

Abstract
The intracellular pathogens Legionella micdadei and Legionella pneumophila are the two most common Legionella species that cause Legionnaires’ disease. Intracellular replication within pulmonary cells is the hallmark of Legionnaires’ disease. In the environment, legionellae are parasites of protozoans, and intracellular bacterial replication within protozoans plays a major role in the transmission of Legionnaires’ disease. In this study, we characterized the initial host signal transduction mechanisms involved during attachment to and invasion of the protozoan host Hartmannella vermiformis by L. micdadei. Bacterial attachment prior to invasion of H. vermiformis by L. micdadei is associated with tyrosine dephosphorylation of multiple host cell proteins, including a 170-kDa protein. We have previously shown that this 170-kDa protein is the galactose N-acetylgalactosamine (Gal/GalNAc)-inhibitable lectin receptor that mediates attachment to and invasion of H. vermiformis by L. pneumophila. Subsequent bacterial entry targets L. micdadei into a phagosome that is not surrounded by the rough endoplasmic reticulum (RER). In contrast, uptake of L. pneumophila mediated by attachment to the Gal/GalNAc lectin is followed by targeting of the bacterium into an RER-surrounded phagosome. These results indicate that despite similarities in the L. micdadei and L. pneumophila attachment-mediated signal transduction mechanisms in H. vermiformis, the two bacterial species are targeted into morphologically distinct phagosomes in their natural protozoan host.
Along with Streptococcus pneumoniae and Haemophilus influenzae, Legionella species are some of the most common etiologic agents of bacterial pneumonia (10). Legionella pneumophila and Legionella micdadei are the two most common Legionella species that are associated with the majority of outbreaks of Legionnaires’ disease (29). Upon inhalation, both of these Legionella species are phagocytosed by alveolar cells (30). After internalization, both Legionella species replicate within pulmonary cells, and this intracellular replication is believed to be the hallmark of Legionnaires’ disease (15, 22). While L. pneumophila is targeted into a rough endoplasmic reticulum (RER)-surrounded phagosome that does not fuse with lysosomes, L. micdadei is targeted into a RER-free phagosome that is thought to fuse to lysosomes in human monocytes (16). Interestingly, within the RER-surrounded phagosome, L. pneumophila undergoes dramatic alterations in gene expression, which are thought to be required for bacterial adaptation to this intracellular niche (2–4, 6, 7). Gene expression by intracellular L. micdadei has never been examined.
Outbreaks of Legionnaires’ disease occur through droplet transmission of the bacteria from an environmental water source, predominantly air conditioning cooling towers and showerheads (16). In the environment, Legionella species are ubiquitous and are parasites of at least 13 species of amoebae and ciliated protozoans (16). Invasion by and intracellular replication of L. pneumophila in protozoans play major roles in the infectivity and transmission of the Legionnaires’ disease bacteria to humans (16). We have recently shown that L. pneumophila possesses genetic loci that are required for survival and replication within both human macrophages and protozoans, and these loci have been designated pmi (protozoan and macrophage infectivity) loci (19, 20). However, there are other distinct bacterial loci that are uniquely required for intracellular replication within macrophages but not within protozoans, and these loci have been designated mil (macrophage-specific infectivity) loci (19, 21).
Uptake of L. pneumophila by monocytes occurs by microfilament-dependent coiling and conventional phagocytosis mechanisms, while uptake of L. micdadei occurs only by conventional phagocytosis (30, 35). In contrast to uptake of L. pneumophila by macrophages, entry of the bacterium into the protozoan host Hartmannella vermiformis occurs by different mechanisms and is not inhibited by the microfilament disrupting agent cytochalasin D (5, 20, 21, 24, 27). Recently, we demonstrated the presence of a 170-kDa galactose N-acetylgalactosamine (Gal/GalNAc)-inhibitable lectin in H. vermiformis (24, 34). We have recently shown that type IV pili, which are filamentous appendages expressed on the bacterial surface, are involved in bacterial adherence to mammalian and protozoan cells (32). Whether the type IV pili are the ligands for the Gal/GalNAc lectin is not known.
Most studies have used L. pneumophila as a model pathogen for studying Legionnaires’ disease, and the mechanism of entry of L. micdadei into H. vermiformis and the intracellular environment in which L. micdadei resides within the host cell are not well-defined. Hence, we sought to address these issues, which may be important for understanding the intracellular life cycle and pathogenesis of L. micdadei in its protozoan host, H. vermiformis.
MATERIALS AND METHODS
Bacterial strain and culture.
Virulent strain AA100 of L. pneumophila is a virulent clinical isolate that has been described previously (3). The virulent strain L. micdadei Rivera was obtained from Barry Fields and Janet Purckler, Centers for Disease Control and Prevention (Atlanta, Ga.). The strains were grown on buffered charcoal yeast extract (BCYE) agar plates.
Protozoan culture.
H. vermiformis CDC-19 (= ATCC 50237) has been cloned and grown in axenic culture and has been used as a model organism to study the pathogenesis of L. pneumophila (1, 17). This strain was isolated from a water source during an outbreak of nosocomial Legionnaires’ disease in a hospital in South Dakota (11, 17). The amoebae were maintained in American Type Culture Collection axenic culture medium 1034 supplemented with 10% fetal calf serum at 37°C (17).
Infection protocol.
H. vermiformis was harvested and infected with L. micdadei as previously described (24, 34). Briefly, amoebae were incubated overnight in culture flasks in serum-free axenic medium. Then amoebae were harvested by centrifugation and resuspended in fresh serum-free axenic medium. Aliquots containing 2 × 107 amoebae/ml were infected with 109 L. micdadei cells. After several intervals of coincubation at 37°C, amoebal cell lysates were prepared for immunoblot analysis as described below.
To examine the ability of some sugars to block tyrosine dephosphorylation of amoebal proteins upon contact with L. micdadei, H. vermiformis cells were preincubated prior to infection in serum-free axenic medium in the presence of different sugars. Incubation was for 15 min on ice and was followed by coincubation for 20 min at 37°C. At the end of the coincubation period, H. vermiformis cell lysates were prepared as described below.
Transmission electron microscopy.
Monolayers were infected with L. micdadei or L. pneumophila by using a multiplicity of infection of 100 for 1 h, followed by extensive washing of extracellular bacteria with American Type Culture Collection axenic medium 1034 supplemented with 10% fetal calf serum. Ultrathin sections were prepared as described previously (21). Briefly, infected amoebae were fixed with 3.5% gluteraldehyde and then with 1% OsO4, dehydrated with ethanol, and embedded in Eponate 12 resin (Ted Pella, Redding, Calif.). Ultrathin sections were stained with uranyl acetate and then with lead citrate and were examined with a model H-7000/STEM electron microscope (Hitachi Inc., Tokyo, Japan) at 75 kV.
Preparation of cell lysates and Western blotting.
After incubation of H. vermiformis with L. micdadei, infection was stopped by using cold stop buffer containing 1× phosphate-buffered saline (pH 7.2) and the phosphatase inhibitors NaF (5 mM) and Na3VO4 (1 mM) (Sigma Chemical Co., St. Louis, Mo.) (34). To examine the effects of methylamine on tyrosine phosphorylation, amoebae were pretreated with methylamine, and cells were also infected in the presence of this inhibitor, which blocks entry of the bacteria (see below). The cells were washed three times with stop buffer and pelleted by low-speed centrifugation at 735 × g for 2 min. The supernatant containing bacteria was discarded, and the amoebae were lysed with cold 1% Triton X-100 lysis buffer (20 mM Tris-HCl [pH 7.6], 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 2 μg of aprotinin per ml, 2 μg of leupeptin per ml) for 30 min on ice (34). The detergent-soluble and -insoluble fractions were separated by centrifugation at 16,000 × g for 30 min at 4°C in a microcentrifuge. Proteins from detergent-soluble fractions were resolved on sodium dodecyl sulfate–10% polyacrylamide gel electrophoresis gels under reducing conditions. The blots were transferred onto Immobilon-P membranes (Millipore, Bedford, Mass.) in a transfer cell (Bio-Rad, Hercules, Calif.) for 1.5 h with 0.2 M Tris–0.025 M glycine buffer containing 20% (vol/vol) methanol. After the proteins were transferred, the membranes were incubated for 30 min in a blocking buffer containing 1.5% bovine serum albumin. The membranes were then probed with a 1:5,000 dilution of horseradish peroxidase-conjugated recombinant anti-phosphotyrosine antibody RC20H (Transduction Laboratories, Lexington, Ky.) for 30 min at room temperature. After extensive washing, the blots were developed with an enhanced chemiluminescence kit (DuPont NEN, Boston, Mass.) according to the manufacturer’s instructions.
Inhibition of L. pneumophila uptake by sugars.
H. vermiformis was infected with L. micdadei as described below (23). To analyze the effects of different sugars on invasion of H. vermiformis by L. micdadei, infection experiments were performed in triplicate in the presence of the following sugars: galactose (Gal), N-acetyl-d-galactosamine (GalNAc), glucose, mannose, and lactose (Sigma). Solutions of these sugars were prepared in assay medium and stored at 4°C (1).
The effects of sugars on invasion of H. vermiformis by L. micdadei were studied by examining the growth kinetics of L. micdadei in H. vermiformis in the presence of sugars. H. vermiformis cells were preincubated for 15 min with various sugars at a concentration of 100 mM prior to infection with L. micdadei. At several times after infection (1, 2, 3, 4, and 5 days) amoebae were lysed by adding a mild detergent (0.04% Triton X-100) (20). Lysis of the amoebae was monitored microscopically and was complete within 1 min. This treatment had no effect on the viability of bacteria (data not shown). Dilutions were plated onto BCYE plates for colony enumeration.
Invasion of amoebae in the presence of methylamine.
Uptake of Legionella strains by amoebae was studied by using gentamicin protection invasion assays (24, 31). Briefly, H. vermiformis was resuspended in axenic medium at a concentration of 107 cells/ml. The amoebae were incubated for 1 h in the presence of 100 mM methylamine (Sigma), an inhibitor of receptor-mediated endocytosis, prior to infection (13, 27). Following infection with 109 L. micdadei cells for 1 h, the extracellular bacteria were killed with 50 μg of gentamicin per ml, and the intracellular bacteria were released following lysis of the amoebae with 0.04% Triton X-100. Lysis of the amoebae was monitored microscopically and was complete within 1 min. Different dilutions of bacteria were plated onto BCYE plates for colony enumeration (34).
RESULTS
Tyrosine dephosphorylation of H. vermiformis proteins after infection by L. micdadei.
In this study we examined the changes in host protein tyrosine phosphorylation status during attachment to and invasion of the protozoan host H. vermiformis by L. micdadei. Amoebae were coincubated with L. micdadei for different periods of time, and cell lysates were immunoblotted with anti-phosphotyrosine antibody (Fig. 1). Infection of H. vermiformis with L. micdadei resulted in prominent time-dependent tyrosine dephosphorylation of several host cell proteins, including 200- to 205-, 170-, 150-, 69-, and 55-kDa proteins (Fig. 1, arrowheads).
FIG. 1.

Attachment to and invasion of H. vermiformis by L. micdadei induces reversible tyrosine dephosphorylation of several host cell proteins. H. vermiformis cell extracts were prepared from uninfected cells (lane 1) or cells infected for 1, 5, 15, and 30 min (lanes 2 through 5), subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and probed with anti-phosphotyrosine antibody. Lane 6 contained cell extracts from uninfected cells incubated at 37°C for 30 min. Lane 7 contained extracts from H. vermiformis infected for 30 min; then the extracellular bacteria were washed off and the amoebae were incubated for 15 min. Arrowheads indicate the positions of proteins that were markedly tyrosine dephosphorylated. The second arrowhead from the top indicates the position of the 170-kDa Gal/GalNAc lectin (34). The results are representative of the results of at least two independent experiments. KD, kilodaltons.
In order to further examine the specificity of the initial signaling events, several control experiments were performed (Fig. 2). H. vermiformis cells were coincubated with dead bacteria (Fig. 2, lane 3) or latex beads (lane 4) for 30 min. Neither of these treatments resulted in any detectable changes in tyrosine phosphorylation of proteins in H. vermiformis. In addition, supernatants from L. micdadei cultures (lane 5) or culture supernatants obtained after infection (lane 6) had no effect on the pattern of protein phosphorylation in H. vermiformis. These results indicated that contact with live bacteria is required to induce host tyrosine dephosphorylation events and that the secretory products of L. micdadei were unable to mediate this effect.
FIG. 2.

Tyrosine dephosphorylation of H. vermiformis proteins induced by L. micdadei is specific and requires attachment of the bacteria to the Gal/GalNAc lectin. H. vermiformis cell extracts were prepared from uninfected cells (lane 1) or cells infected for 30 min with live (lane 2) or dead (lane 3) bacteria. Amoebal extracts were also prepared after stimulation with latex beads (lane 4), bacterial culture supernatants (lane 5), or supernatant collected from an infection (lane 6) as additional controls. Lanes 7 through 9 contained host cell extracts prepared from infections in the presence of Gal, GalNAc, and mannose, respectively. The arrowhead indicates the position of the 170-kDa Gal/GalNAc lectin (34). The results are representative of the results of at least two independent experiments. KD, kilodaltons.
L. micdadei-induced protein tyrosine dephosphorylation in H. vermiformis is reversible.
Next, we examined whether attachment to and invasion of H. vermiformis by L. micdadei resulted in a permanent change in the tyrosine phosphorylation status of cellular proteins or whether the alteration in host cell processes was reversible. H. vermiformis was coincubated with L. micdadei for 30 min, and extracellular bacteria were washed away after this incubation period. H. vermiformis cells were subsequently incubated for an additional 15 min, and the patterns of tyrosine-phosphorylated proteins were determined in immunoblots probed with anti-phosphotyrosine antibody. The levels of all of the tyrosine-dephosphorylated proteins returned to the normal levels observed with uninfected cells (data not shown). Overall, these observations indicated that attachment and invasion by L. micdadei resulted in tyrosine dephosphorylation of multiple proteins in H. vermiformis. This bacterium-induced tyrosine dephosphorylation in H. vermiformis was reversible and dependent on the continuous presence of bacteria.
Invasion of H. vermiformis by L. micdadei can be blocked by Gal and GalNAc.
Previous work in our laboratory showed that a 170-kDa β2 integrin-like Gal/GalNAc lectin is present in H. vermiformis (Fig. 2, arrowhead) (34). This lectin mediates attachment of L. pneumophila and undergoes tyrosine dephosphorylation during this process (34). Invasion and intracellular replication of L. pneumophila in H. vermiformis were effectively blocked by the Gal and Gal/GalNAc monomers (34). To examine the functional significance of the 170-kDa lectin of H. vermiformis in the invasion of L. micdadei, blocking experiments were performed in the presence of Gal and Gal/GalNAc. Amoebae were preincubated with Gal or Gal/GalNAc for 15 min prior to infection with L. micdadei (Fig. 3). Mannose, lactose, and glucose were used as negative controls to verify the specificity of the sugars (data not shown). The presence of GalNAc had a dramatic effect on invasion by L. micdadei. Gal had a less dramatic effect but still sufficiently blocked the invasion of bacteria. Mannose, glucose, and lactose had no effect on bacterial invasion, and the number of intracellular bacteria at the end of the infection period in the presence of these sugars was very similar to the number in the untreated control (data not shown). The inhibition of bacterial invasion by Gal and Gal/GalNAc was dose dependent (data not shown), and these sugars were not toxic to H. vermiformis (34). These results indicated that the 170-kDa Gal/GalNAc lectin was involved in attachment to and invasion of H. vermiformis by L. micdadei.
FIG. 3.

Inhibition of invasion of H. vermiformis by L. micdadei in the presence of different sugar monomers. The growth kinetics of L. pneumophila in cocultures with H. vermiformis were determined in the absence or presence of sugars at a concentration of 100 mM. At several times after infection, the numbers of bacteria in the cocultures were determined following growth on agar plates. The bacteria did not replicate extracellularly in the cocultures, and thus the increase in the number of bacteria was due to intracellular replication.
Attachment of L. micdadei to H. vermiformis is required to induce protein tyrosine dephosphorylation of host cell proteins.
We examined whether bacterial attachment to the Gal/GalNAc lectin was required to cause changes in the pattern of protein tyrosine phosphorylation in H. vermiformis. In these experiments, H. vermiformis cells were incubated with L. micdadei in the presence of Gal, Gal/GalNAc, or mannose (Fig. 2, lanes 7 through 9). Incubation of H. vermiformis with these sugars does not alter the profile of tyrosine phosphorylated proteins in resting amoebae (34). H. vermiformis cell lysates were prepared after 20 min of infection and were analyzed by using immunoblots probed with the anti-phosphotyrosine antibody. The presence of Gal (Fig. 2, lane 7) or Gal/GalNAc (Fig. 2, lane 8) completely blocked bacterium-induced tyrosine dephosphorylation of the 170- and 69-kDa proteins (Fig. 2, lanes 7 and 8). These proteins have been identified as the Gal/GalNAc lectin (170 kDa) (34) and paxillin (69 kDa) (unpublished data). As expected, mannose failed to block tyrosine dephosphorylation of proteins in H. vermiformis (Fig. 2, lane 9). These results indicated that contact of L. micdadei with the Gal/GalNAc lectin on H. vermiformis was required to mediate early changes in the tyrosine phosphorylation of host cell proteins.
Invasion of H. vermiformis by L. micdadei is not required to induce protein tyrosine dephosphorylation.
Since bacterial attachment was essential for induction of tyrosine dephosphorylation of amoebal proteins (Fig. 2), we examined whether entry of L. micdadei into H. vermiformis was also required to mediate these effects. Methylamine has been shown to block the entry of L. pneumophila into H. vermiformis (27). We examined whether methylamine blocks invasion by L. micdadei by using the gentamicin protection assay (see above). In this assay, following a period of infection, extracellular bacteria were killed with the antibiotic gentamicin, while intracellular bacteria were protected and subsequently enumerated (34). The data showed that like inhibition of invasion of H. vermiformis by L. pneumophila, invasion by L. micdadei was inhibited 99% in the presence of 100 mM methylamine (data not shown).
Next, we examined whether inhibition of invasion of H. vermiformis by L. micdadei blocked bacterium-induced tyrosine dephosphorylation. Incubation of resting H. vermiformis with methylamine did not affect the pattern of protein tyrosine phosphorylation (Fig. 4, lanes 1 and 2). Interestingly, inhibition of protozoan invasion by L. micdadei did not block bacterium-induced tyrosine dephosphorylation of H. vermiformis proteins. These results indicated that attachment but not entry of L. micdadei was sufficient to induce protein tyrosine dephosphorylation in H. vermiformis.
FIG. 4.
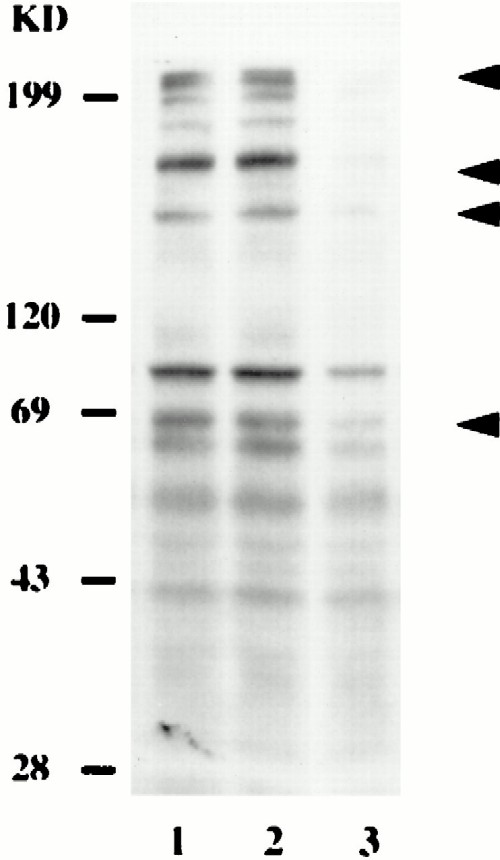
Tyrosine dephosphorylation of H. vermiformis proteins induced by L. micdadei does not require entry of the bacteria. H. vermiformis cell extracts were prepared from uninfected cells in the absence (lane 1) or presence (lane 2) of methylamine. Lane 3 contained cell lysates prepared after 30 min of infection of H. vermiformis with L. micdadei in the presence of methylamine. The arrowheads indicate the positions of the proteins that were tyrosine dephosphorylated. The second arrowhead from the top indicates the position of the 170-kDa Gal/GalNAc lectin (34). The results are representative of the results of at least two independent experiments. KD, kilodaltons.
L. micdadei is surrounded by a smooth phagosome in H. vermiformis.
In H. vermiformis, L. pneumophila is targeted into an RER-surrounded phagosome (1). Transmission electron microscopy showed that after invasion of H. vermiformis by L. micdadei, the bacteria were targeted into RER-free phagosomes (Fig. 5). None of the approximately 100 phagosomes examined that contained L. micdadei was surrounded by RER; in contrast, 83% of the phagosomes containing L. pneumophila were surrounded by RER (1, 20). By 18 h postinfection, the amoebae were heavily infected with both Legionella species. These data showed that despite the fact that L. pneumophila and L. micdadei had similar mechanisms for utilizing the Gal/GalNAc lectin in bacterial attachment and invasion, these two species were targeted into phagosomes that were morphologically distinct, at least at the ultrastructural level.
FIG. 5.

L. micdadei and L. pneumophila are targeted into morphologically distinct phagosomes in H. vermiformis: transmission electron micrographs of H. vermiformis infected with L. micdadei (A and C) or L. pneumophila (B and D). The micrographs were obtained 4 h postinfection (A and B) or 18 h postinfection (C and D). The arrows in panel B indicate the RER-surrounded phagosome. b, bacteria. (A and B) Bars = 0.5 μm. (C and D) Bars = 1 μm.
DISCUSSION
This study focused on the initial signaling events that are activated upon attachment to and invasion of H. vermiformis by the Legionnaires’ disease bacterium L. micdadei. We recently identified the Gal/GalNAc lectin of H. vermiformis as a potential receptor utilized by L. pneumophila for attachment to and invasion of protozoans (34). In this study we examined the role of the Gal/GalNAc lectin in the attachment to and invasion of H. vermiformis by L. micdadei. While entry of several pathogenic bacteria, including Salmonella typhimurium and Shigella flexneri, induces host tyrosine phosphorylation events (18), uptake of L. micdadei by H. vermiformis is associated with rapid and reversible tyrosine dephosphorylation of host cell proteins. These effects of L. micdadei on H. vermiformis are similar to the effects induced by Yersinia pseudotuberculosis during invasion of macrophages, which are mediated by a bacterial tyrosine phosphatase (28). Dephosphorylation of cellular proteins prevents uptake of Yersinia cells (18). In contrast, tyrosine dephosphorylation of H. vermiformis proteins appears to be associated with attachment and uptake of L. micdadei. These observations indicate that there is a novel mechanism for uptake of L. micdadei by H. vermiformis.
The Gal/GalNAc lectin of Entamoeba histolytica is antigenically similar to the human β2 integrin and is involved in cytoskeleton-mediated capping and shedding of antibody and complement from the protozoan surface in order to evade the immune system of the human host (8, 9). Utilization of the Gal/GalNAc lectin on the protozoan host and its tyrosine dephosphorylation upon bacterial attachment and invasion may provide a selective advantage for L. micdadei in the regulation of its uptake process. In contrast to the induction of tyrosine phosphorylation of the receptor and other proteins upon engagement of receptors with their ligands (12), attachment of L. micdadei to the Gal/GalNAc lectin was associated with tyrosine dephosphorylation of the lectin and other host proteins. We offer three hypotheses to explain this observation. First, bacterium-induced tyrosine dephosphorylation of the lectin may disrupt communication of the lectin with the underlying cytoskeleton in order to avoid shedding of bacteria from the protozoan surface. This hypothesis is supported by our recent observations that several lectin-associated proteins are dissociated upon attachment of L. pneumophila to H. vermiformis (unpublished data). Some of these dissociated proteins may represent important cytoskeletal proteins involved in cell shape, motility, and chemotaxis in amoebae. In addition, four protozoan cytoskeletal proteins, including paxillin, vinculin, and pp125FAK, undergo tyrosine dephosphorylation upon attachment to L. pneumophila, indicating that disruption of the cytoskeleton occurs (33). Second, dephosphorylation of H. vermiformis proteins upon attachment to L. micdadei may downregulate host signals in order to prevent activation of phagocytic signals and microbicidal mechanisms that result in degradation of engulfed bacteria. Subversion of the bactericidal protozoan host cell processes by L. micdadei may be supported by observations made with neutrophils (14). Suppression of host cell functions, like superoxide anion production, chemotaxis, and bactericidal activity, by L. micdadei was observed when human neutrophils were allowed to attach to and ingest bacteria (14). Third, dephosphorylation of H. vermiformis proteins, including the Gal/GalNAc lectin, by L. micdadei may increase the efficiency of bacterial uptake. Overall, attachment to and invasion of H. vermiformis by L. micdadei mediated by the Gal/GalNAc lectin may be required to accomplish more than one of the strategies proposed above to ensure efficient uptake and subsequent targeting of the bacteria into a safe phagosome in amoebae.
The Gal/GalNAc lectin of H. vermiformis is important for invasion by L. micdadei, as determined by blocking experiments performed with Gal and Gal/GalNAc. Recent studies in our laboratory have demonstrated the importance of the 170-kDa Gal lectin in the invasion of H. vermiformis by L. pneumophila (24, 34). The sugars Gal and Gal/GalNAc block invasion of H. vermiformis by L. pneumophila (24, 34). It is interesting to note that the two most frequent causative agents of Legionnaires’ disease, L. micdadei and L. pneumophila, utilize a common host cell receptor to invade H. vermiformis.
Intracellular pathogens like L. pneumophila and Mycobacterium tuberculosis utilize the CR3 receptor to enter human macrophages, which prevents activation of oxidative bursts and other microbicidal mechanisms (36, 37). Uptake of many intracellular pathogens through the Fc receptor (instead of CR3) targets the internalized bacteria into lysosomes instead of a replicative niche (25, 26). Although L. pneumophila and L. micdadei utilize the Gal/GalNAc lectin for attachment and invasion of their protozoan host, H. vermiformis, the intracellular fates of the two bacterial species are different, at least at the ultrastructural level. Accordingly, a phagosome that contains L. pneumophila but not L. micdadei is surrounded by RER. These observations are similar to observations made with a phagosome containing both of these Legionella species in macrophages and epithelial cells (unpublished data). These results suggest that additional mechanisms (other than utilizing the Gal/GalNAc lectin) may be involved in the trafficking L. micdadei to the RER-free replicative phagosome. It is possible that L. micdadei resides in a relatively nascent phagosome, while L. pneumophila exists in a more mature phagosome that is surrounded by RER.
In summary, this study and our earlier report (34) revealed novel but similar signaling mechanisms that are triggered in the protozoan host H. vermiformis following attachment and invasion by the two Legionella species that have been most commonly implicated in outbreaks of Legionnaires’ disease. Despite the similarities in what appears to be receptor-mediated signaling events, the two species are targeted into different compartments in the protozoan host. Additional studies should help explain the exact strategies utilized by Legionella spp. to enter and invade two evolutionarily distant hosts, human cells and environmental free-living amoebae.
ACKNOWLEDGMENTS
Y.A. was supported by Public Health Service award R29AI38410. O.S.H. was supported by predoctoral National Research Service award 5T32CA09509.
REFERENCES
- 1.Abu Kwaik Y. The phagosome containing Legionella pneumophila within the protozoan Hartmanella vermiformis is surrounded by the rough endoplasmic reticulum. Appl Environ Microbiol. 1996;62:2022–2028. doi: 10.1128/aem.62.6.2022-2028.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abu Kwaik Y. Induced expression of the Legionella pneumophila gene encoding a 20-kilodalton protein during intracellular infection. Infect Immun. 1998;66:203–212. doi: 10.1128/iai.66.1.203-212.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu Kwaik Y, Eisenstein B I, Engleberg N C. Phenotypic modulation by Legionella pneumophila upon infection of macrophages. Infect Immun. 1993;61:1320–1329. doi: 10.1128/iai.61.4.1320-1329.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abu Kwaik Y, Engleberg N C. Cloning and molecular characterization of a Legionella pneumophila gene induced by intracellular infection and by various in vitro stress stimuli. Mol Microbiol. 1994;13:243–251. doi: 10.1111/j.1365-2958.1994.tb00419.x. [DOI] [PubMed] [Google Scholar]
- 5.Abu Kwaik Y, Fields B S, Engleberg N C. Protein expression by the protozoan Hartmannella vermiformis upon contact with its bacterial parasite, Legionella pneumophila. Infect Immun. 1994;62:1860–1866. doi: 10.1128/iai.62.5.1860-1866.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abu Kwaik Y, Gao L-Y, Harb O S, Stone B J. Transcriptional regulation of the macrophage-induced gene (gspA) of Legionella pneumophila and phenotypic characterization of a null mutant. Mol Microbiol. 1997;24:629–642. doi: 10.1046/j.1365-2958.1997.3661739.x. [DOI] [PubMed] [Google Scholar]
- 7.Abu Kwaik Y, Pederson L L. The use of differential display-PCR to isolate and characterize a Legionella pneumophila locus induced during the intracellular infection of macrophages. Mol Microbiol. 1996;21:543–556. doi: 10.1111/j.1365-2958.1996.tb02563.x. [DOI] [PubMed] [Google Scholar]
- 8.Adams S A, Robson S C, Gathiram V, Jackson T F H G, Pillay T S, Kirsch R E, Makgoba M W. Immunological similarity between the 170 kDa amoebic adherence glycoprotein and human β2 integrins. Lancet. 1993;341:17–19. doi: 10.1016/0140-6736(93)92483-a. [DOI] [PubMed] [Google Scholar]
- 9.Arhets P, Gounon P, Sansonetti P, Guillen N. Myosin II is involved in capping and uroid formation in the human pathogen Entamoeba histolytica. Infect Immun. 1995;63:4358–4367. doi: 10.1128/iai.63.11.4358-4367.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bates J H, Campbell G D, Baron A L. Microbial etiology of acute pneumonia in hospitalized patients. Chest. 1992;101:1005–1012. doi: 10.1378/chest.101.4.1005. [DOI] [PubMed] [Google Scholar]
- 11.Breiman R F, Fields B S, Sanden G N, Volmer L, Meier A, Spika J S. Association of shower use with Legionnaires’ disease: possible role of amoebae. JAMA. 1990;263:2924–2926. [PubMed] [Google Scholar]
- 12.Clark E A, Brugge J S. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- 13.Davies P J A, Davies D R, Levitzki A, Maxfield F R, Milhaud P, Willingham M C, Pastan I H. Transglutaminase is essential in receptor-mediated endocytosis of α2-macroglobulin and polypeptide hormones. Nature. 1980;283:162–167. doi: 10.1038/283162a0. [DOI] [PubMed] [Google Scholar]
- 14.Donowitz G R, Readon I, Dowling J, Rubin L, Focht D. Ingestion of Legionella micdadei inhibits human neutrophil function. Infect Immun. 1990;58:3307–3311. doi: 10.1128/iai.58.10.3307-3311.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dowling J N, Saha A K, Glew R H. Virulence factors of the family Legionellaceae. Microbiol Rev. 1992;56:32–60. doi: 10.1128/mr.56.1.32-60.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fields B S. The molecular ecology of legionellae. Trends Microbiol. 1996;4:286–290. doi: 10.1016/0966-842x(96)10041-x. [DOI] [PubMed] [Google Scholar]
- 17.Fields B S, Nerad T A, Sawyer T K, King C H, Barbaree J M, Martin W T, Morrill W E, Sanden G N. Characterization of an axenic strain of Hartmannella vermiformis obtained from an investigation of nosocomial legionellosis. J Protozool. 1990;37:581–583. doi: 10.1111/j.1550-7408.1990.tb01269.x. [DOI] [PubMed] [Google Scholar]
- 18.Finlay B B, Cossart P. Exploitation of mammalian host cell functions by bacterial pathogens. Science. 1997;276:718–725. doi: 10.1126/science.276.5313.718. [DOI] [PubMed] [Google Scholar]
- 19.Gao, L.-Y., M. Gutzman, J. K. Brieland, and Y. Abu Kwaik. Different fates of Legionella pneumophila pmi and mil mutants within human-derived macrophages and alveolar epithelial cells. Submitted for publication. [DOI] [PubMed]
- 20.Gao L-Y, Harb O S, Abu Kwaik Y. Utilization of similar mechanisms by Legionella pneumophila to parasitize two evolutionarily distant hosts, mammalian and protozoan cells. Infect Immun. 1997;65:4738–4746. doi: 10.1128/iai.65.11.4738-4746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao L-Y, Harb O S, Abu Kwaik Y. Identification of macrophage-specific infectivity loci (mil) of Legionella pneumophila that are not required for infectivity of protozoa. Infect Immun. 1998;66:883–892. doi: 10.1128/iai.66.3.883-892.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gress F M, Myerowitz R L, Pasculle A W, Rinaldo C R, Jr, Dowling J N. The ultrastructural morphologic features of Pittsburgh pneumonia agent. Am J Pathol. 1980;101:63–78. [PMC free article] [PubMed] [Google Scholar]
- 23.Harb O S, Abu Kwaik Y. Identification of the aspartate-β-semiadehyde dehydrogenase gene of Legionella pneumophila and characterization of a null mutant. Infect Immun. 1998;66:1898–1903. doi: 10.1128/iai.66.5.1898-1903.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harb O S, Venkataraman C, Haack B J, Gao L-Y, Abu Kwaik Y. Heterogeneity in the attachment and uptake mechanisms of the Legionnaires’ disease bacterium, Legionella pneumophila, by protozoan hosts. Appl Environ Microbiol. 1998;64:126–132. doi: 10.1128/aem.64.1.126-132.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horwitz M A. The Legionnaires’ disease bacterium (Legionella pneumophila) inhibits phagosome-lysosome fusion in human monocytes. J Exp Med. 1983;158:2108–2126. doi: 10.1084/jem.158.6.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joiner K A, Fuhrman S A, Miettinen H M, Kasper L H, Mellman I. Toxoplasma gondii: fusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- 27.King C H, Fields B S, Shotts E B, Jr, White E H. Effects of cytochalasin D and methylamine on intracellular growth of Legionella pneumophila in amoebae and human monocyte-like cells. Infect Immun. 1991;59:758–763. doi: 10.1128/iai.59.3.758-763.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mecsas J, Strauss E J. Molecular mechanisms of bacterial virulence: type III secretion and pathogenicity islands. Emerg Infect Dis. 1996;2:271–287. doi: 10.3201/eid0204.960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pasculle A W, Feeley J C, Gibson R J, Cordes L G, Myerowitz R L, Patton C M, Gorman G W, Carmack C L, Ezzell J W, Dowling J N. Pittsburgh pneumonia agent: direct isolation from human lung tissue. J Infect Dis. 1980;141:727–732. doi: 10.1093/infdis/141.6.727. [DOI] [PubMed] [Google Scholar]
- 30.Rechnitzer C, Blom J. Engulfment of the Philadelphia strain of Legionella pneumophila within pseudopod coils in human phagocytes. Comparison with the other Legionella strains and species. Acta Pathol Microbiol Immunol Scand Sect B. 1989;97:105–114. [PubMed] [Google Scholar]
- 31.Sadosky A B, Wiater L A, Shuman H A. Identification of Legionella pneumophila genes required for growth within and killing of human macrophages. Infect Immun. 1993;61:5361–5373. doi: 10.1128/iai.61.12.5361-5373.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone B J, Abu Kwaik Y. Expression of multiple pili by Legionella pneumophila: identification and characterization of a type IV pili gene and its role in adherence to mammalian and protozoan cells. Infect Immun. 1998;66:1768–1775. doi: 10.1128/iai.66.4.1768-1775.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Venkataraman C, Gao L-Y, Bondada S, Abu Kwaik Y. Identification of putative cytoskeletal protein homologues in the protozoan Hartmannella vermiformis as substrates for induced tyrosine phosphatase activity upon attachment to the Legionnaires’ disease bacterium, Legionella pneumophila. J Exp Med. 1998;188:1–10. doi: 10.1084/jem.188.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Venkataraman C, Haack B J, Bondada S, Abu Kwaik Y. Identification of a Gal/GalNAc lectin in the protozoan Hartmannella vermiformis as a potential receptor for attachment and invasion by the Legionnaires’ disease bacterium, Legionella pneumophila. J Exp Med. 1997;186:537–547. doi: 10.1084/jem.186.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinbaum D L, Benner R R, Dowling J N, Alpern A, Pasculle A W, Donowitz G R. Interaction of Legionella micdadei with human monocytes. Infect Immun. 1984;46:68–73. doi: 10.1128/iai.46.1.68-73.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilson C B, Tsai V, Remington J S. Failure to trigger the oxidative burst of normal macrophages. Possible mechanisms for survival of intracellular pathogens. J Exp Med. 1980;151:328–346. doi: 10.1084/jem.151.2.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright S D, Silverstein S C. Receptors for C3b and C3bi promote phagocytosis but not the release of toxic oxygen from human phagocytes. J Exp Med. 1983;158:2016–2023. doi: 10.1084/jem.158.6.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]