

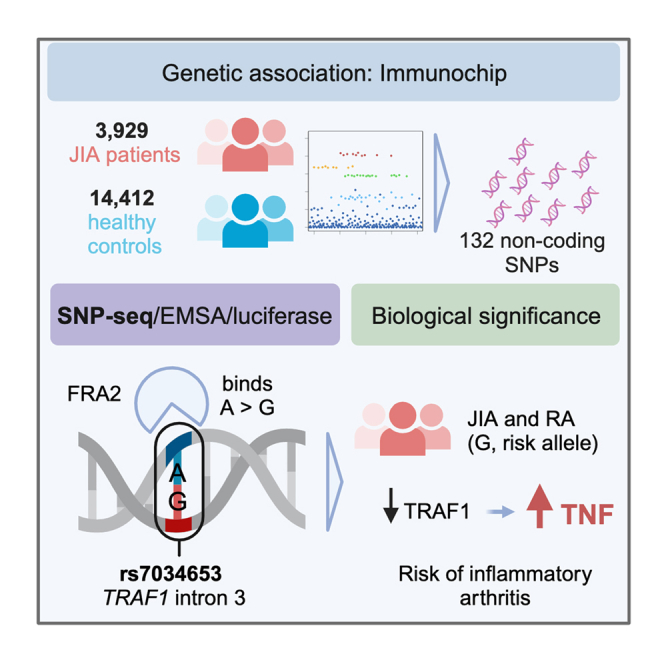
Summary
TRAF1/C5 was among the first loci shown to confer risk for inflammatory arthritis in the absence of an associated coding variant, but its genetic mechanism remains undefined. Using Immunochip data from 3,939 patients with juvenile idiopathic arthritis (JIA) and 14,412 control individuals, we identified 132 plausible common non-coding variants, reduced serially by single-nucleotide polymorphism sequencing (SNP-seq), electrophoretic mobility shift, and luciferase studies to the single variant rs7034653 in the third intron of TRAF1. Genetically manipulated experimental cells and primary monocytes from genotyped donors establish that the risk G allele reduces binding of Fos-related antigen 2 (FRA2), encoded by FOSL2, resulting in reduced TRAF1 expression and enhanced tumor necrosis factor (TNF) production. Conditioning on this JIA variant eliminated attributable risk for rheumatoid arthritis, implicating a mechanism shared across the arthritis spectrum. These findings reveal that rs7034653, FRA2, and TRAF1 mediate a pathway through which a non-coding functional variant drives risk of inflammatory arthritis in children and adults.
Keywords: juvenile idiopathic arthritis, rheumatoid arthritis, genome-wide association study, non-coding variant, TRAF1, C5, monocyte, TNF, FRA2
Graphical abstract
Highlights
-
•
TRAF1/C5 confers risk for juvenile idiopathic arthritis
-
•
SNP-seq, EMSA, and luciferase studies confirm that rs7034653 is functional
-
•
The risk allele reduces TRAF1 expression and thereby increases TNF production
-
•
The same variant drives risk for rheumatoid arthritis, implying a shared mechanism
Qiang and Martínez-Bonet et al. studied 3,939 patients and 14,412 control subjects to establish TRAF1/C5 as a risk locus for juvenile idiopathic arthritis. Using genetic colocalization, SNP-seq, and functional studies, they show that risk is mediated through rs7034653, a non-coding variant that modulates expression of TRAF1 and thereby production of the pro-inflammatory cytokine TNF by monocytes. The same variant predisposes to rheumatoid arthritis, confirming a mechanism shared across children and adults.
Introduction
Translating findings from genome-wide association studies (GWASs) into pathogenic understanding has proven challenging because most implicated loci do not contain coding (exonic) variants.1 Such loci are presumed to carry one or more variants that modulate gene expression, but definitive identification of regulatory variants has proven difficult. Strategies employed have included bioinformatic prediction, fine mapping, and experimental interrogation of plausible non-coding single-nucleotide polymorphisms (SNPs) through methods such as the massively parallel reporter assay and SNP sequencing (SNP-seq).1,2,3,4 The challenges attendant to these strategies, and to downstream experimental confirmation that a particular variant is indeed functional, are such that few non-coding variants are mechanistically defined. Correspondingly, biologic insights obtained through GWASs have often lagged behind those gained from studies of monogenic diseases.5
Among the disease families closely examined by GWASs is inflammatory arthritis. The most common disease in this group, rheumatoid arthritis (RA), has been studied in tens of thousands of patients and control subjects, yielding more than 120 loci associated with disease risk.6,7,8,9,10 One of the first loci identified using a genome-wide approach was a region on chromosome 9 tagged by rs3761847 and containing TRAF1 and C5 (p = 4 × 10−14); unlike other susceptibility loci recognized at that time, including the HLA region and PTPN22, the TRAF1/C5 association did not correspond to any coding variant.11 Small studies in juvenile idiopathic arthritis (JIA) also associated TRAF1/C5 with disease risk, using tagging SNPs rs3761847 (p = 0.035) and rs10818488 (p = 0.012, linkage disequilibrium [LD] with rs3761847 R2 = 0.94), but the largest genetic study of JIA failed to confirm a signal at this locus.12,13,14,15
TRAF1 encodes tumor necrosis factor (TNF) receptor-associated factor 1, expressed in activated myeloid and lymphoid cells.16 TRAF1 mediates pro-survival signaling downstream of TNF receptor superfamily members and negatively regulates Toll-like receptor and Nod-like receptor signaling.17,18 C5 encodes complement factor 5, also implicated in arthritis biology by human and animal studies, rendering both genes plausible candidates.11 Functional data favor TRAF1 as the likely regulatory target since individuals with the high-risk haplotype exhibit lower TRAF1 expression and greater lipopolysaccharide (LPS)-induced cytokine production in monocytes.19 However, the functional variant at TRAF1/C5 remains unknown, as is the pathway by which it reduces TRAF1 expression and whether the same mechanism is relevant for both RA and JIA.
Extending a prior Immunochip study in an expanded cohort of patients and control subjects, we establish a statistically robust disease association of JIA with TRAF1/C5. Colocalization analysis revealed a high likelihood that the same variant modulates expression of TRAF1 in monocytes and macrophages. Using SNP-seq, bioinformatic prediction, and experimental validation, including donors bearing risk and protective alleles, we establish rs7034653 as a functional variant. We identify the transcription factor FRA2 (Fos-related antigen 2; encoded by FOSL2) as the regulatory protein, targeting this protein as orthogonal confirmation of the pathway. Conditioning on rs7034653 eliminates all genetic risk for RA at this locus, implicating a single shared pathway for both JIA and RA. Together, these findings define the mechanism underlying one of the first non-coding variants identified in inflammatory arthritis and model a strategy to transform GWAS findings into biologic understanding.
Results
Identification of rs7039505 as the lead SNP in the TRAF1/C5 locus in JIA
To evaluate whether genetic risk for JIA is conferred by the TRAF1/C5 locus, we studied genotype data from the Immunochip Consortium study of oligoarticular and seronegative polyarticular JIA, including 2,756 published patients and 12,944 published control subjects together with an additional 1,183 patients and 1,468 control subjects.20 The variant rs7039505 achieved statistical significance after correction for multiple comparisons (raw p = 2.39 × 10−7, odds ratio = 1.15, 95% confidence interval 1.09–1.21; Benjamini-Hochberg false discovery rate pFDR = 9.33 × 10−6, adjusted for 119,321 comparisons reflecting SNPs within the Immunochip that passed quality control). rs7039505 resides in LD with lead variants associated previously with risk of JIA (rs10818488, R2 = 0.74) and RA (rs3761847, R2 = 0.74) (Figures S1A and S1B). This result confirms the presence of a functional variant at TRAF1/C5 for JIA (Figure 1).
Figure 1.

Analysis of genetic association at the TRAF1/C5 locus in JIA
To define the non-coding variant within TRAF1/C5, we employed Immunochip data from 3,939 patients with oligoarticular and seronegative polyarticular JIA and 14,412 control subjects. SNP rs7039505 exhibited confirmed significance, p = 2.39 × 10−7 via logistic regression, odds ratio = 1.15, 95% confidence interval 1.09–1.21, Benjamini-Hochberg false discovery rate pFDR = 9.33 × 10−6 adjusted for the number of SNPs that passed quality control, defined as high-quality clustering, call rates >95%, meeting Hardy-Weinberg equilibrium expectations, and no differential missingness between case subjects and control subjects.
To assess the gene driven by this functional variant, we performed colocalization analysis, seeking evidence that rs7039505 colocalizes with an expression quantitative trait locus (eQTL) signal for any of the 11 genes with a transcription start site closer than 250 kb 3′ or 5′ of the tagging SNP. TRAF1 was most commonly identified, including in monocytes stimulated with LPS21,22 as well as in other activated and resting states of monocyte and macrophages23,24 (Figure 2). Colocalization of rs7039505 with a TRAF1 eQTL was evident also in testis, brain, and CD8+ T cells, the latter consistent with published data correlating the arthritis risk haplotype with altered expression of TRAF1 protein in this lymphocyte subset.19 Colocalization signal was identified for C5 in neutrophils; several other genes and lineages also emerged. Together with published experimental data,19 these data provide evidence that the JIA-associated TRAF1/C5 variant modulates TRAF1 in lineages that include monocytes and macrophages.
Figure 2.

Colocalization between JIA GWAS and eQTL signals
We tested for colocalization of the JIA association with eQTLs for 11 genes in the TRAF1/C5 region. Results are shown for eQTL Catalog datasets (y axis) with a probability of colocalization from coloc (PP4, posterior probability of hypothesis 4, that a single variant explains both genetic and eQTL signals, x axis) greater than 0.5. eQTL datasets are ordered by their maximum PP4, and datasets for monocytes and macrophages are highlighted in red. Genes with no PP4 >0.5 are not shown (white points in legend).
Identification of candidate functional variants via SNP-seq
To establish the basis of the association between rs7039505 and JIA, we identified all common variants in LD with this SNP from 1000 Genomes phase 3 (European [EUR] population), filtered on a minor allele frequency (MAF) >1%, a LD R2 >0.7, and a location within a 1 Mb region centered upon the tagging SNP. This strategy yielded 132 candidate SNPs for downstream screening and validation (Table S1).
We then applied SNP-seq to narrow the candidate pool. In this method, a library of double-stranded DNA constructs is generated containing each allele of each candidate variant. A 31 bp segment centered on the SNP of interest is flanked by binding sites for type IIS restriction enzymes (IISREs) that cleave DNA in a sequence-independent manner at 16 bp 5′ to their binding site, i.e., at the SNP itself. Incubation of the library with nuclear extract followed by IISREs and next-generation sequencing (NGS) identifies variants protected from enzymatic restriction by transcription factors (TFs) or other nuclear proteins (Figure 3A).2 For this study, our library contained 271 double-stranded DNA (dsDNA) constructs reflecting each allele of each SNP, including 127 SNPs with two common alleles, 4 with three alleles, and 1 with five alleles (Table S3). The library was incubated with nuclear extract from human monocyte-derived macrophages, washed, and exposed to the IISRE BpmI. As a control, the construct pool was incubated without nuclear extract. Surviving sequences were amplified by PCR and used as the input library for the next round of selection, repeating the procedure for 10 cycles, with barcoding at cycles 4, 6, 8, and 10, to identify overrepresented SNPs. The entire procedure was performed in three independent replicates, with high correlation between sample pairs (Spearman ρ > 0.9) (Figure 3B).
Figure 3.

Screening candidate regulatory variants within TRAF1/C5 by SNP-seq
(A) SNP-seq. A 31 bp sequence centered upon the SNP is flanked by two type IIS restriction enzyme (IISRE) binding sites. A primer is included for high-throughput sequencing. SNPs that fail to bind transcription factors (TFs) or other regulatory proteins are negatively selected. Protected constructs are amplified using primers as per Table S2. Bio, biotin; NGS, next-generation sequencing. Created with BioRender.com.
(B) Correlation analysis of NGS data for each replicate of SNP-seq, using non-linear regression.
(C) Read count ratio between samples and controls at cycle 10 for 24 protected SNPs, with alleles 1 and 2 for each SNP shown in red and blue, respectively. 16 SNPs exhibited allele-specific protection. Statistical analysis was performed using unpaired t tests (n = 3 replicates, ∗p < 0.05).
(D) Top four SNPs showing an increment in allele-specific protection across cycles 4, 6, 8, and 10. A linear model for the ratio of reads in sample vs. control for alleles 1 and 2, with an absolute slope greater than 0.05.
(E) HaploReg annotations for the 11 candidate regulatory SNPs selected from SNP-seq.
We identified SNPs demonstrating allele-specific protection using two different NGS data standards, quality control (QC) 6 and QC12, matching sequencing data for either 6 or 12 nucleotides on either side of the SNP with the original sequences, and then employed two parallel analytical approaches established previously.2 First, we selected SNPs that exhibited a difference in protection between alleles of greater than 20% in cycle 10; of 24 SNPs proportionately enriched after SNP-seq compared with the input library, 16 exhibited such an allelic differential for each of the 2 QC standards (Figure 3C). Second, we sought SNPs with progressive allele-specific protection across cycles, fitting the ratio of reads for alleles 1 and 2 in sample vs. control across cycles 4, 6, 8, and 10 and selecting SNPs with an absolute slope greater than 0.05; 10 SNPs exhibited this pattern for QC12 and 9 for QC6, including 4 that met both criteria for both standards (Figures 3D and S2). In the end, 11 SNPs met both criteria from either QC standard and were considered candidate functional SNPs, a set that did not include either the original tagging SNP, rs7039505, or the lead tagging SNP for RA, rs3761847. All 11 candidates proved plausible based on proximity to epigenetic histone marks, DNase hypersensitivity, sequence preference for TF binding, and association with gene expression as eQTLs (Figure 3E).
Identification of rs7034653 as the likely functional SNP at TRAF1-C5
To confirm allele-specific protein binding, we employed the electrophoretic mobility shift assay (EMSA) using biotinylated DNA fragments of 31 bp centered upon each allele of each candidate SNP, together with nuclear extracts from the monocytic cell line THP-1 and from human monocyte-derived macrophages. The SNPs rs7021880, rs7034653, rs9886724, and rs10985073 exhibited allele-imbalanced binding for both nuclear extracts, while rs758959 and rs1609810 exhibited this property for only THP-1 or macrophage nuclear extract, respectively (Figure 4A, SNPs with allele-imbalanced nuclear protein binding in red, shifted bands marked as red dots). We then tested the allele-specific gene regulatory capacity of these 6 SNPs using a luciferase reporter. We cloned the 31 bp DNA fragment containing either the risk or non-risk allele into the pGL3 vector and then transfected each reporter construct into THP-1, together with control vector pRL to normalize for transfection efficiency. We found that only rs7034653 and rs1609810 exhibited a significant difference between alleles (Figure 4B). Given the focus of prior work on rs3761847,19 we evaluated the alleles of this SNP using both EMSA and the luciferase reporter assay. While the SNP exhibited allele-imbalanced binding in EMSA using both nuclear extracts, we observed no imbalanced luciferase reporter activity (Figure 4C).
Figure 4.

Experimental assessment of candidate regulatory SNPs and impact on inflammatory cytokine production by primary monocytes
(A) EMSA for 11 candidate regulatory SNPs. Allele-specific gel shift/binding is highlighted with red dots. Left: result with nuclear extract from the monocytic cell line THP-1; right: result with nuclear extract from human monocyte-derived macrophages (representative of n = 3 independent biological replicates with similar results).
(B) Luciferase reporter assay showing relative fluorescence in human THP-1 cells between the non-risk (blue) and risk (red) alleles of 6 candidate regulatory SNPs from EMSA (mean ± SEM n = 6 biological replicates, t test with two tails without correction for multiple hypothesis testing); we proceeded with analysis of rs7034653.
(C) EMSA and luciferase reporter assay for SNP rs3761847. M indicates human monocyte-derived macrophages, and T indicates THP-1 cells. For luciferase assays, histogram shows mean ± SEM, n = 6 biological replicates, t test with two tails without correction for multiple hypothesis testing.
(D–F) Purified monocytes from healthy human donors were treated or not with LPS (100 ng/mL for the duration shown).
(D) Histograms by flow cytometry showing the time course of induction of TRAF1 by LPS in purified monocytes from one representative donor (AG genotype at rs7034653).
(E) Mean fluorescence intensity (MFI) of TRAF1 in purified monocytes of subjects with the AA, AG, and GG genotypes at rs7034653.
(F) ELISA measurements of TNF in monocyte supernatants after LPS stimulation. Each symbol represents one donor; n = 13, 13, and 12 human subjects of genotypes AA, AG, and GG, respectively. Statistical analysis was performed using one-way ANOVA with multiple comparisons.
We then employed publicly available data to further assess rs7034653 and rs1609810. Using DICE (Database of Immune Cell Expression, eQTLs, and Epigenomics), we found that both SNPs were associated with expression of TRAF1 as well as of the non-coding transcript PSMD5-AS1. Given our colocalization data, we focused on TRAF1, seeking to identify an eQTL pattern for this gene consistent with our experimental data. Per DICE, the G allele of each SNP is associated with lower TRAF1 expression in monocytes (Figure S3A), consistent with our luciferase result for rs7034653 but opposite that observed for rs1609810. The BIOS QTL browser database also revealed rs7034653 as the best candidate to regulate TRAF1 expression based on methylation QTLs (Figure S3B). Finally, using a bioinformatic model developed to identify sequence-specific binding proteins,25,26 we found that the likely high-binding allele for these SNPs was the A allele for both rs7034653 (top binding candidate TF, MAF BZIP transcription factor G [MAFG]) and rs1609810 (top binding candidate TF, highly divergent homeobox) (Table S4); since only the A allele of rs7034653 exhibited preferential gel shift in EMSA, rs7034653 again appeared the more likely candidate. Considering these data together, we selected rs7034653 and its risk allele G (MAF 32% from TopMed) for further functional study as the best candidate regulatory SNP at TRAF1/C5.
rs7034653 risk allele G is associated with lower TRAF1 expression and increased pro-inflammatory response after LPS stimulus
The arthritis risk haplotype at TRAF1/C5 is believed to reduce expression of TRAF1 and thereby enhance production of inflammatory mediators.19 We therefore tested whether individuals differing in allele carriage at rs7034653 exhibited the predicted variation in TRAF1 expression and TNF release. We recruited healthy donors genotyped through the Mass General Brigham Biobank using the Joint Biology Consortium (JBC) infrastructure (www.jbcwebportal.org). We isolated peripheral blood monocytes by negative selection from 13 donors each of AA and AG genotypes and 12 donors of GG genotypes at rs7034653. TRAF1 is expressed at low levels in resting monocytes but induced via nuclear factor κB (NF-κB), so we selected LPS as the appropriate stimulus. At baseline, TRAF1 protein was similarly low in all groups; however, after LPS exposure, TRAF1 increased more in the AA (protective) than the GG (risk) monocytes, with AG cells exhibiting an intermediate phenotype (Figures 4D and 4E; gating strategy for CD14+HLA-DR+ monocytes shown in Figure S4A). Correspondingly, LPS-stimulated TNF release was greatest for GG monocytes (Figure 4F). Intracellular staining for TNF, interleukin-6 (IL-6), and IL-1β as well as IL-6 release were more variable, such that we could not observe significant differences by genotype, although GG monocytes trended higher across most measures (Figures S4B and S4C).
FRA2 is the TF that binds rs7034653 to regulate TRAF1
rs7034653 is located in the third intron of TRAF1, suggesting that its impact on TRAF1 expression is likely mediated through binding of a TF, a conclusion supported by our SNP-seq, EMSA, and luciferase results. To provide an orthogonal line of evidence implicating rs7034653 in the arthritis risk conferred through TRAF1/C5, we sought to identify the TF and thus the regulatory pathway mediating enhanced pro-inflammatory mediator production. We employed a bioinformatic approach to identify TFs likely to bind this SNP based on protein-binding motif preferences.25,26 Several candidate TFs were identified as having the potential to bind rs7034653 in an allele-imbalanced manner (Table S4). Given the findings from EMSA that the responsible TF likely exhibited preferential binding to the A allele, we performed oligo pull-down with this allele using nuclear extract from THP-1 cells, with or without excess non-biotinylated competitor and employing the incidental TRAF1/C5 SNP rs1609810 as a control. Among the candidate TFs, FRA2, growth factor independent 1 transcriptional repressor (GFI1), and activating transcription factor 3 (ATF3), only FRA2 exhibited the expected specific binding to rs7034653 (Figure 5A). This result was confirmed by EMSA supershift, finding that binding to rs7034653 was weakened by anti-FRA2 antibody (disappearance of the band) compared with isotype control or antibody against another TF candidate, MAFG (Figure 5B). These studies establish that FRA2 can bind the rs7034653 sequence.
Figure 5.

FRA2 binding to rs7034653 regulates TRAF1 expression
(A) Oligonucleotide pull-down western blot assay showing that FRA2 binds specifically to rs7034653-A; binding is eliminated by 30-fold excess non-biotinylated rs7034653-A competitor. GFI1 and ATF3 bind to all samples with or without non-biotinylated competitors.
(B) EMSA supershift showing specific binding of rs7034653-A to FRA2. The addition of antibody to FRA2 in the oligo/nuclear extract (THP-1) mixture caused the disappearance of the shift band (arrow), whereas isotype control antibody and antibody to irrelevant control TF MAFG did not.
(C) ChIP-qPCR showing FRA2 binding to rs7034653 in THP-1 cells (mean ± SEM, n = 3).
(D) Allele discrimination ChIP-qPCR showing that FRA2 preferentially binds to A allele over G allele of rs7034653 in unstimulated human monocytes heterozygous at rs7034653 (mean ± SEM, n = 3).
(E and G) CRISPR-Cas9 FRA2 knockout THP-1 clones show decreased FRA2 and TRAF1 mRNA and protein expression compared with negative single guide (sg) RNA-treated THP-1 control (mean ± SEM, n = 2).
(F) CRISPR-Cas9 FRA2 knockout THP-1 clones show decreased FRA2 binding to rs7034653 compared with negative sgRNA-treated THP-1 control (mean ± SEM, n = 2).
(A), (B), and (E)–(G) are representative of duplicate biological replicates, and (C) and (D) are representative of triplicate biological replicates. Statistical analysis methods used for (C) and (D) were unpaired t test with two tails without correction and for (E) and (F) were one-way ANOVA with multiple comparisons.
To confirm in vivo interaction between rs7034653 and FRA2, we performed chromatin immunoprecipitation (ChIP)-qPCR. In THP-1 cells (genotype AA for rs7034653), anti-FRA2 pulled down rs7034653 more than did the isotype control (Figure 5C). In purified human monocytes from heterozygous donors, we observed more efficient pull-down of the A allele than the G allele by anti-FRA2, consistent with our bioinformatic and experimental predictions (Figure 5D).
To test whether the interaction of FRA2 with rs7034653 modulated TRAF1 expression, we generated two FRA2 knockout clones using CRISPR-Cas9. Both clones showed FRA2 deficiency and reduced TRAF1 expression after LPS stimulation (Figure 5E). As expected, deficiency of FRA2 markedly attenuated the ability of anti-FRA2 to pull down rs7034653 via ChIP (Figure 5F). Gene deletion translated into significantly decreased protein expression of FRA2 and, in parallel, TRAF1 (Figure 5G), confirming the role of this SNP/TF interaction in TRAF1 expression. Of note, FRA2 depletion inhibited, rather than enhanced, LPS-induced TNF production (Figure S5), potentially reflecting either non-TRAF1 effects of this TF or the ability of TRAF1 to promote rather than suppress cytokine production in some contexts.17
Relationship between rs7034653 and RA risk in the TRAF1/C5 locus
Finally, we sought to determine whether the functional SNP identified here could account for some or all of the GWAS signal at this locus for RA.11 Until recently, the lead tagging SNP for RA was rs3761847, which resides in imperfect LD with rs7034653 (R2 = 0.77).11 However, an expanded trans-ancestry GWAS with improved fine-mapping resolution and higher statistical power has now identified rs1953126 as the lead SNP at TRAF1/C5 (p = 3.18 × 10−11; Figure 6A).10 This SNP exhibits much higher LD with rs7034653 (R2 = 0.98). Conditioning on rs7034653 in 25 European RA GWAS datasets resolved all risk signals (Figures 6B and 6C). This finding shows that rs7034653 accounts for all locus-associated disease risk in RA, implicating a shared mechanism at TRAF1/C5 in RA and JIA.
Figure 6.

Conditional analysis on rs7034653 confirms no residual risk in RA
(A) TRAF1/C5 locuszoom plot in trans-ancestry GWAS of RA.
(B) Pre-conditional analysis locuszoom plot in TRAF1/C5 in the European-ancestry cohort, comparable to the JIA Immunochip population.
(A and B) p values for were calculated by a fixed effect meta-analysis of statistics from logistic regression tests.
(C) Post-conditional analysis in the European population detected no residual RA risk. Conditional analysis using a logistic regression model was conducted in each cohort of the 25 European-ancestry GWAS, and the results were meta-analyzed using the inverse-variance weighted fixed-effect model.
Discussion
Inflammatory arthritis encompasses a range of phenotypes, historically dichotomized by age of onset into JIA and adult forms including RA.27 Here, beginning from an Immunochip study of oligoarticular and seronegative polyarticular JIA, we identified the non-coding variant rs7034653 as a functional variant within TRAF1/C5 that mediates risk of both JIA and RA through differential allelic affinity for the TF FRA2. The G risk allele binds FRA2 less strongly than the A allele, resulting in attenuated upregulation of the anti-inflammatory protein TRAF1 and thereby enhanced production of TNF from stimulated monocytes (Figure 7). These findings inform the mechanism through which one of the earliest GWAS “hits” functions in arthritis and model a general approach to the investigation of non-coding functional variants in human polygenic disease.
Figure 7.

Allelic variation at rs7034653 modulates TNF production through differential binding of FRA2 to regulate expression of TRAF1
FRA2 exhibits greater affinity for the protective A allele than the risk G allele of rs7034653, leading to enhanced production of the negative regulatory protein TRAF1 and lower production of the pro-inflammatory mediator TNF. Inhibition of NF-κB by TRAF1 occurs indirectly, mediated through interaction with the linear ubiquitin chain assembly complex (LUBAC) to block linear ubiquitination of IKKγ, preventing the degradation of the IKK complex required for entry of NF-κB into the nucleus.19 Created with BioRender.com.
FRA2 is a member of the activator protein 1 (AP-1) family whose roles involve regulating gene expression in response to a variety of stimuli, including cytokines, growth factors, stress, and bacterial and viral infections.28 FRA2 forms a heterodimer with other AP-1 members to regulate downstream gene expression.28 Expression of FRA2 is induced by LPS but also by cytokines such as IL-1 and TNF, mediators abundant in the inflamed joint and implicated in arthritis pathogenesis both by animal models and by the therapeutic efficacy of cytokine blockade in human patients.29,30,31,32,33,34,35 While FRA2 can, in some contexts, promote inflammation, in other settings, it performs a moderating function, and our results provide a further example of FRA2 as a negative feedback mechanism to help constrain inflammation.36,37 Concordant with earlier investigations of the TRAF1/C5 locus,19 our findings suggest that impaired induction of the counterregulatory factor TRAF1 shifts the balance toward amplified rather than suppressed inflammation to confer excess risk of inflammatory arthritis.
It is notable that many variants at TRAF1/C5 exhibited allele-specific differences in SNP-seq, EMSA, and luciferase steps and passed bioinformatic plausibility filters. It was only by combining them that we could narrow the candidate pool to a single SNP. These results illustrate the complexity intrinsic to experimental study of non-coding variants and the need for orthogonal approaches—in this case, identification of the related TF—to provide additional confidence. Our data do not exclude the possibility that the TRAF1/C5 risk haplotype harbors more than one functional variant. For example, studies of STAT4 identified 2 protein-binding SNPs, tightly linked within one haplotype, that each bound distinct TFs regulating the gene.2 However, the most parsimonious explanation for our findings is that rs7034653 alone drives TRAF1/C5-associated disease risk within the population studied.
Genetic variants can exhibit differential effects on cell function depending on lineage and activation state.38 Both TRAF1 and FRA2 are expressed widely across immune cell types. We selected the monocyte/macrophage lineage for our SNP-seq screen and downstream validation based on colocalization with an eQTL signal in this lineage, but a colocalization signal was also observed in CD8+ T cells, testis, and brain. While it seems most plausible that de-repressed cytokine production in monocytes and macrophages drives the arthritis risk increase associated with TRAF1/C5, we cannot exclude the possibility that effects in these other lineages, or via other mechanisms, can play a role. For example, TRAF1 forms a dimer with TRAF2 to transduce signaling from TNF and related molecules to suppress TNF-induced apoptosis.16,18,39 Correspondingly, mice lacking TRAF1 are highly susceptible to both LPS-induced septic shock and TNF-induced skin necrosis.18,19 In T cells, TRAF1 deficiency enhances susceptibility to TNF-induced activation and proliferation.17,18 In B cells, TRAF1 cooperates with TRAF2 in induction of NF-kB via CD40,40 while B cell chronic lymphocytic leukemia cells show overexpression of TRAF1.17 TRAF1 also enhances classical signaling by NF-κB and mitogen-associated protein (MAP) kinases downstream of TNF receptor (TNFR) to promote lymphocyte survival.39,40,41,42,43 Thus, differential genetic control of TRAF1 by FRA2 could have multiple ramifications for immune-mediated disease, although enhanced monocyte/macrophage-derived cytokine production is a mechanism consistent with the transformative therapeutic impact of TNF blockade in both JIA and RA.
By implicating a shared variant in TRAF1/C5 in both JIA and RA, our findings further support the close relationship between these disease families. The nomenclature divide between childhood and adult arthritis arose in the 1950s out of descriptive convenience, reflecting the age of patients cared for in pediatric centers rather than established biological differences.27 Our Immunochip data reflect children with either oligoarticular- or polyarticular-onset JIA who lack circulating rheumatoid factor, a population that includes three subgroups in the current nomenclature: persistent oligoarticular JIA, extended oligoarticular JIA, and seronegative polyarticular JIA. These patients share HLA and non-HLA associations with adult seronegative arthritis.27,44,45,46 Interestingly, TRAF1/C5 was originally identified in an RA population composed primarily of seropositive patients, a group that also transcends the pediatric/adult boundary but that exhibits HLA associations distinct from those in seronegative disease.11,47,48,49 The fact that the rs7034653/FRA2/TRAF1 mechanism confers risk for both seronegative and seropositive arthritis suggests a role in a shared downstream mechanism, such as synovial inflammation. This possibility is further supported by the fact that TRAF1/C5 is not a GWAS “hit” for autoimmune diseases such as type 1 diabetes and autoimmune thyroid disease, underscoring the potential relevance of excess mediator production by myeloid cells as a key mechanism by which rs7034653 predisposes to arthritis.
Our findings have clinical implications. Identification of a specific functional SNP enables greater ability to test genetic variation as a tool for patient stratification. We identify the FRA2/TRAF1 pathway as a relevant target across the arthritis spectrum, raising the possibility that enhancing the expression of TRAF1, or controlling TRAF1-regulated pathways, could yield important therapeutic benefit. More broadly, the approach modeled here for TRAF1/C5 could be employed across loci and across diseases to define the mechanistic implications of the regulatory variants that are thought to underlie most GWAS “hits,” helping to translate the major public investment in genetic studies of complex polygenic disease into tangible advances in pathogenic understanding and treatment.
Limitations of the study
Patients and controls evaluated by Immunochip were of European ethnicity.20 Variants interrogated by SNP-seq were identified based on European population data. Allele distribution at rs7034653 is comparable across populations, with the frequency of the G minor allele per Ensembl GRCh37: African 33%, American 32%, East Asian 27%, European 36%, and Southeast Asian 20%. Common non-coding variants are conceptually comparable to rare, highly penetrant coding variants, in that both represent “experiments of nature” that illuminate generalizable mechanisms within human biology. Correspondingly, the FRA2/TRAF1/TNF pathway is likely to play a part in JIA and RA pathogenesis even in individuals lacking the risk variant or in populations where the variant is uncommon.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-MAFG antibody | GeneTex | GTX114541 |
| anti-FRA2 antibody | Abcam | ab124830 |
| IgG isotype control antibody | Thermofisher Scientific | 02–6102 |
| anti-HLA-DR-FITC (clone L243) | eBioscience | 11-9952-42 |
| anti-CD14-APC eFluor780 (clone 61D3) | eBioscience | 47-0149-42 |
| anti-TRAF1 antibody | Millipore Sigma | MABC260 |
| anti-IL-1β-PE (clone CRM-56) | eBioscience | 14-7018-81 |
| anti-IL-6-e450 (clone MQ2-13A5) | eBioscience | 14-7069-81 |
| anti-TNF-PerCP-Cy5.5 (clone MAb11) | eBioscience | 45-7349-42 |
| anti-GFI1 antibody | Thermofisher Scientific | PA5-23495 |
| anti-vinculin antibody | Bio-Rad | MCA465GA |
| goat anti-Rabbit IgG (H + L) cross-adsorbed secondary antibody, HRP | Thermofisher Scientific | G-21234 |
| goat anti-mouse IgG-HRP | Santa Cruz | sc-525408 |
| Bacterial and virus strains | ||
| JM109 Competent cells | Promega | L2001 |
| Biological samples | ||
| Human PBMCs | Mass General Brigham Biobank | NA |
| Chemicals, peptides, and recombinant proteins | ||
| 1% Penicillin-Streptomycin | ThermoFisher Scientific | 15140122 |
| 0.05 mM 2-Mercaptoethanol | ThermoFisher Scientific | 21985–023 |
| 1 mM Sodium Pyruvate | ThermoFisher Scientific | 11360070 |
| 10 mM HEPES buffer | ThermoFisher Scientific | 15630130 |
| Ficoll gradient | Cytiva | GE17-1440-02 |
| TransIT-2020 | Mirus | MIR5404 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | 78835 |
| Critical commercial assays | ||
| LightShift Chemiluminescent EMSA Kit | ThermoFisher Scientific | 20148 |
| Dual-Glo Luciferase reporter assay | Promega | E2920 |
| Human TNF ELISA kit | ThermoFisher Scientific | 88-7346-22 |
| Human IL-6 ELISA kit | ThermoFisher Scientific | 88-7066-22 |
| Pierce™ Biotin 3′end DNA labeling kit | Thermo Fisher Scientific | 89818 |
| Deposited data | ||
| Raw data | This paper | Nigrovic, Peter (2023), “2023-WangMartinezBonet-TRAF1C5”, Mendeley Data, V1, https://doi.org/10.17632/vr5f2mbnrm.1 |
| Experimental models: Cell lines | ||
| Human monocytic cell line THP1 | ATCC | TIB-202 |
| Oligonucleotides | ||
| SNP-seq library | Twist Biosciences | NA |
| sgRNAs used targeting FRA2 are: 5′-GGAGAAGCGUCGCAUCCGGC-3′ and 5′-GAACCGACGCCGGGAGCUGA-3′ for k/o clone 1; 5′-CACCGCGGAUCAUGU ACCAG-3′ and 5′-GCGCACGCCG AGUCCUACUC-3′ for k/o clone 2. |
This paper | NA |
| FRA2 qPCR primers: for k/o clone 1, forward, 5′-CAGCCAGCTTGTTCCTCT-3′, reverse, 5′- GATCAAGACCATTGGCA CCA-3’; for k/o clone 2, forward, 5′- CA GCAGAAATTCCGGGTAGAT-3′, reverse, 5′- GGTATGGGTTGGACATGGAG-3′ |
This paper | NA |
| TRAF1 qPCR primers: forward, 5′-GCCCTTCCGGAACAAGGTC-3′, reverse, 5′-CGTCAATGGCGTGCTCAC-3’. | This paper | NA |
| The primers used for THP-1 cell CHIP-qPCR are forward, 5′-CTCCTCCTTTGTCATCATGTT-3′, reverse, 5′- TGGTCAGTTTCCTGGCAAATA-3’; primers and probes used for monocytes CHIP-qPCR are: forward 5′- AGCCTCTCCTCG CTATTC-3′, reverse 5′- GAAGGTGGCAAAGCT GAA-3′, A_Probe 5′- TG + A + C + GA+CAAAG +GA, B_Probe 5′- TG + A + T + GA+CAA+AG+GA. |
This paper | NA |
| Recombinant DNA | ||
| pGL3 promoter vector | Promega | E1761 |
| pRL-TK vector | Promega | E2241 |
| Software and algorithms | ||
| Prism | GraphPad | NA |
| FlowJo | FlowJo | NA |
| ImageLab | Bio-Rad | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter A. Nigrovic, MD (peter.nigrovic@childrens.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and study participant details
For the genetic association study, in total of 18,351 participants of European ancestry with ages younger than 16 years were included for Immunochip analysis.
To study the impact of rs7034653 on TRAF1 expression, we collected primary monocytes from peripheral blood mononuclear cells of 38 healthy individuals (14 males and 24 females) with African American (n = 2), European American (n = 30), Asian American (n = 4) and Indian American (n = 2) ancestry between 23 and 65 years old.
For other in vitro cell culture studies, human monocytic cell line THP1 was purchased from ATCC (catalog number TIB-202) and cultured in RPMI1640 medium (ThermoFisher Scientific, catalog number 11875093) at 37°C.
Genetic association study
Samples are from the Immunochip Consortium study of oligoarticular and seronegative polyarticular JIA, including 2,756 patients and 12,944 controls already published20 together with an additional 1,183 patients and 1,468 controls, totaling 3,939 JIA patients and 14,412 controls. Subjects were of self-declared European ancestry. Samples were genotyped using the Immunochip, a custom Illumina Infinium array, according to Illumina’s protocols at laboratories in Hinxton, UK; Manchester, UK; Cincinnati, USA; Utah, USA; Charlottesville, USA; and New York, USA. The Illumina GenomeStudio GenTrain2.0 algorithm was used to re-cluster all 18,351 samples. Single-nucleotide polymorphisms (SNPs) were initially excluded if they had a call rate <98% and a cluster separation score of <0.4. An SNP was subsequently removed from the primary analysis if it exhibited significant differential missingness between cases and controls (p < 0.05), had significant departure from Hardy-Weinberg equilibrium (p < 0.000001 in cases or p < 0.01 in controls), or had a minor allele frequency (MAF) < 0.01. Then, using only the SNPs that passed the above quality control thresholds, samples were excluded for any of the following: call rate <98%, inconsistencies between reported and genotype-inferred gender, or excess heterozygosity on the autosomes. Duplicates, first-degree, and second-degree relatives were identified using identity-by-descent statistics computed using the program KING.50 From these pairs, the sample with the highest call rate was retained. Admixture estimates were computed on the remaining samples while including the HapMap phase III individuals (CEU, YRI and CHB) as reference populations using the software ADMIXTURE.51 The admixture estimates were then used to identify and remove genetic outliers and included in the statistical models as covariates. Significance was adjudicated as the false discovery rate <0.05 corrected for the number of comparisons performed as per Benjamini-Hochberg.
Colocalization
We performed colocalization analysis for the GWAS variants in the TRAF1-C5 locus against the eQTL Catalogue52 We imported gene expression QTL summary statistics from all RNA-seq studies in eQTL Catalog release 5 (accessed on February 18, 2023). We excluded “GTEx” data to keep only a single GTEx release (“GTEx_V8”), resulting in 109 eQTL datasets. We fetched the summary statistics data using the tabix method with the seqminer R package (v8.5). We included only biallelic SNPs and performed the analysis for genes with a transcription start site within a window of ±250,000 base pairs from the GWAS lead variant, and for which there was at least one eQTL passing the 5 x 10−5 p value threshold. Before merging GWAS and QTL data, the variant coordinates of the GWAS were lifted to the GRCh38 version of the reference genome using liftOver with the UCSC chain file. We used the coloc v5.1.0.1 package53 in R v4.2.0 to test for colocalization at each gene and dataset, assuming a single functional variant at each locus. The code is available at <https://github.com/gutierrez-arcelus-lab/jia_coloc>.
Cells and culture
Human monocytic cell line THP1 was purchased from ATCC (catalog number TIB-202) and cultured in RPMI1640 medium (ThermoFisher Scientific, catalog number 11875093) supplemented with 10% Fetal Bovine Serum (FBS) (ThermoFisher Scientific, catalog number 26140079) and 1% Penicillin-Streptomycin (ThermoFisher Scientific, catalog number 15140122), 0.05 mM 2-Mercaptoethanol (ThermoFisher Scientific, catalog number 21985-023), 1 mM Sodium Pyruvate (ThermoFisher Scientific, catalog number 11360070), 10 mM HEPES buffer (ThermoFisher Scientific catalog number 15630130).
Human PBMC isolation and monocyte purification
For studies in genotyped donors, whole blood was collected from genotyped healthy human subjects recruited from the Mass General Brigham Biobank through Joint Biology Consortium (JBC) recruitment core (www.jbcwebportal.org). Genotype was confirmed in each donor by Sanger sequencing. Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood collected in BD Vacutainer Plastic Blood Collection Tubes with K2EDTA via Ficoll gradient (Cytiva, catalog number GE17-1440-02) and stored in liquid nitrogen in 10% DMSO in complete FBS. After thawing, CD14+ monocytes were isolated using the EasySep Human Monocyte Isolation Kit (Stemcell Technologies, catalog number 19359) according to manufacturer’s instructions. In other experiments, PBMCs were extracted from apheresis leukoreduction collar blood from the Brigham and Women’s blood donation center. Human monocyte derived macrophages were generated by culturing human monocyte purified from collar PBMCs in the presence of 15 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) (PeproTech, catalog number 30003) for 5 days in complete RPMI 1640 medium. Human subjects research with these samples was approved by the Brigham and Women’s Hospital IRB (protocol reference number 2019P003709).
SNP-seq oligos and primers
All primers were purchased from IDT as listed in Table S2. SNP-seq oligo pool was purchased from TwistBiosciences and oligos for EMSA screening and luciferase reporter assay was purchased from EtonBiosciences.
SNP-seq
SNP-seq constructs were built according to Figure 2. The SNP sequence is 31 bp long, centered on the SNP of interest. The sequences of PCR amplification primers for the library oligonucleotides are listed in Table S1. For oligo library amplification, 100 ng of pooled DNA was amplified with bioMagF-G5 and MagR-G3 by PCR for 25 cycles with AccuPrime Taq (Thermo Fisher Scientific, catalog number 12346086) at 94°C for 60 s, 58°C for 60 s and 68°C for 40 s. After gel purification, 10 ng of biotinylated DNA was attached to 4 μL streptavidin-Dynabeads (Invitrogen, catalog number 11205D) according to the manufacturer’s protocol. The DNA-bound beads were then incubated with 60 μg nuclear extract from human monocyte derived macrophages or no nuclear extract for 1 h at room temperature in LightShift Chemiluminescent EMSA Kit reaction buffer (Thermo Fisher Scientific, catalog number 20148X). After washing and separation, the DNA-bound beads were digested with 2 μL BpmI (NEB, catalog number R0565L) for 30 min at 37°C. After another wash and separation, the DNA was amplified again with bioMagF and MagR and reattached to the Dynabeads for the next SNP-seq cycle. Ten cycles were performed in total. DNA from cycle 4, 6, 8 and 10 were used to prepare the Next Generation Sequencing (NGS) library by two consecutive PCRs (PCR with L1seq and R1 primers, 20 cycles of 98°C 1:30 min, 60°C 1:40 min, 72°C 0:40 min; PCR with L2 and R2 primers, 25 cycles of 98°C 1:30 min, 60°C 1:40 min, 72°C 0:40 min) for the independent samples using Herculase polymerase (Agilent, catalog number 600677). The primers used are listed in Table S1. PCR products were run in a 2% agarose gel and the correct band (around 200 bp) was purified with QiaQuick gel extraction Kit (QIAGEN, catalog number 28706) (at least 2 columns) following manufacturerś instructions. The elution was sent for NGS at the Dana Farber Genomics Core, Harvard Medical School.
Electrophoretic mobility shift assay (EMSA)
EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific, catalog number 20148) according to manufacturer’s instructions. For oligo probe, a 31 bp fragment with the SNP centered in the middle was made by annealing two biotinylated oligonucleotides. Nuclear proteins were extracted from either THP-1 cells or human monocyte-derived macrophages using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific, catalog number 78835) per manufacturer instructions. For gel supershifts, 4 μg of antibody was added after for additional 30 min incubation. Antibodies used are anti-MAFG antibody (GeneTex, catalog number GTX114541), anti-FRA2 antibody (Abcam, catalog number ab124830) and IgG isotype control antibody (Thermo Scientific, catalog number 02–6102).
Luciferase reporter assay
The luciferase reporter assay was performed exactly according to the manufacture’s manual (Promega, catalog number E1751). The pGL3 expression vector (0.35 μg) with or without SNPs sequence was co-transfected with the control vector pRLTK (0.25 μg) using TransIT-2020 (Mirus, catalog number MIR 5404) into 1 × 105 THP-1 cells. After 24 h incubation, luciferase activity was measured with the Dual-Glo luciferase assay system (Promega, catalog number E2920).
Transcriptional factors binding prediction
Candidate TFs binding to rs7034653 was analyzed using a published model to identify sequence-specific binding proteins.25,26
Flow cytometry
For monocyte staining, cells were treated with LPS for the indicated times and then blocked with Fc Receptor Binding Inhibitor Polyclonal Antibody (eBioscience, catalog number 14-9161-71) and stained with live/dead-e506 stain (eBioscience, 65-0866-14), followed by staining with anti-HLA-DR-FITC (clone L243), anti-CD14-APC eFluor780 (clone 61D3) and anti-TRAF1-PE for intracellular TRAF1 staining or anti-HLA-DR-FITC (clone L243), anti-CD14-APC eFluor780 (clone 61D3), anti-IL-1β-PE (clone CRM-56), anti-IL-6-e450 (clone MQ2-13A5), anti-TNF-PerCP-Cy5.5 (clone MAb11) for intracellular cytokine staining; all the above antibodies were purchased from eBioscience or labeled manually, used at 1:200 dilution. Purified TRAF1 antibody (Millipore Sigma, catalog number MABC260) was labeled with PE (Biotium, catalog number 92299). For intracellular cytokine staining, purified monocytes were treated with LPS for the indicated times and GolgiPlug (brefeldin A; BD Biosciences, catalog number 00-4506-51) was added in the final 4 h of LPS stimulation; then cells were fixed with Foxp3/Transcription Factor Staining Buffer Set (eBioscience, catalog number 00-5523-00) after surface marker staining. For intracellular TRAF1 staining, supernatants were also collected from each sample and saved for cytokine release measurement by ELISA. Cells were analyzed on a BD FACSCanto II (BD Biosciences).
ELISA
Human TNF (Thermo Scientific, catalog number 88-7346-22) and IL-6 (Thermo Scientific, catalog number 88-7066-22) ELISAs were performed according to manufacturer’s instructions.
Pulldown assay
Pulldown assay is modified from a published protocol for DNA Affinity Purification Assay (DAPA).54 As for SNP-seq, 500 ng of different alleles (31 bp, biotinylated) from a SNPs were attached to 25 μL streptavidin-Dynabeads and then incubated with 100 μg of THP-1 nuclear extract for 1 h with or without non-biotinylated competitor (the same alleles). The incubation mixture was then washed in PBST 5 times before adding elution buffer (2x Laemmli protein sample buffer, Bio-Rad, catalog number 1610747). The elution was then used for Western blot.
Western blot
Antibodies were diluted in PBST according to manufacturer’s suggestion. Antibodies used for Western blot are anti-GFI1 antibody (Thermo Scientific, catalog number PA5-23495); anti-ATF3 antibody (Cell Signaling, catalog number 33593S), anti-FRA2 antibody (Abcam, catalog number ab124830), anti-TRAF1 antibody (Millipore Sigma, catalog number MABC260), anti-vinculin antibody (Bio-Rad, catalog number MCA465GA), goat anti-Rabbit IgG (H + L) cross-adsorbed secondary antibody, HRP (Thermo Scientific, catalog number G-21234) and goat anti-mouse IgG-HRP (Santa Cruz, catalog number sc-525408).
CRISPR-cas9 knockout
FRA2 gene in THP-1 cells was knocked out using CRISPR-cas9 through nucleofection (4D nucleofector from Lonza). 1 μL of 40 μM sgRNAs (two sgRNAs for each reaction to generate a deletion) for FRA2 gene were mixed with 1 μL of 20 μM spCas9 and incubated at 37°C for 15 min to form ribonucleoprotein (RNP). Formed RNP was kept on ice before use. THP-1 cells were then washed with PBS twice and resuspended in SG solution (Lonza, catalog number V4XC-3032), 200k cells per 20 μL solution. Next, RNP was added into each 20 μL solution with THP-1 cells and the whole mixture was transferred into nucleofection strip wells for nucleofection with program code FF100. After nucleofection, cells were transferred into pre-warmed culture medium and incubated at 37°C for 72 h before analyzing knockout efficiency. sgRNAs used are: 5′-GGAGAAGCGUCGCAUCCGGC-3′ and 5′-GAACCGACGCCGGGAGCUGA-3′ for k/o clone 1; 5′-CACCGCGGAUCAUGUACCAG-3′ and 5′-GCGCACGCCGAGUCCUACUC-3′ for k/o clone 2.
RT-qPCR
Total RNA was isolated with Absolutely RNA Miniprep Kit (Agilent, catalog number 400800). cDNA was synthesized with AffinityScript QPCR cDNA Synthesis Kit (Agilent, catalog number 600559). All procedures were performed following the manufacturer’s protocols. RT-qPCR was done with a Agilent AriaMX qPCR machine according to the protocol for Brilliant II SYBR QPCR Low ROX Mstr Mx (Agilent, catalog number 600830). FRA2 qPCR primers: for k/o clone 1, forward, 5′-CAGCCAGCTTGTTCCTCT-3′, reverse, 5′- GATCAAGACCATTGGCACCA-3’; for k/o clone 2, forward, 5′- CAGCAGAAATTCCGGGTAGAT-3′, reverse, 5′- GGTATGGGTTGGACATGGAG-3’. TRAF1 qPCR primers: forward, 5′-GCCCTTCCGGAACAAGGTC-3′, reverse, 5′-CGTCAATGGCGTGCTCAC-3’.
Chromatin immunoprecipitation (ChIP)-qPCR
ChIP was performed for THP-1 cells and human monocytes using CHIP-it kit (Active motif, catalog number 53042) according to manufacturer’s protocols. Antibodies used are isotype control antibody and anti-FRA2 antibody from EMSA. The primers used for THP-1 cell CHIP-qPCR are forward, 5′-CTCCTCCTTTGTCATCATGTT-3′, reverse, 5′- TGGTCAGTTTCCTGGCAAATA-3’; primers and probes used for monocytes CHIP-qPCR are: forward 5′- AGCCTCTCCTCGCTATTC-3′, reverse 5′- GAAGGTGGCAAAGCTGAA-3′, A_Probe 5′- TG + A + C + GA+CAAAG+GA, B_Probe 5′- TG + A + T + GA+CAA+AG+GA.
Allele discrimination CHIP-qPCR
ChIP was performed using human monocytes with rs7034653 A/G genotype. Antibodies used are isotype control antibody and anti-FRA2 antibody from EMSA. Probes and primers used are: G_Probe, 5′- TG + A + C + GA+CAAAG+GA, A_Probe, 5′- TG + A + T + GA+CAA+AG+GA; Forward primer, 5′- AGCCTCTCCTCGCTATTC, Reverse primer, 5′- GAAGGTGGCAAAGCTGAA.
Conditional analysis on RA GWAS
We performed conditional analysis within ±1 Mb from the lead variant rs7034653 in each of the 25 EUR RA GWAS cohort, and meta-analyzed the 25 results using a fixed effect model, using methods and RA cohorts detailed in ref. 55.
Quantification and statistical analysis
All statistical analysis was done using GraphPad software (Prism) using one-way analysis of variance (ANOVA) for comparison of multiple groups, or unpaired t-test (non-parametric Mann-Whitney test) for two groups, with p-values as indicated in the figure legends.
Additional resources
The study did not generate any additional resources.
Acknowledgments
Q.W., M.-M.B., and T.K. were supported by Joint Biology Consortium microgrants off parent grant NIH/NIAMS P30AR070253. J.A.S. was supported by NIH/NIAMS R01 AR077607, P30 AR070253, and P30 AR072577; the R. Bruce and Joan M. Mickey Research Scholar Fund; and the Llura Gund Award for Rheumatoid Arthritis Research and Care. M.T.W. was supported by NIH grants R01NS099068, R01HG010730, U01AI130830, R01AI024717, and R01AR073228. M.S. was supported by P30AR070549. V.A., M.C.H., and M.-G.A. were supported by NIH/NIAMS P30AR069625, Gilead Sciences, the Lupus Research Alliance, and the Arthritis National Research Foundation Vic Braden Family Fellowship. S.P. was supported in part by the Marcus Foundation (Atlanta, GA, USA). S.R. was supported by 5R01AR063759-07. C.D.L. and M.C.M. were supported by R01 HD089928-05. S.D.T. was supported by P30AR070549. P.A.N. was supported by NIH/NIAMS 2R01AR065538, R01AR075906, R01AR073201, and P30AR070253; the Fundación Bechara; and the Arbuckle Family Fund for Arthritis Research. The Trøndelag Health Study (HUNT), which contributed with control samples, is a collaboration between the HUNT Research Centre (Faculty of Medicine and Health Sciences, NTNU - Norwegian University of Science and Technology), Trøndelag County Council, the Central Norway Regional Health Authority, and the Norwegian Institute of Public Health. We acknowledge Nils Thomas Songstad and Nina Moe for patient recruitment in the Norwegian subcohort of the Nordic JIA study; Kristin Rian, NTNU, Trondheim, for technical support; and Helse Nord Research Grants for funding the collection of samples used in this study from Tromsø, Norway.
Author contributions
Q.W., M.M.-B., and P.A.N. conceived and designed the study. Q.W., M.M.-B., T.K., and S.S. performed experiments and data analysis. D.J.C., A.W., and B.W. performed experiments. J.A.S. led recruitment of genotyped human subjects. K.I. and S.R. defined the relationships among SNPs associated with JIA and RA SNPs. Q.W., X.C., and M.T.W. identified candidate TFs. M.S., M.C.M., C.D.L., and S.D.T. provided and analyzed JIA genetic data. V.A., M.C.H., and M.G.-A. performed colocalization analysis. J.B., S.E., E.N., S.P., M.R., and V.V. provided JIA genetic data. Q.W. and P.A.N. drafted the manuscript, and all authors edited and approved the manuscript. P.A.N. supervised the research.
Declaration of interests
The authors declare no competing interests.
Published: October 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100420.
Supplemental information
Data and code availability
-
•
Primary raw data are available at Nigrovic, Peter (2023), “2023-WangMartinezBonet-TRAF1C5”, Mendeley Data, V1, https://doi.org/10.17632/vr5f2mbnrm.1
-
•
Original code for colocalization study is available at https://github.com/gutierrez-arcelus-lab/jia_coloc and via https://doi.org/10.5281/zenodo.8302710.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Farh K.K.H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A., et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li G., Martínez-Bonet M., Wu D., Yang Y., Cui J., Nguyen H.N., Cunin P., Levescot A., Bai M., Westra H.J., et al. High-throughput identification of noncoding functional SNPs via type IIS enzyme restriction. Nat. Genet. 2018;50:1180–1188. doi: 10.1038/s41588-018-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu X., Chen X., Forney C., Donmez O., Miller D., Parameswaran S., Hong T., Huang Y., Pujato M., Cazares T., et al. Global discovery of lupus genetic risk variant allelic enhancer activity. Nat. Commun. 2021;12:1611. doi: 10.1038/s41467-021-21854-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore J.H., Asselbergs F.W., Williams S.M. Bioinformatics challenges for genome-wide association studies. Bioinformatics. 2010;26:445–455. doi: 10.1093/bioinformatics/btp713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Q., Bastard P., Liu Z., Le Pen J., Moncada-Velez M., Chen J., Ogishi M., Sabli I.K.D., Hodeib S., Korol C., et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science. 2020;370:eabd4570. doi: 10.1126/science.abd4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okada Y., Wu D., Trynka G., Raj T., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Yoshida S., et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506:376–381. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A.S., Zhernakova A., Hinks A., et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada Y., Terao C., Ikari K., Kochi Y., Ohmura K., Suzuki A., Kawaguchi T., Stahl E.A., Kurreeman F.A.S., Nishida N., et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012;44:511–516. doi: 10.1038/ng.2231. [DOI] [PubMed] [Google Scholar]
- 9.Eyre S., Bowes J., Diogo D., Lee A., Barton A., Martin P., Zhernakova A., Stahl E., Viatte S., McAllister K., et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012;44:1336–1340. doi: 10.1038/ng.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishigaki K., Sakaue S., Terao C., Luo Y., Sonehara K., Yamaguchi K., Amariuta T., Too C.L., Laufer V.A., Scott I.C., et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 2022;54:1640–1651. doi: 10.1038/s41588-022-01213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plenge R.M., Seielstad M., Padyukov L., Lee A.T., Remmers E.F., Ding B., Liew A., Khalili H., Chandrasekaran A., Davies L.R.L., et al. TRAF1–C5 as a Risk Locus for Rheumatoid Arthritis — A Genomewide Study. N. Engl. J. Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behrens E.M., Finkel T.H., Bradfield J.P., Kim C.E., Linton L., Casalunovo T., Frackelton E.C., Santa E., Otieno F.G., Glessner J.T., et al. Association of the TRAF1-C5 locus on chromosome 9 with juvenile idiopathic arthritis. Arthritis Rheum. 2008;58:2206–2207. doi: 10.1002/art.23603. [DOI] [PubMed] [Google Scholar]
- 13.Albers H.M., Kurreeman F.A.S., Houwing-Duistermaat J.J., Brinkman D.M.C., Kamphuis S.S.M., Girschick H.J., Wouters C., Van Rossum M.A.J., Verduijn W., Toes R.E.M., et al. The TRAF1/C5 region is a risk factor for polyarthritis in juvenile idiopathic arthritis. Ann. Rheum. Dis. 2008;67:1578–1580. doi: 10.1136/ard.2008.089060. [DOI] [PubMed] [Google Scholar]
- 14.Petty R.E., Southwood T.R., Manners P., Baum J., Glass D.N., Goldenberg J., He X., Maldonado-Cocco J., Orozco-Alcala J., Prieur A.-M., et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J. Rheumatol. 2004;31:390–392. [PubMed] [Google Scholar]
- 15.McIntosh L.A., Marion M.C., Sudman M., Comeau M.E., Becker M.L., Bohnsack J.F., Fingerlin T.E., Griffin T.A., Haas J.P., Lovell D.J., et al. Genome-Wide Association Meta-Analysis Reveals Novel Juvenile Idiopathic Arthritis Susceptibility Loci. Arthritis Rheumatol. 2017;69:2222–2232. doi: 10.1002/art.40216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zapata J.M., Reed J.C. TRAF1: lord without a RING. Sci. STKE. 2002;2002:pe27. doi: 10.1126/stke.2002.133.pe27. [DOI] [PubMed] [Google Scholar]
- 17.Edilova M.I., Abdul-Sater A.A., Watts T.H. TRAF1 Signaling in Human Health and Disease. Front. Immunol. 2018;9:2969. doi: 10.3389/fimmu.2018.02969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsitsikov E.N., Laouini D., Dunn I.F., Sannikova T.Y., Davidson L., Alt F.W., Geha R.S. TRAF1 is a negative regulator of TNF signaling. enhanced TNF signaling in TRAF1-deficient mice. Immunity. 2001;15:647–657. doi: 10.1016/s1074-7613(01)00207-2. [DOI] [PubMed] [Google Scholar]
- 19.Abdul-Sater A.A., Edilova M.I., Clouthier D.L., Mbanwi A., Kremmer E., Watts T.H. The signaling adaptor TRAF1 negatively regulates Toll-like receptor signaling and this underlies its role in rheumatic disease. Nat. Immunol. 2017;18:26–35. doi: 10.1038/ni.3618. [DOI] [PubMed] [Google Scholar]
- 20.Hinks A., Cobb J., Marion M.C., Prahalad S., Sudman M., Bowes J., Martin P., Comeau M.E., Sajuthi S., Andrews R., et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 2013;45:664–669. doi: 10.1038/ng.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fairfax B.P., Humburg P., Makino S., Naranbhai V., Wong D., Lau E., Jostins L., Plant K., Andrews R., McGee C., Knight J.C. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. 2014;343:1246949. doi: 10.1126/science.1246949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quach H., Rotival M., Pothlichet J., Loh Y.H.E., Dannemann M., Zidane N., Laval G., Patin E., Harmant C., Lopez M., et al. Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell. 2016;167:643–656.e17. doi: 10.1016/j.cell.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alasoo K., Rodrigues J., Mukhopadhyay S., Knights A.J., Mann A.L., Kundu K., HIPSCI Consortium. Hale C., Dougan G., Gaffney D.J. Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat. Genet. 2018;50:424–431. doi: 10.1038/s41588-018-0046-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen L., Ge B., Casale F.P., Vasquez L., Kwan T., Garrido-Martín D., Watt S., Yan Y., Kundu K., Ecker S., et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell. 2016;167:1398–1414.e24. doi: 10.1016/j.cell.2016.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weirauch M.T., Cote A., Norel R., Annala M., Zhao Y., Riley T.R., Saez-Rodriguez J., Cokelaer T., Vedenko A., Talukder S., et al. Evaluation of methods for modeling transcription factor sequence specificity. Nat. Biotechnol. 2013;31:126–134. doi: 10.1038/nbt.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K., et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014;158:1431–1443. doi: 10.1016/j.cell.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nigrovic P.A., Colbert R.A., Holers V.M., Ozen S., Ruperto N., Thompson S.D., Wedderburn L.R., Yeung R.S.M., Martini A. Biological classification of childhood arthritis: roadmap to a molecular nomenclature. Nat. Rev. Rheumatol. 2021;17:257–269. doi: 10.1038/s41584-021-00590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foletta V.C. Transcription factor AP-1, and the role of Fra-2. Immunol. Cell Biol. 1996;74:121–133. doi: 10.1038/icb.1996.17. [DOI] [PubMed] [Google Scholar]
- 29.Birnhuber A., Crnkovic S., Biasin V., Marsh L.M., Odler B., Sahu-Osen A., Stacher-Priehse E., Brcic L., Schneider F., Cikes N., et al. IL-1 receptor blockade skews inflammation towards Th2 in a mouse model of systemic sclerosis. Eur. Respir. J. 2019;54:1900154. doi: 10.1183/13993003.00154-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granet C., Miossec P. Combination of the pro-inflammatory cytokines IL-1, TNF-alpha and IL-17 leads to enhanced expression and additional recruitment of AP-1 family members, Egr-1 and NF-kappaB in osteoblast-like cells. Cytokine. 2004;26:169–177. doi: 10.1016/j.cyto.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 31.Granet C., Maslinski W., Miossec P. Increased AP-1 and NF-kappaB activation and recruitment with the combination of the proinflammatory cytokines IL-1beta, tumor necrosis factor alpha and IL-17 in rheumatoid synoviocytes. Arthritis Res. Ther. 2004;6:R190–R198. doi: 10.1186/ar1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McInnes I.B., Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007;7:429–442. doi: 10.1038/nri2094. [DOI] [PubMed] [Google Scholar]
- 33.Radner H., Aletaha D. Anti-TNF in rheumatoid arthritis: an overview. Wien Med. Wochenschr. 2015;165:3–9. doi: 10.1007/s10354-015-0344-y. [DOI] [PubMed] [Google Scholar]
- 34.Bradley J.R. TNF-mediated inflammatory disease. J. Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 35.Gabay C., Lamacchia C., Palmer G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. nrrheum.2010.4 [pii] [DOI] [PubMed] [Google Scholar]
- 36.Ucero A.C., Bakiri L., Roediger B., Suzuki M., Jimenez M., Mandal P., Braghetta P., Bonaldo P., Paz-Ares L., Fustero-Torre C., et al. Fra-2-expressing macrophages promote lung fibrosis in mice. J. Clin. Invest. 2019;129:3293–3309. doi: 10.1172/JCI125366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renoux F., Stellato M., Haftmann C., Vogetseder A., Huang R., Subramaniam A., Becker M.O., Blyszczuk P., Becher B., Distler J.H.W., et al. The AP1 Transcription Factor Fosl2 Promotes Systemic Autoimmunity and Inflammation by Repressing Treg Development. Cell Rep. 2020;31:107826. doi: 10.1016/j.celrep.2020.107826. [DOI] [PubMed] [Google Scholar]
- 38.Gutierrez-Arcelus M., Ongen H., Lappalainen T., Montgomery S.B., Buil A., Yurovsky A., Bryois J., Padioleau I., Romano L., Planchon A., et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015;11:e1004958. doi: 10.1371/journal.pgen.1004958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Speiser D.E., Lee S.Y., Wong B., Arron J., Santana A., Kong Y.Y., Ohashi P.S., Choi Y. A regulatory role for TRAF1 in antigen-induced apoptosis of T cells. J. Exp. Med. 1997;185:1777–1783. doi: 10.1084/jem.185.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie P., Hostager B.S., Munroe M.E., Moore C.R., Bishop G.A. Cooperation between TNF receptor-associated factors 1 and 2 in CD40 signaling. J. Immunol. 2006;176:5388–5400. doi: 10.4049/jimmunol.176.9.5388. [DOI] [PubMed] [Google Scholar]
- 41.Wang C.Y., Mayo M.W., Korneluk R.G., Goeddel D.V., Baldwin A.S., Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 42.McPherson A.J., Snell L.M., Mak T.W., Watts T.H. Opposing roles for TRAF1 in the alternative versus classical NF-κB pathway in T cells. J. Biol. Chem. 2012;287:23010–23019. doi: 10.1074/jbc.M112.350538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arron J.R., Pewzner-Jung Y., Walsh M.C., Kobayashi T., Choi Y. Regulation of the subcellular localization of tumor necrosis factor receptor-associated factor (TRAF)2 by TRAF1 reveals mechanisms of TRAF2 signaling. J. Exp. Med. 2002;196:923–934. doi: 10.1084/jem.20020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hinks A., Bowes J., Cobb J., Ainsworth H.C., Marion M.C., Comeau M.E., Sudman M., Han B., Juvenile Arthritis Consortium for Immunochip. Becker M.L., et al. Fine-mapping the MHC locus in juvenile idiopathic arthritis (JIA) reveals genetic heterogeneity corresponding to distinct adult inflammatory arthritic diseases. Ann. Rheum. Dis. 2017;76:765–772. doi: 10.1136/annrheumdis-2016-210025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chistiakov D.A., Savost'anov K.V., Baranov A.A. Genetic background of juvenile idiopathic arthritis. Autoimmunity. 2014;47:351–360. doi: 10.3109/08916934.2014.889119. [DOI] [PubMed] [Google Scholar]
- 46.Prahalad S., Glass D.N. A comprehensive review of the genetics of juvenile idiopathic arthritis. Pediatr. Rheumatol. Online J. 2008;6:11. doi: 10.1186/1546-0096-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Han B., Diogo D., Eyre S., Kallberg H., Zhernakova A., Bowes J., Padyukov L., Okada Y., González-Gay M.A., Rantapää-Dahlqvist S., et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 2014;94:522–532. doi: 10.1016/j.ajhg.2014.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurkó J., Besenyei T., Laki J., Glant T.T., Mikecz K., Szekanecz Z. Genetics of rheumatoid arthritis - a comprehensive review. Clin. Rev. Allergy Immunol. 2013;45:170–179. doi: 10.1007/s12016-012-8346-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terao C., Brynedal B., Chen Z., Jiang X., Westerlind H., Hansson M., Jakobsson P.J., Lundberg K., Skriner K., Serre G., et al. Distinct HLA Associations with Rheumatoid Arthritis Subsets Defined by Serological Subphenotype. Am. J. Hum. Genet. 2019;105:616–624. doi: 10.1016/j.ajhg.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.-M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexander D.H., Novembre J., Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kerimov N., Hayhurst J.D., Peikova K., Manning J.R., Walter P., Kolberg L., Samoviča M., Sakthivel M.P., Kuzmin I., Trevanion S.J., et al. A compendium of uniformly processed human gene expression and splicing quantitative trait loci. Nat. Genet. 2021;53:1290–1299. doi: 10.1038/s41588-021-00924-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller D.E., Patel Z.H., Lu X., Lynch A.T., Weirauch M.T., Kottyan L.C. Screening for Functional Non-coding Genetic Variants Using Electrophoretic Mobility Shift Assay (EMSA) and DNA-affinity Precipitation Assay (DAPA) J. Vis. Exp. 2016:54093. doi: 10.3791/54093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ishigaki K., Sakaue S., Terao C., Luo Y., Sonehara K., Yamaguchi K., Amariuta T., Too C.L., Laufer V.A., Scott I.C., et al. Trans-ancestry genome-wide association study identifies novel genetic mechanisms in rheumatoid arthritis. medRxiv. 2021 doi: 10.1101/2021.12.01.21267132. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Primary raw data are available at Nigrovic, Peter (2023), “2023-WangMartinezBonet-TRAF1C5”, Mendeley Data, V1, https://doi.org/10.17632/vr5f2mbnrm.1
-
•
Original code for colocalization study is available at https://github.com/gutierrez-arcelus-lab/jia_coloc and via https://doi.org/10.5281/zenodo.8302710.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.