

Abstract
Triple-negative breast cancer (TNBC) is one of the most heterogeneous and aggressive breast cancer subtypes with a high risk of death on recurrence. To date, TNBC is very difficult to treat due to the lack of an effective targeted therapy. However, recent advances in the molecular characterization of TNBC are encouraging the development of novel drugs and therapeutic combinations for its therapeutic management. In the present review, we will provide an overview of the currently available standard therapies and new emerging therapeutic strategies against TNBC, highlighting the promises that newly developed small molecules, repositioned drugs, and combination therapies have of improving treatment efficacy against these tumors.
Keywords: triple-negative breast cancer, cancer, inhibitors, drug resistance, PARP, kinases, microtubules, antibody drug conjugates, combination therapy
1. Introduction
Breast cancer (BC) is a serious worldwide disease that threatens women’s health. Globally, 2.3 million new cases of breast cancer have been diagnosed in 2020, with 685,000 estimated deaths.
Triple-negative breast cancer (TNBC) accounts for 15–20% of all invasive breast cancer and is considered as one of the most heterogenous and aggressive breast cancer subtypes. In fact, it is characterized by a high level of cell invasiveness and a tendency to metastasize to various organs, frequently affecting the brain, lungs, and liver, with an average survival time of 18 months.
Notable ethnic and racial disparities in TNBC incidence have been reported so far. African American women have a higher risk of developing TNBC compared to other racial and ethnic groups. Indeed, most of the available evidence shows that black race, younger age (34% were 18–29 years of age), and later tumor stage are associated with the TN subtype compared to other breast cancer subtypes and suggest a positive correlation between increased parity (having more children) and the risk of developing TNBC, while indicating a negative association between the duration of breastfeeding and the risk of developing this disease. Additionally, excess weight represents a factor significantly contributing to TNBC occurrence. In this respect, Trivers et al. demonstrated in their case study that overweight or obese [odds ratio (OR) = 1.89, 95% confidence interval (CI): 1.22–2.92] women were at a higher risk of developing TNBC [1]. In a different study that analyzed clinicopathological data from 112 triple-negative breast cancer patients in a Turkish hospital over a 5-year period, it was reported that, at the time of diagnosis, 30 patients (26.8%) were normal weight or underweight, while 82 (73.2%) were classified as overweight/obese [2]. Although a potential association exists between diabetes and obesity outcomes, the available information on how diabetes may affect the incidence of TNBC is limited.
Furthermore, there is an indication of a potential influence of diet on the risk of developing TNBC. This is based on epidemiological studies that have demonstrated a slightly significant inverse relationship between a high intake of total vegetables (with a relative risk of 0.82 and a 95% confidence interval of 0.74–0.90) [3] or a diet rich in fruits and vegetables (with a relative risk of 0.85 and a 95% confidence interval of 0.76–0.95) [4] and the risk of developing estrogen receptor-negative (ER-negative) breast cancer.
These findings highlight the link between lifestyle behaviors, race, and the probability of developing TNBC. In fact, women, especially of African ancestry, living in areas of low socioeconomic status were more likely to be diagnosed with TNBC.
Triple-negative breast cancer is characterized by the absence of expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER2); therefore, it will not respond to hormonal therapy or medicines that target HER2, rendering targeted therapeutic options limited [5,6]. Triple-negative breast cancers are highly heterogenous tumors and studies from the Lehmann group, based on genomic and transcriptomic analysis, have led to the identification of six distinct and stable molecular subtypes: basal-like 1 (BL1), basal-like 2 (BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR) [5,7].
The primary obstacles in the conventional therapeutic approach to TNBC lie in dealing with intrinsic and acquired chemoresistance. Given the considerable diversity within the TNBC subtypes, multiple lines of evidence suggest that the implementation of molecular stratification for TNBC patients in clinical practice could be a key step in addressing intrinsic chemoresistance effectively. As a proof of concept, these subtypes were replicated in breast cancer cell lines and subsequently used to guide the selection of pharmacological treatments. The BL1 subtype exhibited abnormal levels of gene expression (MYC, PIK3CA, CDK6, AKT2, KRAS, FGFR1, IGF1R, CCNE1, CDKN2A/B, BRCA2, PTEN, MDM2, RB1, and TP53) related to cell proliferation, cell cycle regulation, and DNA repair. Therefore, it is generally considered one of the more aggressive subgroups, with a higher risk of recurrence and metastasis. Santonja et al. demonstrated that BL1 cell lines appeared to be particularly sensitive to chemotherapy regimens including platinum agents [8].
The BL2 subtype has a unique gene expression profile characterized by the upregulation of genes associated with growth factor signaling pathways (EGFR, MET, NGF, Wnt/β-catenin, and IGF-1R) and myoepithelial markers. As well as BL1, BL2 is associated with early recurrence and metastasis. The pathological response (pCR) rate for patients with the BL2 subtype, after neoadjuvant chemotherapy, is reported to be only 10% due to the poor sensitivity to the combination regimen [9].
The IM subtype is composed of immune antigens and genes involved in cytokine and core immune signal transduction pathways. Patients with tumors categorized as the IM subtype tend to have a more favorable prognosis compared to other TNBC subtypes. An examination of the gene expression data within the IM subtype and the identification of transcripts associated with lymphocytes suggest that these IM tumor samples may harbor tumor-infiltrating lymphocytes (TILs). Recent findings from two adjuvant phase III trials have demonstrated a favorable outcome in TNBC patients with higher levels of TILs in their tumors [10]. In addition, given the immune-rich nature of the IM subtype, there is growing interest in exploring immunotherapeutic approaches as a treatment option for these patients.
The M subtype is characterized by a highly activated cell migration-related signaling pathway (regulated by actin), interaction between extracellular matrix and cell receptor, and differentiation pathways (Wnt pathway, anaplastic lymphoma kinase pathway, and transforming growth factor (TGF)-β signaling). Due to these distinctive features, it is often referred to as metaplastic breast cancer. The M subtype exhibits tissue characteristics resembling sarcoma or squamous epithelial cells and tends to develop resistance to chemotherapy. As a result, patients with the M subtype may be considered for treatment with the mammalian target of rapamycin and phosphatidylinositol-3-kinase inhibitors or drugs that target the process of epithelial–mesenchymal transition (EMT) [9].
Both M and MSL share an elevated expression of genes involved in EMT and growth factor pathways; however, the MSL subtype expresses low levels of cell proliferation-related genes and high levels of stemness-related genes, HOX genes, and mesenchymal stem cell-specific markers [9]. MSL subtype patients can be treated with the multifamily tyrosine kinase inhibitor dasatinib that binds to the kinase domain of Bcr-Abl.
The LAR subtype exhibits a notably distinct gene expression profile compared to other TNBC subtypes. Despite the absence of expression of ER, it is characterized by highly activated hormone-related signaling pathways (including steroid synthesis, porphyrin metabolism, and androgen/estrogen metabolism). Notably, the androgen receptor (AR) is highly expressed in LAR subtype breast cancer, with its mRNA level being approximately nine times greater than in other TNBC subtypes [11]. Therefore, anti-AR therapy (e.g., bicalutamide as an AR antagonist) is recommended for patients with LAR subtype breast cancer.
Compared to non-TNBC patients, TNBC patients suffer from worse clinical outcomes, largely due to heterogeneity, lack of treatment options, and frequency of recurrence and metastasis [12,13,14]. Women diagnosed with TNBC between 2012 and 2018 showed a 5-year overall survival rate of 77%, compared with 93% in breast cancer patients of other types [15]. Recently, the main treatment methods for TNBC are mastectomy or breast-conserving surgery, radiation therapy, and chemotherapy. Due to its molecular heterogeneity, poor cell differentiation, high malignancy, lack of molecular targets, and poor prognosis often ascribed to resistance to chemotherapeutic agents, there is no targeted therapy to prevent the recurrence of TNBC [16]. Overall standard neoadjuvant chemotherapy yields a pathological complete response in a limited proportion of patients with TNBC (35–45% in 2020) [17]. Hence, continuous efforts are being pursued to develop more targeted therapies, particularly for patients with advanced TNBC who poorly respond to current chemotherapeutic agents and, even after a good response, show rapid disease progression.
Thus far, TNBC is very difficult to treat. Several novel agents, such as single-target drugs and repurposing drugs, have been developed and evaluated in clinical studies for immuno- and targeted-TNBC therapies. In this review, we will focus on the currently available standard therapies against TNBC and on the new advancement in the development of small molecules for TNBC.
2. Conventional Chemotherapy
Conventional chemotherapy, consisting of different combinations of anthracycline, taxane, cyclophosphamide, and fluorouracil, is the mainstay of adjuvant systemic treatment for most patients with early-stage TNBC, although sometimes schedules are limited by toxicity and tumor response. At present, the 90% of drug failures in metastatic cancers is caused by the development of multidrug resistance by tumor cells. In this respect, tumor cells have given rise to the ability to survive after chemotherapeutic exposure through the employment of several mechanisms, such as APC transporters, β-tubulin III, mutations in DNA repair enzymes such as topoisomerase II and DNA mismatch repair enzymes, alterations in genes involved in apoptosis, ALDH1 and glutathione (GSH)/Glutathione-S-transferase (GST), and NF-kB signaling pathways [18].
In addition to standard chemotherapy, the addition of any other drugs has been regarded as a new regimen [19].
The antimetabolite 5-fluorouracil (5-FU) (1) (Figure 1) is widely employed in treating different cancers, including breast cancer. The chemical structure of this antitumor agent is comprised of a heterocyclic aromatic ring with high similarity to uracil, bearing a fluorine atom in 5C position of the aromatic ring. Continuous progress in comprehending its mechanism of action has led to its extensive anticancer applications [20]. It has been indicated that the anticancer effect of 5-FU, when it is administered as a single agent chemotherapeutic drug, is mainly induced by its conversion to three active metabolites: fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP). In fact, these metabolites cause cell injury by two different mechanisms. Firstly, the binding of FdUMP to the nucleotide binding site of thymidylate synthase (TS), an essential enzyme for catalyzing the reductive methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP), with the folate cofactor 5,10-methylene tetrahydrofolate (CH2THF), as the methyl donor, resulted in the formation of a stable ternary TS–FdUMP–CH2THF complex blocking access of dUMP to the nucleotide binding site and, therefore, in the inhibition of dTMP synthesis. Since dTMP is essential for DNA replication and repair, its depletion causes lethal DNA damage. Furthermore, the cytotoxic effect of 5-FU is also triggered by the incorporation of its active metabolite, FUTP, into RNA. This not only resulted in the inhibition of the processing of pre-rRNA into mature rRNA but also interfered with the post-transcriptional modification of tRNAs and the assembly and activity of snRNA/protein complexes, thereby inhibiting splicing of pre-mRNA. Therefore, the misincorporation of 5-FU has the potential to disrupt various facets of RNA processing, leading to significant effects on cellular metabolism and survival.
Figure 1.

Chemical structures of conventional chemotherapeutic agents.
Moreover, the outbreak of drug resistance, due to the upregulation of ATP-binding cassette (ABC) transporters, is a very common phenomenon among breast cancer patients who were administered 5-FU. In fact, it has been demonstrated that multidrug-resistant protein-1 (ABCC1/MRP1), breast cancer resistance protein (ABCG2/BCRP), and multidrug-resistant protein-8 (ABCC11/MRP8), expressed more frequently in TNBC compared to other breast cancer subtypes, are responsible for the upregulation of ABC transporters that utilize ATP to efflux various compounds—including a wide range of anticancer drugs—across cellular membranes for conferring resistance to 5-FU and other drugs that represent the backbone of current TNBC treatment. The biological effects of developing 5-FU resistance include a decline in apoptosis, disorder in cell cycle, enzyme malfunctioning, etc. [21].
Capecitabine (2) is an oral prodrug, metabolized in vivo to 5-FU by carboxylesterases, cytidine deaminase, and thymidine phosphorylase/uridine phosphorylase sequentially, which has shown effectiveness in treating advanced BC [22] and gastric cancer [23]. In early-stage TNBC patients, capecitabine in combination with standard adjuvant chemotherapy showed significant disease-free survival and overall better survival outcomes than standard chemotherapy with tolerable adverse events [24]. A study showed that the addition of capecitabine-based chemotherapy was the most effective regime [25].
Gemcitabine (2′,2′-difluorodeoxycytidine, dFdC) (3), an efficient chemotherapeutic drug for the treatment of various types of cancer in clinical practice, was also evaluated in the treatment of TNBC in several clinical trials [26]. DFdC is a potent and specific deoxycytidine analog which, once inside the malignant cell, is firstly phosphorylated by deoxycytidine kinase to its monophosphorylated form and subsequently by nucleotide kinases to its active metabolites, dFdC diphosphate (dFdCDP) and dFdC triphosphate (dFdCTP). These active metabolites are nucleosides that mediate antitumor effects. dFdCDP works as an inhibitor of ribonucleotide diphosphate reductases, an enzyme responsible for catalyzing the biosynthesis of deoxycytidine triphosphate (dCTP), which is a precursor necessary for DNA synthesis, from the corresponding ribonucleotide [27]. Therefore, the overexpression of ribonucleotide reductase is associated with the emergence of resistance to dFdC [28]. In addition, the cytotoxicity of dFdC is mainly associated with the cellular accumulation of dFdCTP. dFdCTP competes with dCTP for incorporation into DNA, thereby competitively inhibiting DNA chain elongation. This process is referred to as “masked DNA chain termination” and induces a G0/G1 and S-phase arrest in the cell cycle, which triggers apoptosis [29].
To overcome the drug resistance induced by nucleoside kinase deficiency, various therapeutic approaches have been postulated. In this context, a multisubstrate deoxyribonucleoside kinase of Drosophila melanogaster in the nucleus or cytosol has been reported as a potential candidate suicide gene for reversing acquired dFdC resistance in TNBC cells [30].
Standard anthracycline-based chemotherapy is the treatment of choice as first-line chemotherapy for metastatic breast cancer patients not previously treated with anthracyclines [31]. Its mechanism of action includes DNA intercalation, membrane binding, free radical formation, DNA repair cascade degradation, and cell death. Although anthracyclines represent an important component of adjuvant chemotherapy, they are associated with several short and long-term adverse events, with the major being cardiotoxicity and secondary leukemia [32,33].
Nowadays, in order to avoid unnecessary anthracycline treatment due to its associated cardiotoxicity, taxane-based regimens constitute another standard therapy for breast cancer patients, especially those affected with TNBC. Paclitaxel (PTX), isolated from the bark of the Pacific Yew, and docetaxel, a semisynthetic analog from the renewable and more readily available leaves of the European yew tree, are among the most active agents for metastatic breast cancer. The assembly promoting properties of PTX (4) were firstly reported in 1979 [34]. PTX, through its effects on microtubules, inhibits the growth of a variety of solid tumor cells causing, at high concentration, mitotic arrest at the G2/M phase, whereas, at low concentration, it induces cell apoptosis at the G0 and G1/S phase; however, its clinical application is limited due to poor water solubility.
Many studies have illustrated that PTX combined with 5-FU shows synergic activity and improved tolerability towards some breast carcinoma, ovarian cancer, and gastric cancer [35,36]. Chen et al. [37] have recently reported the development of KLA-modified liposomes co-loaded with 5-FU and PTX (KLA-5-FU/PTX Lps). This new drug formulation was evaluated for its antitumor activity against human breast cancer cells (MDA-MB-231). It showed enhanced cytotoxicity against MDA-MB-231 cells, improved drug delivery to mitochondria, induced mitochondria-mediated apoptosis, and turned out a promising system to target the delivery of antitumor drugs to mitochondria as a treatment for TNBC [37].
For patients unresponsive to the treatment mentioned above, adjuvant capecitabine or platinum-based chemotherapy, such as carboplatin and cisplatin, might be given, although it is still controversial, as researchers are currently leading an ongoing randomized phase III trial to validate the superiority of either adjuvant capecitabine or platinum-based chemotherapy [38].
A drug combination of gemcitabine and cisplatin (cis-diamminedichloroplatinum, cis-DDP (5) has proven to be superior to dFdC/PTX in first-line treatment of metastatic TNBC in terms of progression-free survival (PFS) [39]. The potential effect of nanoparticle albumin-bound (nab)-PTX and cis-DDP was assessed for metastatic mucinous adenocarcinoma [40] and metastatic TNBC [41]. Wang et al. [41] conducted a randomized phase III controlled open-label trial to compare the efficacy of nab-PTX/cis-DDP with dFdC/cis-DDP in metastatic TNBC patients. The study confirmed that the combination of nab-PTX with cis-DDP, as compared to dFdC/PTX, significantly increased the OS and led to a significantly higher objective response rate. The reduction in the risk of death may result from the higher antitumor activity of nab-paclitaxel over gemcitabine when combined with cisplatin.
Eribulin (NSC 707389) (6) is another medication that has been added to the armamentarium of drugs against TNBC after being studied in patients who have received at least two chemotherapeutic regimens, which include an anthracycline and a taxane. It is a non-taxane synthetic analogue of halichondrin B, isolated from the marine sponge Halichondria okadai, which acts as an irreversible inhibitor of microtubule polymerization [42]. Since it does not affect the microtubule depolymerization, it causes less toxicity compared to the previously reported taxanes [43]. This results into apoptosis through the disruption of mitotic spindles and an irreversible block of the cell cycle at the G2-M level. Currently, eribulin is used to treat HER2-negative metastatic or recurrent BC that failed previous exposure to anthracyclines and taxanes, and for treatment of TNBC as well [44]. Eribulin in combination with the PARP-1, PARP-2, and PARP-3 inhibitor olaparib showed good efficacy for advanced TNBC patients and was well tolerated [45].
3. Poly Adenosine Diphosphate-Ribose Polymerase
The integrity and stability of DNA in breast epithelial tissues are key factors in breast homeostasis. However, carcinogenesis is mostly initiated by DNA damage which is an ongoing process resulting from both endogenous (errors in replication) and exogenous (environmental) assaults to the human genome. Usually, cell repair mechanisms ensure that cells with damaged DNA undergo either repair or apoptosis. Therefore, inhibition of these processes can lead to a buildup of damaged DNA in cells, resulting in apoptosis or senescence of the tumor cells. Poly(ADP-ribose) polymerase (PARP) inhibitors have been closely examined as one of the most exciting and promising “targeted” therapeutic strategies to treat advanced TNBC by preventing cancer cells from repairing themselves. Indeed, it has been demonstrated that 10–20% of total patients diagnosed with TNBC have a mutation in breast cancer susceptibility genes (BRCA)1 or (BRCA)2 [46]. BRCA1 and BRCA2 are tumor suppressor genes that are associated with a hereditary predisposition to developing female breast cancer [47], but their pivotal role in the DNA damage response makes cancer cells harboring such mutations more sensitive to drugs eliciting DNA damage or interfering with DNA repair, such as PARP inhibitors.
PARPs are a family of multifunctional enzymes involved in several cellular processes, including DNA repair mechanism and apoptosis [48]. PARP-1, a nuclear, zinc-finger, DNA-binding protein that has been identified as the most abundantly expressed and characterized isoform of the PARP family, localizes to DNA strand breaks as part of the base excision repair process [49]. Upon detection of the DNA damage, PARP-1 catalyzes the addition of a poly-ADP-ribose (PAR) chain to target proteins, thus recruiting repair factors to repair DNA [50]. It is well known that PARP inhibitors are particularly effective against tumors carrying mutations in BRCA. In fact, it has been demonstrated that it is possible to achieve synthetic lethality and increased tumor cell death through the prevention of DNA repair via PARP inhibition, in conjunction with the loss of broken double-strand DNA repair via BRCA-dependent mechanisms. In other words, it has been indicated that the detection of a mutated BRCA gene in some TNBC patients results in the blockage of the repair process of broken single-stranded DNA via PARP inhibitors by obstructing PARP enzyme activity and PARylation reactions through the competition with coenzyme, NAD+, for the interaction with the PARP catalytic domain; in addition, BRCA mutants cannot induce homologues recombination to repair double-stranded DNA, with the consequence of synthetic lethal effects on tumor cells [51]. Therefore, this makes the inhibition of PARP an attractive target for TNBC tumors that are BRCA deficient [52]. The discovery and development of PARP inhibitors began more than 50 years ago using the nicotinamide functional group [53]. Later, PARP inhibitors were designed by introducing nicotinamide and benzamide functional groups into their structure in order to enable them to bind to the catalytic portion of PARPs [54].
3.1. Iniparib (7)
Iniparib was evaluated as the first PARP inhibitor potential candidate agent for TNBC; however, it failed clinical trials and a definitive in vitro study revealed that the agent did not appreciably inhibit PARP [55] (Figure 2).
Figure 2.

Chemical structure of PARP inhibitors.
Here, we have reported several potential PARP inhibitors for TNBC that have been recently discovered and used in clinical progression trials.
3.2. Olaparib (8)
Olaparib was the first small-molecule PARP inhibitor to show clinical efficacy and tolerability in BRCA1/BRCA2-mutated advanced BC [56] (Figure 2). First reported as a PARP-1 and PARP-2 inhibitor, it also showed a potent PARP-3 inhibition. Olaparib and talazoparib (TALA) (9) were approved in 2018 by the Food and Drug Administration as monotherapy for the treatment of metastatic TNBC harboring a germline BRCA1 (gBRCA) or BRCA2 mutation [57,58], based on the results of two OlympiADand EMBRACA clinical trials. Approximately 15% of patients with TNBC have gBRCA mutations, which make them good candidates as PARP inhibitors. Moreover, the use of the PARP inhibitor olaparib in the treatment of TNBC patients without BRCA mutations has recently been shown to be ineffective [59]. Noteworthy, in HER2-negative metastatic BC and BRCA mutation patients, olaparib alone has a remarkable advantage on the standard treatment. However, cancer multidrug resistance (MDR) represents a major challenge for effective cancer treatment, and overexpression of a P-glycoprotein (P-gp) and a breast cancer resistance protein (BCRP) has been hypothesized to be one of the mechanisms responsible for acquired resistance to olaparib. Thus, a second generation of PARP inhibitors has been developed and tested in several clinical trials.
3.3. Talazoparib (9)
Talazoparib shows potent PARP inhibition and trapping potential superior to other PARP inhibitors [60] (Figure 2). It achieved pathologic complete responses in germline BRCA-positive, HER2-negative patients with early breast cancer, including TNBC [61]. Of note, in EMBRACA clinical trials, treatment with single-agent talazoparib in patients with advanced BC and a germline BRCA1/2 mutation, provided a significant PFS and a higher pathological remission rate compared to standard chemotherapy. Patient-reported outcomes were superior with talazoparib [62]. Unfortunately, even with the development of a second-generation PARP inhibitor, the rise of MDR could not be prevented. Eskiler et al. [63] have recently reported that talazoparib resistance is mediated by overexpression of BCRP and multidrug resistance-associated protein 1 (MRP1) genes in BRCA1 mutant TNBC. In this regard, the authors, with the aim to overcome the abovementioned drug-resistance, developed novel talazoparib-solid lipid nanoparticles (SLNs), through a hot homogenization technique, as a promising therapeutic carrier to reverse MDR-mediated resistance in TNBC, showing significantly higher apoptotic rates in vitro than the free drug [63]. In fact, several in vivo and in vitro studies reported increased intracellular drug accumulation in cancer cells and effective overcoming of drug efflux-mediated resistance of SLNs.
3.4. Veliparib (10)
Veliparib is a benzimidazole, having a substitution at C-4 with a carbamoyl cluster and (2R)-2-methylpyrrolidin-2-yl moiety at C-2 (Figure 2). In combination with carboplatin and PTX followed by doxorubicin and cyclophosphamide, it improved the pathological complete response of patients with TNBC (but not when veliparib was added to carboplatin and PTX alone). The addition of carboplatin was considered as a potential component of neoadjuvant chemotherapy for patients with high-risk TNBC [64]. In addition, veliparib is being investigated in combination with radiation to treat patients with advanced TNBC, and thus far, the results are pending (NCT01618357).
3.5. Rucaparib (11)
A small subset of patients with high genomic loss of heterozygosity score or non-germline BRCA1/2 mutation derived benefit from the PARP inhibitor rucaparib [65] (Figure 2). Currently, rucaparib is under investigation in combination with other agents such as immunotherapy, vascular epidermal growth factor receptor (VEGFR) inhibitors, or radiotherapy to treat solid tumors including TNBC (NCT03992131, NCT03542175, and NCT03911453). In 2020, a study assessing the efficacy of radiotherapy in combination with rucaparib was completed, the results of which have not yet been published.
Rucaparib camsylate (camphorsulfonate salt obtained via reaction of rucaparib with one molar equivalent of (1S,4R)-camphorsulfonic acid) has been formulated as film-coated tablets for oral route of administration. It is indicated as a monotherapy treatment of patients with advanced ovarian cancer with germline and/or somatic BRCA mutation who have experienced two or more chemotherapies. Rucaparib camsylate is under clinical development by Clovis Oncology and currently in phase II for TNBC [66].
3.6. Niraparib (12)
Niraparib, in a single-arm, phase II study, demonstrated promising antitumor activity and safety in patients with localized HER2-negative, BRCA-mutated breast cancer (Figure 2). In a pilot, single-arm, phase II study, a neoadjuvant treatment with niraparib as a single-agent demonstrated promising antitumor activity and high levels of tumor penetration in patients with HER2-negative, BRCA-mutated, localized BC. Niraparib showed superior tumor penetration to other PARP inhibitors [67].
3.7. Pamiparib (BGB-290) (13)
A new PARP inhibitor undergoing clinical evaluation in patients with ovarian cancer and TNBC can inhibit PARP1 and PARP2 (NCT03333915) (Figure 2). Moreover, it is being investigated for its efficacy in solid tumors including TNBC as a monotherapy and in combination with the chemotherapy agent temozolomide (NCT03150810). In a recent open-label, phase II, multicenter study in China (NCT03575065), pamiparib showed encouraging efficacy and an acceptable safety profile in patients with locally advanced and metastatic HER2 breast cancer with germline BRCA1/2 mutation [68].
PARP inhibitors represent a valid therapeutical option against the TNBC (Figure 2). Novel PARP inhibitors are currently under investigation for use alone and in combination with established agents. Although BRCA-deficient tumors are more sensitive to PARPi due to its synthetic lethality, nearly 40% of BRCA1/2-deficient patients do not respond to PARPi due to the emergence of drug resistance mechanisms in TNBC. It is worth noting that the administration of PTX and doxorubicin, which are substrates of the MDR1 transporter, prior to PARPi treatment may lead to the upregulation of MDR1 and indirectly induce PARP resistance. In a specific study, the use of paclitaxel before PARPi was found to be significantly linked to the presence of ABCB1 fusion transcripts [69]. However, the use of doxorubicin before PARPi did not show a significant association with ABCB1 fusion transcripts. Other studies have also demonstrated the existence of cross-resistance between MDR1, PARPi, and paclitaxel [70,71]. Therefore, conversely, the occurrence of MDR1 overexpression as a mechanism of resistance to PARPi has implications for the choice of subsequent treatment following PARPi resistance.
An even greater challenge is to decipher the mechanisms of intrinsic PARP inhibitor resistance among patients with gBRCAm. Therefore, there is still a need to improve the therapeutic options for TNBC patients, especially in specific TN molecular subtypes [72].
4. Cyclin-Dependent Kinases
Cyclin-dependent kinases (CDKs) are an evolutionary conserved serine/threonine protein kinases family which do not possess autonomous enzymatic activity but need to be bound to a cyclin partner to function properly. The human genome encodes 20 CDKs, divided into two subfamilies: cell cycle-associated CDKs (CDK1−7 and CDK14−18) and transcription-associated CDKs (CDK7−13, 19, and 20). CDKs are master regulators of cell cycle control, transcription, and pre-mRNA processing, and their proper activity ensures genetic integrity, cell division, and cell cycle regulation [73,74,75]. Hence, it appears clear their relevance in tumor cell proliferation and growth, which has prompted, for more than 25 years so far, the development of small molecules inhibiting their oncogenic activity.
4.1. CDK4 and CDK6 Inhibitors
Within the CDKs family, inhibitors of cyclin-dependent kinase 4 (CDK4) and 6 (CDK6) have been long considered as the most relevant drug candidates for cancer therapy due to their potential to restore control of the cell cycle [76]. CDK4 and CDK6 regulate progression through the cell cycle G1 phase and cell cycle initiation. In detail, both kinases form a complex with cyclin D1 that phosphorylates the tumor suppressor protein retinoblastoma (Rb) (or its homologs, p107 and p130). This activity blocks the growth inhibitory function of Rb; indeed, phospho-Rb (pRb) releases its grip and activates the transcription factors E2F, allowing the transcription of genes which are crucial for cell cycle progression to the S phase [77]. Overexpression of cyclin D1 characterizes almost 50% of BC tumors [78], and nearly 30% of TNBC presents Rb dysfunction [79]. According to The Cancer Genome Atlas, cyclin D1 amplification is preferential to luminal-type tumors, especially luminal B [80].
The first generation of CDK inhibitors (CDKi) (e.g., flavopiridol, olomucine, and roscovitine), which were designed to halt cell cycle and cell proliferation by blocking CDK enzymatic activity, suffered from poor selectivity and high toxicity in normal cells. Successively, a second generation of CDKi (e.g., dinaciclib an CYC065) has been developed with greater selectivity and fewer side effects [81]. Finally, among the third generation of CDKi, in March 2017, selective CDK4/6 inhibitors received FDA approval for the treatment of post-menopausal women with hormone receptor (HR)-positive metastatic breast cancer, in combination with an aromatase inhibitor as initial endocrine-based therapy. Currently, the three FDA- and EMA-approved CDK4/6 inhibitors are palbociclib (14), ribociclib (15), and abemaciclib (16) (Figure 3).
Figure 3.

Chemical structure of CDK4 and CDK6 inhibitors.
4.1.1. Palbociclib (PD-0332991, 14)
Palbociclib is a highly selective cyclin-dependent kinases 4 and 6 (CDK4/6) inhibitor which hampers the phosphorylation of CDK4/6, resulting in hypophosphorylation of Rb and in the blockage of cell cycle progression from the G1 to the S phase, thus preventing DNA synthesis required for cellular replication [82]. Palbociclib has been shown to inhibit the growth of a panel of TNBC cell lines, albeit with a lesser extent than ER-positive cell lines [82].
Although CDK 4/6 inhibitors are a feasible option for BC therapy, resistance to these inhibitors represents a significant barrier to their effective use. It has been reported that non-luminal/basal subtypes were the most resistant to palbociclib [83]. One of the potential drug resistance mechanisms observed in 7–20% of TNBC patients is the decline in Rb1 expression after being exposed to CDK4/6 inhibitors [84]. Therefore, considering the frequent occurrence of Rb downregulation in TNBC, which results in the accumulation of p16ink4a, the use of CDK4/6 inhibitors alone may not be the optimal choice of treatment for TNBC patients [85]. To date, the most significant successes of overcoming resistance in patients with metastatic ER+/HER2 cancer, are represented by the combination of CDK4/6 inhibitors with endocrine therapies [86,87].
A screening on multiple TNBC cell lines, representative of the heterogeneity of this subtype, revealed the luminal androgen receptor subgroup as the most sensitive to palbociclib-mediated CDK4/6 inhibition [84]. In this regard, Tseng et al. found that palbociclib effectively inhibited retinoblastoma-proficient TNBC cell growth, and the expression of androgen receptors might contribute to palbociclib-mediated G1 arrest [88]. Moreover, they showed that treatment with the androgen receptor antagonist enzalutamide enhanced the palbociclib-induced cytostatic effect in androgen receptor-positive/retinoblastoma-proficient TNBC cells.
4.1.2. Ribociclib (LEE011, 15)
Similar observations have been made for ribociclib, another orally accessible, selective, small-molecule inhibitor of CDK4/6 that prevents cell cycle progression and induces G1-phase arrest by inhibiting the phosphorylation of the retinoblastoma protein. Indeed, Hosseini et al. recently evaluated the long-term consequences of the treatment of ribociclib in combination with enzalutamide [89]. They demonstrated that the combination of ribociclib with enzalutamide in an androgen receptor-positive model (MDA-MB-231 and MCF-7 cell lines) results in a more effective improvement in the cell cytotoxicity than the separate treatments, suggesting a potential approach for enhancing antitumor effectiveness. Collectively, these observations suggest that combined inhibition of CDK4/6 and AR is a promising therapeutic strategy for androgen receptor-positive/retinoblastoma-proficient TNBCs.
The activation of alternative proliferative pathways, such as mTOR and PI3K, and the deregulation of D-type cyclins expression represent further mechanisms of resistance to CDK4/6 inhibitors [87,90]. More recently, sequestration of CDK4/6 inhibitors into tumor cell lysosomes was shown to be another evasion mechanism more active in TNBC cells than in other BC cell lines, which might be counteracted by the concomitant administration of lysosome-destabilizing compounds [91]. Remarkably, inhibition of a CDK, cyclin-dependent kinase 2 (CDK2), which, in association with cyclin E, regulates the transition from the G1 to the S phase, synergically with CDK4/6, was shown to sensitize resistant TNBC cells to CDK4/6 inhibition [87,91].
4.1.3. Abemaciclib (16)
As well as all of the new generation selective CDK4/6 inhibitors covered in this review, abemaciclib, an approved monotherapy, also plays an important role in G1–S phase transition in the cell cycle. Although preclinical and clinical studies have shown the therapeutic potential of abemaciclib in pretreated HR+, HER2− breast cancer patients, compared to other CDK4/6 inhibitors, its potential effects on TNBC therapy have not been definitively elucidated. Recently, Sekeroglu et al. demonstrated that abemaciclib caused significant apoptosis in TNBC cells via G0/G1 arrest, chromatin condensation, the upregulation of caspase-3 and Bax levels, and the downregulation of Bcl-2. The formation of a large number of cytoplasmic vacuoles not associated with autophagy suggested that treatment with 16 could be effective for TNBC [92] (Figure 3).
In a study published in 2019 by Petronini et al., it has been also highlighted that the sequential treatment of palbociclib for 24 h followed by PTX for a further 48 h in both MDA-MB-231 and HCC38 TNBC cell lines produced an additive inhibitory effect on cell proliferation associated with a significant increase in apoptotic cell death [93]. The efficacy of this schedule relies on the reversible action of palbociclib on the cell cycle; in fact, upon palbociclib removal, the cells arrested in the G1 phase synchronously re-enter the cell cycle in the S phase, becoming more sensitive to the cytotoxic effect of PTX. On the other hand, it has been demonstrated that the simultaneous combination of palbociclib with increasing concentrations of PTX give rise to an antagonistic effect, highlighting the importance of the successful use of sequential therapy in order to minimize the insurgence of adverse effects and to allow patients to derive benefit from the additional treatment. [93,94]. Indeed, the above-mentioned antagonism could be ascribed to the reduced activity of cytotoxic chemotherapeutic agents, directed against cycling cells, when used in cells already arrested in the G0/G1 phase.
Moreover, the pretreatment of TNBC cells with palbociclib led to an increase in sensitivity to cisplatin therapy, enhancing the anticancer activity. Therefore, these results suggest that the sequential use of CDK4/6 inhibitors with SOC agents might provide an alternative therapeutic approach for TNBC [95].
4.2. CDK2 Inhibitors
4.2.1. Roscovitine (17)
Similarly, sensitizing effects to taxanes in TNBC cells were shown for the CDK2 inhibitor roscovitine [96], which was also shown to interfere with TNBC cell migratory and metastatic properties [97] (Figure 4).
Figure 4.

Chemical structure of CDK2 inhibitors.
4.2.2. Dinaciclib (18)
In addition, CDK2 inhibition via dinaciclib treatment was shown to promote reactivation in TNBC cells of the ER expression, rendering them sensitive to antiestrogen therapy, such as tamoxifen [98] (Figure 4). This latter activity was independent of the prominent role of CDK2 in cell cycle regulation but was correlated to its ability to phosphorylate the epigenetic transcriptional regulator EZH2 [98]. However, clinical trial probing safety and efficacy of the combination of dinaciclib with anthracyclines failed due to its high toxicity and poor response [99]. Thus, the development of novel specific CDK2 inhibitors represents an open field of study, with high potential for TNBC pharmacotherapy.
4.3. CDK7 and CDK12 Inhibitors
Among the transcriptional CDKs ensuring proper progression of the transcription cycle and its accurate coordination with the pre-mRNA processing events, CDK7 is one of the most promising therapeutic targets for TNBC treatment. CDK7 associates with cyclin H and is part of the general transcription factor TFIIH, which regulates transcription initiation by phosphorylating both the C-terminal domain (CTD) of the RNA polymerase II (RNPII) at Ser5 and CDK9, the catalytic subunit of the transcription elongation factor P-TEFb [75]. TNBC was shown to be highly sensitive to chemical inhibition of CDK7 activity via treatment with the inhibitors THZ1 and its derivative THZ2, which significantly display growth inhibitory effects in both TNBC cell lines and xenograft models [100]. Sensitivity to these inhibitors was correlated to a transcriptional addiction of TNBC to CDK7, whose activity ensures proper expression of genes essential for TNBC tumorigenicity [100], including condensin genes guaranteeing chromosome stability [101]. Coherent with CDK7’s essential role for TNBC cell survival, high expression levels of this kinase are a marker of poor prognosis for TNBC patients [102].
4.3.1. Samuraciclib (ICEC0942; CT7001) (19)
A small-molecule, adenosine triphosphate (ATP)-competitive inhibitor of CDK7, orally available, showed tolerable safety and partial clinical benefit for TNBC patients, thus supporting further studies evaluating its potential as a novel therapeutic strategy [103] (Figure 5).
Figure 5.

Chemical structure of CDK7, CDK12, and CDK13 inhibitors.
Although among the other transcriptional kinases only CDK9 was found to be another essential gene for TNBC cell survival [100], CDK12 also represents a therapeutic target of high interest for this tumor subtype. CDK12, together with its highly homologous CDK13 associate with cyclin K, regulates transcriptional elongation by mediating RNPII CTD phosphorylation at Ser2 in the gene body and towards the 3′-end of the transcription units [75].
4.3.2. SR-4835
CDK12 and CDK13 were found to be essential for the proper splicing and the prevention of premature intronic polyadenylation of pre-mRNAs encoded by long genes, thus ensuring their proper expression [104,105] (Figure 5). Notably, expression of DNA damage response (DDR) genes was shown by multiple studies to be highly sensitive to CDK12/13 depletion and/or inhibition [104,105,106,107]. Impaired pre-mRNA processing and consequent downregulation of the expression of genes in the DDR pathway caused by treatment with SR-4835, a selective dual inhibitor of CDK12 and CDK13, was shown to elicit in DNA repair-proficient TNBC cells, such as BRCA1/2 wild-type cells, a “BRCAness” condition which increases their sensitivity to DNA-damaging chemotherapy and PARP inhibitors [108]. By eliciting similar downregulation of DDR genes, profound antiproliferative effects and synergy with PARP inhibitors in TNBC cells were also shown via treatment with PROTAC degraders targeting either both CDK12 and CDK13 [109] or specifically CDK12 (PP-C8) [110]. CDK12/13 inhibitors represent an important innovation in the field of TNBC chemotherapy, as they might allow administration of PARP inhibitors also to tumors displaying either innate (i.e., BRCA1/2 wt) or acquired resistance to these molecules [107,108]. In this regard, particular interest is merited by some recently developed compounds able to simultaneously inhibit CDK12 and PARP1 activity, whose antitumoral activity in TNBC cells has just been preliminarily tested [111]. Nevertheless, as the broad transcriptional and post-transcriptional changes are elicited by CDK12/13 inhibition in TNBC, it is conceivable that such treatment might elicit other actionable vulnerabilities in these tumors, as recently shown by analogous treatment in high-grade serous ovarian cancer [112].
Overall, the pivotal role that CDKs exert in controlling the mammalian cell cycle and transcription and their checkpoints raises the possibility of devising therapeutic strategies based on the druggability of these molecules.
5. Microtubules
In addition to the development of PARP and CDK inhibitors, another strategy for the treatment of patients with TNBC involves the employment of microtubule-targeting agents. Microtubules play critical roles in a wide number of cellular functions, such as motility, division, shape maintenance, and intracellular transport. The major protein component found in microtubules is tubulin. Microtubule-targeting agents (MTAs) inhibit the function of cellular microtubules by promoting polymerization and depolymerization, resulting in the arrest of cell cycle progression and induction of cell death [113,114].
Existing tubulin inhibitors, such as PTX, have shown great effectiveness in treating breast cancer; however, their use is limited by the need for intravenous administration due to poor aqueous solubility. In addition, their clinical use is often limited by drug resistance mediated by ABC transporters and neurotoxic side effects. Extensive preclinical study investigations have suggested that tubulin inhibitors designed to target the colchicine binding site are notably less susceptible to transporter-mediated drug resistance [115]. Although, up to this point, none of these colchicine binding site inhibitors (CBSIs) have gained FDA approval, mainly due their toxicity profiles and limited clinical efficacy; several novel CBSIs have been developed as alternatives to PTX and tested in clinical trials.
5.1. VERU-111
A series of compounds, termed ABI-III chemotypes, were first reported in 2012 by Chen et al. as potent tubulin polymerization inhibitors able to overcome ABC transporter-mediated multidrug resistance [116]. More recently, Deng et al. [117,118] and Kutrilina et al. [119] evaluated in TNBC models the preclinical safety and efficacy of a novel, potent, and orally bioavailable tubulin inhibitor, VERU-111 (also known as sabizabulin, Figure 6) which is currently undergoing clinical trials. The authors reported strong antiproliferative activity in MDA-MB-231 and MDA-MB-468 TNBC cell lines, with an IC50 value in the low nanomolar range, overcoming P-gp-mediated multidrug resistance, which frequently develops in patients treated with conventional taxanes [118,119]. In addition, in vivo assays in two aggressive TNBC xenograft models demonstrated its potent anticancer activity, its antimetastatic potential, and the reduction in adverse side effects relative to PTX [118,119]. Therefore, based on these findings, VERU-111 represents a promising alternative agent to target tubulin in patients with advanced breast cancer, with similar antimetastatic efficacy to PTX but with the advantage of oral bioavailability and lower toxicity than taxanes.
Figure 6.

Chemical structure of microtubule inhibitors.
On the other hand, a synergistic role of PARP and microtubule-targeting agents is also emerging in cancer therapy. Simultaneous targeting of PARP and microtubule polymerization may result in amplified and prolonged DNA damage, an enhanced sensitization of cancers, and a broad antitumor efficacy, with reduced risk for both cancer drug resistance and dose-limiting peripheral neuropathy associated with microtubule-targeting agents [120,121]. In this context, many efforts have been made to design and synthesize, through a medicinal chemistry approach, novel bifunctional small molecules able to synchronously inhibit the catalytic activity of PARP1 and microtubule polymerization.
5.2. AMXI-5001 (20)
AMXI-5001 was therefore engineered as a dual microtubule polymerization and PARP1/2 inhibitor, with favorable metabolic stability, oral bioavailability, and pharmacokinetic properties (Figure 6). AMXI-5001 showed inhibition of both tubulin polymerization, comparable to the vinblastine, and PARP inhibition at the level of PARP inhibitors 7–13. AMXI-5001 showed antitumor activity in a BRCA-mutated TNBC model in vivo as well, achieving complete tumor regression upon oral administration. Compared to the current PARP1 and 2 inhibitors, AMXI-5001 induced antitumor cell responses and elicited DNA repair biomarkers at much lower concentrations. The antitumor effect of AMXI-5001 was superior to PARP or microtubule inhibitors, both alone and a combination of both agents [120].
5.3. Ixabepilone (BMS-247550) (21)
Although there is no apparent mechanistic basis for unique sensitivity of microtubule-stabilizing agents in this subgroup, ixabepilone may have a role in the treatment of TNBC. Ixabepilone is a semisynthetic epothilone B analogue that behaves as microtubule stabilizer (Figure 6). Epothilones are cytotoxic macrolides with a mechanism of action similar to PTX but with the potential advantage of activity in taxane-resistant settings in preclinical models. The antineoplastic effect of epothilones has been ascribed to the stabilization of microtubules, which results in mitotic arrest at the G2/M transition. The effect of ixabepilone in combination with capecitabine in patients with metastatic or locally advanced TNBC was analyzed in two phase III trials (NCT00080301 and NCT00082433). The results showed that the drug combination, compared with capecitabine alone, improved PFS and the objective response rate in patients with advanced TNBC previously treated with anthracyclines and taxanes [122].
6. Mitotic Kinase Inhibitors
Small molecules inhibiting the activity of mitotic kinases have been long studied as alternative antimitotic agents to microtubule-targeting drugs, which still present remarkable toxicity issues. Mitotic kinase inhibitors retain high potential as novel therapies for TNBC, as several of these kinases [i.e., Aurora kinase A (AURKA) and B (AURKB), Polo-like kinase 1 (PLK1), NIMA-related kinase 2 (NEK2), and mitotic checkpoint kinase Mps1/TTK] have been shown to be highly expressed in these tumors, compared to both normal tissue and other BC, and to be prognostic for chemoresistance and worse outcomes [123,124,125,126,127,128].
AURKA is the mitotic kinase that records the largest number of preclinical and clinical studies testing the antitumoral activity of its select inhibitors in TNBC. AURKA is a serine–threonine kinase associated to the centrosome and the spindle microtubules, whose activity is pivotal to the proper progression of mitosis.
6.1. Alisertib
One of the first selective inhibitors of AURKA tested in TNBC was Alisertib (MLN8237, 22) (Figure 7), an orally available molecule showing a potent ad selective activity against its target (IC50 = 1 nM) and 200-fold more selectivity with respect to the close-related AURKB [129]. Preclinical studies showed that the combination of alisertib with either conventional taxanes [130] or with the new generation microtubule inhibitor eribulin (5) [131] reduces the growth and metastatic spreading of TNBC xenograft models. Coherent with these results, different clinical studies showed a significant improvement in PFS in advanced and metastatic BC treated with alisertib in addition to PTX [132,133]. Increased PFS and OS were observed for this therapeutic combination also for TNBC patients, although the cohort tested did not reach an adequate size for a powered statistical analysis [132]. These observations raise hopes that alisertib could be added to currently available therapeutic regimens for TNBC in order to improve their efficacy. In addition to its canonical mitotic role, AURKA was also shown to positively regulate activation of the protumoral mTOR pathway in TNBC cells. This uncanonical function of AURKA underlies the enhancing effects shown by two distinct mTOR inhibitors, rapamycin and TAK-288, on the antitumoral activity of alisertib against TNBC xenograft models [134,135]. Notably, alisertib combination with TAK-288 was proven to be tolerable in patients with advanced solid tumors in a phase I clinical trial [134]. Since other mitosis-unrelated functions, such as regulation of gene expression and RNA splicing, have been described in different cancers for AURKA, as well as for another centrosomal kinase NEK2, [126,136,137,138], further characterization of their activity and of the molecular profile of patients displaying their altered expression could improve the rationale design of novel therapeutic strategies exploiting their inhibitors.
Figure 7.

Chemical structure of AURKA inhibitors.
6.2. ENMD-2076
Another AURKA inhibitor with proven therapeutic efficacy against TNBC, both in preclinical and clinical studies, is the multitarget molecule ENMD-2076 (23, Figure 7) [139,140]. This orally available drug is a multitarget inhibitor, more effective for AURKA IC50 = 14 nM) with respect to AURKB (IC50 = 350 nM), that also targets the receptor-type tyrosine-protein kinase FLT3 (IC50 = 3 nM). A phase II clinical trial enrolling patients with locally advanced or metastatic TNBC previously treated showed 6 months of clinical benefits in 16.7% of patients treated with ENMD-2076 as single agent, with partial response and stable disease. These results encourage further studies investigating the potential therapeutic inhibitors for TNBC patients in combination with other agents. Furthermore, as TP53 gene mutations and overexpression were found to enhance TNBC cell line sensitivity to ENMD-2076 in a preclinical study [140], it is conceivable that the therapeutic efficacy of this drug could be improved via administration to molecularly stratified patients.
6.3. LY3295668
The AURKA inhibitor most recently tested against TNBC tumor is LY3295668 (erbumine, 24, Figure 7), an orally available inhibitor of AURKA, with over 1,000-fold selectivity versus AURKB (Ki of 0.8 nM and 1038 nM for AURKA and AURKB, respectively). This inhibitor was developed following the results of a pharmacogenomics screening identifying synthetic lethality between AURKA inhibition and RB1 mutations in a wide panel of cancer cell lines [141]. Remarkably, LY3295668 displayed a high antitumoral activity and a low toxicity in murine xenograft models of small-cell lung cancer [141]. In addition, a phase 1 study in patients with locally advanced or metastatic solid tumors showed that LY3295668 has a manageable toxicity profile, with 69% of the patients showing a stable disease (NCT03092934, [142]). LY3295668 (24) thus represents a highly promising therapeutic agent for the treatment of TNBC patients who, as mentioned above, display a high rate (~30%) of RB1 dysfunction. Remarkably, a loss of RB1 was demonstrated to also sensitize TNBC cells to the antitumoral activity of several inhibitors for another centrosomal kinase, PLK1 (i.e., BI-2536, HMN-214, and Volasertib [143]). These observations strongly indicate that mitotic kinase inhibitors are a relevant alternative therapeutic strategy for TNBC and other BC which, because of their genetic status, might display intrinsic or acquired resistance to CDK4/6 inhibitors.
Several inhibitors targeting the activity of mitotic kinases displaying aberrant expression and oncogenic activity in TNBC cells have been tested in preclinical studies and showed promising efficacy in interfering with the proliferative, invasive, and chemoresistant phenotype of these cells (Table 1). However, few of these drugs have been clinically tested in TNBC, making their further development and investigation an open field for the unveiling of novel routes of therapeutic intervention against TNBC.
Table 1.
Biological effects of mitotic kinase inhibitors in TNBC cells.
| Targeted Mitotic Kinase |
Cmpd | Biological Effects in TNBC Models | Results from Clinical Studies on TNBC Patients or Other Tumors |
|
|---|---|---|---|---|
| In Vitro | In Vivo | |||
| AURKB | Barasertib (AZD1152) | Reduction in mesenchymal traits [128]. | Inhibition of metastatic spreading of TNBC cells in immunodeficient murine models [128]. | Not available. Adequate levels of tolerance in patients with other advanced solid tumors, with observation of stable disease [144,145]. |
| NEK2 | CMP3A | Induction of mitotic alterations in synergy with PTX [124]. Induction of sensitivity to PTX in resistant cell lines [124]. |
Enhancing antitumoral effects of PTX in TNBC cells xenografts and PDXs [124]. | Not available. |
| JH295 | Inhibition of cells migratory and invasive properties [146]. | Not available. | Not available. | |
| PLK1 | Rigosertib (ON-01910) |
Reactivation of Erα expression [147]. | Reduction in TNBC cell xenograft and PDX growth [147]. | Not available. Moderate toxicity in patients with other advanced solid tumors, with observation of stable disease [148,149]. |
| Volasertib (BI-6727) |
Reactivation of Erα expression [147]. Increased DNA damage, mitotic arrest, and cell death [143,147]. |
Reduction in TNBC cell xenograft and PDX growth [147]. | Tolerable toxicity in patients with other advanced solid tumors, either alone or in combination with other chemotherapy, with partial antitumoral effects [150,151,152,153,154]. | |
| BI-2536 | Induction of mitotic abnormalities and apoptosis [125,143,155]. Reactivation of Erα expression [147]. |
Reduced growth of TNBC PDXs and enhanced activity towards doxorubicin and cyclophosphamide antitumoral effects [125]. | Not available. Moderate toxicity and antitumoral effects in patients with other advanced solid tumors [156]. |
|
| GSK461364 | Synergic antiproliferative and pro-apoptotic effects with docetaxel [157]. | Not available. | Not available. Mild toxicity and moderate antitumoral effects in patients with other solid tumors [158]. |
|
| Onvasertib | Synergic antiproliferative and pro-apoptotic effects with docetaxel [157]. | Synergic antitumoral effects with PTX in TNBC cell xenografts [157]. | Not available. One ongoing trail testing the safety and effectiveness of its combination with PTX in locally advanced and metastatic TNBC (NCT05383196). |
|
| MPS1/ TTK |
CFI-402257 | Inhibition of cell growth and induction of apoptosis and aneuploidy [159,160]. | Antitumoral effects against TNBC cell xenografts either as a single agent or in combination with carboplatin [160]. | Not available. Three ongoing trials testing the safety and antitumoral efficacy in different BC subtypes (NCT02792465, NCT03568422, NCT05251714). |
| BOS172722 | Induction of cell death either as stand-alone treatment or in combination with PTX [161]. | Reduction in the growth of TNBC cell xenografts and PDXs and enhanced activity on the antitumoral effects of PTX [161]. | Not available. One ongoing trial testing its safety and tolerability, either alone or in combination with PTX, in advanced nonhematologic malignancies (NCT03328494). |
|
| NTRC 0066-0 | Reduction in cell growth and induction of mitotic abnormalities [162]. | Reduction in TNBC cell xenograft growth as a single agent. Reduction in the growth of spontaneously developing murine tumors after combination with docetaxel [162]. | Not available. | |
| BAY 1217389 | Inhibition of cell growth [163]. | Reduction in the growth of TNBC cell xenografts and PDXs and enhanced activity on the antitumoral effects of PTX [163]. | Considerable toxicity, without a therapeutic window, in association with PTX [164]. | |
| WEE1 kinase |
Adavosertib (AZD1775) | Reduced cell growth both as a single agent and in association with capecitabine/5FU and/or ATR inhibitor [165,166,167]. | Reduced growth of TNBC xenografts and PDXs both as a single agent and in association with capecitabine/5FU and/or ATR inhibitor [165,166,167]. | Adequate levels of tolerance in TNB and other solid tumors, with stable disease in TNBC patients [168]. Not significant antitumoral effects in combination with cisplatin in metastatic TNBC [169]. |
7. Antibody Drug Conjugates
Antibody–drug conjugates (ACDs) are immunoconjugate agents engineered to selectively deliver small molecules to a cancer cell. This novel approach combines the specificity of a monoclonal antibody (mAb) with the high potency of small molecules with a lower rate of the undesirable side effects of antineoplastics.
In order to design ACDs from a medicinal chemistry point of view, it is necessary to combine three components in the chemical structure: a monoclonal antibody directed to a selective tumor antigen; a highly potent antineoplastic agent; a linker that binds the former two entities. The resulting drug is administrated intravenously to avoid the degradation via gastric acid, and after binding the target antigen expressed on the surface of cancer cells, it subsequently forms a drug–antigen complex which undergoes internalization with the cell via receptor-mediated endocytosis. Hence, an early endosome is firstly formed, and it successively fuses with the cell lysosome, which contains proteases and undergoes lysosomal degradation. The cleavage of the linker due to acidic pH, as a result of the influx of proton ions into the endosome or due to the presence of protease in the lysosome, allows the release of payloads into the cytoplasm and the payloads to take effect.
Recent studies have examined ACDs to potentially replace a chemotherapy backbone as the preferred therapeutic partner for treatment of TNBC [170].
7.1. Mirvetuximab Soravtansine (25)
Mirvetuximab soravtansine is an immunoconjugate composed of the humanized monoclonal antibody M9346A, targeting the folate receptor α (FRα) and conjugated to the cytotoxic maytansinoid DM4 via the disulfide-containing cleavable linker (Figure 8). Maytansinoid DM4 is a cytotoxic molecule which binds to tubulin through a second metabolite called S-methyl-DM4 and inhibits tubulin polymerization and microtubule assembly, resulting in cell cycle arrest and death.
Figure 8.

Chemical structure of mirvetuximab soravtansine.
The anti-FRα monoclonal antibody moiety of mirvetuximab soravtansine targets and binds to the cell surface antigen FRα. FRα has been reported to be expressed in up to 80% of TNBC, with limited expression in normal tissues. Mirvetuximab soravtansine was evaluated in a lead-in cohort to establish its activity in patients with metastatic TNBC [171]. A phase II study of 25 was conducted in TNBC; however, this study was terminated early due to the low rate of FRα positivity in the screened patient population and the lack of disease response in the two patients treated [172].
7.2. Praluzatamab Ravtansine (26)
Praluzatamab ravtansine, also known as CX-2009, is a conditionally activated probody–drug conjugate that demonstrates both translational and clinical activity in a variety of tumor types [173]. A probody therapeutic candidate consists of three components: a monoclonal antibody directed against a specific tumor antigen; a peptide able to mask the complementarity-determining regions; a protease-cleavable substrate that acts as a linker between the former components. Therefore, CX-2009 is composed of praluzatamab, a monoclonal antibody targeting the activated leukocyte cell adhesion molecule (ALCAM/CD116), conjugated to the potent microtubule inhibitor DM4 via a disulfide cleavable linker (Figure 9) [174]. The activated leukocyte cell adhesion molecule (ALCAM), known as CD166, is an attractive target for the activated drug conjugate [175]. CD166 is overexpressed at the outer cell surface of multiple tumor types (breast, prostate, lung, and ovarian cancers) and, to a lesser extent, by healthy tissue (colon, stomach, pancreas, thyroid, uterus, and prostate) [176]. With the aim of avoiding the binding of the monoclonal antibody to the CD116 antigen expressed in healthy cells, the binding area of this antibody is masked by a cleavable peptide (Figure 9). Only the proteases localized in the tumor microenvironment can cleave the linker and cause the release of the complementarity-determining region-masking peptide. Then, the monoclonal antibody binds to the CD116 antigen on the surface of tumoral cells, the ADC is then internalized, and the DM4 payload is released intracellularly.
Figure 9.

Chemical structure of praluzatamab ravtansine.
In preclinical studies, CX-2009 demonstrated selective tumor targeting, with reduced uptake in normal tissue. Moreover, it has been reported in the phase 2 CTMX-2009-002 trial (NCT04596150) that CX-2009 generated responses in HR-positive, HER2-negative breast cancer; however, it did not meet the primary end point in patients with TNBC [177].
7.3. SAR566658
SAR566658 is another humanized DS6 (huDS6) antibody directed against tumor-associated sialoglycotope CA6 (tumor-associated glycosylated MUC-1) and conjugated through a cleavable linker to the cytotoxic maytansinoid-derived drug DM4 [178]. It binds to tumor cells with high affinity, allowing good intracellular delivery of DM4. The CA6 sialoglycotope of MUC1-glycoprotein is highly detected on various tumors of epithelial origin (pancreas, ovary, breast, and bladder) [179]. SAR566658 showed in vivo antitumor efficacy against CA6-positive human pancreas, cervix, bladder, and ovary tumor xenografts and against three breast patient-derived xenografts. A phase I dose expansion was conducted to evaluate the safety and the maximum tolerate dose of SAR566658. A phase II study was then conducted in patients with CA6-positive mTNBC. Preliminary results have shown an unfavorable benefit/risk balance due to a higher-than-expected incidence of ophthalmologic events (e.g., keratitis and keratopathy) [178].
7.4. Sacituzumab Govitecan
Sacituzumab govitecan, also known as IMMU-132 and marketed as Trodelvy®, was approved by the FDA in April 2020 for the treatment of metastatic triple-negative breast cancer that did not respond to at least two prior therapies for metastatic disease [180]. It is an ADC drug composed of SN-38, the active metabolite of irinotecan (CPT-11) with a cytotoxic effect by inhibiting DNA topoisomerase I, that is conjugated via a pH cleavable linker to a humanized IgG1 antibody targeting trophoblastic cell-surface antigen 2 (TROP-2), a transmembrane glycoprotein that participates in the proliferation, survival, and invasion of tumoral cells and whose overexpression has been related to a poor prognosis. Sacituzumab govitecan-bound tumor cells are killed via intracellular SN-38 uptake, while nearby tumor cells are killed by extracellular SN-38 release. In a recent study, the efficacy and safety profile of IMMU-132 as a second-line treatment for refractory mTNBC have been highlighted; however, its utility is limited due to inadequate intratumor exposure and dose-limiting toxicities, as is the case with other topoisomerase-1 inhibitors [181].
8. PI3K
The phosphatidylinositol-3-kinases (PI3Ks) are intracellular signaling enzymes that phosphorylate the free 3-hydroxyl of the phosphoinositides in the cell membrane. Therefore, this signaling pathway is involved in key cellular functions, such as cell proliferation, migration, survival, and metabolism [182]. PI3Ks are formed of a heterodimer composed by a regulatory (p85) and a catalytic (p110) subunit and are usually grouped in different classes, among which class I is the most commonly altered in cancer. In cancer cells, activation of the PI3K pathway is caused by molecular alterations, including gene mutations encoding the PI3K alpha (PIK3CA) and beta (PIK3CB) catalytic subunits, AKT1 mutations, and a loss of expression of phosphatidylinositol-3,4,5 trisphosphate (PIP3), phosphatase and tensin homologue (PTEN), and inositol polyphosphate-4-phosphatase type II B (INPP4B). PIK3CA is the second most frequently mutated gene after TP53 in TNBC, with additional inactivating alterations in PTEN and additional activating mutation in AKT1 [80].
PI3K inhibitors are divided into pan-PI3K inhibitors (buparlisib, pictilisib, and copanlisib) and isoform-specific PI3K inhibitors (alpelisib). Pan-PI3K inhibitors target all four isoforms (alpha, beta, gamma, and delta) of the class-IA PI3K p110 catalytic subunit.
8.1. Buparlisib (BKM120, 27)
Buparlisib is one of the furthest developed pan-class I PI3K inhibitors in combination with chemotherapeutic agents (Figure 10). It is orally bioavailable and acts to inhibit PI3K in an ATP-competitive manner [183]. In the phase 1 study, buparlisib was administered at max dose of 100 mg/die and yielded partial response in a TNBC patient [184,185]. In a phase 2 clinical study, buparlisib monotherapy achieved prolonged stable disease in a small subset of TNBC patients (no strong clinical signal of efficacy was observed). Actually, buparlisib is under clinical development by Adlai Nortye Biopharma and more specifically in phase II for TNBC. According to GlobalData, phase II drugs for TNBC have a 25% phase transition success rate indication benchmark for progressing into phase III [186]. It has also been observed that, compared to selective p119-alpha inhibitors, buparlisib shows a toxicity profile similar to other pan-PI3K inhibitors due to its elevated CNS penetration or to other pharmacokinetic differences [183]. Furthermore, dual PI3K and PARP inhibition with buparlisib and olaparib (8) reduced the tumor growth in patient-derived primary tumor xenografts, displaying BRCA1/2 downregulation following PI3K inhibition in TNBC [187].
Figure 10.

Chemical structure of PI3K inhibitors.
8.2. Alpelisib (28)
Alpelisib is an α-specific PI3K inhibitor that has been evaluated as a monotherapy regimen in patients with advanced PI3K pathway mutant ER+ HER2-negative BC and TNBC (Figure 10). Alpesilib displayed efficacy in heavily pretreated ER+ breast cancer with PIK3CA mutation [188]. Alpelisib in combination with olaparib (8) is tolerable in patients with pretreated TNBC, with evidence of activity in non-BRCA carriers. The results highlight the potential synergistic use of a PI3K inhibitor to sensitize HR-proficient (BRCA wild-type) TNBC to a PARP inhibitor [189]. A phase II trial study has been established to evaluate the drug combination of alpelisib and nab-paclitaxel in treating patients with TNBC with PIK3CA or PTEN alterations who do not respond to anthracycline chemotherapy (anthrocycline refractory) [190].
In addition, it is worth mentioning that nearly 35% of TNBCs have become resistant to PI3K/AKT/mTOR inhibitors due to the loss of PTEN gene expression [191], which encodes a phosphatase protein that returns the PI3K/Akt/mTOR pathway to its inactivated state [192]. The lack of PTEN gene expression has been correlated to the emergence of the glycolytic phenotype, known as the Warburg effect [193]. However, it has been demonstrated that drug combinations may effectively contrast the multidrug resistance [194]; in fact, a positive therapeutic response of PTEN-deficient TNBC was observed by combining PI3K and histone demethylase lysine demethylase 4B (KDM4B) inhibitors and suppression of either the 3-phosphoinositide-dependent kinase 1 or se-rum/glucocorticoid-regulated kinase 1 activity [195]. The drug combination of chemo-therapeutic agents or neoadjuvant chemotherapy with the monoclonal antibody bevacizumab was shown to improve pathological complete response in stage II to III TNBC [196,197].
9. AKT
The serine/threonine kinase AKT, also known as protein kinase B (PKB), is a key component of the phosphatidyl-inositole-3 kinase (PI3K) intracellular pathway that exerts a pivotal role in cell growth, survival, proliferation, and metabolism [198,199,200,201]. The PI3K/AKT signaling pathway has been found to be hyperactivated in BC and is considered a promising target for TNBC [202]. Three isoforms of AKT have been identified so far: AKT1 (also known as PKBα), AKT2 (also known as PKBβ), and AKT3 (also known as PKBγ). In cancer cells, AKT1 is involved in proliferation and growth, promoting tumor initiation and suppressing apoptosis; AKT2 regulates cytoskeleton dynamics, favoring invasiveness and metastatization, whereas the role of the hyperactivation of the AKT3 isoform in cancer is still controversial, although a possible stimulation of cell proliferation has been hypothesized [203].
The PI3K signaling pathway is activated as a result of the binding of a growth factor or ligand to a number of membrane-associated receptor tyrosine kinases (RTKs). Activation of RTK leads to the recruitment of the p85 subunit and a subsequent conformational change that allows the p110 subunit to catalyze the conversion of phosphatidyl-inositol-4,5-bisphosphate (PIP2) to the second messenger phosphatidyl-inositol 3,4,5-trisphosphate (PIP3). Tensin homolog deleted on chromosome 10 (PTEN) phosphatase, together with the tumor suppressor inositol polyphosphate 4-phosphatase type II (INPP4B), negatively controls PI3K by converting PIP3 to PIP2 thorough dephosphorylation, thus regulating PIP3 levels. PIP3 activates the downstream PI3K pathway by colocalization of the serine/threonine kinases PDK1 and AKT to the cell membrane via their pleckstrin homology (PH) domains. Here, AKT undergoes a double phosphorylation, one on the kinase domain (T308, T309, and T305 for AKT1, 2, and 3, respectively) by PDK1 and another on the regulatory domain (S473, S474, and S472 for AKT1, 2, and 3, respectively) by the mToR complex 2 (mTORC2), resulting in its full activation. Once activated, AKT phosphorylates and inhibits its downstream targets, including tuberos sclerosis complex 2 (TSC2), with the consequent RHEB-GTP accumulation that, in turn, activates mTORC1, glycogen synthase kinase-3β (GSK3β), and the forkhead kinase transcription factors (FOXO). The signaling results in the regulation of cell proliferation, survival, and metabolism (Figure 11).
Figure 11.

Schematic presentation of AKT1 signaling cascade. Black arrows represent signaling activation while red bars indicate inhibitory signals.
9.1. Ipatasertib (29)
Ipatasertib is a selective oral ATP-competitive small-molecule AKT inhibitor with anticancer activity in several cancer types, including prostate, breast, ovarian, colorectal, and non-small-cell lung cancers (Figure 12) [204]. Ipatasertib was the first AKT inhibitor to yield positive outcomes for TNBC treatment. PFS was longer in patients who received ipatasertib than in those who received placebo. The LOTUS trial investigated the addition of ipatasertib to PTX as first-line therapy for TNBC [205]. In PIK3CA/AKT/PTEN-altered tumors, the median progression-free survival of ipatasertib-treated patients was higher compared to the untreated patients, with significant improved survival outcomes. However, a significant pathological complete response rate in early TNBC patients was not observed in the FAIRLANE trial [206].
Figure 12.

Chemical structure of AKT inhibitors.
9.2. Capivasertib (AZD5363) (30)
Capivasertib is an orally available inhibitor of all three isoforms, AKT1, AKT2, and AKT3, that belongs to the category of ATP-competitive inhibitors (Figure 12) [207]. It acts by preventing substrate phosphorylation by AKT and downregulates the phosphorylation levels of GSK3β and the proline-rich AKT substrate of 40 kDa (PRAS40) biomarkers present in many cancer cells. Capivasertib achieved dose-dependent growth inhibition of xenografts derived from various tumor types, including HER2(+) breast cancer models resistant to trastuzumab. In breast cancer xenografts, capivasertib demonstrated synergistic activity with docetaxel, apatinib, and trastuzumab [208]. Capivasertib showed preclinical activity in TNBC models; the drug sensitivity has been correlated to PI3K or AKT activation and/or PTEN deletion. The drug combination of capivasertib with PTX in TNBC therapy resulted in significantly longer PFS and OS [209]. In preclinical studies, capivasertib, as a single agent in combination with anti-HER2 agents, effectively inhibited the growth of tumors with PIK3CA or MTOR alterations. Phase I/II trials evaluating the co-administration of capivasertib with PTX, with fulvestrant, an estrogen receptor antagonist, or with olaparib in hormone receptor (HR)-positive and HER2-negative breast cancer, demonstrated greater efficacy with an acceptable toxicity profile [210].
10. Mammalian Target of Rapamycin (mTOR)
mTOR is a serine–threonine kinase activated by both PI3K and MAPK signaling. AKT activation leads to protein synthesis and cell growth by activating the downstream effector mTOR through TSC1/2. Activated mTOR forms two functionally different complexes: mTOR complex mTORC1 and mTORC2 (Figure 13) [211]. mTORC1, on one hand, mediates growth stimulatory effects of mTOR by promoting mRNA translocation and activating protein translation; on the other hand, it is involved in lipid synthesis and metabolism. By contrast, mTORC2 regulates AKT phosphorylation and stimulates further AKT activation and is involved in the organization of the actin cytoskeleton.
Figure 13.

Schematic presentation of PI3K/AKT/mTOR signaling pathway.
Everolimus (31)
Everolimus is an inhibitor of the serine–threonine kinase mammalian target of rapamycin; it is structurally correlated to the natural macrocyclic lactone sirolimus ((-)-Rapamycin) produced by the bacterium Streptomyces hygroscopicus and shows immunosuppressant and anti-angiogenic properties (Figure 14). Several clinical trials have reported the effectiveness of everolimus used in combination with cisplatin in TNBC patients who had residual disease post-standard neoadjuvant chemotherapy. This combination was active in a subset of patients with germline PALB2 mutation or somatic PI3KCA mutation [212].
Figure 14.

Chemical structure of mTOR inhibitor.
11. Vascular Endothelial Growth Factor Receptor (VEGFR)
The vascular endothelial growth factor (VEGF) family consists of five members: EGF-A, VEGF-B, VEGF-C, VEGF-D, and placenta growth factor. Among them, the most extensively studied is the isoform VEGF-A. The latter executes its function through the binding to VEGF receptor. In contrast to individuals with other BCs, TNBC patients exhibited notably elevated levels of intratumoral VEGF. Moreover, they experienced considerably reduced periods of recurrence-free survival and OS, characterized by a shorter time span between diagnosis and relapse, as well as between relapse and mortality [213]. It is noteworthy that VEGF levels demonstrated a consistent association with unfavorable outcomes, regardless of factors such as tumor size, nodal status, histologic grade, patient age, or the nature of relapse. Therefore, the VEGF pathway has been explored as a valuable therapeutic target in TNBC.
11.1. Apatinib (32)
Apatinib is an orally bioavailable tyrosine kinase inhibitor targeting vascular endothelial growth factor receptor-2 (VEGFR-2), a receptor involved in the growth of blood vessels, with promising anti-angiogenesis and antitumor activity for TNBC [214,215] (Figure 15). In the clinical study NCT01176669, apatinib monotherapy showed promising efficacy in heavily pretreated patients with metastatic TNBC in China [216]. In addition, in another study (NCT03254654), patients with advanced TNBC who did not respond to first/second-line treatments received vinorelbine as monotherapy or vinorelbine with apatinib in 28-day treatment cycles. According to the available data on phase III trials enrolling TNBC patients, the PFS was significantly longer in the group treated with the combination of apatinib and vinorelbine than the vinorelbine group (3.9 months vs. 2.0 months; hazard ratio, 1.82; 95% confidence interval [CI], 1.06 to 3.11; p = 0.026) [217]. The combination therapy of apatinib plus etoposide exhibited a promising clinical effect and acceptable toxicity in recurrent or metastatic TNBC as second-or higher-line treatment [187]. The drug combination of apatinib with camrelizumab demonstrated favorable therapeutic effects and a manageable safety profile in patients with advanced TNBC [188].
Figure 15.

Chemical structure of tyrosine kinase inhibitors.
11.2. Lenvatinib (33)
Lenvatinib, a small-molecule tyrosine kinase inhibitor that inhibits VEGFR-1 (FLT1), VEGFR-2 (KDR), and VEGFR-3 (FLT4), in combination with an immune checkpoint inhibitor, may have significant clinical activity in selective patients with heavily pretreated metastatic TNBC [218] (Figure 15). In combination with pembrolizumab, lenvatinib showed promising antitumor activity with manageable toxicity in patients with previously treated advanced TNBC [219].
12. Epidermal Growth Factor Receptor (EGFR)
The epidermal growth factor receptor (EGFR), a member of the ErbB receptor tyrosine kinase (TK) family, is a transmembrane glycoprotein containing an extracellular ligand binding domain and an intracellular RTK domain, and conveys cell proliferation and differentiation extracellular signals from the outer cell surface to the cell interior [220,221]. When activated by its ligands, such as epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α), EGFR forms dimers with another EGFR, leading to self-phosphorylation and initiating a cascade of intracellular signaling events. These events involve the activation of the Ras/Raf/mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription (STAT), and protein kinase C (PKC) pathways, resulting in cell proliferation and survival, invasion, metastasis, and angiogenesis. Recent studies have shown that a subset of TNBC tumors overexpress EGFR, in particular, the basal subtypes of which EGFR is a characteristic marker [222], making it a potential therapeutic target [223]. Overexpression of EGFR can lead to increased cell proliferation and survival, contributing to cancer growth.
12.1. Afatinib (34)
Afatinib is an orally bioavailable EGFR inhibitor with antitumor activity (Figure 16). A study demonstrated that afatinib achieved clinical benefit for at least 4 months in a small number of heavily pretreated unselected patients with TNBC [224]. Preclinical evidence showed that co-administration of EGFR inhibitors may potentiate anti-TNBC chemotherapy [225]. Treatment of metastatic TNBC patients with the anti-EGFR antibody cetuximab plus cisplatin achieved a response superior to the cisplatin monotherapy [226], thus validating EGFR as a target for TNBC [227].
Figure 16.

Chemical structure of EGFR inhibitors.
The drug combination of afatinib with neoadjuvant PTX achieved a modest effect in TNBC. It was suggested that activation of PI3K and JAK2 pathways and mutation of homologous recombination genes reduced the therapeutic response, while amplification of DAXX genes, according to previously reported sensitivity to PTX chemotherapy [228], potentiated the response to afatinib in combination with PTX [229].
12.2. Gefitinib (35)
The antitumor activity of the EGFR tyrosine kinase inhibitor gefitinib was examined in combination with the mTOR inhibitor everolimus in TNBC cells, with or without activating mutations in the PI3K/AKT/mTOR signaling pathway (Figure 16). The drug combination of gefitinib plus everolimus showed synergistic inhibition in the PI3K and PTEN-mutant CAL-51 cell line but not in the PTEN-null HCC-1937 cell line, with significant effects on cell cycle progression and induction of apoptosis. The results suggest this drug combination as an effective treatment for TNBC with activated mutations of PI3K [230].
Recently, Abdelmalek et al., reported the development of a series of hybrid compounds through the combination of tamoxifen, or its active metabolite endoxifen, with the EGFR-inhibitor gefitinib via a covalent linkage [231]. The results revealed that these drug conjugates retained both EGFR inhibition and ER antagonist activities of the parent compounds. Moreover, the most potent analogues showed single digit nanomolar activities at both targets. Two amide-linked endoxifen–gefitinib drug conjugates displayed nanomolar IC50 values in the TNBC cells [231].
13. SRC
The failure of cancer chemotherapy is due to the presence of cancer stem-like cells, a subpopulation of cancer cells that are able to self-renew and to generate heterogeneous tumors [232]. The aggressive phenotype of TNBC is in part attributed to the presence of intrinsically resistant to chemotherapy breast cancer stem cells (BCSCs). Thus, the priority goal of novel therapies for TNBC therapies is overcoming the BCSC drug resistance. SRC kinase mediates cell proliferation, growth, and survival [233], and its aberrant activation has been observed in breast cancers stemness [234] and metastasis [235].
Dasatinib (36)
Dasatinib is a selective SRC tyrosine kinase inhibitor that is used in the therapy of chronic myelogenous leukemia (CML) positive for the Philadelphia chromosome (Figure 17). However, in preclinical studies, dasatinib was shown to inhibit BC cell growth [7]. It has been reported that dasatinib abolished PTX-induced SRC activation as well as BCSC sphere formation capacity and expansion in parental TNBC cells. The drug combination of dasatinib with PTX reduced the drug resistance of PTX-resistant cells in vitro and inhibited breast tumor growth in vivo, suggesting this combination as a potential therapy for overcoming chemotherapy resistance in TNBC patients [236].
Figure 17.

Chemical structure of SRC tyrosine kinase inhibitor.
In addition, a combined treatment with dasatinib and the c-Met inhibitor CpdA (Amgen, Thousand Oaks, CA, USA) significantly inhibited the growth of MDA-MB-231 parental cells and prevented the emergence of dasatinib resistance. The combined treatment with dasatinib and a c-Met inhibitor may block the development of acquired resistance and improve response rates to dasatinib treatment in TNBC [237].
14. c-MET
c-MET, also known as hepatocyte growth factor receptor (HGFR), is a protein encoded by the c-MET gene. It was first identified in 1984 by Cooper et al. as a proto-oncogene [238]. C-Met tyrosine kinase signaling cascades are activated via the binding with its single ligand HGF/SF-hepatocyte growth factor/scatter factor and mediate extracellular signals across the cell membrane into cytoplasm. Therefore, c-Met signaling is required for many physiological processes including proliferation, scattering, morphogenesis, and survival. C-Met is overexpressed in 20–30% of all BC cases and in around 52% of TNBC. This observation, together with recent studies on the c-MET signaling pathway, suggests this protein as a novel target to treat TNBC.
Tivantinib (37)
Tivantinib is an orally bioavailable inhibitor of c-Met receptor tyrosine kinase (Figure 18). In a phase II trial of patients with metastatic TNBC, tivantinib achieved an overall response rate of 5%, the 6-month PFS rate was 5%, and toxicity was negligible. Even if it was well tolerated, it did not fulfill prespecified statistical targets for efficacy [239]. The sensitivity to c-Met inhibition in TNBC cells was increased upon dasatinib treatment [237].
Figure 18.

Chemical structure of c-Met receptor tyrosine kinase inhibitor.
15. Janus Kinase (JAK)
The mammalian JAK-STAT signaling pathway consists of four JK domain-containing proteins, namely, JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2), along with seven signal transducers and activators of transcription abbreviated as STATs (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6) [240]. Dysregulation of this pathway plays a pivotal role in oncogenic processes, including tumorigenesis, proliferation, angiogenesis, oncogenic signaling, cell survival, anti-apoptosis mechanisms, and immune responses [241].
Ruxolitinib (38)
Ruxolitinib is an orally bioavailable inhibitor of Janus-associated kinase (JAK) JAK 1 and 2 with antitumor and anti-inflammatory activity (Figure 19). It has been approved for the treatment of patients with myelofibrosis and those with polycythemia [242]. JAK2 signaling was associated with PTX resistance in TNBC patient-derived xenograft models and regulated PTX-resistant cancer-associated fibroblast phenotype transition [243]. In a non-randomized phase II study enrolling pSTAT3-positive patients with refractory, metastatic TNBC, ruxolitinib, as a single agent, was well tolerated but did not meet the primary efficacy endpoint, despite evidence of on-target activity [244]. Novartis has reported that ruxolitinib phosphate is under clinical development and currently in phase II for TNBC. According to GlobalData, phase II drugs for TNBC have a 25% phase transition success rate indication benchmark for progressing into phase III [245].
Figure 19.
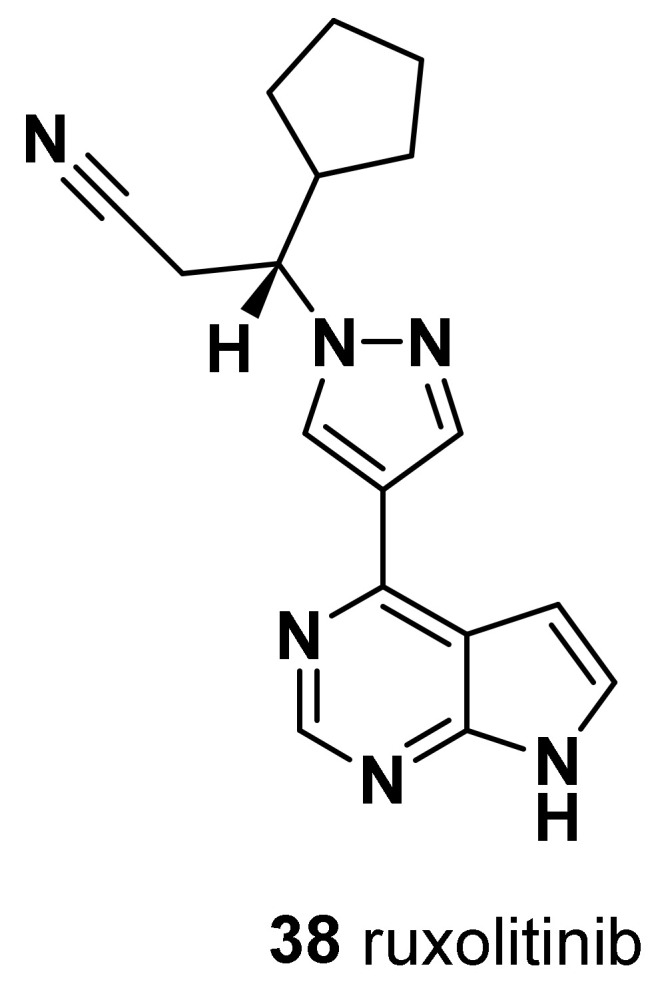
Chemical structure of JAK inhibitor.
16. Inhibitor of Apoptosis Protein (IAP)—Second Mitochondria-Derived Activator of Caspases (SMAC)
Inhibitors of Apoptosis Protein (IAP) are well known for their antiapoptotic activity. Usually, members of the IAP family are characterized by the presence of the baculoviral IAP repeat (BIR) domain, which engages in physical interactions with caspase proteins, consequently inhibiting the functionality of the latter [246]. Second mitochondria-derived activator of caspases (SMAC) mimetics have arisen as a promising category of targeted therapies currently undergoing clinical evaluation for both solid tumors and hematological cancers. These drugs inhibit the activity of inhibitors of apoptosis proteins; therefore, the dysregulation of IAPs promotes tumorigenesis, metastasis, angiogenesis, and therapeutic resistance, including chemotherapy and radiotherapy [246].
LCL161 (39)
LCL161 is an orally bioavailable IAP antagonist and mimics second mitochondria-derived activator of caspases with both pro-apoptotic and antiproliferation effects in cancer cells (Figure 20). At the molecular level, LCL161 binds to the BIR3 domain of cIAP1 and cIAP2 with high affinity and induces cIAP1 and cIAP2 autoubiquitination and proteasome degradation, resulting in the activation of non-canonical NF-κB signaling pathways and the production of tumor necrosis factor α (TNF-α). LCL161 has been shown to induce tumor necrosis factor (TNF)-mediated apoptosis in sensitive cell lines [247], including apoptosis in TNBC xenograft models with TNFα-based gene expression signatures associated with LCL161 [248].
Figure 20.

Chemical structure of IAP-SMAC inhibitor.
In addition, many preclinical studies have demonstrated the synergistic anticancer activity of LCL161 in combination with different anticancer agents such as PTX, vincristine, and obatoclax. A neoadjuvant trial provided evidence supporting a biomarker-driven targeted therapy approach with LCL161 for selected patients with gene expression signature-positive TNBC [249].
17. Splicing Inhibitors
Splicing is the molecular process of introns removal and the joining of exons which mediates maturation of newly transcribed pre-mRNAs into functional mRNA (mRNA). Small molecules inhibiting this process have been shown to exert a potent antitumoral activity, especially in tumors displaying aberrant activity of the oncogenic transcript factor c-MYC [250,251,252,253]. Indeed, the increased activity of this transcription factor dramatically increases the number of synthetized RNA transcripts, creating an overload for the splicing machinery that makes cancer cells vulnerable to its chemical inhibition [254]. In line with the amplification and overexpression of c-MYC which characterizes TNBC, with respect to both normal mammary tissue and other BCs, numerous preclinical studies have demonstrated the antitumoral activity of several splicing inhibitors against cell lines, xenografts, and patient-derived xenograft (PDX) models of this tumoral subtype. The tested inhibitors include a broad list of small molecules acting on different molecular targets. Inhibitors of the SF3b complex, a main spliceosomal component, are the first class of small molecules targeting splicing catalysis to be developed and tested as antitumoral compounds, also in TNBC [250,251]. This class includes sudemycin-D6 [254,255], pladienolide B and its derivatives E7107 [256,257], and H3B-8800, an orally bioavailable analogue of E7107 [255]. These molecules act by interacting with the SF3b complex and preventing U2snRNP interaction with target pre-mRNA, which leads to splicing inhibition and consequent intron retention or exon skipping [250,251]. These agents were found to reduce the growth of TNBC cell lines and xenografts, as well their metastatic spread [254,255,256,257]. Unfortunately, phase I trials testing E7107 in solid tumors revealed severe ocular toxicity, leading to the suspension of the study [258,259]. No similar issues were reported for its derivative H3B-8800 in a trial in leukemia patients [260], which opens up the opportunity for the evaluation of its antitumoral efficacy also for solid tumors, including TNBC.
Another splicing inhibitor with proven efficacy against TNBC preclinical models is the type-I protein arginine methyltransferases (PRMTs) inhibitor MS-023 [261,262]. PRMTs inhibitors interfere with the splicing process by impairing the asymmetric dimethylation of both core spliceosomal proteins and accessory splicing factors [251,263,264]. Similar to the reports for H3B-8800, treatment with MS-023 was shown to induce a global inhibition of the splicing process, which leads to the accumulation of intron retaining transcripts, many of which form cytotoxic double-stranded RNAs (dsRNAs) [255,262]. By mimicking a viral infection, these endogenous dsRNAs activate the cellular antiviral defense pathways, eliciting secretion of inflammatory cytokines and type 1 IFNs and activation of the extrinsic apoptotic pathways [255,262]. Furthermore, at least for H3B-8800, whose efficacy was tested also against TNBC xenografts transplanted in immunocompetent mice, splicing inhibition was shown to enhance the antitumoral activity of the adaptive immune system by promoting the infiltration of cytotoxic CD8+T cells [255]. In this regard, since high levels of infiltrating lymphocytes have shown to be predictive markers for a better response of TNBC patients to chemotherapy [265], it would be interesting to test if splicing inhibitors sequentially combined with conventional chemotherapy might increase TNBC chemosensitivity by enhancing their leukocyte infiltration grade. Furthermore, several splicing inhibitors, with different chemistry and target proteins, were shown, in other tumoral contexts, to elicit T-cell-mediated immune responses by promoting the generation of splicing derived neoantigens [266,267]. Whether this occurs also in TNBC is still unexplored, but it is highly probable that such mechanism might concur to the antitumoral activity of this class of inhibitors also in these tumors. MS023 is still at the preclinical developmental stage, while a phase 1 clinical study testing the safety and efficacy of the PRMTs type I inhibitor GSK3368715 in patients with advanced solid tumors was terminated early because of the risk of thromboembolic events exceeding any potential benefit [268]. Further studies on PRMTs inhibitors are thus needed to translate from the bench to the bedside the putative antitumoral effects of this class of splicing inhibitors.
Another class of small molecules targeting the splicing process, showing promising antiviral effects in TNBC preclinical models, but still far from the clinical testing, are splicing factor kinases inhibitors, for which we guide readers to a specific review on the topic [75].
Overall, several preclinical studies have demonstrated the high potential for multiple splicing inhibitors to be further developed in efficacious therapies against TNBC, both as novel chemotherapeutic agents and pro-immune enhancing agents. Furthermore, the multilayered control of the splicing process offers multiple ways for the development of pharmacological strategies to interfere with it.
18. Recent Promising Biomarkers as Future Therapeutic Approaches
Despite the recent drugs approved for the treatment of TNBC and used in combination with standard chemotherapy, as previously described in the present review, there is still an urgent need to identify alternative molecular targets, along with novel molecular biomarkers involved in cancer progression, to improve the outcome of chemotherapy and to overcome the insurgence of chemotherapy-resistant TNBC.
18.1. Paraoxonase-2 Enzymes
Within this framework, increasing evidence has recently suggested that the enzyme paraoxonase-2 (PON2) could play a key role in various solid malignancies, enhancing cancer cells’ capacity to mitigate the high levels of oxidative stress encountered by these cells [269]. This, in turn, accelerated their rate of proliferation and ultimately contributed to heightened resistance to different chemotherapeutics (imatinib, doxorubicine, staurosporine, or actinomycin) in cell culture models.
PON2, represents one of three highly conserved members of the PON gene family of enzymes consisting of PON1, PON2, and PON3. Their name was inspired by the member PON1, a calcium-dependent esterase that was first described for its capacity to hydrolyze organophosphates and the insecticide parathion, whose active metabolite paraoxon functions as a neurotoxic cholinesterase inhibitor [270]. In humans, PON1 and PON3 are synthesized predominantly in the liver, from where they are secreted into the blood, and are associated with high-density lipoproteins (HDL), whereas human PON2 is ubiquitously expressed in a wide range of tissues and its protein product localizes to the endoplasmic reticulum and nuclear envelope [270,271]. At the plasma membrane level, PON2 is a transmembrane protein with its enzymatic domain facing the extracellular compartment and thus plays an important role in rescuing the peroxidation of membrane components [272,273]. Therefore, it has been hypothesized that PON2 plays a pivotal role in contributing to antioxidant activity against lipoperoxides on HDL particles.
Based on the evidence that, in many types of cancers, PON2 is upregulated in tumor tissues relative to corresponding normal tissues, Campagna et al. firstly evaluated the PON2 levels in BC subtypes and subsequently investigated the role played by the enzyme in TNBC cell metabolism, focusing on the possibility of the enzyme in affecting the tumor cell sensitivity to the chemotherapeutic treatment [274]. The immunohistochemical analysis revealed an upregulation of this enzyme in the infiltrating BC of the subtypes Luminal A, HER2+, and TNBC compared to the healthy tissue. Remarkably, the deficiency of PON2 caused apoptosis of the tumor cells, demonstrating an oncogenic function.
Subsequently, in vitro effects of PON2 downregulation on cell proliferation and response to chemotherapeutics were evaluated clearly highlighting the potential of PON2 to be a promising molecular target capable of improving the efficacy of the chemotherapy in TNBC patients, who, to date, still lack targeted therapies. At present, no PON2 inhibitors have been described in the literature, making the development of efficient drugs a major research challenge.
18.2. Nicotinamide N-Methyltransferase Enzyme
Furthermore, similarly, nicotinamide N-methyltransferase (NNMT), another potential prognostic marker for TNBC, has recently been demonstrated to be overexpressed in TNBC tumors and to be associated with the development of resistance [275,276,277,278,279,280]. Indeed, Wang et al., in 2019, reported that NNMT expression in breast cancer inhibited apoptosis and enhanced chemotherapy resistance, resulting in high metastasis and poor survival, and demonstrated that NNMT expression in tumor tissues of TNBC patients was higher than that in other subtypes [281].
NNMT, a phase II metabolizing enzyme, catalyzes the transfer of the methyl units from S-adenosyl-l-methionine (SAM) to nicotinamide (NAM), the precursor of a large number of energy metabolism-related molecules, such as NAD+ and NADH, producing the stable metabolic products 1-methylnicotinamide (1-MNA) and S-adenosyl-L-homocysteine (AdoHcy); this thereby decreases the overall methyl pools for RNA, histone, and DNA methylation and contributes to a wide range of changes in cancer-associated gene expression [282].
A multitude of NNMT-induced mechanisms, including SAM depletion-induced DNA/histone hypomethylation and the associated upregulation of tumor-promoting genes, have been shown to promote tumorigenic phenotypes in various solid tumors including breast cancer [282].
In a more recent study, Wang et al., investigated the function and mechanism of NNMT on the metastasis of TNBC [283]. Through in vitro assays, they found that NNMT overexpression promoted the migration and invasion of TNBCs by enhancing membrane fluidity and epithelial–mesenchymal transition (which is generally considered to be a major driver of metastasis) of TNBCs through the reduction in cholesterol levels in the cytoplasm and cell membrane. Mechanistically, it has been reported that NNMT repressed protein phosphatase 2A activity to activate the MEK/ERK/c-Jun pathway, leading to the increase in ABCA1 expression in TNBC cells and the reduction in cholesterol levels and EMT activation, which finally enhances migration and invasion in vitro and metastasis capacity in vivo.
18.2.1. NNMT Bisubstrate Inhibitors
Highly potent, selectively targeted, and functional NNMT inhibitors are valuable resources for exploring the intricate regulatory mechanisms governed by NNMT and for investigating various pharmacological hypotheses suggesting NNMT as a viable therapeutic target [284]. Despite the growing interest in its implication in many disease states, few cell-active NNMT inhibitors have been described to date, and none have entered clinical trials. At present, the most powerful NNMT inhibitors reported in the literature are compounds developed through a bisubstrate transition state of the methylation reaction mimic approach [285,286]. Basically, these bisubstrate-like inhibitors consist of two covalently connected moieties mimicking the cofactor and substrate, SAM and NAM, respectively.
In this regard, Gao et al., designed and developed a series of NNMT inhibitors by introducing a nonbenzamide aromatic moiety substituted with different electron-withdrawing groups to mimic the nicotinamide nucleus, which is connected to the SAM unit through a three carbon trans-alkene linker [287]. Among these compounds, the para-cyano-substituted styrene-based inhibitor GYZ-319 (Figure 21) was identified as the most potent NNMT inhibitor with an IC50 value of 3.7 nM. Moreover, ITC experiments confirmed the ability of the inhibitor to strongly bind NNMT, with a dissociation constant of 21 nM. However, the low nanomolar potency exhibited in biochemical assays was not reflected in human cancer cell lines, in which the compound GYZ-319 only showed significant antiproliferative effects at 100 μM. This discrepancy has been explained by the poor cell permeability of GYZ-319, which is due to the presence of two highly polar functional groups present in all potent bisubstrate NNMT inhibitors reported to date.
Figure 21.

Chemical structure of NNMT bisubstrate inhibitors.
Therefore, in a subsequent study [288], the authors focused on a prodrug strategy designed to translate the observed potent biochemical inhibitory activity of NNMT inhibitor GYZ-319 into strong cellular activity. The dual prodrug 14e was thus obtained, bearing the carboxylic acid masked as an isopropyl ester and the amine masked with a trimethyl-lock group in its structure (Figure 21), showing the most promising profile in terms of hydrolytic stability and cellular activity. Indeed, the prodrug exhibited improved cell permeability, which also translated to a significant enhancement of cellular activity compared to the parent compound.
18.2.2. NMMT Macrocyclic Peptides Inhibitors
In another approach, van Haren et al. [289] applied a peptide-mRNA display technology, known as the random nonstandard peptide integrated discovery (RaPID) system, to screen a library of more than 1012 macrocyclic peptides binding to NNMT. A set of macrocyclic peptides were identified with affinity for NNMT. In cell-based assays, administration of the previously synthesized macrocyclic peptides showed a reduction in the concentration of MNA, indicating a target-specific effect. Interestingly, this inhibition was found not to be impacted by elevated concentrations of either NA or SAM substrates, suggesting that these peptides function as allosteric inhibitors, the first to be reported for NNMT.
Taking these findings together, it appears clear that elevated NNMT levels are typically associated with unfavorable clinical parameters such as tumor size and advanced stage and grade, making NNMT an attractive and viable therapeutic target. Moreover, it has been demonstrated that NNMT gene silence promotes proliferation, migration, and therapy resistance. Multiple research groups are currently working on the discovery of novel inhibitors with high selectivity and efficacy which will contribute to elucidating the potential mechanism of NNMT and developing novel therapeutic strategies. Although at the moment is unlikely to use NNMT inhibitors alone to treat TNBC cancer, in the future, combining them with traditional targeted drugs, including EGFR-TKIs and bevacizumab, can improve prognosis through an optimized combination therapy, as well as reduce the possibility for the development of drug resistance. With the advancement of further studies and clinical trials, it is strongly believed that NNMT inhibitors can provide substantial benefits for patients with TNBC cancer.
19. Conclusions
TNBC represents the most aggressive breast cancer subtype characterized by the absence of expression of the estrogen receptor, progesterone receptor, and human epidermal growth factor receptor-2 which are responsible for the failure of common endocrine therapy and chemotherapy. TNBC patients display poorer outcomes and shorter survival compared to other BC subtypes, primarily due to the earlier onset of metastasis and the insurgence of chemoresistance [290,291]. In particular, the poorest prognosis and higher chances of recurrence are shown by the approximate 30% of TNBC patients who do not achieve a pCR to conventional chemotherapy in the neoadjuvant setting [292]. Therefore, the development of a novel, safe, and cost-effective treatment for TNBC is urgently needed. In the present review, we have summarized the current standard therapeutic armamentarium, and we have described novel small molecules and repositioned drugs developed to target the different molecular characteristics of TNBC subtypes. In addition, we have described innovative dual-targeting agents, combinatory treatments, and novel therapeutic strategies in preclinical testing, such as splicing inhibitors, whose further development retains high promises in improving TNBC management.
Intrinsic and acquired chemoresistance represent the major challenges in the current TNBC therapeutic management. Given the high tumoral heterogeneity within the TNBC subtype, several lines of evidence suggest that entry into the clinical practice of molecular stratification of TNBC patients could help defeat intrinsic chemoresistance [293]. Concerning the drugs described in this review, taxanes and mitotic inhibitors were proposed to display higher efficacy in BL1 tumors; PARP inhibitors were candidates for the treatment of BL2 tumors; inhibitors of CDK4/6 and signaling kinases PI3K, AKT, and mTOR were suggested for LAR tumor treatment [293]. Recent results from the FUTURE phase II umbrella clinical trial support the hypothesis that pharmacological treatments matched to TNBC patient molecular subtypes could improve therapeutic responses, as they showed improvements for IM TNBC patients treated with anti-PD1 plus paclitaxel and for basal-like immune suppressed (BLIS) tumors treated with anti-VEGFR therapies [294]. The administration of patient-tailored targeted first-line chemotherapy surely represents a key step forward also for the management of acquired chemoresistance, as it is conceivable that more effective therapies could limit the chances of the evolution/selection of relapsing resistant tumors. Therefore, the improvement of current therapeutic options for TNBC relies on a better molecular characterization of these tumors, which includes, for example, a comparative omics analysis between primary tumors and secondary tumors, shedding light on the molecular determinants of chemoresistance. These studies could unveil actionable vulnerabilities of recurrent and chemoresistant TNBC, paving the way for novel therapies and allowing patients diagnosed with TNBC to be spared from severe adverse side effects of untargeted therapies and have a better outlook on life.
Abbreviations
| 5-FU | 5-fluorouracil |
| AR | androgen receptor |
| BC | breast cancer |
| BL1 | basal-like 1 |
| BL2 | basal-like 2 |
| BRCA | breast cancer susceptibility gene |
| CDK4/6 inhibitors | cyclin-dependent kinase 4/6 inhibitors |
| CDKs | cyclin-dependent kinases |
| EGF | epidermal growth factor |
| ER | estrogen receptor |
| FDA | Food and Drug Administration |
| FoxO | forkhead box class O |
| gBRCA | germline BRCA |
| GSK3β | glycogen synthase kinase-3β |
| HER2 | human epidermal growth factor receptor 2 |
| HER3 | human epidermal growth factor receptor 3 |
| HR | hormone receptor |
| IAP | inhibitor of apoptosis protein |
| IC50 | half-maximal inhibitory concentration |
| LAR | luminal androgen receptor |
| M | mesenchymal (M) |
| mTOR | mammalian target of rapamycin |
| NAM | nicotinamide |
| NNMT | nicotinamide N-methyltransferase |
| ORR | objective response rate |
| OS | overall survival |
| PARP | poly adenosine diphosphate-ribose polymerase |
| pCR | pathological complete response |
| PDXs | patient-derived xenografts |
| PFS | progression free survival |
| PI3K | phosphatidylinositol-3-kinase |
| PKA/B/C | protein kinase A/B/C |
| PON2 | paraoxonase-2 |
| PR | progesterone receptor (PR) |
| PRAS40 | proline rich AKT substrate of 40 kDa |
| PRMTs | type-I protein arginine methyltransferases |
| PTEN | phosphatase and tensin homolog |
| PTX | paclitaxel |
| RTK | receptor tyrosine kinase |
| SAM | S-adenosyl-l-methionine |
| SMAC | second mitochondrial-derived activator of caspases |
| TNBC | triple-negative breast cancer |
| TSC2 | tuberos sclerosis complex 2 |
| ULK1 | Unc-51-like autophagy activating kinase 1 |
| VEGF | vascular endothelial growth factor |
| VEGFA | vascular endothelial growth factor A |
| VEGFR | vascular endothelial growth factor receptor |
Author Contributions
D.M. and C.N.: writing—original draft preparation; M.P.: literature search; A.U. and C.S.: review; G.L.R.: review and editing; R.S.: conceptualization and supervision. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflict of interest.
Funding Statement
This work was supported by grants from the Associazione Italiana Ricerca sul Cancro (MFAG21899), Breast Cancer Now (Catalyst Grant n. 2018NovPCC1283), AIRC IG 2020 n. 24703, MIUR PRIN 2022 2022TPPNTK, and Sapienza University of Rome RG11816428A9B4D5, RM120172A7EAD07C, RM11916B5598E3C4, and RM12218167FD3A37. The Università Cattolica del Sacro Cuore contributed to the funding of this research project and its publication.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Trivers K.F., Lund M.J., Porter P.L., Liff J.M., Flagg E.W., Coates R.J., Eley J.W. The Epidemiology of Triple-Negative Breast Cancer, Including Race. Cancer Causes Control. 2009;20:1071–1082. doi: 10.1007/s10552-009-9331-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cakar B., Muslu U., Erdogan A.P., Ozisik M., Ozisik H., Tunakan Dalgic C., Durusoy R., Karaca B., Sezgin C., Karabulut B., et al. The Role of Body Mass Index in Triple Negative Breast Cancer. Oncol. Res. Treat. 2015;38:518–522. doi: 10.1159/000439551. [DOI] [PubMed] [Google Scholar]
- 3.Jung S., Spiegelman D., Baglietto L., Bernstein L., Boggs D.A., van den Brandt P.A., Buring J.E., Cerhan J.R., Gaudet M.M., Giles G.G., et al. Fruit and Vegetable Intake and Risk of Breast Cancer by Hormone Receptor Status. J. Natl. Cancer Inst. 2013;105:219–236. doi: 10.1093/jnci/djs635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Link L.B., Canchola A.J., Bernstein L., Clarke C.A., Stram D.O., Ursin G., Horn-Ross P.L. Dietary Patterns and Breast Cancer Risk in the California Teachers Study Cohort. Am. J. Clin. Nutr. 2013;98:1524–1532. doi: 10.3945/ajcn.113.061184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehmann B.D., Jovanović B., Chen X., Estrada M.V., Johnson K.N., Shyr Y., Moses H.L., Sanders M.E., Pietenpol J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE. 2016;11:e0157368. doi: 10.1371/journal.pone.0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giuliano A.E., Connolly J.L., Edge S.B., Mittendorf E.A., Rugo H.S., Solin L.J., Weaver D.L., Winchester D.J., Hortobagyi G.N. Breast Cancer-Major Changes in the American Joint Committee on Cancer Eighth Edition Cancer Staging Manual. CA Cancer J. Clin. 2017;67:290–303. doi: 10.3322/caac.21393. [DOI] [PubMed] [Google Scholar]
- 7.Lehmann B.D., Bauer J.A., Chen X., Sanders M.E., Chakravarthy A.B., Shyr Y., Pietenpol J.A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin. Investig. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Santonja A., Sánchez-Muñoz A., Lluch A., Chica-Parrado M.R., Albanell J., Chacón J.I., Antolín S., Jerez J.M., de la Haba J., de Luque V., et al. Triple Negative Breast Cancer Subtypes and Pathologic Complete Response Rate to Neoadjuvant Chemotherapy. Oncotarget. 2018;9:26406–26416. doi: 10.18632/oncotarget.25413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin L., Duan J.-J., Bian X.-W., Yu S. Triple-Negative Breast Cancer Molecular Subtyping and Treatment Progress. Breast Cancer Res. 2020;22:61. doi: 10.1186/s13058-020-01296-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams S., Gray R.J., Demaria S., Goldstein L., Perez E.A., Shulman L.N., Martino S., Wang M., Jones V.E., Saphner T.J., et al. Prognostic Value of Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancers from Two Phase III Randomized Adjuvant Breast Cancer Trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014;32:2959–2966. doi: 10.1200/JCO.2013.55.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayes M.J., Thomas D., Emmons A., Giordano T.J., Kleer C.G. Genetic Changes of Wnt Pathway Genes Are Common Events in Metaplastic Carcinomas of the Breast. Clin. Cancer Res. 2008;14:4038–4044. doi: 10.1158/1078-0432.CCR-07-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anders C.K., Carey L.A. Biology, Metastatic Patterns, and Treatment of Patients with Triple-Negative Breast Cancer. Clin. Breast Cancer. 2009;9:S73–S81. doi: 10.3816/CBC.2009.s.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hong O.-Y., Noh E.-M., Jang H.-Y., Lee Y.-R., Lee B.K., Jung S.H., Kim J.-S., Youn H.J. Epigallocatechin Gallate Inhibits the Growth of MDA-MB-231 Breast Cancer Cells via Inactivation of the β-Catenin Signaling Pathway. Oncol. Lett. 2017;14:441–446. doi: 10.3892/ol.2017.6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury P., Ghosh U., Samanta K., Jaggi M., Chauhan S.C., Yallapu M.M. Bioactive Nanotherapeutic Trends to Combat Triple Negative Breast Cancer. Bioact. Mater. 2021;6:3269–3287. doi: 10.1016/j.bioactmat.2021.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Cancer Society. [(accessed on 4 April 2023)]. Available online: https://www.cancer.org/cancer/breast-cancer/about/types-of-breast-cancer/triple-negative.html.
- 16.Li X., Lewis M.T., Huang J., Gutierrez C., Osborne C.K., Wu M.-F., Hilsenbeck S.G., Pavlick A., Zhang X., Chamness G.C., et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNCI J. Natl. Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 17.Lee J.S., Yost S.E., Yuan Y. Neoadjuvant Treatment for Triple Negative Breast Cancer: Recent Progresses and Challenges. Cancers. 2020;12:1404. doi: 10.3390/cancers12061404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Reilly E.A., Gubbins L., Sharma S., Tully R., Guang M.H.Z., Weiner-Gorzel K., McCaffrey J., Harrison M., Furlong F., Kell M., et al. The Fate of Chemoresistance in Triple Negative Breast Cancer (TNBC) BBA Clin. 2015;3:257–275. doi: 10.1016/j.bbacli.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadi V.K., Davidson N.E. Practical Approach to Triple-Negative Breast Cancer. J. Oncol. Pract. 2017;13:293–300. doi: 10.1200/JOP.2017.022632. [DOI] [PubMed] [Google Scholar]
- 20.Longley D.B., Harkin D.P., Johnston P.G. 5-Fluorouracil: Mechanisms of Action and Clinical Strategies. Nat. Rev. Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 21.Deng J., Wang Y., Lei J., Lei W., Xiong J.P. Insights into the Involvement of Noncoding RNAs in 5-Fluorouracil Drug Resistance. Tumor Biol. 2017;39:101042831769755. doi: 10.1177/1010428317697553. [DOI] [PubMed] [Google Scholar]
- 22.Joensuu H., Kellokumpu-Lehtinen P.-L., Huovinen R., Jukkola-Vuorinen A., Tanner M., Kokko R., Ahlgren J., Auvinen P., Lahdenperä O., Kosonen S., et al. Adjuvant Capecitabine in Combination with Docetaxel, Epirubicin, and Cyclophosphamide for Early Breast Cancer. JAMA Oncol. 2017;3:793. doi: 10.1001/jamaoncol.2016.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noh S.H., Park S.R., Yang H.-K., Chung H.C., Chung I.-J., Kim S.-W., Kim H.-H., Choi J.-H., Kim H.-K., Yu W., et al. Adjuvant Capecitabine plus Oxaliplatin for Gastric Cancer after D2 Gastrectomy (CLASSIC): 5-Year Follow-up of an Open-Label, Randomised Phase 3 Trial. Lancet Oncol. 2014;15:1389–1396. doi: 10.1016/S1470-2045(14)70473-5. [DOI] [PubMed] [Google Scholar]
- 24.Xun X., Cao Q., Hong P., Rai S., Zhou Y., Liu R., Hu H. Efficacy and Safety of Capecitabine for Triple-Negative Breast Cancer: A Meta-Analysis. Front. Oncol. 2022;12:899423. doi: 10.3389/fonc.2022.899423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z., Zheng J., Ji Z., Chen L., Wu J., Zou J., Liu Y., Lin W., Cai J., Chen Y., et al. Addition of Capecitabine to Adjuvant Chemotherapy May Be the Most Effective Strategy for Patients with Early-Stage Triple-Negative Breast Cancer: A Network Meta-Analysis of 9 Randomized Controlled Trials. Front. Endocrinol. 2022;13:939048. doi: 10.3389/fendo.2022.939048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diéras V., Bonnefoi H., Alba E., Awada A., Coudert B., Pivot X., Gligorov J., Jager A., Zambelli S., Lindeman G.J., et al. Iniparib Administered Weekly or Twice-Weekly in Combination with Gemcitabine/Carboplatin in Patients with Metastatic Triple-Negative Breast Cancer: A Phase II Randomized Open-Label Study with Pharmacokinetics. Breast Cancer Res. Treat. 2019;177:383–393. doi: 10.1007/s10549-019-05305-w. [DOI] [PubMed] [Google Scholar]
- 27.Honeywell R.J., Ruiz van Haperen V.W.T., Veerman G., Smid K., Peters G.J. Inhibition of Thymidylate Synthase by 2′,2′-Difluoro-2′-Deoxycytidine (Gemcitabine) and Its Metabolite 2′,2′-Difluoro-2′-Deoxyuridine. Int. J. Biochem. Cell Biol. 2015;60:73–81. doi: 10.1016/j.biocel.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Mitsuno M., Kitajima Y., Ohtaka K., Kai K., Hashiguchi K., Nakamura J., Hiraki M., Noshiro H., Miyazaki K. Tranilast Strongly Sensitizes Pancreatic Cancer Cells to Gemcitabine via Decreasing Protein Expression of Ribonucleotide Reductase 1. Int. J. Oncol. 2010;36:341–349. [PubMed] [Google Scholar]
- 29.Garcia-Diaz M., Murray M.S., Kunkel T.A., Chou K.-M. Interaction between DNA Polymerase Lambda and Anticancer Nucleoside Analogs. J. Biol. Chem. 2010;285:16874–16879. doi: 10.1074/jbc.M109.094391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y., Jiang H., Gu M., Zu C., Zheng X. Gemcitabine Resistance in Triple negative Breast Cancer Cells Can Be Reverted by <Em>Drosophila Melanogaster</Em> Deoxyribonucleoside Kinase in the Nucleus or Cytosol. Oncol. Lett. 2020;20:247. doi: 10.3892/ol.2020.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furlanetto J., Loibl S. Optimal Systemic Treatment for Early Triple-Negative Breast Cancer. Breast Care. 2020;15:217–226. doi: 10.1159/000508759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steinherz L.J., Steinherz P.G., Tan C.T., Heller G., Murphy M.L. Cardiac Toxicity 4 to 20 Years after Completing Anthracycline Therapy. JAMA. 1991;266:1672–1677. doi: 10.1001/jama.1991.03470120074036. [DOI] [PubMed] [Google Scholar]
- 33.Chaplain G., Milan C., Sgro C., Carli P.-M., Bonithon-Kopp C. Increased Risk of Acute Leukemia After Adjuvant Chemotherapy for Breast Cancer: A Population-Based Study. J. Clin. Oncol. 2000;18:2836–2842. doi: 10.1200/JCO.2000.18.15.2836. [DOI] [PubMed] [Google Scholar]
- 34.Schiff P.B., Fant J., Horwitz S.B. Promotion of Microtubule Assembly in Vitro by Taxol. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 35.Ranieri G., Marech I., Porcelli M., Giotta F., Palmiotti G., Laricchia G., Fazio V., Gadaleta C.D. Complete Response in a Patient with Liver Metastases from Breast Cancer Employing Hepatic Arterial Infusion 5-Fluorouracil Based Chemotherapy plus Systemic Nab-Paclitaxel. Oncotarget. 2018;9:8197–8203. doi: 10.18632/oncotarget.23793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Que W.-C., Huang Y.-F., Lin X.-Y., Lan Y.-Q., Gao X.-Y., Wang X.-L., Wu R.-P., Du B., Huang X.-B., Qiu H., et al. Paclitaxel, 5-Fluorouracil, and Leucovorin Combination Chemotherapy as First-Line Treatment in Patients with Advanced Gastric Cancer. Anticancer Drugs. 2019;30:302–307. doi: 10.1097/CAD.0000000000000735. [DOI] [PubMed] [Google Scholar]
- 37.Chen T., Chen H., Jiang Y., Yan Q., Zheng S., Wu M. Co-Delivery of 5-Fluorouracil and Paclitaxel in Mitochondria-Targeted KLA-Modified Liposomes to Improve Triple-Negative Breast Cancer Treatment. Pharmaceuticals. 2022;15:881. doi: 10.3390/ph15070881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elghazaly H., Rugo H.S., Azim H.A., Swain S.M., Arun B., Aapro M., Perez E.A., Anderson B.O., Penault-Llorca F., Conte P., et al. Breast-Gynaecological & Immuno-Oncology International Cancer Conference (BGICC) Consensus and Recommendations for the Management of Triple-Negative Breast Cancer. Cancers. 2021;13:2262. doi: 10.3390/cancers13092262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gradishar W.J., Krasnojon D., Cheporov S., Makhson A.N., Manikhas G.M., Clawson A., Bhar P. Significantly Longer Progression-Free Survival with Nab -Paclitaxel Compared with Docetaxel as First-Line Therapy for Metastatic Breast Cancer. J. Clin. Oncol. 2009;27:3611–3619. doi: 10.1200/JCO.2008.18.5397. [DOI] [PubMed] [Google Scholar]
- 40.Hu X., Wang B., Sun S., Tang L., Zhang J., Lv F., Wang Z., Wang L., Zhang Q., Zheng C., et al. Cisplatin Improves Antitumor Activity of Weekly Nab-Paclitaxel in Patients with Metastatic Breast Cancer. Int. J. Nanomed. 2014;9:1443–1452. doi: 10.2147/IJN.S58275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang B., Sun T., Zhao Y., Wang S., Zhang J., Wang Z., Teng Y.-E., Cai L., Yan M., Wang X., et al. A Randomized Phase 3 Trial of Gemcitabine or Nab-Paclitaxel Combined with CisPlatin as First-Line Treatment in Patients with Metastatic Triple-Negative Breast Cancer. Nat. Commun. 2022;13:4025. doi: 10.1038/s41467-022-31704-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okouneva T., Azarenko O., Wilson L., Littlefield B.A., Jordan M.A. Inhibition of Centromere Dynamics by Eribulin (E7389) during Mitotic Metaphase. Mol. Cancer Ther. 2008;7:2003–2011. doi: 10.1158/1535-7163.MCT-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pizzuti L., Krasniqi E., Barchiesi G., Mazzotta M., Barba M., Amodio A., Massimiani G., Pelle F., Kayal R., Vizza E., et al. Eribulin in Triple Negative Metastatic Breast Cancer: Critic Interpretation of Current Evidence and Projection for Future Scenarios. J. Cancer. 2019;10:5903–5914. doi: 10.7150/jca.35109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yonemori K., Shimomura A., Yasojima H., Masuda N., Aogi K., Takahashi M., Naito Y., Shimizu S., Nakamura R., Hashimoto J., et al. A Phase I/II Trial of Olaparib Tablet in Combination with Eribulin in Japanese Patients with Advanced or Metastatic Triple-Negative Breast Cancer Previously Treated with Anthracyclines and Taxanes. Eur. J. Cancer. 2019;109:84–91. doi: 10.1016/j.ejca.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 45.Hu X.-C., Zhang J., Xu B.-H., Cai L., Ragaz J., Wang Z.-H., Wang B.-Y., Teng Y.-E., Tong Z.-S., Pan Y.-Y., et al. Cisplatin plus Gemcitabine versus Paclitaxel plus Gemcitabine as First-Line Therapy for Metastatic Triple-Negative Breast Cancer (CBCSG006): A Randomised, Open-Label, Multicentre, Phase 3 Trial. Lancet Oncol. 2015;16:436–446. doi: 10.1016/S1470-2045(15)70064-1. [DOI] [PubMed] [Google Scholar]
- 46.Pogoda K., Niwińska A., Sarnowska E., Nowakowska D., Jagiełło-Gruszfeld A., Siedlecki J., Nowecki Z. Effects of BRCA Germline Mutations on Triple-Negative Breast Cancer Prognosis. J. Oncol. 2020;2020:8545643. doi: 10.1155/2020/8545643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diana A., Carlino F., Franzese E., Oikonomidou O., Criscitiello C., De Vita F., Ciardiello F., Orditura M. Early Triple Negative Breast Cancer: Conventional Treatment and Emerging Therapeutic Landscapes. Cancers. 2020;12:819. doi: 10.3390/cancers12040819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoeijmakers J.H.J. Genome Maintenance Mechanisms for Preventing Cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 49.Rouleau M., Patel A., Hendzel M.J., Kaufmann S.H., Poirier G.G. PARP Inhibition: PARP1 and Beyond. Nat. Rev. Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei H., Yu X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016;14:131–139. doi: 10.1016/j.gpb.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Litton J.K., Scoggins M.E., Hess K.R., Adrada B.E., Murthy R.K., Damodaran S., DeSnyder S.M., Brewster A.M., Barcenas C.H., Valero V., et al. Neoadjuvant Talazoparib for Patients with Operable Breast Cancer with a Germline BRCA Pathogenic Variant. J. Clin. Oncol. 2020;38:388–394. doi: 10.1200/JCO.19.01304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tutt A., Robson M., Garber J.E., Domchek S.M., Audeh M.W., Weitzel J.N., Friedlander M., Arun B., Loman N., Schmutzler R.K., et al. Oral Poly(ADP-Ribose) Polymerase Inhibitor Olaparib in Patients with BRCA1 or BRCA2 Mutations and Advanced Breast Cancer: A Proof-of-Concept Trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 53.Clark J.B., Ferris G.M., Pinder S. Inhibition of Nuclear NAD Nucleosidase and Poly ADP-Ribose Polymerase Activity from Rat Liver by Nicotinamide and 5′-Methyl Nicotinamide. Biochim. Biophys. Acta (BBA) Nucleic Acids Protein Synth. 1971;238:82–85. doi: 10.1016/0005-2787(71)90012-8. [DOI] [PubMed] [Google Scholar]
- 54.Steffen J.D., Brody J.R., Armen R.S., Pascal J.M. Structural Implications for Selective Targeting of PARPs. Front. Oncol. 2013;3:301. doi: 10.3389/fonc.2013.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patel A.G., De Lorenzo S.B., Flatten K.S., Poirier G.G., Kaufmann S.H. Failure of Iniparib to Inhibit Poly(ADP-Ribose) Polymerase In Vitro. Clin. Cancer Res. 2012;18:1655–1662. doi: 10.1158/1078-0432.CCR-11-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Menear K.A., Adcock C., Boulter R., Cockcroft X., Copsey L., Cranston A., Dillon K.J., Drzewiecki J., Garman S., Gomez S., et al. 4-[3-(4-Cyclopropanecarbonylpiperazine-1-Carbonyl)-4-Fluorobenzyl]-2 H -Phthalazin-1-One: A Novel Bioavailable Inhibitor of Poly(ADP-Ribose) Polymerase-1. J. Med. Chem. 2008;51:6581–6591. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 57.Peto J., Collins N., Barfoot R., Seal S., Warren W., Rahman N., Easton D.F., Evans C., Deacon J., Stratton M.R. Prevalence of BRCA1 and BRCA2 Gene Mutations in Patients with Early-Onset Breast Cancer. JNCI J. Natl. Cancer Inst. 1999;91:943–949. doi: 10.1093/jnci/91.11.943. [DOI] [PubMed] [Google Scholar]
- 58.Abdel-Razeq H., Al-Omari A., Zahran F., Arun B. Germline BRCA1/BRCA2 Mutations among High Risk Breast Cancer Patients in Jordan. BMC Cancer. 2018;18:152. doi: 10.1186/s12885-018-4079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gelmon K.A., Hirte H.W., Robidoux A., Tonkin K.S., Tischkowitz M., Swenerton K., Huntsman D., Carmichael J., Macpherson E., Oza A.M. Can We Define Tumors That Will Respond to PARP Inhibitors? A Phase II Correlative Study of Olaparib in Advanced Serous Ovarian Cancer and Triple-Negative Breast Cancer. J. Clin. Oncol. 2010;28:3002. doi: 10.1200/jco.2010.28.15_suppl.3002. [DOI] [Google Scholar]
- 60.Murai J., Huang S.-Y.N., Renaud A., Zhang Y., Ji J., Takeda S., Morris J., Teicher B., Doroshow J.H., Pommier Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014;13:433–443. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Litton J.K., Beck J.T., Jones J.M., Andersen J., Blum J.L., Mina L.A., Brig R., Danso M., Yuan Y., Abbattista A., et al. Neoadjuvant Talazoparib in Patients with Germline BRCA1/2 Mutation-Positive HER2-Negative Breast Cancer: Results of a Phase 2 Study. 2021 ASCO Annual Meeting. Abstract 505. Presented 6 June 2021. [(accessed on 23 April 2023)]. Available online: https://ascopost.com/issues/august-10-2021/single-agent-talazoparib-shows-activity-in-the-neoadjuvant-treatment-of-triple-negative-breast-cancer.
- 62.Hurvitz S.A., Gonçalves A., Rugo H.S., Lee K.-H., Fehrenbacher L., Mina L.A., Diab S., Blum J.L., Chakrabarti J., Elmeliegy M., et al. Talazoparib in Patients with a Germline BRCA -Mutated Advanced Breast Cancer: Detailed Safety Analyses from the Phase III EMBRACA Trial. Oncologist. 2020;25:e439–e450. doi: 10.1634/theoncologist.2019-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guney Eskiler G., Cecener G., Egeli U., Tunca B. Talazoparib Nanoparticles for Overcoming Multidrug Resistance in Triple-negative Breast Cancer. J. Cell Physiol. 2020;235:6230–6245. doi: 10.1002/jcp.29552. [DOI] [PubMed] [Google Scholar]
- 64.Loibl S., O’Shaughnessy J., Untch M., Sikov W.M., Rugo H.S., McKee M.D., Huober J., Golshan M., von Minckwitz G., Maag D., et al. Addition of the PARP Inhibitor Veliparib plus Carboplatin or Carboplatin Alone to Standard Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer (BrighTNess): A Randomised, Phase 3 Trial. Lancet Oncol. 2018;19:497–509. doi: 10.1016/S1470-2045(18)30111-6. [DOI] [PubMed] [Google Scholar]
- 65.Kalra M., Tong Y., Jones D.R., Walsh T., Danso M.A., Ma C.X., Silverman P., King M.-C., Badve S.S., Perkins S.M., et al. Cisplatin +/− Rucaparib after Preoperative Chemotherapy in Patients with Triple-Negative or BRCA Mutated Breast Cancer. NPJ Breast Cancer. 2021;7:29. doi: 10.1038/s41523-021-00240-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rucaparib Camsylate by Zr Pharma for Triple-Negative Breast Cancer (Tnbc): Likelihood of Approval. [(accessed on 11 September 2023)]. Available online: https://www.pharmaceutical-technology.com/data-insights/rucaparib-camsylate-zr-pharma-triple-negative-breast-cancer-tnbc-likelihood-of-approval/?cf-view.
- 67.Neoadjuvant Niraparib Demonstrates Promising Antitumour Activity and Safety in Patients with Localised HER2-Negative, BRCA-Mutated Breast Cancer. ESMO Oncology News. [(accessed on 23 April 2023)]. Available online: https://www.esmo.org/oncology-news/neoadjuvant-niraparib-demonstrates-promising-antitumour-activity-and-safety-in-patients-with-localised-her2-negative-brca-mutated-breast-cancer.
- 68.Xu B., Sun T., Shi Y., Cui J., Yin Y., Ouyang Q., Liu Q., Zhang Q., Chen Y., Wang S., et al. Pamiparib in Patients with Locally Advanced or Metastatic HER2-Negative Breast Cancer with Germline BRCA Mutations: A Phase II Study. Breast Cancer Res. Treat. 2023;197:489–501. doi: 10.1007/s10549-022-06785-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christie E.L., Pattnaik S., Beach J., Copeland A., Rashoo N., Fereday S., Hendley J., Alsop K., Brady S.L., Lamb G., et al. Multiple ABCB1 Transcriptional Fusions in Drug Resistant High-Grade Serous Ovarian and Breast Cancer. Nat. Commun. 2019;10:1295. doi: 10.1038/s41467-019-09312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lombard A.P., Liu C., Armstrong C.M., D’Abronzo L.S., Lou W., Chen H., Dall’Era M., Ghosh P.M., Evans C.P., Gao A.C. Overexpressed ABCB1 Induces Olaparib-Taxane Cross-Resistance in Advanced Prostate Cancer. Transl. Oncol. 2019;12:871–878. doi: 10.1016/j.tranon.2019.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaidyanathan A., Sawers L., Gannon A.-L., Chakravarty P., Scott A.L., Bray S.E., Ferguson M.J., Smith G. ABCB1 (MDR1) Induction Defines a Common Resistance Mechanism in Paclitaxel- and Olaparib-Resistant Ovarian Cancer Cells. Br. J. Cancer. 2016;115:431–441. doi: 10.1038/bjc.2016.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barchiesi G., Roberto M., Verrico M., Vici P., Tomao S., Tomao F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol. 2021;11:769280. doi: 10.3389/fonc.2021.769280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morgan D.O. CYCLIN-DEPENDENT KINASES: Engines, Clocks, and Microprocessors. Annu. Rev. Cell Dev. Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 74.Malumbres M. Cyclin-Dependent Kinases. Genome Biol. 2014;15:122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naro C., Bielli P., Sette C. Oncogenic Dysregulation of Pre-mRNA Processing by Protein Kinases: Challenges and Therapeutic Opportunities. FEBS J. 2021;288:6250–6272. doi: 10.1111/febs.16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Malumbres M., Pevarello P., Barbacid M., Bischoff J.R. CDK Inhibitors in Cancer Therapy: What Is Next? Trends Pharmacol. Sci. 2008;29:16–21. doi: 10.1016/j.tips.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 77.Asghar U., Witkiewicz A.K., Turner N.C., Knudsen E.S. The History and Future of Targeting Cyclin-Dependent Kinases in Cancer Therapy. Nat. Rev. Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ishii Y., Pirkmaier A., Alvarez J.V., Frank D.A., Keselman I., Logothetis D., Mandeli J., O’Connell M.J., Waxman S., Germain D. Cyclin D1 Overexpression and Response to Bortezomib Treatment in a Breast Cancer Model. JNCI J. Natl. Cancer Inst. 2006;98:1238–1247. doi: 10.1093/jnci/djj334. [DOI] [PubMed] [Google Scholar]
- 79.Witkiewicz A.K., Cox D., Knudsen E.S. CDK4/6 Inhibition Provides a Potent Adjunct to Her2-Targeted Therapies in Preclinical Breast Cancer Models. Genes. Cancer. 2014;5:261–272. doi: 10.18632/genesandcancer.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.The Cancer Genome Atlas Network Comprehensive Molecular Portraits of Human Breast Tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parry D., Guzi T., Shanahan F., Davis N., Prabhavalkar D., Wiswell D., Seghezzi W., Paruch K., Dwyer M.P., Doll R., et al. Dinaciclib (SCH 727965), a Novel and Potent Cyclin-Dependent Kinase Inhibitor. Mol. Cancer Ther. 2010;9:2344–2353. doi: 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 82.Finn R.S., Dering J., Conklin D., Kalous O., Cohen D.J., Desai A.J., Ginther C., Atefi M., Chen I., Fowst C., et al. PD 0332991, a Selective Cyclin D Kinase 4/6 Inhibitor, Preferentially Inhibits Proliferation of Luminal Estrogen Receptor-Positive Human Breast Cancer Cell Lines in Vitro. Breast Cancer Res. 2009;11:R77. doi: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Feng Y., Spezia M., Huang S., Yuan C., Zeng Z., Zhang L., Ji X., Liu W., Huang B., Luo W., et al. Breast Cancer Development and Progression: Risk Factors, Cancer Stem Cells, Signaling Pathways, Genomics, and Molecular Pathogenesis. Genes. Dis. 2018;5:77–106. doi: 10.1016/j.gendis.2018.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Condorelli R., Spring L., O’Shaughnessy J., Lacroix L., Bailleux C., Scott V., Dubois J., Nagy R.J., Lanman R.B., Iafrate A.J., et al. Polyclonal RB1 Mutations and Acquired Resistance to CDK 4/6 Inhibitors in Patients with Metastatic Breast Cancer. Ann. Oncol. 2018;29:640–645. doi: 10.1093/annonc/mdx784. [DOI] [PubMed] [Google Scholar]
- 85.Pernas S., Tolaney S.M., Winer E.P., Goel S. CDK4/6 Inhibition in Breast Cancer: Current Practice and Future Directions. Ther. Adv. Med. Oncol. 2018;10:1758835918786451. doi: 10.1177/1758835918786451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knudsen E.S., Witkiewicz A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer. 2017;3:39–55. doi: 10.1016/j.trecan.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Asghar U.S., Barr A.R., Cutts R., Beaney M., Babina I., Sampath D., Giltnane J., Lacap J.A., Crocker L., Young A., et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017;23:5561–5572. doi: 10.1158/1078-0432.CCR-17-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu C.-Y., Lau K.-Y., Hsu C.-C., Chen J.-L., Lee C.-H., Huang T.-T., Chen Y.-T., Huang C.-T., Lin P.-H., Tseng L.-M. Combination of Palbociclib with Enzalutamide Shows in Vitro Activity in RB Proficient and Androgen Receptor Positive Triple Negative Breast Cancer Cells. PLoS ONE. 2017;12:e0189007. doi: 10.1371/journal.pone.0189007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Choupani E., Madjd Z., Saraygord-Afshari N., Kiani J., Hosseini A. Combination of Androgen Receptor Inhibitor Enzalutamide with the CDK4/6 Inhibitor Ribociclib in Triple Negative Breast Cancer Cells. PLoS ONE. 2022;17:e0279522. doi: 10.1371/journal.pone.0279522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guarducci C., Bonechi M., Benelli M., Biagioni C., Boccalini G., Romagnoli D., Verardo R., Schiff R., Osborne C.K., De Angelis C., et al. Cyclin E1 and Rb Modulation as Common Events at Time of Resistance to Palbociclib in Hormone Receptor-Positive Breast Cancer. NPJ Breast Cancer. 2018;4:38. doi: 10.1038/s41523-018-0092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fassl A., Brain C., Abu-Remaileh M., Stukan I., Butter D., Stepien P., Feit A.S., Bergholz J., Michowski W., Otto T., et al. Increased Lysosomal Biomass Is Responsible for the Resistance of Triple-Negative Breast Cancers to CDK4/6 Inhibition. Sci. Adv. 2020;6:eabb2210. doi: 10.1126/sciadv.abb2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ozman Z., Guney Eskiler G., Sekeroglu M.R. In Vitro Therapeutic Effects of Abemaciclib on Triple-negative Breast Cancer Cells. J. Biochem. Mol. Toxicol. 2021;35:e22858. doi: 10.1002/jbt.22858. [DOI] [PubMed] [Google Scholar]
- 93.Cretella D., Fumarola C., Bonelli M., Alfieri R., La Monica S., Digiacomo G., Cavazzoni A., Galetti M., Generali D., Petronini P.G. Pre-Treatment with the CDK4/6 Inhibitor Palbociclib Improves the Efficacy of Paclitaxel in TNBC Cells. Sci. Rep. 2019;9:13014. doi: 10.1038/s41598-019-49484-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dean J.L., McClendon A.K., Knudsen E.S. Modification of the DNA Damage Response by Therapeutic CDK4/6 Inhibition. J. Biol. Chem. 2012;287:29075–29087. doi: 10.1074/jbc.M112.365494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Saleh L., Wilson C., Holen I. CDK4/6 Inhibitors: A Potential Therapeutic Approach for Triple Negative Breast Cancer. Medcomm. 2021;2:514–530. doi: 10.1002/mco2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jabbour-Leung N.A., Chen X., Bui T., Jiang Y., Yang D., Vijayaraghavan S., McArthur M.J., Hunt K.K., Keyomarsi K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in P53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016;15:593–607. doi: 10.1158/1535-7163.MCT-15-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tarasewicz E., Rivas L., Hamdan R., Dokic D., Parimi V., Bernabe B.P., Thomas A., Shea L.D., Jeruss J.S. Inhibition of CDK-Mediated Phosphorylation of Smad3 Results in Decreased Oncogenesis in Triple Negative Breast Cancer Cells. Cell Cycle. 2014;13:3191–3201. doi: 10.4161/15384101.2014.950126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nie L., Wei Y., Zhang F., Hsu Y.-H., Chan L.-C., Xia W., Ke B., Zhu C., Deng R., Tang J., et al. CDK2-Mediated Site-Specific Phosphorylation of EZH2 Drives and Maintains Triple-Negative Breast Cancer. Nat. Commun. 2019;10:5114. doi: 10.1038/s41467-019-13105-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitri Z., Karakas C., Wei C., Briones B., Simmons H., Ibrahim N., Alvarez R., Murray J.L., Keyomarsi K., Moulder S. A Phase 1 Study with Dose Expansion of the CDK Inhibitor Dinaciclib (SCH 727965) in Combination with Epirubicin in Patients with Metastatic Triple Negative Breast Cancer. Investig. New Drugs. 2015;33:890–894. doi: 10.1007/s10637-015-0244-4. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y., Zhang T., Kwiatkowski N., Abraham B.J., Lee T.I., Xie S., Yuzugullu H., Von T., Li H., Lin Z., et al. CDK7-Dependent Transcriptional Addiction in Triple-Negative Breast Cancer. Cell. 2015;163:174–186. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Piemonte K.M., Webb B.M., Bobbitt J.R., Majmudar P.R., Cuellar-Vite L., Bryson B.L., Latina N.C., Seachrist D.D., Keri R.A. Disruption of CDK7 Signaling Leads to Catastrophic Chromosomal Instability Coupled with a Loss of Condensin-Mediated Chromatin Compaction. J. Biol. Chem. 2023;299:104834. doi: 10.1016/j.jbc.2023.104834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li B., Ni Chonghaile T., Fan Y., Madden S.F., Klinger R., O’Connor A.E., Walsh L., O’Hurley G., Mallya Udupi G., Joseph J., et al. Therapeutic Rationale to Target Highly Expressed CDK7 Conferring Poor Outcomes in Triple-Negative Breast Cancer. Cancer Res. 2017;77:3834–3845. doi: 10.1158/0008-5472.CAN-16-2546. [DOI] [PubMed] [Google Scholar]
- 103.Coombes R.C., Howell S., Lord S.R., Kenny L., Mansi J., Mitri Z., Palmieri C., Chap L.I., Richards P., Gradishar W., et al. Dose Escalation and Expansion Cohorts in Patients with Advanced Breast Cancer in a Phase I Study of the CDK7-Inhibitor Samuraciclib. Nat. Commun. 2023;14:4444. doi: 10.1038/s41467-023-40061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dubbury S.J., Boutz P.L., Sharp P.A. CDK12 Regulates DNA Repair Genes by Suppressing Intronic Polyadenylation. Nature. 2018;564:141–145. doi: 10.1038/s41586-018-0758-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Panzeri V., Pieraccioli M., Cesari E., de la Grange P., Sette C. CDK12/13 Promote Splicing of Proximal Introns by Enhancing the Interaction between RNA Polymerase II and the Splicing Factor SF3B1. Nucleic Acids Res. 2023;51:5512–5526. doi: 10.1093/nar/gkad258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bajrami I., Frankum J.R., Konde A., Miller R.E., Rehman F.L., Brough R., Campbell J., Sims D., Rafiq R., Hooper S., et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014;74:287–297. doi: 10.1158/0008-5472.CAN-13-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johnson S.F., Cruz C., Greifenberg A.K., Dust S., Stover D.G., Chi D., Primack B., Cao S., Bernhardy A.J., Coulson R., et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep. 2016;17:2367–2381. doi: 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Quereda V., Bayle S., Vena F., Frydman S.M., Monastyrskyi A., Roush W.R., Duckett D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell. 2019;36:545–558.e7. doi: 10.1016/j.ccell.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 109.Yang J., Chang Y., Tien J.C.-Y., Wang Z., Zhou Y., Zhang P., Huang W., Vo J., Apel I.J., Wang C., et al. Discovery of a Highly Potent and Selective Dual PROTAC Degrader of CDK12 and CDK13. J. Med. Chem. 2022;65:11066–11083. doi: 10.1021/acs.jmedchem.2c00384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niu T., Li K., Jiang L., Zhou Z., Hong J., Chen X., Dong X., He Q., Cao J., Yang B., et al. Noncovalent CDK12/13 Dual Inhibitors-Based PROTACs Degrade CDK12-Cyclin K Complex and Induce Synthetic Lethality with PARP Inhibitor. Eur. J. Med. Chem. 2022;228:114012. doi: 10.1016/j.ejmech.2021.114012. [DOI] [PubMed] [Google Scholar]
- 111.Zhang L., Zhen Y., Feng L., Li Z., Lu Y., Wang G., Ouyang L. Discovery of a Novel Dual-Target Inhibitor of CDK12 and PARP1 That Induces Synthetic Lethality for Treatment of Triple-Negative Breast Cancer. Eur. J. Med. Chem. 2023;259:115648. doi: 10.1016/j.ejmech.2023.115648. [DOI] [PubMed] [Google Scholar]
- 112.Cesari E., Ciucci A., Pieraccioli M., Caggiano C., Nero C., Bonvissuto D., Sillano F., Buttarelli M., Piermattei A., Loverro M., et al. Dual Inhibition of CDK12 and CDK13 Uncovers Actionable Vulnerabilities in Patient-Derived Ovarian Cancer Organoids. J. Exp. Clin. Cancer Res. 2023;42:126. doi: 10.1186/s13046-023-02682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sebastiani J., Puxeddu M., Nalli M., Bai R., Altieri L., Rovella P., Gaudio E., Trisciuoglio D., Spriano F., Lavia P., et al. RS6077 Induces Mitotic Arrest and Selectively Activates Cell Death in Human Cancer Cell Lines and in a Lymphoma Tumor in Vivo. Eur. J. Med. Chem. 2023;246:114997. doi: 10.1016/j.ejmech.2022.114997. [DOI] [PubMed] [Google Scholar]
- 114.La Regina G., Bai R., Coluccia A., Naccarato V., Famiglini V., Nalli M., Masci D., Verrico A., Rovella P., Mazzoccoli C., et al. New 6- and 7-Heterocyclyl-1H-Indole Derivatives as Potent Tubulin Assembly and Cancer Cell Growth Inhibitors. Eur. J. Med. Chem. 2018;152:283–297. doi: 10.1016/j.ejmech.2018.04.042. [DOI] [PubMed] [Google Scholar]
- 115.Li L., Jiang S., Li X., Liu Y., Su J., Chen J. Recent Advances in Trimethoxyphenyl (TMP) Based Tubulin Inhibitors Targeting the Colchicine Binding Site. Eur. J. Med. Chem. 2018;151:482–494. doi: 10.1016/j.ejmech.2018.04.011. [DOI] [PubMed] [Google Scholar]
- 116.Chen J., Ahn S., Wang J., Lu Y., Dalton J.T., Miller D.D., Li W. Discovery of Novel 2-Aryl-4-Benzoyl-Imidazole (ABI-III) Analogues Targeting Tubulin Polymerization As Antiproliferative Agents. J. Med. Chem. 2012;55:7285–7289. doi: 10.1021/jm300564b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Deng S., Krutilina R.I., Wang Q., Lin Z., Parke D.N., Playa H.C., Chen H., Miller D.D., Seagroves T.N., Li W. An Orally Available Tubulin Inhibitor, VERU-111, Suppresses Triple-Negative Breast Cancer Tumor Growth and Metastasis and Bypasses Taxane Resistance. Mol. Cancer Ther. 2020;19:348–363. doi: 10.1158/1535-7163.MCT-19-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deng S., Krutilina R.I., Parke D., Chen H., Miller D.D., Li W., Seagroves T.N. VERU-111: An Oral Tubulin Inhibitor That Suppresses Taxane-Sensitive and Taxane-Resistant Breast Cancer. J. Endocr. Soc. 2021;5:A1035. doi: 10.1210/jendso/bvab048.2118. [DOI] [Google Scholar]
- 119.Krutilina R.I., Hartman K.L., Oluwalana D., Playa H.C., Parke D.N., Chen H., Miller D.D., Li W., Seagroves T.N. Sabizabulin, a Potent Orally Bioavailable Colchicine Binding Site Agent, Suppresses HER2+ Breast Cancer and Metastasis. Cancers. 2022;14:5336. doi: 10.3390/cancers14215336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lemjabbar-Alaoui H., Peto C.J., Yang Y.-W., Jablons D.M. AMXI-5001, a Novel Dual Parp1/2 and Microtubule Polymerization Inhibitor for the Treatment of Human Cancers. Am. J. Cancer Res. 2020;10:2649–2676. [PMC free article] [PubMed] [Google Scholar]
- 121.Puxeddu M., Wu J., Bai R., D’Ambrosio M., Nalli M., Coluccia A., Manetto S., Ciogli A., Masci D., Urbani A., et al. Induction of Ferroptosis in Glioblastoma and Ovarian Cancers by a New Pyrrole Tubulin Assembly Inhibitor. J. Med. Chem. 2022;65:15805–15818. doi: 10.1021/acs.jmedchem.2c01457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rugo H.S., Roche H., Thomas E., Chung H.C., Lerzo G.L., Vasyutin I., Patel A., Vahdat L. Efficacy and Safety of Ixabepilone and Capecitabine in Patients with Advanced Triple-Negative Breast Cancer: A Pooled Analysis from Two Large Phase III, Randomized Clinical Trials. Clin. Breast Cancer. 2018;18:489–497. doi: 10.1016/j.clbc.2018.07.024. [DOI] [PubMed] [Google Scholar]
- 123.Martinez R., Blasina A., Hallin J.F., Hu W., Rymer I., Fan J., Hoffman R.L., Murphy S., Marx M., Yanochko G., et al. Mitotic Checkpoint Kinase Mps1 Has a Role in Normal Physiology Which Impacts Clinical Utility. PLoS ONE. 2015;10:e0138616. doi: 10.1371/journal.pone.0138616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roberts M.S., Sahni J.M., Schrock M.S., Piemonte K.M., Weber-Bonk K.L., Seachrist D.D., Avril S., Anstine L.J., Singh S., Sizemore S.T., et al. LIN9 and NEK2 Are Core Regulators of Mitotic Fidelity That Can Be Therapeutically Targeted to Overcome Taxane Resistance. Cancer Res. 2020;80:1693–1706. doi: 10.1158/0008-5472.CAN-19-3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maire V., Némati F., Richardson M., Vincent-Salomon A., Tesson B., Rigaill G., Gravier E., Marty-Prouvost B., De Koning L., Lang G., et al. Polo-like Kinase 1: A Potential Therapeutic Option in Combination with Conventional Chemotherapy for the Management of Patients with Triple-Negative Breast Cancer. Cancer Res. 2013;73:813–823. doi: 10.1158/0008-5472.CAN-12-2633. [DOI] [PubMed] [Google Scholar]
- 126.Naro C., De Musso M., Delle Monache F., Panzeri V., de la Grange P., Sette C. The Oncogenic Kinase NEK2 Regulates an RBFOX2-Dependent pro-Mesenchymal Splicing Program in Triple-Negative Breast Cancer Cells. J. Exp. Clin. Cancer Res. 2021;40:397. doi: 10.1186/s13046-021-02210-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu J., Wu X., Zhou W., Liu A., Wu J., Deng J., Yue C., Yang S., Wang J., Yuan Z., et al. Aurora-A Identifies Early Recurrence and Poor Prognosis and Promises a Potential Therapeutic Target in Triple Negative Breast Cancer. PLoS ONE. 2013;8:e56919. doi: 10.1371/journal.pone.0056919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang J., Lin X., Wu L., Huang J.-J., Jiang W.-Q., Kipps T.J., Zhang S. Aurora B Induces Epithelial-Mesenchymal Transition by Stabilizing Snail1 to Promote Basal-like Breast Cancer Metastasis. Oncogene. 2020;39:2550–2567. doi: 10.1038/s41388-020-1165-z. [DOI] [PubMed] [Google Scholar]
- 129.Görgün G., Calabrese E., Hideshima T., Ecsedy J., Perrone G., Mani M., Ikeda H., Bianchi G., Hu Y., Cirstea D., et al. A Novel Aurora-A Kinase Inhibitor MLN8237 Induces Cytotoxicity and Cell-Cycle Arrest in Multiple Myeloma. Blood. 2010;115:5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huck J.J., Zhang M., Mettetal J., Chakravarty A., Venkatakrishnan K., Zhou X., Kleinfield R., Hyer M.L., Kannan K., Shinde V., et al. Translational Exposure-Efficacy Modeling to Optimize the Dose and Schedule of Taxanes Combined with the Investigational Aurora A Kinase Inhibitor MLN8237 (Alisertib) Mol. Cancer Ther. 2014;13:2170–2183. doi: 10.1158/1535-7163.MCT-14-0027. [DOI] [PubMed] [Google Scholar]
- 131.Kozyreva V.K., Kiseleva A.A., Ice R.J., Jones B.C., Loskutov Y.V., Matalkah F., Smolkin M.B., Marinak K., Livengood R.H., Salkeni M.A., et al. Combination of Eribulin and Aurora a Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Mol. Cancer Ther. 2016;15:1809–1822. doi: 10.1158/1535-7163.MCT-15-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.O’Shaughnessy J., McIntyre K., Wilks S., Ma L., Block M., Andorsky D., Danso M., Locke T., Scales A., Wang Y. Efficacy and Safety of Weekly Paclitaxel with or without Oral Alisertib in Patients with Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Netw. Open. 2021;4:e214103. doi: 10.1001/jamanetworkopen.2021.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Falchook G., Coleman R.L., Roszak A., Behbakht K., Matulonis U., Ray-Coquard I., Sawrycki P., Duska L.R., Tew W., Ghamande S., et al. Alisertib in Combination with Weekly Paclitaxel in Patients with Advanced Breast Cancer or Recurrent Ovarian Cancer: A Randomized Clinical Trial. JAMA Oncol. 2019;5:e183773. doi: 10.1001/jamaoncol.2018.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Davis S.L., Ionkina A.A., Bagby S.M., Orth J.D., Gittleman B., Marcus J.M., Lam E.T., Corr B.R., O’Bryant C.L., Glode A.E., et al. Preclinical and Dose-Finding Phase I Trial Results of Combined Treatment with a TORC1/2 Inhibitor (TAK-228) and Aurora A Kinase Inhibitor (Alisertib) in Solid Tumors. Clin. Cancer Res. 2020;26:4633–4642. doi: 10.1158/1078-0432.CCR-19-3498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang W., Xia D., Li Z., Zhou T., Chen T., Wu Z., Zhou W., Li Z., Li L., Xu J. Aurora-A/ERK1/2/MTOR Axis Promotes Tumor Progression in Triple-Negative Breast Cancer and Dual-Targeting Aurora-A/MTOR Shows Synthetic Lethality. Cell Death Dis. 2019;10:606. doi: 10.1038/s41419-019-1855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang X., Zhang G., Tang T., Gao X., Liang T. One Shoot, Three Birds: Targeting NEK2 Orchestrates Chemoradiotherapy, Targeted Therapy, and Immunotherapy in Cancer Treatment. Biochim. Biophys. Acta Rev. Cancer. 2022;1877:188696. doi: 10.1016/j.bbcan.2022.188696. [DOI] [PubMed] [Google Scholar]
- 137.Naso F.D., Boi D., Ascanelli C., Pamfil G., Lindon C., Paiardini A., Guarguaglini G. Nuclear Localisation of Aurora-A: Its Regulation and Significance for Aurora-A Functions in Cancer. Oncogene. 2021;40:3917–3928. doi: 10.1038/s41388-021-01766-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Moore M.J., Wang Q., Kennedy C.J., Silver P.A. An Alternative Splicing Network Links Cell-Cycle Control to Apoptosis. Cell. 2010;142:625–636. doi: 10.1016/j.cell.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diamond J.R., Eckhardt S.G., Pitts T.M., van Bokhoven A., Aisner D., Gustafson D.L., Capasso A., Sams S., Kabos P., Zolman K., et al. A Phase II Clinical Trial of the Aurora and Angiogenic Kinase Inhibitor ENMD-2076 for Previously Treated, Advanced, or Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2018;20:82. doi: 10.1186/s13058-018-1014-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Diamond J.R., Eckhardt S.G., Tan A.C., Newton T.P., Selby H.M., Brunkow K.L., Kachaeva M.I., Varella-Garcia M., Pitts T.M., Bray M.R., et al. Predictive Biomarkers of Sensitivity to the Aurora and Angiogenic Kinase Inhibitor ENMD-2076 in Preclinical Breast Cancer Models. Clin. Cancer Res. 2013;19:291–303. doi: 10.1158/1078-0432.CCR-12-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gong X., Du J., Parsons S.H., Merzoug F.F., Webster Y., Iversen P.W., Chio L.-C., Van Horn R.D., Lin X., Blosser W., et al. Aurora A Kinase Inhibition Is Synthetic Lethal with Loss of the RB1 Tumor Suppressor Gene. Cancer Discov. 2019;9:248–263. doi: 10.1158/2159-8290.CD-18-0469. [DOI] [PubMed] [Google Scholar]
- 142.Chu Q.S.-C., Bouganim N., Fortier C., Zaknoen S., Stille J.R., Kremer J.D., Yuen E., Hui Y.-H., de la Peña A., Lithio A., et al. Aurora Kinase a Inhibitor, LY3295668 Erbumine: A Phase 1 Monotherapy Safety Study in Patients with Locally Advanced or Metastatic Solid Tumors. Investig. New Drugs. 2021;39:1001–1010. doi: 10.1007/s10637-020-01049-3. [DOI] [PubMed] [Google Scholar]
- 143.Witkiewicz A.K., Chung S., Brough R., Vail P., Franco J., Lord C.J., Knudsen E.S. Targeting the Vulnerability of RB Tumor Suppressor Loss in Triple-Negative Breast Cancer. Cell Rep. 2018;22:1185–1199. doi: 10.1016/j.celrep.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schwartz G.K., Carvajal R.D., Midgley R., Rodig S.J., Stockman P.K., Ataman O., Wilson D., Das S., Shapiro G.I. Phase I Study of Barasertib (AZD1152), a Selective Inhibitor of Aurora B Kinase, in Patients with Advanced Solid Tumors. Investig. New Drugs. 2013;31:370–380. doi: 10.1007/s10637-012-9825-7. [DOI] [PubMed] [Google Scholar]
- 145.Boss D.S., Witteveen P.O., van der Sar J., Lolkema M.P., Voest E.E., Stockman P.K., Ataman O., Wilson D., Das S., Schellens J.H. Clinical Evaluation of AZD1152, an i.v. Inhibitor of Aurora B Kinase, in Patients with Solid Malignant Tumors. Ann. Oncol. 2011;22:431–437. doi: 10.1093/annonc/mdq344. [DOI] [PubMed] [Google Scholar]
- 146.Naro C., Barbagallo F., Caggiano C., De Musso M., Panzeri V., Di Agostino S., Paronetto M.P., Sette C. Functional Interaction Between the Oncogenic Kinase NEK2 and Sam68 Promotes a Splicing Program Involved in Migration and Invasion in Triple-Negative Breast Cancer. Front. Oncol. 2022;12:880654. doi: 10.3389/fonc.2022.880654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Vulin M., Jehanno C., Sethi A., Correia A.L., Obradović M.M.S., Couto J.P., Coissieux M.-M., Diepenbruck M., Preca B.-T., Volkmann K., et al. A High-Throughput Drug Screen Reveals Means to Differentiate Triple-Negative Breast Cancer. Oncogene. 2022;41:4459–4473. doi: 10.1038/s41388-022-02429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jimeno A., Li J., Messersmith W.A., Laheru D., Rudek M.A., Maniar M., Hidalgo M., Baker S.D., Donehower R.C. Phase I Study of ON 01910.Na, a Novel Modulator of the Polo-like Kinase 1 Pathway, in Adult Patients with Solid Tumors. J. Clin. Oncol. 2008;26:5504–5510. doi: 10.1200/JCO.2008.17.9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bowles D.W., Diamond J.R., Lam E.T., Weekes C.D., Astling D.P., Anderson R.T., Leong S., Gore L., Varella-Garcia M., Vogler B.W., et al. Phase I Study of Oral Rigosertib (ON 01910.Na), a Dual Inhibitor of the PI3K and Plk1 Pathways, in Adult Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2014;20:1656–1665. doi: 10.1158/1078-0432.CCR-13-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lin C.-C., Su W.-C., Yen C.-J., Hsu C.-H., Su W.-P., Yeh K.-H., Lu Y.-S., Cheng A.-L., Huang D.C.-L., Fritsch H., et al. A Phase I Study of Two Dosing Schedules of Volasertib (BI 6727), an Intravenous Polo-like Kinase Inhibitor, in Patients with Advanced Solid Malignancies. Br. J. Cancer. 2014;110:2434–2440. doi: 10.1038/bjc.2014.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schöffski P., Awada A., Dumez H., Gil T., Bartholomeus S., Wolter P., Taton M., Fritsch H., Glomb P., Munzert G. A Phase I, Dose-Escalation Study of the Novel Polo-like Kinase Inhibitor Volasertib (BI 6727) in Patients with Advanced Solid Tumours. Eur. J. Cancer. 2012;48:179–186. doi: 10.1016/j.ejca.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 152.Awada A., Dumez H., Aftimos P.G., Costermans J., Bartholomeus S., Forceville K., Berghmans T., Meeus M.-A., Cescutti J., Munzert G., et al. Phase I Trial of Volasertib, a Polo-like Kinase Inhibitor, plus Platinum Agents in Solid Tumors: Safety, Pharmacokinetics and Activity. Investig. New Drugs. 2015;33:611–620. doi: 10.1007/s10637-015-0223-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.de Braud F., Cascinu S., Spitaleri G., Pilz K., Clementi L., Liu D., Sikken P., De Pas T. A Phase I, Dose-Escalation Study of Volasertib Combined with Nintedanib in Advanced Solid Tumors. Ann. Oncol. 2015;26:2341–2346. doi: 10.1093/annonc/mdv354. [DOI] [PubMed] [Google Scholar]
- 154.Machiels J.-P., Peeters M., Herremans C., Surmont V., Specenier P., De Smet M., Pilz K., Strelkowa N., Liu D., Rottey S. A Phase I Study of Volasertib Combined with Afatinib, in Advanced Solid Tumors. Cancer Chemother. Pharmacol. 2015;76:843–851. doi: 10.1007/s00280-015-2860-2. [DOI] [PubMed] [Google Scholar]
- 155.Ueda A., Oikawa K., Fujita K., Ishikawa A., Sato E., Ishikawa T., Kuroda M., Kanekura K. Therapeutic Potential of PLK1 Inhibition in Triple-Negative Breast Cancer. Lab. Investig. 2019;99:1275–1286. doi: 10.1038/s41374-019-0247-4. [DOI] [PubMed] [Google Scholar]
- 156.Schöffski P., Blay J.-Y., De Greve J., Brain E., Machiels J.-P., Soria J.-C., Sleijfer S., Wolter P., Ray-Coquard I., Fontaine C., et al. Multicentric Parallel Phase II Trial of the Polo-like Kinase 1 Inhibitor BI 2536 in Patients with Advanced Head and Neck Cancer, Breast Cancer, Ovarian Cancer, Soft Tissue Sarcoma and Melanoma. The First Protocol of the European Organization for Research and Treatment of Cancer (EORTC) Network of Core Institutes (NOCI) Eur. J. Cancer. 2010;46:2206–2215. doi: 10.1016/j.ejca.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 157.Giordano A., Liu Y., Armeson K., Park Y., Ridinger M., Erlander M., Reuben J., Britten C., Kappler C., Yeh E., et al. Polo-like Kinase 1 (Plk1) Inhibition Synergizes with Taxanes in Triple Negative Breast Cancer. PLoS ONE. 2019;14:e0224420. doi: 10.1371/journal.pone.0224420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Olmos D., Barker D., Sharma R., Brunetto A.T., Yap T.A., Taegtmeyer A.B., Barriuso J., Medani H., Degenhardt Y.Y., Allred A.J., et al. Phase I Study of GSK461364, a Specific and Competitive Polo-like Kinase 1 Inhibitor, in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2011;17:3420–3430. doi: 10.1158/1078-0432.CCR-10-2946. [DOI] [PubMed] [Google Scholar]
- 159.Thu K.L., Silvester J., Elliott M.J., Ba-Alawi W., Duncan M.H., Elia A.C., Mer A.S., Smirnov P., Safikhani Z., Haibe-Kains B., et al. Disruption of the Anaphase-Promoting Complex Confers Resistance to TTK Inhibitors in Triple-Negative Breast Cancer. Proc. Natl. Acad. Sci. USA. 2018;115:E1570–E1577. doi: 10.1073/pnas.1719577115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mason J.M., Wei X., Fletcher G.C., Kiarash R., Brokx R., Hodgson R., Beletskaya I., Bray M.R., Mak T.W. Functional Characterization of CFI-402257, a Potent and Selective Mps1/TTK Kinase Inhibitor, for the Treatment of Cancer. Proc. Natl. Acad. Sci. USA. 2017;114:3127–3132. doi: 10.1073/pnas.1700234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Anderhub S.J., Mak G.W.-Y., Gurden M.D., Faisal A., Drosopoulos K., Walsh K., Woodward H.L., Innocenti P., Westwood I.M., Naud S., et al. High Proliferation Rate and a Compromised Spindle Assembly Checkpoint Confers Sensitivity to the MPS1 Inhibitor BOS172722 in Triple-Negative Breast Cancers. Mol. Cancer Ther. 2019;18:1696–1707. doi: 10.1158/1535-7163.MCT-18-1203. [DOI] [PubMed] [Google Scholar]
- 162.Maia A.R.R., de Man J., Boon U., Janssen A., Song J.-Y., Omerzu M., Sterrenburg J.G., Prinsen M.B.W., Willemsen-Seegers N., de Roos J.A.D.M., et al. Inhibition of the Spindle Assembly Checkpoint Kinase TTK Enhances the Efficacy of Docetaxel in a Triple-Negative Breast Cancer Model. Ann. Oncol. 2015;26:2180–2192. doi: 10.1093/annonc/mdv293. [DOI] [PubMed] [Google Scholar]
- 163.Wengner A.M., Siemeister G., Koppitz M., Schulze V., Kosemund D., Klar U., Stoeckigt D., Neuhaus R., Lienau P., Bader B., et al. Novel Mps1 Kinase Inhibitors with Potent Antitumor Activity. Mol. Cancer Ther. 2016;15:583–592. doi: 10.1158/1535-7163.MCT-15-0500. [DOI] [PubMed] [Google Scholar]
- 164.Elmaleh C., Dubuisson C., Fouret C., Thibaut J. Ophthalmic Migraine Causing Definitive Homonymous Lateral Hemianopsia in a 19-Year-Old Girl. Bull. Soc. Ophtalmol. Fr. 1987;87:1223–1226. [PubMed] [Google Scholar]
- 165.Chen X., Low K.-H., Alexander A., Jiang Y., Karakas C., Hess K.R., Carey J.P.W., Bui T.N., Vijayaraghavan S., Evans K.W., et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin. Cancer Res. 2018;24:6594–6610. doi: 10.1158/1078-0432.CCR-18-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jin J., Fang H., Yang F., Ji W., Guan N., Sun Z., Shi Y., Zhou G., Guan X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia. 2018;20:478–488. doi: 10.1016/j.neo.2018.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Pitts T.M., Simmons D.M., Bagby S.M., Hartman S.J., Yacob B.W., Gittleman B., Tentler J.J., Cittelly D., Ormond D.R., Messersmith W.A., et al. Wee1 Inhibition Enhances the Anti-Tumor Effects of Capecitabine in Preclinical Models of Triple-Negative Breast Cancer. Cancers. 2020;12:719. doi: 10.3390/cancers12030719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bauer T.M., Moore K.N., Rader J.S., Simpkins F., Mita A.C., Beck J.T., Hart L., Chu Q., Oza A., Tinker A.V., et al. A Phase Ib Study Assessing the Safety, Tolerability, and Efficacy of the First-in-Class Wee1 Inhibitor Adavosertib (AZD1775) as Monotherapy in Patients with Advanced Solid Tumors. Target. Oncol. 2023;18:517–530. doi: 10.1007/s11523-023-00965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Keenan T.E., Li T., Vallius T., Guerriero J.L., Tayob N., Kochupurakkal B., Davis J., Pastorello R., Tahara R.K., Anderson L., et al. Clinical Efficacy and Molecular Response Correlates of the WEE1 Inhibitor Adavosertib Combined with Cisplatin in Patients with Metastatic Triple-Negative Breast Cancer. Clin. Cancer Res. 2021;27:983–991. doi: 10.1158/1078-0432.CCR-20-3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nagayama A., Vidula N., Ellisen L., Bardia A. Novel Antibody–Drug Conjugates for Triple Negative Breast Cancer. Ther. Adv. Med. Oncol. 2020;12:175883592091598. doi: 10.1177/1758835920915980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Damodaran S., Symmans F., Helgason T., Mittendorf E.A., Tripathy D., Hess K., Litton J., Moulder S. A Phase II Trial of Mirvetuximab Soravtansine in Patients with Localized Triple-Negative Breast Cancer (TNBC) with Tumors Predicted Insensitive to Standard Neoadjuvant Chemotherapy (NACT) Including a Lead-in Cohort to Establish Activity in Patients with Metastatic TNBC. Ann. Oncol. 2017;28:v106. doi: 10.1093/annonc/mdx365.083. [DOI] [Google Scholar]
- 172.Yam C., Rauch G.M., Rahman T., Karuturi M., Ravenberg E., White J., Clayborn A., McCarthy P., Abouharb S., Lim B., et al. A Phase II Study of Mirvetuximab Soravtansine in Triple-Negative Breast Cancer. Investig. New Drugs. 2021;39:509–515. doi: 10.1007/s10637-020-00995-2. [DOI] [PubMed] [Google Scholar]
- 173.Boni V., Fidler M.J., Arkenau H.-T., Spira A., Meric-Bernstam F., Uboha N., Sanborn R.E., Sweis R.F., LoRusso P., Nagasaka M., et al. Praluzatamab Ravtansine, a CD166-Targeting Antibody–Drug Conjugate, in Patients with Advanced Solid Tumors: An Open-Label Phase I/II Trial. Clin. Cancer Res. 2022;28:2020–2029. doi: 10.1158/1078-0432.CCR-21-3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chomet M., Schreurs M., Nguyen M., Howng B., Villanueva R., Krimm M., Vasiljeva O., van Dongen G.A.M.S., Vugts D.J. The Tumor Targeting Performance of Anti-CD166 Probody Drug Conjugate CX-2009 and Its Parental Derivatives as Monitored by 89 Zr-Immuno-PET in Xenograft Bearing Mice. Theranostics. 2020;10:5815–5828. doi: 10.7150/thno.44334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Weidle U.H., Eggle D., Klostermann S., Swart G.W.M. ALCAM/CD166: Cancer-Related Issues. Cancer Genom. Proteom. 2010;7:231–243. [PubMed] [Google Scholar]
- 176.Swart G.W.M. Activated Leukocyte Cell Adhesion Molecule (CD166/ALCAM): Developmental and Mechanistic Aspects of Cell Clustering and Cell Migration. Eur. J. Cell Biol. 2002;81:313–321. doi: 10.1078/0171-9335-00256. [DOI] [PubMed] [Google Scholar]
- 177.CytomX Therapeutics Announces Phase 2 Results for Praluzatamab Ravtansine in Breast Cancer. News Release. CytomX Therapeutics. 6 July 2022. [(accessed on 29 April 2023)]. Available online: https://bit.ly/3yno2ec.
- 178.Nicolazzi C., Caron A., Tellier A., Trombe M., Pinkas J., Payne G., Carrez C., Guérif S., Maguin M., Baffa R., et al. An Antibody–Drug Conjugate Targeting MUC1-Associated Carbohydrate CA6 Shows Promising Antitumor Activities. Mol. Cancer Ther. 2020;19:1660–1669. doi: 10.1158/1535-7163.MCT-19-0826. [DOI] [PubMed] [Google Scholar]
- 179.Smith N.L., Halliday B.E., Finley J.L., Kellogg Wennerberg A.E. Immunohistochemical Distribution of Tumor-Associated Antigen CA6 in Gynecological Neoplasms as Detected by Monoclonal Antibody DS6. Int. J. Gynecol. Pathol. 2001;20:260–266. doi: 10.1097/00004347-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 180.Syed Y.Y. Sacituzumab Govitecan: First Approval. Drugs. 2020;80:1019–1025. doi: 10.1007/s40265-020-01337-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kathpalia M., Sharma A., Kaur N. Sacituzumab Govitecan as a Second-Line Treatment in Relapsed/Refractory Metastatic Triple-Negative Breast Cancer Patients: A Systematic Review and Meta-Analysis. Ann. Pharmacother. 2023:1060028023116410. doi: 10.1177/10600280231164110. [DOI] [PubMed] [Google Scholar]
- 182.Thorpe L.M., Yuzugullu H., Zhao J.J. PI3K in Cancer: Divergent Roles of Isoforms, Modes of Activation and Therapeutic Targeting. Nat. Rev. Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Garrido-Castro A.C., Saura C., Barroso-Sousa R., Guo H., Ciruelos E., Bermejo B., Gavilá J., Serra V., Prat A., Paré L., et al. Phase 2 Study of Buparlisib (BKM120), a Pan-Class I PI3K Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. Breast Cancer Res. 2020;22:120. doi: 10.1186/s13058-020-01354-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ando Y., Inada-Inoue M., Mitsuma A., Yoshino T., Ohtsu A., Suenaga N., Sato M., Kakizume T., Robson M., Quadt C., et al. Phase I Dose-escalation Study of Buparlisib (BKM 120), an Oral Pan-class I PI 3K Inhibitor, in Japanese Patients with Advanced Solid Tumors. Cancer Sci. 2014;105:347–353. doi: 10.1111/cas.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bendell J.C., Rodon J., Burris H.A., de Jonge M., Verweij J., Birle D., Demanse D., De Buck S.S., Ru Q.C., Peters M., et al. Phase I, Dose-Escalation Study of BKM120, an Oral Pan-Class I PI3K Inhibitor, in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2012;30:282–290. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 186.Buparlisib Hydrochloride by Adlai Nortye Biopharma for Triple-Negative Breast Cancer (TNBC): Likelihood of Approval. Pharm. [(accessed on 30 August 2023)]. Available online: https://www.pharmaceutical-technology.com/data-insights/buparlisib-hydrochloride-adlai-nortye-biopharma-triple-negative-breast-cancer-tnbc-likelihood-of-approval/
- 187.Ibrahim Y.H., García-García C., Serra V., He L., Torres-Lockhart K., Prat A., Anton P., Cozar P., Guzmán M., Grueso J., et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012;2:1036–1047. doi: 10.1158/2159-8290.CD-11-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Savas P., Lo L.L., Luen S.J., Blackley E.F., Callahan J., Moodie K., van Geelen C.T., Ko Y.-A., Weng C.-F., Wein L., et al. Alpelisib Monotherapy for PI3K-Altered, Pretreated Advanced Breast Cancer: A Phase II Study. Cancer Discov. 2022;12:2058–2073. doi: 10.1158/2159-8290.CD-21-1696. [DOI] [PubMed] [Google Scholar]
- 189.Batalini F., Xiong N., Tayob N., Polak M., Eismann J., Cantley L.C., Shapiro G.I., Adalsteinsson V., Winer E.P., Konstantinopoulos P.A., et al. Phase 1b Clinical Trial with Alpelisib plus Olaparib for Patients with Advanced Triple-Negative Breast Cancer. Clin. Cancer Res. 2022;28:1493–1499. doi: 10.1158/1078-0432.CCR-21-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Damodaran S., Litton J.K., Hess K.R., Eppig C.T., Grzegorzewski K.J., Meric-Bernstam F., Wistuba I.I., White J.B., Rauch G.M., Candelaria R.P., et al. Abstract OT2-06-01: A Phase-2 Trial of Neoadjuvant Alpelisib and Nab-Paclitaxel in Anthracycline Refractory Triple Negative Breast Cancers with PIK3CA or PTEN Alterations. Cancer Res. 2020;80:OT2-06-01. doi: 10.1158/1538-7445.SABCS19-OT2-06-01. [DOI] [Google Scholar]
- 191.Juric D., Castel P., Griffith M., Griffith O.L., Won H.H., Ellis H., Ebbesen S.H., Ainscough B.J., Ramu A., Iyer G., et al. Convergent Loss of PTEN Leads to Clinical Resistance to a PI(3)Kα Inhibitor. Nature. 2015;518:240–244. doi: 10.1038/nature13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yehia L., Keel E., Eng C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020;71:103–116. doi: 10.1146/annurev-med-052218-125823. [DOI] [PubMed] [Google Scholar]
- 193.Cordero-Espinoza L., Hagen T. Increased Concentrations of Fructose 2,6-Bisphosphate Contribute to the Warburg Effect in Phosphatase and Tensin Homolog (PTEN)-Deficient Cells. J. Biol. Chem. 2013;288:36020–36028. doi: 10.1074/jbc.M113.510289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang W., Oguz G., Lee P.L., Bao Y., Wang P., Terp M.G., Ditzel H.J., Yu Q. KDM4B-Regulated Unfolded Protein Response as a Therapeutic Vulnerability in PTEN -Deficient Breast Cancer. J. Exp. Med. 2018;215:2833–2849. doi: 10.1084/jem.20180439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Li Y., Zhang H., Merkher Y., Chen L., Liu N., Leonov S., Chen Y. Recent Advances in Therapeutic Strategies for Triple-Negative Breast Cancer. J. Hematol. Oncol. 2022;15:121. doi: 10.1186/s13045-022-01341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sikov W.M., Berry D.A., Perou C.M., Singh B., Cirrincione C.T., Tolaney S.M., Kuzma C.S., Pluard T.J., Somlo G., Port E.R., et al. Impact of the Addition of Carboplatin and/or Bevacizumab to Neoadjuvant Once-per-Week Paclitaxel Followed by Dose-Dense Doxorubicin and Cyclophosphamide on Pathologic Complete Response Rates in Stage II to III Triple-Negative Breast Cancer: CALGB 40603 (Alliance) J. Clin. Oncol. 2015;33:13–21. doi: 10.1200/JCO.2014.57.0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Barton M.K. Bevacizumab in Neoadjuvant Chemotherapy Increases the Pathological Complete Response Rate in Patients with Triple-Negative Breast Cancer. CA Cancer J. Clin. 2014;64:155–156. doi: 10.3322/caac.21223. [DOI] [PubMed] [Google Scholar]
- 198.Manning B.D., Cantley L.C. AKT/PKB Signaling: Navigating Downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Cantley L.C. The Phosphoinositide 3-Kinase Pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 200.LoRusso P.M. Inhibition of the PI3K/AKT/MTOR Pathway in Solid Tumors. J. Clin. Oncol. 2016;34:3803–3815. doi: 10.1200/JCO.2014.59.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Bhaskar P.T., Hay N. The Two TORCs and Akt. Dev. Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 202.Di Cosimo S., Baselga J. Management of Breast Cancer with Targeted Agents: Importance of Heterogenicity. Nat. Rev. Clin. Oncol. 2010;7:139–147. doi: 10.1038/nrclinonc.2009.234. [DOI] [PubMed] [Google Scholar]
- 203.Pascual J., Turner N.C. Targeting the PI3-Kinase Pathway in Triple-Negative Breast Cancer. Ann. Oncol. 2019;30:1051–1060. doi: 10.1093/annonc/mdz133. [DOI] [PubMed] [Google Scholar]
- 204.Lin J., Sampath D., Nannini M.A., Lee B.B., Degtyarev M., Oeh J., Savage H., Guan Z., Hong R., Kassees R., et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013;19:1760–1772. doi: 10.1158/1078-0432.CCR-12-3072. [DOI] [PubMed] [Google Scholar]
- 205.Kim S.-B., Dent R., Im S.-A., Espié M., Blau S., Tan A.R., Isakoff S.J., Oliveira M., Saura C., Wongchenko M.J., et al. Ipatasertib plus Paclitaxel versus Placebo plus Paclitaxel as First-Line Therapy for Metastatic Triple-Negative Breast Cancer (LOTUS): A Multicentre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol. 2017;18:1360–1372. doi: 10.1016/S1470-2045(17)30450-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Oliveira M., Saura C., Nuciforo P., Calvo I., Andersen J., Passos-Coelho J.L., Gil Gil M., Bermejo B., Patt D.A., Ciruelos E., et al. FAIRLANE, a Double-Blind Placebo-Controlled Randomized Phase II Trial of Neoadjuvant Ipatasertib plus Paclitaxel for Early Triple-Negative Breast Cancer. Ann. Oncol. 2019;30:1289–1297. doi: 10.1093/annonc/mdz177. [DOI] [PubMed] [Google Scholar]
- 207.Addie M., Ballard P., Buttar D., Crafter C., Currie G., Davies B.R., Debreczeni J., Dry H., Dudley P., Greenwood R., et al. Discovery of 4-Amino-N-[(1 S)-1-(4-Chlorophenyl)-3-Hydroxypropyl]-1-(7 H-Pyrrolo[2,3-d]Pyrimidin-4-Yl)Piperidine-4-Carboxamide (AZD5363), an Orally Bioavailable, Potent Inhibitor of Akt Kinases. J. Med. Chem. 2013;56:2059–2073. doi: 10.1021/jm301762v. [DOI] [PubMed] [Google Scholar]
- 208.Davies B.R., Greenwood H., Dudley P., Crafter C., Yu D.-H., Zhang J., Li J., Gao B., Ji Q., Maynard J., et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012;11:873–887. doi: 10.1158/1535-7163.MCT-11-0824-T. [DOI] [PubMed] [Google Scholar]
- 209.Schmid P., Abraham J., Chan S., Wheatley D., Brunt A.M., Nemsadze G., Baird R.D., Park Y.H., Hall P.S., Perren T., et al. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin. Oncol. 2020;38:423–433. doi: 10.1200/JCO.19.00368. [DOI] [PubMed] [Google Scholar]
- 210.Andrikopoulou A., Chatzinikolaou S., Panourgias E., Kaparelou M., Liontos M., Dimopoulos M.-A., Zagouri F. The Emerging Role of Capivasertib in Breast Cancer. Breast. 2022;63:157–167. doi: 10.1016/j.breast.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Khan M.A., Jain V.K., Rizwanullah M., Ahmad J., Jain K. PI3K/AKT/MTOR Pathway Inhibitors in Triple-Negative Breast Cancer: A Review on Drug Discovery and Future Challenges. Drug Discov. Today. 2019;24:2181–2191. doi: 10.1016/j.drudis.2019.09.001. [DOI] [PubMed] [Google Scholar]
- 212.Anand K., Patel T., Niravath P., Rodriguez A., Darcourt J., Belcheva A., Boone T., Ensor J., Chang J. Targeting MTOR and DNA Repair Pathways in Residual Triple Negative Breast Cancer Post Neoadjuvant Chemotherapy. Sci. Rep. 2021;11:82. doi: 10.1038/s41598-020-80081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Linderholm B.K., Hellborg H., Johansson U., Elmberger G., Skoog L., Lehtiö J., Lewensohn R. Significantly Higher Levels of Vascular Endothelial Growth Factor (VEGF) and Shorter Survival Times for Patients with Primary Operable Triple-Negative Breast Cancer. Ann. Oncol. 2009;20:1639–1646. doi: 10.1093/annonc/mdp062. [DOI] [PubMed] [Google Scholar]
- 214.Fathi Maroufi N., Rashidi M.R., Vahedian V., Akbarzadeh M., Fattahi A., Nouri M. Therapeutic Potentials of Apatinib in Cancer Treatment: Possible Mechanisms and Clinical Relevance. Life Sci. 2020;241:117106. doi: 10.1016/j.lfs.2019.117106. [DOI] [PubMed] [Google Scholar]
- 215.Gao Z., Shi M., Wang Y., Chen J., Ou Y. Apatinib Enhanced Anti-Tumor Activity of Cisplatin on Triple-Negative Breast Cancer through Inhibition of VEGFR-2. Pathol. Res. Pract. 2019;215:152422. doi: 10.1016/j.prp.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 216.Hu X., Cao J., Hu W., Wu C., Pan Y., Cai L., Tong Z., Wang S., Li J., Wang Z., et al. Multicenter Phase II Study of Apatinib in Non-Triple-Negative Metastatic Breast Cancer. BMC Cancer. 2014;14:820. doi: 10.1186/1471-2407-14-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Li D.-D., Tao Z., Wang B.-Y., Wang L.-P., Cao J., Hu X.-C., Zhang J. Apatinib plus Vinorelbine versus Vinorelbine for Metastatic Triple-Negative Breast Cancer Who Failed First/Second-Line Treatment: The NAN Trial. NPJ Breast Cancer. 2022;8:110. doi: 10.1038/s41523-022-00462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lee J.S., Yost S.E., Yuan Y. Case Report: Significant Response to the Combination of Lenvatinib and Immune Checkpoint Inhibitor in a Patient with Heavily Pretreated Metastatic Triple Negative Breast Cancer. Front. Oncol. 2021;10:582185. doi: 10.3389/fonc.2020.582185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chung H.C., Saada-Bouzid E., Muñoz F.L., Yanez E., Im S.-A., Castanon E., Graham D.M., Garcia-Corbacho J., Lopez J., Ghori R., et al. Abstract PS12-07: Lenvatinib plus Pembrolizumab for Previously Treated, Advanced Triple-Negative Breast Cancer: Early Results from the Multicohort Phase 2 LEAP-005 Study. Cancer Res. 2021;81:PS12-07. doi: 10.1158/1538-7445.SABCS20-PS12-07. [DOI] [Google Scholar]
- 220.Ceresa B.P., Peterson J.L. Cell and Molecular Biology of Epidermal Growth Factor Receptor. Int. Rev. Cell Mol. Biol. 2014;313:145–178. doi: 10.1016/B978-0-12-800177-6.00005-0. [DOI] [PubMed] [Google Scholar]
- 221.Masuda H., Zhang D., Bartholomeusz C., Doihara H., Hortobagyi G.N., Ueno N.T. Role of Epidermal Growth Factor Receptor in Breast Cancer. Breast Cancer Res. Treat. 2012;136:331–345. doi: 10.1007/s10549-012-2289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Livasy C.A., Karaca G., Nanda R., Tretiakova M.S., Olopade O.I., Moore D.T., Perou C.M. Phenotypic Evaluation of the Basal-like Subtype of Invasive Breast Carcinoma. Mod. Pathol. 2006;19:264–271. doi: 10.1038/modpathol.3800528. [DOI] [PubMed] [Google Scholar]
- 223.Zakaria Z., Zulkifle M.F., Wan Hasan W.A.N., Azhari A.K., Abdul Raub S.H., Eswaran J., Soundararajan M., Syed Husain S.N.A. Epidermal Growth Factor Receptor (EGFR) Gene Alteration and Protein Overexpression in Malaysian Triple-Negative Breast Cancer (TNBC) Cohort. OncoTargets Ther. 2019;12:7749–7756. doi: 10.2147/OTT.S214611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Schuler M., Awada A., Harter P., Canon J.L., Possinger K., Schmidt M., De Grève J., Neven P., Dirix L., Jonat W., et al. A Phase II Trial to Assess Efficacy and Safety of Afatinib in Extensively Pretreated Patients with HER2-Negative Metastatic Breast Cancer. Breast Cancer Res. Treat. 2012;134:1149–1159. doi: 10.1007/s10549-012-2126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Corkery B., Crown J., Clynes M., O’Donovan N. Epidermal Growth Factor Receptor as a Potential Therapeutic Target in Triple-Negative Breast Cancer. Ann. Oncol. 2009;20:862–867. doi: 10.1093/annonc/mdn710. [DOI] [PubMed] [Google Scholar]
- 226.Baselga J., Gómez P., Greil R., Braga S., Climent M.A., Wardley A.M., Kaufman B., Stemmer S.M., Pêgo A., Chan A., et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab with Cisplatin Versus Cisplatin Alone in Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013;31:2586–2592. doi: 10.1200/JCO.2012.46.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Costa R., Shah A.N., Santa-Maria C.A., Cruz M.R., Mahalingam D., Carneiro B.A., Chae Y.K., Cristofanilli M., Gradishar W.J., Giles F.J. Targeting Epidermal Growth Factor Receptor in Triple Negative Breast Cancer: New Discoveries and Practical Insights for Drug Development. Cancer Treat. Rev. 2017;53:111–119. doi: 10.1016/j.ctrv.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 228.Giovinazzi S., Lindsay C.R., Morozov V.M., Escobar-Cabrera E., Summers M.K., Han H.S., McIntosh L.P., Ishov A.M. Regulation of Mitosis and Taxane Response by Daxx and Rassf1. Oncogene. 2012;31:13–26. doi: 10.1038/onc.2011.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Lin P.-H., Tseng L.-M., Lee Y.-H., Chen S.-T., Yeh D.-C., Dai M.-S., Liu L.-C., Wang M.-Y., Lo C., Chang S., et al. Neoadjuvant Afatinib with Paclitaxel for Triple-Negative Breast Cancer and the Molecular Characteristics in Responders and Non-Responders. J. Formos. Med. Assoc. 2022;121:2538–2547. doi: 10.1016/j.jfma.2022.05.015. [DOI] [PubMed] [Google Scholar]
- 230.El Guerrab A., Bamdad M., Bignon Y.-J., Penault-Llorca F., Aubel C. Co-Targeting EGFR and MTOR with Gefitinib and Everolimus in Triple-Negative Breast Cancer Cells. Sci. Rep. 2020;10:6367. doi: 10.1038/s41598-020-63310-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Abdelmalek C.M., Hu Z., Kronenberger T., Küblbeck J., Kinnen F.J.M., Hesse S.S., Malik A., Kudolo M., Niess R., Gehringer M., et al. Gefitinib-Tamoxifen Hybrid Ligands as Potent Agents against Triple-Negative Breast Cancer. J. Med. Chem. 2022;65:4616–4632. doi: 10.1021/acs.jmedchem.1c01646. [DOI] [PubMed] [Google Scholar]
- 232.Magee J.A., Piskounova E., Morrison S.J. Cancer Stem Cells: Impact, Heterogeneity, and Uncertainty. Cancer Cell. 2012;21:283–296. doi: 10.1016/j.ccr.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Yeatman T.J. A Renaissance for SRC. Nat. Rev. Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 234.Wheeler D.L., Iida M., Dunn E.F. The Role of Src in Solid Tumors. Oncologist. 2009;14:667–678. doi: 10.1634/theoncologist.2009-0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Thakur R., Trivedi R., Rastogi N., Singh M., Mishra D.P. Inhibition of STAT3, FAK and Src Mediated Signaling Reduces Cancer Stem Cell Load, Tumorigenic Potential and Metastasis in Breast Cancer. Sci. Rep. 2015;5:10194. doi: 10.1038/srep10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tian J., Raffa F.A., Dai M., Moamer A., Khadang B., Hachim I.Y., Bakdounes K., Ali S., Jean-Claude B., Lebrun J.-J. Dasatinib Sensitises Triple Negative Breast Cancer Cells to Chemotherapy by Targeting Breast Cancer Stem Cells. Br. J. Cancer. 2018;119:1495–1507. doi: 10.1038/s41416-018-0287-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gaule P., Mukherjee N., Corkery B., Eustace A., Gately K., Roche S., O’Connor R., O’Byrne K., Walsh N., Duffy M., et al. Dasatinib Treatment Increases Sensitivity to C-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers. 2019;11:548. doi: 10.3390/cancers11040548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Cooper C.S., Park M., Blair D.G., Tainsky M.A., Huebner K., Croce C.M., Vande Woude G.F. Molecular Cloning of a New Transforming Gene from a Chemically Transformed Human Cell Line. Nature. 1984;311:29–33. doi: 10.1038/311029a0. [DOI] [PubMed] [Google Scholar]
- 239.Phase II Trial of Tivantinib in Metastatic Triple-Negative Breast Cancer. Univadis from Medscape. [(accessed on 4 August 2023)]. Available online: https://www.univadis.it/viewarticle/phase-ii-trial-of-tivantinib-in-metastatic-triple-negative-breast-cancer-284443.
- 240.Shuai K., Liu B. Regulation of JAK–STAT Signalling in the Immune System. Nat. Rev. Immunol. 2003;3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 241.Balko J.M., Schwarz L.J., Luo N., Estrada M.V., Giltnane J.M., Dávila-González D., Wang K., Sánchez V., Dean P.T., Combs S.E., et al. Triple-Negative Breast Cancers with Amplification of JAK2 at the 9p24 Locus Demonstrate JAK2-Specific Dependence. Sci. Transl. Med. 2016;8:334ra53. doi: 10.1126/scitranslmed.aad3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Harrison C., Kiladjian J.-J., Al-Ali H.K., Gisslinger H., Waltzman R., Stalbovskaya V., McQuitty M., Hunter D.S., Levy R., Knoops L., et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 243.Han J., Yun J., Quan M., Kang W., Jung J.-G., Heo W., Li S., Lee K.J., Son H.-Y., Kim J.H., et al. JAK2 Regulates Paclitaxel Resistance in Triple Negative Breast Cancers. J. Mol. Med. 2021;99:1783–1795. doi: 10.1007/s00109-021-02138-3. [DOI] [PubMed] [Google Scholar]
- 244.Stover D.G., Gil Del Alcazar C.R., Brock J., Guo H., Overmoyer B., Balko J., Xu Q., Bardia A., Tolaney S.M., Gelman R., et al. Phase II Study of Ruxolitinib, a Selective JAK1/2 Inhibitor, in Patients with Metastatic Triple-Negative Breast Cancer. NPJ Breast Cancer. 2018;4:10. doi: 10.1038/s41523-018-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ruxolitinib Phosphate by Novartis for Triple-Negative Breast Cancer (TNBC): Likelihood of Approval. Pharmaceutical Technology, 24 February 2023. [(accessed on 4 August 2023)]. Available online: https://www.pharmaceutical-technology.com/data-insights/ruxolitinib-phosphate-novartis-triple-negative-breast-cancer-tnbc-likelihood-of-approval/
- 246.Chang Y.-C., Cheung C.H.A. An Updated Review of Smac Mimetics, LCL161, Birinapant, and GDC-0152 in Cancer Treatment. Appl. Sci. 2020;11:335. doi: 10.3390/app11010335. [DOI] [Google Scholar]
- 247.Firestone B., Conway C., Yang G., Gao H., Porter D., Slisz J., He D., Mosher R., Monahan J., Straub C., et al. Abstract B27: Correlation between TNFα and LCL161 Anti-Tumor Activity in Patient Derived Xenograft Models of Human Cancer. Mol. Cancer Ther. 2009;8:B27. doi: 10.1158/1535-7163.TARG-09-B27. [DOI] [Google Scholar]
- 248.Bardia A., Cameron J.S., Venkatesan K., Robinson D., Porter D., Kim S.B., Parton M., Kümmel S., Estevez L.G., Huang C.S., et al. 1977 Synergy of LCL161, an Antagonist of Inhibitor of Apoptosis Proteins (IAPs), with Paclitaxel in a Gene Expression Signature-Enriched Cohort of Triple-Negative Breast Cancer (TNBC) Eur. J. Cancer. 2015;51:S326. doi: 10.1016/S0959-8049(16)30925-X. [DOI] [Google Scholar]
- 249.Bardia A., Parton M., Kümmel S., Estévez L.G., Huang C.-S., Cortés J., Ruiz-Borrego M., Telli M.L., Martin-Martorell P., López R., et al. Paclitaxel with Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J. Clin. Oncol. 2018;36:3126–3133. doi: 10.1200/JCO.2017.74.8392. [DOI] [PubMed] [Google Scholar]
- 250.Stanley R.F., Abdel-Wahab O. Dysregulation and Therapeutic Targeting of RNA Splicing in Cancer. Nat. Cancer. 2022;3:536–546. doi: 10.1038/s43018-022-00384-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Sette C., Paronetto M.P. Somatic Mutations in Core Spliceosome Components Promote Tumorigenesis and Generate an Exploitable Vulnerability in Human Cancer. Cancers. 2022;14:1827. doi: 10.3390/cancers14071827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Koh C.M., Sabò A., Guccione E. Targeting MYC in Cancer Therapy: RNA Processing Offers New Opportunities. Bioessays. 2016;38:266–275. doi: 10.1002/bies.201500134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Iwai K., Yaguchi M., Nishimura K., Yamamoto Y., Tamura T., Nakata D., Dairiki R., Kawakita Y., Mizojiri R., Ito Y., et al. Anti-Tumor Efficacy of a Novel CLK Inhibitor via Targeting RNA Splicing and MYC-Dependent Vulnerability. EMBO Mol. Med. 2018;10:e8289. doi: 10.15252/emmm.201708289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hsu T.Y.-T., Simon L.M., Neill N.J., Marcotte R., Sayad A., Bland C.S., Echeverria G.V., Sun T., Kurley S.J., Tyagi S., et al. The Spliceosome Is a Therapeutic Vulnerability in MYC-Driven Cancer. Nature. 2015;525:384–388. doi: 10.1038/nature14985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bowling E.A., Wang J.H., Gong F., Wu W., Neill N.J., Kim I.S., Tyagi S., Orellana M., Kurley S.J., Dominguez-Vidaña R., et al. Spliceosome-Targeted Therapies Trigger an Antiviral Immune Response in Triple-Negative Breast Cancer. Cell. 2021;184:384–403.e21. doi: 10.1016/j.cell.2020.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Koedoot E., van Steijn E., Vermeer M., González-Prieto R., Vertegaal A.C.O., Martens J.W.M., Le Dévédec S.E., van de Water B. Splicing Factors Control Triple-Negative Breast Cancer Cell Mitosis through SUN2 Interaction and Sororin Intron Retention. J. Exp. Clin. Cancer Res. 2021;40:82. doi: 10.1186/s13046-021-01863-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Chan S., Sridhar P., Kirchner R., Lock Y.J., Herbert Z., Buonamici S., Smith P., Lieberman J., Petrocca F. Basal-A Triple-Negative Breast Cancer Cells Selectively Rely on RNA Splicing for Survival. Mol. Cancer Ther. 2017;16:2849–2861. doi: 10.1158/1535-7163.MCT-17-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hong D.S., Kurzrock R., Naing A., Wheler J.J., Falchook G.S., Schiffman J.S., Faulkner N., Pilat M.J., O’Brien J., LoRusso P. A Phase I, Open-Label, Single-Arm, Dose-Escalation Study of E7107, a Precursor Messenger Ribonucleic Acid (Pre-MRNA) Splicesome Inhibitor Administered Intravenously on Days 1 and 8 Every 21 Days to Patients with Solid Tumors. Investig. New Drugs. 2014;32:436–444. doi: 10.1007/s10637-013-0046-5. [DOI] [PubMed] [Google Scholar]
- 259.Eskens F.A.L.M., Ramos F.J., Burger H., O’Brien J.P., Piera A., de Jonge M.J.A., Mizui Y., Wiemer E.A.C., Carreras M.J., Baselga J., et al. Phase I Pharmacokinetic and Pharmacodynamic Study of the First-in-Class Spliceosome Inhibitor E7107 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013;19:6296–6304. doi: 10.1158/1078-0432.CCR-13-0485. [DOI] [PubMed] [Google Scholar]
- 260.Steensma D.P., Wermke M., Klimek V.M., Greenberg P.L., Font P., Komrokji R.S., Yang J., Brunner A.M., Carraway H.E., Ades L., et al. Phase I First-in-Human Dose Escalation Study of the Oral SF3B1 Modulator H3B-8800 in Myeloid Neoplasms. Leukemia. 2021;35:3542–3550. doi: 10.1038/s41375-021-01328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Eram M.S., Shen Y., Szewczyk M., Wu H., Senisterra G., Li F., Butler K.V., Kaniskan H.Ü., Speed B.A., Dela Seña C., et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016;11:772–781. doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Wu Q., Nie D.Y., Ba-Alawi W., Ji Y., Zhang Z., Cruickshank J., Haight J., Ciamponi F.E., Chen J., Duan S., et al. PRMT Inhibition Induces a Viral Mimicry Response in Triple-Negative Breast Cancer. Nat. Chem. Biol. 2022;18:821–830. doi: 10.1038/s41589-022-01024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Fong J.Y., Pignata L., Goy P.-A., Kawabata K.C., Lee S.C.-W., Koh C.M., Musiani D., Massignani E., Kotini A.G., Penson A., et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell. 2019;36:194–209.e9. doi: 10.1016/j.ccell.2019.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Yamauchi H., Nishimura K., Yoshimi A. Aberrant RNA splicing and therapeutic opportunities in cancers. Cancer Sci. 2022;113:373–381. doi: 10.1111/cas.15213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Denkert C., von Minckwitz G., Darb-Esfahani S., Lederer B., Heppner B.I., Weber K.E., Budczies J., Huober J., Klauschen F., Furlanetto J., et al. Tumour-Infiltrating Lymphocytes and Prognosis in Different Subtypes of Breast Cancer: A Pooled Analysis of 3771 Patients Treated with Neoadjuvant Therapy. Lancet Oncol. 2018;19:40–50. doi: 10.1016/S1470-2045(17)30904-X. [DOI] [PubMed] [Google Scholar]
- 266.Matsushima S., Ajiro M., Iida K., Chamoto K., Honjo T., Hagiwara M. Chemical Induction of Splice-Neoantigens Attenuates Tumor Growth in a Preclinical Model of Colorectal Cancer. Sci. Transl. Med. 2022;14:eabn6056. doi: 10.1126/scitranslmed.abn6056. [DOI] [PubMed] [Google Scholar]
- 267.Lu S.X., De Neef E., Thomas J.D., Sabio E., Rousseau B., Gigoux M., Knorr D.A., Greenbaum B., Elhanati Y., Hogg S.J., et al. Pharmacologic Modulation of RNA Splicing Enhances Anti-Tumor Immunity. Cell. 2021;184:4032–4047.e31. doi: 10.1016/j.cell.2021.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.El-Khoueiry A.B., Clarke J., Neff T., Crossman T., Ratia N., Rathi C., Noto P., Tarkar A., Garrido-Laguna I., Calvo E., et al. Phase 1 Study of GSK3368715, a Type I PRMT Inhibitor, in Patients with Advanced Solid Tumors. Br. J. Cancer. 2023;129:309–317. doi: 10.1038/s41416-023-02276-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Devarajan A., Su F., Grijalva V., Yalamanchi M., Yalamanchi A., Gao F., Trost H., Nwokedi J., Farias-Eisner G., Farias-Eisner R., et al. Paraoxonase 2 Overexpression Inhibits Tumor Development in a Mouse Model of Ovarian Cancer. Cell Death Dis. 2018;9:392. doi: 10.1038/s41419-018-0395-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Lucas S.A.M., Graham A.M., Presnell J.S., Clark N.L. Highly Dynamic Gene Family Evolution Suggests Changing Roles for PON Genes Within Metazoa. Genome Biol. Evol. 2023;15:evad011. doi: 10.1093/gbe/evad011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Kulka M. A Review of Paraoxonase 1 Properties and Diagnostic Applications. Pol. J. Vet. Sci. 2016;19:225–232. doi: 10.1515/pjvs-2016-0028. [DOI] [PubMed] [Google Scholar]
- 272.Hagmann H., Kuczkowski A., Ruehl M., Lamkemeyer T., Brodesser S., Horke S., Dryer S., Schermer B., Benzing T., Brinkkoetter P.T. Breaking the Chain at the Membrane: Paraoxonase 2 Counteracts Lipid Peroxidation at the Plasma Membrane. FASEB J. 2014;28:1769–1779. doi: 10.1096/fj.13-240309. [DOI] [PubMed] [Google Scholar]
- 273.Devarajan A., Bourquard N., Hama S., Navab M., Grijalva V.R., Morvardi S., Clarke C.F., Vergnes L., Reue K., Teiber J.F., et al. Paraoxonase 2 Deficiency Alters Mitochondrial Function and Exacerbates the Development of Atherosclerosis. Antioxid. Redox Signal. 2011;14:341–351. doi: 10.1089/ars.2010.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Campagna R., Pozzi V., Giorgini S., Morichetti D., Goteri G., Sartini D., Serritelli E.N., Emanuelli M. Paraoxonase-2 Is Upregulated in Triple Negative Breast Cancer and Contributes to Tumor Progression and Chemoresistance. Hum. Cell. 2023;36:1108–1119. doi: 10.1007/s13577-023-00892-9. [DOI] [PubMed] [Google Scholar]
- 275.Zhang J., Wang Y., Li G., Yu H., Xie X. Down-Regulation of Nicotinamide N-Methyltransferase Induces Apoptosis in Human Breast Cancer Cells via the Mitochondria-Mediated Pathway. PLoS ONE. 2014;9:e89202. doi: 10.1371/journal.pone.0089202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wu Y., Siadaty M.S., Berens M.E., Hampton G.M., Theodorescu D. Overlapping Gene Expression Profiles of Cell Migration and Tumor Invasion in Human Bladder Cancer Identify Metallothionein 1E and Nicotinamide N-Methyltransferase as Novel Regulators of Cell Migration. Oncogene. 2008;27:6679–6689. doi: 10.1038/onc.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Xu Y., Liu P., Zheng D.-H., Wu N., Zhu L., Xing C., Zhu J. Expression Profile and Prognostic Value of NNMT in Patients with Pancreatic Cancer. Oncotarget. 2016;7:19975–19981. doi: 10.18632/oncotarget.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Zhou W., Gui M., Zhu M., Long Z., Huang L., Zhou J., He L., Zhong K. Nicotinamide N-Methyltransferase Is Overexpressed in Prostate Cancer and Correlates with Prolonged Progression-Free and Overall Survival Times. Oncol. Lett. 2014;8:1175–1180. doi: 10.3892/ol.2014.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Sartini D., Santarelli A., Rossi V., Goteri G., Rubini C., Ciavarella D., Muzio L.L., Emanuelli M. Nicotinamide N-Methyltransferase Upregulation Inversely Correlates with Lymph Node Metastasis in Oral Squamous Cell Carcinoma. Mol. Med. 2007;13:415–421. doi: 10.2119/2007-00035.Sartini. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Ulanovskaya O.A., Zuhl A.M., Cravatt B.F. NNMT Promotes Epigenetic Remodeling in Cancer by Creating a Metabolic Methylation Sink. Nat. Chem. Biol. 2013;9:300–306. doi: 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Wang Y., Zeng J., Wu W., Xie S., Yu H., Li G., Zhu T., Li F., Lu J., Wang G.Y., et al. Nicotinamide N-Methyltransferase Enhances Chemoresistance in Breast Cancer through SIRT1 Protein Stabilization. Breast Cancer Res. 2019;21:64. doi: 10.1186/s13058-019-1150-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Li X.-Y., Pi Y.-N., Chen Y., Zhu Q., Xia B.-R. Nicotinamide N-Methyltransferase: A Promising Biomarker and Target for Human Cancer Therapy. Front. Oncol. 2022;12:894744. doi: 10.3389/fonc.2022.894744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Wang Y., Zhou X., Lei Y., Chu Y., Yu X., Tong Q., Zhu T., Yu H., Fang S., Li G., et al. NNMT Contributes to High Metastasis of Triple Negative Breast Cancer by Enhancing PP2A/MEK/ERK/c-Jun/ABCA1 Pathway Mediated Membrane Fluidity. Cancer Lett. 2022;547:215884. doi: 10.1016/j.canlet.2022.215884. [DOI] [PubMed] [Google Scholar]
- 284.Parsons R.B., Facey P.D. Nicotinamide N-Methyltransferase: An Emerging Protagonist in Cancer Macro(r)Evolution. Biomolecules. 2021;11:1418. doi: 10.3390/biom11101418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Van Haren M.J., Taig R., Kuppens J., Sastre Toraño J., Moret E.E., Parsons R.B., Sartini D., Emanuelli M., Martin N.I. Inhibitors of Nicotinamide N-Methyltransferase Designed to Mimic the Methylation Reaction Transition State. Org. Biomol. Chem. 2017;15:6656–6667. doi: 10.1039/C7OB01357D. [DOI] [PubMed] [Google Scholar]
- 286.Gao Y., van Haren M.J., Moret E.E., Rood J.J.M., Sartini D., Salvucci A., Emanuelli M., Craveur P., Babault N., Jin J., et al. Bisubstrate Inhibitors of Nicotinamide N -Methyltransferase (NNMT) with Enhanced Activity. J. Med. Chem. 2019;62:6597–6614. doi: 10.1021/acs.jmedchem.9b00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Gao Y., van Haren M.J., Buijs N., Innocenti P., Zhang Y., Sartini D., Campagna R., Emanuelli M., Parsons R.B., Jespers W., et al. Potent Inhibition of Nicotinamide N -Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021;64:12938–12963. doi: 10.1021/acs.jmedchem.1c01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Van Haren M.J., Gao Y., Buijs N., Campagna R., Sartini D., Emanuelli M., Mateuszuk L., Kij A., Chlopicki S., Escudé Martinez de Castilla P., et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules. 2021;11:1357. doi: 10.3390/biom11091357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Van Haren M.J., Zhang Y., Thijssen V., Buijs N., Gao Y., Mateuszuk L., Fedak F.A., Kij A., Campagna R., Sartini D., et al. Macrocyclic Peptides as Allosteric Inhibitors of Nicotinamide N -Methyltransferase (NNMT) RSC Chem. Biol. 2021;2:1546–1555. doi: 10.1039/D1CB00134E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Foulkes W.D., Smith I.E., Reis-Filho J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 291.Bianchini G., De Angelis C., Licata L., Gianni L. Treatment Landscape of Triple-Negative Breast Cancer—Expanded Options, Evolving Needs. Nat. Rev. Clin. Oncol. 2022;19:91–113. doi: 10.1038/s41571-021-00565-2. [DOI] [PubMed] [Google Scholar]
- 292.Cortazar P., Zhang L., Untch M., Mehta K., Costantino J.P., Wolmark N., Bonnefoi H., Cameron D., Gianni L., Valagussa P., et al. Pathological Complete Response and Long-Term Clinical Benefit in Breast Cancer: The CTNeoBC Pooled Analysis. Lancet. 2014;384:164–172. doi: 10.1016/S0140-6736(13)62422-8. [DOI] [PubMed] [Google Scholar]
- 293.Asleh K., Riaz N., Nielsen T.O. Heterogeneity of Triple Negative Breast Cancer: Current Advances in Subtyping and Treatment Implications. J. Exp. Clin. Cancer Res. 2022;41:265. doi: 10.1186/s13046-022-02476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Liu Y., Zhu X.-Z., Xiao Y., Wu S.-Y., Zuo W.-J., Yu Q., Cao A.-Y., Li J.-J., Yu K.-D., Liu G.-Y., et al. Subtyping-Based Platform Guides Precision Medicine for Heavily Pretreated Metastatic Triple-Negative Breast Cancer: The FUTURE Phase II Umbrella Clinical Trial. Cell Res. 2023;33:389–402. doi: 10.1038/s41422-023-00795-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are contained within the article.