

Key Points
-
•
In normal and malignant human B cells, CD20 mRNA is alternatively spliced to yield 4 5′ untranslated region isoforms, some of which are translation-deficient.
-
•
The balance between translation-deficient and -competent isoforms modulates CD20 protein levels and responses to CD20-directed immunotherapies.
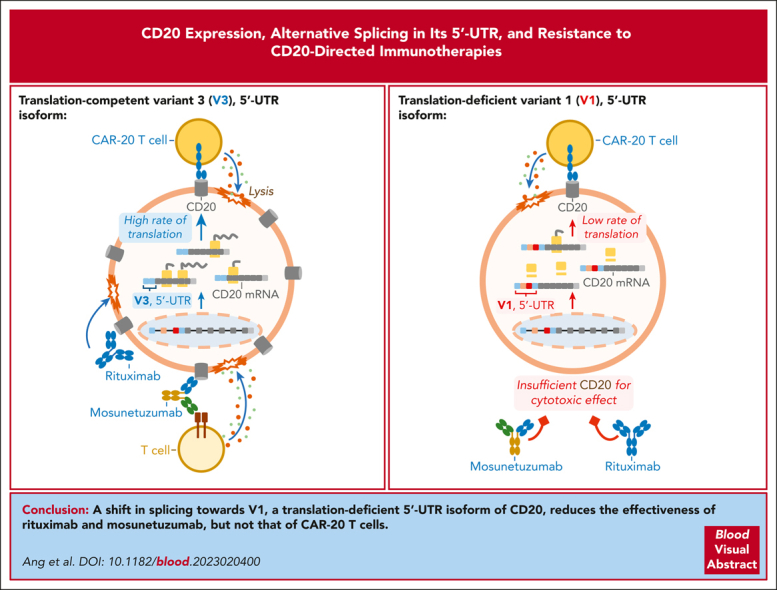
Visual Abstract
Abstract
Aberrant skipping of coding exons in CD19 and CD22 compromises the response to immunotherapy in B-cell malignancies. Here, we showed that the MS4A1 gene encoding human CD20 also produces several messenger RNA (mRNA) isoforms with distinct 5′ untranslated regions. Four variants (V1-4) were detected using RNA sequencing (RNA-seq) at distinct stages of normal B-cell differentiation and B-lymphoid malignancies, with V1 and V3 being the most abundant. During B-cell activation and Epstein-Barr virus infection, redirection of splicing from V1 to V3 coincided with increased CD20 positivity. Similarly, in diffuse large B-cell lymphoma, only V3, but not V1, correlated with CD20 protein levels, suggesting that V1 might be translation-deficient. Indeed, the longer V1 isoform contained upstream open reading frames and a stem-loop structure, which cooperatively inhibited polysome recruitment. By modulating CD20 isoforms with splice-switching morpholino oligomers, we enhanced CD20 expression and anti-CD20 antibody rituximab-mediated cytotoxicity in a panel of B-cell lines. Furthermore, reconstitution of CD20-knockout cells with V3 mRNA led to the recovery of CD20 positivity, whereas V1-reconstituted cells had undetectable levels of CD20 protein. Surprisingly, in vitro CD20-directed chimeric antigen receptor T cells were able to kill both V3- and V1-expressing cells, but the bispecific T-cell engager mosunetuzumab was only effective against V3-expressing cells. To determine whether CD20 splicing is involved in immunotherapy resistance, we performed RNA-seq on 4 postmosunetuzumab follicular lymphoma relapses and discovered that in 2 of them, the downregulation of CD20 was accompanied by a V3-to-V1 shift. Thus, splicing-mediated mechanisms of epitope loss extend to CD20-directed immunotherapies.
While CD20 is a long-established therapeutic target, the regulation of its expression has not been well understood, nor has the basis for the significant interindividual variation observed. Ang and colleagues advance our knowledge by revealing that CD20 mRNA undergoes alternative splicing to generate distinct 5′-UTR variants which are variably translated and collectively determine the cell-surface CD20 levels in malignant B cells. Alternative splicing explains some therapeutic resistance and can be manipulated to enhance CD20-directed immunotherapy.
Introduction
The plasma membrane protein CD20, encoded in humans by the MS4A1 gene, is a clinically significant target for multiple monoclonal antibody (mAb) therapies because of its lineage-specific expression in mature B cells and the malignancies derived therefrom.1 For adult patients, CD20-directed therapies are administered alone or in combination with chemotherapy as the standard of care for mature B-cell neoplasms, such as diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), Burkitt lymphoma (BL), and high-grade B-cell lymphoma.2,3 To date, the most widely prescribed anti-CD20 mAb is rituximab, which is listed on the World Health Organization's List of Essential Medicines.4 For pediatric patients, rituximab and chemotherapy combinations have been approved for previously untreated advanced-stage CD20+ DLBCL, BL, and high-grade B-cell lymphomas.5 In the European Union and other countries, rituximab is also used to treat children and adults with CD20+ B-cell acute lymphoblastic leukemia (B-ALL).6 Newer CD20-directed immunotherapies include mosunetuzumab, a CD20 × CD3 bispecific mAb that redirects T cells to engage and eliminate malignant B cells.7,8 In 2022, mosunetuzumab was granted accelerated approval in the European Union and by the Food and Drug Administration for the treatment of relapsed or refractory FL as a third-line or later therapy.9
Although CD20-targeted immunotherapies have increased the median overall survival of patients with B-cell malignancies, de novo or acquired immunotherapy resistance due to antigen loss remains a challenge.2,10 For patients with relapsed or refractory B-cell non-Hodgkin lymphomas (B-NHLs), low levels of CD20 at baseline (16 of 293, or 5.5% of cases) were associated with a lack of response to mosunetuzumab monotherapy. In chronic lymphocytic leukemia (CLL), CD20 levels are generally lower than those in other mature B-cell neoplasms.11, 12, 13, 14 This is believed to contribute to the relatively low efficacy of rituximab monotherapy against CLL.15 Beyond this intrinsic resistance, CD20 protein was lost in 7 of 26 patients with B-NHL (27%) who relapsed after initial responses to mosunetuzumab.16 Overall, up to a third of all adult patients with DLBCL and most patients with FL and CLL are not cured by rituximab ± chemotherapy combinations.17,18 This resistance is commonly associated with the downmodulation of CD20.19, 20, 21, 22, 23, 24
Despite the clear role of CD20 antigen loss in immunotherapy resistance, significant gaps in our understanding of the underlying mechanism(s) still exist. A recent report demonstrated that in a BL cell line, the loss of CD20 reduced CD19 expression over time,25 although this was not an immediate effect, and CD19 loss in CD20− relapses has not been reported. We previously reported that the Myc oncoprotein downregulates CD20 messenger RNA (mRNA) expression in human B cells and renders them partly resistant to rituximab in vitro.26 However, the role this regulation plays, if any, in patients with B-cell lymphomas remains to be determined. The loss of CD20 protein in 3 of 4 of the mosunetuzumab-resistant B-NHL tumors analyzed using next-generation sequencing could not be explained by the disappearance of CD20 mRNA or the emergence of MS4A1 genetic variants, as recently reported.16 A lack of concordance between CD20 mRNA and protein was also observed in CLL samples, which were found to be low in CD20 protein despite a near-normal level of CD20 mRNA relative to that in healthy B cells.27 Here, we addressed the post-transcriptional mechanisms of CD20 dysregulation, with a focus on alternative splicing.
Materials and methods
Data set usage
The endemic BL2 (eBL2[PRJNA292327]), eBL3(PRJNA374464),28 DLBCL2(PRJNA531552), DLBCL3 (PRJNA752102), DLBCL4 (PRJNA373954),29 FL1(PRJNA596663),30 FL2(PRJNA263567),31 CLL2(PRJNA376727),32 CLL3(PRJEB4498), CLL4(PRJNA792609),33 CLL5(PRJNA450999),34 bone marrow subsets (PRJNA475684),35, 36, 37 and peripheral blood B-cell (PRJNA418779)38 data sets were obtained from the BioProject databases of the National Center for Biotechnology Information (NCBI). The eBL1(phs001282.V2.p1) and CLL1(phs000767.V1.p1)39 data sets were from the dbGaP database of the NCBI. For The Cancer Genome Atlas (TCGA) DLBCL samples, CD20 isoform RNA-Seq by expectation maximization (RSEM) and protein reverse-phase protein array data were obtained from the TCGA Splicing Variants Database (TSVdb)40 and cBioPortal41 websites, respectively. Controlled access data sets were downloaded through the database of Genotypes and Phenotypes (dbGaP) project no. 11199: “Post-transcriptional regulation in B-lymphoid malignancies.”
RNA-seq analysis
RNA sequencing (RNA-seq) reads were first trimmed to remove adapters (BBTools version 38.96) and then aligned using Spliced Transcripts Alignment to a Reference (STAR) version 2.7.9a to the hg38 reference genome while providing known gene isoforms through the GENCODE annotation V32. In addition, we used STAR flags “–quantMode GeneCounts” and “–alignSJoverhangMin 8” to quantify genes and ensure that spliced reads had an overhang of at least 8 bases. Junction-spanning reads were obtained from the STAR “∗_SJ.out.tab” result files, and each entry was normalized by dividing by the total number of junction-spanning reads and multiplying by a factor of 1 million to obtain the junctions per million. Visualization and downstream analyses were conducted via R using the ggplot2 and tidyverse packages. Transcripts per million (TPMs) for all samples were calculated using TPMCalculator42 version 0.0.3, followed by ComBat-seq43 batch correction as implemented in the R Bioconductor package sva (version 3.42.0).
MAJIQ analysis
For RNA splicing quantification, Modeling Alternative Junction Inclusion Quantification (MAJIQ) v2.4.dev+g85d07819 was used. The bam files from STAR and the GENCODE.v37 annotation file were processed using the MAJIQ-build functionality to generate splicegraph and MAJIQ files. Next, for each patient sample, the files from the corresponding prerelapse and postrelapse samples were compared using the MAJIQ deltapsi tool to estimate the changes in splicing (deltaPSI). The results were exported with voila tsv into 2 different files: 1 including all local splice variations (flag –show-all) and another file with the flag “–changing-between-group-dpsi 0.2” to select for those local splice variations with a change in inclusion of at least 20% between prerelapse and postrelapse samples.
Nanopore long-read direct RNA sequencing
Total RNA was isolated from whole Raji cells using a Maxwell RSC simplyRNA Cell Kit (Promega). Approximately 500 ng of mRNA was isolated from total RNA using the Dynabeads mRNA DIRECT kit (Invitrogen) and used for direct RNA (SQK-RNA002; Oxford Nanopore Technologies [ONT]) library preparation. Subsequently, each library was loaded into a Spot-ON Flow Cell R9 version (FLO-MIN106D; ONT) and sequenced using a MinION Mk1B device (ONT) for 48 hours. Raw Fast5 files were converted to fastq with guppy (version 3.4.5), followed by alignment to the GENCODE version of hg38 (version 30) using minimap2 (version 2.18); the resulting bam file was visualized using the Integrative Genomics Viewer (version 2.11.0).
Transient expression of CD20 mRNA isoforms
The full-length sequence of MS4A1 V1 (NM_152866), V2 (NM_152867), V3 (NM_021950), and V4 were cloned into the MXS_CMV::PuroR-bGHpA plasmid, replacing the PuroR open reading frame (ORF) in the process, to generate the V1, V2, V3, or V4 expression plasmids. MXS_CMV::PuroR-bGHpA was a gift from Pierre Neveu (Research Resource Identification Portal ID: Addgene_62439).44 Subsequent deletions and mutations were generated using Gibson assembly. Plasmids were transfected into HEK293T cells using the ViaFect Transfection Reagent (Promega) according to the manufacturer’s instructions.45
Stable expression of CD20 mRNA isoforms
The T2A sequence within the pCDH-EF1α-MCS-T2A-Puro plasmid (System Biosciences) was replaced with the internal ribosome entry site (IRES) sequence from pMXs-IRES-Puro (Cell Biolabs) to generate the pCDH-EF1α-MCS-IRES-Puro lentiviral transfer plasmid. The 5′-UTR and coding sequence (CDS) of MS4A1 V1, V2, and V3 were cloned into the pCDH-EF1α-MCS-IRES-Puro vector to generate the V1-, V2-, and V3-Puro plasmids. The CD20-targeting guide RNA (gRNA)–resistant V1-, V2-, and V3-rCD20 lentiviral transfer plasmids were generated by mutating the CCTGGGGGGTCTTCTGATGATCC sequence, found within the CD20 CDS of the V1-, V2-, and V3-Puro plasmids, to GTTGGGCGGACTACTTATGATTC and replacing the puromycin resistance gene with a blasticidin resistance gene. Subsequent deletions and mutations were generated via Gibson assembly.
CD20 knockout
A gRNA sequence targeting the GGATCATCAGAAGACCCCCC sequence within the CD20 CDS and a nonspecific sequence targeting GTTCCGCGTTACATAACTTA were cloned into the lentiCRISPR v2 lentiviral vector for coexpression with Cas9. The lentiCRISPR v2 plasmid was a gift from Feng Zhang (Addgene plasmid no. 52961).46 We caused the stable expression of these plasmids in OCI-Ly8 cells and used fluorescence-activated cell sorting to enrich for cells that stained negative with phycoerythrin (PE)-conjugated anti-human CD20 antibodies (clone 2H7, BioLegend). The sorted CD20− cell population were the OCI-Ly8 CD20KO cells.
Morpholino treatments
The Ex2-1, Ex2-2, and 5ex3 morpholino sequences were as follows: AGTAGAGATTTTGTTCTCTCTTGTT, ATTGTCAGTCTCTTCCCCACAGAAT, and CTGCTGAGTTCTGAGAAAGGAGATG, respectively. Standard morpholinos (Gene Tools LLC) were electroporated into Raji and OCI-Ly8 cells using the Neon Transfection System 10 μL Kit (Thermo Fisher Scientific). Briefly, for every 10 μL tip, half a million live cells were electroporated in buffer R containing 10 mM morpholino at 1350 V and 30 ms pulse width. The Random Control 25-N morpholino (Gene Tools) was electroporated as a control (Ctrl) phosphorodiamidate morpholino oligomer (PMO). Raji, OCI-Ly8, and MEC-1 cells were incubated in cell culture media containing 10 mM Vivo-Morpholinos (viMO) for 3 hours before replacing with fresh media. For Ctrl-viMO, the reverse sequence of 5ex3, GTAGAGGAAAGAGTCTTGAGTCGTC, was used. The cells were tested for rituximab sensitivity on day 2 after morpholino treatment.
Evaluation of rituximab sensitivity
Rituximab (Roche) was added to cell cultures containing 1 × 106 live cells per mL. After 15 minutes, healthy human serum (Complement Technology) was added at 20% concentration (volume-to-volume ratio) as a source of complement, for complement-dependent cytotoxicity. After another 2-hour incubation, cell viability was evaluated by propidium iodide staining before acquisition on a BD Accuri C6 Cytometer (BD Biosciences) or by the WST-1 cell proliferation reagent, according to the manufacturer’s protocol (Sigma-Aldrich). The WST-1 absorbance signal (A450nm-A690nm) was measured using a BioTek Synergy 2 instrument. The blank control well contained the same volume of culture medium and WST-1, but no cells. The percentage of cell viability was calculated as (absorbance at X concentration of rituximab − absorbance of blank control well)/(absorbance with no rituximab – absorbance of blank control well). Dose-response curves and 50% inhibitory concentration (IC50) values were calculated using the nonlinear regression curve fit function in GraphPad Prism 5 (version 5.01), with the top and bottom of the curve constrained to 100% and 0%, respectively.
Manufacturing of CART-20
The CD20-directed chimeric antigen receptor (CAR20) plasmid was custom-generated by TWIST Bioscience and encompassed the 1F5 single-chain variable fragment (scFv),47, 48, 49 CD8A hinge, 4-1bb costimulatory domain, and CD3ζ stimulatory domains. Healthy primary human T cells were obtained from the Human Immunology Core at the University of Pennsylvania. To produce CAR20 T cells (CART-20), CD4+ and CD8+ cells were combined at a 1:1 ratio and activated using anti-human CD3/CD28 Dynabeads (Invitrogen, no. 40203D) at a 3:1 ratio. Twenty-four hours (day 2) after activation, T cells were infected with a lentiviral vector containing the CAR20 transgene (multiplicity of infection ∼1.5, viral particles/T cells). Magnetic beads were removed from T cells on day 6, and the transduction efficiency of CAR (percentage of CAR+) was measured by flow cytometry using a PE anti-G4S antibody (Cell Signaling Technology no. 38907S). T cells were expanded, counted every other day, and cryopreserved at a cell volume ≤350 fL. Before functional assays, T cells were thawed and rested overnight at 37°C.
Mosunetuzumab cytotoxicity assays
OCI-Ly8 was transduced to express green click beetle luciferase and ZsGreen using pHIV-Luc-ZsGreen (a gift from Bryan Welm [Addgene plasmid no. 39196]). These cells were cultured with healthy donor T cells (E:T ratio of 5:1) and different concentrations of mosunetuzumab (Genentech and Roche): 0, 1, 10, and 100 ng/ml. After 24 hours, cancer cell survival was monitored by luciferase bioluminescence using a BioTek Synergy H4 Imager (560 nm). Cell viability (%) was calculated relative to that of the Ctrl cancer cells and T cells without mosunetuzumab.
CART-20 cytotoxicity assays
OCI-Ly8 were cocultured with CART-20 or untransduced T cells at a 1:4 effectors-to-target ratio for 24 hours. Cell survival was analyzed using bioluminescent quantification, as described earlier. Cell viability (%) was calculated relative to that of the Ctrl cancer cells alone.
Results
Up to 4 distinct 5′-UTRs of CD20 (V1, V2, V3, and V4) can be detected and quantified using long-read direct RNA sequencing, illumina RNA sequencing, and RT-qPCR
We and others have implicated aberrant splicing as a recurrent mechanism of resistance to CD19- and CD22-directed immunotherapies (reviewed previously50,51). Here, we hypothesized that the CD20-encoding gene MS4A1 may also undergo alternative splicing, during which exons from the same gene are joined in different combinations, leading to the generation of different mRNA isoforms. To identify full-length cap-to-poly(A) mRNA isoforms of CD20 and rule out reverse transcription artifacts that are common in complementary DNA-seq approaches,52 we performed long-read Oxford Nanopore direct RNA sequencing on Raji cell line. Using this approach, we found 3 mRNA isoforms of CD20 corresponding to annotated transcripts, dubbed variants 1 to 3, or V1, V2, and V3 (NCBI annotation release 109.20211119 for the MS4A1 gene; Figure 1A). These mRNA isoforms have distinct 5′-UTRs but identical CDSs. We also detected a fourth, albeit rare, variant with an unannotated permutation of the 5′-UTR exons that we dubbed V4 (Figure 1A, bottom). For each 5′-UTR variant, at least 2 mRNA isoforms existed because of 2 potential alternative polyadenylation sites within the 3′-UTR of CD20 (Figure 1A).
Figure 1.

Healthy and malignant B cells express 4 CD20 transcript variants (V1-V4) with distinct 5′-UTRs. (A) Oxford Nanopore long-read direct RNA sequencing of mRNA from Raji cells. Sequence alignments corresponding to full-length, cap-to-poly(A) CD20 mRNAs with 4 5′-UTR splice variants and 2 alternative 3′-UTRs are shown. Alignments were extracted and visualized using an Integrative Genomics Viewer. (B) Diagram depicting the CD20 pre-mRNA and 4 distinct 5′-UTR splice variants, from V1 to V4. (C-D) Relative abundance of the V1 (red), V2 (green), V3 (blue), and V4 (yellow) isoforms in OCI-Ly8, Raji, and MEC-1 cells. Panel C is based on RNA-seq data, with the stack plot showing the ratio of sequencing reads mapped to the unique exon junctions found in each 5′-UTR variant of CD20. Here and below, these variants are color-coded as shown in the Reads spanning panel. Here and below, pan-isoform reads mapping to any exon of CD20 are shown on the right for comparison as TPM. Panel D shows RT-qPCR-mediated quantification of pan- and 5′-UTR variant–specific CD20 levels in OCI-Ly8, Raji, and MEC-1 cells. RNA levels are normalized to the reference gene RPL27. Each bar represents the average from each repeated experiment (N = 4). (E-I) Relative abundance of the V1 (red), V2 (green), V3 (blue), and V4 (yellow) isoforms in primary samples corresponding to healthy and malignant B cells. Each bar represents data from a single donor. The corrected TPM (cTPM) values at the top of panel H are the TPM values corrected for potential batch effects between the different data sets. Healthy B-cell subsets were fluorescence-activated cell sorting–enriched from the human bone marrow and tonsils (E) and peripheral blood (F). In panel E, CD19+IgM+IgD+ naïve tonsillar B cells were treated with anti-IgM or an isotype control. Western blots of phosphorylated SYK are displayed at the bottom right corner as a marker of B-cell activation, with actin serving as a loading control.
Although 4 different 5′-UTR variants of CD20 (summarized in Figure 1B) were detected via long-read direct RNA sequencing, we were unable to confidently quantify the relative abundance of these variants because of the low throughput and, hence, low read depth of direct RNA sequencing. To determine the relative abundance of each 5′-UTR variant, we analyzed the available high-depth short-read RNA-seq data (>50 million reads of 2 × 150 bp) corresponding to Raji BL, OCI-Ly8 DLBCL, and MEC-1 CLL cell lines. Our custom pipeline quantified RNA-seq reads that mapped to the unique exon-exon junctions found in each of the 4 5′-UTR variants (V1 to V4). In all 3 cell lines, V3 was the predominant isoform, making up at least 50% of all reads, followed by V1, V2, and V4 (Figure 1C). To validate the RNA-seq results, the different 5′-UTRs of CD20 were also quantified using an isoform-specific quantitative real-time polymerase chain reaction (RT-qPCR) assay, as described in supplemental Methods and supplemental Figure 1, available on the Blood website. In Raji, OCI-Ly8, and MEC-1 cells, RT-qPCR assay identified V1 and V3 as the 2 most abundant 5′-UTR variants (Figure 1D), which was consistent with our RNA-seq findings (Figure 1C).
The 5′-UTRs of CD20 are alternatively spliced in healthy and malignant human B cells
Having determined that our RNA-seq pipeline broadly reflects the diversity of CD20 5′-UTR isoforms, we used it to analyze several in-house35, 36, 37 and publicly available38 data sets. Similar to B-lymphoid cell lines (Figure 1C-D), the V1 and V3 isoforms accounted for the bulk of CD20 transcripts in both healthy B cells and malignancies derived therefrom (ie, precursor B-ALL, eBL, CLL, DLBCL, and FL; Figure 1E-I). In most healthy B-cell subsets from the bone marrow, tonsils (Figure 1E), and peripheral blood (Figure 1F), V1 transcripts outnumbered V3 transcripts. The few exceptions included class-switched B cells from the tonsils (Figure 1E, middle) and germinal center (GC) B cells (Figure 1G), in which V3 predominated. Because B-cell receptor activation occurs in GCs before class switching, we analyzed RNA-seq data from naïve tonsillar B cells that were activated with antibodies directed against immunoglobulin M (IgM), as described previously.53,54 As expected, B-cell receptor ligation led to SYK phosphorylation and an increase in the relative abundance of V3 (Figure 1E, right). These findings suggest that alternative splicing in the 5′-UTR of CD20 is tightly modulated during normal B-cell development. Compared with this relatively homogenous distribution in healthy B-cell samples from different donors (Figure 1E-G), a high degree of intertumoral variability in the relative abundance of the V1 and V3 isoforms was observed for CLL, DLBCL, FL (Figure 1H), and precursor B-ALL (Figure 1I).
EBV infection increases the abundance of V3 and V4
Among the B-cell neoplasms, eBLs were found to have relatively high levels of V3, per RNA-seq (Figure 1H; supplemental Figure 2A). High V3 may be a consequence of Epstein-Barr virus (EBV) infection, because up to 95% of eBL cases are associated with this human pathogen.55 To test this hypothesis, we analyzed lymphoblastoid cell cultures (LCLs) derived from EBV-infected mononuclear cells. We observed a clear increase in V3, even as the total CD20 mRNA levels were lower than those in DLBCL and FL (Figure 2A). RT-qPCR assays also detected a similar increase in V3 and V4 in B cells 3 days after exposure to EBV, relative to the untreated controls (Figure 2B, top). Similar results were obtained using the published RNA-seq data set,56 in which B cells rapidly upregulated V3 and V4 as early as 1 day after exposure to EBV (supplemental Figure 2B). Thus, alternative splicing in the 5′-UTR of CD20 was affected by EBV infection.
Figure 2.

CD20 protein levels positively correlate with the abundance of V3 and V4 but not V1, V2, or pan-isoform CD20 mRNA. (A-C) Levels of pan-isoform and 5′-UTR variant–specific mRNAs and CD20 protein (expressed as molecules of soluble fluorochrome or MESF) in primary or immortalized human samples. They were quantified using RT-qPCR and flow cytometry, respectively. RNA levels are normalized to those of 3 reference genes: RPL27, B-actin (ACTB), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Each bar or point represents data from a single donor. Panel A displays data from EBV-transformed lymphoblastoid B-cell lines (LCLs) and peripheral blood B cells from healthy donors and patients with DLBCL, CLL, or FL. In panel B, peripheral blood B cells from 4 different donors (N = 4) were cultured with the B95.8 strain of EBV (EBV+) or without EBV (EBV−) for 3 days before CD20 expression quantification. (C) Regression analysis of 7 LCL samples from panel A, in which CD20 protein levels are independently correlated with each of the known CD20 mRNA isoforms. (D) A similar regression analysis of 27 DLBCL samples from TCGA consortium for which reverse phase protein array (RPPA) and RNA-seq data are available. The shaded area around the regression line indicates the standard error. The Spearman rank correlation coefficient, R, and the 2-tailed P value, P, as calculated using GraphPad Prism, are shown. APC, allophycocyanin.
An increase in V3 and V4 coincides with elevated CD20 protein level
The increase in V3 and V4 after EBV infection of B cells (supplemental Figure 2B; Figure 2B, top) was notable because it occurred in tandem with elevated levels of cell surface CD20, as measured by flow cytometry. EBV infection increased CD20 protein levels despite also downregulating total CD20 mRNA, as observed in our experiments (Figure 2B) and as described in the literature.56 This trend was preserved in LCLs, in which elevated levels of V3 coincided with elevated levels of the surface CD20 protein relative to those in CLL, DLBCL, and FL. In contrast, pan-isoform CD20 mRNA levels did not correlate with CD20 protein levels, as the levels in LCLs were among the lowest (Figure 2A). When we compared individual LCL samples, we again found that CD20 protein levels did not positively correlate with the levels of V1, V2, or total CD20 mRNA but instead correlated with the levels of V3 or V4 mRNA isoforms (Figure 2C). The same correlations were found in the DLBCL samples from TCGA consortium, which had reverse-phase protein array–based measures of CD20 protein (Figure 2D). Taken together, these findings suggest that CD20 protein levels are controlled exclusively by the abundant V3 isoform.
The extended 5′-UTRs variants (V1 and V2) have increased transcript half-life but reduced ribosome recruitment
Our CD20 data mirrored a similar expression pattern established by us for CD22, in which its protein levels positively correlated with the abundance of productive CD22 mRNA isoforms but not with total CD22 mRNA (which included a large fraction of AUG-lacking noncoding transcripts).37 This led us to hypothesize that a similar mechanism was at play for CD20. Although V1 and V3 CD20 mRNA isoforms have identical CDSs, they possess distinct 5′-UTRs that can affect CD20 protein production by altering mRNA turnover and/or translation rates. To investigate whether there was a difference in mRNA turnover, we used our isoform-specific RT-qPCR assay to measure the rate of turnover or decay for each CD20 mRNA isoform in OCI-Ly8 and Raji cells after inhibiting transcription in these cells with actinomycin D. Relative to V1 and V2, the mRNA decay rates for V3 and V4 were only slightly higher (supplemental Figure 3). The RT-qPCR assay also allowed us to measure the rate of translation of each 5′-UTR isoform in Raji and OCI-Ly8 cells using polysome profiling, as described previously.57,58 Relative to V3 and V4, a smaller fraction of V1 and V2 molecules was detected in the high-density sucrose gradient fractions (Figure 3A). This pattern is indicative of a lower rate of translation for V1 and V2 than for V3 and V4.
Figure 3.

CD20 mRNA isoforms V3 and V4, but not V1 and V2, are efficiently translated into protein. (A) Polysome profiling of OCI-Ly8 and Raji cells. The top panel shows the ribosomal content measured at an absorbance of 254 nm, whereas the bottom panels show the relative distribution of specific transcripts across sucrose gradient fractions, as measured by RT-qPCR (supplemental Methods). (B) CD20 mRNA and protein measurements in HEK293T cells transiently transfected to express V1, V2, V3, or V4 mRNA isoforms. Pan-isoform CD20 transcripts were measured using RT-qPCR. Total CD20 protein was measured by flow cytometry and expressed as delta median fluorescence intensity (ΔMFI) (supplemental Methods). Representative scatter plots (bottom). ∗∗∗P < .001 per 1 way analysis of variance test (pairwise comparison [pwc]: Bonferroni). (C) Immunoblotting analysis of CD20 protein levels in cells from the previous panel. ACTB served as a loading control. Data from 2 independent experiments are shown. (D) CD20 mRNA and protein measurements in HEK293T stably expressing the empty vector (Ctrl-Puro) or the 5′-UTR and CDSs of V1, V2, or V3 (V1-, V2-, or V3-Puro). The kz-Puro Ctrl has the GCCACC Kozak consensus as its sole 5′-UTR element. The levels of CD20 mRNA were quantified by RT-qPCR. The MFI of total cellular PE-CD20 were determined by flow cytometry. The representative histograms are shown. (E) The same experiment was performed using OCI-Ly8 cells. (F) CD20 mRNA and protein measurements in OCI-Ly8 cells stably expressing the V1 or V3 5′-UTR sequences followed by a green fluorescence reporter (GFP) reporter (V1- and V3-GFP). The kz-GFP Ctrl has the GCCACC Kozak consensus as its sole 5’-UTR element. The GFP mRNA levels were measured using RT-qPCR. The MFI of GFP was measured using flow cytometry. Representative histograms are shown. In panels B,D-F, RNA levels are normalized to the reference gene RPL27. Each bar or dot represents the average of repeated experiments (N ≥ 2). RNA and MFI values are expressed as log2 fold change relative to the Ctrl or Ctrl-Puro samples, as indicated. LTR, lentiviral tong terminal repeats.
Heterologous expression of V3, but not V1, enhances CD20 protein expression
Inefficient translation of V1 and V2 transcripts could be rate-limiting for CD20 protein levels. To investigate this scenario, we engineered HEK293T and OCI-Ly8 cells to express each 5′-UTR variant (from V1 to V4) of CD20 and then investigated whether these cells had increased levels of CD20 mRNA (measured via RT-qPCR) and protein (measured via flow cytometry and/or western blotting) relative to those in Ctrl cells transfected with an empty vector (Ctrl or Ctrl-Puro). In HEK293T cells, which do not express CD20 endogenously, transient transfection with full-length V1, V2, V3, and V4 cassettes led to uniformly elevated levels of CD20 mRNA (Figure 3B, top). However, these increases in mRNA levels were accompanied by increased CD20 protein production only in V3- and V4-transfected cells, whereas V1- and V2-transfected cells remained CD20 protein–negative and were indistinguishable from Ctrl cells (Figure 3B-C). This 5′-UTR–specific difference in CD20 protein levels was independent of the 3′-UTR because cells transfected with V3:del-3′ (the 3′-UTR deletion–mutant version of V3) still showed markedly more CD20 protein staining than those transfected with V1:del-3′ or V2:del-3′ (supplemental Figure 4A). In these and subsequent experiments, the GCCACC Kozak consensus alone was used as a positive Ctrl 5′-UTR element (supplemental Figure 4B).
Similar results were obtained when HEK293T or OCI-Ly8 cells were stably transduced with lentiviral vectors to express these variants lacking the 3′-UTRs (V1-Puro, V3-Puro, etc; Figure 3D-E). Unlike HEK293T cells, OCI-Ly8 cells expressed endogenous CD20 protein, which was detected in the empty vector-transduced cells (Ctrl-Puro), and transduction with only V3-Puro, but not V1-Puro, increased the total CD20 protein levels (Figure 3E, right). To determine whether the 5′-UTRs alone were responsible for the difference in expression, we generated green fluorescent protein reporters lacking CD20 CDSs. Again, only V3 and not V1 constructs yielded robust expression of the reporter gene, as evidenced by flow cytometry (Figure 3F).
The extended 5′-UTRs (V1 and V2) contain uORFs and an RNA stem-loop structure that repress translation
The inefficient translation of V1 and V2 implied the presence of repressive 5′-UTR elements. We reasoned that such elements would be localized in exon 3a based on 2 observations. First, exon 3a was included in V1 and V2 but not in V3 and V4. Second, a truncated mutant construct that retained exon 3a but lacked exons 1 and 2 (ie, Δex1-2) retained the ability to repress CD20 protein levels (supplemental Figure 4C). Within the exon 3a sequence, we found multiple upstream ORFs (uORFs). At the exon 3a-b boundary immediately upstream of the CD20 start codon, a stem-loop secondary structure is predicted using the RNAfold algorithm59 (Figure 4A). Because translational repression by uORFs and stem loops has been observed in other genes,60,61 we hypothesized that CD20 could be regulated in the same manner. To investigate this possibility, we mutated all start codons of the uORFs in V1 from AUGs to AUCs to generate V1AUC-Puro. We also generated a V1DelStem-Puro variant with disrupted base pairing in the stem-loop, alone or in combination with the AUC variant, and V1Stem-Puro, in which the hairpin was stabilized via additional base pairing (Figure 4B). We then measured CD20 RNA and protein levels by RT-qPCR and flow cytometry, respectively, and compared them with those of the Ctrl-Puro empty vector.
Figure 4.

An RNA stem-loop structure and uORFs repress translation of the CD20 V1 isoform. (A) Diagram depicting the uORFs and the stem-loop in the 5′-UTR of the CD20 V1 isoform. The stem loop was predicted by the RNAfold web server. (B) Diagrams of mRNA products corresponding to various derivatives of V1-Puro. All AUG start codons found in the 5′-UTR of V1-Puro were mutated to AUC in the V1AUC-Puro and V1AUCDel-Stem-Puro constructs. Stem and Del-Stem refer to mutations stabilizing and destabilizing the predicted secondary structure, respectively. (C-F) Expression of V1- and V3-Puro and the V1-Puro mutants in stably transduced HEK293T cells. Pan-isoform CD20 RNA levels were quantified using RT-qPCR. The MFI of the total cellular CD20 protein was measured using flow cytometry. Representative histograms are shown on the right. Each bar or dot represents the average of repeated experiments (N = 2). All values are expressed as log2 fold change relative to the Ctrl-Puro sample. (G) Diagram depicting putative Sam68 binding sites found within exons 2 and 3 of the V1. (H-I) Sam68 and CD20 expression measurements in Raji cells electroporated with a mixture of 2 Cas9-gRNA ribonucleoproteins targeting the CDS of Sam68. Shown in panel H is immunoblotting analysis of Sam68 protein expression, with ACTB serving as the loading control. Shown on the left side of panel I are Sam68 and CD20 mRNA levels, as quantified using RT-qPCR. Two PCR primer pairs measured nonoverlapping regions within the Sam68 transcript. The second primer pair is denoted with an asterisk (∗). Shown on the right side of panel I are MFI of total CD20 protein, as determined using flow cytometry. Representative histograms are shown on the far right. In both panels H and I, each row in the immunoblotting image and the heat map represent an independent experiment (N = 4).
We found that V1AUC-Puro cells remained CD20 protein–negative (Figure 4C), as did V1Stem-Puro cells (Figure 4D). There was a minor recovery in CD20 protein levels with the V1DelStem-Puro variant (Figure 4E). Notably, disrupting both uORFs and the stem-loop (V1ATCDelStem-Puro) led to a near-complete recovery of CD20 protein levels (Figure 4F). These results suggest that these 2 elements are responsible for the negligible CD20 protein being produced from V1 and V2.
Sam68 contributes to suppressed CD20 protein levels
We searched for RNA-binding proteins that could be responsible for the high V1-to-V3 ratios and translational repression of CD20. Using the SpliceAid algorithm,62 we identified within exon 2 (Figure 4G) and notably within the loop structure of exon 3a (Figure 4A) several putative binding sites for Sam68 (also known as KHDRBS1), a splicing factor known to bind to AU-rich sequences.63 To determine whether Sam68 regulates CD20 splicing, we electroporated Raji cells with a mixture of 2 Cas9-gRNA ribonucleoproteins targeting the CDS of Sam68. Relative to Ctrl gRNAs targeting the irrelevant EMX1 locus and untransfected Ctrl cells, Sam68 gRNAs successfully reduced Sam68 protein levels (western blots in Figure 4H). This was predictably accompanied by a reduction in Sam68 mRNA levels, as measured by RT-qPCR (heat map in Figure 4I, 2 leftmost columns). We also observed a redistribution of all 4 5′-UTR variants: a reduction in V2 mRNA levels and increases in V1, V3, and V4, and total CD20 mRNA (Figure 4I, 5 rightmost columns). We also observed a modest increase in CD20 protein levels (flow cytometry in Figure 4I, right). The same shifts in CD20 mRNA splicing and protein levels were evident when we stably knocked down Sam68 in Raji and OCI-Ly8 cells with 2 different short hairpin RNAs (supplemental Figure 5A-B). Taken together, our findings suggest that Sam68 is at least partly responsible for the selection of CD20 5′-UTR isoforms and for suppressed CD20 protein levels. However, we could not distinguish between its effects on splicing and overall mRNA levels.
Morpholinos redirect splicing toward V3 and V4 to augment rituximab-mediated cytotoxicity
To modulate CD20 splicing more directly, we tested 3 different PMO64 sequences that target exons 2 to 3 in CD20 pre-mRNA (supplemental Figure 5C, top right). These PMOs were electroporated into Raji and OCI-Ly8 cells. Among the PMOs tested against a nonspecific Ctrl sequence pool (Ctrl-PMO), the 5ex3-PMO sequence (targeting the junction between CD20 intron 2 and exon 3a) was the most effective at reducing V1 and V2 mRNA while increasing V3 and, particularly, V4 mRNA (approximately threefold change, as measured by RT-qPCR). It was also superior to Ex2-1 and Ex2-2 in terms of increasing CD20 protein levels, as measured using flow cytometry; supplemental Figure 5C).
Next, we obtained 5ex3 in the viMO format (Figure 5A, top left), which could be added directly to the culture media for efficient delivery into cells.65 We tested 5ex3-viMO in OCI-Ly8, Raji, and MEC-1 cell cultures and measured the levels of CD20 mRNA (by RT-qPCR and RNA-seq) and total cellular CD20 protein (by flow cytometry). As a control, we used the oligomer with the reverse order of 5ex3 nucleotides (Ctrl-viMO), which did little to affect V1, V2, and V3 variants but induced nonspecific increases in V4 and CD20 protein levels relative to the no-Morpholino treatment cells (Figure 5A, top rows). To account for these sequence-independent effects, we normalized the 5ex3-viMO–treated samples to Ctrl-viMO treatment. Even after normalization, 5ex3-viMO treatment induced clear shifts in CD20 splicing, namely, reducing V1 and V2 mRNA levels while increasing V3 and V4 variants (Figure 5A, bottom rows). Visual analysis of the sashimi plots corresponding to the RNA-seq data confirmed that 5ex3-viMO had a noticeably greater effect on reducing exon 2 and 3a usage (Figure 5B, exons within the red boxes) without affecting the usage of the other exons (Figure 5B, exons outside of the red boxes). Most importantly, relative to the Ctrl-viMO treatment, 5ex3-viMO-treated cells showed increased CD20 protein staining (Figure 5A, bottom right corner) across multiple experiments (supplemental Figure 5D).
Figure 5.

Shifting CD20 splicing toward V3 and V4 increases CD20 protein expression and rituximab-mediated cytotoxicity. (A-C) CD20 mRNA and protein measurements in OCI-Ly8, Raji, and MEC-1 cells treated with either the solvent control (no-Morpholino treatment [NT]), Ctrl-viMO, or 5ex3-viMO. (D) The same experiment performed on OCI-Ly8 variants with the intact MS4A1 gene (WT, top), knocked out MS4A1 gene (CD20-KO, middle), or knocked out MS4A1 gene replaced with the V3 expression cassette (V3-rCD20, bottom). CD20 RNA levels were quantified using RNA-seq (leftmost heat map in panel A and sashimi plot in panel B) and RT-qPCR (middle heatmaps in panel A). For the RNA-seq data in panel A, V1∗-4∗ are reads that mapped to the unique exon junctions found in each 5′-UTR variant of CD20, color-coded as shown in the RNA-seq reads panel. The MFI of total CD20 protein was determined using flow cytometry (rightmost heat map in panel A, top panel in panel C, and left and middle panels in panel D). Representative histograms are shown on the far right of panel A and far left of panel D. At the bottom panel of panel C and the right panel of panel D are shown cells that were additionally treated with increasing concentrations of rituximab (RTX), and cell viability (plotted) was measured using the WST-1 assay. All values are normalized to the “no rituximab Ctrl.” In panels A,C-D, each dot in a graph or value in a heat map represents independent experiments (N ≥ 2), with mean values indicated by red lines in the graphs.
To determine whether this increase was therapeutically relevant, we used an anti-CD20 antibody–mediated cytotoxicity assay. In a pilot experiment, we treated OCI-Ly8 cells overexpressing Ctrl-, V1-, and V3-Puro with a wide range of rituximab concentrations and measured the cell viability. We achieved 50% killing of cells expressing the V3 isoform with 4.3 μg/mL rituximab but failed to reach IC50 for their Ctrl and V1 counterparts (supplemental Figure 6A). We then repeated the assay using OCI-Ly8, Raji, and MEC-1 cells treated with Ctrl or 5ex3-viMO. In 2 out of the 3 lines (OCI-Ly8 and Raji), we observed a >10-fold difference in IC50 between Ctrl- and 5ex3-viMO, as measured using the WST-1 cell viability reagent (Figure 5C). This difference in sensitivity was further verified by propidium iodide assays, in which cell death, rather than cell survival, was measured (supplemental Figure 6B).
To confirm that the 5ex3-viMO effect is dependent on CD20 splicing, we generated MS4A1-knockout OCI-Ly8 cells (CD20KO) that, unlike parental cells (Figure 5D, top row), lacked CD20 expression and were insensitive to rituximab (Figure 5D, middle row). We then reconstituted them with an intron-less V3-rCD20 cassette. This resensitized the cells to rituximab but also made them refractory to 5ex3-viMO (Figure 5D, bottom row), attesting to the specificity of this reagent and its potential clinical utility.
V1 generates sufficient CD20 protein levels to trigger killing by CARTs but not mosunetuzumab
Our RNA-seq analysis of FL, DLBCL, CLL, and pre–B-ALL helped identify a subset of samples in which the entire CD20 mRNA pool consisted almost entirely of V1, with little to no V3 (Figure 1H-I). To model B-cell neoplasms with such a lopsided CD20 splicing pattern and to test their responses to CD20-directed immunotherapeutics, we generated an OCI-Ly8 derivative that simultaneously expressed the ZsGreen and luciferase reporters. These cells were then transduced with MS4A1-specific CRISPR-Cas9 lentiviral vectors (lentiCRISPR v2) to knock out endogenous CD20 expression before being reconstituted with V1-, V2-, or V3-rCD20 constructs or an empty vector Ctrl (CD20KO). A separate batch of parental cells was transduced with the lentiCRISPR v2 vector containing a nonspecific gRNA; these wild-type (WT) cells retained the expression of endogenous CD20 (Figure 6A). We evaluated the efficiency of CD20 knockouts and reconstitutions using isoform-specific CD20 RT-qPCR (Figure 6B) and flow cytometry for cell surface and total CD20 protein (Figure 6C). Interestingly, in the RT-qPCR assay, CD20KO cells showed roughly the same CD20 transcript levels as WT Ctrl cells (Figure 6B), but these transcripts were apparently noncoding, because the CD20KO cells were completely CD20 protein–negative (Figure 6C). Furthermore, relative to WT cells, V1-, V2-, and V3-rCD20 cells had elevated levels of the cognate 5′ UTR isoforms (Figure 6B). Thus, these cells could act as models for the complete shift in CD20 splicing toward a specific 5′-UTR variant. As expected, reconstitution with only V3-rCD20 led to a recovery in CD20 protein expression, which remained undetectable by flow cytometry in V1- and V2-rCD20 cells (Figure 6C).
Figure 6.

Shifting CD20 splicing from V3-to-V1 increases resistance to mosunetuzumab but not CART-20. (A) Diagram depicting the lentiviral vector transductions performed on OCI-Ly8 cells expressing a luciferase reporter. The MS4A1 gene was knocked out (CD20KO) and reconstituted with V1-, V2-, or V3-rCD20. The resistant CD20 (or rCD20) constructs have silent mutations in their CDS to avoid recognition by the CD20-targeting gRNA used in the knockout. (B) Isoform-specific RT-qPCR measurements of CD20 mRNA isoform relative to the RPL27 reference gene. (C) Measurement of CD20 protein levels by flow cytometry. On the left, it is quantitated as a log2 fold change in the MFI of total APC-CD20 relative to the unstained Ctrls. On the right, it is quantitated as the cell surface CD20 levels expressed as MESF. Each spot is an independent experiment. Representative histograms are included in each subpanel. (D) Viability of OCI-Ly8 cells after 24 hours of coculturing with CART-20 or untransduced donor T cells at 1:4 effector-to-target ratio. Cell viability (in percentage) is shown relative to that of Ctrl OCI-Ly8 cells that were not cocultured with either CART-20 or donor T cells. (E) OCI-Ly8 cell viability after 24 hours of coculturing with donor T cells and the indicated concentrations of mosunetuzumab. Cell viability (in percentage) is shown relative to that of the NT Ctrl. For panel D, data from 2 independent experiments using T cells from a single donor are shown. For panel E, data from 4 independent experiments with 2 unrelated donors are shown. In panel D and on the right of panel E, each spot represents the results from the replicate wells, with different colors denoting independent experiments. Horizontal bars represent the mean of all replicates. ∗∗P < .01; ∗∗∗P < .001 per the Kruskal-Wallis test (pwc: the Dunn test). (F) CD20 and PAX5 status per immunohistochemistry (IHC) (top) and transcript read abundance per RNA-seq (bar graphs) in paired pre- and postmosunetuzumab FL. TPM values quantify RNA-seq reads mapping to any exon in CD20 and PAX5. The relative abundance of V1 (red), V2 (green), V3 (blue), and V4 (yellow) is the ratio of sequencing reads mapping to the unique exon-exon junctions found in each 5′-UTR variant of CD20, as color-coded in the Reads spanning panel. (G) Micrographs corresponding to P29 pre and postmosunetuzumab FL samples. Formalin-fixed paraffin-embedded sections were IHC stained for CD20 (brown colorimetric detection with 3, 3'-diaminobenzidine [DAB]) using hematoxylin nuclear counterstain (blue). (H) Changes in ΔPSI values or in the ratio between reads including or excluding CD20 exons in the P29-post sample relative to the P29-pre sample. RNA-seq data were analyzed using the MAJIQ algorithm for all possible splicing changes in CD20, but only ΔPSI values above or below 0.05 are shown. (I) Sashimi plots depicting the density of exon-including and exon-skipping reads in the RNA-seq data corresponding to P29-pre and -post samples. FACS, fluorescence-activated cell sorting; H&E, hematoxylin and eosin.
We then performed in vitro assays using 1F5 single-chain variable fragment–based CARTs (CART-20), as described in our earlier studies on B-ALL.37,66 As expected, CART-20 spared CD20KO- and V2-rCD20–expressing cells. Surprisingly, CART-20 was able to kill V3-rCD20– and V1-rCD20–expressing cells equally well despite the apparent lack of detectable CD20 expression in the latter (Figure 6D). We also tested the sensitivity of these cells to the bispecific CD3/CD20 antibody mosunetuzumab in the presence of donor T cells. We found that unlike CART-20, mosunetuzumab was only effective against V3-rCD20– and spared V1-rCD20–expressing cells, just as it did CD20KO cells (Figure 6E, left). This difference was statistically significant across 2 independent experiments, with multiple technical replicates (Figure 6E, right). Taken together, our in vitro results suggested that B-cell neoplasms in which the V1 variant predominates could still generate sufficient levels of the CD20 protein to trigger CART-mediated cytotoxicity, but these levels would be insufficient for mosunetuzumab to be effective. This implied that the V3-to-V1 shift could underlie resistance to mosunetuzumab in patients.
CD20 antigen loss in FL after mosunetuzumab treatment coincides with a V3-to-V1 shift
It was previously found that in 3 of 4 of mosunetuzumab-resistant B-NHL tumors that were CD20 protein–negative by immunohistochemistry, the whole exome sequencing and RNA-seq failed to detect any MS4A1 genetic variants or loss of CD20 mRNA, which could have explained the apparent loss of CD20 protein.16 For this study, we obtained formalin-fixed paraffin-embedded tumor samples from a similar cohort of 4 additional patients relapsing after mosunetuzumab and performed RNA-seq to look for changes in the splicing pattern of CD20. All 4 patients had paired pretreatment tumor samples that were CD20+ by immunohistochemistry and mosunetuzumab relapse (“post”) samples that had lost CD20 protein expression. In 3 of 4 mosunetuzumab relapse samples, the loss in CD20 protein expression coincided with either the complete loss of CD20 mRNA (for P195 and P100; Figure 6F) or an out-of-frame mutation (for P73; supplemental Figure 7). The P29-post sample, in contrast, retained 300 TPM of the CD20 mRNA (Figure 6F) that contained no deleterious mutations that could explain the complete lack of CD20 protein immunoreactivity (Figure 6G). Using the previously developed MAJIQ package,67 the RNA-seq data of P29 were analyzed for any splicing changes that could explain the loss of the CD20 protein. Changes in splicing of the P29-post sample relative to the P29-pre sample were quantified as delta percentage spliced-in (ΔPSI) values. Predictably, no significant changes were found to affect the Ctrl CD19 mRNA because no loss of CD19 expression was observed, which is consistent with the data in the literature.68 Within the CD20 transcript, only 2 alternative splicing events exceeded the ΔPSI value of ±0.05. The first event corresponded to a reduction (ΔPSI = −0.2251) in the use of an alternative donor site that extended to exon 5 (Figure 6H, top). Because the alternative donor site leads to a frameshift and premature stop codon, a reduction in its usage is expected to increase the expression of the full-length CD20 protein and would not explain the loss of CD20 expression. The second event corresponded to a decrease in splicing from exon 1 to 3b (ΔPSI = −0.2622) that occurred during the V3-to-V1 shift (diagram in Figure 6H, bottom, and sashimi plots in Figure 6I). A similar shift was observed in P73 relapse (Figure 6F), which could have predated the inactivating mutation. Thus, the loss of the CD20 protein in the post-mosunetuzumab P29 sample coincided with a shift in CD20 splicing from productive V3 to translation-deficient V1.
Discussion
Here, we demonstrate that in healthy and malignant B cells, human CD20 mRNA undergoes alternative splicing to generate up to 4 distinct 5′-UTRs variants, from V1 to V4 (Figure 1B). Among them, V1 and V3 were the most abundant and together comprised >90% of the total CD20 transcript pool in most cell lines and primary samples (Figure 1). Although V1 and V3 contained the same ORF, only V3 was efficiently translated, accounting for the bulk of the CD20 protein. In contrast, V1 generates negligible amounts of CD20 protein because of translational inhibition by a prominent stem-loop and multiple uORFs (Figure 2., Figure 3.).
We also found that by shifting splicing away from V1 and toward V3, B cells undergoing GC reactions and class switching could sustain or even enhance CD20 protein levels despite the reduction in total CD20 mRNA (Figure 1F-G). This model is in agreement with multiple reports of elevated CD20 protein in GC cells, relative to those in memory and naïve B cells.69,70 We further showed that this V1-to-V3 shift is mediated at least partly by B-cell receptor signaling because activating this pathway in tonsillar B cells by ligating surface IgM is sufficient to shift splicing toward V3 (Figure 1E). Increases in the V3 isoform and CD20 protein can also be observed during EBV infection (Figure 2B), perhaps as part of the GC-like differentiation program that occurs in EBV-infected naïve B cells.56
Regarding cancer, V3 upregulation persisted after peripheral blood B cells were fully transformed into LCLs (Figure 2A). The same appears to be the case for eBL (Figure 1H), for which 95% of the cases are associated with EBV infection.55 Apart from eBL, both V1 and V3 can be detected in most precursor B-ALL and mature B-cell neoplasms (eg, DLBCL; Figure 1H), but CD20 protein levels correlate solely with the V3 isoform (Figure 2). Conversely, a subset of neoplasms predominantly express V1 at diagnosis, which could account for the inferior responses of CLL11, 12, 13, 14,27 and precursor B-ALL71 to existing anti-CD20 antibody–based therapies. This suggests that the bulk of the CD20 protein is produced by V3 and that in cells with active CD20 transcription, the V3-to-V1 ratio is a key determinant of CD20 positivity. It remains to be determined whether the redirection of splicing toward V3 mediates the enhanced CD20 expression and rituximab sensitivity observed in vitro following treatment with interleukin-4, protein kinase C agonists, histone deacetylase inhibitors, or knockdown of the FOXO1 transcription factor.72, 73, 74, 75
To determine whether the V3-to-V1 shift is a relevant mechanism of resistance to CD20-directed immunotherapies, we generated OCI-Ly8 variants that express either V1 or V3 individually and tested their responses to CART-20 and mosunetuzumab. We found that V1-expressing cells had no CD20 protein detectable by flow cytometry (Figure 6C), and it took a much more sensitive 5′-UTR–green fluorescent protein reporter assay to observe baseline translation from the V1 mRNA (Figure 3F). Nevertheless, the V1-expressing cells remained sensitive to CART-20 (Figure 6D). Although unexpected, this finding is in agreement with the large body of literature attesting to the unique efficacy of CAR T cells against lymphoid malignancies with very low expression of the target antigens.76 This situation might contrast with the use of rituximab and bispecific CD3/CD20 T-cell engagers, such as mosunetuzumab, which, in our experimental system, required high levels of CD20 expression only afforded by the V3 cassette (Figure 6E). It was no surprise that the V3-to-V1 shift was apparent in 2 postmosunetuzumab FL relapses, P73 and P29 (Figure 6F). Moreover, in the latter case, the shift was the only molecular event known to reduce CD20 expression because no concomitant MS4A1 mutations could be identified in that sample.16
The propensity of B-cell malignancies to exploit alternative splicing mechanisms for antigen escape is now well documented, with CD19 and CD22 serving as prime examples.50,51 With respect to rituximab treatment, both mutational77 and nonmutational20 mechanism of CD20 loss have been reported, but the former is generally not considered a significant source of rituximab resistance in DLBCL.78 Data on postmosunetuzumab relapse are just beginning to emerge.16 As more paired pre- and postrelapse samples from patients on CD20-directed therapies become available, the extent to which acquired mutations and aberrant splicing (of both coding and noncoding exons79,80) supplement each other will become clearer.
At least in theory, targeting aberrant splicing in cancer with small-molecule inhibitors and antisense oligonucleotides (ASOs)81, 82, 83, 84 is more feasible than reversing hardwired mutations in DNA. Although previous studies have used ASOs to increase translation by blocking inhibitory elements within the 5′-UTR,85,86 our results demonstrated that ASOs could also enhance translation by modulating the alternative splicing of noncoding exons. Besides CD20, at least a few dozen other genes regulate mRNA translation via alternative splicing of their 5′-UTRs,87 and >50% of human genes use uORFs to regulate protein output in both diseased and healthy tissues.88 Harnessing this mechanism to achieve better treatment outcomes will be the next challenge for the cancer immunotherapy research community.
Conflict-of-interest disclosure: S.J.S. provides consulting to and receives research funding from Genentech/Roche. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Martin P. Carroll, Craig W. Freyer, and Mitchell Hughes of the University of Pennsylvania for their kind assistance with the rituximab and mosunetuzumab assays. The authors are grateful to all members of the A.T.-T. laboratory, in particular Priyanka Sehgal, for the helpful discussions.
This work was supported by grants from the National Institutes of Health, National Cancer Institute (U01CA232563 and U01 CA232563-S3 [A.T.-T., Y.B.], U01CA232486 and U01CA243072 [S.K.T.], and T32CA009615 [S.Z.]), United States Department of Defense (CA180683P1) (S.K.T.), The V Foundation for Cancer Research (T2018-014) (A.T.-T.), The Emerson Collective (886246066) (A.T.-T.). S.K.T. is a Scholar of the Leukemia & Lymphoma Society. S.K.T. has a Joshua Kahan Endowed Chair in Pediatric Leukemia Research, and A.T.-T. has a Mildred L. Roeckle Endowed Chair in Pathology at Children’s Hospital of Philadelphia. S.K.T. and A.T.T. are investigators of the St. Baldrick's Foundation EPICC Team.
Authorship
Contribution: Z.A., L.P., C.S., M.T.D., F.X., U.Z., Y.Z., S.S., S.Z., C.D.F., J.P.L., S.Y.Y., M.A., and P.K.S. performed the research; Z.A., K.E.H., M.M.L., and Y.B. analyzed the sequencing data; V.P., E.C., and S.J.S. analyzed the clinical data; S.K.T., P.M.L., M.R., and A.T.-T. supervized the laboratory experiments; Z.A. and A.T.-T. wrote the manuscript; and all other authors reviewed the manuscript.
Footnotes
Next-generation sequencing data are available in the Gene Expression Omnibus (GEO) repository with the following accession numbers: GSE212312 (primary human B cells), GSE243919 (paired NHL tumor samples, collected pretreatment and after mosunetuzumab relapse), and GSE243920 (direct long-read RNA sequencing of Raji cells).
Information regarding renewable reagents are available to all investigators on request from the corresponding author, Andrei Thomas-Tikhonenko (andreit@pennmedicine.upenn.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Pavlasova G, Mraz M. The regulation and function of CD20: an "enigma" of B-cell biology and targeted therapy. Haematologica. 2020;105(6):1494–1506. doi: 10.3324/haematol.2019.243543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pierpont TM, Limper CB, Richards KL. Past, present, and future of rituximab-the world's first oncology monoclonal antibody therapy. Front Oncol. 2018;8:163. doi: 10.3389/fonc.2018.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grillo-Lopez AJ, Hedrick E, Rashford M, Benyunes M. Rituximab: ongoing and future clinical development. Semin Oncol. 2002;29(1 suppl 2):105–112. doi: 10.1053/sonc.2002.30145. [DOI] [PubMed] [Google Scholar]
- 4.Marks VA, Latham SR, Kishore SP. On essentiality and the World Health Organization's model list of essential medicines. Ann Glob Health. 2017;83(3-4):637–640. doi: 10.1016/j.aogh.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Minard-Colin V, Auperin A, Pillon M, et al. Rituximab for high-risk, mature B-cell non-Hodgkin's lymphoma in children. N Engl J Med. 2020;382(23):2207–2219. doi: 10.1056/NEJMoa1915315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maury S, Chevret S, Thomas X, et al. Rituximab in B-lineage adult acute lymphoblastic leukemia. N Engl J Med. 2016;375(11):1044–1053. doi: 10.1056/NEJMoa1605085. [DOI] [PubMed] [Google Scholar]
- 7.Sun LL, Ellerman D, Mathieu M, et al. Anti-CD20/CD3 T cell-dependent bispecific antibody for the treatment of B cell malignancies. Sci Transl Med. 2015;7(287) doi: 10.1126/scitranslmed.aaa4802. [DOI] [PubMed] [Google Scholar]
- 8.Budde LE, Sehn LH, Matasar M, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022;23(8):1055–1065. doi: 10.1016/S1470-2045(22)00335-7. [DOI] [PubMed] [Google Scholar]
- 9.Cao Y, Marcucci EC, Budde LE. Mosunetuzumab and lymphoma: latest updates from 2022 ASH annual meeting. J Hematol Oncol. 2023;16(1):69. doi: 10.1186/s13045-023-01462-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glennie MJ, French RR, Cragg MS, Taylor RP. Mechanisms of killing by anti-CD20 monoclonal antibodies. Mol Immunol. 2007;44(16):3823–3837. doi: 10.1016/j.molimm.2007.06.151. [DOI] [PubMed] [Google Scholar]
- 11.Almasri NM, Duque RE, Iturraspe J, Everett E, Braylan RC. Reduced expression of CD20 antigen as a characteristic marker for chronic lymphocytic leukemia. Am J Hematol. 1992;40(4):259–263. doi: 10.1002/ajh.2830400404. [DOI] [PubMed] [Google Scholar]
- 12.Ginaldi L, De Martinis M, Matutes E, Farahat N, Morilla R, Catovsky D. Levels of expression of CD19 and CD20 in chronic B cell leukaemias. J Clin Pathol. 1998;51(5):364–369. doi: 10.1136/jcp.51.5.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perz J, Topaly J, Fruehauf S, Hensel M, Ho AD. Level of CD 20-expression and efficacy of rituximab treatment in patients with resistant or relapsing B-cell prolymphocytic leukemia and B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2002;43(1):149–151. doi: 10.1080/10428190210178. [DOI] [PubMed] [Google Scholar]
- 14.Olejniczak SH, Stewart CC, Donohue K, Czuczman MS. A quantitative exploration of surface antigen expression in common B-cell malignancies using flow cytometry. Immunol Invest. 2006;35(1):93–114. doi: 10.1080/08820130500496878. [DOI] [PubMed] [Google Scholar]
- 15.Golay J, Lazzari M, Facchinetti V, et al. CD20 levels determine the in vitro susceptibility to rituximab and complement of B-cell chronic lymphocytic leukemia: further regulation by CD55 and CD59. Blood. 2001;98(12):3383–3389. doi: 10.1182/blood.v98.12.3383. [DOI] [PubMed] [Google Scholar]
- 16.Schuster SJ, Huw L-Y, Bolen CR, et al. Characterization of CD20 expression loss as a mechanism of resistance to mosunetuzumab in patients with relapsed/refractory B-cell non-Hodgkin lymphomas. J Clin Oncol. 2022;40(16 suppl):7526. [Google Scholar]
- 17.Nowakowski GS, Blum KA, Kahl BS, et al. Beyond RCHOP: a blueprint for diffuse large B cell lymphoma research. J Natl Cancer Inst. 2016;108(12):djw257. doi: 10.1093/jnci/djw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown JR, Cymbalista F, Sharman J, Jacobs I, Nava-Parada P, Mato A. The role of rituximab in chronic lymphocytic leukemia treatment and the potential utility of biosimilars. Oncologist. 2018;23(3):288–296. doi: 10.1634/theoncologist.2017-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haidar JH, Shamseddine A, Salem Z, et al. Loss of CD20 expression in relapsed lymphomas after rituximab therapy. Eur J Haematol. 2003;70(5):330–332. doi: 10.1034/j.1600-0609.2003.00007.x. [DOI] [PubMed] [Google Scholar]
- 20.Hiraga J, Tomita A, Sugimoto T, et al. Down-regulation of CD20 expression in B-cell lymphoma cells after treatment with rituximab-containing combination chemotherapies: its prevalence and clinical significance. Blood. 2009;113(20):4885–4893. doi: 10.1182/blood-2008-08-175208. [DOI] [PubMed] [Google Scholar]
- 21.Miyoshi H, Arakawa F, Sato K, et al. Comparison of CD20 expression in B-cell lymphoma between newly diagnosed, untreated cases and those after rituximab treatment. Cancer Sci. 2012;103(8):1567–1573. doi: 10.1111/j.1349-7006.2012.02307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D'Auria F, Guariglia R, Villani O, et al. Modulation of CD20 antigen expression after rituximab treatment: a retrospective study in patients with chronic lymphocytic leukemia. Clin Ther. 2010;32(11):1911–1916. doi: 10.1016/j.clinthera.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 23.Yu H, Sotillo E, Harrington C, et al. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am J Hematol. 2017;92(1):E11–E13. doi: 10.1002/ajh.24594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson NA, Boyle M, Bashashati A, et al. Diffuse large B-cell lymphoma: reduced CD20 expression is associated with an inferior survival. Blood. 2009;113(16):3773–3780. doi: 10.1182/blood-2008-09-177469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klasener K, Jellusova J, Andrieux G, et al. CD20 as a gatekeeper of the resting state of human B cells. Proc Natl Acad Sci U S A. 2021;118(7) doi: 10.1073/pnas.2021342118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu D, Dews M, Park A, Tobias JW, Thomas-Tikhonenko A. Inactivation of Myc in murine two-hit B lymphomas causes dormancy with elevated levels of interleukin 10 receptor and CD20: implications for adjuvant therapies. Cancer Res. 2005;65(12):5454–5461. doi: 10.1158/0008-5472.CAN-04-4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarro SM, Unruh TL, Zuccolo J, et al. Quantification of CD20 mRNA and protein levels in chronic lymphocytic leukemia suggests a post-transcriptional defect. Leuk Res. 2010;34(12):1670–1673. doi: 10.1016/j.leukres.2010.06.031. [DOI] [PubMed] [Google Scholar]
- 28.Abate F, Ambrosio MR, Mundo L, et al. Distinct viral and mutational spectrum of endemic Burkitt lymphoma. PLoS Pathog. 2015;11(10) doi: 10.1371/journal.ppat.1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park HY, Lee SB, Yoo HY, et al. Whole-exome and transcriptome sequencing of refractory diffuse large B-cell lymphoma. Oncotarget. 2016;7(52):86433–86445. doi: 10.18632/oncotarget.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parsa S, Ortega-Molina A, Ying HY, et al. The serine hydroxymethyltransferase-2 (SHMT2) initiates lymphoma development through epigenetic tumor suppressor silencing. Nat Cancer. 2020;1:653–664. doi: 10.1038/s43018-020-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koues OI, Kowalewski RA, Chang LW, et al. Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity. 2015;42(1):186–198. doi: 10.1016/j.immuni.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agrawal AA, Seiler M, Brinton LT, et al. Novel SF3B1 in-frame deletions result in aberrant RNA splicing in CLL patients. Blood Adv. 2017;1(15):995–1000. doi: 10.1182/bloodadvances.2017007062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghia EM, Rassenti LZ, Choi MY, et al. High expression level of ROR1 and ROR1-signaling associates with venetoclax resistance in chronic lymphocytic leukemia. Leukemia. 2022;36(6):1609–1618. doi: 10.1038/s41375-022-01543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wernig-Zorc S, Yadav MP, Kopparapu PK, et al. Global distribution of DNA hydroxymethylation and DNA methylation in chronic lymphocytic leukemia. Epigenetics Chromatin. 2019;12(1):4. doi: 10.1186/s13072-018-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Black KL, Naqvi AS, Asnani M, et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018;46(21):11357–11369. doi: 10.1093/nar/gky946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang SY, Hayer KE, Fazelinia H, et al. FBXW7beta isoform drives transcriptional activation of the proinflammatory TNF cluster in human pro-B cells. Blood Adv. 2023;7(7):1077–1091. doi: 10.1182/bloodadvances.2022007910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng S, Gillespie E, Naqvi AS, et al. Modulation of CD22 protein expression in childhood leukemia by pervasive splicing aberrations: implications for CD22-directed immunotherapies. Blood Cancer Discov. 2022;3(2):103–115. doi: 10.1158/2643-3230.BCD-21-0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monaco G, Lee B, Xu W, et al. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26(6):1627–1640.e7. doi: 10.1016/j.celrep.2019.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaymaz Y, Oduor CI, Yu H, et al. Comprehensive transcriptome and mutational profiling of endemic Burkitt lymphoma reveals EBV type-specific differences. Mol Cancer Res. 2017;15(5):563–576. doi: 10.1158/1541-7786.MCR-16-0305-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun W, Duan T, Ye P, et al. TSVdb: a web-tool for TCGA splicing variants analysis. BMC Genomics. 2018;19(1):405. doi: 10.1186/s12864-018-4775-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vera Alvarez R, Pongor LS, Marino-Ramirez L, Landsman D. TPMCalculator: one-step software to quantify mRNA abundance of genomic features. Bioinformatics. 2019;35(11):1960–1962. doi: 10.1093/bioinformatics/bty896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Parmigiani G, Johnson WE. ComBat-seq: batch effect adjustment for RNA-seq count data. NAR Genom Bioinform. 2020;2(3):lqaa078. doi: 10.1093/nargab/lqaa078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sladitschek HL, Neveu PA. MXS-chaining: a highly efficient cloning platform for imaging and flow cytometry approaches in mammalian systems. PLoS One. 2015;10(4) doi: 10.1371/journal.pone.0124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodbin SE, Denning C, Mosqueira D. Transfection of hPSC-cardiomyocytes using Viafect transfection reagent. Methods Protoc. 2020;3(3):57. doi: 10.3390/mps3030057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shan D, Press OW, Tsu TT, Hayden MS, Ledbetter JA. Characterization of scFv-Ig constructs generated from the anti-CD20 mAb 1F5 using linker peptides of varying lengths. J Immunol. 1999;162(11):6589–6595. [PubMed] [Google Scholar]
- 48.Budde LE, Berger C, Lin Y, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8(12) doi: 10.1371/journal.pone.0082742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rufener GA, Press OW, Olsen P, et al. Preserved activity of CD20-specific chimeric antigen receptor-expressing T cells in the presence of rituximab. Cancer Immunol Res. 2016;4(6):509–519. doi: 10.1158/2326-6066.CIR-15-0276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng S, Asnani M, Thomas-Tikhonenko A. Escape from ALL-CARTaz: leukemia immunoediting in the age of chimeric antigen receptors. Cancer J. 2019;25(3):217–222. doi: 10.1097/PPO.0000000000000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bourcier J, Abdel-Wahab O. Splicing-mediated antigen escape from immunotherapy for B-cell malignancies. Blood Cancer Discov. 2022;3(2):87–89. doi: 10.1158/2643-3230.BCD-21-0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulz L, Torres-Diz M, Cortes-Lopez M, et al. Direct long-read RNA sequencing identifies a subset of questionable exitrons likely arising from reverse transcription artifacts. Genome Biol. 2021;22(1):190. doi: 10.1186/s13059-021-02411-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, Thomas-Tikhonenko A. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest. 2012;122(6):2257–2266. doi: 10.1172/JCI45851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Psathas JN, Doonan PJ, Raman P, Freedman BD, Minn AJ, Thomas-Tikhonenko A. The Myc-miR-17-92 axis amplifies B-cell receptor signaling via inhibition of ITIM proteins: a novel lymphomagenic feed-forward loop. Blood. 2013;122(26):4220–4229. doi: 10.1182/blood-2012-12-473090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brady G, MacArthur GJ, Farrell PJ. Epstein-Barr virus and Burkitt lymphoma. J Clin Pathol. 2007;60(12):1397–1402. doi: 10.1136/jcp.2007.047977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mrozek-Gorska P, Buschle A, Pich D, et al. Epstein-Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc Natl Acad Sci U S A. 2019;116(32):16046–16055. doi: 10.1073/pnas.1901314116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Asnani M, Hayer KE, Naqvi AS, et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia. 2020;34(4):1202–1207. doi: 10.1038/s41375-019-0580-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ang Z, Koean RAG, Er JZ, et al. Novel AU-rich proximal UTR sequences (APS) enhance CXCL8 synthesis upon the induction of rpS6 phosphorylation. PLoS Genet. 2019;15(4) doi: 10.1371/journal.pgen.1008077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lorenz R, Bernhart SH, Honer Zu Siederdissen C, et al. ViennaRNA package 2.0. Algorithms Mol Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20(23):8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol. 1989;9(11):5134–5142. doi: 10.1128/mcb.9.11.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Piva F, Giulietti M, Nocchi L, Principato G. SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics. 2009;25(9):1211–1213. doi: 10.1093/bioinformatics/btp124. [DOI] [PubMed] [Google Scholar]
- 63.Itoh M, Haga I, Li QH, Fujisawa J. Identification of cellular mRNA targets for RNA-binding protein Sam68. Nucleic Acids Res. 2002;30(24):5452–5464. doi: 10.1093/nar/gkf673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morcos PA. Achieving targeted and quantifiable alteration of mRNA splicing with Morpholino oligos. Biochem Biophys Res Commun. 2007;358(2):521–527. doi: 10.1016/j.bbrc.2007.04.172. [DOI] [PubMed] [Google Scholar]
- 65.Morcos PA, Li Y, Jiang S. Vivo-Morpholinos: a non-peptide transporter delivers Morpholinos into a wide array of mouse tissues. Biotechniques. 2008;45(6):613–614. doi: 10.2144/000113005. 616, 618 passim. [DOI] [PubMed] [Google Scholar]
- 66.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vaquero-Garcia J, Barrera A, Gazzara MR, et al. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife. 2016;5 doi: 10.7554/eLife.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horna P, Nowakowski G, Endell J, Boxhammer R. Comparative assessment of surface CD19 and CD20 expression on B-cell lymphomas from clinical biopsies: implications for targeted therapies [abstract] Blood. 2019;134(suppl 1):5345. [Google Scholar]
- 69.Reichard KK, McKenna RW, Kroft SH. Comparative analysis of light chain expression in germinal center cells and mantle cells of reactive lymphoid tissues. A four-color flow cytometric study. Am J Clin Pathol. 2003;119(1):130–136. doi: 10.1309/9MYM-D68F-U8YE-843D. [DOI] [PubMed] [Google Scholar]
- 70.Polikowsky HG, Wogsland CE, Diggins KE, Huse K, Irish JM. Cutting edge: redox signaling hypersensitivity distinguishes human germinal center B cells. J Immunol. 2015;195(4):1364–1367. doi: 10.4049/jimmunol.1500904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jeha S, Behm F, Pei D, et al. Prognostic significance of CD20 expression in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2006;108(10):3302–3304. doi: 10.1182/blood-2006-04-016709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sandova V, Pavlasova GM, Seda V, et al. IL4-STAT6 signaling induces CD20 in chronic lymphocytic leukemia and this axis is repressed by PI3Kdelta inhibitor idelalisib. Haematologica. 2021;106(11):2995–2999. doi: 10.3324/haematol.2021.278644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pyrzynska B, Dwojak M, Zerrouqi A, et al. FOXO1 promotes resistance of non-Hodgkin lymphomas to anti-CD20-based therapy. Oncoimmunology. 2018;7(5) doi: 10.1080/2162402X.2017.1423183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bobrowicz M, Dwojak M, Pyrzynska B, Stachura J, Muchowicz A. HDAC6 inhibition upregulates CD20 levels and increases the efficacy of anti-CD20 monoclonal antibodies. Blood. 2017;130(14):1628–1638. doi: 10.1182/blood-2016-08-736066. [DOI] [PubMed] [Google Scholar]
- 75.Wojciechowski W, Li H, Marshall S, Dell'Agnola C, Espinoza-Delgado I. Enhanced expression of CD20 in human tumor B cells is controlled through ERK-dependent mechanisms. J Immunol. 2005;174(12):7859–7868. doi: 10.4049/jimmunol.174.12.7859. [DOI] [PubMed] [Google Scholar]
- 76.Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 77.Rushton CK, Arthur SE, Alcaide M, et al. Genetic and evolutionary patterns of treatment resistance in relapsed B-cell lymphoma. Blood Adv. 2020;4(13):2886–2898. doi: 10.1182/bloodadvances.2020001696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson NA, Leach S, Woolcock B, et al. CD20 mutations involving the rituximab epitope are rare in diffuse large B-cell lymphomas and are not a significant cause of R-CHOP failure. Haematologica. 2009;94(3):423–427. doi: 10.3324/haematol.2008.001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Henry C, Deschamps M, Rohrlich PS, et al. Identification of an alternative CD20 transcript variant in B-cell malignancies coding for a novel protein associated to rituximab resistance. Blood. 2010;115(12):2420–2429. doi: 10.1182/blood-2009-06-229112. [DOI] [PubMed] [Google Scholar]
- 80.Gamonet C, Bole-Richard E, Delherme A, et al. New CD20 alternative splice variants: molecular identification and differential expression within hematological B cell malignancies. Exp Hematol Oncol. 2015;5:7. doi: 10.1186/s40164-016-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang E, Lu SX, Pastore A, et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell. 2019;35(3):369–384.e7. doi: 10.1016/j.ccell.2019.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–694. doi: 10.1038/s41573-020-0075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bradley RK, Anczukow O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. 2023;23(3):135–155. doi: 10.1038/s41568-022-00541-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liang XH, Sun H, Shen W, et al. Antisense oligonucleotides targeting translation inhibitory elements in 5' UTRs can selectively increase protein levels. Nucleic Acids Res. 2017;45(16):9528–9546. doi: 10.1093/nar/gkx632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liang XH, Shen W, Sun H, Migawa MT, Vickers TA, Crooke ST. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat Biotechnol. 2016;34(8):875–880. doi: 10.1038/nbt.3589. [DOI] [PubMed] [Google Scholar]
- 87.Floor SN, Doudna JA. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016;5 doi: 10.7554/eLife.10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee DSM, Park J, Kromer A, et al. Disrupting upstream translation in mRNAs is associated with human disease. Nat Commun. 2021;12(1):1515. doi: 10.1038/s41467-021-21812-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.