

Engineered neuronal microtissue with bundled axons was grown, controlled using light, and implanted as a living neural interface.
Abstract
For implantable neural interfaces, functional/clinical outcomes are challenged by limitations in specificity and stability of inorganic microelectrodes. A biological intermediary between microelectrical devices and the brain may improve specificity and longevity through (i) natural synaptic integration with deep neural circuitry, (ii) accessibility on the brain surface, and (iii) optogenetic manipulation for targeted, light-based readout/control. Accordingly, we have developed implantable “living electrodes,” living cortical neurons, and axonal tracts protected within soft hydrogel cylinders, for optobiological monitoring/modulation of brain activity. Here, we demonstrate fabrication, rapid axonal outgrowth, reproducible cytoarchitecture, and simultaneous optical stimulation and recording of these tissue engineered constructs in vitro. We also present their transplantation, survival, integration, and optical recording in rat cortex as an in vivo proof of concept for this neural interface paradigm. The creation and characterization of these functional, optically controllable living electrodes are critical steps in developing a new class of optobiological tools for neural interfacing.
INTRODUCTION
Most devices for neuromodulation (e.g., deep brain stimulation electrodes for Parkinson’s disease) and neural recording [commonly called brain-computer interfaces (BCIs)] work by electrically stimulating or capturing neuronal activity within the brain (1). These neural interfaces have been developed across a range of medical applications; two notable milestones include cochlear prostheses for people with hearing loss and thought-driven computer control for people with neuromuscular disorders (1). Despite these achievements, the clinical impact of more advanced neural interfaces is beset by several underlying functional challenges. Implantable BCIs primarily use inorganic microelectrodes, which often exhibit diminished recording quality over time due to a host of biological factors (e.g., inflammation, neuronal loss, and glial scarring) and abiotic biostability issues (including decreasing impedance due to loss of insulation and mechanical failure) (1–5). In parallel, the effectiveness of electrical neuromodulation is limited by an inability to target specific neurons or neuronal subtypes (e.g., excitatory versus inhibitory neurons) within the volume of charge injection, as well as the thresholds for both safe and functional therapeutic stimulation (6). Specificity in neuromodulation may be improved with optogenetics, where inducing the expression of light-sensitive proteins in specific neuronal populations allows these subgroups to be controlled on a wavelength-specific basis through photostimulation. However, the longevity and immune response to viral optogenetic transduction in humans is currently unknown, with nonhuman primate studies suggesting an elevated immunogenic response (7). Further, light scattering properties of tissue block precise photostimulation of neurons more than a few hundred microns deep (8). Deeper tissue is accessible with implantable optical fibers, lenses, or micro–LEDs (light-emitting diodes), yet chronic performance must also address complications from the foreign body response to these materials and overheating of surrounding tissue (9, 10). Across electric and/or optical input-output paradigms, the information transfer bandwidth limits the quality of the neural interface. The ability to address these design challenges—compatibility with the brain, target specificity, and long-term stability—will direct the utility and clinical translation of future neuromodulation and neural recording technologies.
We are advancing a potential solution to these limitations through our existing platform, the microtissue engineered neural network (μTENN). μTENNs are discrete population(s) of neurons connected by long bundles of axons protected within a microscopic hydrogel cylinder (“microcolumn”) (Fig. 1) (11, 12). μTENNs were originally developed to reconstruct lost or injured neuroanatomy by approximating the brain’s network-level structure—locally connected neurons spanned by dense axonal tracts—and previously demonstrated neuronal survival, maintenance of axonal architecture, and synaptic integration with host neurons following cortical microinjection in rats (11–14). Experimental evidence and computational network analyses using fluorescent calcium imaging have already demonstrated that bidirectional μTENNs exhibit functional connectivity and the potential for directed information transfer by at least 10 days in vitro (DIV) (15). Here, we advance the development of μTENNs into a putative “living electrode,” that is, a self-contained, implantable, axon- and synapse-based conduit for optically controlled neuronal activity and information transfer to/from the brain. In this approach, the μTENN is implanted at a predetermined depth to form synapses with local neural circuitry and propagate information along μTENN axons to/from an externalized apparatus at the brain surface (Fig. 1, I and J). Transduction of the μTENN neurons to express optogenetic proteins before implant would thus enable light-driven neuromodulation (through photostimulation of the μTENN neurons to influence downstream cortical activity) or monitoring (by recording μTENN neurons as a representation of multiple cortical synaptic inputs) (Fig. 1, I and J).
Fig. 1. Aggregate μTENN fabrication and living electrode concept.
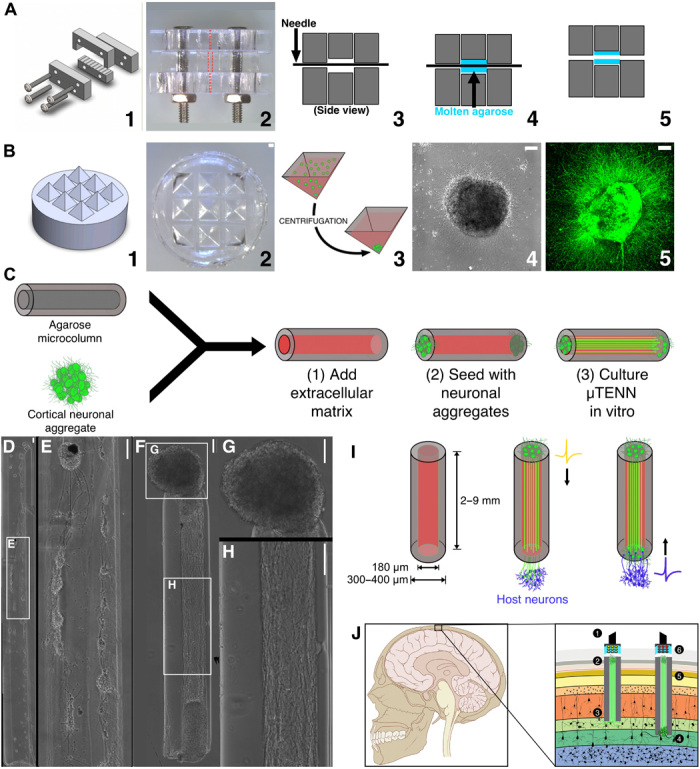
μTENNs comprise a hydrogel microcolumn, living neuronal aggregates, and an extracellular matrix lumen. (A) 1: A customizable acrylic mold for generating microcolumns. 2: Top view of the mold dashed lines indicate the outer diameter (OD; middle) and the inner diameter (ID; top and bottom). 3: Needles of the desired inner diameter are inserted into the mold. 4: Microcolumns are cast in agarose (blue). 5: Microcolumns are removed after needle removal and mold disassembly. (B) 1: A three-dimensional (3D) printed mold for square pyramidal wells. 2: Pyramidal wells cast in polydimethylsiloxane (PDMS). 3: Dissociated neurons (green) centrifuged in the wells to form spheroidal aggregates. 4: Phase image of an aggregate 24 hours after plating. 5: Confocal reconstruction of aggregate at 72 hours, labeled with green fluorescent protein (GFP). (C) Microcolumns (gray) are filled with an extracellular collagen-laminin matrix (red). Neuronal aggregates are then placed at the microcolumn terminal(s) and grown in vitro. (D) Early-generation μTENNs fabricated with dissociated neurons yielded limited control over the final network structure (E). Aggregate μTENNs (F) exhibit robust axonal growth and more controllable architecture, with discrete regions of cell bodies (G) and neuritic projections (H). (I) Left: Current μTENN dimensions for implantable living electrodes. Middle: Unidirectional μTENNs synapse host neurons (purple) to relay external inputs to targeted cortical regions. Right: Host neurons synapse bidirectional μTENNs, relaying activity from host cortex for monitoring via the dorsal aggregate. (J) Optogenetically active μTENNs as transplantable input/output channels. Inputs: An LED array (1) optically stimulates a unidirectional, channelrhodopsin-positive μTENN (2) to activate layer IV neurons (3). Outputs: Layer V neurons (4) synapse a bidirectional μTENN (5); relayed neuronal activity is recorded by a photodiode array on the brain surface (6). Scale bars, 100 μm. Photo credit: Dayo O. Adewole, University of Pennsylvania.
This living electrode approach as described may address multiple functional challenges in current neuromodulation and neural recording strategies. As engineered neural microtissue, living electrodes provide a natural biological substrate for signal transfer between the neural target and the brain surface through synapse formation with host neurons. Further, the wholly organic μTENN may ameliorate the chronic foreign body response as any inorganic materials (e.g., photodiode arrays) are relegated to the brain surface, potentially improving long-term stability (Fig. 1J). Combined with the targeting specificity of optogenetic neuromodulation, light-driven living electrodes could provide access to deep neuronal circuitry without the light penetration limitations or potential viral delivery risks of existing optogenetic approaches (as viral transduction is restricted to μTENN neurons before implant).
Motivated by these potential benefits, the present work details the biofabrication and characterization of a novel optically controlled living electrode approach to neural interface. Here, we present the following advancements in μTENN technology as a medium for neural interface: (i) the fabrication of next-generation, neuronal aggregate-based unidirectional (single-aggregate) and bidirectional (dual-aggregate) μTENNs; (ii) characterization of their growth and viability across multiple construct parameters; (iii) validation of their network formation and maturation over time; (iv) optical control and monitoring of optogenetically active μTENNs in vitro; (v) implantation and optical readout of aggregate-based μTENNs in rodent cortex as proof of concept for living electrodes; and (vi) histological evidence of a modest gliotic response to μTENNs, as well as evidence of neuronal survival, neurite outgrowth, and synaptogenesis with host. Through the successful creation and validation of implantable, optically controlled, and functional neuron- and axon-based microtissue, these developments represent a notable milestone toward the long-term goal of achieving a biologically based neural interface that offers improved selectivity and longevity compared to alternative nonorganic approaches.
MATERIALS AND METHODS
All procedures were approved by the Institutional Animal Care and Use Committees at the University of Pennsylvania and the Michael J. Crescenz Veterans Affairs Medical Center and adhered to the guidelines set forth in the National Institutes of Health Public Health Service Policy on Humane Care and Use of Laboratory Animals (2015).
Cortical neuron isolation and culture
Neural cell isolation and culture protocols are similar to that of published work (11, 12, 16). Briefly, timed-pregnant rats were euthanized, and the uterus was removed. Embryonic day 18 fetuses were transferred from the uterus to cold Hanks’ balanced salt solution (HBSS), wherein the brains were extracted and the cerebral cortical hemispheres were isolated under a stereoscope via microdissection. Cortical tissue was dissociated in 0.25% trypsin + 1 mM EDTA at 37°C for 10 to 12 min, after which the trypsin/EDTA was removed and replaced with deoxyribonuclease (DNase) (0.15 mg/ml) in HBSS. Dissociated tissue + DNase were centrifuged for 3 min at 3000 rpm before the DNase was removed and the cells were resuspended in serum-free neuronal culture media, composed of Neurobasal + B27 + Glutamax (Thermo Fisher Scientific) and 1% penicillin-streptomycin.
μTENN fabrication
μTENNs were constructed in a three-phase process (Fig. 1, A to C). First, agarose microcolumns of a specified geometry [outer diameter (OD), inner diameter (ID), and length] were formed in a custom-designed acrylic mold as described in earlier work (Fig. 1A) (13). The mold is an array of cylindrical channels that allow for the insertion of acupuncture needles (Seirin, Weymouth, MA) to form a void with the annular dimensions (OD × ID) of the microcolumn once assembled. The mold has been fabricated with more precise machining equipment relative to earlier work to improve coaxial alignment of the needles and channels. Molten agarose in Dulbecco’s phosphate-buffered saline (DPBS) was poured into the mold-needle assembly and allowed to cool (agarose, 3% weight/volume). Once the agarose solidified, the needles were removed and the mold was disassembled, yielding hollow agarose microcolumns with a specific OD equal to the size of the channels and ID equal to the OD of the needles. Microcolumns were sterilized via ultraviolet light for 30 min and stored in DPBS to prevent dehydration until needed. For these studies, the mold channels were 398 μm in diameter and the acupuncture needles were 180 μm, resulting in microcolumns with a 398-μm OD and a 180-μm ID. Microcolumns were cut to the desired length for each cohort as described below.
Next, primary cortical neurons were forced into spheroidal aggregates (Fig. 1C). Early-generation μTENNs were created by seeding microcolumns with dissociated primary cortical neurons, resulting in random clustering and multidirectional axonal growth (Fig. 1, D and E). The formation of neuronal aggregates before seeding provides more precise control over μTENN architecture, enabling the growth of long axonal fascicles spanning the length of the microcolumn (Fig. 1, F to H). To create aggregates, dissociated cortical neurons were suspended at a density of 1.0 million to 2.0 million cells/ml and transferred to an array of inverted pyramidal wells made in polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning) cast from a custom-designed, three-dimensional (3D) printed mold (Fig. 1B). Neuron suspensions were then centrifuged in the wells at 200g for 5 min before being incubated overnight at 37°C, 5% CO2. This centrifugation resulted in forced aggregation of neurons with precise control of the number of neurons per aggregate/sphere (12 μl of cell suspension per well). Pyramidal wells and forced aggregation protocols were adapted from Ungrin et al. (17).
Last, microcolumns were removed from DPBS and excess DPBS were removed from the microcolumn channels via micropipette. Microcolumns were then filled with extracellular matrix (ECM) composed of rat tail collagen (1.0 mg/ml) + mouse laminin (1.0 mg/ml) (Reagent Proteins, San Diego, CA) (Fig. 1C). Unidirectional or bidirectional μTENNs were seeded by carefully placing an aggregate at one (unidirectional) or both (bidirectional) ends of a microcolumn using fine forceps under a stereoscope and confirming aggregate adherence for 45 min at 37°C, 5% CO2. Early-generation dissociated μTENNs were fabricated by transferring dissociated cortical neurons into the ECM-filled microcolumn via micropipette as detailed in prior work (11, 12). All μTENNs were grown in neuronal culture media with fresh media replacements every 2 DIV.
Growth characterization
Several groups of aggregate-based μTENNs were fabricated to assess growth across construct polarities (unidirectional or bidirectional) and lengths (from 2 to 9 mm). In addition, early-generation dissociated μTENNs were generated to compare growth of aggregate-based μTENNs to those generated using that previous methodology. In total, eight experimental groups were generated: dissociated/2 mm long (LEDISS,2) (n = 7), unidirectional/2 mm long (LEUNI,2) (n = 6), unidirectional/5 mm long (LEUNI,5) (n = 3), bidirectional/2 mm long (LEBI,2) (n = 15), bidirectional/3 mm long (LEBI,3) (n = 12), bidirectional/5 mm long (LEBI,5) (n = 17), bidirectional/7 mm long (LEBI,7) (n = 8), and bidirectional/9 mm long (LEBI,9) (n = 3). Low sample size for LEBI,9 was due to cessation of neurite outgrowth before 10 DIV without network formation (i.e., not forming interaggregate connections).
Phase-contrast microscopy images of μTENNs in culture were taken at 1, 3, 5, 8, and 10 DIV at ×10 magnification using a Nikon Eclipse Ti-S microscope, paired with a QIClick camera and NIS Elements BR 4.13.00. Growth rates for each group at specific time points were quantified as the change in the length of the longest identifiable neurite divided by the number of days between the current and preceding time point. The longest neurites were manually identified within each phase image using ImageJ (National Institutes of Health, MD), and length was measured from the edge of the source aggregate to the neurite tip. To standardize measurements, the edge of the source aggregate identified at 1 DIV was used as the reference point across subsequent time points. Growth was measured until axons crossed the length of the column (for unidirectional constructs) or axons crossed the distance between aggregates (for bidirectional constructs). Growth rates were averaged for each time point, with the average maximum and minimum growth rates and average crossing time compared across aggregate-based μTENNs with one-way analysis of variance (ANOVA). Post hoc analyses were performed where necessary with Bonferroni-corrected pairwise comparisons. For reference, planar cultures of cortical neurons (n = 10) were grown in parallel with μTENN cultures, with the longest identifiable neurites measured at 1, 3, and 5 DIV. Single neurites could not be identified at later time points due to culture maturation. Axonal outgrowth in planar cultures was taken as the average growth rate across time points, which were compared via unpaired t test.
To identify aggregate-specific growth across the microcolumns, cortical neuronal aggregates were labeled with either green fluorescent protein (GFP) or the red fluorescent protein mCherry via adeno-associated virus 1 (AAV1) transduction (Penn Vector Core, Philadelphia, PA). Briefly, after centrifuging aggregates in the pyramid wells, 1 μl of AAV1 packaged with the human synapsin-1 promoter was added to the aggregate wells (final titer, ~3 × 1010 genomic copies/ml). Aggregates were incubated at 37°C, 5% CO2 overnight before the medium was replaced twice, after which transduced aggregates were plated in microcolumns as described above, each with one GFP+ and one mCherry+ aggregate (n = 6). Over multiple DIV, images of the μTENNs were taken on a Nikon A1RSI laser scanning confocal microscope paired with NIS Elements AR 4.50.00. Sequential slices of 10 to 20 μm in the z plane were acquired for each fluorescent channel. All confocal images presented are maximum intensity projections of the confocal z-slices.
Viability assessment
To assess neuronal viability in both newly formed and mature networks, unidirectional/5 mm (LEUNI) and bidirectional/5 mm (LEBI) μTENNs were grown to 10 (immature) and 28 (mature) DIV before staining with a calcein-AM/ethidium homodimer-1 (EthD-1) assay (Thermo Fisher Scientific). Metabolically active cells convert the membrane-permeable calcein-AM to calcein, which fluoresces green (λexc, ~495 nm; λem, ~515 nm), while EthD-1 enters membrane-compromised cells and fluoresces red upon binding to nucleic acids (λexc, ~495 nm; λem, ~635 nm). Age-matched, 2D cortical cultures were plated on polystyrene and stained to identify differences in survival due to the aggregate culture method.
Briefly, cultures were gently rinsed in DPBS at the selected time points. A solution of calcein-AM (1:2000 dilution; final concentration of ~2 μM) and ethidium homodimer-1 (1:500; ~4 μM) in DPBS was added to each culture, followed by incubation at 37°C, 5% CO2 for 30 min. Following incubation, cultures were rinsed twice in fresh DPBS and imaged at ×10 magnification on a Nikon A1RSI laser scanning confocal microscope paired with NIS Elements AR 4.50.00. Viability was quantified as the ratio of the total area of calcein-AM+ cells to the total area of both calcein-AM+ and ethidium-homodimer+ cells using ImageJ (National Institutes of Health, MD). Sample sizes for each group were as follows: LEUNI,5mm (n = 4, 4); LEBI,5mm (n = 7, 4); planar cultures (n = 9, 5) for 10 and 28 DIV, respectively.
Optical stimulation, calcium imaging, and optical recording analysis
As proof-of-concept validation for the optically controlled living electrode approach, we established an “all-optical” paradigm enabling the simultaneous optogenetic control and optical monitoring of aggregate-based μTENNs in vitro. Cortical neuronal aggregates were transduced with either channelrhodopsin-2 (ChR2) for light-based neuronal activation (“input”) or the genetically encoded fluorescent calcium reporter RCaMP1b for optical readout of neuronal activity (“output”) via AAV1 transduction as described above (Penn Vector Core). Five- to 6-mm-long bidirectional μTENNs were then plated with one input/ChR2+ aggregate and one output/RCaMP1b+ aggregate (n = 5). ChR2 and RCaMP have been investigated and used for all-optical electrophysiology in vitro with minimal spectral overlap, reducing the likelihood of false-positive responses in RCaMP+ neurons due to photostimulation of ChR2+ neurons (18). At 10 DIV, μTENNs were stimulated via an LED optical fiber positioned approximately 1 to 3 mm above the input aggregate, such that the entire aggregate was illuminated. A Plexon Optogenetic Stimulation System with LED modules for each desired wavelength was used to stimulate the μTENNs (Plexon Inc.). Stimulation consisted of a train of 10 100-ms pulses (1 Hz) at 465 nm, within the excitation spectra of ChR2. Each train was repeated three times for a given LED current amplitude (50, 100, 200, 250, 300 mA); amplitudes corresponded to approximate stimulation intensities of 106, 211, 423, 528, and 634 mW/mm2 from the tip of the optical fiber and 4.7, 9.3, 18.7, 23.3, and 28.0 mW/mm2 at the aggregate, respectively. As a control, μTENNs were stimulated as above at 620 nm (outside of the excitation spectra of ChR2) at 300 mA/28.0 mW/mm2. Recordings of the μTENNs’ output aggregates were acquired at 25 to 30 frames per second on a Nikon Eclipse Ti microscope paired with an ANDOR Neo/Zyla camera and Nikon Elements AR 4.50.00 (Nikon Instruments).
Following optical stimulation and/or recording, calcium imaging acquisitions were manually reviewed against phase images of the same μTENNs to identify regions of interest (ROIs) containing neurons and background ROIs. Neuronal ROIs were identified as areas clearly containing one or more neuronal cell bodies that exhibited repeated synchronous changes in pixel intensity, although because of the dense packing of the aggregate, these ROIs also contained axons. Background ROIs were empty square areas containing no neurons (i.e., only culture media). The mean pixel intensities for each ROI were imported into MATLAB for further analysis via custom scripts (MathWorks Inc.). Within MATLAB, the background ROI intensity for each recording was subtracted from active ROIs. Ten such ROIs were randomly selected and averaged to obtain a representative fluorescence intensity trace across each output aggregate. Subsequently, the percent change in fluorescence intensity over time (∆F/Fo) was calculated for each mean ROI, where ∆F equals (FT − Fo), FT is the mean ROI fluorescent intensity at time T, and Fo is the average of the lower half of the preceding intensity values within a predetermined sampling window (19). The peak ∆F/Fo for each train was averaged per μTENN for each of the given stimulation intensities. The average maximum ∆F/Fo was then compared across stimulation intensities with a one-way ANOVA, with post hoc analysis performed where necessary with the Tukey procedure. In addition, the peak ∆F/Fo of the output aggregate under 620-nm stimulation was used as a control (620 nm being outside of ChR2’s activation spectra) and compared to that under 465-nm stimulation at 300 mA/28 mW/mm2 using an unpaired t test.
Immunocytochemistry
To determine whether aggregate-based μTENNs matured and attained the desired network architecture over time, μTENNs were grown and fixed in 4% formaldehyde for 35 min at 4, 10, and 28 DIV (n = 6, 4, and 8, respectively). μTENNs were then rinsed in 1× PBS and permeabilized with 0.3% Triton X-100 + 4% horse serum in PBS for 60 min before being incubated with primary antibodies overnight at 4°C. Primary antibodies were Tuj-1/β-tubulin III (1:500; T8578, Sigma-Aldrich) to label axons and synapsin-1 (1:500; A6442, Invitrogen) to label presynaptic specializations. Following primary antibody incubation, μTENNs were rinsed in PBS and incubated with fluorescently labeled secondary antibodies (1:500; sourced from Life Technologies and Invitrogen) for 2 hours at 18° to 24°C. Last, Hoechst (1:10,000; 33342, Thermo Fisher Scientific) was added for 10 min at 18° to 24°C before rinsing in PBS. μTENNs were imaged on a Nikon A1RSI laser scanning confocal microscope paired with NIS Elements AR 4.50.00. Sequential slices of 10 to 20 μm in the z plane were acquired for each fluorescent channel. All confocal images presented are maximum intensity projections of the confocal z-slices.
Cortical implantation and intravital calcium imaging
As proof-of-concept validation for optical living electrodes in vivo, bidirectional, approximately 1.5-mm-long (n = 6) or 5.5-mm-long (n = 4) μTENNs expressing GCaMP were delivered into the brain via stereotaxic microinjection using similar methodology to that described in prior work (11, 12). Male Sprague-Dawley rats weighing 325 to 350 g were anesthetized with isoflurane at 1.0 to 2.0 liters/min (induction, 5.0%; maintenance, 1.5-2.0%) and mounted in a stereotactic frame. Meloxicam (2.0 mg/kg) and bupivacaine (2.0 mg/kg) were given subcutaneously at the base of the neck and along the incision line, respectively. The area was shaved and cleaned with betadine solution, after which a small craniotomy was made over the primary visual cortex (V1) [coordinates: −5.0-mm anterior-posterior (AP), ±4.0-mm medial-lateral (ML) relative to bregma]. μTENNs were carefully loaded into a needle under a dissecting microscope by using fine forceps to gently manipulate the microcolumn into the needle shaft. The needle containing the μTENN was then coupled to a Hamilton syringe and mounted onto a stereotactic arm. To deliver the constructs into the brain without forcible expulsion, the needle was mounted on a micromanipulator and slowly inserted into the cortex to a depth of 1.0 mm such that the dorsal μTENN terminal was left ~500 μm above the brain surface. The plunger of the Hamilton syringe was then immobilized while the needle containing the μTENN was slowly raised. This method effectively deposited the μTENN in the wake of the needle withdrawal to minimize forces on the preformed neural network. Upon needle removal from the brain, the dorsal aggregate of the μTENN was immersed in artificial cerebrospinal fluid warmed to 37°C. To protect the dorsal μTENN terminal and enable imaging of the μTENN and surrounding tissue, two custom-made PDMS rings (OD, 5.0 mm; ID, 2.0 mm; thickness, 0.35 mm) were placed over the craniotomy/μTENN and secured to the skull with cyanoacrylate glue. A 3.0-mm-diameter glass coverslip was sandwiched between the two rings.
To assess whether μTENN neurons could deliver optical readout following microinjection, animals were anesthetized and mounted on a stereotactic frame for multiphoton calcium imaging of the μTENN neurons following a recovery period, i.e., at 5 and 10 days after implant. μTENNs were imaged on a Nikon A1RMP+ multiphoton confocal microscope paired with NIS Elements AR 4.60.00 and a 16× immersion objective. Recordings of the μTENNs’ dorsal aggregates were taken at 3 to 5 frames per second, similarly to other intravital work (20). After recording, ROIs of μTENN neurons were manually identified, with the mean pixel intensity of each ROI plotted over time as an aggregate-level measure of neuronal activity. To distinguish neuronal activity from the animal breathing artifact, the fast Fourier transform (FFT) of the mean pixel intensity averaged across 10 to 15 ROIs was used to identify the frequency peak(s) associated with the observed breathing rate during imaging. Peaks were identified as frequencies whose amplitudes were 2 SDs or more than the average amplitude of the Fourier spectra.
Tissue harvest and histology
At 1 week and 1 month after implant, rats were anesthetized with euthasol (150 mg/kg) (Midwest) and transcardially perfused with cold heparinized saline and 10% formalin. After postfixation of the head overnight, the brain was harvested and stored in PBS to assess μTENN survival and host/μTENN synaptic integration (n = 10). Histology was performed via traditional immunohistology (IHC) and the Visikol clearing method to resolve thicker tissue sections where appropriate.
For traditional IHC, brains were blocked (sagittally for longitudinal μTENN visualization and obliquely for axial μTENN visualization) and cut in 20- or 40-μm slices for cryosectioning. For frozen sections, slices were air-dried for 30 min, twice treated with ethanol for 3 min, and rehydrated in PBS twice for 3 min. Sections were blocked with 5% normal horse serum (ABC Universal Kit, Vector Labs, catalog no. PK-6200) in 0.1% Triton X-100/PBS for 30 to 45 min. Primary antibodies were applied to the sections in 2% normal horse serum/Optimax buffer for 2 hours at room temperature. In longitudinal sections, primary antibodies labeled neurons/axons (1:1000; rabbit anti-NF200), neurons/dendrites (1:1000; chicken anti-MAP2), neurons/axons (1:1000; mouse anti-Tuj1), and/or presynaptic terminals (1:1000; mouse anti-synapsin). In axial sections, primary antibodies labeled microglia/macrophages (1:1000; rabbit anti–Iba-1) and astrocytes [1:1000; goat anti-glial fibrillary acidic protein (GFAP)]. Sections were rinsed with PBS three times for 5 min, after which secondary antibodies (1:1000) were applied in 2% normal horse serum/PBS for 1 hour at room temperature. Sections were counterstained with DNA-specific fluorescent Hoechst 33342 for 10 min and then rinsed with PBS. After immunostaining, slides were mounted on glass coverslips with Fluoromount-G mounting media.
In the Visikol method, brains were glued to a vibratome mounting block directly in front of a 5% low electroendoosmosis (EEO) agarose post (Sigma-Aldrich, A-6103) and placed in PBS surrounded by ice. The brain was cut in 100- to 200-μm coronal segments with a Leica VT-1000S vibratome until the μTENN implantation site was approximately 1 mm from the cutting face. Subsequently, a single 2-mm section containing the μTENN was cut and placed in PBS (frequency setting, 9; speed, 10). The 2-mm brain section was treated at 4°C with 50, 70, and 100% tert-butanol, each for 20 min. After the ascending tert-butanol steps, the tissue was removed and placed on a Kimwipe to carefully remove any excess reagent. Visikol HISTO-1 was applied to the sample for 2 hours at 4°C followed by Visikol HISTO-2 for 2 hours at 4°C to complete the clearing process. The sample was placed in a petri dish, and a hydrophobic well was drawn around the tissue. Fresh Visikol HISTO-2 was applied to completely submerge the tissue, which was then covered by a glass coverslip.
Coverslips containing brain slices were imaged on a Nikon A1RMP+ multiphoton confocal microscope paired with NIS Elements AR 4.60.00 and a 16× immersion objective. A 960-nm laser was used to visualize the μTENNs containing neurons expressing GFP/GCaMP for qualitative observations of neuronal presence, location, and construct architecture after implant.
General statistical methodology
The Shapiro-Wilk test was used to test data for normality before statistical comparisons. Unless otherwise specified, ANOVA was used with post hoc analyses as appropriate. The threshold for statistical significance was defined in all cases as P < 0.05. All data are presented as means ± SEM.
RESULTS
The objectives of our current efforts were threefold: (i) to reproducibly fabricate living electrode aggregate–based μTENNs and characterize their growth, viability, and network architecture; (ii) to demonstrate the ability to control and monitor μTENNs via light; and (iii) to determine whether living electrode neurons survive in vivo and remain viable for optical monitoring once transplanted in the host cortex.
Fabrication and axonal outgrowth
A fundamental step in the creation of μTENN-based living electrodes is a fabrication method that enables control and consistency of their structure across preparations. In earlier work, μTENNs were seeded with dissociated cortical neurons suspended in growth media, which, in many cases, formed clusters at random throughout the microcolumn interior (Fig. 1, D and E). By instead inducing the neurons to form spheroidal aggregates before their plating, these newly developed aggregate-based μTENNs consistently generated the desired cytoarchitecture of discrete somatic and axonal zones in vitro (Fig. 1, F to H). This macro-level aggregate-based μTENN structure was demonstrably reproducible across both unidirectional and bidirectional polarities and a range of microcolumn parameters (e.g., 2 to 9 mm in length), although, as described below, growth dynamics varied across these design parameters.
In practice, implanted μTENNs may need to integrate with targets several millimeters below the brain surface; hence, a critical milestone was the establishment of long axons in a new 3D microenvironment, as well as early characterization of these axonal growth dynamics to begin exploring the practical constraints of the aggregate culture method. Qualitatively, phase microscopy revealed healthy and rapid axonal outgrowth across all aggregate-based μTENNs through the ECM core; observed growth rates generally peaked at 3 to 5 DIV before slowing as unidirectional μTENN axons reached the opposing end (Fig. 2A) or bidirectional μTENN axons from opposing aggregates grew toward and along each other (Fig. 2, B and C). For both μTENN polarities, the maximal growth rate increased with the construct length (Fig. 2D), with one-way ANOVA of the average maximum growth rate implicating the LE cohort as a significant effect (F-statistic = 14.1, P < 0.0001). The fastest growth rate was observed in bidirectional 9-mm (LEBI,9) μTENNs, which reached 1101.8 ± 81.1 μm/day at 3 DIV—nearly 17× the rate of early-generation μTENNs (Fig. 2, D and F, and table S1).
Fig. 2. Axonal growth in aggregate μTENNs over time.

Aggregate μTENNs grow rapidly over 1 to 8 DIV; unidirectional μTENNs (A) project axons to the opposing terminal, while bidirectional μTENNs (B) axons cross the microcolumn and synapse with the opposing aggregate. Representative 2-mm μTENNs shown at 1, 3, 5, and 8 DIV. (C) Representative 5-mm μTENN shown at 1, 3, and 5 DIV. (D) Average maximum growth rates across μTENN lengths. In general, longer bidirectional μTENNs displayed higher peak growth rates. Symbols indicate a significantly lower maximum growth rate than a specific group: 9-mm bidirectional (*), 7-mm bidirectional (#), and 5-mm bidirectional (+) μTENNs, respectively. Symbol count denotes significance level (1: P < 0.05; 2: P < 0.01; 3: P < 0.001). (E) Average crossing times across μTENN groups. Similarly, longer constructs tended to take more time to develop. Unidirectional (5 mm) μTENNs did not fully cross the microcolumn by 10 DIV and were not included. Symbols and symbol counts match those described in (D), with the addition of significance versus 8-mm bidirectional (^). (F) Growth rates for unidirectional, bidirectional, and dissociated μTENNs at 1, 3, 5, 8, and 10 DIV. Growth rates were quantified by identifying the longest neurite from an aggregate in phase microscopy images (×10 magnification) at the specified time points. Crosses indicate axons crossing the length of the microcolumn (unidirectional) or connecting between aggregates (bidirectional). Error bars denote SEM. Scale bars, 200 μm.
Since the cohort was a statistically significant factor, subsequent Bonferroni-corrected pairwise comparisons showed that the average maximum growth rates of the longest cohorts (LEBI, 5, LEBI,7, and LEBI,9) were indeed statistically higher than those of the shortest cohorts LEBI,2 and LEBI,3 (P < 0.001), while only LEBI,9 surpassed the unidirectional cohorts with significance (LEUNI,2: P < 0.01; LEUNI,5: P < 0.05) (Fig. 2D). In addition, both dissociated μTENNs and control planar neuronal cultures exhibited similar growth rates that were an order of magnitude slower than their aggregate counterparts; peak growth rates were 61.7 ± 5.01 (LEDISS) and 39.1 ± 20.6 (CTRL) μm/day, respectively (Fig. 2F and table S1).
While the longest μTENNs exhibited the fastest peak growth rates, they took the most time to cross the microcolumn length. In addition, several constructs in the LEBI,7 and LEBI,9 cohorts exhibited a decline and eventual cessation of measurable outgrowth before fully forming networks. Similarly, one-way ANOVA and Bonferroni post hoc analysis of the average crossing time showed that LEBI,7 and LEBI,9 axons crossed the length of the microcolumn significantly later than those of LEUNI,2, LEBI,2, LEBI,3, and LEBI,5 (F-statistic = 12.99, P < 0.0001 to 0.05) (Fig. 2E). LEUNI,5 axons did not, on average, fully cross the construct length by 10 DIV (Fig. 2F and table S1). Notably, one-way ANOVA of the average minimum growth rate did not detect any significant differences across aggregate-based LE groups (F-statistic = 1.17, P = 0.332).
μTENN viability
Neuronal maturation and network development are denoted by a complex interplay of growth, pruning (elimination), and remodeling of synaptic connections as individual neurons form larger networks (21). For μTENNs, phase/fluorescent images and calcium imaging analyses of bidirectional aggregate-based μTENNs provide structural and functional evidence for the presence of an initial interaggregate network by 10 DIV (15). To assess the impact of network maturation and pruning on neuronal survival in these constructs, unidirectional and bidirectional μTENNs were grown to 10 and 28 DIV. Survival was quantified as the ratio of the summed area of calcein-AM+ (live) cells to that of all (i.e., both calcein-AM+ and ethidium homodimer+) cells at the specified time points, with age-matched planar cultures on polystyrene as controls for the aggregate culture method (fig. S1, A to F). μTENN neurons survived up to at least 28 DIV, with qualitative observation of survival out to 40 DIV (fig. S1I). Although ANOVA identified the DIV as a significant main effect (F-statistic = 32.21, P < 0.0001), the LE/culture group was not a significant factor (P > 0.84). The interaction effect was significant (P < 0.01), so Bonferroni analysis was used to compare groups at each time point (fig. S1G). Survival of planar cultures at 28 DIV (53.6%) was found to be statistically lower than that of LEUNI (80.3%) (P < 0.05), LEBI (84.8%) (P < 0.001), and planar cultures (97.7%) (P < 0.0001) at 10 DIV (fig. S1G). Moreover, planar culture viability at 10 DIV surpassed those of both LEUNI (68.1%) and LEBI (69.0%) at 28 DIV (P < 0.01). Overall, planar cultures exhibited an average 45% reduction in viability from 10 to 28 DIV, while LEUNI and LEBI showed a 15.2 and 18.6% drop over time, respectively (fig. S1H).
μTENN architecture and synaptogenesis
To characterize changes in μTENN architecture over time, bidirectional μTENNs were engineered to express GFP and mCherry to track cross-aggregate neurite outgrowth and integration. μTENNs were fixed and immunolabeled to determine the spatial distribution of cell somata/nuclei, axons, and synapses at set time points before and during network formation (Fig. 3). Transducing each aggregate before seeding enabled the consistent identification of aggregate-specific projections over time (Fig. 3A), even within dense axonal bundles (Fig. 3, B to D). Confocal images of GFP/mCherry μTENNs revealed that projections from each aggregate made contact and grew along opposing axons toward the opposite aggregate (Fig. 3, E to G); qualitatively, no gross trends were observed in the extent of physical interaction and integration between the two neuronal populations labeled with either reporter. Immunolabeling of cell nuclei with Hoechst revealed that neuronal somata remained localized almost exclusively to the aggregates across time points, which were spanned by long bundles of Tuj-1 expressing axonal projections through the microcolumn lumen (Fig. 3H). Synapsin-1 is expressed at the presynaptic terminals of mature neurons and is involved in the regulation of myriad developmental processes, including the formation and maintenance of synapses—making it a suitable proxy for characterizing the neuronal maturity in culture (22–25). Immunolabeling revealed highly dense intra-aggregate clusters of axons and synapses within μTENNs at all time points, presumably formed upon or shortly after plating (Fig. 3, I and J). No statistically significant differences in synapsin distribution were identified; however, qualitative observations of synapsin-1 expression showed a moderate increase in synapsin-1+ puncta within the μTENN aggregates as well as along the axonal tracts within the microcolumn lumen over time (fig. S2). Immunolabeling thus suggests that neurons within bidirectional μTENNs reach a mature state, i.e., are capable of both intra-aggregate and interaggregate synaptogenesis following axonal growth.
Fig. 3. Aggregate μTENN architecture.
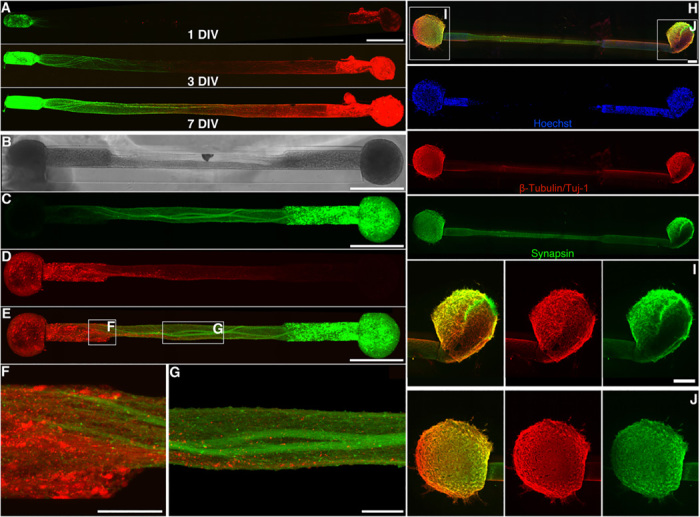
Bidirectional μTENNs were labeled with GFP (green) and mCherry (red) to observe aggregate-specific axonal growth and structure in vitro. (A) Confocal reconstructions of a bidirectional, GFP/mCherry-labeled μTENN at 1, 3, and 7 DIV. (B) Phase image of a bidirectional, GFP/mCherry-labeled μTENN at 5 DIV. (C to E) Confocal reconstruction of the μTENN from (B) at 7 DIV, with insets showing axons from each aggregate making contact with the opposing population (F) and growing along each other (G) in the microcolumn lumen. (H) Confocal reconstruction of a representative bidirectional μTENN at 10 DIV, immunolabeled for cell nuclei (Hoechst, blue), axons (β-tubulin III/Tuj-1, red), and synapses (synapsin, green). Cell bodies are localized to the microcolumn terminals with axonal tracts spanning the distance. Insets in (H) refer to callout boxes (I) and (J) showing aggregate zoom-ins of synapses, axonal networks, and their overlay. Scale bars, 500 μm (A to C and E); 100 μm (F and G); and 200 μm (H and I).
Calcium imaging and optical stimulation
Bidirectional μTENNs expressing the calcium reporter GCaMP6f exhibited spontaneous oscillations at and below the delta band (0.5 to 5 Hz) in the absence of external stimulation (Fig. 4, A and B; see movies S1 to S3). These observations reflect earlier findings wherein GCaMP+ μTENNs demonstrated functional connectivity with strongly correlated oscillations in the delta and theta bands, or 1 to 8 Hz, across aggregates (15). The abundance of synapsin expression coupled with synchronicity of oscillation between aggregates further support the conclusion that aggregate-based μTENNs form functional synaptic networks with a coherent structure that may be observed at both the neuron and aggregate levels.
Fig. 4. Calcium imaging and optical stimulation in aggregate μTENNs.
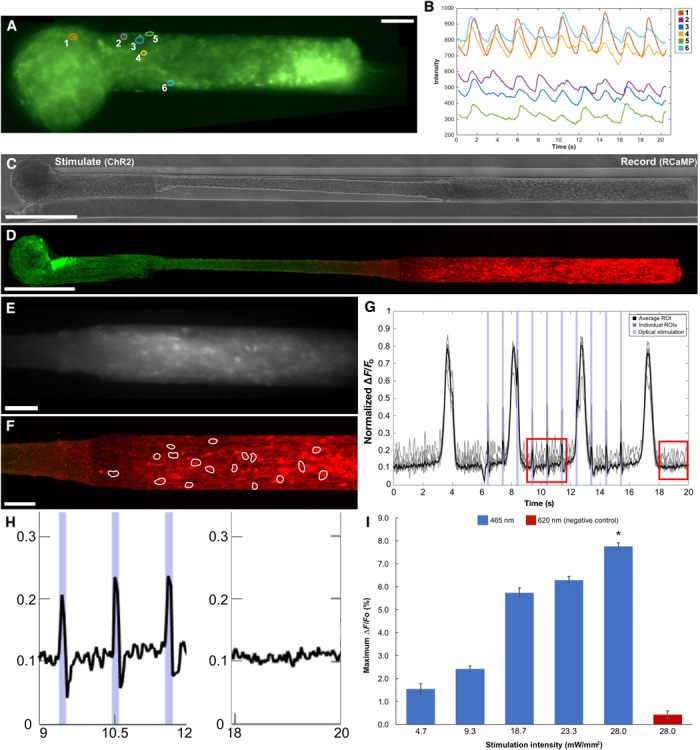
(A) Representative GCaMP+ μTENN at 10 DIV with single-neuron ROIs outlined. (B) Average fluorescent intensities of the ROIs from (A) recorded over time indicate neuronal activity similar to constructs in earlier work (15). Intensity traces are normalized to a background (empty) region. Phase image (C) and confocal reconstruction (D) of a μTENN at 10 DIV in vitro, virally transduced such that the left aggregate expresses ChR2 (optical actuator) and the right aggregate expresses the calcium reporter RCaMP, enabling simultaneous control and monitoring with light. (E) The RCaMP+ aggregate from (C) and (D) under fluorescent microscopy during recording (16 frames per second). (F) Confocal image of the RCaMP+ aggregate poststimulation. ROIs containing single neurons were manually defined (white outlines). (G) Normalized pixel intensity of ROIs within the RCaMP+ aggregate from (A) to (C) during stimulation. Gray lines indicate representative, user-defined ROIs randomly selected for analysis, which were averaged to obtain a mean ROI of the aggregate (solid black line). A single train of 1-Hz, 100-ms stimulation pulses is shown as blue bands along the abscissa. The changes in pixel intensity due to stimulation of the input aggregate can be seen as sharp spikes occurring within the endogenous, large-amplitude slow-wave activity. (H) Zoom-ins of the red insets from (D) showing μTENN activity during (left) stimulation and after (right) optical stimulation. (I) Average maximum ∆F/Fo across stimulation intensities. Although the maximum ∆F/Fo trended upward, the differences were not significant across intensities. Statistical comparison revealed that stimulation with the control wavelength (620 nm) yielded significantly lower maximum ∆F/Fo than with 465 nm (*P < 0.05). Scale bars, 100 μm.
Bidirectional μTENNs were also engineered to enable light-based stimulation and concurrent calcium imaging in vitro by transducing one aggregate with ChR2 and the opposing aggregate with RCaMP (Fig. 4, C and D). Upon illumination of ChR2+ (input) aggregates with 465-nm light (within the excitation spectrum of ChR2), the opposing RCaMP+ (output) aggregates exhibited time-locked changes in fluorescence intensity in response (Fig. 4, E to H). All-optical μTENNs were fabricated 5 mm in length with wavelength-separated vectors to reduce the probability of photostimulation artifact. To further reduce the potential for confound from photostimulation artifact, constructs’ input aggregates were illuminated with 620-nm light (outside of the ChR2 activation spectrum) of equal intensity to serve as a negative stimulation control. Photostimulation at 620 nm induced no readily observable responses; the mean peak ∆F/Fo of the output aggregate was significantly greater under 465-nm stimulation than 620-nm stimulation at 28.0 mW/mm2 (P < 0.05) (Fig. 4I). Collectively, these findings suggest that the changes in ∆F/Fo under 465-nm stimulation reflected synaptically mediated firing of neurons in the output aggregate in response to light-based activation of neurons within the input aggregate. Although there was high variability in ∆F/Fo between μTENNs, the percent change in the fluorescence of the output aggregate relative to baseline under optical stimulation could be reproducibly distinguished from endogenous activity across all the μTENNs studied; further, the average maximum ∆F/Fo positively correlated with the stimulation intensity (Fig. 4I). Overall, these results suggest that photostimulation of the input aggregates resulted in controllable signal propagation and modulation of activity in the associated output aggregates.
Implantation and intravital calcium imaging
Following in vitro characterization, 1.0- to 1.2-mm aggregate-based μTENNs were fabricated as described above, transduced to express GCaMP6, and implanted in rodent V1 via stereotactic microinjection as a proof of concept for living electrode survival and function in vivo. Following μTENN delivery, a cranial window was affixed to a custom-build PDMS ring assembly adhered to the surrounding skull to permit repeated noninvasive monitoring of the dorsal μTENN aggregate and surrounding V1 (Fig. 5A). This noninvasive imaging was performed using multiphoton microscopy in animals anesthetized using controlled isoflurane delivery with carefully monitored, voluntary respiration. Acute postoperative fluorescent imaging confirmed that the bulk of the dorsal aggregate remained intact at the original implant location (Fig. 5, B and C). Multiphoton calcium imaging of the dorsal aggregate was performed at 5 and 10 days to avoid potential confounds from the microtrauma of the immediate posttransplant environment affecting neuronal activity. This calcium imaging revealed surviving and active GCaMP+ μTENN neurons in vivo, with transients in GCaMP intensity in the delta band (1 to 5 Hz) (Fig. 5, D to F). Putative activity was also present at frequencies below 1 Hz (Fig. 5F, inset). Of note, the respiration of the animals caused a consistent artifact in the recorded fluorescence signals resembling a low-frequency oscillation in the baseline fluorescence across all ROIs; however, this respiratory artifact could be readily identified and isolated within the FFT of the time-lapse recordings as a ~0.5- to 0.7-Hz peak distinct from both the delta band and putative sub–1 Hz activity (red band, Fig. 5F). The intravital μTENN neuronal activity patterns were similar to those recorded from age-matched nonimplanted μTENNs in vitro, suggesting that aggregate-based μTENNs retain their network structure over days after transplant. Longer-term studies will evaluate the utility of μTENNs as an output for neuronal activity as host neurons integrate with the ventral terminus of the network, which would presumably alter the network dynamics.
Fig. 5. Living electrode function in vivo.
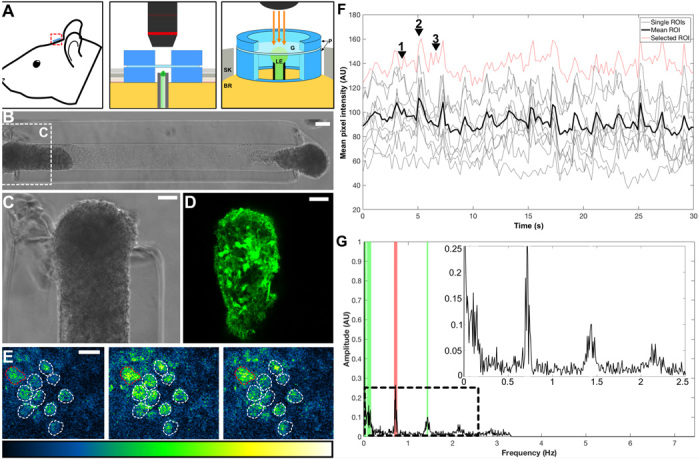
(A) Conceptual schematic of the living electrode and cranial window, with adjacent zoom-ins (middle and right) showing the PDMS rings (P) sized to the skull craniotomy (SK), securing the glass coverslip (G), and protecting the implanted living electrode (LE) and underlying brain (BR), which may then be imaged chronically (orange arrows). (B) Phase image of a bidirectional GCaMP+ μTENN before implant in rodent cortex with a zoom-in (C) of the dorsal aggregate. (D) Multiphoton image of the same dorsal μTENN aggregate acquired immediately after implant. (E) Single frames from multiphoton recording of the living electrode from (B) to (D) at 10 days after implant during low activity (left), breathing (middle), and nonartifact neuronal activity (right). ROIs containing single neurons are outlined. The Lookup Table (LUT) scale (0 to 4096) is provided below. (F) Time course of calcium fluorescence acquisition from (D), showing the individual (gray/red) and mean (black) ROIs. The red trace represents the ROI outlined in red in (E). Numbered arrowheads denote timestamps from (D). (G) Frequency analysis via Fourier transform of the data from (E), showing spectral peaks due to breathing (red) and neuronal activity (green). Inset shows low-frequency activity similar to that observed in vitro. Scale bars, 50 μm (B and C) and 20 μm (D). AU, arbitrary units.
At 1 week and 1 month after injection in the rodent brain, immunohistochemistry in optically cleared as well as sectioned tissue revealed that μTENN neurons survived and maintained the preformed somatic-axonal architecture, with GFP+/GCaMP+ cell bodies predominantly localized to the microcolumn terminals and spanned by axonal tracts (Fig. 6). Staining for markers of astrogliosis and microgliosis in axial sections revealed only a modest host inflammatory reaction to the μTENNs (Fig. 6C). Large, dense clusters of GCaMP+ cell bodies were found at the dorsal regions of implantation (Fig. 6B), with axons and dendrites within the lumen spanning the microcolumn (Fig. 6D). Dorsal aggregates were observed spreading along the brain surface or further along the microcolumn, putatively due to a cell migration away from the aggregate to increase interactions with host tissue (Fig. 6B). In some cases, there was also neuronal migration up to several millimeters from the ventral implant location, although the presence and extent of migration varied across implants. In general, there was widespread neurite outgrowth from the ventral aggregate of the living electrode, with structural evidence of synapse formation with host neurons (Fig. 6, E and F).
Fig. 6. Living electrode survival and integration in vivo.

(A) Phase image of a bidirectional μTENN before implantation; aggregates have been internalized to the microcolumn. (B) Multiphoton image of the μTENN from (F) at 1 month after implant, showing GCaMP+ μTENN neurons and processes within and immediately surrounding the construct. At 1 month, the dorsal aggregate had descended into the microcolumn, suggesting that externalized aggregates may be required to maintain a cohesive neuronal population at the surface. (C) Axial view of implant showing μTENN neurons/axons in the lumen at 1 month after implant. To visualize the extent of the inflammation response at 1 month following delivery, sections orthogonal to the implant site were stained for microglia/macrophages (Iba-1, red) and astrocytes (GFAP, far red). Minimal host neuroinflammatory response was observed at this time point surrounding the implanted construct. Dashed lines denote the host brain-μTENN interface. (D to F) Longitudinal view of implanted μTENN within the corticothalamic tract at 1 month after implant, with evidence that the construct retained its axonal tracts and overall axosomatic architecture (GFP, green). (E) μTENN neurons and axons (GFP, green) were found projecting ventrally with neurons and neurites interfacing with host tissue at the (deep) ventral end. GFP+ μTENN neurons were visualized in discrete regions with axons extending within the lumen parallel to the cortical-thalamic axis. These findings demonstrate that following stereotaxic microinjection, aggregate μTENNs survive, with neurite extension and integration out to at least 1 month in vivo. Arrows denote μTENN neurites penetrating the host brain and putative synapse formation, with dashed inset representing zoom-in (F). Scale bars, 100 μm (A to C); 50 μm (D and E); and 10 μm (F). HST, Hoechst.
DISCUSSION
Microelectrodes—the current gold standard for recordings—have functioned in neural interfaces successfully on the order of months and, less frequently, years, in rodents, nonhuman primates, and human patients (1, 26–28). However, microelectrode-based BCIs generally succumb to a complex combination of abiotic and biological factors (including host neuronal loss, gliosis, biofouling, electrode movement, and/or mechanical failure) that impede stability, specificity, and clinical deployment (1–5, 29). Optogenetic strategies for neuromodulation enable more selective stimulation but must restrict the vector of interest to targeted cells, address the scattering and limited tissue penetration of light, and activate transduced cells without overheating brain tissue (30–33). Ongoing efforts to minimize inflammation have yielded more compliant electrodes and electrode coatings/cofactors; however, the chronic foreign body response, consequent signal drop, and increase in stimulation thresholds continue to negatively affect current neural interface systems.
μTENNs as living electrodes may present an alternative to conventional microelectrode- or optogenetic-based strategies for neuromodulation/recording with improved biocompatibility, selectivity, and longevity. The integration of living cells as functional device elements is not a new concept, for instance, other groups have exploited electroactive bacteria for biosensing applications (34). Similar efforts to develop biohybrid electronics include the introduction of living neural progenitors (or neural-like PC12 cells) and/or glial support cells into biocompatible hydrogels or hydrogel-polymer composites; these “biosynthetic hydrogels” are then layered to coat metallic electrodes (35–37). However, the living electrode approach presented in this work is distinct as an implantable, anatomically inspired, 3D engineered microtissue preformed in vitro before delivery into the brain. This approach uses aggregate-based μTENNs, which are designed to form a relay for biological (i.e., synaptic) interface with existing neuronal circuitry while remaining accessible at the brain surface for optical stimulation (input) or imaging (output). By enabling the segregation of cell bodies (aggregates) and axons in a single structure, these μTENNs may be considered a physical abstraction of the mammalian neural connectome, wherein locally connected neuronal populations are spanned by long distance axonal pathways.
The work presented here sets a critical foundation in developing aggregate-based μTENNs toward a viable neural interface and provides insight into the experimental milestones needed to scale more long-term translational challenges for similar “living” interfaces. In addition, these constructs consistently generate the connectome architecture described above based on the few design parameters (length and polarity) explored in this work. In reality, the biofabrication method developed for aggregate-based μTENNs provides several degrees of control over their microenvironment through systematic variation of its material (e.g., microcolumn curvature, stiffness, and bioactivity), chemical (ECM composition and growth factor gradients), and cellular [phenotype(s) and expression] properties. There are thus unexplored opportunities for aggregate-based μTENNs to serve as a multifaceted and scalable investigative tool both in vitro and in vivo for the neuroscience field.
Early growth characterization demonstrated that these next-generation μTENNs, created using forced neuronal aggregation and improved microfabrication techniques, can grow to at least 9 mm while maintaining the desired cytoarchitecture. Further, they exhibit accelerated axonal outgrowth and greater longitudinal “bundling” of axons than in early-generation dissociated μTENNs (Fig. 1, E and H). The observed growth rates for axons in dissociated μTENNs averaged ~60 μm/day over the first 3 days, reflecting growth trends in literature for cortical axons (38, 39). In comparison, the peak axonal growth rates from aggregate-based μTENNs exceeded the rate exhibited by dissociated neurons by two orders of magnitude, with the LEBI,9 group attaining the remarkable axon extension rate of more than 1000 μm/day (Fig. 2). Neuronal aggregation likely induced bundled and directed neurite extension and may positively influence axon-ECM interactions with the collagen and laminin comprising the microcolumn lumen. Other potential factors for such accelerated axonal outgrowth from the neuronal aggregates include the restriction of axonal growth to the microcolumn interior and the lack of synaptic targets within the microcolumn, which may reduce nonlinear axon branching between aggregates and permit more efficient growth cone migration within the lumen (38, 40–43). Although peak growth rates across aggregate-based μTENN groups were positively correlated with μTENN length, their initial growth rates did not vary significantly. This may implicate a critical distance between aggregates below which neuronal processes meet before reaching their maximum growth rates. This distance may be required for the neurons to establish sufficient neurotrophic support and/or up-regulate the growth machinery required to reach these speeds.
With respect to neuronal health in aggregates, neuronal attrition from 10 to 28 DIV was ~2.4 to 3× lower in unidirectional and bidirectional μTENNs than in control planar cultures (fig. S1, G to H). Established work has shown that neurons exhibit better growth and survival in 3D environments, which more accurately approximate conditions in vivo (44). Similarly, the connectome-inspired 3D microstructure of spheroidal aggregates and axonal bundles appears to provide cortical neurons with the requisite conditions for rapid axonal growth and prolonged viability. μTENNs also demonstrated survival out to at least 40 DIV (fig. S1I), enabling more chronic in vitro investigations of these dynamics and their mechanisms. Potential subsequent steps include the measuring of neuron health and functionality over time across various cell preparations, neuronal phenotypes, and alternative design parameters.
Beyond axonal growth and survival, another notable outcome was the structural and functional connectivity between aggregates for bidirectional μTENNs. Our biofabrication methodology enabled for aggregate-specific transduction and labeling of individual μTENNs such that we could track structural integration between neuronal populations as the constructs matured (Fig. 3, A to G). Immunocytochemistry provided additional evidence of network maturation with the presence and proliferation of synapsin+ puncta from as early as 4 DIV to at least 28 DIV (fig. S2). As the primary points of contact and communication between neurons, synapses are often used to determine the functional maturity of neuronal networks (25, 45). These results suggest that μTENN neurons in aggregates begin to form synaptic networks soon after plating, which mature and expand over time, findings consistent with those in planar cortical cultures (25). Intra-aggregate synapses presumably dominate the synaptic population before axons span the aggregates to form interaggregate synapses. Future structural connectivity studies may build on the aggregate-specific labeling here to characterize intra- and interaggregate connectivity with greater resolution.
With respect to functional connectivity, we measured spontaneously generated calcium reporter activity in and across the GCaMP+ aggregates of bidirectional μTENNs (Fig. 4 and movies S1 and S2), which consisted predominantly of delta-band oscillations (1 to 5 Hz). High synchronicity across aggregates has been identified in delta-band oscillations through concurrent network analyses of 1.0- to 1.2-mm bidirectional GCaMP+ μTENNs in vitro, confirming their functional connectivity and capacity for information transfer (15). Within all-optical 5- to 6-mm μTENNs, neurons in the RCaMP+ aggregate for optical output displayed similar high-amplitude delta-band activity, with the addition of smaller transients corresponding to photostimulation delivered at the ChR2+ aggregate for light input (Fig. 4, G to I, and movie S3). The evoked activity was wavelength dependent, with a positive correlation to stimulus intensity that strongly suggested increased activation of ChR2+ neurons (Fig. 4I). These observations validated the presence of functional, long-distance axonal tracts and synaptic integration between two aggregate populations and, crucially, demonstrate the ability of these constructs to serve as an optically driven input-output platform for the transmission of information. Variability in the input-output response behavior may be attributed to the stimulation source size and position relative to the aggregate, which as a densely packed 3D sphere would inherently experience nonuniform illumination. Techniques for more uniform spheroidal photostimulation (e.g., with multiple light sources) or sufficient single-source stimulation to approximate the same (e.g., through depolarization of a critical fraction of ChR2+ neurons) remain an ongoing objective.
After transplant into rat cortex, we observed both delta-band oscillations (similar to those measured in vitro) and slow-wave oscillations below 1 Hz in GCaMP+ μTENN neurons (Fig. 5). Slow-wave oscillations have been recorded under anesthesia and during slow-wave sleep, as well as in cortical neuronal cultures in vitro (46, 47). Whether the detected <1 Hz of activity represents a response to cortical synaptic inputs remains beyond the scope of the current manuscript but seems unlikely, given the relatively acute imaging time points. However, the detection of calcium transients in μTENN neurons demonstrates they remain viable posttransplant, while sustained delta-band activity suggests active maintenance of the required network structure for synaptic transmission. A more direct assessment of functional integration over time would comprise longitudinal comparative measurements of intravital μTENN activity and host circuitry.
Histological observations at 1 week and 1 month revealed gross preservation of construct architecture and neurite outgrowth at the μTENN terminals (Fig. 6), reflecting similar findings with early-generation μTENNs in prior work. Further, colocalization of GCaMP+ processes with synapsin+ puncta suggests the structural formation of synapses with the cortex, although more functional validation of these histological implications in vivo is required as described above. The information transfer bandwidth of a living electrode-based interface is shaped by the extent of its synaptic integration with the brain. While this may enable highly dense, long-term stability as observed with native synaptic connections, controllability over the targeting of synaptic integration and neuronal migration remains a complex, nontrivial translational challenge that may be addressed through one or more strategies. Potential physical methods include a porous membrane at the ventral microcolumn terminal to restrict neuronal migration while permitting axonal projections between the μTENN and host brain. The μTENN itself may be fabricated with a neuronal phenotype known for its desired synaptogenetic behaviors or genetically engineered for more controllable synaptogenesis versus widespread outgrowth through the up-/down-regulation of specific developmental proteins (23, 48). Last, more direct regulation of the transplant environment may also promote more targeted integration, for instance, by introducing or promoting expression of trophic factors implicated in axonal guidance and/or synaptic pruning during development (21, 49). Another key concern for implantable interfaces is the invasiveness of delivery. Combined with well-established stereotactic neurosurgical techniques, the living electrode microcolumns may be further minimized to reduce the microinjection footprint. In addition, their material properties (e.g., stiffness) and potential cofactors (e.g., anti-inflammatory/growth factor cues) may be tailored against any subsequent foreign body response.
In summary, we have used optogenetic and tissue engineering techniques to create so-called living electrodes—cylindrical, hydrogel-encapsulated neuronal populations linked by functional axonal tracts—and demonstrated their biofabrication and functional validation in vitro, as well as their targeted delivery, survival, and continued function posttransplant. The results described indicate that aggregate-based μTENNs quickly and consistently form similar functional architecture—important for the biofabrication and scale-up of experimentally useful constructs—which is maintained over weeks in vitro. Thus, μTENNs may serve as an ideal system for studying neuronal growth, maturation and network dynamics due to their “abstraction” of mammalian brain connectome into individual units (44). While the constructs in this study were composed predominantly of glutamatergic neurons, μTENNs may be seeded with aggregates of different neuronal phenotypes or sources. Within the living electrode paradigm, this may enable the delivery of light-driven, synaptically transduced neuromodulation in vivo for specific clinical effects, for instance, neural circuit inhibition or diffuse neurotransmitter release. Multiple distinct μTENNs could also be grown in proximity to form connected assemblies, comprising a modular “building-block” approach to model, manipulate, and characterize systems-level network dynamics across neuronal circuits of mixed cell population and increased pathway complexity in vitro. Last, aggregate-based μTENNs were able to survive, grow, and provide GCaMP-based readout following implantation as proof-of-concept validation for preformed, implantable, biological pathways to synaptically integrate with, probe, and modulate neuronal targets through optical interface on the brain surface. These studies lay the groundwork for more in-depth investigations of the utility of aggregate-based μTENNs as both an experimental tool for in vitro modeling and a translational tool for neural interface following targeted transplant in the cerebral cortex or other anatomical targets.
Acknowledgments
Funding: Financial support was primarily provided by the NIH [BRAIN Initiative U01-NS094340 (D.K.C.), T32-NS043126 (J.P.H.), and T32-NS091006 (L.A.S.)] and the NSF [Graduate Research Fellowship DGE-1321851 (D.O.A.)], with additional support from the Penn Medicine Neuroscience Center (D.K.C.), American Association of Neurological Surgeons and Congress of Neurological Surgeons [Codman Fellowship in Neurotrauma and Critical Care (D.P.)], and the Department of Veterans Affairs [Merit Review I01-BX003748 (D.K.C.), Merit Review I01-RX001097 (D.K.C.), Career Development Award #IK2-RX001479 (J.A.W.), and Career Development Award #IK2-RX002013 (H.I.C.)]. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors(s) and do not necessarily reflect the views of the NIH, NSF, or Department of Veterans Affairs. Author contributions: Conceptualization: D.K.C., J.A.W., M.D.S., and H.I.C. Methodology: D.K.C., D.O.A., L.A.S., J.P.H., A.D.N., J.C.B., D.P., H.I.C., and J.A.W. Formal analysis: D.O.A. Investigation: D.O.A. and J.C.B. Resources: R.H.K. Visualization: D.O.A. Writing (original draft): D.O.A. Writing (review and editing): D.O.A., D.K.C., L.A.S., J.P.H., A.D.N., J.C.B., D.P., R.H.K., H.I.C., J.A.W., and M.D.S. Supervision: D.K.C., J.A.W., M.D.S., R.H.K., and H.I.C. Project administration: D.K.C. Funding acquisition (primary): D.K.C. Competing interests: D.K.C. is a co-founder of two University of Pennsylvania spin-out companies related to nervous system regeneration and restoration: INNERVACE Inc. and Axonova Medical LLC. J.P.H. is currently a paid consultant and an equity holder in INNERVACE Inc. There are two patent applications related to the methods, composition, and use of μTENNs. D.K.C. is an inventor on a U.S. patent application filed by the University of Pennsylvania (no. 15/032,677, filed 4 November 2013, published 1 September 2016) titled “Neuronal replacement and reestablishment of axonal connections.” D.K.C., J.P.H., J.A.W., H.I.C., and M.D.S. are inventors on a U.S. patent application filed by the University of Pennsylvania (no. 16/093,036, filed 14 April 2016, published 2 May 2019) titled “Implantable living electrodes and methods for use thereof.” The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eaay5347/DC1
REFERENCES AND NOTES
- 1.Adewole D. O., Serruya M. D., Harris J. P., Burrell J. C., Petrov D., Chen H. I., Wolf J. A., Cullen D. K., The evolution of neuroprosthetic interfaces. Crit. Rev. Biomed. Eng. 44, 123–152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tresco P. A., Winslow B. D., The challenge of integrating devices into the central nervous system. Crit. Rev. Biomed. Eng. 39, 29–44 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Harris J. P., Tyler D. J., Biological, mechanical, and technological considerations affecting the longevity of intracortical electrode recordings. Crit. Rev. Biomed. Eng. 41, 435–456 (2013). [PubMed] [Google Scholar]
- 4.Grill W. M., Norman S. E., Bellamkonda R. V., Implanted neural interfaces: Biochallenges and engineered solutions. Annu. Rev. Biomed. Eng. 11, 1–24 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Polikov V. S., Tresco P. A., Reichert W. M., Response of brain tissue to chronically implanted neural electrodes. J. Neurosci. Methods 148, 1–18 (2005). [DOI] [PubMed] [Google Scholar]
- 6.Cogan S. F., Neural stimulation and recording electrodes. Annu. Rev. Biomed. Eng. 10, 275–309 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Mendoza S. D., El-Shamayleh Y., Horwitz G. D., AAV-mediated delivery of optogenetic constructs to the macaque brain triggers humoral immune responses. J. Neurophysiol. 117, 2004–2013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aravanis A. M., Wang L.-P., Zhang F., Meltzer L. A., Mogri M. Z., Schneider M. B., Deisseroth K., An optical neural interface: In vivo control of rodent motor cortex with integrated fiberoptic and optogenetic technology. J. Neural Eng. 4, S143–S156 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Pashaie R., Anikeeva P., Lee J. H., Prakash R., Yizhar O., Prigge M., Chander D., Richner T. J., Williams J., Optogenetic brain interfaces. IEEE Rev. Biomed. Eng. 7, 3–30 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Fan B., Li W., Miniaturized optogenetic neural implants: A review. Lab Chip 15, 3838–3855 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Harris J. P., Struzyna L. A., Murphy P. L., Adewole D. O., Kuo E., Cullen D. K., Advanced biomaterial strategies to transplant preformed micro-tissue engineered neural networks into the brain. J. Neural Eng. 13, 016019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Struzyna L. A., Wolf J. A., Mietus C. J., Chen I. H., Smith D. H., Cullen D. K., Chen H. I., Smith D. H., Cullen D. K., Adewole D. O., Chen H. I., Smith D. H., Cullen D. K., Rebuilding brain circuitry with living micro-tissue engineered neural networks. Tissue Eng. 21, 2744–2756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Struzyna L. A., Adewole D. O., Gordián-Vélez W. J., Grovola M. R., Burrell J. C., Katiyar K. S., Petrov D., Harris J. P., Cullen D. K., Anatomically inspired three-dimensional micro-tissue engineered neural networks for nervous system reconstruction, modulation, and modeling. J. Vis. Exp. 2017, 55609 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Struzyna L. A., Harris J. P., Katiyar K. S., Chen H. I., Cullen D. K., Restoring nervous system structure and function using tissue engineered living scaffolds. Neural Regen. Res. 10, 679–685 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dhobale A. V., Adewole D. O., Chan A. H. W., Marinov T., Serruya M. D., Kraft R. H., Cullen D. K., Assessing functional connectivity across three-dimensional tissue engineered axonal tracts using calcium fluorescence imaging. J. Neural. Eng. 15, 056008 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen D. K., Wolf J. A., Smith D. H., Pfister B. J., Neural tissue engineering for neuroregeneration and biohybridized interface microsystems in vivo (part 2). Crit. Rev. Biomed. Eng. 39, 243–262 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Ungrin M. D., Joshi C., Nica A., Bauwens C., Zandstra P. W., Reproducible, ultra high-throughput formation of multicellular organization from single cell suspension-derived human embryonic stem cell aggregates. PLOS ONE 3, e1565 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akerboom J., Carreras Calderón N., Tian L., Wabnig S., Prigge M., Tolö J., Gordus A., Orger M. B., Severi K. E., Macklin J. J., Patel R., Pulver S. R., Wardill T. J., Fischer E., Schüler C., Chen T., Sarkisyan K. S., Marvin J. S., Bargmann C. I., Kim D. S., Kügler S., Lagnado L., Hegemann P., Gottschalk A., Schreiter E. R., Looger L. L., Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 6, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patel T. P., Man K., Firestein B. L., Meaney D. F., Automated quantification of neuronal networks and single-cell calcium dynamics using calcium imaging. J. Neurosci. Methods 243, 26–38 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mank M., Santos A. F., Direnberger S., Mrsic-Flogel T. D., Hofer S. B., Stein V., Hendel T., Reiff D. F., Levelt C., Borst A., Bonhoeffer T., Hübener M., Griesbeck O., A genetically encoded calcium indicator for chronic in vivo two-photon imaging. Nat. Methods 5, 805–811 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Low L. K., Cheng H.-J., Axon pruning: An essential step underlying the developmental plasticity of neuronal connections. Philos. Trans. R. Soc. B Biol. Sci. 361, 1531–1544 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferreira A., Chin L. S., Li L., Lanier L. M., Kosik K. S., Greengard P., Distinct roles of synapsin i and synapsin II during neuronal development. Mol. Med. 4, 22–28 (1998). [PMC free article] [PubMed] [Google Scholar]
- 23.Ferreira A., Rapoport M., The synapsins: Beyond the regulation of neurotransmitter release. Cell. Mol. Life Sci. 59, 589–595 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikolaev M., Heggelund P., Functions of synapsins in corticothalamic facilitation: Important roles of synapsin I. J. Physiol. 593, 4499–4510 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullen D. K., Gilroy M. E., Irons H. R., Laplaca M. C., Synapse-to-neuron ratio is inversely related to neuronal density in mature neuronal cultures. Brain Res. 1359, 44–55 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hochberg L. R., Serruya M. D., Friehs G. M., Mukand J. A., Saleh M., Caplan A. H., Branner A., Chen D., Penn R. D., Donoghue J. P., Neuronal ensemble control of prosthetic devices by a human with tetraplegia. Nature 442, 164–171 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Gilja V., Pandarinath C., Blabe C. H., Nuyujukian P., Simeral J. D., Sarma A. A., Sorice B. L., Perge J. A., Jarosiewicz B., Hochberg L. R., Shenoy K. V., Henderson J. M., Clinical translation of a high-performance neural prosthesis. Nat. Med. 21, 1142–1145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krüger J., Caruana F., Volta R. D., Rizzolatti G., Seven years of recording from monkey cortex with a chronically implanted multiple microelectrode. Front. Neuroeng. 3, 6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prasad A., Xue Q.-S., Sankar V., Nishida T., Shaw G., Streit W. J., Sanchez J. C., Comprehensive characterization and failure modes of tungsten microwire arrays in chronic neural implants. J. Neural Eng. 9, 056015 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Towne C., Montgomery K. L., Iyer S. M., Deisseroth K., Delp S. L., Optogenetic control of targeted peripheral axons in freely moving animals. PLOS ONE 8, e72691 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scharf R., Tsunematsu T., Mcalinden N., Dawson M. D., Sakata S., Mathieson K., Depth-specific optogenetic control in vivo with a scalable, high-density μled neural probe. Sci. Rep. 6, 28381 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Llewellyn M. E., Thompson K. R., Deisseroth K., Delp S. L., Orderly recruitment of motor units under optical control in vivo. Nat. Med. 16, 1161–1165 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Favre-Bulle I. A., Preece D., Nieminen T. A., Heap L. A., Scott E. K., Rubinsztein-Dunlop H., Scattering of sculpted light in intact brain tissue, with implications for optogenetics. Sci. Rep. 5, 11501 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zajdel T. J., Baruch M., Méhes G., Stavrinidou E., Berggren M., Maharbiz M. M., Simon D. T., Ajo-Franklin C. M., PEDOT:pss-based multilayer bacterial-composite films for bioelectronics. Sci. Rep. 8, 15293 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goding J., Gilmour A., Robles U. A., Poole-Warren L., Lovell N., Martens P., Green R., A living electrode construct for incorporation of cells into bionic devices. MRS Commun. 7, 487–495 (2017). [Google Scholar]
- 36.Richardson-Burns S. M., Hendricks J. L., Foster B., Povlich L. K., Kim D. H., Martin D. C., Polymerization of the conducting polymer poly(3,4-ethylenedioxythiophene) (pedot) around living neural cells. Biomaterials 28, 1539–1552 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aregueta-Robles U. A., Martens P. J., Poole-Warren L. A., Green R. A., Tissue engineered hydrogels supporting 3d neural networks. Acta Biomater. 95, 269–284 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Szebenyi G., Callaway J. L., Dent E. W., Kalil K., Interstitial branches develop from active regions of the axon demarcated by the primary growth cone during pausing behaviors. J. Neurosci. 18, 7930–7940 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meberg P. J., Miller M. W., Culturing hippocampal and cortical neurons. Methods Cell Biol. 71, 111–127 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Kalil K., Szebenyi G., Dent E. W., Common mechanisms underlying growth cone guidance and axon branching. J. Neurobiol. 44, 145–158 (2000). [PubMed] [Google Scholar]
- 41.Halloran M. C., Kalil K., Dynamic behaviors of growth cones extending in the corpus callosum of living cortical brain slices observed with video microscopy. J. Neurosci. 14, 2161–2177 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang F., Dent E. W., Kalil K., Spontaneous calcium transients in developing cortical neurons regulate axon outgrowth. J. Neurosci. 23, 927–936 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalil K., Li L., Hutchins B. I., Signaling mechanisms in cortical axon growth, guidance, and branching. Front. Neuroanat. 5, 62 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LaPlaca M. C., Vernekar V. N., Shoemaker J. T., Cullen D. K., Three-dimensional neuronal cultures. Methods Bioeng. 3D Tissue Eng. 2010, 187–204 (2010). [Google Scholar]
- 45.Harrill J. A., Chen H., Streifel K. M., Yang D., Mundy W. R., Lein P. J., Ontogeny of biochemical, morphological and functional parameters of synaptogenesis in primary cultures of rat hippocampal and cortical neurons. Mol. Brain 8, 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franken P., Dijk D. J., Tobler I., Borbely A. A., Sleep deprivation in rats: Effects on eeg power spectra, vigilance states, and cortical temperature. Am. J. Physiol. 261, R198–R208 (1991). [DOI] [PubMed] [Google Scholar]
- 47.Steriade M., Nuñez A., Amzica F., A novel slow (< 1 hz) oscillation of neocortical neurons in vivo: Depolarizing and hyperpolarizing components. J. Neurosci. 13, 3252–3265 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fornasiero E. F., Bonanomi D., Benfenati F., Valtorta F., The role of synapsins in neuronal development. Cell. Mol. Life Sci. 67, 1383–1396 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vanderhaeghen P., Cheng H.-J., Guidance molecules in axon pruning and cell death. Cold Spring Harb. Perspect. Biol. 2, a001859 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/4/eaay5347/DC1