

Summary
Chromosome-level design-build-test-learn cycles (chrDBTLs) allow systematic combinatorial reconfiguration of chromosomes with ease. Here, we established chrDBTL with a redesigned synthetic Saccharomyces cerevisiae chromosome XV, synXV. We designed and built synXV to harbor strategically inserted features, modified elements, and synonymously recoded genes throughout the chromosome. Based on the recoded chromosome, we developed a method to enable chrDBTL: CRISPR-Cas9-mediated mitotic recombination with endoreduplication (CRIMiRE). CRIMiRE allowed the creation of customized wild-type/synthetic combinations, accelerating genotype-phenotype mapping and synthetic chromosome redesign. We also leveraged synXV as a “build-to-learn” model organism for translation studies by ribosome profiling. We conducted a locus-to-locus comparison of ribosome occupancy between synXV and the wild-type chromosome, providing insight into the effects of codon changes and redesigned features on translation dynamics in vivo. Overall, we established synXV as a versatile reconfigurable system that advances chrDBTL for understanding biological mechanisms and engineering strains.
Keywords: Saccharomyces cerevisiae, synthetic yeast, synthetic chromosome, CRISPR-Cas9, mitotic recombination, synthetic biology, design-build-test-learn, synthetic genomics, chromosome reconfiguration
Graphical abstract
Highlights
-
•
A 1.05-Mbp redesigned synthetic Saccharomyces cerevisiae chromosome is constructed
-
•
The synthetic chromosome accelerates chromosomal design-build-test-learn cycles
-
•
Synthetic yeast is a “build-to-learn” model organism for biological investigations
Foo et al. accelerated chromosome-level design-build-test-learn cycles (chrDBTLs) through a synthetic Saccharomyces cerevisiae chromosome XV, synXV. synXV was strategically recoded, which enabled systematic combinatorial reconfiguration of chromosomes, hence facilitating genotype-phenotype mapping. synXV also served as a “build-to-learn” model organism for ribosome profiling studies, allowing translation dynamics comparison with the wild-type chromosome.
Introduction
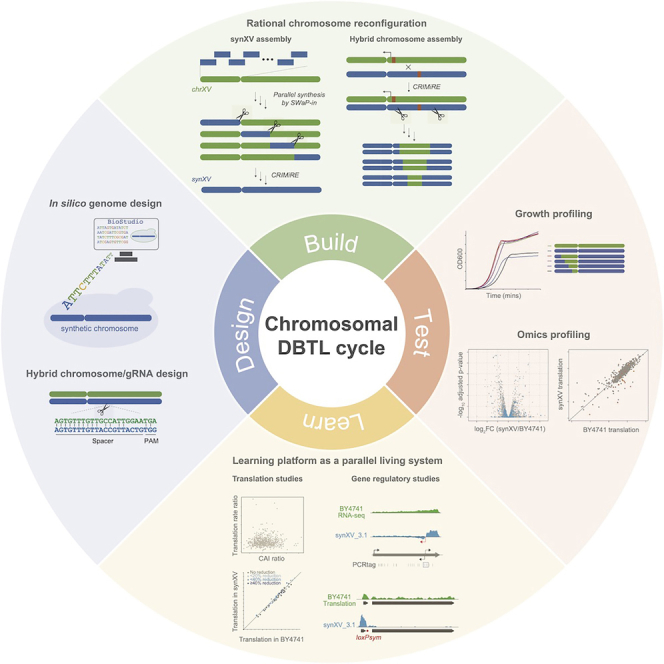
The design-build-test-learn cycle (DBTL) has been instrumental for modifying living organisms to investigate biological systems.1 To date, the DBTL framework has been limited mainly to episomal gene expression or chromosomal gene deletion, insertion, and mutation to study and understand the effects on cellular characteristics. Such specific genetic modifications often allow only isolated, localized effects to be studied, and the difficulty in generating wide genotypic variations makes the elucidation of intergenic interactions or global genome-wide effects challenging.2 Moreover, episomal systems may not provide an accurate representation of the in vivo processes and interactions.3 To overcome these limitations, chromosome-level DBTL cycles (chrDBTLs) are essential, where chromosomes can be systematically reconfigured combinatorially to generate genetic diversity for characterization and learning the factors that contribute to phenotypic differences (Figure 1).
Figure 1.

Establishing chromosome-scale design-build-test-learn cycles (chrDBTLs) with synXV
We established chrDBTLs by developing a rational chromosome reconfiguration method, CRISPR-Cas9-mediated mitotic recombination with endoreduplication (CRIMiRE). CRIMiRE was applied to accelerate the construction of synXV and to generate a rationally designed semisynthetic synXV library. The ease of generating chromosome libraries by CRIMiRE provided systematic approaches to advance chrDBTL and facilitate the identification of causal variants. We also attested that the redesigned synXV functions as a “build-to-learn” platform and serves as a parallel living system for probing and answering fundamental biological questions.
The emergence of synthetic genomics has contributed to advancements in establishing chrDBTL by enabling the redesign of chromosomal sequences and construction of synthetic chromosomes. Early efforts in synthetic genomics mainly focused on “design-build-test” and overcame major technical aspects of synthetic chromosome design and construction.4,5 However, the “learn” part of DBTL was still lacking because it was difficult to manipulate the synthetic chromosomes to create customized versions of the chromosomes to set up chrDBTLs for characterization and analysis. A breakthrough in synthetic genomics came when the Sc2.0 consortium modified the chromosomes of Saccharomyces cerevisiae with strategic and extensively recoded sequences and additional features, notably the insertion of symmetrical loxP (loxPsym) sites.6,7 These redesigned elements of Sc2.0 confer the synthetic chromosomes ease of reconfiguration, creating a reprogrammable synthetic model yeast system to realize chrDBTLs, and facilitate “build-to-learn” biological research.
Here, we report the establishment of chrDBTL with a redesigned synthetic chromosome XV of S. cerevisiae, synXV (Figure 1). We first designed and built a synthetic chromosome for chrDBTL, with genes synonymously recoded extensively throughout the chromosome, forming watermark sequences with high sequence specificity. Leveraging the recoded sequences of synXV, we then developed a method to enable chrDBTL: CRISPR-Cas9-mediated mitotic recombination with endoreduplication (CRIMiRE). Using CRIMiRE, we demonstrated chrDBTL by rationally generating synXV variants harboring different synthetic/wild-type combinations for subsequent characterization through genotype-phenotype mapping, identifying the causes of phenotypic defects and refining the synXV design to improve the phenotype of synXV. Furthermore, we demonstrated that synXV could function as a “comparative synthetic genomics” platform by systematically testing hypotheses on the biological effects because of the differences between the recoded and wild-type sequences; knowledge learned through the testing enabled the debugging and redesigning of synXV. Overall, we designed, built, and established synXV as a pliable synthetic model system for implementing chrDBTL through systematic combinatorial chromosome reconfiguration.
Results
Design of synXV and assembly of “hyperchunks”
Because chromosome XV (chrXV) is the second largest chromosome in S. cerevisiae (after chrIV), its substantial size offers ample genetic space for a redesign aimed at creating its synthetic analog, synXV, which serves as a platform for demonstrating chrDBTL. We redesigned chrXV in silico according to the specifications of the Sc2.0 project using BioStudio;6 the design principles of Sc2.0 are detailed in the literature.7 The recoded synXV sequence included several major changes, including (1) recoding with synonymous mutations to create highly specific watermark sequences as unique polymerase chain reaction tags (PCRTags) (1,408 sites); (2) modifications to create unique restriction sites (308 sites); (3) insertion of loxPsym sites (395 sites); (4) changing of TAG stop codons to TAA (143 sites); (5) deletion of tRNAs, introns, and long terminal repeats (56 sites); and (6) replacement of the telomeres with universal telomere caps (UTCs) (2 sites). The length of synXV was reduced by 3.9% from 1,091,291 bp in the wild type to 1,048,343 bp.7 To facilitate assembly, synXV was segmented into 42 regions (termed “megachunks”), each comprising 4–5 fragments (named “chunks”) (Table S1) of chemically synthesized DNA. The megachunks were then integrated sequentially by switching auxotrophies progressively by integration (SwAP-In).7 Because synXV was over 1 Mbp in size, the megachunks were grouped into four “hyperchunks” for concurrent construction to accelerate synXV assembly (Figure S1A). The semisynthetic strains harboring the hyperchunks were of appropriate MATa and MATα mating types to allow subsequent crossing of the hyperchunks for consolidation into a complete synXV sequence by mitotic and meiotic recombination-based methods (Figures S1A–S1E; Table S2). Based on the incorporation of the recoded sequences, features, and elements in synXV, we hypothesized that synXV could be reconfigured combinatorially and that synXV and the resultant reconfigured variants could be harnessed to elucidate the roles of genetic variations in transcription and translation and generate strains with desired phenotypes, thereby enabling chrDBTL.
CRIMiRE enabled targeted recombination of chromosomes and facilitated synXV assembly
To test our hypothesis that the recoded sequences of synXV could allow combinatorial reconfiguration to create chromosomes with customized combinations of synthetic/wild-type sections, we developed the CRIMiRE method. CRIMiRE fuses desired segments of chromosomes rationally by CRISPR-Cas9-mediated mitotic recombination and, crucially, simplifies the isolation of the desired recombinant chromosome. As a test bed, we consolidated hyperchunks to generate synXV with CRIMiRE (Figure 2A). Currently, meiotic recombination is a mechanism that is commonly used to recombine chromosomes. The process has been harnessed to consolidate several partially synthetic strains into a contiguous synthetic chromosome.8 It was also employed to progressively recombine hyperchunk 2 (HC2), HC3, and HC4, ultimately forming HC234 (as depicted in Figure S1A), all preceding the development of CRIMiRE. However, the recombination was random, and extensive PCRTag screening was required to identify colonies without cross-mixed wild-type sequences, a major bottleneck in constructing long synthetic chromosomes such as synXV (Figure 2A). CRISPR-Cas9 has been employed previously for mitotic recombination,9 and CRISPR-Cas9-induced gene conversion assembly (CiGa) was developed for accelerated assembly of synthetic chromosomes using CRISPR-Cas9;10 CiGa was utilized for the construction of HC4 prior to the development of CRIMiRE. In parallel, the CRISPR directed biallelic URA3-assisted genome scan (CRISPR-D-BUGS) was developed to create chromosomes with synthetic/wild-type combinations to efficiently identify sources of phenotypic defects in Sc2.0 strains.11 Although these studies generated the desired recombined chromosomes, the resultant diploids were still partially heterozygous. Thus, these methods are not ideal for the purpose of isolating a synXV haploid; the issue of having cross-mixed wild-type sequences still remains because of meiotic recombination during sporulation. Through CRIMiRE, many of these challenges are effectively addressed. CRIMiRE leverages the recoded chromosomal sequences, which serve as distinctive cleavage sites for CRISPR-Cas9, facilitating chromosome recombination at precise locations. The semisynthetic chromosomes incorporate strategically positioned URA3 markers and a GAL1 promoter situated upstream of one of the semisynthetic chromosome’s centromeres. Through galactose induction, missegregation and subsequent loss of the “undesired” chromosome occur.12 Simultaneously, counterselection with 5-fluoroorotic acid enables the isolation of Ura− strains carrying the desired synXV chromosome. Consequently, through spontaneous endoreduplication, a homozygous synXV diploid configuration is achieved, effectively eliminating the potential for synXV to intermingle with wild-type sequences during sporulation (Figure 2A). Hence, CRIMiRE could significantly expedite the screening and isolation of a strain with the desired chromosomal combination and was therefore applied to accelerate synXV’s construction.
Figure 2.

Assembly of synXV by CRIMiRE
(A) CRIMiRE requires strategic modifications to the semisynthetic chromosomes as follows: (1) URA3 markers near the respective telomere and centromere within the wild-type segments to be removed and (2) a galactose-inducible promoter (PGAL1) upstream of the centromere within the wild-type segment to be removed. CRISPR-Cas9-mediated mitotic recombination commences upon mating of the two strains carrying plasmids for expressing Cas9 and a single guide RNA (sgRNA) separately. Assembled synXV coexists in a mixed population of heterozygous diploids. Galactose induction missegregates chromosomes with PGAL1 upstream of the centromere.13 Under 5-fluoroorotic acid (5-FOA) selection, hemizygous synXV diploids are the only Ura– cells within the missegregated population that can grow. Subsequent endoreduplication of the synXV chromosome in the 2n-1 strain results in a 2n homozygous synXV diploid, which is then sporulated to obtain a haploid synXV. In contrast, meiotic recombination would lead to a myriad of chimeric chromosomes, which would be difficult to screen to identify a strain with complete synXV.
(B) HC1A and HC1B were modified with a KlURA3-PGAL1-CEN15 cassette and URA3 from chunk A5 near the left terminus, respectively. These derived strains were consolidated into HC1 by CRIMiRE by cleaving between megachunks I and J. The resulting clones were screened by PCRTags (Figures S1F–S1H) to identify clones with homozygous HC1.
(C) Analysis of HC1 with synthetic and wild-type PCRTags (SYN and WT, respectively). A strain (Figures S1G–S1H, clone 7) showed only synthetic PCRTags in megachunks I and J. Further analysis with the first and last PCRTags of each megachunk showed the clone to be synthetic throughout HC1.
(D) HC1 was modified with a KlURA3-PGAL1-CEN15 cassette and a LEU2 from chunk NN4 near the right terminus. HC234 was modified with a URA3 from chunk A5 near the left terminus. These derived strains were consolidated into synXV by CRIMiRE by cleaving between megachunks M and N. The resulting clones were screened by auxotrophy and PCRTags (Figures S1J–S1N) to identify clones with homozygous synXV.
(E) A synXV candidate (Figures S1L–S1N, clone L6) was found to be synthetic and homozygous in megachunks M and N, which flanked the cleavage site.
(F) Two representative PCRTags from each megachunk showing that the synXV clone was synthetic and homozygous throughout the chromosome. A complete PCRTag analysis was performed (Figure S2).
To evaluate CRIMiRE, we employed this technique to construct HC1 from HC1A and HC1B, which are partial HC1s containing megachunks A–I and J–M, respectively, by cleaving at the junction of HC1A and HC1B at a unique wild-type sequence on HC1B that was recoded in HC1A (Figure 2B). A primary PCRTag screening of 10 isolated diploid HC1 candidates showed only amplicons specific to synthetic sequences at the termini (Figures S1F–S1G), indicating that CRIMiRE successfully selected homozygous strains that underwent crossover and possessed synthetic sequences from HC1A and HC1B. Further PCRTag screening of megachunks I and J flanking the cleavage site revealed several wild-type PCRTags (Figures S1H), consistent with a previous report on mitotic recombination events occurring within 20 kb of the targeted site.9 Nevertheless, a strain with all synthetic PCRTags in megachunks I and J was identified from the 10 candidates and further verified by PCRTag screening to harbor a synthetic sequence throughout HC1 (Figure 2C). These findings clearly illustrate the significantly enhanced efficiency of CRIMiRE compared with previously developed methods for consolidating semisynthetic chromosomes. For instance, the success rate of isolating a clone with 10 megachunks consolidated through CiGA was approximately 1%.10 Moreover, while preparing the chromosomes for CRIMiRE necessitates supplementary steps involving established techniques to introduce URA3 markers and a conditional centromere, the resultant strain becomes a homozygous diploid. This characteristic simplifies subsequent processes, such as PCRTag screening and overall strain identification, rendering them less labor intensive and time consuming and more cost effective. Additionally, the isolation of haploids that originate from the homozygous diploid can be accomplished through random spore analysis, eliminating the necessity for specialized equipment to conduct intricate tetrad dissection—a requirement in conventional meiotic recombination approaches for further screening. Consequently, CRIMiRE emerges as a potent tool for facilitating the combinatorial reconfiguration of chromosomes.
Encouraged by the results, we performed CRIMiRE to consolidate HC1 and HC234 to create a complete synXV (Figure 2D). Ultimately, a clone was verified to possess synthetic sequences throughout chrXV (Figures 2E, 2F, S1I–S1N, S2A, and S2B). Whole-genome sequencing of the strain confirmed the successful construction of an initial synXV isolate, synXV_3.1. Taken together, we validated the hypothesis that the recoded sequences of synXV can enable the rational reconfiguration of chromosomes with customized combinations using the CRIMiRE method we developed.
ChrDBTL with CRIMiRE identified the origins of growth defects
We then tested whether CRIMiRE could be employed to establish chrDBTL by rationally generating homozygous synXV variants that harbor different synthetic/wild-type combinations for subsequent characterization through genotype-phenotype mapping, refining the synXV design to improve the phenotype of synXV and learning the causes of the phenotypic defects (“bugs”). In particular, although synXV_3.1 was viable, two distinct defects were observed. First, in YPD (yeast extract-peptone-dextrose), the doubling time of synXV_3.1 was 39.7% longer than that of BY4741 (118.3 vs. 84.6 min, respectively; Figure S3A). Second, sporulation of the synXV_3.1 diploid was much slower than that of BY4743 (no asci were observed after 10 days; Figure S3B). A major defect likely existed in HC1 because the hyperchunk had a long doubling time (132.7 min), but that of HC234 (77.7 min) was closer to that of BY4741 (Figure S3A). Furthermore, the HC1A diploid sporulated poorly, akin to synXV_3.1, while diploids of HC1B and HC234 sporulated similarly as BY4743. With CRIMiRE, we can generate multiple versions of chromosomes with increasing lengths of the synthetic HC1 region added to HC234, thereby launching the “debugging” chrDBTL for pinpointing the positions of the bugs underlying the defects (Figures 3A and 3B; Table S3). Notably, because the resulting CRIMiRE-generated strains are homozygous for semisynthetic synXV, it can be said that CRIMiRE is suitable for identifying and simultaneously correcting dominant and recessive bugs.
Figure 3.

ChrDBTL with CRIMiRE identified the origin of growth defects in the left arm
(A) Schematic of CRIMiRE-driven chrDBTL for bug identification in the left arm of synXV. HC1 and HC234 were modified with a KlURA3-PGAL1-CEN15 cassette and URA3 at chunk A5, respectively. CRIMiRE generated rationally designed semisynthetic diploid strains. The regions responsible for the bug were identified by phenotyping assays and PCRTag screening. Then, a second CRIMiRE-driven chrDBTL was performed by designing new WT-specific sgRNAs to cleave within the responsible region identified in the previous cycle to pinpoint the bug locations.
(B) Growth profiles of BY4743 and four semisynthetic strains with varying lengths of synthetic sequence in the left arm.
(C) Growth profiles of the semisynthetic strains obtained by CRIMiRE upon cleavage within megachunks H and I.
(D) PCRTag maps of semisynthetic strains obtained by CRIMiRE upon cleavage within megachunks H and I. Each block denotes a pair of PCRTags. White blocks indicate the absence of PCRTag amplicons, possibly because of mixed synthetic/wild-type primer pairs or deletion of the regions. sgRNA-H3 and sgRNA-I1 indicate cleavage sites in chunks H3 and I1, which generated the strains mr-H3-1 and mr-I1-x, respectively. The red box shows the region where a fitness defect may reside.
(E) The doubling time and PCRTag maps of strains generated for debugging synXV_3.1 with various combinations of synthetic and wild-type PCRTags in IRA2 and REX4.
(F) RNA-seq mapping of IRA2. Read depth was normalized by total mapped reads. Positive and negative values denote read depth on sense and antisense strands, respectively.
(G and H) Volcano plot comparing transcript profiles between BY4741 and synXV_3.1 (n = 3) or synXV_3.2 (n = 3).
(I) Scatterplot showing the correlation of expression changes between synXV_3.1 and the ira2Δ mutant14 for genes in the TCA cycle, oxidative phosphorylation pathway, and starch and sucrose metabolism.
A comparison of the series of CRIMiRE-generated semisynthetic synXV strains shows that the addition of megachunks H–I has detrimental effects on growth rate (strain mr-I; Figure 3B). Additional hybrid strains were created with CRIMiRE by directing cleavage within the two megachunks. These strains revealed that having wild-type PCRTags in the region encompassing IRA2 and REX4 improved the growth rate (Figures 3C and 3D), with sporulation also occurring within 5 days. To identify the exact location of the bugs, we reverted the PCRTags within the IRA2-REX4 region in synXV_3.1 back to wild type (strains dbg-1.1, 1.2, and 1.3; Figure 3E) and found that restoring only IRA2 back to wild type (strain dbg-1.2) was sufficient for improving doubling time (Figures 3E and S3C). Through a series of PCRTag replacements in IRA2, we discovered the eighth pair of synthetic PCRTags in IRA2 to be a determinant in lengthening doubling time (Figures 3E, S3D, and S3E), with its reversion to wild type in synXV_3.1 markedly improving doubling time (dbg-1.4;Figures 3E and S3F). The enhanced growth fitness of dbg-1.4 indicated the resolution of a major defect in synXV, and the strain is hereafter referred to as synXV_3.2.
IRA2 encodes a guanosine triphosphate hydrolase (GTPase)-activating protein that negatively regulates Ras to reduce intracellular cyclic AMP (cAMP) levels and repress the cAMP/protein kinase A (PKA) pathway. Its deletion is known to increase cAMP levels and cause abnormal phenotypes, such as slow cell growth and sporulation defects,15 consistent with the traits of synXV_3.1. To investigate the effects the problematic IRA2 PCRTags have on local and global gene expression and elucidate how the PCRTag bugs in IRA2 caused phenotypic defects in synXV_3.1, we performed RNA sequencing (RNA-seq) of BY4741, synXV_3.1, and synXV_3.2. Upon analyzing the RNA-seq reads mapped around IRA2, we observed two additional peaks in synXV_3.1 on the sense and antisense strands starting around the problematic PCRTags. We also observed 42% fewer reads mapped on the sense strand upstream of these PCRTags in synXV_3.1 compared with synXV_3.2 (Figure 3F). Notably, the antisense peak was not observed in synXV_3.2. These results indicate that the defective PCRTag region of IRA2 in synXV_3.1 may have created divergent promoter regions internal to the gene that led to reduced levels of IRA2 mRNA, possibly because of RNA polymerase collision16 and/or transcription interference.17 The potential creation of divergent promoter regions was supported by transcription factor binding site prediction with YEASTRACT+18 (Figure S3G). To validate the reduction of IRA2 expression because of antisense transcription, we employed CRISPR interference (CRISPRi). We introduced dCas9-Mxi19 along with a single guide RNA (sgRNA) targeting the divergent promoter region into synXV_3.1 to suppress the antisense transcription. Transcription analysis clearly demonstrates the reduction of the antisense IRA2 transcript upon application of CRISPRi. Additionally, IRA2 expression increased compared with the negative control employing a mock sgRNA (as shown in Figure S3H). Moreover, synXV_3.1 exhibited a significantly shorter doubling time with the targeting sgRNA compared with the control with the mock sgRNA, as evidenced in Figure S3I. Taken together, these data further substantiate the hypothesis that the PCRTag bug introduced a divergent promoter region within IRA2, which led to the expression of the antisense transcript and subsequent repression of IRA2 expression, ultimately contributing to the growth defect in synXV_3.1. Interestingly, the introduction of the PCRTag bug into the wild-type BY4741 strain did not result in a growth defect (Figure S3J), suggesting that the manifestation of the bug is specific to the synthetic genomic background, possibly arising from epistatic interactions with other synthetic regions.
Next, we analyzed the global gene expression changes among the three strains. Compared with BY4741, there were 479 and 180 differentially expressed genes (DEGs) in synXV_3.1 and synXV_3.2, respectively (Figures 3G, 3H, and S3K). Gene Ontology analysis revealed that the 324 synXV_3.1-specific DEGs were enriched in the TCA cycle, starch and sucrose metabolism, and oxidative phosphorylation (Figures S3K–S3N; Table S4), a trend that has been reported in an IRA2-deleted strain14 (Figure 3I). Inferring from these data and prior knowledge about IRA2 deletion, we hypothesize that the IRA2 bugs perturbed the Ras-PKA pathway, further disrupting other native metabolic pathways and leading to the growth defects in synXV_3.1. Via the debugging chrDBTL, resolving the PCRTag bugs in IRA2 restored normal IRA2 transcription and the Ras-PKA pathway, thus improving growth fitness. Assuming this hypothesis is correct, this would be the first instance of a bug of this type in the Sc2.0 project.
Despite the correction of the bugs in IRA2, synXV_3.2 still exhibited discernibly slower growth than BY4741. Because epistasis is known to cause growth defects when constructing synthetic chromosomes,8,10 we applied CRIMiRE to the right arm of synXV_3.2 for further debugging (Figures 4A–4C). Upon generating a series of CRIMiRE’d strains, including one with targeted sectional replacement (mr-wtAAGG; Figures 4B and S3O), a comparison of the growth profiles showed that restoring megachunks AA–GG to wild type improved the growth rate. Analysis of the RNA-seq data of BY4741, synXV_3.1, and synXV_3.2 (Table S5) revealed that two genes, DCS2 and OSW1, in the region of megachunks AA–GG had significantly altered transcriptional levels (Figure 4D). Reverting these genes to the wild type did not reduce the doubling time (Figure S3P), but the strain with wild-type OSW1 (dbg-2O) displayed fitness similar to that of BY4741 on YPD and YPG (yeast extract-peptone-glycerol) medium plates (Figure 4E). Transcriptional analysis shows the overexpression of OSW1, a gene linked to sporulation, within synXV_3.2 (as depicted in Figure 4F). Upon restoring OSW1 to its wild-type sequence in synXV_3.3, there was a noticeable reduction in gene expression. This decline in expression, accompanied by an enhancement in growth, strongly suggests that the anomalous ectopic expression of OSW1 potentially underlies the growth defect observed in YPG at 37°C. Moreover, the reversion of OSW1 to its wild-type configuration appears to mitigate this effect.
Figure 4.

ChrDBTL with CRIMiRE identified the origin of bugs in the right arm
(A) Schematic illustrating the reconfiguration of the terminal region of the synXV right arm by CRIMiRE. BY4742 and synXV_3.2 were modified with a KlURA3-PGAL1-CEN15 cassette and URA3 at chunk OO4, respectively. CRIMiRE enabled the swapping of the terminal region of the synXV right arm to WT sequences to generate mr-Z and mr-GG. The scissors represent CRISPR-Cas9 cleavage.
(B) Schematic illustrating the targeted reconfiguration of an internal segment in the synXV right arm by CRIMiRE. BY4742-cCEN15 and synXV_3.2 were modified with LEU2 at chunk NN4 and URA3 at chunk CC4, respectively. The strategic marker placement and dual-site cleavage enabled CRIMiRE to swap a targeted internal segment in the right arm of synXV to WT sequences, which enabled generation of mr-wtAAGG.
(C) Growth profiles of BY4743 and strains with semisynthetic right arm.
(D) RNA-seq data show that OSW1 and DCS2 were differentially expressed in synXV_3.1 and synXV_3.2 compared with BY4741.
(E) PCRTags in DCS2 and OSW1 were reverted to wild type in strains dbg-2D and dbg-2O, respectively. Spotting assays revealed that dbg-2O had growth similar to BY4741.
(F) qRT-PCR results depict a reduction in OSW1 expression in synXV_3.3 upon the reversion of synthetic OSW1. The error bars represent standard errors (biological triplicates). The p values were calculated using a t test.
Hereafter, the improved strain dbg-2O is denoted by synXV_3.3. Taken together, we demonstrated that synXV can function as a platform that enables chrDBTL, where CRIMiRE can be employed to (1) design, build, and characterize homozygous strains carrying customized versions of chromosomes; (2) learn the effects of genetic variations on phenotypes through genotype-phenotype mapping; and (3) apply this knowledge to improve the phenotype of synXV by refining its design, thus establishing synXV (and Sc2.0) as a versatile, reprogrammable synthetic model yeast for investigating biological questions.
Learning from synXV: tRNA loss is tolerated by synXV and does not increase translational arrest
In this light, we used synXV to test biological hypotheses through comparative synthetic genomics, focusing on the more extensive modifications in synXV as exemplars; i.e., tRNA removals, PCRTag recoding, and loxPsym site insertion. Specifically, we tested the biological hypotheses that (1) reductions in tRNA copy number might decrease translational efficiency; (2) changes in codon usage might alter translation elongation; and (3) the recoded sequences and elements of synXV might alter gene transcription and translation.
First, by using synXV, we tested the hypothesis that tRNA loss might decrease the translation rate at the codons that correspond to deficient tRNAs. In synXV, 7.3% (20 of 275) of tRNA genes were deleted, which includes deletion of one copy from two-copy tRNAs (tG(CCC)O and tS(GCU)O; Table S6A–S6C). This hypothesis was based on the fact that synXV was redesigned to remove all tRNAs to reduce chromosomal instability.7,20,21 Therefore, the synthetic strain served as a model system to understand the effect of tRNA loss on global translation. The reduced copy numbers of tRNA genes may reduce the level of the ternary complex of aminoacylated tRNA, eEF1A, and guanosine triphosphate (GTP),22 potentially causing a translational pause at the A site, where the tRNA complex enters to recognize a corresponding codon and impede translation. To investigate the global effects of tRNA loss on translation elongation, we performed ribosome profiling (ribo-seq) and RNA-seq on synXV to monitor ribosome occupancy at codon-level resolution and estimate the A-site locations to predict the locations where translational arrest occurs.23 We elucidated the A-site locations from the analysis of 27- to 32-nt ribosome footprint reads24 and confirmed that the peaks corresponded to the codons following the start codons (Figures S4A and S4B). This result is consistent with the fact that initiated methionine tRNAs are loaded at the P site for translation initiation, and the A sites of the ribosomes are at the codons succeeding the start codons, thus validating the reliability of the A-site prediction dataset.
To examine whether tRNA removal from synXV causes translational arrest, we sought to identify ribosomal pauses in synXV_3.1 at positions where the relevant isoacceptor tRNAs would be expected to recognize the codons. Ribosomal pauses because of insufficiency of a particular tRNA deleted from synXV would manifest as peaks in ribo-seq analysis, and the corresponding codon in the genome would be enriched in the ribosome footprint, indicating the sites of translational arrest and the deficient tRNAs. To this end, we computed the relative ribosome occupancies (RROs) on all codons in BY4741 and synXV_3.1 (Figure 5A). Higher RROs indicate that more ribosome footprints were mapped on the codons and signify longer ribosomal pauses at the sites. Comparison of the RROs of the same codons between BY4741 and synXV showed that none of the codons, affected or unaffected, exhibited significant changes. Therefore, our analysis indicates that tRNA loss in synXV did not significantly reduce translational efficiency at the corresponding codons globally (Figures 5B and S4C).
Figure 5.

Translatomic analysis of the synthetic yeast
(A) Schematic of relative ribosome occupancy (RRO). The blue boxes represent codons. The bars on the blue boxes represent the number of ribosome footprints (RFs) whose A site was mapped onto the codons.
(B) Scatterplot showing that loss of tRNA did not affect the translation elongation rate. Each dot represents the median value of the RROs for a codon sequence in all coding DNA sequences (CDSs) encoded on non-chrXV. The percentage reduction in the tRNA copy number in synXV relative to BY4741 was calculated for every corresponding codon sequence and is represented in different colors.
(C) Differentially translated genes were enriched in the synthetic chromosome in synXV_3.1. Translated genes were defined as genes with reads per kilobase per million mapped (RPKM) > 1 in BY4741 and synXV_3.1. FC, fold change.
(D) Schematic of the 30-nt regions on and downstream of each PCRTag analyzed in (E) and (F).
(E and F) Scatterplots showing a lower correlation of the translation elongation rate on PCRTags between BY4741 and synXV_3.1 (E) but not on 30-nt regions downstream of the PCRTags (F). RFs on PCRTags (or 30 nt downstream)/CDSs were defined as the number of RFs mapped onto PCRTag regions (or 30 nt downstream of the PCRTags) divided by those onto the corresponding CDSs. Sample numbers are indicated after the strain names (biological duplicates).
(G) Scatterplot showing no correlation between codon adaptation index (CAI) changes on the PCRTags and translation elongation changes on the PCRTags. CAIs of all synthetic PCRTags were divided by the wild-type ones in BY4741 and compared with the ratio of RROs between PCRTags in BY4741 and synXV_3.1.
Previous Sc2.0 studies showed that tRNA loss can cause reduced fitness that is recoverable by tRNA complementation, indicating that a reduction in levels of certain tRNAs can cause translational changes deleterious to cell fitness.8,25,26 Nevertheless, our study demonstrates that global translation in S. cerevisiae is tolerant to a certain amount of tRNA loss and does not always suffer from reduced translational efficiency. A possible reason why translational efficiency did not increase is that the loss of tRNAs in synXV was less severe than that observed in synX and synXII. While synX lost the sole copy of tR(CCU) and synXII lost two of three copies of tL(UAG) in the genome, the maximum reduction observed in synXV was that of one of two copies of tS(GCU) (Table S6). This is consistent with a previous study showing that tS(GCU) loss did not cause growth defects in rich medium.27 Another possible explanation for the tolerance of synXV to tRNA removal is adaptative evolution of existing tRNAs;28 no mutation was found on anticodons in any existing tRNAs (or in their vicinity) in the synXV_3.1 strain. Therefore, we conclude that the loss of tRNA in synXV has no significant impact on the translation elongation rate in rich medium and that translation elongation in S. cerevisiae is tolerant to reduced tRNA copy number.
Learning from synXV: Codon usage is not a determinant of translation elongation in S. cerevisiae
We then tested, using synXV, the hypothesis that changes in codon usage could alter translation elongation efficiency. Because proteomics examines only the protein expression level and not translation itself,29 we analyzed ribo-seq and RNA-seq data of synXV to investigate the influence of genomic sequence on protein translation level and dynamics. One prominent aspect of synXV is 6,005 codons synonymously recoded as PCRTags and unique restriction sites (at 1,716 sites in total). Importantly, all such segments of the recoded ORFs were positioned after the first 100 bases, a part of the open reading frame (ORF) that numerous studies have implicated as being particularly sensitive to codon usage.30,31,32 However, it is possible that, even downstream of this sensitive region, there might be recoded sequences with undetected effects on translation efficiency. The synthetic yeast strain is essentially a parallel living system for comparative synthetic genomics, in which locus-to-locus comparison with the wild type can be made to learn about the effects of codon changes on protein translation.
The general assumption is that ribosomes translate faster on frequently used codons because these codons have a higher copy number of the corresponding tRNAs and that the rate-limiting step of translation is the incorporation of the ternary complex of tRNA. However, several conflicting studies have been reported regarding the correlation between codon usage and translation elongation rate. For example, a series of studies has shown that there is no correlation between translation elongation rate and codon usage in in vivo models.23,33,34,35 In contrast, Yu et al.36 demonstrated in vitro, with wild-type and mutant luciferase genes having varying codon adaptation indexes (CAIs), that lower CAIs can cause translational pauses. However, these studies had certain drawbacks. In the former studies, the deduction was based on the comparison of the average translation elongation rates on a specific codon, where many factors, such as the mRNA structure and nascent peptide, could influence the translation dynamics and conceal the effects of codon usage. In the latter study by Yu et al.,36 an in vitro system was used, and only a single gene was evaluated. It is therefore unclear whether the correlation is applicable to other genes in vivo. Given the extensive codon modifications in chromosome XV, synXV serves as an ideal system for direct comparison of codon usage and translation dynamics at the corresponding loci in the wild-type and synthetic chromosomes, overcoming the inadequacies in previous studies regarding the relationship between the translation elongation rate and codon usage.
To investigate the effects of PCRTag recoding on translation, we acquired data on ribosome footprints and mRNA levels of BY4741 and synXV_3.1. Translational efficiency (i.e., the ratio of the abundance of ribosome footprints to mRNA) was altered for 45 (fold change [FC] > 2) or 13 (FC > 4) genes in synXV_3.1 (Figure 5C). While most of the genes in the synXV_3.1 strain were not influenced by the redesigned sequence, the differentially translated genes were enriched in the synXV chromosome by 2.0- or 2.6-fold (FC > 2 or 4, respectively) compared with the other 15 native chromosomes (Figure 5C), consistent with the hypothesis that the altered sequences of mRNAs from synXV directly influenced the translation of genes encoded in the synthetic chromosome. However, the remaining 439 genes were not affected despite containing recoded regions of a similar length. Subsequently, we analyzed the ribosome occupancies on the PCRTags to determine whether the recoded 18,015-bp region had an influence on the translation elongation rate at each locus.
We discovered that, between BY4741 and synXV_3.1, the ribosome occupancy correlation was lower between the synthetic PCRTags and the corresponding wild-type sequences compared with that between the unmodified regions downstream of the PCRTags. Moreover, the ribosome occupancy correlation between the regions downstream of wild-type and synthetic PCRTags was similar to that between wild-type replicates, indicating that the altered codons in the PCRTags indeed affected the translation elongation rate at the PCRTags but not downstream regions (Figures 5D–5F). To examine whether these changes in translational dynamics were related to codon usage, we analyzed the association between translational changes and CAI37,38 on each PCRTag. After sequence recoding, the median CAIs of the synthetic PCRTags were biased toward slightly higher values than those of the corresponding wild-type sequences. Nevertheless, combining the data on ribosome occupancy and CAI revealed no correlation between ribosome occupancy on PCRTags and CAI (Figure 5G). Therefore, our results show that although codon usage does cause certain relatively minor translational changes in some reading frames, it is not a dominant factor that determines the translation elongation rate in S. cerevisiae. One possible explanation for the minor influence from codon usage is that the supply and demand of tRNAs in our strains is well balanced under the test conditions; hence, CAI did not greatly affect the translation elongation rate.33,35 Another possibility is that factors other than CAI, such as mRNA secondary structure,32,39 can potentially influence the translation elongation rate in recoded chromosomes. For example, PCRTag recoding of PRE4 in the Sc2.0 synVI chromosome altered the predicted mRNA secondary structure of the gene and profoundly decreased protein accumulation.39 Overall, the results demonstrate that synXV can provide a synthetic homologous in vivo environment that allows holistic investigation of biological processes.
Learning from synXV: loxPsym insertion can create novel upstream ORFs to alter gene translation
To test the hypothesis that synXV design and elements might alter transcription and translation processes and determine whether learning from this testing could assist in the debugging and redesigning of synXV, we performed RNA-seq and ribo-seq analysis. One notable modification in synXV is the insertion of 395 loxPsym sites 3 bp downstream of the stop codons in nonessential genes. Importantly, as noted in other studies of Sc2.0, the addition of loxPsym sites downstream of dubious ORFs (which were not annotated as such when the chromosomes were designed) led to the inadvertent/unintended insertion of loxPsym sites in the promoters or even 5′ UTRs of real genes.11,25 Undoubtedly, these insertions can have unintended consequences for fitness. Therefore, we sought to investigate the influence of loxPsym, which originated from bacteriophage P1,40 on yeast translation, using synXV as a model system.
We based our hypothesis on prior studies suggesting that the extensive insertion of the heterologous loxPsym site deployed in the Sc2.0 project may have profound effects on 3′ UTRs (Figures S5A–S5C) and 5′ UTRs, especially when inadvertently inserted into a 5′ UTR to potentially alter gene translation.39,41 Indeed, when comparing the mapping pattern around the genes that were translated at a lower level in synXV_3.1 than in BY4741, we found that these genes in synXV tended to have loxPsym sites in their 5′ UTR, either from insertion after an upstream dubious ORF or overlapping dubious ORF encoded on the opposite strand (Figure 6A). Ribo-seq analysis showed a strong trend for the translation of genes with loxPsym sites within 100 bp from the start codons being repressed in synXV_3.1 compared with that of the corresponding genes in BY4741 (Figures 6B and 6C; Table S7). Notably, the translation efficiencies of REV1, REX4, CPA1, and SFL1 in synXV_3.1 were 34.1, 18.1, 5.4, and 4.1 times lower, respectively, than those in BY4741 (Figures 6B and 6C; Table S7). All four of those genes contain or are immediately adjacent to dubious ORFs. Ribosome footprint peaks were observed in the 5′ UTRs of the genes (Figures 6D and S5D–S5F), suggesting that insertion of loxPsym activated translation from the 5′ UTR of these genes. Moreover, these translation products appear to be initiated from AUG and non-AUG start codons. These observations are consistent with the fact that the loxPsym sequence (ATAACTTCGTATAATGTACATTATACGAAGTTAT) has an ATG and an AT at the end and that the strong secondary structure of the palindromic loxPsym might activate translation from near-cognate non-AUG start codons,42 hence creating upstream ORFs (uORFs). Consequently, translation of these newly created uORFs in the 5′ UTRs can, in turn, repress the translation of the downstream genes because of release of the ribosomes from the mRNAs after translation of the uORFs.43 Thus, these data show evidence that loxPsym insertion can repress translation of protein-coding regions in a uORF-mediated manner.
Figure 6.

Effects of loxPsym insertion on translation
(A) Schematic of genes with loxPsym sites in their 5′ UTRs and enrichment analysis showing that genes with loxPsym sites in their 5′ UTRs are enriched in differentially translated genes. FC, fold change in translation level.
(B and C) Scatterplot showing that the translation level (B) and translation efficiency (TE) of genes (C) with loxPsym in the 5′ UTRs were repressed in synXV_3.1.
(D) Ribo-seq results showing RFs mapped around CPA1 and loxPsym.
(E) Spotting assay showing improved tolerance to hygromycin B upon deleting the loxPsym from the 5′ UTRs of REX4 and CPA1 but not REV1 and SFL1.
(F) The result of pulse field gel electrophoresis displays the size reduction of synXV_3.4.
(G) The dot-plot provides a visualization confirming the absence of duplications, deletions, and insertions in synXV_3.4.
(H) Spot plating assay of synXV_3.4. The conditions evaluated were (1) YPD, (2) synthetic complete medium (SC), (3) YPG (2% glycerol), (4) YPD at pH 4, (5) YPD at pH 9, (6) YPD with benomyl, (7) YP with sorbitol, (8) YPD with camptothecin, (9) SC with 6-azauracil (6-AU), (10) YPD with hygromycin B (HygB), and (11) cycloheximide (CHX).
To assess the effects of the uORF-generating loxPsym sites, we removed the loxPsym sites from the 5′ UTRs of REV1, REX4, CPA1, and SFL1 individually from synXV_3.2 and evaluated strain fitness. Interestingly, although the slow growth of synXV_3.2 was not resolved, removal of the 5′ UTR loxPsym from REX4 and CPA1 diminished the sensitivity to hygromycin B (Figure 6E). While REX4 is a putative gene with inconclusive function, CPA1 is known to encode a small subunit of carbamoyl phosphate synthetase, and deletion of CPA1 increased the sensitivity to various chemicals, including hygromycin B.44 Hence, the 5′ UTR loxPsym of CPA1 was removed from synXV_3.3 to generate the final synXV strain, synXV_3.4. Pulse field electrophoresis and de novo sequence assembly demonstrated the absence of significant deletions or duplications in synXV_3.4 (Figures 6F and 6G). This strain was characterized and exhibited marked improvements in phenotype and growth compared with the initial synXV_3.1 strain; its features were similar to those of BY4741 (Figures 6H and S5G). Taken together, we revealed that loxPsym insertion can alter gene translation by creating uORFs and successfully removed the associated bugs to redesign synXV for improved phenotype. Future genome designs should carefully consider the placement of loxPsym insertions to avoid disrupting promoters and introducing unintended 5′ UTRs, which might alter gene transcription and translation.
Discussion
In this study, we demonstrated the establishment of chrDBTL with a redesigned synthetic chromosome XV of S. cerevisiae, synXV. By exploiting the vastly recoded sequence of synXV, we achieved systematic combinatorial chromosome reconfiguration with CRIMiRE, thus enabling chrDBTL for genotype-phenotype mapping. CRIMiRE could potentially be employed to multiplex the reconfiguration of chromosomes and recombine chromosomes of S. cerevisiae with other Saccharomyces species to create desired phenotypes. However, depending on the homology between the chromosomes and species, these endeavors may pose challenges, such as off-target cleavage and non-specific recombination, which will need to be evaluated and resolved to extend the application of CRIMiRE beyond two homologous chromosomes.
Upon comprehensive debugging through chrDBTL with CRIMiRE and omics analysis, the synXV strain demonstrates fitness comparable with the wild type across a broad spectrum of tested media and conditions (Figure 6H). While it does exhibit slightly diminished fitness in certain stress tests, the synXV strain’s robustness is evident in its ability to grow alongside the wild type on YPD, synthetic complete (SC), and YPG media at 37C. This robust performance in common media containing fermentable and non-fermentable sugars, even under heat stress, is of notable significance. In view of the extensive recoding made to the 1.05-Mb chromosome, the reduced growth of synXV under certain conditions is within reason. Being the second-largest chromosome in S. cerevisiae, the redesigned synXV sequence has a greater possibility than smaller chromosomes to have minor bugs, which can be due to a range of factors, including altered mRNA secondary structures and synthetic epistatic interactions. Although there is a possibility of these bugs hindering the integration of synXV with other synthetic chromosomes, it is important to emphasize that the Sc2.0 consortium has adeptly consolidated synthetic chromosomes displaying minor fitness defects into a single strain.11 For instance, synIII and synVI manifest slower growth compared with the wild type.39 Similarly, synII exhibits slight growth defects under specific conditions tested.26 Despite these minor imperfections, the consolidation of synIII, synVI, and synII into a single strain has been effectively accomplished.
Furthermore, insights gleaned from reports by the Sc2.0 consortium11,39 underscore the challenge inherent in predicting the fitness outcomes when consolidating multiple synthetic chromosomes. The amalgamation of synthetic chromosomes may either alleviate pre-existing growth defects or introduce new ones. Given the overarching objective of the Sc2.0 consortium to achieve strains with near-wild-type phenotypes upon combining all synthetic chromosomes, it becomes prudent to address any persisting design flaws post consolidation. Notably, this strategy has proven successful within the Sc2.0 consortium, as demonstrated by a team’s accomplishments using CRISPR-D-BUGS,11 further highlighting its efficacy.
Besides enabling CRIMiRE, the extensively recoded synXV sequence allows the synthetic strain to also function as a parallel living system for comparative synthetic genomics that empowered us to answer fundamental questions regarding the effects of codon usage, tRNA copy number, and loxPsym insertion on transcription and translation. Overall, by unlocking the ability to execute chrDBTL, we anticipate synXV (and Sc2.0) to have a far-reaching impact as a synthetic eukaryotic model system for probing biological hypotheses and engineering strains.
Limitations of the study
In our study, we utilized the synXV synthetic yeast model with deletion of 20 tRNAs and 6,005 synonymously replaced codons to investigate the effects on translation efficiency. Interestingly, ribo-seq revealed that the tRNA deletions did not significantly affect translation efficiencies at the modified codons and showed no correlation between CAI and translation efficiency. These conclusions hold true in the context of the synXV strain, but as more tRNAs are removed and codons recoded with the consolidation of several synthetic chromosomes, the effects of these changes on translation efficiency may become apparent. Therefore, the study of a multisynthetic chromosome yeast strain would provide a more holistic understanding of the impact of tRNA copy number and CAI on translation efficiency. Furthermore, we note that we conducted ribo-seq exclusively in rich medium during the exponential phase. Because tRNA abundance in S. cerevisiae is known to fluctuate based on environmental factors,45,46 the specific condition in our study might have obscured the effects of tRNA deletion and codon alterations in synXV. Under environments constraining the tRNA pool, translational stalling may be more likely to occur. Hence, future studies should include ribosome profiling under various stress conditions to gain deeper insights into the relationship between translation rates and the balance of tRNA supply and demand.
Consortia
This work is part of the international Synthetic Yeast Genome (Sc2.0) consortium. The chromosome design and building consortium includes research groups worldwide: the Boeke lab at Johns Hopkins University and New York University (led chromosomes I, III, IV, VI, VIII, and IX); the Chandrasegaran lab at Johns Hopkins (led chromosomes III and IX); the Cai lab at University of Edinburgh and University of Manchester (led chromosomes II and VI and tRNA neochromosome); Yue Shen’s team at BGI-Research SHENZHEN (led chromosomes II, VII, and XIII); Y.J. Yuan’s team at Tianjin University (led chromosomes V and X); the Dai lab at Tsinghua University and Shenzhen Institute of Advanced Technology, CAS (led chromosome XII); the Ellis lab at Imperial College London (led chromosome XI); Sakkie Pretorius’s team at Macquarie University (led chromosomes XIV and XVI); Matthew Wook Chang’s team at National University of Singapore (led chromosome XV); the Bader and Boeke labs at Johns Hopkins University (led design and workflow); and the Build-A-Genome undergraduate teams at Johns Hopkins University and Loyola University Maryland (contributed to chromosomes I, III, IV, VIII, and IX). The Sc2.0 consortium includes numerous other participants, and they are acknowledged on the project website (www.syntheticyeast.org).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| AMPure XP 5mL | Beckman Coulter | A63880 |
| 15% Mini-PROTEAN® TBE-Urea Gel, 10 well, 30 μL | Bio-Rad Laboratories | 4566053 |
| 10% Mini-PROTEAN® TBE-Urea Gel, 10 well, 30 μL | Bio-Rad Laboratories | 566033 |
| CircLigase | Epicentre | CL4111K |
| illustra MicroSpin S-400 HR Columns | GE Healthcare | 27-5140-01 |
| 20/100 Oligo ladder | IDT | 51-05-15-02 |
| RNA Loading Dye (2X) | New England Biolabs | B0363S |
| T4 PNK | New England Biolabs | M0201S |
| T4 RNA ligase 2, truncated | New England Biolabs | M0242S |
| Cycloheximide | Sigma Aldrich | C1988-1G |
| Chloroform: isoamyl alcohol (24:1) | Sigma Aldrich | C0549-1PT |
| Nuclease-Free Water (not DEPC-Treated) | Thermo Fisher Scientific | AM9932 |
| Triton™X-100, 98%, for molecular biology, DNase, RNase and Protease free, ACROS Organics™ | Thermo Fisher Scientific | AC327371000 |
| Tris (1 M), pH 8.0, RNase-free | Thermo Fisher Scientific | AM9855G |
| KCl (2 M), RNase-free | Thermo Fisher Scientific | AM9640G |
| MgCl2 (1 M), RNase-free | Thermo Fisher Scientific | AM9530G |
| Ammonium Acetate (5 M), RNase-free | Thermo Fisher Scientific | AM9070G |
| Turbo DNase (2 U/ul) | Thermo Fisher Scientific | AM2238 |
| RNase I (100 U/ul) | Thermo Fisher Scientific | AM2294 |
| GlycoBlue | Thermo Fisher Scientific | AM9515 |
| SUPERase⋅InTM RNase Inhibitor | Thermo Fisher Scientific | AM2696 |
| Acid Phenol:Chloroform | Thermo Fisher Scientific | AM9720 |
| RiboMinus™ Transcriptome Isolation Kit, yeast | Thermo Fisher Scientific | K155003 |
| SuperScript III | Thermo Fisher Scientific | 18080044 |
| RNA Clean & ConcentratorTM-5 kit | Zymo Research | R1013 |
| Nextra DNA Flex Library Prep Kit | illumina | 20018704 |
| iSeq 100 i1 Reagent | illumina | 20021533 |
| CHEF Yeast Genomic DNA Plug Kit | Bio-Rad Laboratories | 1703593 |
| Deposited data | ||
| WGS, RNA-seq and Ribo-seq data | National Institute of Health, Sequence Read Archive | SRA: PRJNA821366 |
| Experimental models: Organisms/strains | ||
| Yeast strains used in this study are summarized in Figure S2 | This paper | N/A |
| Oligonucleotides | ||
| Primers in this study, see Table S3 | IDT | N/A |
| Upper size marker RNA (34 nt) | IDT | AUGUACACGGAGUCGAGCUCAACCCGCAA CGCGA/3Phos/ |
| Lower size marker RNA (26 nt) | IDT | AUGUACACGGAGUCGACCCAACGCGA/3Phos/ |
| Adaptor RNA | IDT | /5rApp/CTGTAGGCACCATCAAT/3ddC/ |
| RT DNA primer for RNA-seq and Ribo-seq | IDT | /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAG TGTAGATCTCGGTGGTCGC/iSp18/CACTCA/iSp18/TTCAGACGTGTGCTCTTCCGATCTATTGATG GTGCCTACAG |
| Forward PCR primer for RNA-seq and Ribo-seq | IDT | AATGATACGGCGACCACCGAGATCTACAC |
| Reverse Index Primer for RNA-seq WT1 | IDT | CAAGCAGAAGACGGCATACGAGATACATCGGTG ACTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for RNA-seq WT2 | IDT | CAAGCAGAAGACGGCATACGAGATGCCTAAGTG ACTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for RNA-seq SYN1 | IDT | CAAGCAGAAGACGGCATACGAGATCACTGTGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for RNA-seq SYN2 | IDT | CAAGCAGAAGACGGCATACGAGATATTGGCGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for Ribo-seq WT1 | IDT | CAAGCAGAAGACGGCATACGAGATTCAAGTGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for Ribo-seq WT2 | IDT | CAAGCAGAAGACGGCATACGAGATCTGATCGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for Ribo-seq SYN1 | IDT | CAAGCAGAAGACGGCATACGAGATGTAGCCGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Reverse Index Primer for Ribo-seq SYN2 | IDT | CAAGCAGAAGACGGCATACGAGATTACAAGGTGA CTGGAGTTCAGACGTGTGCTCTTCCG |
| Software and algorithms | ||
| BWA (0.7.17-r1188) | Heng Li et al. | https://github.com/lh3/bwa |
| SAMtools (1.15) | Heng Li et al. | https://github.com/samtools/samtools |
| GATK (4.2.6.1) | Ryan Poplin et al. | https://gatk.broadinstitute.org/hc/en-us |
| STAR (2.5.3) | Alexander Dobin et al. | https://github.com/alexdobin/STAR |
| DESeq2 | Michael I Love et al. | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DAVID | Da Wei Huang et al. | https://david.ncifcrf.gov/ |
| CAI | Benjamin D. Lee | https://github.com/Benjamin-Lee/CodonAdaptationIndex |
| BEDTools (2.30.0) | Aaron R. Quinlan et al. | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| RNAfold (2.4.18) | Ronny Lorenz et al. | https://www.tbi.univie.ac.at/RNA/ |
| Minimap2 (2.22-r1101) | Heng Li et al. | https://github.com/lh3/minimap2 |
| D-Genies | Floréal Cabanettes et al. | https://dgenies.toulouse.inra.fr/ |
| FASTX-Toolkit | Gregory Hannon | http://hannonlab.cshl.edu/fastx_toolkit/ |
| Bowtie (1.0.0) | Ben Langmead et al. | https://bowtie-bio.sourceforge.net/manual.shtml |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Matthew Wook Chang (bchcmw@nus.edu.sg).
Materials availability
Synthetic yeast strains generated in this study are available from the lead contact upon request.
Experimental model and study participant Details
Details of yeast strains used in this study are summarized in Table S2. Growth condition of each experiment are described in method details section.
Method details
Yeast transformation
For transforming plasmids and linear integrative cassettes, yeast competent cell preparation and DNA transformation were performed using the LiOAc/PEG method.47 This method was adapted for megachunk transformation. A yeast colony was grown in 5 mL YPD medium (containing 1% yeast extract, 2% peptone and 2% glucose) overnight at 30°C with shaking at 220 rpm. The overnight culture was diluted in 20 mL fresh YPD to A600 = 0.1 and grown under the same conditions to A600 = 0.4. The cells were centrifuged (3000 x g, 5 min), and the pellet was resuspended in 10 mL 0.1 M lithium acetate. The lithium acetate-treated cells were concentrated by centrifugation (3000 x g, 5 min) and resuspended in 200 μL 0.1 M lithium acetate. To each 100 μL of competent cells, 10 μL denatured salmon sperm DNA (2 mg/mL, Invitrogen) and 30 μL megachunk ligation mixture were added and mixed gently. The mixture was incubated at room temperature for 30 min before a transformation mix [containing 600 μL 50% PEG 3350, 90 μL 1 M lithium acetate, 100 μL DMSO and 98 μL deionized water (dH2O)] was added and mixed thoroughly by gentle pipetting. After incubation at room temperature for 30 min and heat shocking at 42°C for 14 min, the chemically transformed cells were centrifuged (3000 x g, 5 min). The pellet was resuspended in 1 mL 5 mM CaCl2 and incubated at room temperature for 10 min. The cells were then plated in 250-μL portions onto the appropriate solid selective medium and incubated at 30°C.
Replacement of wild-type chromosome XV with synthetic DNA by SwAP-In
synXV was designed using BioStudio to segment the chromosome into 42 megachunks, each consisting of 4–5 chunks (Table S1). The chunks were synthesized by GenScript, WuXi Qinglan Biotechnology Inc. and BioBasic and were delivered as plasmids, with each chunk flanked by the restriction sites as designed. The chunks were digested with the respective restriction enzymes according to the manufacturer’s recommendation and gel purified with the Promega Wizard SV Gel and PCR Clean-Up System. To ligate the chunks into a megachunk, the first to last chunks were mixed in the ratio 5.0:2.5:1.0:0.4 ng (or 10.0 : 5.0: 2.5 : 1.0: 0.4 ng, if the megachunk contained 5 chunks), respectively, and the DNA mixture was pelleted by ethanol precipitation. The DNA pellet was resuspended in a 30-μL ligation mix consisting of 1x T4 buffer and 1.2 μL T4 ligase (2000 units/μL, New England BioLabs) and incubated for 18 h at 16°C. The ligated megachunk was transformed directly without gel purification into the respective yeast strain and colonies with the expected auxotrophy based on the URA3 or LEU2 marker present in the rightmost chunk, as detailed in Table S1. The clones were selected for subsequent PCRTag analysis to verify successful integration of the synthetic sequences. As adjacent megachunks have the selection marker alternating between URA3 and LEU2, semisynthetic chromosomes and hyperchunks were assembled by iteratively integrating the megachunks and selecting them sequentially based on uracil and leucine prototrophy prior to PCRTag verification.
PCRTag verification
The genomic DNA of each colony was extracted as described48 and dissolved in 50 μL of dH2O. Premixed pairs of PCRTag oligonucleotides were synthesized by Integrated DNA Technologies (Singapore) in a 96-well format and diluted with dH2O to 10 μM. PCRTag reaction mixtures were prepared using a Labcyte Echo 525 Acoustic Liquid Handler (Labcyte, USA). Each 10-μL PCR mixture consisted of 1x Q5 buffer, Q5 polymerase (0.02 U/μL, New England Biolabs), 0.5 μM PCRTag oligonucleotide pair, 200 μM dNTPs and 0.5 μL genomic DNA. The PCRs were run using the following thermocycling program: 98°C for 30 s; 35 cycles of 98°C for 10 s, 68°C for 20 s and 72°C for 30 s; and 72°C for 2 min. Agarose gel electrophoresis was performed to visualize PCRTag bands.
Assembly of synXV by CiGA and meiotic recombination
HC4 was constructed by integrating the megachunks GG-PP into several semisynthetic strains and recombining them with CiGA10 (Figure S1E). HC234 was assembled by mating and sporulation to sequentially combine HC2, HC3 and HC4 via meiotic recombination8; HC3 and HC4 were mated, sporulated and screened by PCRTag analysis to isolate HC34, which was subsequently combined with HC2 to create HC234. Details are illustrated in Figure S1.
CRISPR/Cas9 genome editing
Genome editing by CRISPR/Cas9 was based on plasmids and methods described by Foo et al.49 pHCas9-H, a Cas9-expressing plasmid, was constructed by replacing the LEU2 marker of pHCas9-L with a hygromycin B resistance gene (hph). pgRNA-K, a plasmid for transcribing single guide RNA (sgRNA), was constructed by replacing the URA3 marker of pgRNA with a G418 resistance gene (KanMX) and inserting the cassette for cloning an sgRNA sequence from pBS-gRNA1 with BamHI/PstI. The desired sgRNA sequences were cloned into pgRNA-K using one-pot digestion and ligation of annealed oligonucleotides (Integrated DNA Technologies, Singapore). pHCas9-H, pgRNA-K with cloned sgRNA sequence and a donor DNA (Integrated DNA Technologies, Singapore), where applicable, were cotransformed to achieve genome editing. For editing using NG PAM sites, pWZ401 (unpublished) expressing spCas9-NG50 was used, and sgRNA was cloned into pgRNA; transformants were selected on SC–Leu–Ura plates. The oligonucleotides used for cloning sgRNAs and creating donor DNAs are listed in Table S3.
Correction of sequences that deviated from the synXV design
During the integration of chunks and assembly of synXV, several unexpected sequence deviations from the intended design occurred, particularly (1) duplicated or triplicated regions, (2) missing loxPsym sites and (3) residual wild-type sequences (Figure S6). To remove the triplicated segment in chunks Z4-AA1 and a complex combination of duplication, triplication and inversions in chunks JJ1-KK1 (Figures S6B and S6D), a two-step correction approach was adopted using CRISPR/Cas9 (Figure S6A). Each replicated region was fragmented by directing cleavage at a PCRTag within the region, followed by homologous recombination using a 90-bp donor oligonucleotide to join the ends and remove the replicated segment. To prevent cleavage of the corrected region, the donor oligonucleotide contained a partial wild-type sequence in the PCRTag, which was reverted to the synthetic PCRTag sequence by a subsequent round of CRISPR/Cas9 using a donor containing the desired sequence (Figure S6A). Regions in chunks DD2 and PP3 that were duplicated along with the insertion of the plasmid backbone that harbored the chunks (likely due to incomplete digestion of the chunks from the plasmids) were corrected by CRISPR/Cas9 (Figures S6C and S6E). Cleavage was directed at the unique sequences at the junctions of the chunks and plasmid backbone with concurrent homologous recombination with the respective donor DNAs. Aberrations involving missing loxPsym sites and residual wild-type sequences were corrected by single or multiplexed CRISPR/Cas949 (Figures S6F–S6L) using the sgRNAs and donor DNAs described in Table S3. These sequence corrections were performed separately in HC1 and HC234, which were then consolidated by CRIMiRE to obtain synXV_3.1, as mentioned above. Deviations from the designed synXV sequence that remained in the chromosome are listed in Table S2C.
Recombination of chromosomes by CRIMiRE
HC1A and HC1B had the URA3 markers removed by CRISPR/Cas9 using the sgRNA shown in Table S3. To integrate the KlURA3-PGAL1-CEN15 cassette, pCEN15-UG13 was linearized by NotI digestion and transformed into HC1A to generate HC1A-U. Chunk A5 was transformed into HC1B to create HC1B-U. pgRNA-IJ-K was created by cloning an sgRNA sequence into pgRNA-K to enable CRISPR-mediated cleavage of a unique wild-type sequence between megachunks I and J that was recoded. pHCas9-H and pgRNA-IJ-K were transformed into HC1A-U and HC1B-U, respectively, plated on appropriate solid YPD medium containing hygromycin B (200 μg/mL) or G418 (200 μg/mL) and incubated at 30°C. A single colony from each of the resulting strains, HC1A-U-Cas9 and HC1b-U-gRNA, was mixed in 10 μL sterile water and spotted on a YPD plate to mate overnight at 30°C. Cells were scraped from the patch, inoculated into YPD-HG and grown overnight at 30°C with shaking at 220 rpm to propagate diploid cells containing both pHCas9-H and pgRNA-IJ-K. The overnight culture was spotted on a YPGR plate (containing 1% yeast extract, 2% peptone, 2% galactose and 2% raffinose) and grown overnight at 30°C to induce mis-segregation. Cells from the patch were streaked on a selective plate containing 5-fluoroorotic acid and grown for 2–3 days at 30°C until colonies appeared to select for ura− strains. The colonies were subjected to PCRTag verification to isolate homozygous diploid strains with consolidated HC1. CRIMiRE of other strains was performed similarly using the respective haploid strains.
Generation of homozygous synXV or semisynthetic chromosome XV diploids from haploid strains
The URA3-PGAL1-CEN15 cassette obtained by NotI digestion of pCEN15-UG13 was transformed into BY4741 and BY4742 to generate the BY4741-cCEN15 and BY4742-cCEN15 strains, respectively, with a conditional centromere. Depending on the mating types and auxotrophy (Met− or Lys−), a single colony of the haploid strain and BY4741-cCEN15 or BY4742-cCEN15 were resuspended in 100 μL YPD to mate at 30°C for 8 h without shaking. The cell pellets were washed and streaked on SC–Lys–Met plates to select for heterozygous diploids. After 2–3 days of incubation at 30°C, colonies on the SC–Lys–Met plate were spotted on YPGR and grown overnight at 30°C to induce mis-segregation. Ura– strains were selected by streaking on 5-FOA selective plates and grown for 2–3 days at 30°C. The colonies were verified by PCRTag to isolate homozygous diploids of synXV or semisynthetic chromosome XV.
Sporulation
Diploid strains were grown overnight at 30°C in 5 mL YPD medium. The overnight culture was diluted to OD600 = 1.0 in 5 mL presporulation GNA medium (containing 5% glucose, 3% Difco Nutrient Broth and 1% yeast extract) and grown until the A600 reached at least 4.0. The cells were thoroughly washed three times by repeated centrifugation (2000 x g, 2 min) and resuspended in sterile water. The washed cells were diluted to A600 = 1 in 20 mL sporulation medium (containing 1% potassium acetate, 0.005% zinc acetate, 0.3% yeast extract and appropriate amino acids for which the diploids are auxotrophic) and grown in baffled flasks at 25°C with shaking at 150 rpm until sporulation occurred. Spores were isolated either by tetrad dissection or random spore isolation.
Tetrad dissection
Cells from 0.5 mL sporulation culture were washed in sterilized dH2O and resuspended in 0.2 mL zymolyase cocktail [0.5 mg/mL Zymolyase-100T, 0.1 M potassium phosphate (pH 7.4), 1.2 M sorbitol and 5% beta-mercaptoethanol]. After incubation at room temperature for 10 min, 200 μL 0.1 M potassium phosphate (pH 7.4)/1.2 M sorbitol was added, and the tubes were placed on ice to stop the zymolyase reaction. Ten microliters of zymolyase-treated spores were spotted on one side of the YPD plate and spread by tilting the YPD plates. After the YPD plates were dried, tetrads were dissected and isolated with SporePlay or MSM (Singer Instruments). Isolated haploid cells were grown on YPD plates at 30°C for 2–3 days.
Random spore isolation method and karyotype analysis by flow cytometry
Cells from 0.5 mL sporulation culture were washed in sterilized dH2O twice and resuspended in 0.5 mL sterilized dH2O with 0.5 mg/mL Zymolylase-100T and 1% beta-mercaptoethanol. After overnight incubation at 30 °C at 200 rpm, 0.2 mL 1.5% Triton X-100 was added, and the samples were mixed vigorously by vortexing for 60 s. After centrifuging at 1,200 x g for 15 min at room temperature and resuspending the cell pellet in 0.1 mL sterilized dH2O, the separated spores were plated onto YPD plates and incubated for 2–3 days at 30°C until colonies appeared.
The karyotypes of the isolated haploids were verified by flow cytometry. The colonies were grown in YPD overnight at 30°C with shaking at 200 rpm. The overnight culture was diluted in 5 mL fresh YPD to A600 = 0.4 and grown under the same conditions until the A600 reached 1.0. After the cells from 0.5 mL culture were washed with sterilized dH2O, they were fixed in 0.5 mL 70% ethanol for 1 h at room temperature. After washing with sterilized dH2O, RNase digestion was performed on the cells in 0.2 mL 50 mM Tris-HCl (pH 8.0) with 0.4 mg/mL RNaseA (Sigma, R6148) at 37°C for 2 h followed by proteinase K treatment in 0.2 mL 50 mM Tris-HCl (pH 7.5) with 2 mg/mL proteinase K at 50°C for 1 h. The cells were then resuspended in 0.2 mL FACS buffer [0.2 M Tris (pH 7.5), 0.2 M NaCl, 78 mM MgCl2]. The chromosomal DNA was stained by adding 0.2 mL FxCycle PI/RNase Staining Solution (Thermo Fisher Scientific). The cells were analyzed using a BD Accuri C6 Flow Analyzer (BD Biosciences), and the ploidy of the strains was determined by comparing the fluorescence histograms of the strains to those of BY4741 and BY4743.
Growth profiling of strains
A single colony was inoculated into 5 mL YPD medium and grown overnight at 30°C with shaking at 220 rpm. The seed culture was diluted to OD600 = 0.1 in 500 μL fresh YPD. One hundred microliters of the diluted cell culture were aliquoted into a 96-well flat-bottom microplate and grown at 30°C for 24 h with double-orbital shaking at maximum speed in a BioTek Synergy H1M microplate reader. The OD600 was measured every 15 min, and the doubling time was calculated based on the growth curve when the cells were in the exponential phase between 240 and 540 min using Microsoft Excel. The function used for calculating the doubling time was LN(2)/SLOPE(ARRAY_TIME, ARRAY_OD600), where ARRAY_TIME and ARRAY_OD600 represent an array of the time when OD600 was measured and an array of background-subtracted OD600, respectively.
Phenotyping of the strains under various growth conditions by a spot plating assay
BY4741 and the strains of interest were inoculated in 5 mL YPD and grown overnight at 30°C with shaking. The overnight cultures were diluted to OD600 = 0.1 in 5 mL fresh YPD and regrown for 4–5 h. The cells in exponential phase were centrifuged (4000 x g, 5 min) and washed twice with sterile dH2O. The cells were resuspended in sterile dH2O to OD600 = 0.5 and serially diluted 10-fold stepwise. Then, 5 μL of the diluted cells was spotted on various solid medium plates and incubated at 30 or 37°C, as required (Figure 6H). The media used were as follows: (i) YPD, (ii) YPG (YP with 3% glycerol), (iii) synthetic complete medium (SC), (iv) YP with sorbitol (0.5, 1.0, 1.5 or 2.0 M), (v) YPD at pH 4 (buffered with 50 mM citrate buffer), (vi) YPD at pH 9 (adjusted with NaOH), (vii) YPD with benomyl (15 μg/mL), (viii) YPD with camptothecin (0.1, 0.5, 1.0 μg/mL), (ix) YPD with hygromycin B (50 μg/mL), and (x) SC with 6-azauracil (100 μg/mL). The tolerance of the strains to cycloheximide (10 μg/mL) was tested by pretreatment of the cells in exponential phase with the chemical for 2 h. The cells were washed, serially diluted and spotted on YPD, as described for the other media.
Whole-genome sequencing
Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega) and lyticase (Sigma–Aldrich) following the manufacturer’s instructions. For in-house MiSeq and iSeq 100 (Illumina) analysis, whole-genome sequencing samples were prepared with the Nextra DNA Flex Library Prep Kit (Illumina) following the manufacturer’s instructions. Library quality control was conducted with a QIAxel system and the QIAxel DNA High Resolution Kit (Qiagen). The molar concentration of the library DNA was determined using the Collibri Library Quantification Kit (Thermo Fisher Scientific) and CFX Connect Real-Time PCR Detection System (Bio–Rad Laboratories). The samples were sequenced with MiSeq or iSeq 100 in 150-bp paired-end mode. For HiSeq analysis, sample preparation was performed by NovogeneAIT (Singapore). The genomic DNA was randomly fragmented by sonication followed by end repair, A-tailing and adaptor ligation. The DNA was amplified by PCR with P5 and indexed P7 oligos. The FASTQ files were mapped to the synXV reference genome, which is the S288C genome (GCF_000146045.2) with chromosome XV replaced by the designed synXV, using BWA with the mem -M option.51 Structural variants and short variants were detected by SAMtools mpileup52 or GATK HaplotypeCaller.53
RNA-seq analysis and gene ontology analysis
Three unique colonies each from BY4741, synXV_3.1 and synXV_3.2 were inoculated into 5 mL of YPD and grown at 30°C overnight with shaking at 200 rpm. The overnight cultures were diluted to OD600 = 0.4 in 5 mL fresh YPD and shaken and grown at 30°C for 4 h with 200 rpm shaking. After washing in sterile dH2O, the pellets were frozen in liquid nitrogen and kept at −80°C until RNA extraction. The frozen cell pellets were suspended in 600 μL RLT buffer (Qiagen) containing 1% (v/v) 2-mercaptoethanol. The cell suspensions were transferred into FastPrep tubes (MP Biomedicals) containing 250 mg of 425–600 μm acid-washed beads (Sigma–Aldrich). The cells were homogenized by FastPrep-24 5G (MP Biomedicals) following the recommended protocol for S. cerevisiae. The cell lysates were transferred into RNase-free 1.5-mL tubes and centrifuged (20,000 x g, 1 min) to remove the cell debris. The RNAs were extracted from 350 μL of the supernatant using the Qiagen RNeasy Kit with DNase I (Qiagen) following the manufacturer’s instructions. RNA quality control, depletion of rRNA with a Ribo-zero kit and library preparation were performed by NovogeneAIT (Singapore). rRNA-depleted RNAs were fragmented randomly and reverse transcribed with random hexamer primers followed by RNase H treatment and second-strand synthesis using deoxyuridine triphosphate. After end repair and A-tailing, the adaptor was ligated to the reverse transcribed DNA followed by uracil-DNA glycosylase degradation of second-strand DNA and PCR amplification. The prepared libraries were sequenced by HiSeq using 150-bp paired-end mode.
The FASTQ files were mapped to the S288C reference genome or synXV reference genome using STAR (version 2.5.3) with the parameters --outFilterType BySJout --outFilterMismatchNmax 2 --outSAMtype BAM SortedByCoordinate --quantMode TranscriptomeSAM GeneCounts --outFilterMultimapNmax 1 --outFilterMatchNmin 16 --alignEndsType EndToEnd.54 Statistical tests to identify differentially expressed genes (DEGs) were performed using DESeq2.55 FPKM values were calculated in each sample based on the total uniquely mapped reads and averaged among the same strains. Gene ontology analysis was performed using DAVID.56,57 Transcriptomic data of an IRA2-deleted strain were obtained from a previous study using a microarray.14
Ribo-seq sample preparation and data analysis
RNA-seq and ribo-seq samples were prepared as described in previous studies,29,58 except that the cells were ground in liquid nitrogen using a mortar and pestle. A single colony was inoculated into 5 mL of YPD and grown at 30°C overnight with shaking at 200 rpm. The overnight cultures were diluted to A600 = 0.03 in 750 mL fresh YPD in a 2-L baffled flask and grown at 30°C with 200 rpm shaking until A600 reached 0.6–0.7. 1.5 mL of 50 mg/mL cycloheximide was added to the culture and the flask was shaken at 30°C for another 2 min. Cells were harvested using 0.2 μm filter and a pump. Filtered cells were collected by a spatula and suspended in 2 mL of lysis buffer (20 mM Tris-HCl pH 8.0, 140 mM potassium chloride, 1.5 mM magnesium chloride, 1% Triton X-100, 0.1 mg/mL cycloheximide). To freeze the cells, the cell slurry was dripped into 50 mL conical tube containing liquid nitrogen with a vented cap. The tubes were placed in a −80°C freezer to allow the liquid nitrogen to evaporate. The cell pellet was ground using a pre-chilled mortar and pestle. The ground cells were collected and placed into a 50 mL conical tube with a vented cap containing liquid nitrogen. The tubes were placed in a −80°C freezer to allow the liquid nitrogen to evaporate. The cell powder was thawed by gently swirling in a 30°C water bath. As soon as the cell powder was fully thawed, the tube was centrifuged for 5 min at 3,000 x g at 4°C. The supernatant was transferred into a pre-chilled 1.5 mL tube and centrifuged for 10 min at 20,000 x g and 4°C. The supernatant was recovered, avoiding the pellet and the lipid layer. The A260 value of the lysate was measured after 1:200 dilution to estimate the RNA content in the lysate. The lysate was stored at −80°C before proceeding to ribosome footprint preparation.
For ribosome footprint preparation, 150 units of RNaseI (Thermo Fisher Scientific, AM2294) were added to 20 A260 units of lysate to digest RNA regions not protected by ribosomes. The lysate was incubated at 25°C with gentle agitation for 60 min. To stop RNA digestion, 2.5 μL of SUPERase⋅In RNase Inhibitor (Thermo Fisher Scientific, AM2696) was added to the lysate and the tube was immediately placed on ice. Concurrently, during the RNaseI reaction, the Sephacryl S400 (GE healthcare, 27-5140-01) resin was equilibrated by passing through 3 mL of 1x polysome buffer (20 mM Tris-HCl pH 8.0, 140 mM potassium chloride, 1.5 mM magnesium chloride) under gravity flow. The column was attached to a collection tube and centrifuged for 4 min at 600 x g. The flow-through was discarded and 200 μL of the RNaseI/SUPERase⋅In treated lysate was applied to the column. The column was centrifuged for 2 min at 600 x g. Twenty μL of 10% SDS and 200 μL Acid Phenol:Chloroform were added to the flow-through. The sample was mixed by vortex for 30 s then centrifuged for 5 min at 20,000 x g at 4°C for phase separation. The aqueous (upper) phase was transferred into a fresh 1.5 mL tube and one volume of Chloroform:Isoamyl Alcohol (24:1) was added. After vortexing for 30 s, the aqueous phase was transferred into a fresh 1.5 mL tube. For RNA precipitation, 1 μL of GlycoBlue, 1/10th volume of 5M RNase-free Ammonium Acetate and 1.5 volumes of 100% Isopropyl Alcohol were added to the RNA solution. The mixture was placed at −20°C for 1 h then centrifuged at 14,000 rpm for 20 min to pellet RNA. The RNA pellet was washed with 80% ethanol and dried for 10 min. RNA was resuspended in 25 μL of nuclease-free water. Ten μL of RNA was mixed with 10 μL of 2 x RNA loading dye (New England Biolabs, B0363S) and denatured at 80°C for 90 s. After pre-run of 15% TBE/Urea gel in 1xTBE for 15 min at 200 V, the denatured RNA and 26/34 nt marker (IDT) were loaded to the gel. After running at 200 V for about 50 min, the gel was stained in 50 mL 1x TBE buffer containing Gel Star for 30 min at room temperature with gentle shaking. A gel section containing 26–34 nt size of ribosome protected fragment was cut into small pieces and transferred into a fresh 1.5 mL tube. After addition of 400 μL of RNA extraction buffer (300 mM sodium acerate (NaOAc) pH5.5, 1 mM EDTA and 0.1U/μL SUPERase⋅In), the sample was incubated at −80°C for 30 min followed by incubation at 25°C overnight with gentle shaking to elute RNA from gels. The supernatant was transferred into a fresh 1.5 mL tube and 1.5 μL of GlycoBlue and 500 μL of isopropanol were added. After incubation at −80°C for 30 min, RNA was pelleted by centrifugation at 20,000 rpm for 30 min at 4°C. The RNA pellet was washed by 80% ethanol and air-dried at room temperature for 10 min. The ribosome-protected RNA was resuspended in 10 μL of 10 mM Tris-HCl. 13 μL of nuclease-free water and 5 μL of 10X T4 PNK buffer (New England Biolabs, M0201S) were added to the ribosome-protected RNA. The mixture was placed on ice until fragmentation of rRNA-depleted RNA was finished.
For rRNA-depleted RNA preparation, 20 μL of 10% SDS was added to 200 μL of the lysate prepared in the first paragraph followed by addition of 220 μL of Acid Phenol:Chloroform pre-heated to 65°C. After vigorously vortexing the samples for 1 min and centrifugation at 14,000 rpm for 5 min, the aqueous phase (upper) was transferred into a fresh 1.5 mL tube. Repeat the Acid Phenol:Chloroform extraction step once more. Two hundred μL of Chloroform:Isoamyl Alcohol (24:1) was added to the RNA solution to extract excess phenol. After vigorously vortexing the samples for 1 min and centrifugation at 14,000 rpm for 5 min, the aqueous phase (upper) was transferred into a fresh 1.5 mL tube. To the solution, 2 μL of glycogen, 1/10th volume of 5 M NH4OAc, and 1.5 volumes of 100% Isopropyl alcohol were added. The mixture was then incubated at −20°C for 1 h. Subsequently, the total RNA was pelleted by centrifugation at 14,000 rpm for 20 min. After washing in ice-cold 80% ethanol, RNA pellet was air-dried at room temperature for 10 min and resuspended in 25 μL of nuclease-free water. rRNA was removed from the total RNA using RiboMinus Transcriptome Isolation Kit, yeast (Thermo Fisher Scientific, K155003). rRNA-depleted RNA was resuspended in 10 μL of 10 mM Tris-HCl pH8.
For fragmentation and end repair of the rRNA-depleted RNA, after addition of 13 μL of nuclease-free water and 5 μL of 10X T4 PNK buffer (New England Biolabs, M0201S), the RNA was fragmented by incubation at 94°C for 25 min followed by incubation at 4°C for 1 min. Both the ribosome-protected RNA and the rRNA-depleted RNA were denatured by incubation at 80°C for 90 s and then kept at 37°C. After addition of 20 μL of nuclease-free water, 1 μL SUPERase⋅In and 1 μL of T4 PNK (New England Biolabs, M0201S) into the denatured RNA, the denatured RNA was incubated at 37°C for 1 h for end repair and at 70°C for 10 min for heat-inactivation. The end repaired RNA was purified using RNA Clean & Concentrator-5 kit (Zymo Research, R1013) following the manufacturer’s instruction. The RNA was eluted in 10 μL of nuclease-free water.
For adaptor ligation, after addition of 1.5 μL of pre-adenylated RNA (0.5 μg/μL) listed in key resources table to the end-repaired RNA, the mixture was denatured at 80°C for 90 s. Two μL of 10x T4 Rnl2 buffer, 6 μL of 50% PEG8000, 1 μL of SUPERase⋅In and 1 μL of T4 Rnl2 ligase were added to the denatured RNA followed by incubation at 25°C for 2.5 h for adaptor ligation. After adaptor ligation, 338 μL of nuclease-free water, 40 μL of 3 M NaOAc pH5.5, 1.5 μLof Glycoblue and 500 μL of isopropanol were added. Isopropanol precipitation was performed as previously described. The pelleted RNA was resuspended in 10 μL of 10 mM Tris-HCl (pH 8).
For reverse transcription (RT), 2 μL of 1.25 μM RT primer listed in key resources table was added to the adaptor-ligated RNA followed by incubation at 80°C for 2 min for denaturation and incubation on ice. After addition of 4 μL of 5x First-strand Buffer, 1 μL of 10 mM dNTP, 1 μL of 0.1 M DTT, 1 μL of SuperScript III (Thermo Fisher Scientific, 18080044) and 1 μL of SUPERase⋅In, the mixture was incubated at 48°C for 30 min. To hydrolyze RNA, 2.2 μL of 1 M NaOH was added to the cDNA sample followed by incubation at 98°C for 20 min. For isopropanol precipitation, 156 μL of nuclease-free water, 20 μL of 3 M NaOAc pH 5.5, 2 μL of GlycoBlue and 300 μL of isopropanol were added. Isopropanol precipitation was performed as previously described. The pelleted cDNA was resuspended in 10 μL of 10 mM Tris-HCl (pH 8) and stored at −80°C until proceeding to size selection.
For size selection, 5 μL of cDNA or RT primer and 5 μL of 2x RNA loading dye were mixed and incubated at 80°C for 90 s. The denatured cDNA and RT primer were loaded into 10% Mini-PROTEAN TBE-Urea Gel (Bio-Rad Laboratories, 566033) and run at 200 V until the lower dye reached the bottom of the gel. After staining in 1xTBE containing 1x GelStar for 30 min at room temperature with gentle shaking, a gel section containing cDNA which was longer than RT primer was excised. The excised gel was placed in 400 μL of extraction buffer (40 μL of 3 M NaOAc pH 5.5, 8 μL of 50 mM EDTA and 352 μL of nuclease-free water) and incubated at −80°C for 30 min followed by incubation at 25°C for overnight with gentle shaking. The elution was mixed with 1.5 μL of Glycoblue and 500 μL of isopropanol and isopropanol precipitation was performed following the same method mentioned in the second paragraph in this section. The pelleted size-elected cDNA was resuspended in 8 μL of 10 mM Tris-HCl (pH 8). 7.5 μL of size-elected cDNA was mixed with 1 μL of 10x CircLigase Buffer, 1 μL of 1 mM ATP, 50 mM MnCl2 and 1 μL of CircLigase (Epicentre, CL4111K) followed by incubation at 60°C for 1 h for circularization and 80°C for 10 min for heat inactivation. After addition of 179 μL of nuclease-free water, 1 μL of Glycoblue, 20 μL of 3M NaOAc pH5.5 and 300 μL of isopropanol, isopropanol precipitation was performed as previously described. The pelleted circularized cDNA was resuspended in 10 μL of nuclease-free water.
For PCR optimization, both non-diluted circularized cDNA and 1:5 diluted circularized cDNA were amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs, M0491L). Each 50-μL PCR mixture consisted of 1x Q5 buffer, 0.5 μL of Q5 polymerase (0.02 U/μL, New England Biolabs), 0.5 μM primer mixtures listed in key resources table, 200 μM dNTPs and 5 μL diluted or non-diluted circularized cDNA. The PCRs were run using the following thermocycling program: 98°C for 30 s; 20 cycles of 98°C for 20 s and 72°C for 20 s. Half of the PCR reaction was collected at the end of 10th cycle. The size distribution of amplicons was analyzed using Qiaxel Advanced Instrument (Qiagen, 9001941) and Qiaxel DNA High Resolution kit (Qiagen, 929002) following the manufacturer’s instruction with QX DNA Size Marker 50 bp – 1.5 kb (Qiagen, 929554) and OL500 setting. Samples with PCR conditions that yielded a single peak were chosen for analysis. The chosen DNA samples were purified using AMPure XP (Beckman Coulter, A63880), following the manufacturer’s instructions. The libraries were eluted in 15 μL of nuclease-free water.
The RNA-seq and ribo-seq libraries were sequenced according to the SE50 protocol using NovaSeq (Illumina). Quality controls and rRNA-derived reads removal were applied to the FASTQ files following the published methods.58 The original FASTQ files underwent quality filtering with fastq_quality_filter using the options -Q33 -q 20 -p 80. Additionally, fastx_clipper was employed with the options -Q33 -a $adaptor -l 25 -c -n -v, and fastx_trimmer was used with -Q33 -f 2, where $adaptor denotes the adaptor sequence utilized for the ribosome profiling experiment. From these quality-filtered FASTQ files, rRNA-derived reads were excluded by mapping the files to a FASTA file containing rDNA sequences, utilizing bowtie59 with the parameters -p 1 --seedlen = 23 --un. The filtered reads were mapped to either the S288C genome or the synXV genome, and reads on each gene were counted using STAR with the parameters --outFilterMismatchNmax 2 --quantMode TranscriptomeSAM GeneCounts --outFilterMultimapNmax 1. RPKM values were computed based on the ReadsPerGene.out.tab files generated by STAR. For calculation of translation efficiency, the RPKM values of the ribo-seq data were divided by the RPKM values of the RNA-seq data.
For A-site prediction, the ribo-seq reads were remapped to the genome sequences using STAR with the –sjdbGTFfile option and a GTF file, in which gene loci were extended by 45 nt from both the start codons and stop codons to compute the distance from regions mapped to corresponding genes. After mapping, ribo-seq reads shorter than 27 or longer than 32 were discarded from the BAM files that contained information on the positions of mapped reads relative to start codons. A-sites were predicted as described in a previous study,24 in which the 16th position of 27- or 28-nt reads and 17th position of 30-, 31- or 32-nt reads were assigned as the A-sites. After A-site prediction, BED files describing the A-positions of each mapped read were generated. To verify the accuracy of the A-site prediction, reads mapped around start codons or stop codons on non-chrXV were extracted from the BED files, and distances to predicted A-positions from start codons and stop codons were computed based on the relative positions to the start codons and transcript length. After accuracy verification, new BED files describing genomic coordinates of A-positions were generated for correlation analysis, readthrough analysis and RRO analysis.
For the correlation analysis between the CAI and translation elongation rate, CAIs of each PCRTag sequence were calculated using the Python program CAI.38 Reads mapped on each PCRTag and coding DNA sequence (CDS) were counted by intersectBED.60 The number of reads mapped onto each PCRTag was normalized by the number of reads mapped onto the corresponding CDS for comparison between BY4741 and synXV_3.1. The correlation coefficients were computed by the cor.test() function of R.
For secondary structure analysis of 3′UTRs, sequences of 50 nt after the stop codons of all genes encoded on chrXV and synXV were used. The 50 nt sequences were analyzed by RNAfold61 to compute the free energy of the sequences.
For the readthrough analysis, BED files of 99-nt downstream regions from stop codons (for BY4741) or loxPsym sites (for synXV_3.1) were generated. Reads mapped onto the downstream regions and the corresponding CDSs were computed by insersectBED60 using the BED files of the downstream regions and CDSs. The number of mapped reads was normalized by the length of the regions. The readthrough efficiencies were computed by dividing the normalized number of reads mapped to the downstream regions by those to the corresponding CDS.
For identification of genes with loxPsym sites in their 5′ UTRs, a BED file of 5′ UTRs of genes on synXV (−1 to −100 position from the start codons) was generated. By using intersectBED with the BED file of 5′ UTRs and loxPsym sites, the genes with loxPsym sites in 5′ UTRs were identified.
For the RRO analysis, BED files describing all codons on CDSs of BY4741 or synXV were generated. IntersectBED was used to count the reads mapped on each codon and the corresponding CDSs. The number of reads mapped onto the codons and CDSs was normalized by their length. RRO values were computed by dividing the normalized read counts on codons by those on CDSs. RRO values were classified by the codon sequences, and the median values were calculated by R. Because RRO values in low-expression genes can easily fluctuate randomly, we computed the values for only higher-expression genes (the average RPKM of ribo-seq on CDS in BY4741 is higher than 100).
Sample preparation and procedure for pulse field gel electrophoresis
Agarose plugs containing yeast chromosomes were prepared using the CHEF Yeast Genomic DNA Plug Kit (Bio–Rad Laboratories, 1703593) following the procedure recommended by the manufacturer. The agarose plugs were applied to 1% agarose (Bio–Rad, 1620137)/0.5x TBE buffer (1st Base) and run in 0.5x TBE buffer using a Chromatic DNA Electrophoresis Device BS-80 (Bio Craft) at 9°C for 5 h with 120-s pulse time and 19 h with 90-s pulse time. The DNA was stained in 0.5x TBE buffer containing 1x SYBR safe (Thermo Fisher Scientific) for 30 min at room temperature with gentle shaking. After washing the gel in 0.5x TBE for 30 min to remove residual SYBR safe, gel images were taken with iBright 1500 (Thermo Fisher Scientific).
De novo assembly and dot-plot analysis
The genomic DNA was prepared using the method described in “whole genome sequencing”. The genomic DNA was sent to Macrogen for their de novo assembly and error correction service, which includes both sequencing and data analysis. In their service, the genomic DNA was sequenced by PacBio RSII to generate HiFi reads and NovaSeq 6000 to generate short reads. The HiFi reads were produced using the circular consensus sequencing (CCS) application, a tool included in SMRT Link (https://www.pacb.com/support/software-downloads/). Genome assembly, utilizing the HiFi reads, was executed using the tools provided by the SMRT Link application. Firstly, Pancake overlapped the reads and Nighthawk phased the overlapped reads. After chimeras and duplicates were removed from these overlapped reads, a string graph was constructed, producing primary contigs and haplotigs. The primary contigs and haplotigs were polished using Racon with phased reads. To eliminate haplotype duplications in the primary contig set, purge_dups was deployed to identify potential haplotype duplications, which were then relocated to the haplotig set. Picon was used for error correction, refining the draft assemblies using Illumina reads.
To create the dot-plot, the reference sequence of synXV and the contig sequence assembled by de novo assembly were aligned using Minimap262 with the `-ax asm5` option, resulting in a PAF alignment file. The PAF file was uploaded to D-Genies63 (https://dgenies.toulouse.inra.fr/) to create the dot-plot using their default setting.
Quantitative reverse transcription polymerase chain reaction (qRT–PCR)
For qRT-PCR targeting OSW1, a single colony was inoculated into 5 mL YPG medium (1% yeast extract, 2% peptone and 3% glycerol) and grown overnight at 30°C with shaking at 220 rpm. The seed culture was diluted to A600 of 0.4 in 5 mL of fresh YPG media and incubated at 37°C with shaking at 220 rpm for 8 h. After extracting total RNA as previously described for RNA-seq, 1 μg of total RNA was reverse transcribed in a 20 μL reaction using the qScript cDNA Synthesis Kit (Quanta Biosciences) following the manufacturer’s procedure. For negative control, Milli-Q water was used in place of 5x cDNA SuperMix to ensure that genomic DNA contamination was limited. After a 10-fold dilution of the cDNA in Milli-Q water, quantitative PCR was conducted using the Luna Universal qPCR Master Mix (New England Biolabs) and CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories) with the primers listed in Table S3E. Relative RNA expression was calculated by the ΔΔCt method. The ALG9 gene served as the standard control for normalization.
CRISPRi experiments
pTDH3-dCas9-Mxi119 was obtained from Addgene (Cat. 46921). The backbone of the plasmid was replaced with pHCas9-H plasmid backbone. pHCas9-H backbone was PCR-amplified with SK3728 (CTGCAAATCGCTCCCCATTTCTCTAGAGGATCCTTGCGTTGCGCTCACTGCC) and SK3729 (TCGAACTGACTAGTAGACTGAATTCCACATTATCTCGAGAGCTCG) using KOD One Master Mix (Toyobo) following their instruction. pTDH3-dCas9-Mxi1 was digested with XbaI (New England Biolabs) and EcoRI-HF (New England Biolabs). Both the PCR-amplified backbone and the digested insert were purified using the Qiagen Gel Extraction kit (Qiagen) and following their instruction. The purified DNAs were joined by Gibson Assembly using NEBuilder HiFi DNA Assembly (New England Biolabs) to construct pTDH3-dCas9-Mxi1-HphMX.
To change the truncated promoter of the KanMX cassette in pgRNA-KB to the full-length promoter, pgRNA-KB and pRS42K were digested by MfeI (New England Biolabs) and NcoI-HF (New England Biolabs). The digested DNAs were purified using the Qiagen Gel Extraction kit (Qiagen) and following their instruction. The purified DNAs were ligated by T4 DNA Ligase (Thermo Fisher Scientific) following their instruction to construct pRS42K-gRNA-KB. sgRNA was cloned into pRS42K-gRNA-KB using the method described in “CRISPR/Cas9 genome editing” section with the oligonucleotide DNAs listed in Table S3D.
The synXV_3.1 strain was transformed with pTDH3-dCas9-Mxi1-HphMX and either pRS42K-gRNA-KB or pRS42K-gRNA-KB encoding sgRNA targeting the divergent promoter following the method in Yeast Transformation section. A yeast colony was grown in 5 mL YPD medium containing G418 (200 μg/mL) and hygromycin B (200 μg/mL) overnight at 30°C with shaking at 220 rpm. Growth assay was conducted following the method described in “Growth profiling of strains section”. For qRT-PCR, the overnight culture was diluted in 5 mL fresh YPD medium containing G418 (200 μg/mL) and hygromycin B (200 μg/mL) to A600 = 0.4 and grown under the same conditions for 8 h. RNA was extracted from the cultures following the method described in “RNA-seq analysis and gene ontology analysis”. The RNA was reverse transcribed using specific primers targeting the anti-sense transcript of IRA2, IRA2 and TFC1 listed in the Table S3E with SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) following their manual. After the cDNA was diluted 10-fold in dH2O, quantitative PCR was conducted using the Luna Universal qPCR Master Mix (New England Biolabs) and CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories) with the primers listed in Table S3E. Relative RNA expression was calculated by the ΔΔCt method. TFC1 was used as a standard control for normalization.
Quantification and statistical analysis
Homoscedastic t test was used for p value computation for Figures 4F, S3H, and S3I using Microsoft Excel. These experiments were conducted with three biological replicates. The error bars on the figures represent standard errors computed by Microsoft Excel.
Wald test was used for adjusted p value computation of RNA-seq data of Figures 3G and 3H and 4D using DESeq2. The RNA-seq experiment was conducted with three biological replicates.
Acknowledgments
pCEN15-UG was a gift from Professor Rodney Rothstein. We gratefully acknowledge financial support from the Synthetic Biology Translational Research Program of the Yong Loo Lin School of Medicine of the National University of Singapore (NUHSRO/2020/077/MSC/02/SB and NUHS RO/2020/046/T1/3), the Summit Research Program of the National University Health System (NUHSRO/2016/053/SRP/05), the Synthetic Biology Initiative of the National University of Singapore (DPRT/943/09/14), the Synthetic Biology R&D Program of the National Research Foundation of Singapore (SBP-P2, SBP-P7, and SBP-P9), the Investigatorship of the National Research Foundation of Singapore (NRF-NRFI05-2019-0004), the Industry Alignment Fund-Industry Collaboration Project (I1701E0011), and the Ministry of Education of Singapore (MOE/2014/T2/2/128). Work in the J.D. lab was supported by the National Natural Science Foundation of China (31725002 and 32150025), the Shenzhen Science and Technology Program (KQTD20180413181837372), and the Shenzhen Outstanding Talents Training Fund and Bureau of International Cooperation, Chinese Academy of Sciences (172644KYSB20180022). Work in the J.D.B. lab was supported by US NSF grants MCB-1026068, MCB-1443299, MCB-1616111, and MCB-1921641. This work used the resources of the Singapore BioFoundry, a bio-manufacturing research facility located at the National University of Singapore.
Author contributions
J.L.F., S.K., and M.W.C. conceptualized the manuscript. L.A.M., K.Y., J.S.B., and J.D.B. designed synXV. J.L.F., S.K., A.V.S., Z.J., and A.W. constructed the hyperchunks HC1, HC2, and HC3. Y.L., Z. Luo., L.H., Z. Liang, and J.D. constructed the hyperchunk HC4. J.L.F., S.K., A.V.S., and Z.J. consolidated the hyperchunks and created synXV. J.L.F., S.K., and A.V.S. developed and performed CRIMiRE. S.K., A.V.S., and Z.J. performed the editing and characterization of synXV and its derivatives. J.L.F., S.K., A.V.S., and M.W.C. analyzed the data and wrote the manuscript. G.S., Y.C., and J.C. verified the sequencing data and assisted with the data submission to NCBI. M.W.C. supervised the study. All authors contributed to review and editing of the manuscript.
Declaration of interests
J.D.B. is a founder and director of CDI Labs, Inc.; a founder of and consultant to Neochromosome, Inc.; and a founder of, scientific advisory board member of, and consultant to ReOpen Diagnostics, LLC and serves or served on the scientific advisory boards of the following: Sangamo, Inc.; Logomix, Inc.; Modern Meadow, Inc.; Rome Therapeutics, Inc.; Sample6, Inc.; Tessera Therapeutics, Inc.; and the Wyss Institute.
Published: November 8, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100435.
Contributor Information
Junbiao Dai, Email: junbiao.dai@siat.ac.cn.
Matthew Wook Chang, Email: bchcmw@nus.edu.sg.
Supplemental information
Details on the location, length of chunks, selection markers used for SwAP-In and restriction enzymes used to digest chunks from the synthesized plasmids.
(A) List of the strains used in this study.
(B) Record of the synXV versions generated.
(C) Deviations from the designed synXV sequence identified in synXV_3.1.
Oligonucleotides used to construct gRNA expression plasmids and donor DNAs for CRIMiRE (A), debugging (B), fixing deviations (C), CRISPRi (D), qRT-PCR (E), and PCRTag primers (F).
Gene Ontology analysis (GO analysis) was performed to the differentially expressed genes specific to synXV_3.1 with DAVID.
RNA expression levels and fold changes in synXV strains compared to BY4741 and the adjusted p values. FPKM stands for fragments per kilobase per million reads mapped. Adjusted p values were computed by DESeq2.
(A) Deleted tRNAs in synXV and their anticodon sequences.
(B) The copy number of tRNAs grouped by anticodon sequences in BY4741 and synXV. The highlighted anticodons have reduced tRNA copy number in synXV.
(C) The copy number of tRNAs grouped by the corresponding codons in BY4741 and synXV.
Translation level and translation efficiency of genes encoded on (A) chrXV or synXV and (B) all chromosomes.
Data and code availability
Deep sequencing data have been deposited at NCBI SRA under BioProject, SRA: PRJNA821366 as an umbrella project of PRJNA351844, and are publicly available as of the date of publication.
No original code was generated in this study except analysis related to A-site prediction. All software used in this study are summarized in key resources table. The code for A-site prediction is available on Zenodo (https://doi.org/10.5281/zenodo.8336863). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Chao R., Mishra S., Si T., Zhao H. Engineering biological systems using automated biofoundries. Metab. Eng. 2017;42:98–108. doi: 10.1016/j.ymben.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rao S., Yao Y., Bauer D.E. Editing GWAS: experimental approaches to dissect and exploit disease-associated genetic variation. Genome Med. 2021;13:41. doi: 10.1186/s13073-021-00857-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inoue F., Kircher M., Martin B., Cooper G.M., Witten D.M., McManus M.T., Ahituv N., Shendure J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017;27:38–52. doi: 10.1101/gr.212092.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibson D.G., Glass J.I., Lartigue C., Noskov V.N., Chuang R.Y., Algire M.A., Benders G.A., Montague M.G., Ma L., Moodie M.M., et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. doi: 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- 5.Hutchison C.A., 3rd, Chuang R.Y., Noskov V.N., Assad-Garcia N., Deerinck T.J., Ellisman M.H., Gill J., Kannan K., Karas B.J., Ma L., et al. Design and synthesis of a minimal bacterial genome. Science. 2016;351:aad6253. doi: 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- 6.Dymond J.S., Richardson S.M., Coombes C.E., Babatz T., Muller H., Annaluru N., Blake W.J., Schwerzmann J.W., Dai J., Lindstrom D.L., et al. Synthetic chromosome arms function in yeast and generate phenotypic diversity by design. Nature. 2011;477:471–476. doi: 10.1038/nature10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson S.M., Mitchell L.A., Stracquadanio G., Yang K., Dymond J.S., DiCarlo J.E., Lee D., Huang C.L.V., Chandrasegaran S., Cai Y., et al. Design of a synthetic yeast genome. Science. 2017;355:1040–1044. doi: 10.1126/science.aaf4557. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W., Zhao G., Luo Z., Lin Y., Wang L., Guo Y., Wang A., Jiang S., Jiang Q., Gong J., et al. Engineering the ribosomal DNA in a megabase synthetic chromosome. Science. 2017;355 doi: 10.1126/science.aaf3981. [DOI] [PubMed] [Google Scholar]
- 9.Sadhu M.J., Bloom J.S., Day L., Kruglyak L. CRISPR-directed mitotic recombination enables genetic mapping without crosses. Science. 2016;352:1113–1116. doi: 10.1126/science.aaf5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Y., Zou X., Zheng Y., Cai Y., Dai J. Improving Chromosome Synthesis with a Semiquantitative Phenotypic Assay and Refined Assembly Strategy. ACS Synth. Biol. 2019;8:2203–2211. doi: 10.1021/acssynbio.8b00505. [DOI] [PubMed] [Google Scholar]
- 11.Zhao Y., Coelho C., Hughes A.L., Lazar-Stefanita L., Yang S., Brooks A.N., Walker R.S.K., Zhang W., Lauer S., Hernandez C., et al. Debugging and consolidating multiple synthetic chromosomes reveals combinatorial genetic interactions. bioRxiv. 2022 doi: 10.1101/2022.04.11.486913. Preprint at. [DOI] [PubMed] [Google Scholar]
- 12.Hill A., Bloom K. Genetic manipulation of centromere function. Mol. Cell Biol. 1987;7:2397–2405. doi: 10.1128/mcb.7.7.2397-2405.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid R.J.D., Sunjevaric I., Voth W.P., Ciccone S., Du W., Olsen A.E., Stillman D.J., Rothstein R. Chromosome-scale genetic mapping using a set of 16 conditionally stable Saccharomyces cerevisiae chromosomes. Genetics. 2008;180:1799–1808. doi: 10.1534/genetics.108.087999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carter G.W., Rupp S., Fink G.R., Galitski T. Disentangling information flow in the Ras-cAMP signaling network. Genome Res. 2006;16:520–526. doi: 10.1101/gr.4473506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tanaka K., Nakafuku M., Tamanoi F., Kaziro Y., Matsumoto K., Toh-e A. IRA2, a second gene of Saccharomyces cerevisiae that encodes a protein with a domain homologous to mammalian ras GTPase-activating protein. Mol. Cell Biol. 1990;10:4303–4313. doi: 10.1128/mcb.10.8.4303-4313.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobson D.J., Wei W., Steinmetz L.M., Svejstrup J.Q. RNA Polymerase II Collision Interrupts Convergent Transcription. Mol. Cell. 2012;48:365–374. doi: 10.1016/j.molcel.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hongay C.F., Grisafi P.L., Galitski T., Fink G.R. Antisense transcription controls cell fate in Saccharomyces cerevisiae. Cell. 2006;127:735–745. doi: 10.1016/j.cell.2006.09.038. [DOI] [PubMed] [Google Scholar]
- 18.Monteiro P.T., Oliveira J., Pais P., Antunes M., Palma M., Cavalheiro M., Galocha M., Godinho C.P., Martins L.C., Bourbon N., et al. YEASTRACT+: a portal for cross-species comparative genomics of transcription regulation in yeasts. Nucleic Acids Res. 2020;48:D642–D649. doi: 10.1093/nar/gkz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji H., Moore D.P., Blomberg M.A., Braiterman L.T., Voytas D.F., Natsoulis G., Boeke J.D. Hotspots for unselected Ty1 transposition events on yeast chromosome III are near tRNA genes and LTR sequences. Cell. 1993;73:1007–1018. doi: 10.1016/0092-8674(93)90278-x. [DOI] [PubMed] [Google Scholar]
- 21.Admire A., Shanks L., Danzl N., Wang M., Weier U., Stevens W., Hunt E., Weinert T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–173. doi: 10.1101/gad.1392506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dever T.E., Dinman J.D., Green R. Translation Elongation and Recoding in Eukaryotes. Cold Spring Harb. Perspect. Biol. 2018;10 doi: 10.1101/cshperspect.a032649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome Profiling of Mouse Embryonic Stem Cells Reveals the Complexity and Dynamics of Mammalian Proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu C.C.C., Zinshteyn B., Wehner K.A., Green R. High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell. 2019;73:959–970.e5. doi: 10.1016/j.molcel.2018.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu Y., Li B.Z., Zhao M., Mitchell L.A., Xie Z.X., Lin Q.H., Wang X., Xiao W.H., Wang Y., Zhou X., et al. Bug mapping and fitness testing of chemically synthesized chromosome X. Science. 2017;355 doi: 10.1126/science.aaf4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen Y., Wang Y., Chen T., Gao F., Gong J., Abramczyk D., Walker R., Zhao H., Chen S., Liu W., et al. Deep functional analysis of synII, a 770-kilobase synthetic yeast chromosome. Science. 2017;355 doi: 10.1126/science.aaf4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bloom-Ackermann Z., Navon S., Gingold H., Towers R., Pilpel Y., Dahan O. A comprehensive tRNA deletion library unravels the genetic architecture of the tRNA pool. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yona A.H., Bloom-Ackermann Z., Frumkin I., Hanson-Smith V., Charpak-Amikam Y., Feng Q., Boeke J.D., Dahan O., Pilpel Y. tRNA genes rapidly change in evolution to meet novel translational demands. Elife. 2013;2 doi: 10.7554/eLife.01339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingolia N.T., Ghaemmaghami S., Newman J.R.S., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pechmann S., Frydman J. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol. 2013;20:237–243. doi: 10.1038/nsmb.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010;141:344–354. doi: 10.1016/j.cell.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 32.Goodman D.B., Church G.M., Kosuri S. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013;342:475–479. doi: 10.1126/science.1241934. [DOI] [PubMed] [Google Scholar]
- 33.Charneski C.A., Hurst L.D. Positively Charged Residues Are the Major Determinants of Ribosomal Velocity. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li G.W., Oh E., Weissman J.S. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 2012;484:538–541. doi: 10.1038/nature10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qian W., Yang J.-R., Pearson N.M., Maclean C., Zhang J. Balanced Codon Usage Optimizes Eukaryotic Translational Efficiency. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu C.-H., Dang Y., Zhou Z., Wu C., Zhao F., Sachs M.S., Liu Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell. 2015;59:744–754. doi: 10.1016/j.molcel.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharp P.M., Li W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee B.D. Python Implementation of Codon Adaptation Index. J. Open Source Softw. 2018;3:905. doi: 10.21105/joss.00905. [DOI] [Google Scholar]
- 39.Mitchell L.A., Wang A., Stracquadanio G., Kuang Z., Wang X., Yang K., Richardson S., Martin J.A., Zhao Y., Walker R., et al. Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science. 2017;355 doi: 10.1126/science.aaf4831. [DOI] [PubMed] [Google Scholar]
- 40.Hoess R.H., Wierzbicki A., Abremski K. The role of the loxP spacer region in P1 site-specific recombination. Nucleic Acids Res. 1986;14:2287–2300. doi: 10.1093/nar/14.5.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rodnina M.V., Korniy N., Klimova M., Karki P., Peng B.Z., Senyushkina T., Belardinelli R., Maracci C., Wohlgemuth I., Samatova E., Peske F. Translational recoding: canonical translation mechanisms reinterpreted. Nucleic Acids Res. 2020;48:1056–1067. doi: 10.1093/nar/gkz783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kearse M.G., Wilusz J.E. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev. 2017;31:1717–1731. doi: 10.1101/gad.305250.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinnebusch A.G., Ivanov I.P., Sonenberg N. Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–1416. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banuelos M.G., Moreno D.E., Olson D.K., Nguyen Q., Ricarte F., Aguilera-Sandoval C.R., Gharakhanian E. Genomic analysis of severe hypersensitivity to hygromycin B reveals linkage to vacuolar defects and new vacuolar gene functions in Saccharomyces cerevisiae. Curr. Genet. 2010;56:121–137. doi: 10.1007/s00294-009-0285-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Torrent M., Chalancon G., de Groot N.S., Wuster A., Madan Babu M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aat6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagai A., Mori K., Shiomi Y., Yoshihisa T. OTTER, a new method quantifying absolute amounts of tRNAs. RNA. 2021;27:628–640. doi: 10.1261/rna.076489.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gietz R.D., Schiestl R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007;2:31–34. doi: 10.1038/nprot.2007.13. [DOI] [PubMed] [Google Scholar]
- 48.Lõoke M., Kristjuhan K., Kristjuhan A. Extraction of genomic DNA from yeasts for PCR-based applications. Biotechniques. 2011;50:325–328. doi: 10.2144/000113672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foo J.L., Rasouliha B.H., Susanto A.V., Leong S.S.J., Chang M.W. Engineering an Alcohol-Forming Fatty Acyl-CoA Reductase for Aldehyde and Hydrocarbon Biosynthesis in Saccharomyces cerevisiae. Front. Bioeng. Biotechnol. 2020;8 doi: 10.3389/fbioe.2020.585935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv. 2013 doi: 10.48550/arXiv.1303.3997. Preprint at. [DOI] [Google Scholar]
- 52.Danecek P., Bonfield J.K., Liddle J., Marshall J., Ohan V., Pollard M.O., Whitwham A., Keane T., McCarthy S.A., Davies R.M., Li H. Twelve years of SAMtools and BCFtools. GigaScience. 2021;10 doi: 10.1093/gigascience/giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Auwera G.A.V.d., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D., et al. Scaling Accurate Genetic Variant Discovery to Tens of Thousands of Samples. bioRxiv. 2018 doi: 10.1101/201178. Preprint at. [DOI] [Google Scholar]
- 54.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 57.Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lorenz R., Bernhart S.H., Höner Zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L. ViennaRNA Package 2.0. Algorithm Mol. Biol. 2011;6:26. doi: 10.1186/1748-7188-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cabanettes F., Klopp C. D-GENIES: dot plot large genomes in an interactive, efficient and simple way. PeerJ. 2018;6 doi: 10.7717/peerj.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details on the location, length of chunks, selection markers used for SwAP-In and restriction enzymes used to digest chunks from the synthesized plasmids.
(A) List of the strains used in this study.
(B) Record of the synXV versions generated.
(C) Deviations from the designed synXV sequence identified in synXV_3.1.
Oligonucleotides used to construct gRNA expression plasmids and donor DNAs for CRIMiRE (A), debugging (B), fixing deviations (C), CRISPRi (D), qRT-PCR (E), and PCRTag primers (F).
Gene Ontology analysis (GO analysis) was performed to the differentially expressed genes specific to synXV_3.1 with DAVID.
RNA expression levels and fold changes in synXV strains compared to BY4741 and the adjusted p values. FPKM stands for fragments per kilobase per million reads mapped. Adjusted p values were computed by DESeq2.
(A) Deleted tRNAs in synXV and their anticodon sequences.
(B) The copy number of tRNAs grouped by anticodon sequences in BY4741 and synXV. The highlighted anticodons have reduced tRNA copy number in synXV.
(C) The copy number of tRNAs grouped by the corresponding codons in BY4741 and synXV.
Translation level and translation efficiency of genes encoded on (A) chrXV or synXV and (B) all chromosomes.
Data Availability Statement
Deep sequencing data have been deposited at NCBI SRA under BioProject, SRA: PRJNA821366 as an umbrella project of PRJNA351844, and are publicly available as of the date of publication.
No original code was generated in this study except analysis related to A-site prediction. All software used in this study are summarized in key resources table. The code for A-site prediction is available on Zenodo (https://doi.org/10.5281/zenodo.8336863). Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.