

Abstract
Near-infrared photoimmunotherapy (NIR-PIT) selectively kills tumor cells to which the photo-absorber dye IR700DX-conjugated antibodies are bound and induces a systemic anti-tumor immune response. NIR-PIT induces immunogenic cell death (ICD), releases damage-associated molecular patterns (DAMPs) molecules from dying tumor cells, and activates dendritic cells (DCs). However, it is unclear whether NIR-PIT affects migration of tumor-infiltrating (Ti)-DCs to draining lymph nodes (dLNs), where a systemic anti-tumor response is induced. Here, we utilized in vivo photolabeling of Ti-DCs in tumors in photoconvertible protein Kikume Green-Red (KikGR) mice to show that NIR-PIT enhanced migration of Ti-DCs including cDC1s, cDC2s, and CD326+ DCs to dLNs. This effect was abolished by blocking adenosine triphosphate (ATP), one of the DAMPs molecules, as well as by inhibition of Gαi signaling by pertussis toxin. Thus, ICD induction by NIR-PIT stimulates Ti-DC migration to dLNs via ATP-P2X7 receptor and Gαi protein-coupled receptor signaling pathways and may augment tumor antigen presentation to induce anti-tumor T cells in dLNs.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00262-022-03216-2.
Keywords: Near-infrared photoimmunotherapy, Immunogenic cell death, Tumor-infiltrating dendritic cells, Tumor, Cellular migration, Photo labeling
Introduction
The various approaches to treating cancer include surgery, radiation, chemotherapy, or a combination of several treatments to provide the most effective treatment. In order to eradicate tumor cells, these therapies sacrifice normal cells, including immune cells that can contribute to tumor regression. Near-infrared photoimmunotherapy (NIR-PIT) is newly established cancer therapy that uses monoclonal antibodies to specifically induce cell death in target cells. NIR-PIT uses a complex of antibodies and InfraRedDye700DX (IR700), a light-absorbing substance that is activated by NIR light (near 690 nm) irradiation [1].
Exposure of the tumor coated with antibody-photoabsorber conjugate (APC) to NIR light deforms the antigen-APC complexes, causing holes in the cell membrane and an influx of water, which ruptures the cells and induces cell death. In NIR-PIT, APCs bind predominantly and specifically to the target tumor cells, and only the APCs present in the light-exposed area are activated, thus killing the tumor cells with minimal damage to the neighboring normal cells [2].
Induction of tumor cell death by some anticancer drugs results in enhanced systemic anti-tumor immune response, which has been termed immunogenic cell death (ICD) [3]. Enhancement of the systemic anti-tumor immune response by ICD is promoted by activation of the innate and adaptive immune response. In the initial stage, damage-associated molecular patterns (DAMPs) molecules such as adenosine triphosphate (ATP) and high-mobility group box (HMGB)1 released from dead tumor cells activate tumor-infiltrating dendritic cells (Ti-DCs) [4]. Subsequently, Ti-DCs phagocytose dying tumor cells, mature and migrate to the draining lymph nodes (dLNs) where they present antigen to T cells, thus initiating an anti-tumor adaptive immune response. In fact, we have shown that ICD induced by diphtheria toxin (DT) in the DT receptor expressing tumor cell line releases ATP and HMGB1 and increases Ti-DC migration to dLNs in ATP-P2X7 receptor and HMGB1-toll-like receptor 4 dependent manner. This leads to enhancement of antigen-specific anti-tumor immunity [5].
NIR-PIT-induced cell death is immunogenic and enhances the systemic immune response [2]. NIR-PIT mediated by cetuximab-IR700 in A431 epidermoid carcinoma and MDA-MB-468 triple negative breast cancer cell line showed enhanced release of DAMPs molecules such as ATP, HMGB1, and calreticulin (CRT). In addition, co-culture of immature DCs with cells killed by NIR-PIT enhanced the expression of CD80 and CD86 in DCs [6]. In mice with CD44-expressing MC38 carcinoma, NIR-PIT induced by IR700 conjugated anti-CD44 antibody (anti-CD44-IR700) together with anti-programmed death-1 (PD-1) immune checkpoint inhibition, increased Ti- CD8+ and CD4+ T cells in non-treated tumors and suppressed tumor growth [7]. This indicates that NIR-PIT may enhance the adaptive immune response to inducing DC maturation, while the effect on DC migration has not yet been assessed.
Migratory CD103+ DCs (cDC1) have been reported to play a critical role in induction of anti-tumor CD8+ T cells in dLNs [8]. After phagocytosis of dying tumor cells, cDC1 migrate to dLNs, present tumor antigens via major histocompatibility complex (MHC) class I pathway and prime anti-tumor CD8+ T cells. On the other hand, CD103– CD11b+ DCs (cDC2) induce interleukin-17 producing CD4+ T cells, which also contribute to tumor growth regression [9]. The pivotal role of these DC subsets in induction of anti-tumor immunity provides strong impetus to understand the effect of NIR-PIT in modulating DC migration to dLNs.
In this study, we utilized in vivo photolabeling by using a mouse line which expresses the photoconvertible protein Kikume Green-Red (KikGR mouse) and thus allows us to label tumor immune cells and analyze their movement [5, 10–13]. Taking advantage of this in vivo photolabeling system, we evaluated the effect of NIR-PIT induced ICD on Ti-DC migration and underlying signaling pathways.
Materials and methods
Mice
Knock-in mice carrying KikGR cDNA under the CAG promoter (KikGR mice) were made previously [10]. KikGR mice were bred in specific pathogen–free facilities at Osaka Ohtani University. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Osaka-Ohtani University Faculty of Pharmacy.
Cell lines
MC38/TfROVA-DTR cells were established using the colon adenocarcinoma cell line MC38 [14] as described previously [5]. MC38/TfROVA-DTRs cell line was maintained in culture medium consisting of Roswell Park Memorial Institute-1640 medium (RPMI; Sigma) supplemented with 10% (v/v) heat-inactivated bovine calf serum (BCS; Hyclone), 1% (v/v) Penicillin–Streptomycin-Glutamine mixed solution (NACALAI TESQUE, INC.), 10 mmol/L HEPES Buffer Solution (NACALAI TESQUE, INC.) and 1% (v/v) non-essential amino acids solution (NACALAI TESQUE, INC.). For tumor inoculation, MC38/TfROVA-DTR cells were harvested and single-cell suspensions of 2 × 106 cells/100 µL were inoculated intradermally in the abdominal skin on the left and right sides of the abdomen across the midline that approximately connects both inguinal LNs. At this tumor location, the migration of cells from the tumor to the inguinal LN confirms that the inguinal LN is the draining LN of these tumors.
Synthesis of IR700-conjugated anti-CD44 antibody
Anti-mouse/human-CD44 antibody (1 mg, 6.7 nmol; clone IM7; Bio X Cell, West Lebanon, NH, USA) was incubated with IR700 NHS ester (65.1 μg, 33.3 nmol, 10 mmol/L in DMSO; LI-COR Bioscience, Lincoln, NE, USA) and phosphate buffer (pH 8.5; Teknova, Hollister, CA, USA) at room temperature for 1 h. The mixture was purified with a gel filtration column (Sephadex G 25 column, PD-10, GE Healthcare, Piscataway, NJ, USA). Protein concentration was determined with Coomassie Plus protein assay kit (Thermo Fisher Scientific Inc, Rockford, IL, USA) by measurement of the absorption at 595 nm with spectroscopy (8453 Value System; Agilent Technologies, Santa Clara, CA, USA). We abbreviate IR700-conjugated anti-CD44 antibody as anti-CD44-IR700.
NIR-PIT, photoconversion, and surgical procedures
Tumor-bearing mice were intravenously injected with anti-CD44-IR700 (50 µg) 4 days after tumor inoculation, and 24 h later, NIR-PIT and tumor-infiltrating cell photoconversion were performed, as described previously [10, 15]. Briefly, KikGR mice were anesthetized and luminal side of tumors was exposed to NIR light (25 J/cm2) and violet light (120 s, 436 nm, 200 mW/cm2) using a red light-emitting diode which emits at 670 to 710 nm wavelength (L690-66-60; Marubeni America Co.) and SP500 spot UV curing equipment with a 436 nm bandpass filter (USHIO), respectively. During photoconversion, skin around tumors was protected from light with aluminum foil. After NIR and violet light irradiation, the abdominal wall was closed by suturing. To keep exposed tissues moist during exposure to light, warmed phosphate buffered saline (PBS) was applied to the region of photoconversion.
Cell isolation
Cell suspensions from tumors and LNs were described previously [10]. For tumor cells, tumors were finely dissected in RPMI 1640-medium (Sigma-Aldrich) containing 300 U/mL of collagenase from C. Histolyticum (Sigma-Aldrich), and 1 mg/mL of dispase II (Roche). After mincing with scissors, tumors were incubated in the same solution for 30 min at 37 °C. After adding 2000 U/mL of DNase I (Calbiochem), cell suspensions were incubated for 15 min at 37 °C and filtered through a cell strainer. For LNs, cells were extruded into RPMI 1640-medium containing 100 U/mL of collagenase type IV (Worthington) and 2000 U/mL of DNase I and incubated for 15 min at 37 °C. After adding 0.5 M EDTA, cell suspensions were filtered through a cell strainer.
Antibodies and flow cytometry
Fluorochrome-conjugated monoclonal antibodies (mAbs) were obtained from BD Bioscience, eBioscience, or BioLegend as specified in Supplementary Table 1. For flow cytometric analysis, cells were washed with Dulbecco’s PBS containing 2% FCS and 0.02% sodium azide. Next, cells were incubated with 2.4G2 hybridoma culture supernatant to block Fc binding, then stained with fluorochrome-conjugated mAbs for 15 min at 4 °C, and then, cells were analyzed by flow cytometry analysis with SP6800 (SONY). Flow cytometry data were analyzed using FlowJo software (Tree Star). The number of cells in each cell subset of the LNs was calculated by multiplying the total number of cells by the frequency of each cell subset obtained by flow cytometric analysis.
Reagents and treatment
In some experiments, tumor-bearing mice were intratumorally treated with 300 µg of oATP (Calbiochem), or 500 ng of PTX (Wako), or vehicle (PBS(-)) 5 days after tumor inoculation.
Statistical analysis
Data were tested by Mann–Whitney’s U test, one-way ANOVA with Holm-Sidak’s multiple comparisons test, which was selected depending on comparison group, and all tests were done by using GraphPad Prism version 9.0 (GraphPad Software). Data in bar graphs represent means ± SEM. P-values of less than 0.05 were considered to be statistically significant and labeled: *, P < 0.05; **, P < 0.01.
Results
NIR-PIT accelerates migration of migratory Ti-DCs but not MoDCs from tumors to dLNs
We have reported that tumor cell ICD enhances phagocytosis and accelerates Ti-DC turn-over in tumors [5]. To determine whether NIR-PIT has a similar effect on immune cell turn-over in tumors and emigration to dLNs, we took advantage of the KikGR photoconvertible transgenic mice. These mice express KikGR photoconvertible protein, which is initially green (KikGR-Green) but can be irreversibly photoconverted to red (KikGR-Red) by exposure to violet light [10] (Fig. 1A). Thus, cells that were present in the tumor at the time of photoconversion and either remained in the tumor or migrated to other anatomical sites can be identified as KikGR-Red, while cells that infiltrated the tumor after photoconversion are KikGR-Green. MC38 cells express cell surface antigen CD44, and anti-CD44-IR700 binds to these cells, allowing NIR light irradiation to specifically induce cell death of MC38 tumor cells [16]. Thus, we subcutaneously inoculated MC38 cells in KikGR mice and administered anti-CD44-IR700 intravenously 4 days later. Twenty-four hours following anti-CD44-IR700 administration, tumor cell death was induced by NIR light irradiation. This was immediately followed by photoconversion of tumor-infiltrating KikGR+ cells using violet light as previously [5]. Prior to photoconversion, all tumor-infiltrating KikGR+ cells were of the KikGR-Green phenotype. Irradiation with violet light, but not NIR light, photoconverted tumor-infiltrating cells from KikGR-Green to KikGR-Red. Importantly, none of the cells in dLNs were KikGR-Red immediately after photoconversion (Fig. 1B).
Fig. 1.

Experimental setup for the analysis of Ti-DC migration to dLNs after NIR-PIT induction. A Time course and flow diagram of NIR and violet light irradiation of tumor. Twenty-four hours after anti-CD44-IR700 intravenous injection, tumor cell death was induced by NIR irradiation and at the same time, tumor infiltrating KikGR+ cells were photoconverted by exposure to violet light. Cells in dLNs were analyzed 24 h later. B KikGR cells were detected in photoconverted and non-photoconverted tumors and inguinal dLNs in KikGR mice immediately after NIR and/or violet light irradiation of tumors. Data are representative of two independent experiments
After another 24 h we sacrificed the mice and analyzed DCs in tumors and dLNs by flow cytometry. DCs in the dLNs are subdivided into CD11cint MHC class IIhigh migratory DCs and CD11chigh MHC class IIint lymph node-resident DCs (LNDCs). As reported previously [5], photoconverted tumor-egressing KikGR-Red cells were exclusively detected in the migratory DC population, but not in LNDC population (Supplementary Fig. 1A). When we evaluated the effect of NIR-PIT on Ti-DC migration, we observed that the frequency of KikGR-Red+ total migratory DCs increased to 27% in NIR-PIT group compared to 12% in the vehicle group and the number of total tumor-egressing Ti-DCs increased almost threefold following NIR-PIT (Fig. 2). Migratory Ti-DCs in dLNs are made up of cDC1, cDC2, and CD326+ Langerhans linage cells and all of these DC subsets included tumor-derived KikGR-Red DCs (Fig. 2, Supplementary Fig. 1A, and [5]). When tumor cell death was induced using NIR-PIT, the number of KikGR-Red cells in all of these DC subsets increased significantly (Fig. 2).
Fig. 2.

NIR-PIT enhances Ti-DC migration to dLNs but not MoDCs. Anti-CD44-IR700 or vehicle was intravenously administered into tumor-bearing KikGR mice 24 h before the time of NIR light and violet light irradiation of tumor and another 24 h later, cells in dLNs were analyzed. A Representative flow cytometry plots of total migratory, CD103+, CD103–, CD326+ and MoDCs in dLNs of Anti-CD44-IR700 or vehicle-treated KikGR mice. B Number of KikGR-Red cells in total migratory DCs, and DC subsets in dLNs. Numbers in parentheses indicate increased multiples to vehicle. Bar graph shows means ± SEM of pooled data from two independent experiments (n = 4–5). *P < 0.05, (Mann–Whitney U test)
A type of ICD induced by anthracycline can increase intratumoral CD11c+CD11b+Ly6Chigh monocyte-derived DCs (MoDCs). Since this DC subset plays a critical role in ICD-induced anti-tumor immunity and particularly in mediating tumor regression [17], understanding their migration in steady state and in response to ICD is of considerable importance. Analysis of MoDC migration to dLNs (Supplementary Fig. 1B) showed that although we could detect KikGR-Red MoDCs in dLNs, their migration was not affected by NIR-PIT (Fig. 2).
NIR-PIT enhanced Ti-DC migration depends on ATP-P2X7 and Gαi signaling
NIR-PIT-induced tumor cell death causes release of DAMPs molecules such as ATP from dying tumor cells and activates DCs [6]. We reported that ICD-induced acceleration of Ti-DC migration from tumors to dLNs depends on ATP-P2X7 signaling pathway [5]. Here, we examined whether the ATP-P2X7 signaling pathway is similarly involved in the enhancement of Ti-DC migration by NIR-PIT. Four days after tumor inoculation in KikGR mice, anti-CD44-IR700 was injected intravenously and 24 h later, selective inhibitor of ATP-P2X7 receptor, oxidized ATP (oATP) [18] or vehicle was injected intratumorally and tumors were exposed to NIR and violet light. Twenty-four hours later, NIR-PIT induced increase in KikGR-Red total migratory DCs, cDC1s, cDC2s, and CD326+ DC subsets in the dLNs was abrogated completely in mice treated with the inhibitor (Fig. 3 and Supplementary Fig. 2).
Fig. 3.

NIR-PIT enhanced Ti-DC migration to dLNs is inhibited by ATP-P2X7R, Gαi-protein blockade. Anti-CD44-IR700 or vehicle was intravenously administered 24 h before intratumoral administration of inhibitors at the time of NIR light and violet light irradiation of tumor and another 24 h later, cells in dLNs were analyzed. Number of KikGR-Red cells in total migratory DCs, and DC subsets in dLNs. Bar graph shows means ± SEM of pooled data from at least two independent experiments (n = 5–11). *P < 0.05, **P < 0.01, ***P < 0.001. (one-way ANOVA with Holm-Sidak’s multiple comparisons test)
DC activation leads to maturation and upregulation of the Gαi-protein-coupled chemokine receptor CCR7, which allows mature Ti-DCs migrate to dLNs. CCR7-dependent Ti-DC migration can be blocked by pertussis toxin (PTX), which is an inhibitor of Gαi signaling [8]. Therefore, we evaluated whether NIR-PIT-induced migration is mediated by Gαi-protein-coupled chemokine receptor signaling. Intratumoral administration of PTX prior to NIR and violet light irradiation of tumors significantly reduced cell number of KikGR-Red total migratory DCs, cDC1s, cDC2s, and CD326+ DC subsets compared to NIR-PIT induced vehicle-treated group (Fig. 3). These results indicate that NIR-PIT enhances Ti-DC migration to dLNs via ATP-P2X7 and Gαi signaling-dependent pathways. Taken together, our results indicate the NIR-PIT can enhance DC migration from tumors via mechanisms that parallel those for other types of ICD.
Discussion
NIR-PIT induces ICD in targeted tumor cells and leads to the enhancement of systemic anti-tumor immunity [2]. Tumor antigen presentation to naive T cells in the tumor dLNs plays a key role in inducing systemic anti-tumor immunity and cDC1s [8] and cDC2s [19] are proposed to be the main antigen-presenting subsets. In this study, we demonstrated using KikGR-photoconvertible protein expressing mice that ICD induction by NIR-PIT increased Ti-DC migration (and specifically cDC1 and cDC2 subsets) from tumors to dLNs, and this acceleration of Ti-DC migration was blocked by the intratumoral administration of oATP and PTX (Fig. 4). In addition to the ATP-P2X7 pathway, it is likely that the HMGB1-TLR4 signaling pathway also contributes to ICD-induced acceleration of Ti-DC migration to dLNs since NIR-PIT stimulates the release of HMGB1 from dead tumor cells [6]. In addition, we previously showed that the enhanced migration of Ti-DCs induced by ICD in MC38 cells expressing DTR by DT administration was inhibited by blocking the HMGB1-TLR4 signaling pathway [5].
Fig. 4.
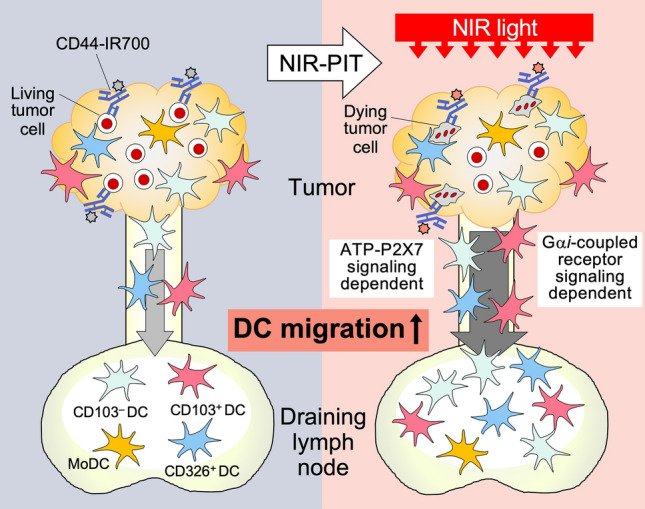
NIR-PIT induces immunogenic tumor cell death and increases Ti-DC migration from tumor to dLN via ATP-P2X7R and Gαi-protein-coupled receptor signaling pathways. cDC1s, cDC2s, CD326+ DCs, and MoDCs migrate from tumor to dLNs. When ICD was induced by NIR-PIT, DAMPS molecules such as P2X7 released from dying tumor cells stimulate maturation and increase cDC1s, cDC2s, CD326+ DCs migration. NIR-PIT enhanced migration of Ti-DCs including cDC1s, cDC2s, and CD326+ DCs to dLNs via Gαi protein-coupled receptor signaling pathways
Our previous work indicates that inducing ICD in as few as one-third of total tumor cells is sufficient to induce a twofold increase in Ti-DC migration as well as systemic tumor antigen-specific T cells and suppress ectopic tumor growth. In this study, NIR-PIT induction led to a threefold increase in KikGR-Red DCs in the dLN 24 h after treatment, indicating that the enhancement of Ti-DC migration by NIR-PIT is on par with our previous results.
We previously demonstrated that tumor-egressing DCs in dLNs have higher expression of CD80 and CD86 molecules [5]. Further, ICD induction by NIR-PIT in A431 cell line enhanced expression level of CD40, CD80, and CD86 in co-cultured bone marrow-derived DCs via DAMPs molecules, such as ATP and HMGB1 released from dying A431 cells [6]. In addition, we observed ICD induced by DTR increased proliferating tumor antigen-specific CD8+ T cells in dLN [5]. Since NIR-PIT enhanced DC migration in similar fashion, we hypothesize that the downstream effect would be a similar induction of tumor antigen-specific CD8+ T cell proliferation and this contributes to the anti-tumor effect of NIR-PIT.
Induction of ICD in tumor cells induces phagocytosis of dying tumor cells by Ti-DCs including cDC1s [5]. Phagocytosis and DAMPs molecules accelerate Ti-DC maturation and upregulation of chemokine receptor CCR7. Ti-DCs migrate to dLNs in response to CCR7 ligands [8]. Administration of PTX to the tumor, concurrently with NIR-PIT and photoconversion of tumor-infiltrating cells, abolished NIR-PIT enhancement of Ti-DC migration, confirming that Gαi protein-coupled receptors are involved in this migration. Additional pathways that are important for this step likely include the S1P1-S1PR1 pathway since it has previously been shown to be critical for ICD-induced enhancement of Ti-DC migration [5].
A previous report has suggested that Mo-DCs play an essential role in anti-tumor immunity at the site of ICD induction in the tumor [17], however, it was unclear whether this influences Mo-DC migration from tumors to dLNs. Our study shows that a proportion of Mo-DCs migrates from tumors to dLNs, although this migration was not altered by ICD. On the other hand, we previously demonstrated that enhanced Ti-DC, especially cDC1 migration at the time of ICD induction is essential [5] to stimulate systemic anti-tumor immunity based on the induction of anti-tumor CD8+ T cells. Since NIR-PIT stimulated cDC1 and cDC2 migration to dLNs, where antigen-specific T cell response is initiated, it is likely that these Ti-DC subsets rather than Mo-DCs play an important role in NIR-PIT-induced systemic anti-tumor immunity as antigen-carrying or -presenting cells.
In this study, we utilized CD44, which is abundantly expressed by MC38 cells, as a target molecule of NIR-PIT [7, 20, 21]. Since CD44 is also expressed on activated effector T cells and DCs, NIR-PIT targeting this molecule may also induce damage to cells important for anti-tumor immunity. However, even if there is temporary damage to immune cells infiltrating the tumor at the time of treatment, NIR-PIT will induce systemic anti-tumor immunity successfully, since increased infiltration of antigen-specific CD4 and CD8+ T cells into the tumor, and regression of untreated distant tumors in addition to NIR-PIT treated tumors were observed after combination therapy of anti-CD44-IR700 NIR-PIT and anti-PD1 antibody treatment [7]. Our report suggests that NIR-PIT-induced enhancement of Ti-DC migration contributes to improved systemic anti-tumor immunity following treatment. Based on the results in this report and previously published studies, we suggest that NIR-PIT induced ICD would induce phagocytosis of dying tumor cells by Ti-DCs and release of DAMPs molecules such as ATP and HMGB1 from dying tumor cells. This in turn would lead to maturation and activation of Ti-DCs, migration to dLNs, and subsequent tumor-antigen specific T cell proliferation. This sequential process would elicit strong systemic anti-tumor immunity [7].
Traditional cancer therapies, such as chemotherapy and radiation therapy damage not only tumor cells but also normal cells, and suppress the immune response. In addition, surgical therapy frequently involves lymph node dissection, especially when metastasis is suspected. In contrast, NIR-PIT uses the internal foreign body exclusion mechanism of the tumor and maintains the regional LNs, which are important as a site of induction of systemic anti-tumor immunity. The establishment of a next-generation treatment method that combines conventional cancer treatment with NIR-PIT may lead to the development of a promising cancer treatment approach for patients with cancer that has spread throughout the body, as well as a recurrence-free cure for cancer.
Supplementary Information
Below is the link to the electronic supplementary material.
Author contribution
TM, RO, AF, DF, HW, TK, PLC, YK and HK designed experiments. TM, MH, HM, SM, RY and MT, performed experiments. TM and MT analyzed data. TM, YK, TC, HK and MT wrote the manuscript.
Funding
This work was supported in part by JSPS Grants-in-Aid for Scientific Research in Innovative Areas ‘‘Analysis and Synthesis of Multidimensional Immune Organ Network’’ (#24111007); JSPS Grants-in-Aid for Scientific Research (B) (#16H05087); JSPS Grants-in-Aid for Scientific Young Scientists (#18K14598); Special Coordination Funds for Promoting Science and Technology of the Japanese Government; National Health and Medical Research Council Australia project grants GNT1106043 (T.C.), National Breast Cancer Foundation grants IIRS-19–027 and IIRS-22–053 (T.C.), Pankind Foundation grant (T.C.), Perpetual Impact grant (T.C.) and Astellas Pharma Inc. through the Formation of Innovation Centers for the Fusion of Advanced Technologies Program. H.K. was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (ZIA BC 011513).
Declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mitsunaga M, Ogawa M, Kosaka N, et al. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685. doi: 10.1038/NM.2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kobayashi H, Furusawa A, Rosenberg A, Choyke PL. Near-infrared photoimmunotherapy of cancer: a new approach that kills cancer cells and enhances anti-cancer host immunity. Int Immunol. 2021;33:7–15. doi: 10.1093/intimm/dxaa037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krysko DV, Garg AD, Kaczmarek A, et al. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 4.Fucikova J, Kepp O, Kasikova L, et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1–13. doi: 10.1038/s41419-020-03221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moriya T, Kitagawa K, Hayakawa Y, et al. Immunogenic tumor cell death promotes dendritic cell migration and inhibits tumor growth via enhanced T cell immunity. iScience. 2021 doi: 10.1016/j.isci.2021.102424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ogawa M, Tomita Y, Nakamura Y, et al. Immunogenic cancer cell death selectively induced by near infrared photoimmunotherapy initiates host tumor immunity. Oncotarget. 2017;8:10825–10436. doi: 10.18632/oncotarget.14425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagaya T, Friedman J, Maruoka Y, et al. Host immunity following near-infrared photoimmunotherapy is enhanced with PD-1 checkpoint blockade to eradicate established antigenic tumors. Cancer Immunol Res. 2019;7:401–413. doi: 10.1158/2326-6066.CIR-18-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts EW, Broz ML, Binnewies M, et al. Critical role for CD103+/CD141+ dendritic cells bearing ccr7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30:324–336. doi: 10.1016/j.ccell.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laoui D, Keirsse J, Morias Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. 2016;7:1–17. doi: 10.1038/ncomms13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomura M, Hata A, Matsuoka S, et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep. 2014;4:1–11. doi: 10.1038/srep06030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Torcellan T, Hampton HR, Bailey J, et al. In vivo photolabeling of tumor-infiltrating cells reveals highly regulated egress of T-cell subsets from tumors. Proc Natl Acad Sci. 2017;114:5677–5682. doi: 10.1073/pnas.1618446114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomura M, Ikebuchi R, Moriya T, Kusumoto Y. Tracking the fate and migration of cells in live animals with cell-cycle indicators and photoconvertible proteins. J Neurosci Methods. 2021;355:109127. doi: 10.1016/j.jneumeth.2021.109127. [DOI] [PubMed] [Google Scholar]
- 13.Ikebuchi R, Moriya T, Ueda M, et al. Cutting edge: recruitment, retention, and migration underpin functional phenotypic heterogeneity of regulatory T cells in tumors. J Immunol. 2021;207:771–776. doi: 10.4049/jimmunol.2001083. [DOI] [PubMed] [Google Scholar]
- 14.Spiess PJ, Yang JC, Rosenberg SA. In vivo antitumor activity of tumor-infiltrating lymphocytes expanded in recombinant interleukin-2. J Natl Cancer Inst. 1987;79:1067–1075. [PubMed] [Google Scholar]
- 15.Nakanishi Y, Ikebuchi R, Chtanova T, et al. Regulatory T cells with superior immunosuppressive capacity emigrate from the inflamed colon to draining lymph nodes. Muc Immunol. 2017 doi: 10.1038/mi.2017.64. [DOI] [PubMed] [Google Scholar]
- 16.Maruoka Y, Furusawa A, Okada R, et al. Combined CD44- And CD25-targeted near-infrared photoimmunotherapy selectively kills cancer and regulatory T cells in syngeneic mouse cancer models. Cancer Immunol Res. 2020;8:345–355. doi: 10.1158/2326-6066.CIR-19-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y, Adjemian S, Mattarollo SR, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38:729–741. doi: 10.1016/j.immuni.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 18.Mutini C, Falzoni S, Ferrari D, et al. Mouse dendritic cells express the P2X7 purinergic receptor: characterization and possible participation in antigen presentation. J Immunol. 1999;163:1958–1965. [PubMed] [Google Scholar]
- 19.Binnewies M, Mujal AM, Pollack JL, et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4+ T cell immunity. Cell. 2019;177:556–571.e16. doi: 10.1016/J.CELL.2019.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maruoka Y, Furusawa A, Okada R, et al. Near-infrared photoimmunotherapy combined with CTLA4 checkpoint blockade in syngeneic mouse cancer models. Vaccines. 2020;8:1–14. doi: 10.3390/vaccines8030528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maruoka Y, Furusawa A, Okada R, et al. Interleukin-15 after near-infrared photoimmunotherapy (Nir-pit) enhances t cell response against syngeneic mouse tumors. Cancers (Basel) 2020;12:1–13. doi: 10.3390/cancers12092575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.