

Abstract
Cellular communication is essential for cell-cell interactions, maintaining homeostasis and progression of certain disease states. While many studies examine extracellular proteins, the holistic extracellular proteome is often left uncaptured, leaving gaps in our understanding of how all extracellular proteins may impact communication and interaction. We used a cellular-based proteomics approach to more holistically profile both the intracellular and extracellular proteome of prostate cancer. Our workflow was generated in such a manner that multiple experimental conditions can be observed with the opportunity for high throughput integration. Additionally, this workflow is not limited to a proteomic aspect, as metabolomic and lipidomic studies can be integrated for a multi-omics workflow. Our analysis showed coverage of over 8000 proteins while also garnering insights into cellular communication in the context of prostate cancer development and progression. Identified proteins covered a variety of cellular processes and pathways, allowing for investigation of multiple aspects into cellular biology. This workflow demonstrates advantages for integrating intra- and extracellular proteomic analyses as well as potential for multi-omics researchers. This approach possesses great value for future investigations into the systems biology aspects of disease development and progression.
Keywords: Extracellular profiling, intracellular profiling, proteomic analysis, prostate cancer, network analysis
Introduction
Cellular interaction and communication via proteins is integral in the maintenance of normal cell function. Many types of secreted proteins and peptides play critical roles in maintaining cellular homeostasis, with disruptions in these processes contributing to disease development and progression.[1-4] A greater understanding of these secreted proteins and their roles in cellular communication allows for better comprehension of their roles in disease development and progression.
Many studies have examined the role of secreted proteins using different methodologies and sample types. Some focus on screening biofluids derived from patients or animal models, such as urine, serum, or cerebrospinal fluid, depending on disease type and state.[5-7] Multiple studies have been described profiling exosomes and other secretory vesicles released by cells that interact with neighboring cell populations to modulate activity.[8-10] However, some proteins – such as cytokines and growth factors – are not always confined to these secretory vesicles and may be overlooked in analyses that isolate these proteins.[11] Examination of the entire secreted proteome allows for a more holistic look into how cellular communication drives certain pathologies. Other strategies focus instead on the effects of conditioned media on intracellular processes to determine the crosstalk occurring between two cell types.[12] Yet these studies only investigate the effects of secretory proteins, leaving the secretory proteins unidentified and thus their potential as new therapeutic targets is lost. Thus, there is critical need for additional studies that apply a holistic profiling strategy when examining secreted proteins.
Global proteomic profiling approaches are commonly executed using mass spectrometry (MS)-based methods. High sensitivity allows for identification of thousands of proteins within a single complex sample, demonstrating the utility of MS in global proteomic workflows.[13] Additionally, various modes of multiplexing help to reduce spectral complexity and instrument time without significant losses in proteomic coverage while also allowing for relative quantitation.[14] Global investigations of intracellular proteomes are routinely performed with great success, allowing for increased insight into protein interactions and cellular processes in various disease states.[15-17] These overarching examinations take a discovery-based approach, allowing for the generation of new hypotheses in elucidating disease mechanism and progression. Yet these examinations would be benefited further with a complementary secretory proteome screening, allowing for intracellular and extracellular interactions to be encapsulated within a single study for a given biological focus.
One such disease state that would benefit greatly from this complementary analysis is prostate cancer. The current standard for diagnosis initially requires a digital rectal examination coupled with measuring serum prostate specific antigen (PSA) levels; however, this biomarker is not robust and reliable across patient populations. [18] While multiple panels for prostate cancer biomarkers have been developed, many are not FDA-approved and most require tissue-based biopsy samples which can be invasive for the patient. [19] Additionally, many studies focusing on prostate cancer biomarker detection utilize only the intracellular or extracellular protein fractions alone rather than examining them simultaneously to gain deeper insights into how these proteomes interact and impact each other. [20-22] Complementary analyses of prostate cancer cell models are crucial to understanding how signaling networks contribute to disease progression and metastasis, especially in later-stage castration resistant cancers.
Here, we demonstrate the utility of a complementary profiling strategy of the intracellular and secreted proteomes from cultured prostate cancer cells that allows for in-depth analysis of cellular communication networks. This analysis was performed using a model of castration resistant prostate cancer (CRPC) to increase our understanding of how this lethal subtype progresses. [23] Our strategy utilizes a simplistic workflow so that future researchers can easily adapt and optimize based on their specific experimental needs. The integration of therapeutic treatments in cell culture allows for determination of therapeutic mechanisms relative to normal conditions, furthering the applicability of this complementary analysis in assessing efficacy and mechanisms of therapeutic treatments. Further, multiplexing comparative quantitation strategies – such as stable isotope labeling in cell culture (SILAC) or isobaric labeling – can be easily integrated into our workflow, allowing for a reduction in instrument usage and variation. The complementary coverage obtained allows for the understanding of how the two proteomes communicate and modulate one another for a more holistic view of cellular mechanisms. These deeper insights into cellular communication will mold the way we view cellular interactions and shall yield a better understanding of our cellular prostate cancer model interactions.
Materials and Methods
Cell Culture.
BCaPMT10 lines were produced from our own stocks as described previously.[23] Cells were maintained in DMEM/F-12 (Gibco, Framingham, MA) containing 5% fetal bovine serum (HyClone) and 1% penicillin-streptomycin (Gibco) in 75 cm2 flasks at 37°C in an atmosphere of 5% CO2 and 98% humidity. Once cells reached full confluency, each flask was passaged into two 175 cm2 flasks and allowed to grow overnight. After overnight incubation, DMEM/F-12 was removed and cells were washed in 1x phosphate-buffered saline (PBS, Cytiva). Cells were treated with 0.1% DMSO in DMEM/F-12 supplemented with 1X insulin-transferrin-selenium (ITS, Gibco) and 1% penicillin-streptomycin for 48 hours with a fresh media change at 24 hours. As a control, BCaPMT10 cells grown in media containing fetal bovine serum were harvested and processed for analysis as described to ensure no loss in proteome coverage was observed. For these cells, only the intracellular proteome was examined, as its variable nature and lack of known composition would confound extracellular investigations.
Extracellular Proteome Preparation.
At 48 hours, conditioned media was collected and centrifuged at 4,000rpm for 5 minutes to remove dead cells and debris. The supernatant was immediately placed into cold 80% acetone for overnight precipitation at a ratio of 4:1 acetone to sample by volume. After overnight precipitation, proteins were centrifuged at 20,000rpm for 20 minutes at 4°C. The supernatant was discarded, and the pellets were allowed to dry for 15 minutes before resuspension in 1x PBS. Samples were lyophilized and resuspended in lysis buffer containing 8M urea (Sigma) in 50mM Tris-HCl (Sigma) with 0.1X protease and phosphatase inhibitors (Roche). Proteins were reduced and alkylated prior to overnight trypsin digestion (1:100, Promega) at 37°C. Digested peptides were desalted via Sep-Pak Vac 1cc C18 cartridges (Waters) and dried in vacuo. Peptide concentrations were estimated via Pierce Peptide Assay.
Cell Sample Preparation.
Once conditioned media was collected, cells were harvested using a cell scraper in 12mL PBS before centrifugation at 1,100rpm for 5 minutes. This process of aspiration and pellet washing with PBS was repeated once more before freezing on dry ice and subsequent storage at −80°C. Cell pellets were briefly thawed on ice before resuspension in 2-4 volumes of lysis buffer used for conditioned media samples. Cells were lysed via ultrasonication then centrifuged at 14,000rpm for 15 minutes to pellet cell debris. The remainder of cell sample preparation was carried out as mentioned above, starting with reduction and alkylation prior to overnight trypsin digestion. Digested peptides were desalted, dried in vacuo and quantified prior to instrumental analysis as described above.
LC-MS/MS analysis.
Tryptic peptides were resuspended in 0.1% formic acid (Sigma) at a concentration of 0.5mg/mL for instrumental analysis. Samples were analyzed using a Dionex UltiMate 3000 UPLC coupled to a Thermo Scientific QE-HF mass spectrometer. Solvents consisted of 0.1% formic acid in water as buffer A and 0.1% formic acid in 80% acetonitrile as buffer B. LC gradients consisted of a trapping phase from 0 to 18 minutes at 4% B moving to 40% at 120 minutes, 75% B from minutes 120.5 to 130, 97% B from minutes 130.5 to 140, and 4% B from minutes 140.5 to 155. Survey scans of peptide precursors from 300 to 1500 m/z were performed using a resolving power of 60,000 with an AGC target of 1 x 106 and maximum injection time of 150 ms. The top 20 precursors were then selected for higher energy collisional dissociation fragmentation with a normalized collision energy of 30, an isolation width of 2.0 Da, resolving power of 15,000, an AGC target of 5 x 104, a maximum injection time of 150 ms and a lower mass limit of 120.0 m/z. Precursors were subject to dynamic exclusion for 15 seconds with a 10-ppm mass tolerance. Each sample was acquired in technical triplicate.
Database searching and data analysis.
MS analysis of BCaPMT10 intracellular and secreted peptide digests were identified using Proteome Discoverer (version 2.5, Thermo Scientific). Raw files were searched against the UniProt reviewed human proteome using Sequest HT algorithm with trypsin selected as the enzyme and an allowance of two missed cleavages. Precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.2 Da were set for the searching. Carbamidomethylation of cysteine residues (+57.021 Da) was chosen as a static modification, while dynamic modifications selected consisted of oxidation of methionine residues (+15.995 Da). Search results were filtered to 1% FDR at both peptide and protein levels. The INFERYS 1.0 rescoring algorithm (MSAID) was used after initial search to increase peptide identifications and scoring confidence.[24] Functional annotation analyses were performed using DAVID bioinformatics resources and visualized using Cytoscape (version 3.8.2).[25] Further statistical analyses and generation of figures and plots was performed using Perseus and R.[26]
Results
We sought to examine the utility of a workflow that allows for simultaneous examination of the intracellular and extracellular proteomes for deeper insights into systems biology and cellular crosstalk. Our aim was to provide a framework method so that future investigations can build upon the base method and adjust as their investigations require. Figure 1 shows the overall workflow utilized for these preliminary examinations. Cells were cultured for 48 hours in media with a defined serum supplement to allow for maximal secretion of extracellular proteins while also providing a therapeutic window for future pharmacological analyses. Although the cell line used in these analyses is normally cultured in RPMI-1640, we selected DMEM/F-12 to help alleviate any detrimental effects that may occur when using a defined serum supplement over FBS.[27] Harvested media was centrifuged to ensure removal of any dead cells and debris that may conflate the extracellular proteome identifications.
Figure 1.
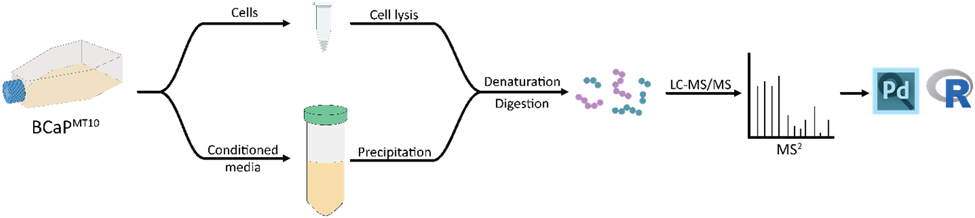
Brief outline depicting the general workflow for sample preparation. Cells were grown in serum-free media containing a defined serum supplement for 48 hours, at which point cells and media were harvested for preparation. Once proteins were collected and quantified for each sample type, a bottom-up sample preparation was implemented using trypsin as the enzyme of choice for digestion. Unlabeled peptides were analyzed in technical triplicate on a Q Exactive HF with database searching of raw spectra in Proteome Discoverer.
Overall proteome coverage remained relatively constant across all three sample groups (Figure 2, Table S1). Both intracellular fractions had over 90% proteome overlap despite the differences in growth media, a good indicator that these experiments are both reproducible and can be compared with cells grown with FBS. Intracellular protein fractions from our FBS and defined serum supplemented cells were compared to look for any detrimental effects on cellular homeostasis (Figure S1). While pathway enrichment showed that cell death pathways were not enriched in the significantly upregulated proteins, it did show an enrichment in cell cycle regulation pathways within the significantly downregulated proteins (Tables S2-S4). Though this may affect cellular proliferation rates, no change in cell viability or morphology was noted between the two sample sets at the conclusion of treatment (Figure S2). The majority of identified proteins were located across all three sample types (Figure 2B). The number of proteins overlapping between the intracellular and extracellular proteome fractions is quite high. Secreted proteins should only make up approximately 13% of the total human proteome, indicating that intracellular proteins from dead and/or lysed cells may be influencing the overall protein number.[28] Additionally, any extracellular proteins must be transcribed intracellularly and then transported to the extracellular environment during either translation or post-translational processing, so there should be fewer proteins unique to this sample group. Although all proteins originate intracellularly, the final protein sequence may not be contained intracellularly for the active conformation of the protein, as there may only be proprotein sequences intracellularly. Thus, for the extracellular fraction analyses, we focused on proteins that were uniquely identified within our extracellular fraction to reduce any conflicting identifications.
Figure 2.
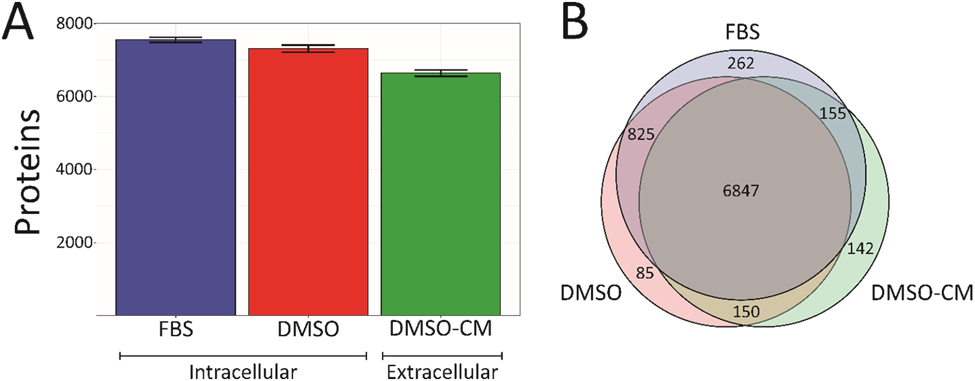
Results detailing the number of proteins identified across sample types. A. Proteins identified across intracellular fractions with or without FBS supplementation as well as the extracellular proteome fraction. Error bars denote standard deviation of biological replicates. While there is a slight decrease in total number of proteins identified in the extracellular fraction, there is little change between the two intracellular fractions, indicating good proteomic coverage. B. Overlap of identified proteins across the three proteomic fractions. As expected, the majority of identified proteins are shared across all three groups, indicating that our proposed method provides adequate coverage and does not lead to a reduction of protein identifications.
Confirmation of experimental reproducibility and sample homogeneity across growth conditions was necessary, so principal component analysis (PCA) was performed for each biological replicate across all treatment types (Figure 3A). Biological replicates from each sample type were found to cluster together, indicating high sample reproducibility and robustness of our sample workflow for high throughput studies. Normalized protein expression values for each replicate were also plotted and showed small variations in median and abundance (Figure 3B). This consistency across both biological replicates and sample types further validates the utility of this methodology in future proteomics studies. To verify that our harvested conditioned media was capturing more of the extracellular proteome, a t-test was performed comparing protein expressions in harvested conditioned media versus the intracellular proteome of its corresponding cell fraction in addition to a heatmap comparing shared protein abundances (Figure S3, S4). From these analyses, the fraction of significantly upregulated proteins was processed using Gene Ontology (GO) enrichment analyses via the DAVID bioinformatics and Metascape software (Table S5, S6). Figure 4A shows the top five enriched cellular components for these significantly upregulated proteins. The top three components all relate to the extracellular proteome, indicating that our extracellular sample fraction significantly enriches proteins normally located within this region. These top three components had both the highest percentage of mapped identifiers as well as the largest log-transformed p-values after Benjamini-Hochberg correction, with the remaining two processes having p-values half as large in magnitude. Metascape software was used to analyze the entirety of the significantly upregulated proteins and protein interaction networks were generated to yield greater insight into how these various processes intersect (Figure 4B). From this mapping, we can see how processes such as cell growth, regulation and morphogenesis are all interconnected and significantly upregulated within our extracellular proteome.
Figure 3.
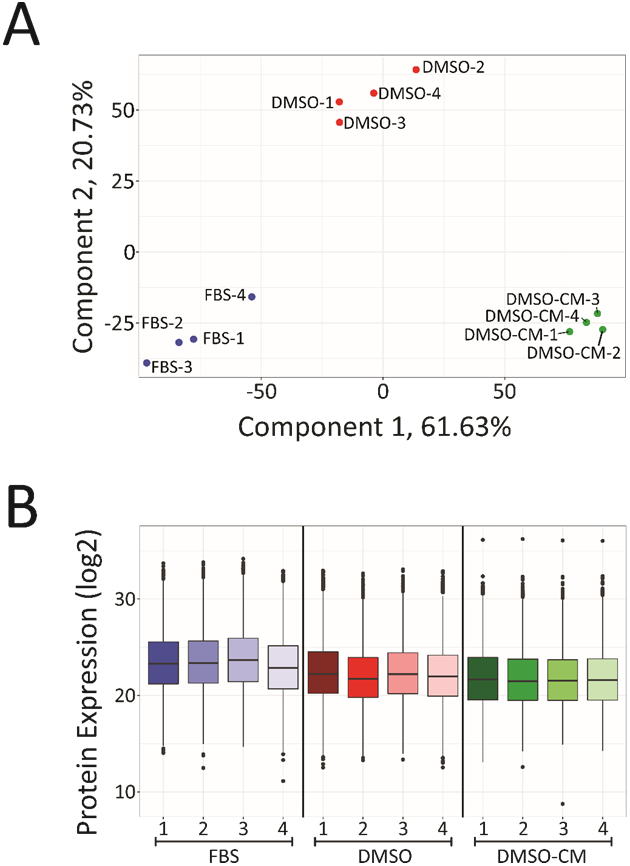
Examination of sample coverage and overlap within and across sample types. A. Principal component analysis (PCA) of all biological replicates for each of the three sample types. All biological replicates within a treatment group were found to cluster together, indicating reproducibility of our method. B. Box plots showing overall proteome coverage in terms of normalized protein expression levels. Looking across all three treatment groups, the majority of expression levels remain relatively constant against one another, demonstrating that serum replacement and analysis of secreted proteins does not substantially alter protein intensities.
Figure 4.
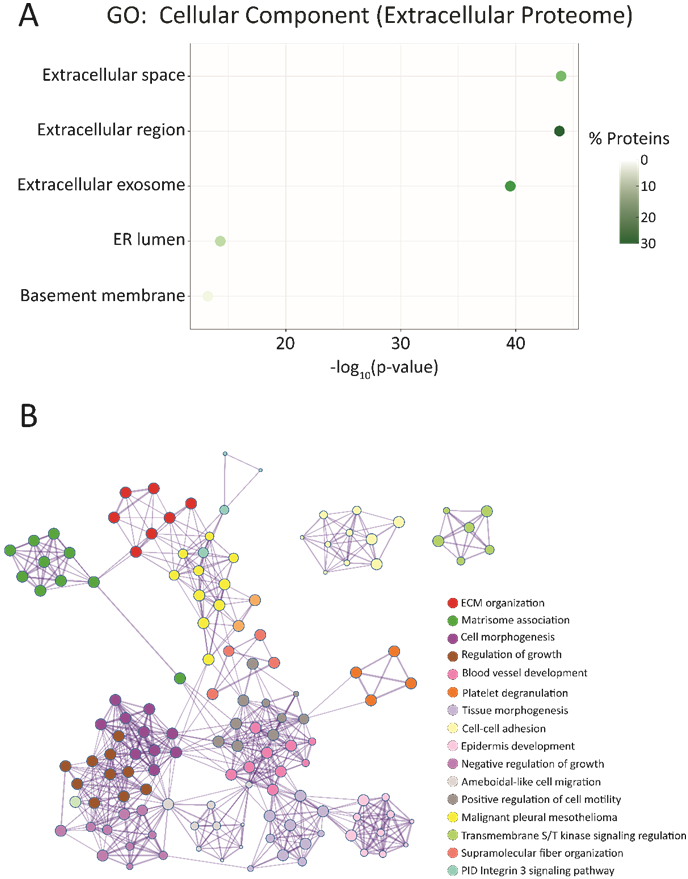
Functional annotation and enrichment analyses for the significantly upregulated proteins in the extracellular proteome fraction relative to their intracellular counterpart. A. Gene Ontology (GO) analysis of the significantly upregulated proteins within the extracellular proteome. The top 5 enriched cellular components are plotted above, with plot colors indicating the percentage of proteins mapped to a particular process out of the 647 total upregulated proteins. B. Visualization of enrichment analyses and how these various components interconnect to impact the entire system. All mapping was performed using Metascape, and proteins used to generate the protein interaction network pertain to those significantly enriched within the extracellular proteome samples. Listed processes and annotations are in order from most to least significant by Benjamini-Hochberg corrected p-value.
We further examined the 142 proteins identified to be unique to the extracellular fraction. Proteins were searched using both Human Protein Atlas and UniProt to confirm their subcellular localization or potential for secretion (Table S7). According to Human Protein Atlas, approximately 20% of the proteins unique to the extracellular fraction have a predicted subcellular localization denoting them as secreted proteins. An additional 31% of proteins that did not meet this criteria are either detected within the blood by mass spectrometry or membrane associated and potentially shed into the extracellular environment, indicating almost 51% of our unique proteins can be found extracellularly and thus confirming increased coverage of these proteins. UniProt had a higher percentage of proteins with listed extracellular localizations at 30%, while those found to be membrane associated comprised just under 29% of this unique fraction. Using both of these sources, we filtered out any proteins that had zero evidence of extracellular localization in either source or whose localization information was unknown. This filtering yielded a finalized list of 93 proteins, which is just over 68% of our total proteins in the unique fraction (Table S7).
Using the filtered list of unique extracellular proteins, pathway enrichment was performed using Metascape software (Figure 5, Table S8). Cell morphogenesis was the top associated pathway, indicating that our extracellular proteins may act on neighboring cells to promote proliferation, invasion, or mobilization. Post-translational phosphorylation was the second most enriched pathway which points towards activation of signaling cascades. Particularly, some of the associated member processes of phosphorylation were directly tied to transforming growth factor beta (TGFβ) signaling, which is also involved in regulating cell migration, proliferation and plasticity.[29] Hair follicle morphogenesis was the third most enriched pathway, which contains the subcategories of epithelial generation and development. The fourth and fifth most enriched pathways were extracellular matrix organization or matrisome-associated, indicating a high level of enrichment for extracellular content. Coupling these pathways together, we see a clearer picture of what processes and pathways these metastatic prostate cancer cells may be utilizing that are unique to cancers with little androgen receptor protein expression, thus opening up therapeutic opportunities for these AR-negative cancers.
Figure 5.
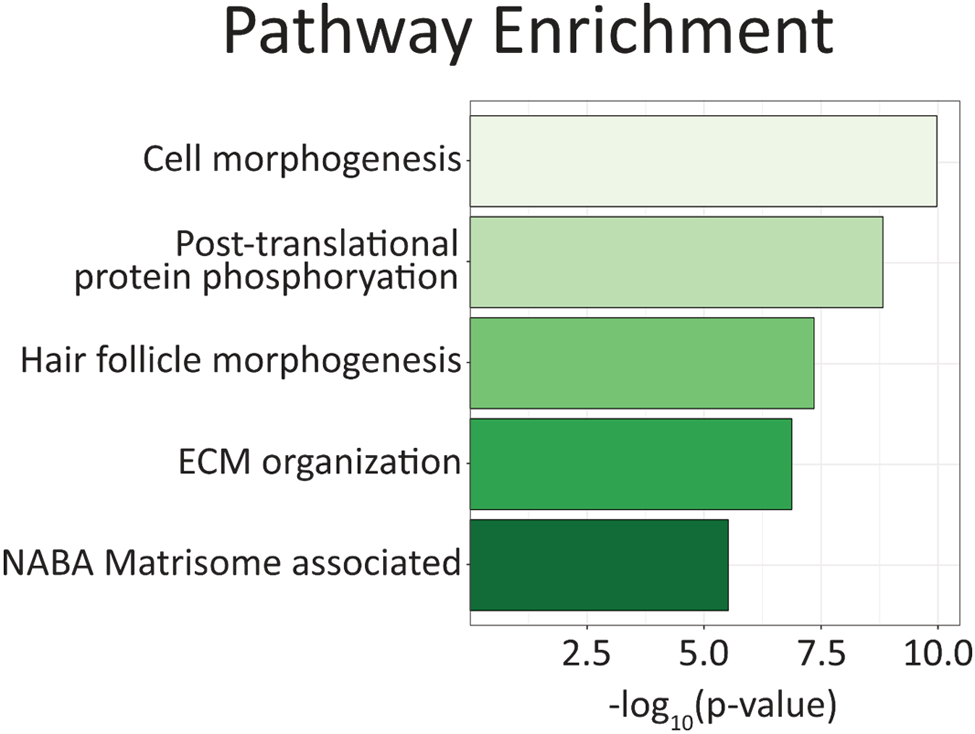
The top five enriched pathways and processes for the 142 proteins identified exclusively within the extracellular proteome fraction. Enrichment analysis was performed using open source Metascape software.
Discussion
A critical aspect of systems biology research is understanding how cellular communication and signaling networks operate across model types.[30] While cellular based models may seem contradictory to a systems-based approach, they allow for deeper investigations into the roles that cellular communication networks play in maintaining homeostasis across tissue and organ types. Utility of cellular-based models allows for integration of isotopic and/or isobaric labeling, increasing throughput as needed. We utilized a serum-defined based approach to study both the intracellular and extracellular proteomes to help determine how both aspects contribute to cellular communication and system function. Cells were cultured in media containing a small percentage of DMSO to demonstrate that future pharmacological analyses can be performed without significant hindrances on proteome coverage and depth. Additionally, coculture experiments can be conducted, allowing a further glimpse into how cell heterogeneity plays a role in system homeostasis.
These analyses were performed using an aggressive metastatic prostate cancer model that has yet to be well defined on the proteomic level.[23] We selected such a model due to its unique characteristics of modeling CRPC that has little to no AR protein expression as well as no neuroendocrine differentiation. [31] The vast majority of prostate cancers are dependent on androgen receptor (AR) expression and will develop resistance to therapies that attempt to target AR expression and availability.[32] Clinically, a CRPC subtype known as double negative prostate cancer (DNPC) also shares these two characteristics, giving our cellular model a unique edge over other more commonly used cell lines. [33,34] Our model allows us to delve deeper into the DNPC phenotype that has slowly increased in emergence as a response to anti-androgen therapeutics used clinically. By studying the intracellular and extracellular proteomes of this cellular model, we can further investigate the signaling networks and communication that occurs between cancer cells that may contribute to DNPC progression. Further investigations comparing enriched pathways between our cancer progression model and other commonly utilized prostate cancer cellular models need to be performed to allow for further elucidation of signaling pathways in AR dependent and independent cancer subtypes.
As expected, both AR protein and a downstream secreted protein, prostate specific antigen (PSA), were not identified across any of our biological and technical replicates. Other androgen-regulated genes, such as kallikreins 2 and 4 (KLK2/4), homeobox protein Nkx-3.1 (NKX3-1) and zinc-α-glycoprotein 1 (AZGP1), were all not identified across any sample runs.[35] Interestingly, transforming growth factor beta 1 (TGFβ1) was strongly expressed across all sample sets. This particular protein has been demonstrated to be overexpressed in advanced prostate cancers and is not androgen dependent.[36] Our overlap with these previous findings indicate that TGFβ1 may be an interesting target for advanced prostate cancers.
Analysis of our extracellular proteome fraction indicates potential pathways that require investigation in models of androgen independent prostate cancer. Post-translational phosphorylation is known to play a role in promoting prostate cancer survival and evasion of current therapeutics, though studies targeting extracellular molecules that promote these signaling cascades have been seldom performed.[37-39] Enrichment of post-translational phosphorylation within the extracellular proteome indicates that neighboring benign cells may experience an increase in phosphorylation events due to the large amount of phosphorylation-promoting proteins secreted from primary cancer cells. Such communication may encourage tumor growth and/or crosstalk from neighboring stroma that benefits tumor progression and metastasis. Extracellular matrix (ECM) remodeling is a common hallmark of carcinogenesis and metastasis, as communication between the extracellular matrix and primary tumor is essential for cancer development.[40] Future investigations into these specific remodeling mechanisms – such as coculture experiments with prostate stroma – will be necessary to determine the specific pathways involved. These experiments can be performed using the same workflow described here and allow for determination of specific proteins that promote remodeling and tumor progression.
Though our workflow does allow for analysis of multiple treatment groups, integration of multiplexing capabilities and the ability to study other groups of interest – such as metabolites and lipids – it is limited to a cellular-based analysis method. While tissue-based analyses could potentially provide more translational information, the cost to separate extracellular and intracellular proteins for tissue samples would far outweigh the benefits. On-tissue analyses could potentially be performed in a similar manner using mass spectrometry imaging (MSI)-based techniques and still allow for multi-omics investigations in a high throughput manner. Coupling such imaging techniques to the methodology used here would provide robust and quantitative profiling across multiple model types that would deepen our knowledge of how disease works across multiple systems.
The workflow outlined here allows for the analysis of both intracellular and extracellular proteomes and can be further expanded to include investigations of metabolites and lipids, allowing researchers to answer fundamental systems biology questions. We demonstrated that this approach is robust and does not result in significant loss of intracellular proteome coverage for our prostate cancer model. The methodology can and is currently being used in high throughput studies with equal success, indicating its utility in future proteomic investigations. Additionally, insights into cellular communication and interaction were unearthed that generate new testable hypotheses, including the role that extracellular communicators may play in phosphorylation status of both primary tumor and surrounding tissues. Our method is a broadly applicable tool for culture-based experimentation and possesses immense value to future multi-omics and systems biology researchers in understanding how cellular communication influences disease progression.
Supplementary Material
Significance of Study.
This research presents a flexible cellular workflow allowing for simultaneous observation of intracellular and extracellular proteins. The outlined method can easily enable and integrate multiplexing studies, allows for testing of pharmacological agents, and can be further expanded to include metabolomic and lipidomic investigations for high throughput multi-omics. Observation of extracellular protein changes in relation to their intracellular counterparts allows researchers to draw conclusions about how these signaling and secretory pathways interact between cells in order to promote disease progression that cannot otherwise be concluded in studies that observe these proteomes separately. Thus, more complete pictures and a better understanding about these diseases and the impact they have on multiple systems can be obtained.
Acknowledgements
The authors would like to acknowledge D.G. Delafield of the Li Research Group for their technical assistance. Support for this research is provided in part by the National Institutes of Health (NIH) grants RF1AG052324 (LL), R01DK071801 (LL), U54DK104310 (WAR, LL), and the UW Comprehensive Carbone Cancer Center P30CA014520. The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. WAR acknowledges NIH grant support R01DK127081 and R01DK13117. LL acknowledges NIH grant support R01 AG078794, R21AG065728, S10OD028473, and S10OD025084 as well as a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Abbreviations:
- AGC
automatic gain control
- AR
androgen receptor
- CM
conditioned media
- DNPC
double negative prostate cancer
- GO
gene ontology
- PCA
principal component analysis
- PSA
prostate specific antigen
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD037806.
References
- [1].Simeone P, Bologna G, Lanuti P, Pierdomenico L, Guagnano MT, Pieragostino D, … Mariani-Costantini R (2020). Extracellular Vesicles as Signaling Mediators and Disease Biomarkers across Biological Barriers. International Journal of Molecular Sciences, 21(7), 2514. doi: 10.3390/ijms21072514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schubert A, & Boutros M (2021). Extracellular vesicles and oncogenic signaling. Molecular oncology, 15(1), 3–26. doi: 10.1002/1878-0261.12855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].DeLaney K, Hu M, Hellenbrand T, Dickinson PS, Nusbaum MP, & Li L (2021). Mass Spectrometry Quantification, Localization, and Discovery of Feeding-Related Neuropeptides in Cancer borealis. ACS Chemical Neuroscience, 12(4), 782–798. doi: 10.1021/acschemneuro.1c00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yu Q, Zhong X, Chen B, Feng Y, Ma M, Diamond CA, … Li L (2020). Isobaric Labeling Strategy Utilizing 4-Plex N,N-Dimethyl Leucine (DiLeu) Tags Reveals Proteomic Changes Induced by Chemotherapy in Cerebrospinal Fluid of Children with B-Cell Acute Lymphoblastic Leukemia. Journal of Proteome Research, 19(7), 2606–2616. doi: 10.1021/acs.jproteome.0c00291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hao L, Thomas S, Greer T, Vezina CM, Bajpai S, Ashok A, … Ricke WA (2019). Quantitative proteomic analysis of a genetically induced prostate inflammation mouse model via custom 4-plex DiLeu isobaric labeling. American Journal of Physiology - Renal Physiology, 316(6), F1236–F1243. doi: 10.1152/ajprenal.00387.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ye H, Wang J, Tian Z, Ma F, Dowell J, Bremer Q, … Li L (2017). Quantitative Mass Spectrometry Reveals Food Intake-Induced Neuropeptide Level Changes in Rat Brain: Functional Assessment of Selected Neuropeptides as Feeding Regulators *. Molecular & Cellular Proteomics, 16(11), 1922–1937. doi: 10.1074/mcp.RA117.000057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wang J, Cunningham R, Zetterberg H, Asthana S, Carlsson C, Okonkwo O, & Li L (2016). Label-free quantitative comparison of cerebrospinal fluid glycoproteins and endogenous peptides in subjects with Alzheimer's disease, mild cognitive impairment, and healthy individuals. PROTEOMICS – Clinical Applications, 10(12), 1225–1241. doi: 10.1002/prca.201600009 [DOI] [PubMed] [Google Scholar]
- [8].Correll VL, Otto JJ, Risi CM, Main BP, Boutros PC, Kislinger T, … Yang L (2022). Optimization of small extracellular vesicle isolation from expressed prostatic secretions in urine for in-depth proteomic analysis. Journal of Extracellular Vesicles, 11(2), e12184. doi: 10.1002/jev2.12184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sequeiros T, Rigau M, Chiva C, Montes M, Garcia-Grau I, Garcia M, … Olivan M (2017). Targeted proteomics in urinary extracellular vesicles identifies biomarkers for diagnosis and prognosis of prostate cancer. Oncotarget, 8(3), 4960–4976. doi: 10.18632/oncotarget.13634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Barnabas GD, Bahar-Shany K, Sapoznik S, Helpman L, Kadan Y, Beiner M, … Levanon K (2019). Microvesicle proteomic profiling of uterine liquid biopsy for ovarian cancer early detection. Molecular and Cellular Proteomics, 18(5), 865–875. doi: 10.1074/mcp.RA119.001362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stanley AC, & Lacy P (2010). Pathways for Cytokine Secretion. Physiology, 25(4), 218–229. doi: 10.1152/physiol.00017.2010 [DOI] [PubMed] [Google Scholar]
- [12].Chen S.-j., Lian G.-d., Li J.-j., Zhang Q.-b., Zeng L.-j., Yang K.-g., … Huang K.-h. (2018). Tumor-driven like macrophages induced by conditioned media from pancreatic ductal adenocarcinoma promote tumor metastasis via secreting IL-8. Cancer Medicine, 7(11), 5679–5690. doi: 10.1002/cam4.1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Macklin A, Khan S, & Kislinger T (2020). Recent advances in mass spectrometry based clinical proteomics: applications to cancer research. Clinical Proteomics, 17(1), 17. doi: 10.1186/s12014-020-09283-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Frost DC, Greer T, & Li L (2015). High-Resolution Enabled 12-Plex DiLeu Isobaric Tags for Quantitative Proteomics. Analytical Chemistry, 87(3), 1646–1654. doi: 10.1021/ac503276z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhong X, Lietz CB, Shi X, Buchberger AR, Frost DC, & Li L (2020). Highly multiplexed quantitative proteomic and phosphoproteomic analyses in vascular smooth muscle cell dedifferentiation. Analytica Chimica Acta, 1127, 163–173. doi: 10.1016/j.aca.2020.06.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Li Z, Tremmel DM, Ma F, Yu Q, Ma M, Delafield DG, … Li, L. (2021). Proteome-wide and matrisome-specific alterations during human pancreas development and maturation. Nature Communications, 12(1), 1020. doi: 10.1038/s41467-021-21261-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lapek JD, McGrath JL, Ricke WA, & Friedman AE (2012). LC/LC–MS/MS of an innovative prostate human epithelial cancer (PHEC) in vitro model system. Journal of Chromatography B, 893-894, 34–42. doi: 10.1016/j.jchromb.2012.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, … Coltman CA (2004). Prevalence of Prostate Cancer among Men with a Prostate-Specific Antigen Level ≤4.0 ng per Milliliter. New England Journal of Medicine, 350(22), 2239–2246. doi: 10.1056/NEJMoa031918 [DOI] [PubMed] [Google Scholar]
- [19].Sartori DA, & Chan DW (2014). Biomarkers in prostate cancer: what's new? Curr Opin Oncol, 26(3), 259–264. doi: 10.1097/cco.0000000000000065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chang L, Ni J, Beretov J, Wasinger VC, Hao J, Bucci J, … Li Y (2017). Identification of protein biomarkers and signaling pathways associated with prostate cancer radioresistance using label-free LC-MS/MS proteomic approach. Scientific Reports, 7(1), 41834–41834. doi: 10.1038/srep41834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Höti N, Shah P, Hu Y, Yang S, & Zhang H (2017). Proteomics analyses of prostate cancer cells reveal cellular pathways associated with androgen resistance. PROTEOMICS, 17(6), 1600228–1600228. doi: 10.1002/pmic.201600228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee JK, Bangayan NJ, Chai T, Smith BA, Pariva TE, Yun S, … Witte ON (2018). Systemic surfaceome profiling identifies target antigens for immune-based therapy in subtypes of advanced prostate cancer. Proceedings of the National Academy of Sciences of the United States of America, 115(19), E4473–E4482. doi: 10.1073/pnas.1802354115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu TT, Ewald JA, Ricke EA, Bell R, Collins C, & Ricke WA (2019). Modeling human prostate cancer progression in vitro. Carcinogenesis, 40(7), 893–902. doi: 10.1093/carcin/bgy185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zolg DP, Gessulat S, Paschke C, Graber M, Rathke-Kuhnert M, Seefried F, … Frejno M (2021). INFERYS rescoring: Boosting peptide identifications and scoring confidence of database search results. Rapid Commun Mass Spectrom, e9128. doi: 10.1002/rcm.9128 [DOI] [PubMed] [Google Scholar]
- [25].Huang DW, Sherman BT, & Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- [26].Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, … Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods, 13(9), 731–740. doi: 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- [27].van der Valk J, Brunner D, De Smet K, Fex Svenningsen Å, Honegger P, Knudsen LE, … Gstraunthaler G (2010). Optimization of chemically defined cell culture media – Replacing fetal bovine serum in mammalian in vitro methods. Toxicology in Vitro, 24(4), 1053–1063. doi: 10.1016/j.tiv.2010.03.016 [DOI] [PubMed] [Google Scholar]
- [28].Uhlén M, Karlsson MJ, Hober A, Svensson A-S, Scheffel J, Kotol D, … Sivertsson Å (2019). The human secretome. Science Signaling, 12(609), eaaz0274. doi: doi: 10.1126/scisignal.aaz0274 [DOI] [PubMed] [Google Scholar]
- [29].Huang F, & Chen Y-G (2012). Regulation of TGF-β receptor activity. Cell & Bioscience, 2(1), 9. doi: 10.1186/2045-3701-2-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aderem A (2005). Systems Biology: Its Practice and Challenges. Cell, 121(4), 511–513. doi: 10.1016/j.cell.2005.04.020 [DOI] [PubMed] [Google Scholar]
- [31].Vellky JE, McSweeney ST, Ricke EA, & Ricke WA (2020). RNA-binding protein DDX3 mediates posttranscriptional regulation of androgen receptor: A mechanism of castration resistance. Proceedings of the National Academy of Sciences, 117(45), 28092–28101. doi: 10.1073/pnas.2008479117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lonergan PE, & Tindall DJ (2011). Androgen receptor signaling in prostate cancer development and progression. Journal of carcinogenesis, 10, 20–20. doi: 10.4103/1477-3163.83937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, … Nelson PS (2017). Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell, 32(4), 474–489.e476. doi: 10.1016/j.ccell.2017.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Formaggio N, Rubin MA, & Theurillat J-P (2021). Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene, 40(7), 1205–1216. doi: 10.1038/s41388-020-01598-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, … Lin B (2002). The program of androgen-responsive genes in neoplastic prostate epithelium. Proceedings of the National Academy of Sciences, 99(18), 11890–11895. doi: doi: 10.1073/pnas.182376299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Karantanos T, Corn PG, & Thompson TC (2013). Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene, 32(49), 5501–5511. doi: 10.1038/onc.2013.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stope MB, Weiss M, Preuss M, Streitbörger A, Ritter CA, Zimmermann U, … Burchardt M (2014). Immediate and transient phosphorylation of the heat shock protein 27 initiates chemoresistance in prostate cancer cells. Oncol Rep, 32(6), 2380–2386. doi: 10.3892/or.2014.3492 [DOI] [PubMed] [Google Scholar]
- [38].Eerola SK, Santio NM, Rinne S, Kouvonen P, Corthals GL, Scaravilli M, … Koskinen PJ (2019). Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Communication and Signaling, 17(1), 148. doi: 10.1186/s12964-019-0463-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Smith AJ, Konduru S, Prokopovich S, Karpova Y, & Kulik G (2005). Role of BAD phosphorylation in prostate cancer xenografts. Cancer Research, 65(9_Supplement), 826–827. [Google Scholar]
- [40].Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, & Werb Z (2020). Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nature Communications, 11(1), 5120. doi: 10.1038/s41467-020-18794-x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with dataset identifier PXD037806.