, increase hepatic fat metabolism and reduce lipotoxicity and have recently emerged as leading pharmacological candidates for the treatment of MASLD56. Indeed, resmetirom57 has advanced to phase 3 trials58, and a MASLD cirrhosis-outcomes trial is underway. Our data reinforce the importance of THRB as a therapeutic target with the potential to affect hard clinical endpoints and illustrate how adding transcriptomic information to histology might further stratify prognosis and enable tailoring of treatment.
The creation of SteatoSITE was facilitated by the Scottish demographics and healthcare infrastructure. Multimodal data were drawn from three of the four regional Safe Havens (trusted research environments), covering 12 of the 14 territorial Health Boards, and thus sampling most of the Scottish population. Scotland has a well-established ecosystem for precision medicine consisting of a stable population base of ~5.5 million people, a significant incidence of major chronic disease (death rates from chronic liver disease in Scotland are 70% higher than the UK average59), a single healthcare provider (National Health Service (NHS) Scotland) and an EHR system operating on a national scale with a unique common field identifier (Community Health Index (CHI) number) allowing characterization and longitudinal follow-up of well-defined patient cohorts; these key elements differentiate Scotland from many other countries.
Given the scale and complexity of SteatoSITE and its use of routine retrospective multicentric clinical data, several technical and organizational challenges and limitations are acknowledged. Leveraging EHR data for MASLD research is recognized as potentially powerful, but consensus guidelines have only emerged over the past three years21,60. Using three different NHS Safe Havens necessitated an exacting and systematic data-cleaning process, including data duplication, human error (for example, incorrect data entry, typographical errors, sample mislabeling), issues with data standardization, errors due to different delimiters/encoding in input files, data formatting and missing data/completeness. Notably, some source data (for example, FibroScan results) are inconsistently recorded across Health Boards and were therefore hard to obtain, even using laborious manual methods. Additionally, there are specific limitations about the cohort itself. Inevitably, using a secondary-care tissue-first selection process introduces inherent spectrum bias. This is a strength in terms of outcome enrichment but means that SteatoSITE will have less value for modeling the population-level natural history of MASLD. Indeed, patient characteristics (higher age and body mass index (BMI)), more frequent comorbidities and baseline disease severity likely explain the higher incidence of outcomes in SteatoSITE compared with other published MASLD cohorts11,61,62. The demographic makeup of the SteatoSITE cohort also lacks ethnic diversity (majority white Scottish), so caution is advised about the generalizability of findings to other geographical areas and ethnic populations. Finally, confounding due to unsuspected63 or poorly recorded alcohol use is a perennial issue in SLD research. To exclude erroneous cases of alcohol-related liver disease in SteatoSITE, we manually reviewed every individual clinical history section of the pathology request forms and each subsequent diagnostic report from the biorepository databases and carefully checked the cause-of-death data. We also collected International Classification of Diseases (ICD)-10 codes for alcohol-use disorders as indicated by consensus guidelines21. However, data on alcohol consumption (in grams per week) and drinking patterns are limited by this study’s retrospective routine/real-world nature and the lack of detailed and standardized recording of alcohol-related data in historical EHRs. Yet even in prospective studies, alcohol is a likely confounder, as highlighted by the measurement of ethyl glucuronide in hair that detected harmful alcohol consumption in 29% of patients with ‘presumed NAFLD’63. SteatoSITE has laid the groundwork for a variety of important research themes in MASLD, providing a rich multimodal database to inform drug and biomarker development64 and a catalog of high-resolution whole-slide images as a valuable testbed for novel digital pathology algorithms. Here we describe SteatoSITE v.1, but our intention is that this resource will be added to and refined, following further research and evolving disease classifications, to enhance its content and operability.
Methods
Regulatory approvals
Anonymized tissue was supplied after approval by the National Health Service Research Scotland (NRS) Biorepository Network (Reference: SR1032; 2 August 2018). Unified transparent approval for unconsented data inclusion in this pan-Scotland project was provided by the West of Scotland Research Ethics Committee 4 (Reference: 20/WS/0002; 18 February 2020), Public Benefit and Privacy Panel for Health and Social Care (PBPP; Reference: 1819-0091; 4 June 2021), Institutional Research & Development departments and Caldicott Guardians.
Study cohort and sample collection
Initial case selection was on the basis of the availability of archival liver tissue (from biopsies, resections or explants that were surplus to diagnosis) in FFPE blocks available in the NRS Biorepository Network, with the clinical and/or histological diagnosis of NAFLD (MASLD) and meeting the following inclusion/exclusion criteria:
(1) Inclusion criteria—men or women; >18 years of age at the time of tissue sampling; all ethnic groups, socioeconomic backgrounds and health status; dead or alive at the time of inclusion into the data commons.
(2) Exclusion criteria—cases were excluded if any of the following applied: documented history of chronic liver disease of any non-MASLD etiology, including alcohol-related liver disease, chronic viral hepatitis, hemochromatosis, Wilson’s disease, autoimmune hepatitis, primary biliary cholangitis, or primary sclerosing cholangitis, and patients with excessive alcohol use documented in the clinical data supplied on the specimen request form (>21 units per week for men, >14 units per week for women) or histological features indicating a secondary non-MASLD diagnosis.
FFPE blocks of normal non-lesional liver from 39 control cases, defined as liver samples without fat deposition or any active primary parenchymal disease, were identified from resection specimens.
Clinical data extraction and processing
After approval from the NHS Scotland PBPP, we obtained relevant unconsented clinical data from three of the four regional Safe Havens that cover 12 of the 14 territorial health boards in Scotland, including demographics (age, gender, ethnicity, Scottish Index of Multiple Deprivation (SIMD)), diagnostic coding (ICD-9, ICD-10) for inpatient and outpatient episodes and procedures (OPCS Classification of Interventions and Procedures (OPCS-4)), routine laboratory blood tests (biochemistry and hematology), liver transient elastography (FibroScan) where available, prescribing information and cancer and death registry data. The comprehensive list of variables (including ICD codes) is shown in Supplementary Table 1. To maximize future comparability and generalizability of results across studies, we followed recent expert consensus guidelines for using administrative coding in EHR-based research of MASLD21.
The EHR data was uploaded to the Precision Medicine Scotland-Innovation Centre (PMS-IC) Healthcare Landing Zone from the NHS Safe Havens as comma-separated value (.csv) files. In areas where the Safe Havens had poor coverage from their national or regional datasets, further data files were provided by the Biorepository Network. In total, 38 .csv files containing 1,450 columns and 1,055,618 rows were supplied for processing. On upload, data was quality controlled, processed and further minimized to comply with regulatory requirements and ethical approvals.
A Python (v.3.9) script was developed to carry out these data-processing tasks. These included data cleaning, data restructuring and the standardization of file inputs to resolve differences in file encoding, text delimitation and formatting. Where appropriate, differences in data labeling were resolved by adopting the naming conventions used by the NHS Greater Glasgow and Clyde Safe Haven. After standardizing case and spacing, 962 distinct columns remained from the original 1,450 columns. After a more detailed review, columns containing overlapping clinical information were relabeled and merged. This further reduced the dataset to 534 distinct columns, which are present in the final project database.
A final key requirement of the pseudonymization processing was to ensure that all event dates were obfuscated to minimize reidentification risk while allowing for clinically meaningful patient timelines to be established. To achieve this, the duration of time between the specimen acquisition date and the clinical event recorded in the EHR was calculated as Δt in seconds and stored.
Histopathology methods
Blocks for which sufficient tissue was available were assigned a study ID, and samples were cut for histology (3 × 4 µm) and, where possible, RNA extraction (4 × 10 µm). Unstained sections were sent for histological staining by the NHS Lothian pathology laboratory and curls for RNA extraction at the Genetics Core in the Edinburgh Clinical Research Facility. Only NRS biorepository staff independent of the study group held the participant key; they passed data-linkage documents to the local NHS Safe Havens, which were also independent of the study group. No information from the original clinical pathology report was supplied with the sections.
One section from each case was H&E-stained, and one section was PSR-stained. Stained sections of control cases were reviewed to ensure that no histopathological abnormalities were present. Stained sections of MASLD-spectrum cases were scanned at ×20 depth on a Hamamatsu NanoZoomer whole-slide scanner. Whole-slide image files are available as part of the SteatoSITE resource to allow further or repeat subjective or computational analysis by users.
Raw .ndpi files of H&E- and PSR-stained MASLD-spectrum cases were uploaded to a custom server installation of OMERO65 hosted by PMS-IC (Glasgow, UK).
In June 2019, before SteatoSITE case scoring, three consultant pathologists and participants in the UK National Liver External Quality Assurance scheme (T.J.K., G.K., P.K.) undertook an in-person scoring-harmonization exercise during which examples of histological features scored in the NAS18 and SAF66 scoring systems and for staging using NASH-CRN and modified Ishak67 systems were reviewed. To assess inter-rater performance following this meeting, scans of H&E- and PSR-stained sections from 20 MASLD-spectrum cases including biopsies, explants and resections were each scored by the three pathologists. For ordinal variables, Light’s kappa (square weighted, for >2 raters) was calculated using the ‘psy’68 package (v.1.2), and intraclass correlation coefficient, Krippendorf’s alpha and Kendall’s W were calculated using the ‘irr’69 package (v.0.84.1). For the single scored categorical variable (histological diagnosis of NASH), Light’s kappa and Krippendorf’s alpha were calculated.
Each pathologist was randomly allocated one-third of the cases and scored the whole-slide images in the OMERO platform, entering the scores as key–value pairs associated with each image (NAS and SAF features for H&E, NASH-CRN and modified Ishak scores for PSR).
A duplexed classification script was applied to the raw .ndpi scans of PSR-stained sections. An initial ‘whole tissue’ classifier was applied, and small artifacts on the image were removed from the mask on the basis of size. A second pixel classifier of random trees (‘RTrees’) type with ‘gaussian’ and ‘weighted deviation’ features selected, pretrained by one pathologist (T.J.K.), was applied to classify pixels into the histological classes ‘fat’, ‘psr’ and ‘other tissue’ in the ‘whole tissue’ masked area, and the total number of pixels for each class and the percentage fat and PSR-positive pixels were returned. Whole-slide scan processing was undertaken using command-line execution of a QuPath (v.0.2.3)70 script on the University of Edinburgh’s Edinburgh Compute and Data Facility Linux compute cluster (Eddie). Classified images were reviewed by one pathologist (T.J.K.), independent of all other data, and cases were excluded if large amounts of liver capsule or other artifacts had been included in the classification.
Time-to-event analysis
Analysis using clinical and histopathological data only was undertaken in R71 (v.4.1.0) using the packages ‘survival’ (v.3.3-1)72, ‘survminer’ (v.0.4.9)73 and ‘finalfit’ (v.1.0.5)74. Only needle biopsy cases were used for time-to-event analysis. Decompensation events in the clinical data extract were those defined by a combination of ICD codes and UK OPCS-4 codes identifying activity relating to cirrhosis-related hospital admissions activity21,22. Decompensation and HCC event analysis was only undertaken on biopsy cases for which the first decompensation or HCC-related coding was present in the clinical data extract after the biopsy date; and analysis was undertaken using death as a competing risk using Cox regression on the cause-specific hazard and Fine–Gray regression for proportional-hazards modeling of the subdistribution hazard as a recommended comprehensive approach75. Kaplan–Meier estimator curves of all-cause mortality or event for assigned NASH-CRN fibrosis stages were compared by log-rank testing corrected for multiple comparisons using the Benjamini and Hochberg method in ‘survminer’72. Clustering of survival/event curves was determined using a k-means bootstrapped method20. Hazard ratios and 95% confidence intervals were derived from standard Cox regression models comparing mortality and rates of new-onset decompensation events with NASH-CRN stage, age at biopsy date and gender as covariates. The assumption of proportional hazards for fitted Cox regression models was verified by examining plots of scaled Schoenfeld residuals against time for each covariate.
Multiplex immunofluorescence
mIF staining was performed using the MultiOmyx platform according to ref. 76. This step was performed using a single 4 μm FFPE slide: for each staining round, two cyanine dye-labeled (Cy3, Cy5) antibodies were paired. A custom multiplex panel was created consisting of 17 markers, for a total of nine rounds of antibody staining performed in sequence on the FFPE slides. In the cases of triggering receptor expressed on myeloid cells 2 (TREM2) and CD9 antigen (CD9), which were applied as primary–secondary antibodies, samples were incubated with primary TREM2 and CD9 antibody followed by incubation with a species-specific secondary antibody conjugated to cyanine 5 or 3 (Cy5 or Cy3). The staining signal was then imaged and followed by a dye-inactivation step, enabling repeated rounds of staining. The proprietary deep learning–based workflow NeoLYTX (v.2.0) was subsequently applied to identify individual cells and perform cell classification for each marker, and the phenotype of each cell was determined through coexpression analysis.
Antibodies for MultiOmyx analysis, by staining order, were rabbit anti-TREM2 (polyclonal, ProteinTech, Catalog no. 13483-1-AP, Vendor Lot ID NG) mouse anti-MNDA (253A, Abcam, Catalog no. ab270556, Vendor Lot ID GR3326911), rabbit anti-CD9 (EPR2949, Abcam, Catalog no. ab195422, Vendor Lot ID GR3282696), mouse anti-CD66b (G10F5, BioLegend, Catalog no. 93231, Vendor Lot ID B276347), mouse anti-CD11B (238439, R&D Systems, Catalog no. MAB16992, Vendor Lot ID KGZ0418101), rabbit anti-DC-SIGN (D7F5C, Cell Signaling Technology, Catalog no. 13193, Vendor Lot ID 2), rabbit anti-Ki67 (SP6, Abcam, Catalog no. ab231172, Vendor Lot ID GR3277378), rabbit anti-IDO1 (SP260, Abcam, Catalog no. ab228468, Vendor Lot ID GR3208566), rabbit anti-CD11c (D3V1E, Cell Signaling Technology, Catalog no. 45581BF, Vendor Lot ID 2), rabbit anti-PD-L1 (SP142, Abcam, Catalog no. ab236238, Vendor Lot ID GR3246745), rabbit anti-CD14 (EPR3652, Abcam, Catalog no. ab209971, Vendor Lot ID GR316076), mouse anti-CD16 (DJ130c, Thermo Fisher Scientific, Catalog no. MA1-84008, Vendor Lot ID TK2673378), mouse anti-CD68 (KP1, BioLegend, Catalog no. 98998, Vendor Lot ID B297229), mouse anti-CD163 (EDHu-1, Bio-Rad, Catalog no. MCA1853, Vendor Lot ID 149022A), mouse anti-HLA DQ/DR/DP (WR18, Novus Biologicals, Catalog no. NB100-64358, Vendor Lot ID 1808), mouse anti-CD33 (44M12D3, Novus Biologicals Catalog no. NBP2-22377, Vendor Lot ID 1127455612D3) and mouse anti-SMA (1A4, Sigma-Aldrich, Catalog no. A5228, Vendor Lot ID 037M4805V).
RNA-seq methods
RNA extraction was performed by the Genetics Core (Clinical Research Facility, Western General Hospital, Edinburgh, UK) from 4 × 10 µm curls of FFPE archival human NAFLD liver, using the Qiagen miRNeasy FFPE Kit according to the manufacturer’s instructions. Quality control was by Qubit to measure RNA yield (and any potential DNA contamination) and Agilent Bioanalyzer to assess DV200. Samples with DV200 below 30% were not progressed for sequencing.
Libraries were prepared at PMS-IC using the low-input Takara Bio SMARTer Stranded Total RNA-Seq Kit v.2 (Pico Input Mammalian).
Sequencing data were generated by Edinburgh Genomics (University of Edinburgh, UK) using the Illumina NovaSeq 6000 platform. Libraries were sequenced over a total of 22 S2 flow cells. Reads were trimmed using ‘Cutadapt’ (v.cutadapt-1.9.dev2)77 and aligned to the reference genome (GRCh38) using ‘STAR’ (v.2.5.2b)78. Reads were assigned to features using ‘featureCounts3’ (v.1.5.1)79 with a .gtf file from Ensembl (annotation v.84).
For further analyses, R (v.4.1.2) was used. Samples with fewer than one million counts or 70% mapped reads were removed. Any specimen found to include HCC on histological review was excluded from further RNA-seq analyses.
Reads were normalized using the weighted trimmed mean of M values method80. Differential analysis was carried out with ‘limma-voom’ (v.3.28.14) with the protein-coding genes. Statistical significance of genes was determined by an adjusted P value according to the Benjamini–Hochberg procedure of P < 0.05 and absolute FC ≥1 (ref. 81).
PCA was performed to identify covariates that significantly correlated with the main principal components so they could be controlled for downstream analyses (Extended Data Fig. 9). For this reason, gender was included as an additive effect in the linear model used for differential expression when comparing fibrotic stages and NAS score. GSEA82 was performed with ‘clusterProfiler’ (v.4.0.5)83. Data were visualized with ‘ggplot2’ (v.3.3.5)84 and ‘clusterProfiler’.
Extended Data Fig. 9. Correlations between the main principal components and the clinical data.
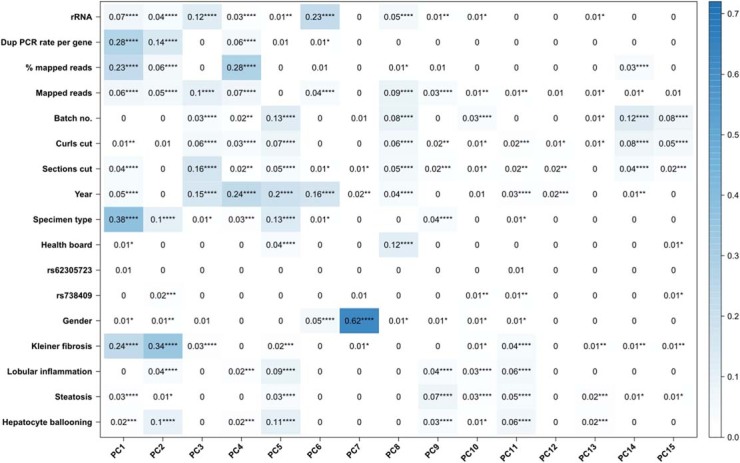
P-values of Spearman correlation test. * P < 0.05, ** P < 0.01, ** P < 0.001, **** P < 0.0001.
For genes PNPLA3, HSD17B13, TM6SF2 and MBOAT7 containing prespecified variants (rs738409, rs72613567, rs58542926 and rs641738, respectively), the Genome Analysis Toolkit (v.4.0.1.2)85 was used to call genotypes at all sites. The ‘HaplotypeCaller’ tool from the Genome Analysis Toolkit was used to produce a single genomic variant call format (GVCF) file per sample, with the following parameters: ‘--standard-min-confidence-threshold-for-calling 20’; ‘-dont-use-soft-clipped-bases’; ‘-L genes.bed’; ‘-ERC GVCF’, where ‘genes.bed’ is a bed file containing the genome coordinates of the genes of interest (padded by 1 KB in both directions to cover upstream and downstream SNPs). The ‘GenotypeGVCFs’ tool was then run with the parameter ‘--include-non-variant-sites’ to produce a single VCF file per sample. ‘SnpEff’ was used to functionally annotate the variants in the VCF files.
The Multi-subject Single-cell Deconvolution tool (v.0.1.1)86 was run using R (v.3.6.3) to compare the counts from each bulk RNA-seq sample to reference scRNA-seq data from five healthy and five cirrhotic livers30 to estimate the proportions of each subtype of various lineages of non-parenchymal cells. This was done separately for each of the macrophage (non-circulating), B cells, mesenchyme, endothelial and T-cell lineages. In each analysis, only the scRNA-seq data from cells annotated as belonging to the lineage were used as a reference in the deconvolution, so the sum of estimated proportions of subtypes in each lineage equals one.
To assess the association between histological scores and proportions of various cell subtypes, the Spearman’s partial rank correlation coefficient was calculated using the R package ‘ppcor’ (v.1.1) between the proportion of each subtype and the histological score, accounting for age and gender. Gender was converted to a numerical value by setting ‘male’ to ‘1’ and ‘female’ to ‘2’. The histological data contained the ‘fibrosis stage’, which includes scores ‘1a’, ‘1b’ and ‘1c’, with ‘1a’ being the least severe and ‘1c’ being the most severe. To perform a rank correlation, these scores were converted to 1, 1.25 and 1.5, respectively. The association between proportion of each subtype and patient outcome (all-cause mortality or a decompensation event) was also assessed using Pearson’s correlation. For any given timepoint, samples were given a ‘0’ if the event had not occurred or a ‘2’ if the event had occurred and were excluded if the patient was censored at that time.
Samples with fewer than one million reads assigned to genes in the bulk RNA-seq analysis, that lacked a patient age or that did not come from a biopsy were omitted.
DEGs between F3/F4 and F0/F1 biopsy cases were identified with ‘limma-voom’ in R. The criteria for statistical significance were adjusted P < 0.05 and absolute FC ≥ 1. Univariate Cox analysis was carried out to determine the DEGs associated with decompensation events according to an adjusted P < 0.01. Next, lasso-penalized Cox regression was used to filter features using ‘glmnet’ . Patients were divided into high- and low-risk groups according to the median score. Kaplan–Meier analysis was performed using ‘survival’ (v.3.4-0) and ‘survminer’ (v.0.4.9) packages. A time-dependent ROC curve was created with the ‘timeROC’ package (v.0.4) to evaluate the predictive ability of the signature.
Transcriptional network inference and regulon analysis was undertaken in R (v.4.1.0) using the ‘RTN’ package (v.2.16.0)87. Normalized gene expression of all sequenced SteatoSITE samples was used to infer regulons corresponding to all TFs compiled by ref. 33 by permutation analysis with the non-parametric estimator of mutual information and a P value cut-off of 1.75 × 10−7 to correct for multiple hypothesis testing. Unstable gene–TF interactions were removed by bootstrap analysis to produce a consensus network. The ARACNe algorithm88 using the data-processing inequality theorem to enrich regulons with direct TF–gene target interactions was used to remove the weakest interaction in any triplet composed of two TFs with a common gene target; the threshold for filtering was determined from the null distribution derived in the permutation and bootstrapping steps.
To determine regulon activity scores for each sample, the tni.gsea2() function was used to calculate differential expression of each gene relative to the expression in the entire cohort, and the ranked list of genes was used to form a differential expression signature that was used alongside the TRN to determine differential sample-specific regulon activity.
Regulon activity profiles across disease stage were placed into four clusters by unsupervised soft clustering using the ‘Mfuzz’ package (v.2.52.0). The fuzzifier parameter, ‘m’, was directly estimated from the data using mestimate() to apply the method of ref. 89.
The ‘RTNsurvival’ package (v.1.16.0)90 was used to undertake time-to-event analysis with THRB regulon activity. The cutpoint for THRB regulon activity was calculated using surv_cutpoint() from the ‘survminer’ package, applying maximally selected rank statistics of the ‘maxstat’ package (v.0.7-25) with a minimum proportion of 0.25.
We developed an open-access gene browser on the basis of the START app91, which will ultimately be hosted on the SteatoSITE website (https://steatosite.com/). The ‘shiny’ R package allows the production of interactive plots and charts and is already widely used to allow users to explore RNA-seq data and create their own customized figures. A ‘shiny’ interface was created to allow scientists to visualize the results of the analysis of differential gene expression according to NASH-CRN fibrosis stage.
Inclusion and ethics statement
SteatoSITE was a consortium project involving the University of Edinburgh, University of Glasgow, NHS Greater Glasgow and Clyde and Eagle Genomics; roles and responsibilities were agreed on among collaborators ahead of the research. The study has included local researchers throughout the research process: study design, study implementation, data ownership, intellectual property and authorship.
Reporting summary
Further information on the research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-023-02602-2.
Supplementary information
Supplementary Tables 1–11.
Acknowledgements
We are grateful to the following for assistance: L. McMahon, M. McNeil (PMS-IC)—project management, data compliance and security, workspace provision; B. Hartzoulakis (Eagle Genomics) and D. Harbison (PMS-IC)—acquisition of funding; Y. Staeva (Eagle Genomics)—project management; M. Jones (PMS-IC)—RNA-seq libraries; E. Coulston (PMS-IC)—administrative support; Edinburgh Clinical Research Facility Genetics Core/L. Murphy—RNA extraction; Edinburgh Genomics/M. Arno—RNA-seq; electronic Data Research and Innovation Service (eDRIS)/B. Bruce—clinical data access approval; Safe Havens: Lothian (C. Duncan, P. Linksted), Grampian (K. Wilde, J. Lumsden), GG&C (J. Burnie, S. McKenna)—EHR data retrieval; Lothian Bioresource (L. McLeod, C. Marshall)—human liver tissue access; E. McDowall (Institute of Genetics and Cancer)—‘shiny’ app hosting; SteatoSITE group (Chairperson L. Laidlaw: visiting fellow, University of Bournemouth). This work was funded by Innovate UK ((Precision medicine: impacting through innovative technology (Reference: TS/R017581/1; J.A.F. and T.J.K.)), Innovate UK Eureka (Reference: 105976; J.A.F. and T.J.K.), Guts UK Development Grant (Reference: DGO2019_16; J.A.F., T.J.K., P.R. and F.T.), industrial Collaborative Awards in Science and Engineering (iCASE) PhD studentship funded by the Medical Research Council Precision Medicine Doctoral Training Programme (Reference: MR/R01566X/1; M.J.-R.) and Galecto Biotech (M.J.-R.), Engineering and Physical Sciences Research Council (EPSRC) Innovation Fellowship (Reference: EP/S001921/1; F.M.), EPSRC New Investigator Award (Reference: EP/R035350/1; F.M.), UK Research and Innovation Engineering Biology Transition Award (Reference: BB/W014610/1; F.M.), Wellcome Trust-University of Edinburgh Institutional Strategic Support Fund (Reference: 204804/Z/16/Z; L.B.), EPSRC Postdoctoral Fellowship (Reference: EP/P017134/1; L.B.) and MRC Senior Clinical Fellowship (Reference: MR/W015919/1; P.R.).
Extended data
Author contributions
J.A.F., T.J.K. and M.D.M. contributed to the development of the study concept and design, and J.A.F., T.J.K., M.J.-R., F.T., H.E., G.K., P.K., P.R., M.D.M., D.R.D., L.B., F.M., A.J.-J., J.M. and I.N.G. contributed to data analysis and interpretation. J.A.F. and T.J.K. drafted the initial manuscript. T.J.K., G.K., P.K., K.A.O., C.M., D.A., D.R.D., M.A. and M.D.M. provided technical and/or material support for the project. J.A.F. is the guarantor of the work. Each author contributed important intellectual content during manuscript drafting or revision and accepts accountability for the overall work by ensuring that questions pertaining to the accuracy or integrity of any portion of the work are appropriately investigated and resolved. All authors approved the final version of the manuscript. The corresponding author attests that all the listed authors meet the authorship criteria and that no others meeting the criteria have been omitted.
Peer review
Peer review information
Nature Medicine thanks Maja Thiele, Silvia Sookoian and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Jennifer Sargent, in collaboration with the Nature Medicine team.
Data availability
Hepatic bulk RNA-seq data are deposited in the European Nucleotide Archive (https://www.ebi.ac.uk/ena; study accession number: PRJEB58625). Gene expression data are also freely available for user-friendly interactive browsing online at https://shiny.igc.ed.ac.uk/SteatoSITE_gene_explorer/. SteatoSITE has delegated ethics from West of Scotland Research Ethics Committee 4 (Reference: 20/WS/0002; 18 February 2020) allowing the granting of access to the full dataset (histopathology scoring, hepatic bulk RNA-seq data, EHR data) only in the PMS-IC secure environment to third parties by application (full details at https://steatosite.com/researchers/), overseen and reviewed by the SteatoSITE Scientific Advisory Board.
Code availability
The codes used for the data analyses in this study can be made available by contacting the corresponding author. Access to codes will be granted for requests for academic use within four weeks of application. R is required software to use the code.
Competing interests
T.J.K. serves as a consultant for or has received speakers’ fees from Resolution Therapeutics, Clinnovate Health, Perspectum and Incyte Corporation. P.R. serves as a consultant for Merck and has received research grant funding from Genentech. J.A.F. serves as a consultant or advisory board member for Resolution Therapeutics, Kynos Therapeutics, Ipsen, Redx Pharma, River 2 Renal Corp., Stimuliver, Galecto Biotech, Global Clinical Trial Partners and Guidepoint and has received research grant funding from Intercept Pharmaceuticals and Genentech. A.J.-J. is an employee and stock owner at NeoGenomics. I.N.G. serves as an advisory board member for Resolution Therapeutics and has received research grant funding from Gilead Sciences. M.D.M. and D.R.D. have a controlling shareholder interest in Biodev Ltd. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Timothy J. Kendall, Maria Jimenez-Ramos.
Extended data
is available for this paper at 10.1038/s41591-023-02602-2.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-023-02602-2.
References
- 1.Rinella, M. E. et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. Hepatology10.1097/HEP.0000000000000520 (2023). [DOI] [PubMed]
- 2.WHO European Region. SDR, chronic liver disease and cirrhosis, all ages, per 100 000. European Health Information Gatewayhttps://gateway.euro.who.int/en/indicators/hfa_236-1860-sdr-chronic-liver-disease-and-cirrhosis-all-ages-per-100-000/ (2021).
- 3.Lazarus JV, et al. The global NAFLD policy review and preparedness index: are countries ready to address this silent public health challenge? J. Hepatol. 2022;76:771–780. doi: 10.1016/j.jhep.2021.10.025. [DOI] [PubMed] [Google Scholar]
- 4.NHS Blood and Transplant Annual Report and Accounts 2018/19 (NHS Blood and Transplant, 2019).
- 5.Le MH, et al. Forecasted 2040 global prevalence of nonalcoholic fatty liver disease using hierarchical Bayesian approach. Clin. Mol. Hepatol. 2022;28:841–850. doi: 10.3350/cmh.2022.0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184:2537–2564. doi: 10.1016/j.cell.2021.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Cai J, Zhang X-J, Li H. The role of innate immune cells in nonalcoholic steatohepatitis. Hepatology. 2019;70:1026–1037. doi: 10.1002/hep.30506. [DOI] [PubMed] [Google Scholar]
- 8.Anstee QM, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 2020;73:505–515. doi: 10.1016/j.jhep.2020.04.003. [DOI] [PubMed] [Google Scholar]
- 9.Loomba R, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 2017;25:1054–1062.e5. doi: 10.1016/j.cmet.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meijnikman AS, et al. Microbiome-derived ethanol in nonalcoholic fatty liver disease. Nat. Med. 2022;28:2100–2106. doi: 10.1038/s41591-022-02016-6. [DOI] [PubMed] [Google Scholar]
- 11.Sanyal AJ, et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 2021;385:1559–1569. doi: 10.1056/NEJMoa2029349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Angulo P, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015;149:389–397.e10. doi: 10.1053/j.gastro.2015.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vilar-Gomez E, et al. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology. 2018;155:443–457.e17. doi: 10.1053/j.gastro.2018.04.034. [DOI] [PubMed] [Google Scholar]
- 14.Rowe IA, Parker R. The placebo response in randomized trials in nonalcoholic steatohepatitis simply explained. Clin. Gastroenterol. Hepatol. 2022;20:e564–e572. doi: 10.1016/j.cgh.2021.05.059. [DOI] [PubMed] [Google Scholar]
- 15.Ratziu, V. & Friedman, S. L. Why do so many nonalcoholic steatohepatitis trials fail? Gastroenterology10.1053/j.gastro.2020.05.046 (2020). [DOI] [PubMed]
- 16.Chopra H, Baig AA, Gautam RK, Kamal MA. Application of artificial intelligence in drug discovery. Curr. Pharm. Des. 2022 doi: 10.2174/1381612828666220608141049. [DOI] [PubMed] [Google Scholar]
- 17.Asiimwe R, et al. From biobank and data silos into a data commons: convergence to support translational medicine. J. Transl. Med. 2021;19:493. doi: 10.1186/s12967-021-03147-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleiner DE, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 19.Jung ES, et al. Interobserver agreement on pathologic features of liver biopsy tissue in patients with nonalcoholic fatty liver disease. J. Pathol. Transl. Med. 2016;50:190–196. doi: 10.4132/jptm.2016.03.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villanueva NM, Sestelo M, Meira-Machado L. A method for determining groups in multiple survival curves. Stat. Med. 2019;38:866–877. doi: 10.1002/sim.8016. [DOI] [PubMed] [Google Scholar]
- 21.Hagström H, et al. Administrative coding in electronic health care record-based research of NAFLD: an expert panel consensus statement. Hepatology. 2021;74:474–482. doi: 10.1002/hep.31726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Innes H, et al. Performance of routine risk scores for predicting cirrhosis-related morbidity in the community. J. Hepatol. 2022;77:365–376. doi: 10.1016/j.jhep.2022.02.022. [DOI] [PubMed] [Google Scholar]
- 23.Piscaglia F, et al. Clinical patterns of hepatocellular carcinoma in nonalcoholic fatty liver disease: a multicenter prospective study. Hepatology. 2016;63:827–838. doi: 10.1002/hep.28368. [DOI] [PubMed] [Google Scholar]
- 24.Stine JG, et al. Systematic review with meta-analysis: risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018;48:696–703. doi: 10.1111/apt.14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evaluating RNA Quality from FFPE Samples (Illumina, 2015).
- 26.Govaere O, et al. Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 2020;12:eaba4448. doi: 10.1126/scitranslmed.aba4448. [DOI] [PubMed] [Google Scholar]
- 27.Suppli MP, et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019;316:G462–G472. doi: 10.1152/ajpgi.00358.2018. [DOI] [PubMed] [Google Scholar]
- 28.Holmer M, et al. Effect of common genetic variants on the risk of cirrhosis in non-alcoholic fatty liver disease during 20 years of follow-up. Liver Int. 2022;42:2769–2780. doi: 10.1111/liv.15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vandel J, et al. Hepatic molecular signatures highlight the sexual dimorphism of nonalcoholic steatohepatitis (NASH) Hepatology. 2021;73:920–936. doi: 10.1002/hep.31312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramachandran P, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. 2019;575:512–518. doi: 10.1038/s41586-019-1631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu X, et al. Polygenic transcriptome risk scores for COPD and lung function improve cross-ethnic portability of prediction in the NHLBI TOPMed program. Am. J. Hum. Genet. 2022;109:857–870. doi: 10.1016/j.ajhg.2022.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang Y, et al. Polygenic transcriptome risk scores (PTRS) can improve portability of polygenic risk scores across ancestries. Genome Biol. 2022;23:23. doi: 10.1186/s13059-021-02591-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lambert SA, et al. The human transcription factors. Cell. 2018;172:650–665. doi: 10.1016/j.cell.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 34.Carro MS, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanyal, A. J. et al. Diagnostic performance of circulating biomarkers for non-alcoholic steatohepatitis. Nat. Med.10.1038/s41591-023-02539-6 (2023). [DOI] [PMC free article] [PubMed]
- 36.Friedman SL, Pinzani M. Hepatic fibrosis 2022: unmet needs and a blueprint for the future. Hepatology. 2022;75:473–488. doi: 10.1002/hep.32285. [DOI] [PubMed] [Google Scholar]
- 37.Ng, C. H. et al. Mortality outcomes by fibrosis stage in nonalcoholic fatty liver disease: a systematic review and meta-analysis. Clin. Gastroenterol. Hepatol.10.1016/j.cgh.2022.04.014 (2023). [DOI] [PMC free article] [PubMed]
- 38.Burra P, et al. Clinical impact of sexual dimorphism in non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) Liver Int. 2021;41:1713–1733. doi: 10.1111/liv.14943. [DOI] [PubMed] [Google Scholar]
- 39.Ramachandran P, Matchett KP, Dobie R, Wilson-Kanamori JR, Henderson NC. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020;17:457–472. doi: 10.1038/s41575-020-0304-x. [DOI] [PubMed] [Google Scholar]
- 40.Chu AL, Schilling JD, King KR, Feldstein AE. The power of single-cell analysis for the study of liver pathobiology. Hepatology. 2021;73:437–448. doi: 10.1002/hep.31485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace SJ, Tacke F, Schwabe RF, Henderson NC. Understanding the cellular interactome of non-alcoholic fatty liver disease. JHEP Rep. 2022;4:100524. doi: 10.1016/j.jhepr.2022.100524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Buisseret L, et al. Clinical significance of CD73 in triple-negative breast cancer: multiplex analysis of a phase III clinical trial. Ann. Oncol. 2018;29:1056–1062. doi: 10.1093/annonc/mdx730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bosisio FM, et al. Next-generation pathology using multiplexed immunohistochemistry: mapping tissue architecture at single-cell level. Front. Oncol. 2022;12:918900. doi: 10.3389/fonc.2022.918900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilie M, et al. Automated chromogenic multiplexed immunohistochemistry assay for diagnosis and predictive biomarker testing in non-small cell lung cancer. Lung Cancer. 2018;124:90–94. doi: 10.1016/j.lungcan.2018.07.037. [DOI] [PubMed] [Google Scholar]
- 45.D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J. Hepatol. 2006;44:217–231. doi: 10.1016/j.jhep.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 46.Bianco C, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J. Hepatol. 2021;74:775–782. doi: 10.1016/j.jhep.2020.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Vincentis A, et al. A polygenic risk score to refine risk stratification and prediction for severe liver disease by clinical fibrosis scores. Clin. Gastroenterol. Hepatol. 2022;20:658–673. doi: 10.1016/j.cgh.2021.05.056. [DOI] [PubMed] [Google Scholar]
- 48.Marigorta UM, et al. Transcriptional risk scores link GWAS to eQTLs and predict complications in Crohn’s disease. Nat. Genet. 2017;49:1517–1521. doi: 10.1038/ng.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu I, Moses MA. BNF-1, a novel gene encoding a putative extracellular matrix protein, is overexpressed in tumor tissues. Gene. 2003;311:105–110. doi: 10.1016/S0378-1119(03)00563-8. [DOI] [PubMed] [Google Scholar]
- 50.Huang G, Brigstock DR. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012;17:2495–2507. doi: 10.2741/4067. [DOI] [PubMed] [Google Scholar]
- 51.Chan KK-S, et al. Stanniocalcin 1 is a serum biomarker and potential therapeutic target for HBV-associated liver fibrosis. J. Pathol. 2022;257:227–238. doi: 10.1002/path.5880. [DOI] [PubMed] [Google Scholar]
- 52.Tao L, et al. Glial cell line-derived neurotrophic factor (GDNF) mediates hepatic stellate cell activation via ALK5/Smad signalling. Gut. 2019;68:2214–2227. doi: 10.1136/gutjnl-2018-317872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu Z, et al. Wnt-TCF7-SOX9 axis promotes cholangiocarcinoma proliferation and pemigatinib resistance in a FGF7-FGFR2 autocrine pathway. Oncogene. 2022;41:2885–2896. doi: 10.1038/s41388-022-02313-x. [DOI] [PubMed] [Google Scholar]
- 54.Gerhard GS, et al. AEBP1 expression increases with severity of fibrosis in NASH and is regulated by glucose, palmitate, and miR-372-3p. PLoS ONE. 2019;14:e0219764. doi: 10.1371/journal.pone.0219764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bobowski-Gerard M, et al. Functional genomics uncovers the transcription factor BNC2 as required for myofibroblastic activation in fibrosis. Nat. Commun. 2022;13:5324. doi: 10.1038/s41467-022-33063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wirth EK, Puengel T, Spranger J, Tacke F. Thyroid hormones as a disease modifier and therapeutic target in nonalcoholic steatohepatitis. Expert Rev. Endocrinol. Metab. 2022;17:425–434. doi: 10.1080/17446651.2022.2110864. [DOI] [PubMed] [Google Scholar]
- 57.Harrison SA, et al. Effects of resmetirom on noninvasive endpoints in a 36-week phase 2 active treatment extension study in patients with NASH. Hepatol. Commun. 2021;5:573–588. doi: 10.1002/hep4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harrison, S. A. et al. Resmetirom for nonalcoholic fattyliver disease: a randomized, double-blind, placebo controlled phase 3 trial. Nat. Med. 10.1038/s41591-023-02603-1 (2023). [DOI] [PMC free article] [PubMed]
- 59.Chronic Liver Disease: International Comparisons (The Scottish Public Health Observatory, 2022); https://www.scotpho.org.uk/health-conditions/chronic-liver-disease/data/international-comparisons
- 60.Shearer JE, et al. Systematic review: development of a consensus code set to identify cirrhosis in electronic health records. Aliment. Pharm. Ther. 2022;55:645–657. doi: 10.1111/apt.16806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hagström H, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017;67:1265–1273. doi: 10.1016/j.jhep.2017.07.027. [DOI] [PubMed] [Google Scholar]
- 62.Boursier J, et al. Non-invasive tests accurately stratify patients with NAFLD based on their risk of liver-related events. J. Hepatol. 2022;76:1013–1020. doi: 10.1016/j.jhep.2021.12.031. [DOI] [PubMed] [Google Scholar]
- 63.Staufer K, et al. Ethyl glucuronide in hair detects a high rate of harmful alcohol consumption in presumed non-alcoholic fatty liver disease. J. Hepatol. 2022;77:918–930. doi: 10.1016/j.jhep.2022.04.040. [DOI] [PubMed] [Google Scholar]
- 64.Carlessi R, et al. Single-nucleus RNA sequencing of pre-malignant liver reveals disease-associated hepatocyte state with HCC prognostic potential. Cell Genom. 2023;3:100301. doi: 10.1016/j.xgen.2023.100301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allan C, et al. OMERO: flexible, model-driven data management for experimental biology. Nat. Methods. 2012;9:245–253. doi: 10.1038/nmeth.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bedossa P, et al. Histopathological algorithm and scoring system for evaluation of liver lesions in morbidly obese patients. Hepatology. 2012;56:1751–1759. doi: 10.1002/hep.25889. [DOI] [PubMed] [Google Scholar]
- 67.McDonald N, et al. Multiparametric magnetic resonance imaging for quantitation of liver disease: a two-centre cross-sectional observational study. Sci. Rep. 2018;8:9189. doi: 10.1038/s41598-018-27560-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Falissard, B. psy: various procedures used in psychometrics. R package version 1.2 (2022).
- 69.Gamer, M., Lemon, J., Fellows, I. & Singh, P. irr: various coefficients of interrater reliability and agreement. R package version 0.84.1 (2019).
- 70.Bankhead P, et al. QuPath: open source software for digital pathology image analysis. Sci. Rep. 2017;7:16878. doi: 10.1038/s41598-017-17204-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
- 72.Kassambara, A., Kosinski, M. & Biecek, P. survminer: drawing survival curves using ‘ggplot2’. R package version 0.4.9 (2021).
- 73.Therneau, T. M. survival: a package for survival analysis in R. R package version 3.5-7 https://CRAN.R-project.org/package=survival (2023).
- 74.Harrison, E., Drake, T. & Ots, R. finalfit: quickly create elegant regression results tables and plots when modelling. R package version 1.0.6 (2022).
- 75.Latouche A, Allignol A, Beyersmann J, Labopin M, Fine JP. A competing risks analysis should report results on all cause-specific hazards and cumulative incidence functions. J. Clin. Epidemiol. 2013;66:648–653. doi: 10.1016/j.jclinepi.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 76.Gerdes MJ, et al. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl Acad. Sci. USA. 2013;110:11982–11987. doi: 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 78.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 80.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. doi: 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu T, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, 2016).
- 85.Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics. 2013;43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X, Park J, Susztak K, Zhang NR, Li M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 2019;10:380. doi: 10.1038/s41467-018-08023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Castro MAA, et al. Regulators of genetic risk of breast cancer identified by integrative network analysis. Nat. Genet. 2016;48:12–21. doi: 10.1038/ng.3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Margolin AA, et al. Reverse engineering cellular networks. Nat. Protoc. 2006;1:662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- 89.Schwämmle V, Jensen ON. A simple and fast method to determine the parameters for fuzzy c–means cluster analysis. Bioinformatics. 2010;26:2841–2848. doi: 10.1093/bioinformatics/btq534. [DOI] [PubMed] [Google Scholar]
- 90.Groeneveld CS, et al. RTNsurvival: an R/Bioconductor package for regulatory network survival analysis. Bioinformatics. 2019;35:4488–4489. doi: 10.1093/bioinformatics/btz229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nelson JW, Sklenar J, Barnes AP, Minnier J. The START app: a web-based RNAseq analysis and visualization resource. Bioinformatics. 2017;33:447–449. doi: 10.1093/bioinformatics/btw624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1–11.
Data Availability Statement
Hepatic bulk RNA-seq data are deposited in the European Nucleotide Archive (https://www.ebi.ac.uk/ena; study accession number: PRJEB58625). Gene expression data are also freely available for user-friendly interactive browsing online at https://shiny.igc.ed.ac.uk/SteatoSITE_gene_explorer/. SteatoSITE has delegated ethics from West of Scotland Research Ethics Committee 4 (Reference: 20/WS/0002; 18 February 2020) allowing the granting of access to the full dataset (histopathology scoring, hepatic bulk RNA-seq data, EHR data) only in the PMS-IC secure environment to third parties by application (full details at https://steatosite.com/researchers/), overseen and reviewed by the SteatoSITE Scientific Advisory Board.
The codes used for the data analyses in this study can be made available by contacting the corresponding author. Access to codes will be granted for requests for academic use within four weeks of application. R is required software to use the code.