

Abstract
Humans are constantly exposed to fungi, either in the form of commensals at epithelial barriers or inhaled spores. Innate immune cells play a pivotal role in maintaining commensal relationships and preventing skin, mucosal or systemic fungal infections due to the expression of pattern recognition receptors that recognize fungal cell wall components and modulate both their activation status and the ensuing adaptive immune response. Commensal fungi also play critical role in the modulation of homeostasis and disease susceptibility at epithelial barriers. This review will outline cellular and molecular mechanisms of anti-fungal innate immunity focusing on C-type lectin receptors and their relevance in the context of host-fungi interactions at skin and mucosal surfaces in murine experimental models as well as patients susceptible to fungal infections.
Keywords: C-type lectin receptors, dectin-1, dectin-2, Candida albicans, CARD9
Graphical Abstract
C-type lectin receptor (CLR)-mediated recognition of fungi by innate immune cells at the epithelial barriers prevents recurrent fungal infections and establishes host-fungi commensal relationships. These concepts have been demonstrated in mouse models and patients with primary immunodeficiencies affecting the function of CLRs (e.g. dectin-1) or their signaling molecules (e.g. CARD9).
Introduction
Fungi are ubiquitous in the environment but only a few species are considered true or opportunistic pathogens. Nevertheless, fungal diseases represent a major healthcare problem due to the high rate of mortality of invasive fungal infections despite available antifungal drugs [1]. In addition, there are several reports on the emergence of multi-drug resistant fungal species (e.g. Candida auris) as well as development of drug resistance in previously sensitive fungal species (e.g. Aspergillus fumigatus) [2]. The interaction between fungi and humans does not only result in fungal diseases. The analysis of endogenous fungal communities (collectively defined as mycobiota) has identified several fungal species at epithelial barriers, with Malassezia spp. and Candida spp. being predominant in the skin and the intestine, respectively. Multiple lines of evidence support a critical role for the mycobiota in modulating homeostasis and disease susceptibility [3].
The innate immune system plays a pivotal role in the modulation of host-fungi interaction and protection against fungal diseases by direct killing of fungi, modulation of microbial communities that influences fungal commensalism, and polarization of the adaptive immune response (e.g. Th1 and Th17 polarization). This host of functions is accomplished mainly through receptor-mediated recognition of fungal determinants and also by sensing geometric cues (e.g. fungal size) [4]. Fungal ligands activate a variety a pattern recognition receptors (PRRs) including C-type lectin receptors (CLRs), Toll-like receptors (TLRs) and NOD-like receptors (NLRs) [5]. Interestingly, primary immunodeficiencies of TLR and NLR pathways do not increase the risk of fungal infections, while deficiencies of the CLR CLEC7A (dectin-1) or the signaling molecule CARD9 downstream to many CLRs are linked to chronic mucocutaneous candidiasis (although a causative connection for CLEC7A has not been fully established) and, in the case of CARD9 deficiency, also to systemic fungal diseases [6]. Altogether, an in-depth knowledge of the innate immune response to fungi will be key to understand the pathophysiological implications of host-fungi interaction and define novel immunotherapeutic approaches to fungal diseases.
In this review we will outline the cellular and molecular basis of fungal recognition by the innate immune system at epithelial barriers with a particular focus on CLRs, their role in shaping host-fungi interaction and fungal disease susceptibility.
CLR signaling and anti-fungal innate immunity
Human clinical and mouse experimental results support a critical role for CLRs in mediating anti-fungal innate immune recognition and defense. CLRs are a superfamily of more than 1000 secreted and transmembrane proteins that contain one or more C-type lectin domains (CTLDs). Although CTLDs are responsible for the binding of carbohydrates by CLRs, they can also bind several chemically unrelated molecules including proteins, lipids and inorganic materials [7]. In particular, some CLRs expressed mainly by myeloid cells recognize fungal cell wall polysaccharides. It is worth noting that CLR-activated signaling pathways and cellular responses can be cell-specific and context dependent. Moreover, out of the many CLRs identified so far relatively few are recognized as key players of anti-fungal immunity, leaving open the opportunity for the identification of novel CLR-fungal ligand pairs [8] (Fig. 1A). For example, it has recently been shown that the fungal cell wall component naphthalene-diol unit of 1,8-dihydroxynaphthalene (DHN)-melanin is recognized by the melanin-sensing CLR MelLec. This receptor is expressed by endothelial cells in mice as well as myeloid cells in humans and is required for protection against experimental infections with A. fumigatus (whose cell wall contains melanin).
Figure 1. CLRs and signaling pathways activated by fungal cell wall polysaccharides.
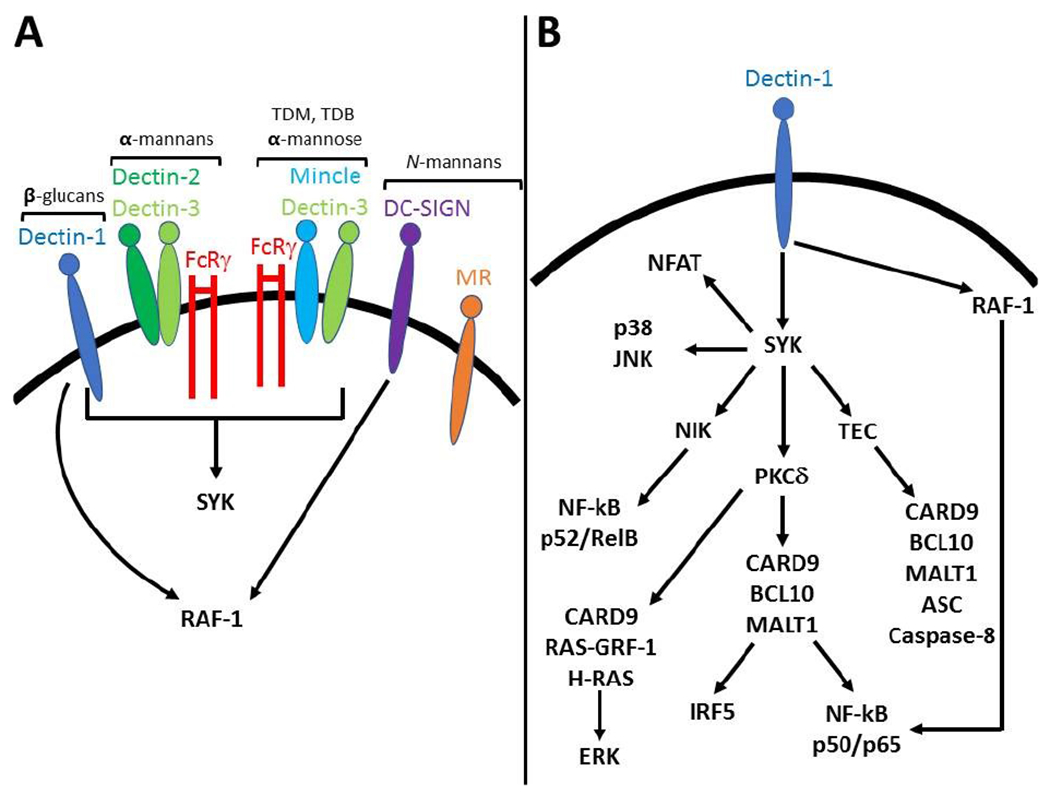
A Fungal cell wall polysaccharides activate several CLRs including dectin-1, dectin-2, mincle, DC-SIGN and MR. Dectin-1 and dectin-2 respectively bind β-(1,3) glucans andα-mannans. Mincle binds the glycolipid trehalose-6,6-dimycolate (TDM), its analogue trehalose-6,6-dibehenate (TDB) as well as α-mannose residues. DC-SIGN and the mannose receptor bind N-inked mannans. Dectin-1, dectin-2 and mincle relay intracellular signals through the kinase SYK. Dectin-2 and mincle couple with dectin-3 and FcRγ in order to signal. DC-SIGN as well as dectin-1 activate RAF-1, while no signaling pathway has been described for MR so far. B Dectin-1 activates multiple signaling pathways and most of them are SYK-dependent with the exception of RAF-1 which modulates NF-κB activation. SYK-dependent PKC-δ activation leads to the formation of the CARD9/BCL10/MALT1 complex that is required for canonical NF-κB and IRF5 activation. SYK also activates NIK which control non-canonical NF-κB activation. The CARD9/BCL10/MALT1 complex also induces non-canonical caspase 8 inflammasome assembly in a TEC kinase-dependent manner. CARD9 coupling with RAS-GRF-1 and H-RAS leads to ERK activation while the other MAPKs p38 and JNK are activated in a CARD9-independent manner. Finally, SYK activates PLCγ2/calcium/calcineurin pathways that are required for NFAT nuclear translocation.
The fungal cell wall surrounds the plasma membrane and is composed of different layers [9, 10]: an innermost layer of the N-acetylglucosamine polymer chitin; an adjacent external layer of β-(1,3) and β-(1,6) glucans which elicits a pro-inflammatory response and is usually masked by an outermost layer whose composition varies among different fungal species. For example, A. fumigatus covers the β-glucans with a proteinaceous hydrophobin layer in the conidial form and galactomannan and galactosaminogalactan in the hyphal form. Criptococcus neoformans surrounds the β-glucan layer with a capsule composed of glucuronoxylomannan and galactoxylomannan. Finally, Candida albicans outer layer consists of mannans N- and O-linked to cell wall proteins.
β-(1,3) glucans from several fungi bind to and activate the CLR dectin-1, one of the best characterized receptors for fungal cell wall components [5, 11]. The current model of dectin-1 activation states that only β-glucans in a particulate form can activate downstream signaling by inducing clustering of dectin-1, displacement of regulatory phosphatases CD45 and CD148, and SRC-dependent phosphorylation of the intracellular ITAM-like motif of dectin-1 [12]. The complex interplay between membrane dynamics and dectin-1 signaling is highlighted by recent evidence supporting that pharmacologic impairment of phagocytosis (e.g. by using actin polymerization inhibitors cytochalasin D and latrunculin) enhances human and mouse cell activation upon stimulation with β-glucans (but not intact C. albicans) [13, 14]. A critical event in dectin-1 signaling is the activation of SYK which in turn activates CARD9 via PKC-δ and the VAV family of GEFs [15–17]. CARD9 form the CBM complex with BCL10 and MALT1 and leads to IKK activation, IκB phosphorylation and nuclear translocation of the canonical NF-κB subunits p50, p65 and c-REL [18]. These events are critical for the induction of pro-inflammatory genes. SYK also activates NIK and the non-canonical NF-κB subunits p52 and RELB in a CARD9-independent manner [19]. In addition, SYK-independent activation of RAF-1 modulates the activity of NF-κB subunits for example by inducing the formation of p65-RELB dimers [19]. Although the functional consequences of these events in the context of dectin-1 signaling and innate anti-fungal immunity are not completely understood, there is evidence suggesting an important role for RAF-1- and NIK-dependent modulation of NF-κB subunit activity in eliciting a Th1 response through the induction of IL-12p40 and IL-1β. Dectin-1-elicited IL-1β requires inflammasome activation. Interestingly, some fungi trigger dectin-1-dependent canonical caspase 1 inflammasome activation which also requires fungal internalization, reactive oxygen species production and potassium efflux [20–22]. Alternatively, dectin-1 can induce non-canonical caspase 8 inflammasome assembly independently of fungal internalization in a process that requires SYK-dependent activation of TEC kinase [23, 24] (Fig. 1B).
MAPKs are also activated downstream of dectin-1 in a SYK-dependent manner. CARD9 activates ERK through RAS-GRF-1 and H-RAS, while activation JNK and p38 is CARD9-independent [25]. Of note, ERK cooperates with NF-κB to modulate the pro-inflammatory response and Th1/Th17 polarization upon fungal infection downstream of the dectin-1 (and also dectin-2)/SYK/CARD9 axis, therefore exerting a critical role in protection against fungal insults [19, 26, 27]. On the other hand, JNK1 negatively regulates innate antifungal immunity by suppressing the expression of CD23, a novel identified receptor for α-mannans and β-glucans that induces the antifungal effector NOS2 [28]. Another negative regulator of dectin-1 as well as dectin-2 signaling is the E3 ubiquitin ligase CBLB that ubiquitinates dectin-1, dectin-2 and SYK and targets them for degradation [29–31]. Dectin-1 signaling also activates the transcription factors IRF1, IRF5 and NFAT. IRF5 is activated in a CARD9-dependent manner and induces IFNβ production whose role in anti-fungal defense is still controversial [32, 33]. NFAT is activated in a PLCγ2/calcium/calcineurin-dependent manner and regulates the expression of a subset of genes like IL-2 that plays a critical role in NK and T cell activation [34–36] (Fig. 1B).
It is worth noting that dectin signaling might exhibit context- and cell-dependent features. For example, in vitro exposure to GM-CSF and IFNγ is required for eliciting a pro-inflammatory response upon dectin activation [37]. The functional significance of this phenomenon and its relevance to in vivo anti-fungal immunity still have to be clarified. Finally, it is important to mention that fungal cell wall glucans comprise both β-(1,3) and β-(1,6) glucans. While the former ones bind to dectin-1, no specific receptor has been identified for β-(1,6) glucans so far [38].
Mannans and mannosylated moieties are recognized by a variety of CLRs, including dectin-2, mincle, mannose receptor (MR) and DC-SIGN [5, 11]. Of these, MR does not seem to signal upon binding to its ligand (N-linked Candida mannans) nor does it seem to be required in experimental models of fungal infection [5, 11], although it might play a role in the polarization of human Th17 cells [39]. DC-SIGN recognizes a variety of carbohydrate including N-linked mannosyl residues of Candida [40]. DC-SIGN activates RAF-1 which in turn modulates NF-κB transcriptional program [41]. The outcome of DC-SIGN signaling seems to vary according to the triggering stimulus and concurrent activation of other PRRs as well [5, 11]. At variance with MR and DC-SIGN, mincle and dectin-2 signal through the SYK/CARD9 pathway. However, they do not have ITAM or ITAM-like motifs in their cytoplasmic tails like dectin-1 [5, 11]. Instead, mincle and dectin-2 couple with FcRγ chains that provide ITAM sequences required to activate the SYK/CARD9 pathway and also with dectin-3 which modulates ligand recognition and signal transduction [5, 11]}. The glycolipid trehalose-6,6-dimycolate found in the cell wall of Mycobacteria and its analogue trehalose-6,6-dibehenate are prototypical ligands of mincle [5, 11]. This receptor also binds cell wall components of Malassezia, namely bipolar glycolipids with disaccharide composed of glucose or mannose attached to fatty acids, and unknown components of Fonsecaea spp. and Cladophialophora carrionii [42, 43]. Interestingly, while mincle activation leads to SYK/CARD9-dependent NF-κB activation and cytokine production in mice, no NF-κB activation nor cytokine gene expression are detected in human cells [43]. Instead, mincle activation leads to SYK/CARD9-dependent activation of the PI3K/AKT pathway which is required for activation of the E3 ubiquitin ligase MDM2 and proteasomal degradation of IRF1 [43]. Dectin-2 recognizes α-mannans and α-1,2-mannosylated residues of several fungal species [5, 11]. Although the responses elicited by dectin-1 and dectin-2 may exhibit some degree of overlap, signaling pathways activated by dectin-2 have been less intensively investigated compared to dectin-1 and require further studies.
It is worth noting that anti-fungal innate immunity is often determined by cooperation between CLRs and other receptors. Fungal cell walls contain multiple CLR ligands that can exert synergistic or sometimes even antagonist effects on the innate immune system. Concurrent activation of dectin-1 and TLRs enhances the expression of some cytokines (e.g. TNF) while downregulating others (e.g. IL-12p70) [44]. Dectin-1 activates complement receptor 3 (CR3) in a VAV-dependent manner to enhance neutrophil phagocytosis and killing of C. albicans [45]. Moreover, simultaneous activation of dectin-1 and CR3 by Histoplasma capsulatum increases cytokine production by enhancing signaling through the SYK/JNK/AP1 pathway [46]. Engagement of both mincle and dectin-1 by fungi (e.g. Fonsecaea spp.) or purified ligands induces IRF1 degradation through MDM2 and impairs IL-12p70 expression [43].
Overall, CLR activation by fungi and their cell wall components triggers several signaling pathways whose contribution to anti-fungal innate and adaptive immunity is still only partially understood. Several factors may confound the functional analysis of CLR signaling pathways and will need to be taken into account in future studies, including fungal species-, organ- and cell-specific CLR activation and signaling.
Anti-fungal innate immunity in the lungs
The respiratory tract is constantly exposed to fungal spores [47]. Fungal infections and diseases are prevented by a hydrophobic layer produced by airborne fungal spores that masks immunogenic moieties in the fungal cell wall as well as competent innate and adaptive immune responses. Therefore, immunosuppressed patients are susceptible to pulmonary fungal infections, and some species exacerbates pre-existing lung diseases such as asthma and cystic fibrosis.
Alveolar macrophages represent an important innate line of defense against fungal infection. It has been shown for several fungal species that alveolar macrophages recognize and phagocytose them mainly through binding to dectin-1 [47]. Some fungi such as A. fumigatus are readily killed by alveolar macrophages [48] and requires fungal recognition by soluble receptors like the long pentraxin 3 [49] which is produced by myeloid cells under the control of the transcription factor NFAT [50]. Conidial spores of A. fumigatus are also recognized by MelLec which is expressed in the lung and is required for early neutrophil recruitment at the infection site [8]. Interestingly, lung immunity against A. fumigatus infection involves a complex cross-talk between lung epithelial cells and immune cells [51, 52]. In this context, IL-1α is required for early neutrophil recruitment most likely by inducing CXC chemokine production by epithelial cells in a MYD88-dependent manner. A second phase of neutrophil recruitment is driven by CARD9-dependet CXC chemokine production. The main chemokine source in this second phase is represented by neutrophils themselves. It is worth noting that there is no intrinsic role for MYD88 and CARD9 in neutrophil uptake and killing of A. fumigatus [52].
Other fungi have developed strategies to evade the lung innate immune response. For example, Blastomyces dermatitidis inhibits iNOS and therefore escapes oxidative killing upon phagocytosis by alveolar macrophages [53]. This fungus also produces the protease DppIVA that cleaves GM-CSF, CC and CXC chemokines, therefore blunting monocyte recruitment to the lung, myeloid cell activation and promoting Blastomyces dermatitidis virulence. Interestingly, cleaved CXC chemokines have greater chemoattractant properties but fail to prime neutrophils for ROS production [54]. This might explain the pyogranulomatous lesions of blastomycosis (i.e. neutrophil recruitment at the infection site with inability to control fungal growth).
Lung anti-fungal T cell responses are driven mainly by conventional and monocyte-derived dendritic cells (DCs). The specific DC subset required to generate anti-fungal Th1 or Th17 responses varies depending on the fungal species. Overall, Th17 responses seem to play a critical role in protecting against fungal lung infections [47]. Interestingly, in a mouse model of lung aspergillosis NFAT-dependent production of IL-2 by CD103+ lung DCs curbs the Th17 response that would otherwise lead to hyperinflammation [55]. Again, the molecular signals required to generate such responses are only partially defined and can be fungal species-specific. For example, several CLRs (e.g. dectin-1, dectin-2 and MR) as well as CARD9 and MYD88 have been involved in driving Th17 polarization upon lung infection with different fungal species [47]. It should be noted that this evidence has been mostly generated in animal models while patients with primary immunodeficiencies affecting CLRs, CARD9 or IL-17 receptor subunits do not have increased risk of developing fungal lung infections [6]. Nevertheless, polymorphisms in CLEC7A and CLEC1A that respectively diminish dectin-1 and MelLec activities increase susceptibility to invasive pulmonary aspergillosis in patients receiving hematopoietic stem cell transplantation [8, 56].
Anti-fungal innate immunity in the skin
Human skin is colonized by commensal fungi, especially Malassezia species [57]. Although they have been suggested to modulate skin diseases such as alopecia and psoriasis, no clear evidence supporting this claim has been found so far [47]. Human fungal skin infections are usually superficial and most frequently caused by dermatophytes. Occasionally, some fungal species can also lead to extensive skin and subcutaneous tissue infections such as chromoblastomycosis (most often caused by Fonsecaea pedrosoi), phaeohyphomycosis (caused by dematiaceous fungi) and eumycetoma (most often caused by Madurella species). Skin resident immune cells play a pivotal role in anti-fungal innate immunity. Fungi are recognized by a complex web of myeloid cells that can be divided according to their locations and functions into Langerhans cells (LCs) residing in the epidermal layer and dermal dendritic cells. In experimental models of epicutaneous C. albicans infection LCs recognize the yeast form and drive Th17 polarization through dectin-1-mediated IL-6 production. Conversely, dermal DCs recognize the filamentous form and promote Th1 differentiation. In this experimental model Th17 and Th1 lymphocytes are respectively required for protection against cutaneous and systemic C. albicans infections [58, 59]. Th17 cells are not the only immune subset that produces IL-17 in response to epicutaneous C. albicans infection. For example, nociceptive neurons are activated upon infection and secrete the neuropeptide CGRP, which induces the release of IL-23 by CD301b+ dermal DCs which in turn promotes IL-17 production by γδ T cells [60]. Interestingly, optogenetic activation of nociceptive neurons (i.e. in the absence of damage- or pathogen-associated molecular patterns) is sufficient to elicit CGRP-mediated IL-17 production at the stimulation site as well as adjacent sites through a nerve reflex arc. These events lead to enhanced protection against C. albicans and Staphylococcus aureus skin infections [61]. Heterologous protection against C. albicans can also be promoted by colonization of the skin with other commensals. Upon skin colonization with Staphylococcus epidermidis CD103+ DCs prime S. epidermidis-specific CD8 T cells in the draining lymph node. Then, CD8 T cells migrate to the skin where they are further activated by CD11b+ DC-derived IL-1 and produce IL-17 which in turn leads to production of antimicrobial peptides by keratinocytes and protection against C. albicans infection [62]. Experimental models of intradermal C. albicans infection have uncovered critical roles for NK cells in the modulation of skin inflammation. In the absence of LCs, CXCR6+ NK cells are recruited to skin and promote exaggerated inflammation [63]. We have recently shown that intradermal C. albicans infection leads to abscess formation in which the pyogenic membrane is formed in a TGFβ-dependent manner. Abscess elimination requires NK cell-derived IFNγ that blocks differentiation of fibroblasts into myofibroblasts and also promotes the formation of plasmin which leads to abscess capsule digestion. Interestingly, in this experimental model NK cells are activated by NFATc2-mediated production of IL-2 by DCs [64]. The molecular signals that lead to neutrophil recruitment and abscess formation following infection and how IFNγ influences the activation of the fibrinolytic system have to be defined.
Human CARD9 deficiency is associated with the development of uncommon and debilitating fungal skin infections. Although a critical role for IL-17 in skin anti-fungal immunity seems to emerge from experimental models, we are far from understanding whether this holds true also for patients with CARD9 deficiency. Indeed, reduction in peripheral Th17 cells has not been observed in all patients with CARD9 deficiency [65]. Further studies are required to define the cellular and molecular mechanisms of susceptibility to skin fungal infections in these patients.
Anti-fungal innate immunity in the oral mucosa and gastrointestinal tract
C. albicans is the most common etiologic agent of oral fungal infections that occur in infants, HIV/AIDS patients if CD4+ T cell counts are low, patients with inborn errors of IL-17 immunity or APECED, and patients with immunodeficiency secondary to chemotherapy [47, 66]. Clinical and experimental evidence has shown a critical role for IL-17 in protecting against oropharyngeal candidiasis (OPC) [67]. Upon C. albicans infection of the oral mucosa IL-17 is produced mainly by natural Th17 cells, γδ T cells and ILC3, while conventional Th17 contribute to IL-17 production after re-infection [68–70]. Interestingly, CARD9 is required for conventional Th17 differentiation but dispensable for innate IL-17 production [70]. IL-17-mediated immunity against OPC is independent of neutrophil recruitment and correlates with the induction of antimicrobial peptides in the epithelium [71]. Instead, another mechanism promoting neutrophil recruitment during OPC has recently been described. C. albicans produces the toxin Candidalysin that induces oral epithelial cell damage and activation leading to the release of pro-inflammatory cytokines such as IL-1 [72, 73]. IL-1 also induces G-CSF production by endothelial cells that induces neutrophil recruitment to the oral mucosa, and it also promotes proper IL-17 innate immunity [74].
Commensal fungi are now considered an integral part of the gut microbiome partly due to their detection using culture-independent methodologies and there is mounting evidence for their involvement in health and disease [3, 47]. Although some fungal species are only transiently present in the gut as part of the diet or environmental exposure, others like Saccharomyces and Candida spp. stably colonize the gut [3, 47]. Clinical evidence has pointed to a role for commensal fungi in inflammatory bowel disease (IBD) pathogenesis [75]. Genome-wide association studies have linked polymorphisms in CLEC7A and CARD9 to IBD development and severity. Anti-Saccharomyces cerevisiae antibodies specific for fungal cell wall mannans can be found in patients with Chron’s disease. Moreover, fungal dysbiosis has been observed in gut biopsies and fecal samples obtained from IBD patients. Experimental models have been employed to investigate the cellular and molecular mechanisms of host-fungi interaction in the gut and their contribution to health and disease, sometimes yielding contrasting results. Dectin-1 KO mice experience more severe DSS-induced colitis [76]. Symptoms are further aggravated by administration of C. tropicalis while they are alleviated by treatment with the anti-fungal drug fluconazole [76]. Nevertheless, a separate study has shown that dectin-1 KO mice are less susceptible to DSS-induced colitis due to expansion of lactobacilli that promote an increase in Treg cells in the colon [77]. Differences in gut microflora and colonization by opportunistic fungal commensals probably account for these discrepancies. Recently, gut-resident CX3CR1+ mononuclear phagocytes (MNPs) have been identified as the main cell subset responsible for fungal recognition and required for innate and adaptive immune responses to fungi in the gut [78]. Interestingly, deletion of CX3CR1+ MNPs alters the composition of intestinal fungal communities and aggravates DSS-induced colitis. The latter phenomenon is reversed by anti-fungal treatment with fluconazole. Other cells and factors have been involved in the modulation of host-fungi interactions. Mast cells and IL-9 have been implicated in defining the pathogenic versus commensal nature of C. albicans [79]. They promote both mucosal immune tolerance to C. albicans challenge and barrier function loss and inflammation in experimental models of leaky gut. Fungal species other that Candida have also been associated with the pathogenesis of IBD. The skin-resident fungus Malassezia restricta is particularly abundant in the gut of IBD patients carrying the CARD9 risk allele [80]. This polymorphism is associated with increased pro-inflammatory response to M. restricta in vitro. Interestingly, M. restricta also exacerbates disease severity in mouse models of IBD in a CARD9-dependent manner. CARD9 has also been implicated in the development of colitis-associated colorectal cancer using CARD9 KO mice, although conflicting results have been reported so far regarding its role in increasing or reducing susceptibility and also the role of intestinal fungi in this process [81–83]. To further complicate the scenario, CARD9 KO mice display altered composition not only of fungal but also bacterial communities in the gut [84]. Defects in tryptophan metabolism and generation of aryl hydrocarbon receptor ligands by gut bacteria in CARD9 KO mice may be accountable for increased susceptibility to colitis. Finally, altered gut fungal communities have also been implicated in the modulation of peripheral immune responses, for example by exacerbating allergic airway disease [85, 86].
Conclusions
Recent years have witnessed increasing interest in understanding the biology of host-fungi interaction and its consequences in health and disease. Innate immune pathways responsible for sensing fungi and their cell wall components have been better characterized by combining in vitro and in vivo experimental results with clinical data of inborn errors of anti-fungal immunity. Fungi are now considered integral constituents of the microbiota at epithelial barriers and seem to have specific roles in modulating tissue homeostasis and disease susceptibility. However, several questions still remain unanswered. Compared to the number of identified CLRs, relatively few of them have been investigated in the context of anti-fungal immunity. Although CLRs can activate several signaling pathways, their contribution to anti-fungal innate and adaptive immunity as well as their regulatory mechanisms have been marginally investigated so far. Important concepts that will need to be taken into account are fungal species-specific and also organ-specific requirements of distinct CLRs and signaling pathways in anti-fungal immunity. Moreover, as culture-independent methodologies for detecting fungal communities at epithelial barriers improves, novel insights into how commensal fungi contribute to tissue homeostasis and disease susceptibility will be generated. Therefore, it will be critical to understand the interplay between fungal and bacterial communities also accounting for variabilities among different vivaria by using appropriate experimental controls [87]. Altogether, improving our knowledge of anti-fungal innate immunity will be instrumental to develop immune-based therapies for fungal diseases and define the contribution of fungi to homeostasis and pathophysiology.
Acknowledgments
I. Z. is supported by the National Institutes of Health (R01AI121066, R01DK115217, NIAID-DAIT-NIHAI201700100).
F. G. is supported by the Associazione Italiana per la Ricerca sul Cancro (IG 2016Id. 18842), the Cariplo Foundation (Grant 2014-0655), and the Fondazione Regionale per la Ricerca Biomedica, FRRB.
Footnotes
Conflict of interest:
F.B. has signed a consulting agreement with Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc. This commercial relationship is unrelated to the current study. The rest of the authors declare no commercial or financial conflict of interest.
References
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG and White TC, Hidden killers: human fungal infections. Sci Transl Med 2012. 4: 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Lockhart SR, Etienne KA, Vallabhaneni S, Farooqi J, Chowdhary A, Govender NP, Colombo AL et al. , Simultaneous Emergence of Multidrug-Resistant Candida auris on 3 Continents Confirmed by Whole-Genome Sequencing and Epidemiological Analyses. Clin Infect Dis 2017. 64: 134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iliev ID and Leonardi I, Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat Rev Immunol 2017. 17: 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD and Papayannopoulos V, Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 2014. 15: 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lionakis MS, Iliev ID and Hohl TM, Immunity against fungi. JCI Insight 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lionakis MS, Netea MG and Holland SM, Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb Perspect Med 2014. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown GD, Willment JA and Whitehead L, C-type lectins in immunity and homeostasis. Nat Rev Immunol 2018. 18: 374–389. [DOI] [PubMed] [Google Scholar]
- 8.Stappers MHT, Clark AE, Aimanianda V, Bidula S, Reid DM, Asamaphan P, Hardison SE et al. , Recognition of DHN-melanin by a C-type lectin receptor is required for immunity to Aspergillus. Nature 2018. 555: 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gow NAR, Latge JP and Munro CA, The Fungal Cell Wall: Structure, Biosynthesis, and Function. Microbiol Spectr 2017. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Netea MG, Brown GD, Kullberg BJ and Gow NA, An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 2008. 6: 67–78. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, Lin G, Langdon WY, Tao L and Zhang J, Regulation of C-Type Lectin Receptor-Mediated Antifungal Immunity. Front Immunol 2018. 9: 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, Bose N et al. , Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011. 472: 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Camilli G, Eren E, Williams DL, Aimanianda V, Meunier E and Quintin J, Impaired phagocytosis directs human monocyte activation in response to fungal derived beta-glucan particles. Eur J Immunol 2018. 48: 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hernanz-Falcon P, Joffre O, Williams DL and Reis e Sousa C, Internalization of Dectin-1 terminates induction of inflammatory responses. Eur J Immunol 2009. 39: 507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogers NC, Slack EC, Edwards AD, Nolte MA, Schulz O, Schweighoffer E, Williams DL et al. , Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005. 22: 507–517. [DOI] [PubMed] [Google Scholar]
- 16.Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP et al. , Syk kinase-coupled C-type lectin receptors engage protein kinase C-sigma to elicit Card9 adaptor-mediated innate immunity. Immunity 2012. 36: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth S, Bergmann H, Jaeger M, Yeroslaviz A, Neumann K, Koenig PA, Prazeres da Costa C et al. , Vav Proteins Are Key Regulators of Card9 Signaling for Innate Antifungal Immunity. Cell Rep 2016. 17: 2572–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I et al. , Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 2006. 442: 651–656. [DOI] [PubMed] [Google Scholar]
- 19.Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC and Geijtenbeek TB, Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol 2009. 10: 203–213. [DOI] [PubMed] [Google Scholar]
- 20.Cheng SC, van de Veerdonk FL, Lenardon M, Stoffels M, Plantinga T, Smeekens S, Rizzetto L et al. , The dectin-1/inflammasome pathway is responsible for the induction of protective T-helper 17 responses that discriminate between yeasts and hyphae of Candida albicans. J Leukoc Biol 2011. 90: 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G et al. , Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 2009. 459: 433–436. [DOI] [PubMed] [Google Scholar]
- 22.Kankkunen P, Teirila L, Rintahaka J, Alenius H, Wolff H and Matikainen S, (1,3)-beta-glucans activate both dectin-1 and NLRP3 inflammasome in human macrophages. J Immunol 2010. 184: 6335–6342. [DOI] [PubMed] [Google Scholar]
- 23.Gringhuis SI, Kaptein TM, Wevers BA, Theelen B, van der Vlist M, Boekhout T and Geijtenbeek TB, Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1beta via a noncanonical caspase-8 inflammasome. Nat Immunol 2012. 13: 246–254. [DOI] [PubMed] [Google Scholar]
- 24.Zwolanek F, Riedelberger M, Stolz V, Jenull S, Istel F, Koprulu AD, Ellmeier W et al. , The non-receptor tyrosine kinase Tec controls assembly and activity of the noncanonical caspase-8 inflammasome. PLoS Pathog 2014. 10: e1004525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jia XM, Tang B, Zhu LL, Liu YH, Zhao XQ, Gorjestani S, Hsu YM et al. , CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J Exp Med 2014. 211: 2307–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LeibundGut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, Schweighoffer E et al. , Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 2007. 8: 630–638. [DOI] [PubMed] [Google Scholar]
- 27.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS et al. , Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med 2009. 206: 2037–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao X, Guo Y, Jiang C, Chang Q, Zhang S, Luo T, Zhang B et al. , JNK1 negatively controls antifungal innate immunity by suppressing CD23 expression. Nat Med 2017. 23: 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu LL, Luo TM, Xu X, Guo YH, Zhao XQ, Wang TT, Tang B et al. , E3 ubiquitin ligase Cbl-b negatively regulates C-type lectin receptor-mediated antifungal innate immunity. J Exp Med 2016. 213: 1555–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wirnsberger G, Zwolanek F, Asaoka T, Kozieradzki I, Tortola L, Wimmer RA, Kavirayani A et al. , Inhibition of CBLB protects from lethal Candida albicans sepsis. Nat Med 2016. 22: 915–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xiao Y, Tang J, Guo H, Zhao Y, Tang R, Ouyang S, Zeng Q et al. , Targeting CBLB as a potential therapeutic approach for disseminated candidiasis. Nat Med 2016. 22: 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.del Fresno C, Soulat D, Roth S, Blazek K, Udalova I, Sancho D, Ruland J et al. , Interferon-beta production via Dectin-1-Syk-IRF5 signaling in dendritic cells is crucial for immunity to C. albicans. Immunity 2013. 38: 1176–1186. [DOI] [PubMed] [Google Scholar]
- 33.Majer O, Bourgeois C, Zwolanek F, Lassnig C, Kerjaschki D, Mack M, Muller M et al. , Type I interferons promote fatal immunopathology by regulating inflammatory monocytes and neutrophils during Candida infections. PLoS Pathog 2012. 8: e1002811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodridge HS, Simmons RM and Underhill DM, Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol 2007. 178: 3107–3115. [DOI] [PubMed] [Google Scholar]
- 35.Xu S, Huo J, Lee KG, Kurosaki T and Lam KP, Phospholipase Cgamma2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J Biol Chem 2009. 284: 7038–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granucci F, Zanoni I, Pavelka N, Van Dommelen SL, Andoniou CE, Belardelli F, Degli Esposti MA et al. , A contribution of mouse dendritic cell-derived IL-2 for NK cell activation. J Exp Med 2004. 200: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodridge HS, Shimada T, Wolf AJ, Hsu YM, Becker CA, Lin X and Underhill DM, Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J Immunol 2009. 182: 1146–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noss I, Ozment TR, Graves BM, Kruppa MD, Rice PJ and Williams DL, Cellular and molecular mechanisms of fungal beta-(1-->6)-glucan in macrophages. Innate Immun 2015. 21: 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng SC, Joosten I, van den Berg WB et al. , The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 2009. 5: 329–340. [DOI] [PubMed] [Google Scholar]
- 40.Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, Kullberg BJ et al. , Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J Biol Chem 2008. 283: 20590–20599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y and Geijtenbeek TB, C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity 2007. 26: 605–616. [DOI] [PubMed] [Google Scholar]
- 42.Ishikawa T, Itoh F, Yoshida S, Saijo S, Matsuzawa T, Gonoi T, Saito T et al. , Identification of distinct ligands for the C-type lectin receptors Mincle and Dectin-2 in the pathogenic fungus Malassezia. Cell Host Microbe 2013. 13: 477–488. [DOI] [PubMed] [Google Scholar]
- 43.Wevers BA, Kaptein TM, Zijlstra-Willems EM, Theelen B, Boekhout T, Geijtenbeek TB and Gringhuis SI, Fungal engagement of the C-type lectin mincle suppresses dectin-1-induced antifungal immunity. Cell Host Microbe 2014. 15: 494–505. [DOI] [PubMed] [Google Scholar]
- 44.Dennehy KM, Willment JA, Williams DL and Brown GD, Reciprocal regulation of IL-23 and IL-12 following co-activation of Dectin-1 and TLR signaling pathways. Eur J Immunol 2009. 39: 1379–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Utomo A, Cullere X, Choi MM, Milner DA Jr., Venkatesh D, Yun SH et al. , The beta-glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe 2011. 10: 603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang JH, Lin CY, Wu SY, Chen WY, Chu CL, Brown GD, Chuu CP et al. , CR3 and Dectin-1 Collaborate in Macrophage Cytokine Response through Association on Lipid Rafts and Activation of Syk-JNK-AP-1 Pathway. PLoS Pathog 2015. 11: e1004985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drummond RA and Lionakis MS, Organ-specific mechanisms linking innate and adaptive antifungal immunity. Semin Cell Dev Biol 2019. 89: 78–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhatia S, Fei M, Yarlagadda M, Qi Z, Akira S, Saijo S, Iwakura Y et al. , Rapid host defense against Aspergillus fumigatus involves alveolar macrophages with a predominance of alternatively activated phenotype. PLoS One 2011. 6: e15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Garlanda C, Hirsch E, Bozza S, Salustri A, De Acetis M, Nota R, Maccagno A et al. , Non-redundant role of the long pentraxin PTX3 in anti-fungal innate immune response. Nature 2002. 420: 182–186. [DOI] [PubMed] [Google Scholar]
- 50.Bendickova K, Tidu F, De Zuani M, Kohoutkova MH, Andrejcinova I, Pompeiano A, Belaskova S et al. , Calcineurin inhibitors reduce NFAT-dependent expression of antifungal pentraxin-3 by human monocytes. J Leukoc Biol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Caffrey AK, Lehmann MM, Zickovich JM, Espinosa V, Shepardson KM, Watschke CP, Hilmer KM et al. , IL-1alpha signaling is critical for leukocyte recruitment after pulmonary Aspergillus fumigatus challenge. PLoS Pathog 2015. 11: e1004625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jhingran A, Kasahara S, Shepardson KM, Junecko BA, Heung LJ, Kumasaka DK, Knoblaugh SE et al. , Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog 2015. 11: e1004589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rocco NM, Carmen JC and Klein BS, Blastomyces dermatitidis yeast cells inhibit nitric oxide production by alveolar macrophage inducible nitric oxide synthase. Infect Immun 2011. 79: 2385–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lorenzini J, Scott Fites J, Nett J and Klein BS, Blastomyces dermatitidis serine protease dipeptidyl peptidase IVA (DppIVA) cleaves ELR(+) CXC chemokines altering their effects on neutrophils. Cell Microbiol 2017. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zelante T, Wong AY, Ping TJ, Chen J, Sumatoh HR, Vigano E, Hong Bing Y et al. , CD103(+) Dendritic Cells Control Th17 Cell Function in the Lung. Cell Rep 2015. 12: 1789–1801. [DOI] [PubMed] [Google Scholar]
- 56.Cunha C, Di Ianni M, Bozza S, Giovannini G, Zagarella S, Zelante T, D’Angelo C et al. , Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 2010. 116: 5394–5402. [DOI] [PubMed] [Google Scholar]
- 57.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, Schoenfeld D et al. , Topographic diversity of fungal and bacterial communities in human skin. Nature 2013. 498: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kashem SW, Igyarto BZ, Gerami-Nejad M, Kumamoto Y, Mohammed JA, Jarrett E, Drummond RA et al. , Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 2015. 42: 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Igyarto BZ, Haley K, Ortner D, Bobr A, Gerami-Nejad M, Edelson BT, Zurawski SM et al. , Skin-resident murine dendritic cell subsets promote distinct and opposing antigen-specific T helper cell responses. Immunity 2011. 35: 260–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L and Kaplan DH, Nociceptive Sensory Fibers Drive Interleukin-23 Production from CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity 2015. 43: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cohen JA, Edwards TN, Liu AW, Hirai T, Jones MR, Wu J, Li Y et al. , Cutaneous TRPV1(+) Neurons Trigger Protective Innate Type 17 Anticipatory Immunity. Cell 2019. 178: 919–932 e914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S et al. , Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015. 520: 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scholz F, Naik S, Sutterwala FS and Kaplan DH, Langerhans Cells Suppress CD49a+ NK Cell-Mediated Skin Inflammation. J Immunol 2015. 195: 2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santus W, Barresi S, Mingozzi F, Broggi A, Orlandi I, Stamerra G, Vai M et al. , Skin infections are eliminated by cooperation of the fibrinolytic and innate immune systems. Sci Immunol 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drummond RA, Franco LM and Lionakis MS, Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front Immunol 2018. 9: 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Naglik JR, Konig A, Hube B and Gaffen SL, Candida albicans-epithelial interactions and induction of mucosal innate immunity. Curr Opin Microbiol 2017. 40: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW et al. , Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 2009. 206: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Conti HR, Peterson AC, Brane L, Huppler AR, Hernandez-Santos N, Whibley N, Garg AV et al. , Oral-resident natural Th17 cells and gammadelta T cells control opportunistic Candida albicans infections. J Exp Med 2014. 211: 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gladiator A, Wangler N, Trautwein-Weidner K and LeibundGut-Landmann S, Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J Immunol 2013. 190: 521–525. [DOI] [PubMed] [Google Scholar]
- 70.Bishu S, Hernandez-Santos N, Simpson-Abelson MR, Huppler AR, Conti HR, Ghilardi N, Mamo AJ et al. , The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect Immun 2014. 82: 1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P and LeibundGut-Landmann S, IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol 2015. 8: 221–231. [DOI] [PubMed] [Google Scholar]
- 72.Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Hofs S et al. , Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 2016. 532: 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, Huppler AR et al. , Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci Immunol 2017. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Altmeier S, Toska A, Sparber F, Teijeira A, Halin C and LeibundGut-Landmann S, IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa. PLoS Pathog 2016. 12: e1005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Limon JJ, Kershaw KM and Underhill DM, Mucosal immune responses to fungi and the implications for inflammatory bowel disease. Curr Opin Gastroenterol 2018. 34: 398–403. [DOI] [PubMed] [Google Scholar]
- 76.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J et al. , Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012. 336: 1314–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang C, Kamiya T, Liu Y, Kadoki M, Kakuta S, Oshima K, Hattori M et al. , Inhibition of Dectin-1 Signaling Ameliorates Colitis by Inducing Lactobacillus-Mediated Regulatory T Cell Expansion in the Intestine. Cell Host Microbe 2015. 18: 183–197. [DOI] [PubMed] [Google Scholar]
- 78.Leonardi I, Li X, Semon A, Li D, Doron I, Putzel G, Bar A et al. , CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science 2018. 359: 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Renga G, Moretti S, Oikonomou V, Borghi M, Zelante T, Paolicelli G, Costantini C et al. , IL-9 and Mast Cells Are Key Players of Candida albicans Commensalism and Pathogenesis in the Gut. Cell Rep 2018. 23: 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, Funari V, Gargus M et al. , Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe 2019. 25: 377–388 e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Malik A, Sharma D, Malireddi RKS, Guy CS, Chang TC, Olsen SR, Neale G et al. , SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 2018. 49: 515–530 e515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang T, Fan C, Yao A, Xu X, Zheng G, You Y, Jiang C et al. , The Adaptor Protein CARD9 Protects against Colon Cancer by Restricting Mycobiota-Mediated Expansion of Myeloid-Derived Suppressor Cells. Immunity 2018. 49: 504–514 e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bergmann H, Roth S, Pechloff K, Kiss EA, Kuhn S, Heikenwalder M, Diefenbach A et al. , Card9-dependent IL-1beta regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur J Immunol 2017. 47: 1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, Da Costa G, Bridonneau C et al. , CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat Med 2016. 22: 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li X, Leonardi I, Semon A, Doron I, Gao IH, Putzel GG, Kim Y et al. , Response to Fungal Dysbiosis by Gut-Resident CX3CR1(+) Mononuclear Phagocytes Aggravates Allergic Airway Disease. Cell Host Microbe 2018. 24: 847–856 e844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wheeler ML, Limon JJ, Bar AS, Leal CA, Gargus M, Tang J, Brown J et al. , Immunological Consequences of Intestinal Fungal Dysbiosis. Cell Host Microbe 2016. 19: 865–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robertson SJ, Lemire P, Maughan H, Goethel A, Turpin W, Bedrani L, Guttman DS et al. , Comparison of Co-housing and Littermate Methods for Microbiota Standardization in Mouse Models. Cell Rep 2019. 27: 1910–1919 e1912. [DOI] [PubMed] [Google Scholar]