

Abstract
We report a new positively charged azidoamino acid for strain-promoted azide–alkyne cycloaddition (SPAAC) applications that overcomes possible solubility limitations of commonly used azidolysine, especially in systems with numerous ligation sites. The residue is easily synthesized, is compatible with Fmoc-based solid-phase peptide synthesis employing a range of coupling conditions, and offers efficient second-order rate constants in SPAAC ligations employing DBCO (0.34 M−1 s−1) and BCN (0.28 M−1 s−1).
Graphical Abstract
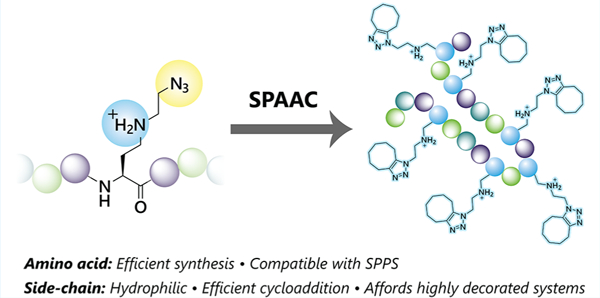
Strain-promoted azide–alkyne cycloaddition has become a widely used tool for selective modifications of biomolecules in vitro and in vivo for numerous chemical biology and materials applications. This reaction is advantageous because of its chemoselectivity, reaction rates, and compatibility with aqueous physiological environments.1 However, the modification of peptides, proteins, and other macromolecules with azide or strained alkyne groups for SPAAC reactions can affect the macromolecule’s physical, structural, and functional properties, particularly when residues in the native primary sequence are altered or when multiple modifications are required. For example, incorporating ligation handles can dramatically increase the macromolecular hydrophobicity, which affects the solubility and aggregation propensity. In some cases, organic solvents are needed to aid solubility, but at the risk of influencing the macromolecular structure.2,3
A case in point is azidolysine (), which is commonly used to introduce an azide into peptides and proteins by solid-phase peptide synthesis (SPPS), often replacing a Lys residue in the native primary sequence. However, this replacement eliminates the Lys side-chain amine group that provides positive charge, which in turn facilitates solubility. Furthermore, side-chain positive charge may also be required for molecular interactions relevant to structure and function. In our own work, we experienced a problematic decrease in the solubility of peptides when was incorporated at the expense of Lys.
One strategy to improve on commonly used SPAAC-enabling amino acids is to design unnatural residues that not only contain the necessary click chemistry handles but also functional groups that mimic those found in naturally occurring side chains. In this work, we designed the positively charged azido-containing amino acid 1 to address the issue of decreased macromolecule solubility that often accompanies replacment of Lys with (Scheme 1). Residue 1 contains the required azido group but also a secondary amine in its side chain, which is protonated at physiological pH, greatly aiding solubility. The synthesis of its Nα-Fmoc-Nδ-Boc-protected analogue 2 is simple, high-yielding, and scalable and uses inexpensive starting materials. Protected 2 is compatible with standard Fmoc SPPS, and numerous copies of the residue can be incorporated into a single peptide while minimally affecting its solubility. Importantly, this new side chain offers similar reaction kinetics with strained alkynes compared to other azides.
Scheme 1. Structure of Residue 1 and Synthesis of Nα-Fmoc-Nδ-Boc-Protected 2.

With respect to synthesis, several approaches have been reported for azido-containing amino acids. For example, Hoffmann rearrangement of Fmoc-protected Gln or Asn followed by diazo transfer affords the corresponding azido side chain.4,5 Another common strategy involves the mesylation or bromination of a hydroxyl side chain and subsequent substitution by sodium azide.6,7 Although useful, this approach requires Boc-to-Fmoc protecting group swapping. Furthermore, the multistep preparation of residues containing aromatic azides and branched azido side chains has been reported.8–10 Here we developed a simple two-step synthesis of 2 starting from commercially available Fmoc-allyl-Gly-OH (Scheme 1). Ozonolysis of the allyl group quantitatively yields aldehyde 3 after reductive workup using thiourea. Reductive amination of 3 employing 2-azidoethan-1-amine (4) yields azidoamine 5, which is directly Boc-protected in situ to afford Nα-Fmoc-Nδ-Boc-protected 2. The overall synthesis can be performed on a multigram scale with only one flash chromatography purification, affording 2 in 60% overall yield.
Residue 2 is compatible with both conventional manual and microwave-assisted Fmoc-based SPPS. We first investigated its coupling efficiency via the preparation of a family of tri-, tetra-, and octapeptides where residue 2 was coupled to the bulky β-branched residues Ile and Val (peptides 6, 8, 9, 10, and 11), small aliphatic Ala (peptide 12), and the secondary amine of Pro (peptide 7) (Scheme 2). We also coupled bulky Trt-protected His and Gln, Pbf-protected Arg, and Boc-protected Ser to residue 2 in a growing chain (peptides 8–11). The purities of peptides 6–12 after TFA cleavage and standard workup ranged from 80% to 91% as determined by HPLC, indicating that residue 2 allows efficient coupling. Next, we evaluated the compatibility of residue 2 toward four widely used sets of coupling conditions in SPPS, including HATU–DIPEA, DIC–HOAt, PyBOP–DIPEA, and microwave-assisted Oxyma–DIC, via the synthesis of tetrapeptide 9. The different coupling conditions resulted in similar peptide purities (91%, 81%, 87%, and 82%, respectively), indicating broad tolerance to different coupling conditions.
Scheme 2. Peptides Incorporating Residue 1, Exploring the Scope of Fmoc-Based SPPSe.

aCoupling conditions: 4 equiv of Fmoc-protected amino acid, 4 equiv of HATU, 6 equiv of DIPEA, DMF, r.t., 0.5 h. bCoupling conditions: 5 equiv of HOAt, 5 equiv of DIC, DMF, r.t., 1 h. cCoupling conditions: 5 equiv of PyBOP, 10 equiv of DIPEA, DMF, r.t., 1 h. dCoupling conditions: 5 equiv of Oxyma, 5 equiv of DIC, DMF, microwave, 90 °C, 4 min. eA full list of all peptides used in this study is provided in Figure S1. Peptide purity was determined by C18 RP-HPLC monitoring at 220 nm. X represents residue 1.
We then determined the intrinsic hydrophilicity/hydrophobicity coefficients (ΔtR(Gly)) for residue 1, , and Lys according to the procedure of Hodges et al.11 to gain comparative insight into the hydrophilic nature of the new residue. This method compares the RP-HPLC retention time of a peptide containing guest residues at a single position within a sequence to that of a Gly-containing standard. Amino acids with side chains more hydrophobic than Gly have positive ΔtR(Gly) values, and vice versa. Figure 1a shows chromatograms of peptides 12, 13, 14 (the Lys control), and 15 (the Gly standard), from which the coefficients were derived. The value of ΔtR(Gly) for residue 1 (1.4) is substantially smaller than that for (7.25) and closer to those for Gly and Lys (−0.04). This indicates that residue 1 is significantly more hydrophilic than and only slightly more hydrophobic than Lys.
Figure 1.

(a) RP-HPLC elution profiles of peptides 12, 13, control 14, and Gly standard 15. Inset: retention times and ΔtR(Gly) values. (b) UV–vis spectra of peptide solutions in water before (solid line) and after (dashed line) centrifugation. Peptides 16 and 18 at 20 mg/mL 1:300 dilutions and peptide 17 at 1 mg/mL.
Residue 1’s ability to enhance solubility was tested via the amphiphilic peptide 16 [(VX)4VpPTXVEVXVXV-NH2; X = residue 1], which contains seven copies of 1 along with nine hydrophobic Val residues. Here we directly measured the solubility via UV–vis spectroscopy of aqueous preparations of peptides before and after centrifugation (Figure 1b). Peptide 16 (Figure S1) was fully soluble in water at 20 mg/mL, the highest concentration tested, as was its Lys-containing control peptide 18 [(VK)4VpPTKVEVKVKV-NH2].12 Each peptide showed similar absorbance values at 220 nm independent of centrifugation status and no light scattering at longer wavelengths (Figure 1b). Furthermore, peptides 16 and 18 had nearly identical RP-HPLC chromatograms (Figure S2), indicating similar hydrophilic character. In contrast, -containing peptide 17 was completely insoluble in water at only 1 mg/mL, showing extensive light scattering before centrifugation and no scattering after, due to sedimentation of aggregates. Taken together, the data show that residue 1 significantly enhances the solubility.
The reaction kinetics of amino acid 1 in SPAAC with two strained cyclooctynes, DBCO and BCN, was determined in comparison with . First, peptides 12 and 13, which contain residue 1 and , respectively, were independently ligated to PEG-functionalized DBCO. Rates were determined under pseudo-first-order conditions by UV–vis spectroscopy at 308 nm (Figures 2a–c, S3, and S4). The calculated second-order rate constant for peptide 12 was 0.34 M−1 s−1 in HBS buffer (pH 7.4) at 25 °C, which is similar to previously reported rate constants for other azides.13 The ligation rate of peptide 12 is slightly lower than that of peptide 13 (2.6 M−1 s−1), which may be due to hydrophobic interactions between the side chain and aromatic DBCO, facilitating ligation. However, the reaction is fast and completed in about 15 min with concentrations of 0.1 mM DBCO and 14 mM peptide 12 (Figure S3). We then determined the reaction rate of Boc-protected amino acid 2 itself with BCN in methanol at 25 °C (Figures 2d,e and S5) under second-order conditions using NMR spectroscopy and eq S1, resulting in a k2 of 0.28 M−1 s−1, which is comparable with the literature14 and slightly larger than that of Fmoc- (0.037 M−1 s−1) (Figures 2f and S6). Again, the higher rate for residue 2 may occur because its hydrophobic Boc group promotes BCN association. Taken together, these experiments demonstrate that amino acid 1 is a useful residue for the modification of peptides via SPAAC reactions in water or organic media.
Figure 2.

Reaction kinetics of residue 1 and with DBCO and BCN. (a) Ligation of peptide 12, which contains residue 1, with DBCO in HBS buffer at pH 7.4 and 25 °C. The ligation of control peptide 13, which contains , was also performed. (b, c) Determination of second-order rate constants (k2) for (b) peptide 12 and (c) peptide 13 with DBCO calculated based on the values of kobs for different peptide concentrations. kobs was calculated using pseudo-first-order kinetics conditions by monitoring the absorbance decay at 308 nm. (d) Ligation of compound 2 with BCN in MeOD at 25 °C. The ligation of control Fmoc- was also performed. (e, f) Determination of second-order rate constants (k2) for (e) 2 and (f) Fmoc- calculated by linear regression between k2t and time. k2t was calculated under second-order kinetics conditions by monitoring the concentration of the product using NMR spectroscopy and eq S1.
One of the main advantages of residue 1 is its increased hydrophilicity compared to , and ability to impart solubility, which should prove useful in the preparation of highly functionalized peptides and other biomolecules containing multiple ligation sites. To test this assertion, we evaluated the reaction of peptide 16, containing seven copies of residue 1, with BCN. Although the peptide is soluble in water, 10% DMSO was added to solubilize the BCN. The ligation efficiency was dependent on the BCN concentration, with 1.5 equiv needed to drive the reaction to completion at room temperature (Figure 3a). However, when the reaction was performed at 37 °C, 1.1 equiv of BCN was sufficient to obtain the heptamodified peptide product (Figure 3b).
Figure 3.

ESI(+) mass spectra of SPAAC reactions between peptide 16 and different numbers of equivalents of BCN at (a) room temperature and (b) 37 °C. For clarity, only the [M + 5H]5+ charge state manifold is shown. Complete spectra are provided in Figures S18 and S19.
In summary, we have designed a new unnatural amino acid that contains both a secondary amine and azide in its side chain to impart charge and solubility while allowing efficient SPAAC ligation. The synthesis of 2 is efficient and scalable, resulting in an amino acid that is compatible with Fmoc-SPPS. Numerous azide residues 1, capable of efficient ligation, can be incorporated into a single peptide without sacrificing solubility or affecting the peptide’s primary charge state. This new amino acid is a new tool in the SPAAC toolbox to facilitate macromolecular ligation where solubility and maintenance of primary charge are of concern, especially in highly decorated systems.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Biophysical Resource Laboratory at the Center of Structural Biology, Center for Cancer Research for acquiring the high-resolution mass spectroscopy data for residue 2. This work was funded by the Center for Cancer Research, National Cancer Institute.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c02906.
Experimental methods, compound characterization, peptide structures, RP-HPLC chromatograms, LC-MS mass spectra, kinetic data, and NMR data (PDF)
The authors declare no competing financial interest.
Contributor Information
Yixin Xie, Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, United States.
Tania L. Lopez-Silva, Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, United States.
Joel P. Schneider, Chemical Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Frederick, Maryland 21702, United States
REFERENCES
- (1).Scinto SL; Bilodeau DA; Hincapie R; Lee W; Nguyen SS; Xu M; am Ende CW; Finn MG; Lang K; Lin Q; Pezacki JP; Prescher JA; Robillard MS; Fox JM Bioorthogonal chemistry. Nat. Rev. Methods Primers 2021, 1 (1), 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pickens CJ; Johnson SN; Pressnall MM; Leon MA; Berkland CJ Practical Considerations, Challenges, and Limitations of Bioconjugation via Azide–Alkyne Cycloaddition. Bioconjugate Chem 2018, 29 (3), 686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nessen MA; Kramer G; Back J; Baskin JM; Smeenk LEJ; de Koning LJ; van Maarseveen JH; de Jong L; Bertozzi CR; Hiemstra H; de Koster CG Selective Enrichment of Azide-Containing Peptides from Complex Mixtures. J. Proteome Res 2009, 8 (7), 3702–3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lau YH; Spring DR Efficient Synthesis of Fmoc-Protected Azido Amino Acids. Synlett 2011, 2011 (13), 1917–1919. [Google Scholar]
- (5).Pícha J; Buděšínský M; Macháčková K; Collinsová M; Jiráček J Optimized syntheses of Fmoc azido amino acids for the preparation of azidopeptides. J. Pept. Sci 2017, 23 (3), 202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schmidt U; Mundinger K; Riedl B; Haas G; Lau R Synthesis of Enantiomerically Pure and Compatibly Protected (2S,3R)- and (2S,3S)-Diaminobutyric Acids. Synthesis 1992, 1992 (12), 1201–1202. [Google Scholar]
- (7).Roth S; Thomas NR A Concise Route to l-Azidoamino Acids: l-Azidoalanine, l-Azidohomoalanine and l-Azidonorvaline. Synlett 2010, 2010 (04), 607–609. [Google Scholar]
- (8).Tookmanian EM; Fenlon EE; Brewer SH Synthesis and protein incorporation of azido-modified unnatural amino acids. RSC Adv 2015, 5 (2), 1274–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Richardson MB; Brown DB; Vasquez CA; Ziller JW; Johnston KM; Weiss GA Synthesis and Explosion Hazards of 4-Azido-l-phenylalanine. J. Org. Chem 2018, 83 (8), 4525–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pitteloud J-P; Bionda N; Cudic P Direct access to side chain N,N′-diaminoalkylated derivatives of basic amino acids suitable for solid-phase peptide synthesis. Amino Acids 2013, 44 (2), 321–333. [DOI] [PubMed] [Google Scholar]
- (11).Kovacs JM; Mant CT; Hodges RS Determination of intrinsic hydrophilicity/hydrophobicity of amino acid side chains in peptides in the absence of nearest-neighbor or conformational effects. Pept. Sci 2006, 84 (3), 283–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Haines-Butterick L; Rajagopal K; Branco M; Salick D; Rughani R; Pilarz M; Lamm MS; Pochan DJ; Schneider JP Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U. S. A 2007, 104 (19), 7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Davis DL; Price EK; Aderibigbe SO; Larkin MXH; Barlow ED; Chen R; Ford LC; Gray ZT; Gren SH; Jin Y; Keddington KS; Kent AD; Kim D; Lewis A; Marrouche RS; O’Dair MK; Powell DR; Scadden M. l. H. C.; Session CB; Tao J; Trieu J; Whiteford KN; Yuan Z; Yun G; Zhu J; Heemstra JM Effect of Buffer Conditions and Organic Cosolvents on the Rate of Strain-Promoted Azide–Alkyne Cycloaddition. J. Org. Chem 2016, 81 (15), 6816–6819. [DOI] [PubMed] [Google Scholar]
- (14).Dommerholt J; Rutjes FPJT; van Delft FL Strain-Promoted 1,3-Dipolar Cycloaddition of Cycloalkynes and Organic Azides. Top. Curr. Chem 2016, 374 (2), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.