

Abstract
Over the last decades, PCR and molecular cloning have profoundly impacted various biological areas, from basic to pharmaceutical sciences. Presented in this study is a simple and step-by-step protocol that uses PCR to recover a poor-quality ligase product. In fact, a classic step that can be problematic in typical recombinant DNA manipulations can be the recovery of a product from a T4 DNA ligase reaction between two or more suitably prepared DNA fragments (sticky ends, blunt ends, TA cloning, etc.). This reaction can result in poor yields of the ligation product, due to various causes, mainly the preparation of the DNA fragments, and the poor yield can severely invalidate all subsequent steps. To overcome this problem, we designed a pair of PCR primers to amplify the entire ligase product into satisfactory amount. Of course, high-fidelity DNA polymerase must be used to obtain a faithful copy of the DNA of interest. The fragment thus amplified can then be inserted into a suitable vector and propagated by bacterial transformation.
We applied this procedure to modify a synthetic gene by adding a His-Tag to its 5’ end, and to insert this new construct into an expression cassette. This last step was achieved by employing a PCR cloning system. In our practical example, comprehensive PCR-based protocol with important tips were introduced. This methodological paper can serve as a roadmap for biologists who want to quickly/fully exploit the potential of the PCR-cloning to get desired constructs.
Keywords: T4 DNA ligase, Polymerase Chain Reaction (PCR), cloning, recombinant DNA, educational guide
BACKGROUND
In molecular biology, T4 DNA ligation is an important tool mostly used during cloning to ligate either cohesive or blunt ends of a DNA insert into a vector, where it can be replicated and expressed [1]. This process allows for the joining of two DNA fragments through the formation of a phosphodiester bond, attained by using T4 DNA ligase. This enzyme requires Mg2+ and ATP to work, and sometimes needs 5’-phosphorylation of one or both of the fragments. A great many factors can cause a decrease in efficiency and accuracy during ligation, including those related to the concentration or the stability of the enzyme, presence of inhibitors such as EDTA or salts, insert-to-vector molar ratio, temperature or buffer composition [2]. All these issues can lead to a poor-quality ligase product. Over time, researchers have tried to implement various techniques to overcome the ligase-related problems, such as accelerating the enzymatic processes (for example modifying ligation buffer) [3] or increasing the probability of incorporating insert in blunt-end ligation [4]. In this trail, our difficulty lied in that the ligase product during our cloning process was of poor quality, which would compound all downstream experiments. Our project consisted of the cloning of the APEH cDNA from the Antarctic fish Chionodraco hamatus into the pET-28a(+)vector, to express the recombinant protein. APEH (acyl peptide hydrolase, also known as acylaminoacyl-peptide hydrolase, acylamino acid releasing protein [5] and oxidized protein hydrolase [6]) is a member of the prolyl oligopeptidase (POP) family of serine peptidases [7]. It has been studied in a multitude of organisms: ranging from Eukarya [8] to Archaea [9] and to bacteria [10]. APEH is a bifunctional enzyme, which is mostly studied as an exopeptidase but endowed with an interesting endopeptidase activity [11]. To better investigate the latter activity, we selected an organism where it is more accentuated [12], and we decided to express the recombinant protein in order to obtain a higher amount of it. We purchased the C. hamatus APEH synthetic gene inserted into an expression construct. Unfortunately, due to the difficulty in expressing a psychrophilic protein [13], the initial experiments of expression and purification of the enzyme failed. So, we elected to modify the expression construct by inserting a His-Tag to facilitate the purification of the protein via an affinity column. So, we needed to transfer our synthetic gene in an expression vector with a poly His-Tag, such as pET28a(+). After appropriate enzyme restrictions of the synthetic gene from the original construct and of the pET28a(+) vector, we then combined the two DNA fragments by means of a T4 DNA ligation. After trials with various concentration ratios between the two DNAs, the ligase product was consistently of poor quality. In the end, we devised a procedure that could overcome this impasse and help other reasearchers who need to recover a poor ligase product.
MATERIALS
Chemicals/Reagents/Others
✓ Cloning cells: DH5 α (Solopac) (Agilent)
✓ Strataclone Midi Prep DNA Kit (Stratagene)
✓ pET Vector: pET28a(+) (Addgene)
✓ Restriction Enzymes: Nde I; HindIII (New England Biolabs)
✓ Agarose powder (Sigma)
✓ Antibiotics: Ampicillin; Kanamycine (Sigma)
✓ Marker: GeneRuler 1 kb PlusDNA Ladder (Thermo Scientific)
✓ LB powder (Sigma)
✓ T4 DNA ligase (New England Biolabs)
✓ Gel extraction kit (Promega)
✓ PFU Taq Polimerase (Agilent)
✓ Primers: T7 forward and T7 reverse (Eurofins)
✓ Plastics: 0.2 ml, 1.5 ml, 15 ml and 50 ml tube (Sarstedt)
Equipment
✓ Centrifuge: 5415 R (Eppendorf)
✓ Incubator: KS 4000 (IKA)
✓ Pipettors: 10, 20, 200, 1000 μl (Finpipette)
✓ Spectrophotometer: Nanodrop (Thermo Scientific)
✓ Elettrophoretic system: Mupid One (Mupid-One)
✓ Imaging system: ChemiDoc XRS+ (Biorad)
✓ Water bath: 1003, 14 L (GFL)
PROCEDURES
Preparation of a ligase reaction: : Preparation of vector pET28a(+) and APEH synthetic gene DNAs
1. pET28a (+) vector and APEH synthetic gene were purchased from Eurofins.
2. Plate cells transformed with pET28a(+) vector on LB + kanamycin (30 mg/ml) and plate cells transformed with a plasmid containing the synthetic APEH gene on LB + ampicillin (100 mg/ml). Grow at 37 °C O/N in the incubator.
3. The day after, pick separately one colony of cells containing pET28a(+) in 5 ml of LB + kanamycin (30 mg/ml) and one colony of cells containing a plasmid with APEH gene in 5 ml of LB + ampicillin (100 mg/ml). Grow them at 37 °C O/N.
4. Then, extract the DNAs of pET28a(+) and of the vector containing APEH synthetic gene by using a Strataclone DNA midiprep kit. Measure DNA concentrations with a Nanodrop instrument. Control the quality of DNA by electrophoresis on agarose gel at 1%.
Digestion with restriction enzymes
5. Digest 300 ng of both DNAs for 2 hours at 37 °C with 1 μl restriction enzymes Nde I and Hind III, which have compatible sticky ends for a ligation reaction. (Fig. 1A)
6. Check digestions on 1% agarose gel.
7. Cut from the gel the Nde I/Hind III DNA fragments from pET28a (+) and APEH plasmids.
8. Use an extraction kit to recover the DNA. (Fig. 1B)
Figure 1.

A. Plasmid maps with sites of restriction enzymes B. Agarose gel. Lane 1: Marker (Thermo Scientific); Lane 2: digestion of APEH gene; Lane 3: digestion of pET28 a (+).
T4 DNA ligase reaction
9. Ligate the two NdeI/HindIII DNA fragments with T4 DNA ligase in the following reaction mixture: 50 ng of each DNA fragments, enzyme (400.000 U/ml), buffer 50 mM Tris-HCl, 10 mM MgCl2, 10 mM Dithiothreitol 1 mM ATP, pH 7.5, total volume 20 μL. The reaction is carried out at room temperature for 2 hours.
10. Heat-inactivate at 65°C for 10 minutes. Chill on ice.
11. Transform into competent cells.
Cloning of the ligase product
Unfortunately, having obtained a very poor ligase yield, the transformation plates were negative in the screening of clones containing the ligase product.
If this happens, overcome the problem by using a highly efficient and one-step cloning strategy for the direct insertion of a PCR-amplified product into a plasmid vector.
PCR Amplification
12. Set up a PCR amplification of the APEH synthetic gene by using Pfu DNA polymerase and a pair of primers at the two ends of the region of interest, containing the two ligated species. Here we used T7 forward and T7 reverse primers (see Fig. 2). The sequences of T7 forward and T7 reverse primers were: 5’-TAATACGACTCACTATAGGG-3’ (Tm 53.2 °C) and 5’-GCTAGTTATTGCTCAGC-3’ (Tm 50.4 °C), respectively. Primers were purchased from Eurofins. Moreover, it is necessary to use a high-fidelity polymerase to preserve the sequence of the region to clone.
Figure 2.

Primers used for PCR amplification of a fragment containing a His-Tag sequence at the 5’ terminal of the APEH synthetic gene.
Prepare a mix, in a final volume of 20 μl, containing Pfu polymerase buffer 1X, MasterMix PFU Taq polymerase, DNTP mix, T7 forward and T7 reverse primers each one at the concentration of 0.5 μM, 1 μl of T4 DNA ligase reaction.
Perform PCR using optimized conditions:
Denaturation phase: 10’’ at 95°C
Annealing phase: 10” at 50°C
Extension phase: 1’30’’ at 72°C
Repeat for 34 cycles.
13. Check the PCR reaction on 1% agarose gel (Fig. 3)
Figure 3.
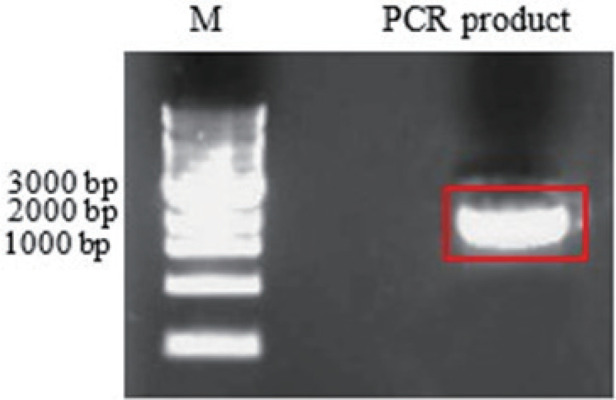
Lane 1: 1 Kb Plus DNA Ladder (Thermo Scientific); Lane 2: amplification product
Gel extraction
14. Cut the gel fragment containing the band at 2532 bp, compatible with the length of HisTag-APEH gene.
15. Extract the DNA with a suitable kit (Promega).
Blunt PCR Cloning
Various kits are available for cloning a Pfu-amplified product. We used the method of StrataClone Blunt PCR Cloning Kit.
The StrataClone Blunt PCR Cloning Kit allows for high-efficiency, 5-minute cloning of blunt-ended PCR products, using the efficient DNA rejoining activity of DNA topoisomerase I and the DNA recombination activity of Cre recombinase (Fig. 4).
Figure 4.
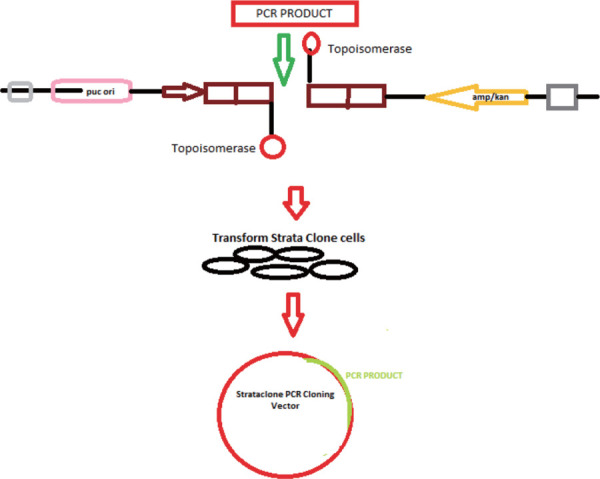
Representation of the system utilized, a figure modified from the PSCB blunt PCR cloning kit.
Prepare the ligation reaction mixture by following the suitable protocol.
Trasformation in SoloPack cells
16. Use 5 μl of ligase to transform the SoloPack cells.
17. On ice for 30 minutes.
18. Heat shock for 60 seconds at 42°C in a water bath.
19. Incubate 1 hour at 220 rpm at 37 °C.
-
20. Put 200 μl of cells onto a plate of LB + Kanamycine O/N at 37°C.
The day after, at least 20 clones will be on the plate.
PCR colony
PCR colony is a convenient high-throughput method for determining the presence or absence of insert DNA in plasmid constructs. About 20 individual transformants were added directly to the 25 μl of final volume PCR reaction and lysed for 5 min at 95°C during the initial heating step. Then:
21. Denaturation phase at 95°C for 1 minute.
22. Annealing phase at 45°C for 1 minute.
-
23. Extension phase for 1.5 minutes (this step depends on the length of the base pairs involved).
All steps are repeated for 30 cycles.
24. Load 5 μl of each reaction into a 1% agarose gel, allowing a lane for an appropriate ladder, and perform electrophoresis.
25. Choose the positive clones.
Sequencing of DNA by Sanger method
26. Check the positivity of the DNA clone by sending it to a sequencing service (we used Eurofins Scientific).
The Sanger’s method generates nested set of labelled fragments from a template strand of DNA to be sequenced by replicating that template strand and interrupting the replication process at one of the four bases.
The DNA sequence was correct.
ANTICIPATED RESULTS
Having provided the sequence of our gene of interest with a His-Tag allowed us to use an affinity chromatography step for the purification of the recombinant protein, using a column containing a resin with nickel ions (Fig. 5).
Figure 5.

Representation of nickel ions chain and hystidine contained in the His-Tag at N-terminal of APEH recombinant protein.
This step allows to facilitate the purification of a protein.
As shown in the following figure, the passage with affinity chromatography allowed for a notable enrichment of the recombinant protein, compared to the whole extract. (Fig. 6). The idea is that, starting from this point, we could utilize this clone, obtained with the use of PCR and the PSCB strategy, to increase the level of expression changing some variables in the growth conditions. (for example, pH, salinity and temperature)
Figure 6.
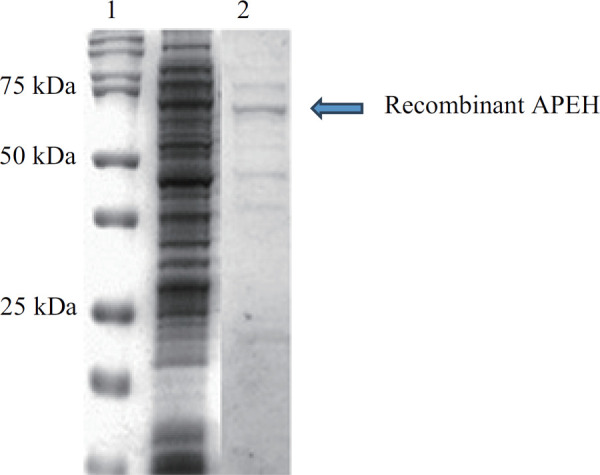
Comparison between the whole extract and the enrichment of recombinant APEH obtained by the chromatographic step. Lane 1: Marker (Bio-Rad Pecision Plus Dual Standard) Lane 2: Total extract Lane 3: elution fraction containing the recombinant APEH (75 kDa).
TROUBLESHOOTING
| Steps | Problems | Causes | Suggestions |
|---|---|---|---|
| 5 | Null or partial digestions of DNAs |
|
|
| 22 | No positive colonies |
|
|
References
- 1.Rossi R, Montecucco A, Ciarrocchi G, Biamonti G. Functional characterization of the T4 DNA ligase: a new insight into the mechanism of action. Nucleic Acids Res. 1997. Jun 01;25(11):2106-13. https://doi.org/10.1093/nar/25.11.2106 10.1093/nar/25.11.2106 PMID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ichiro M. Why Johnny can’t clone: Common pitfalls and not so common solutions. Biotechniques 2015. Sep 1;59(3): IV-XIII. eCollection 2015 Sep. https://doi.org/10.2144/000114324 10.2144/000114324 PMID: [DOI] [PubMed] [Google Scholar]
- 3.Yoshino Y, Ishida M, Horii A. A new 10-min ligation method using a modified buffer system with a very low amount of T4 DNA ligase: the “Coffee Break Ligation” technique. Biotechnol Lett. 2007;29(10):1557-60. https://doi.org/10.1007/s10529-007-9429-z 10.1007/s10529-007-9429-z PMID: [DOI] [PubMed] [Google Scholar]
- 4.Kristie N. DNA and General PCR Methods: Blunt-end Ligation. BioTechniques 48:273-275 (April 2010). https://doi.org/10.1016/B978-0-12-418687-3.00024-0 10.1016/B978-0-12-418687-3.00024-0 PMID: [DOI] [PubMed] [Google Scholar]
- 5.Nguyen KT, Pei D. Purification and characterization of enzymes involved in the degradation of chemotactic N-formyl peptides, Biochemistry 44(23) (2005) 8514–8522. https://doi.org/10.1021/bi050191o 10.1021/bi050191o PMID: [DOI] [PubMed] [Google Scholar]
- 6.Fujino T, Watanabe K, Beppu M, Kikugawa K, Yasuda H. Identification of oxidized protein hydrolase of human erythrocytes as acylpeptide hydrolase, Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 1478(1) (2000) 102–112. https://doi.org/10.1016/s0167-4838(00)00004-2 10.1016/s0167-4838(00)00004-2 PMID: [DOI] [PubMed] [Google Scholar]
- 7.Polgár L. The prolyl oligopeptidase family. Cell Mol Life Sci. 2002;59(2):349-62. https://doi.org/10.1007/s00018-002-8427-5. 10.1007/s00018-002-8427-5 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma KK, Ortwerth BJ. Bovine lens acylpeptide hydrolase. Purification and characterization of a tetrameric enzyme resistant to urea denaturation and proteolytic inactivation. Eur J Biochem (1993), 216, 631–637. https://doi.org/10.1111/j.1432-1033.1993.tb18183.x 10.1111/j.1432-1033.1993.tb18183.x PMID: [DOI] [PubMed] [Google Scholar]
- 9.Kiss AL, Hornung B, Radi Gengeliczki Z, Sztaray B, Juhasz T, Szeltner Z, Harmat V, Polgar L. The acylaminoacyl peptidase from Aeropyrum pernix K1 thought to be an exopeptidase displays endopeptidase activity. J Mol Biol (2007) 368, 509–520. https://doi.org/10.1016/j.jmb.2007.02.025 10.1016/j.jmb.2007.02.025 PMID: [DOI] [PubMed] [Google Scholar]
- 10.Gogliettino M, Balestrieri M, Cocca E, Mucerino S, Rossi M, Petrillo M, Mazzella E, Palmieri G. Identification and characterisation of a novel acylpeptide hydrolase from Sulfolobus solfataricus: structural and functional insights. PLOS ONE 7 (2012), e37921. https://doi.org/10.1371/journal.pone.0037921 10.1371/journal.pone.0037921 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gogliettino M, Riccio A, Balestrieri M, Cocca E, Facchiano A, D’Arco TM, Tesoro C, Rossi M, Palmieri G. A novel class of bifunctional acylpeptide hydrolases--potential role in the antioxidant defense systems of the Antarctic fish Trematomus bernacchii. FEBS J. 2014;281(1):401-15. https://doi.org/10.1111/febs.12610 10.1111/febs.12610 PMID: [DOI] [PubMed] [Google Scholar]
- 12.Riccio A, Gogliettino M, Palmieri G, Balestrieri M, Facchiano A, Rossi M, Palumbo S, Monti G, Cocca E. A New APEH Cluster with Antioxidant Functions in the Antarctic Hemoglobinless Icefish Chionodraco hamatus. PLOS One. 2015; 10(5): e0125594. https://doi.org/10.1371/journal.pone.0125594 10.1371/journal.pone.0125594 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feller G, Le Busy O, Gerday C. Expression of Psychrophilic Genes in Mesophilic Hosts: Assessment of the Folding State of a Recombinant α-Amylase. Applied and environmental microbiology, Mar.1998, p. 1163–1165. https://doi.org/10.1128/AEM.64.3.1163-1165.1998 10.1128/AEM.64.3.1163-1165.1998 PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]