

Abstract
Objective:
Cathodal direct current stimulation (cDCS) induces long-term depression (LTD)-like reduction of cortical excitability (DCS-LTD), which has been tested in the treatment of epilepsy with modest effects. In part, this may be due to variable cortical neuron orientation relative to the electric field. We tested, in vivo and in vitro, whether DCS-LTD occurs throughout the cortical thickness, and if not, then whether drug–DCS pairing can enhance the uniformity of the cortical response and the cDCS antiepileptic effect.
Methods:
cDCS-mediated changes in cortical excitability were measured in vitro in mouse motor cortex (M1) and in human postoperative neocortex, in vivo in mouse somatosensory cortex (S1), and in a mouse kainic acid (KA)-seizure model. Contributions of N-methyl-D-aspartate–type glutamate receptors (NMDARs) to cDCS-mediated plasticity were tested with application of NMDAR blockers (memantine/D-AP5).
Results:
cDCS reliably induced DCS-LTD in superficial cortical layers, and a long-term potentiation (LTP)-like enhancement (DCS-LTP) was recorded in deep cortical layers. Immunostaining confirmed layer-specific increase of phospho-S6 ribosomal protein in mouse M1. Similar nonuniform cDCS aftereffects on cortical excitability were also found in human neocortex in vitro and in S1 of alert mice in vivo. Application of memantine/D-AP5 either produced a more uniform DCS-LTD throughout the cortical thickness or at least abolished DCS-LTP. Moreover, a combination of memantine and cDCS suppressed KA-induced seizures.
Interpretation:
cDCS aftereffects are not uniform throughout cortical layers, which may explain the incomplete cDCS clinical efficacy. NMDAR antagonists may augment cDCS efficacy in epilepsy and other disorders where regional depression of cortical excitability is desirable.
Introduction
Transcranial direct current stimulation (tDCS) is a noninvasive brain stimulation technique where weak constant electrical current is conducted to the cortex via scalp electrodes. Prolonged tDCS exposure in vivo1 (or, given absent cranium, DCS in vitro2–4) produces effects that are dependent on the direction of electric field (E-field) relative to specific neuronal populations.5 Loosely, this is termed “polarity.” Thus, under the cathode cortical excitability is reduced to resemble long-term depression (LTD) of excitatory synaptic strength in both human1,6–8 and animal studies.3,4,9 Previously,5 in vitro, we demonstrated that modulation of cortical excitability by DCS is dependent on the orientation of the axonal components of synaptic pathways relative to the E-field vector. This raises the possibility that DCS effects may be nonuniform within the neocortex given variable orientations of neurons across layers. Such inhomogeneity of tDCS effects on cortical excitability may explain the relatively modest clinical tDCS effects.10
One extension of our hypothesis is that DCS-mediated changes in synaptic strength may be facilitated by selective pharmacology. We previously identified that DCS-mediated LTD (DCS-LTD) induced under the cathode in mouse primary motor cortex (M1) requires activation of the metabotropic glutamate receptor 5 (mGluR5) signaling pathway,4 and others demonstrated that the DCS-mediated long-term potentiation (DCS-LTP) under the anode requires activation of the N-methyl-D-aspartate–type glutamate receptor (NMDAR).2 Thus, we test in vivo and in vitro (1) whether cathodal DCS (cDCS)-mediated modulation of excitatory synaptic strength is uniform throughout the cortical thickness, (2) whether a uniform cDCS aftereffect can be achieved by combination of cDCS and NMDAR blockade, and (3) whether such drug–DCS coupling enhances cathodal tDCS (ctDCS) efficacy in a mouse epilepsy model.
Materials and Methods
Mice
Adult male mice (C57BL/6J, Jackson Laboratory, Bar Harbor, ME; and C57BL/6, University of Seville, Seville, Spain) were used for all experiments. Mice were housed in a 12/12-hour light–dark cycle in a climate-controlled environment and fed ad libitum. All procedures were in accordance with institutional animal care and use committees of Boston Children’s Hospital and Pablo de Olavide University.
Mouse and Human Cortical Slice Preparation
As previously described,4 mouse brains were removed after decapitation and sectioned in ice-cold treatment artificial cerebrospinal fluid (tACSF). Four hundred-micrometer coronal slices were transferred to oxygenated tACSF for 90 minutes at 30°C before recording. Slices were then transferred to the MED64 chamber (MED-P5155, AutoMate Scientific, Berkeley, CA) with oxygenated recording ACSF (rACSF) at 30°C.
Human cortical tissues (from temporal lobe, insula, or occipital lobe) were transferred into oxygenated ice-cold tACSF after surgical removal of the epileptogenic zone of patients (1–14 years old) with focal cortical dysplasia. Human brain slice preparation was identical to mouse protocols described above. Postoperative tissue analysis was approved under Boston Children’s Hospital Institutional Review Board Protocol 09-02-0043.
Drugs
NMDAR antagonists D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5; Cat# 0106) and 3,5-dimethyl-tricyclo-(3.3.1.13,7) decan-1-amine hydrochloride (memantine, Cat# 0773), group 1 mGluR agonist (RS)-3,5-dihydroxyphenylglycine (DHPG; Cat# 0342), and kainate (KA; Cat# 0222) were purchased from Tocris Bioscience (Bristol, UK).
In vitro, drugs were applied to the slice perfusion bath, as previously described.4 In vivo, drug or vehicle control was administered by intraperitoneal (IP) injection or, for experiments requiring cortical drug microinjection (1 hour prior to ctDCS) by a 30G injection tube coupled to a 10μl Hamilton syringe and a perfusion pump (NE-1000, New Era Pump Systems, Farmingdale, NY).
In Vitro cDCS and Microelectrode Array Recording
In vitro cDCS was delivered via 2 cylindrical Ag/AgCl electrodes (1mm diameter, 3mm length; EP1, WPI, Sarasota, FL) connected to a constant-current stimulus isolator (A365R, WPI) as previously described.4,5 DCS electrodes were half-submerged in rACSF and positioned external to the M1 slice. The direct current (DC) field was oriented orthogonal to the pia and parallel to the vertical interlayer M1 projections. For cDCS, the negative polarity electrode (cathode) was positioned proximal to the cortical pial surface, and the reference electrode (anode) was positioned beneath the subcortical white matter. cDCS was applied at 400μA (corresponding to average estimated11 E-field strength of 9.2V/m in the ACSF and 2.3V/m at the recording surface) for 25 minutes.
To monitor in vitro cDCS-induced cortical excitability changes, field excitatory postsynaptic potentials (fEPSPs) were recorded by a MED-P5155 probe as previously.4 After incubation, a mouse or human neocortical slice was positioned in the center of the 8 × 8 microelectrode array and perfused with oxygenated rACSF. To measure pharmacologic effects on cDCS-mediated changes in synaptic strength, D-AP5 or memantine was introduced into the recording chamber before baseline recording (>20 minutes prior to DCS). No baseline fEPSP changes were induced by D-AP54 or memantine (data not shown). Data were collected by system software (Mobius 0.4.2; 1Hz–10kHz filter; 20kHz sampling rate).
In Vivo ctDCS in Anesthetized Mice
ctDCS (1mA/25 minutes) in urethane (2g/kg, IP) anesthetized mice was delivered via an Ag/AgCl scalp electrode (6mm diameter, Cat# SPIDER100626, Rhythmlink International, Columbia, SC) connecting to the same DC stimulator as for in vitro cDCS. As described previously,4,12 the edge of the cathodal electrode was glued to the dorsal scalp centered on the interocular line, whereas the anode was a saline-saturated (0.9% NaCl) sponge (1.5 × 1.5cm) placed under the ventral torso. ctDCS was administrated before seizure induction. No current was delivered during sham ctDCS.
Immunofluorescence
Mice were transcardially perfused with 4% paraformaldehyde (PFA), 1 hour after in vivo ctDCS or sham treatment. Free-floating sections (40μm13) were sliced after overnight post-fixation of brains in 4% PFA at 4°C. pS6 Ser 240/244 (Cat# 5364, Cell Signaling Technology, Danvers, MA) was used as a primary antibody at 1:500 dilution overnight at 4°C. DyLight 488 conjugated goat antirabbit IgG (Cat# 111–485-144, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) was used as secondary antibody at 1:500 dilution for 1 hour at room temperature. Sections were mounted with Vecta Shield mounting media with 4,6-diamidino-2-phenylindole and imaged using a Zeiss (Oberkochen, Germany) LSM 710 confocal microscope. pS6 density was analyzed by Volocity (Quorum Technologies, Lewes, UK) software and normalized by the pS6 density in the deep layer of sham ctDCS-treated slices. Superficial cortical layer pS6 density was measured from layer 2/3 to layer 5, whereas deep cortical pS6 density was measured from layer 5/6.
Scalp Electroencephalography and KA-Induced Seizures
Scalp electroencephalogram (EEG) was acquired by 2 thin Ag/AgCl Teflon-coated subdermal wire electrodes (Ives EEG Solutions, Newburyport, MA), with a reference contact positioned over the animal’s dorsal snout at midline olfactory bulb, and the active contact over the parietal region,14 under urethane anesthesia. EEG was sampled at 400Hz (Cadwell Laboratories, Kennewick, WA) and bandpass filtered at 1 to 70Hz for offline review.
KA dose (20mg/kg, IP) was determined prior to start of the experiment to reliably produce electrographic seizures 20 minutes after dosing, but not convulsions, which would confound the recordings.15–18 KA injection was followed by 60 minutes of EEG. Seizure was operationally defined as a train of epileptic spikes occurring at ≥1Hz and lasting ≥5 seconds. To test the impact of NMDAR block on ctDCS effects, mice were administered 1 memantine dose (2.5mg/kg,19 IP, in saline) or saline, 20 minutes prior to ctDCS or sham treatment before KA injection. Primary outcomes were latency to first seizure, seizure frequency, and total ictal time (calculated from the point of first detected seizure).
In Vivo ctDCS in Alert Mice and Somatosensory Evoked Potential Recording
Surgery.
Mice were anesthetized with ketamine–xylazine (100mg/kg, 10mg/kg). Under aseptic conditions, a ring Ag/AgCl electrode (3mm diameter; A-M Systems, Sequim, WA) was placed over the skull above the right somatosensory cortex (S1), and a 2mm-diameter hole was drilled in the middle of the ring centered on the S1 vibrissa area (anteroposterior = 0.9mm posterior to bregma, lateral = 3.0mm from midline). A silver electrode (1mm diameter) in contact with the dura mater was attached to the left parietal bone as a reference. A head-holding system, consisting of 1 bolt cemented to the skull perpendicular to the stereotaxic plane, was implanted.
Recording and Stimulation Procedures.
One week after surgery, each mouse was placed on a treadmill with the head restrained. For somatosensory evoked potential (SSEP) recordings during ctDCS, a glass micropipette was first inserted into S1 areas corresponding to the whiskers. Once the S1 was mapped, the glass micropipette was substituted by a chronically implanted linear array of 4 electrodes made from 50μm Teflon-coated tungsten wire (Advent Research Materials, Eynsham, UK) with recording tips separated by 500μm. Superficial and deeper characteristic SSEPs were simultaneously recorded in response to whisker stimulation. The recording array was attached to the skull, and the craniotomy was covered with dental cement. SSEPs were band-pass filtered 0.3Hz–10kHz (3600 model, A-M Systems) and sampled at 25kHz. Facial dermatomes of the whisker regions were electrically stimulated (Cibertec CS-420, Madrid, Spain) with a pair of cutaneous needles inserted under the skin (interelectrode distance = 3–4mm; square pulses, 0.2 milliseconds, <2mA). Based on maximal peak-to-peak SSEP amplitude, final current intensity applied to the whiskers was adjusted to obtain submaximal (50% maximum) responses.
ctDCS in Alert Mice.
ctDCS (200μA, 20 minutes) was delivered by a current-controlled stimulator (A395, WPI) through the ring Ag/AgCl electrode previously placed over the skull and a saline-soaked sponge (6cm2) attached to the back of the animal.
Postexperiment Electrode Placement Confirmation.
Mice were deeply anesthetized with sodium pentobarbital and perfused transcardially with saline and 4% PFA. To confirm the electrode location in the S1 area, the brain was removed and sectioned (50μm), and relevant cortical areas were processed for toluidine blue staining.
Data Analysis and Statistics
Slice electrophysiological data were analyzed by MED64 Mobius software. To detect cDCS or chemically induced LTD/LTP, fEPSP slopes (average of last 10 minutes of the 60-minute post-DCS recording) after cDCS or drug application (DHPG or high Ca2+) were normalized and expressed as fold change relative to averaged baseline (average of first 10-minute recording). fEPSP slope fold change obtained from 3 × 7 (mouse M1 slices) or 3 × 4 or 3 × 5 (human cortical slices) microelectrodes surrounding the test stimulus site were first log-transformed, then converted into a finer 2-dimensional meshgrid, followed by linear interpolation of the gridded data into a 2-dimensional surface plot using a custom algorithm in MATLAB (R2017a; MathWorks, Natick, MA). Values for the channel that delivered the test stimuli were interpolated as an average of the fEPSP slope changes from the 8 adjacent channels.
For mouse slice data, statistics were performed using the number of mice as n value, with the data from 1 to 3 slices per mouse. Latency from KA injection to EEG seizure onset, seizure frequency, and ictal time were scored by blinded reviewers (Y.S. and S.C.D.; Neuroscore, Data Sciences International, St Paul, MN) per laboratory protocol.20 Latency to seizure onset was compared between groups using Kaplan–Meier analysis and log-rank (Mantel–Cox) test. Statistical significance between >2 groups was determined by 1-way analysis of variance (ANOVA) or Kruskal–Wallis test based on normality of data, followed by pairwise post hoc comparisons. Paired t test, as necessary, was used to compare within-group baseline and poststimulation means.
For human tissue data, given the unknown cortical layer orientation and absent control of the slice angle relative to the pial surface, we did not average the data to either make a composite figure or analyze the data as if recorded from a group. Instead, a representative color map of cDCS aftereffects with or without memantine treatment obtained from 2 separate human cortical slices is shown in the fifth figure. Repeated-measure ANOVA and post hoc tests were used to compare the baseline and last 10-minute fEPSP slopes recorded from each channel as well as to compare between 2 channels.
For SSEP studies in alert mice, signals were analyzed offline (1401plus, Cambridge Electronic Design, Cambridge, UK). SPIKE2 analysis software (Cambridge Electronic Design) was used to quantify peak N1 SSEP component amplitude. N1 amplitudes were normalized to baseline, and the 20-minute baseline average was compared to the final 20-minute average after stimulation. In-group comparisons were performed by paired t test, unpaired t test, and the nonparametric Friedman test for repeated measures on ranks. Multiple comparisons were performed using post hoc Dunn test.
All statistical analyses were performed using either Prism (GraphPad Software, San Diego, CA), SPSS, or SigmaPlot 14.0 (Systat Software, San Jose, CA). Differences with p < 0.05 were considered statistically significant, and all data are presented as mean ± standard error of the mean.
Results
cDCS Induces Nonuniform fEPSP Changes across Mouse M1 Layers In Vitro
Consistent with our previous work,4 cDCS in M1 slices resulted in reliable DCS-LTD at the superficial layers (layers 2/3–5). fEPSP slopes were reduced to 49.5 ± 8.8% of baseline 1 hour after cDCS (p < 0.001; see Fig 1). Notably, an LTP-like change of fEPSP slopes was detected in the deep layers (layers 5/6), where fEPSP slopes increased to 164.7 ± 28.4% of baseline 1 hour after cDCS (p < 0.05). cDCS aftereffects in superficial M1 layers were significantly different from those in deep layers (p < 0.01). Thus, we identify that a constant DC field induces divergent cDCS aftereffects across cortical layers, resulting in simultaneous depression and potentiation of excitatory synaptic strength.
FIGURE 1:
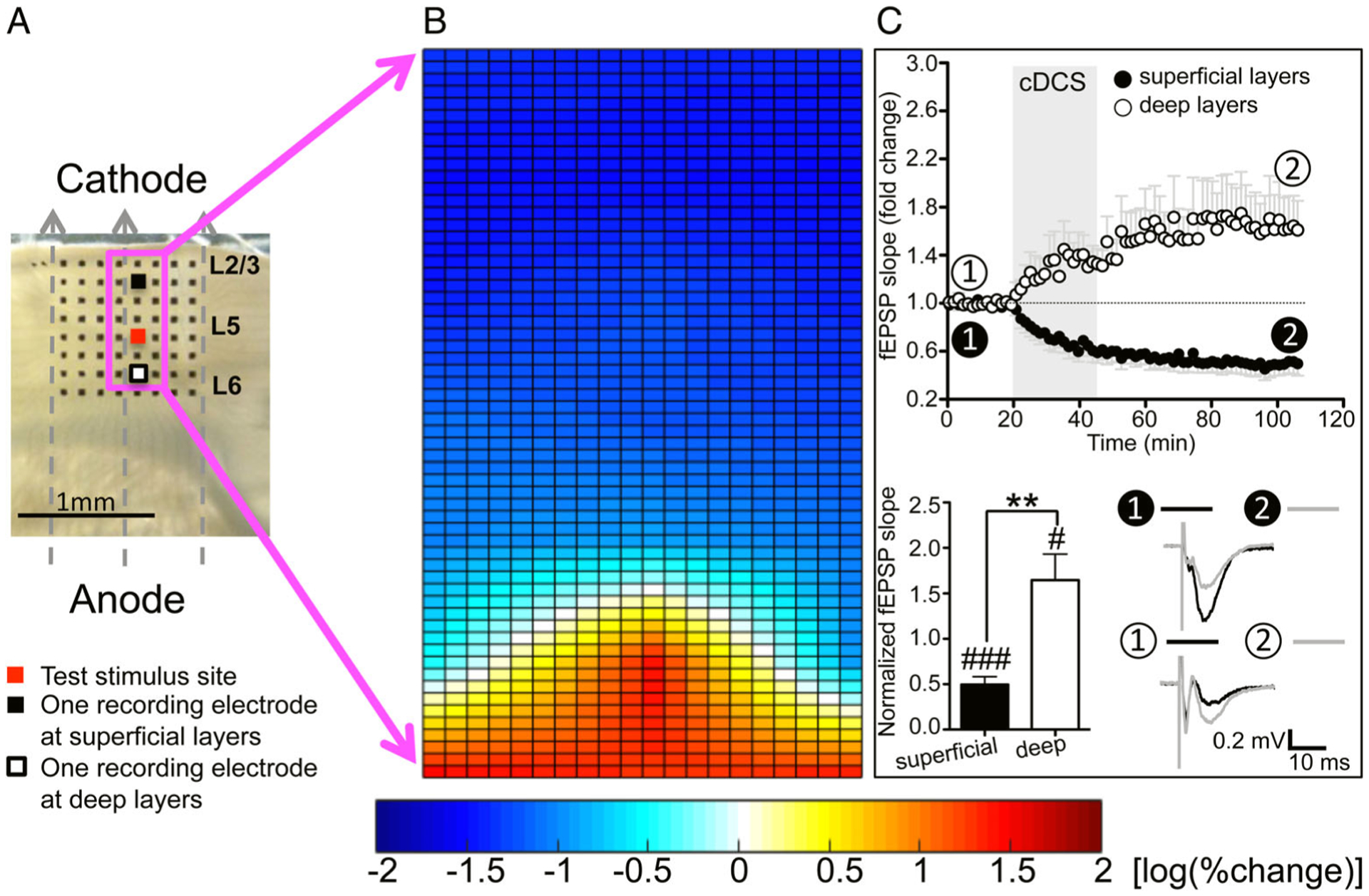
Cathodal direct current stimulation (cDCS) modulation of cortical excitability is inhomogeneous across mouse primary motor cortex (M1) layers in vitro. (A) Orientation of the cathodal direct current (DC) field over a mouse M1 slice. Dashed arrows indicate the electrical field orientation. The 8 × 8 electrode array (black dots) fully covered the M1 area. We stimulated at layer 5 (marked as red) and recorded field excitatory postsynaptic potentials (fEPSPs) from the channels indicated by the magenta square to create color maps. Two channels selected to draw the time course of fEPSP changes were marked as black (superficial to the test stimulus site) and white (deep to the test stimulus site). Scale bar = 1mm. The DC field orientation, the M1 slice placement, fEPSP stimulating and recording sites, and the location of the two channels selected to draw the time course of fEPSP changes are same for the subsequent color maps in Figures 3 (no cDCS) and 4. (B) Percentage change of fEPSP slopes (n = 10) 1 hour after cDCS was log transferred and plotted as a 2-dimensional color map. The color scale bar for fEPSP changes is shown on the bottom in this and subsequent color map figures. More red indicates more potentiation, and more blue indicates more depression. (C) Time course of fEPSP slope changes recorded from 1 channel at superficial layers (closed circles in this and subsequent electrophysiology figures) and one channel at deep layers (open circles in this and subsequent electrophysiology figures) before, during, and after cDCS (400μA/25 minutes, n = 10; top). Time courses of averaged fEPSP slopes recorded from superficial and deep layers show a decrease or increase immediately after cDCS onset and continued to decrease or increase after cDCS cessation for about 30 minutes before stabilization, respectively. Transparent gray rectangle indicates DCS duration in this and Figures 4 to 7. Representative fEPSP traces (bottom right in C) were taken at times indicated by numerals in this and subsequent electrophysiology figures. Scale bars are equal to 0.2mV and 10 milliseconds in this and subsequent electrophysiology figures. Statistical analysis (bottom left in C) shows significant differences of fEPSP slope changes recorded from 1 channel at superficial layers and 1 channel at deep layers (###p < 0.001 and #p < 0.05 indicate paired t test between the last 10 minutes of recording from each channel and their baseline; **p < 0.01 indicates paired t test compared between the 2 channels; n = 10).
Nonuniform Changes of pS6 Labeling Are Detected across Mouse M1 Layers after in vivo ctDCS
We previously showed that DCS-LTD in our preparation requires mGluR5-mTOR-pS6 pathway activation.4 Consistent with the electrophysiology data (see Fig 1) demonstrating DCS-LTD in superficial M1 layers (layers 2/3–5), pS6 immunostaining in M1 after in vivo ctDCS also revealed a layer-specific increase of pS6 protein (Fig 2A); pS6 increased 1 hour after ctDCS only in superficial (layers 2/3–5), but not in the deep (layers 5–6) cortical layers (as compared to sham stimulation). Comparison of pS6 density (in arbitrary units) between sham and active ctDCS groups indicate increase from 7.1 ± 0.2. to 13.8 ± 1.3 in superficial layers (p < 0.001) and absent change (1.0 ± 0.1 v 1.4 ± 0; not significant [n.s.]) in deep layers (see Fig 2B). The layer-specific pS6 staining change after ctDCS confirms a mechanistically heterogeneous modulation of cortical excitability across cortical layers by ctDCS.
FIGURE 2:
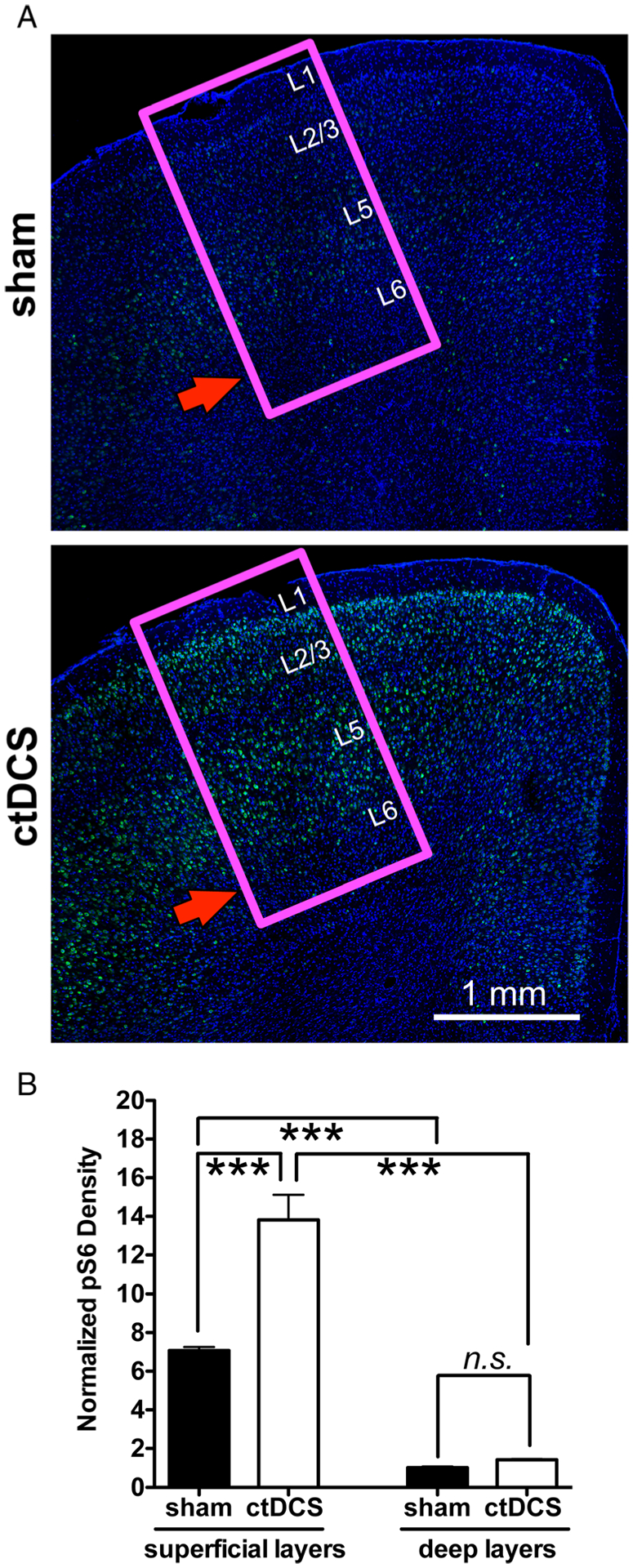
Confocal immunofluorescent images of phospho-S6 ribosomal protein (pS6) labeled across mouse primary motor cortex (M1) layers also shows a nonuniform pS6 increase after cathodal transcranial direct current stimulation (ctDCS). (A) Representative images of pS6 labeling in mouse M1 sections (indicated by magenta square) dissected out 1 hour after sham tDCS (n = 3, top) and ctDCS (1 mA/25 minutes; n = 3; bottom). The red arrow indicates that there is no significant increase of pS6 staining in M1 layer 5/6 after ctDCS. Scale bar = 1mm. The approximate M1 layers are indicated by L1 to L6. (B) Statistical analysis shows a significant increase of pS6 labeling in superficial layers (***p < 0.001, 1-way analysis of variance [ANOVA] post-test) after ctDCS, but not in deep layers (not significant [n.s.], p > 0.05, 1-way ANOVA post-test). All pS6 densities are normalized by the pS6 density of sham deep layers.
cDCS-Induced Inhomogeneous Patterns of Synaptic Strength Are Specific to cDCS and Absent in Pharmacologically Induced LTD or LTP
We tested whether inhomogeneous patterns of regulating cortical excitability by cDCS are also present in conventional LTD or LTP that is pharmacologically, rather than electrically, induced. Consistent with reports that group 1 mGluR activation by its agonist DHPG reliably induces LTD in vitro,21–23 bath application of DHPG (50μM, 5 minutes)22 induced LTD uniformly throughout M1 slices (Fig 3A, left). In contrast to results pursuant to electrical stimulation, differences between superficial and deep layers in the DHPG-induced LTD of the fEPSP are absent (n.s.; see Fig 3B, left column).
FIGURE 3:
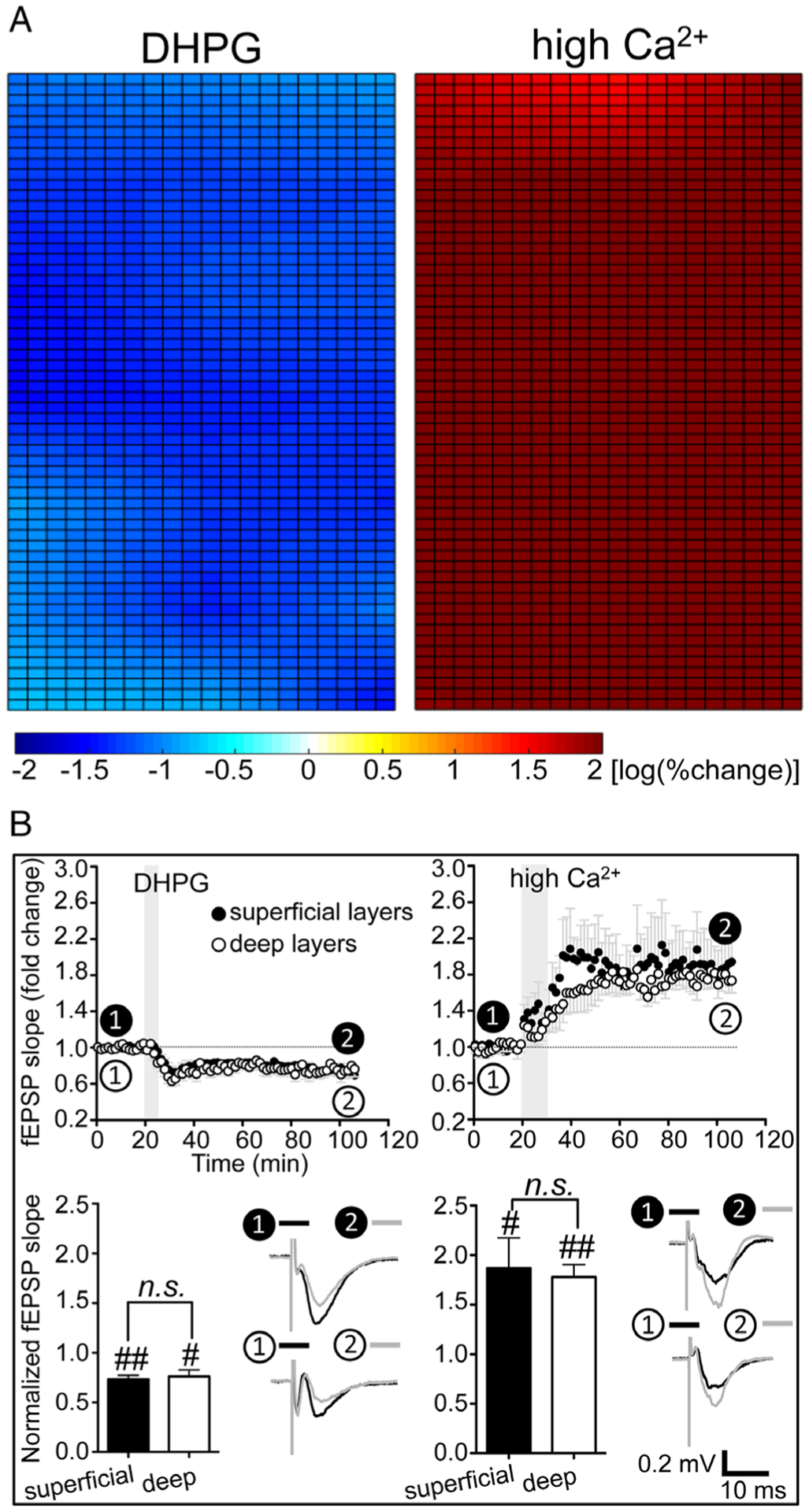
The inhomogeneous modulatory patterns of the synaptic strength were absent after pharmacologically induced long-term depression or long-term potentiation. (A) Application of group I metabotropic glutamate receptor agonist (RS)-3,5-dihydroxyphenylglycine (DHPG; 50μM, 5 minutes, n = 5; left) or high Ca2+ (4mM, 10 minutes, n = 5; right) induces uniform depression or potentiation of cortical excitability throughout mouse primary motor cortex slices. (B) The time course of averaged field excitatory postsynaptic potential (fEPSP) slopes recorded from 1 channel at superficial layers and 1 channel at deep layers both showed a decrease of fEPSP slope: 73.5 ± 3.8% (p < 0.01, n = 5; closed circles) and 76.2 ± 6.8% (p < 0.05, n = 5; open circles) of their baseline 80 minutes after DHPG application, respectively (left column). The fEPSP slopes recorded from 1 channel at superficial layers and 1 channel at deep layers both increased to 186.8 ± 30.5% (p < 0.05, n = 5; closed circles) and 177.9 ± 12.6% (p < 0.01, n = 5; open circles) of their baseline 75 minutes after high Ca2+, respectively (right column). Transparent gray rectangles indicate the application of DHPG or high Ca2+. Statistical analysis in B shows no significant differences of fEPSP slope changes recorded from 1 channel at superficial layers and 1 channel at deep layers (#p < 0.05 and ##p < 0.01 indicate paired t test between the last 10 minutes of recording from each channel and their baseline; not significant [n.s.], p > 0.05 indicates paired t test between the 2 channels; n = 5 for both DHPG [bottom left] and high Ca2+ [bottom right]).
Complementary to pharmacologically induced LTD, and to detect the anatomic pattern of chemical LTP, we exposed M1 slices to high Ca2+ (4mM) rACSF for 10 minutes,24,25 and uniform LTP response of fEPSP was elicited throughout M1 layers (see Fig 3A, right). As with pharmacologic LTD, and also in contrast to DCS, no differences in fEPSP changes at superficial and deep layers are detected after Ca2+-induced LTP (n.s.; see Fig 3B, right column).
Taken together, these results reveal that, in contrast to DCS-induced fEPSP change, pharmacologically induced LTD and LTP are uniform throughout the neocortical slice thickness in vitro, indicating that the depth heterogeneity produced by cDCS is not due to inherent tissue plasticity differences across layers.
cDCS Coupled with NMDAR Block Produces LTD throughout the Cortical Thickness
Given the distinct molecular pathways underlying DCS-LTD (mGluR5-dependent4) and DCS-LTP (NMDARs2), we asked whether cDCS induces uniform DCS-LTD across M1 layers in setting of pharmacologic NMDAR blockade. With 100μM D-AP5, cDCS induced uniform DCS-LTD throughout the M1 layers (Fig 4A, left). The time courses of averaged fEPSPs slopes from 1 channel at superficial layers (decreased to 71.5 ± 4.4% of baseline, p < 0.01) and 1 channel at deep layers (decreased to 68.3 ± 4.2% of baseline, p < 0.01; see Fig 4B, left column) both demonstrated LTD-like changes of cortical excitability, 1 hour after cDCS in the presence of D-AP5. No difference (n.s.) was found between the slope changes of superficial and deep layers (see Fig 4B, left bottom).
FIGURE 4:
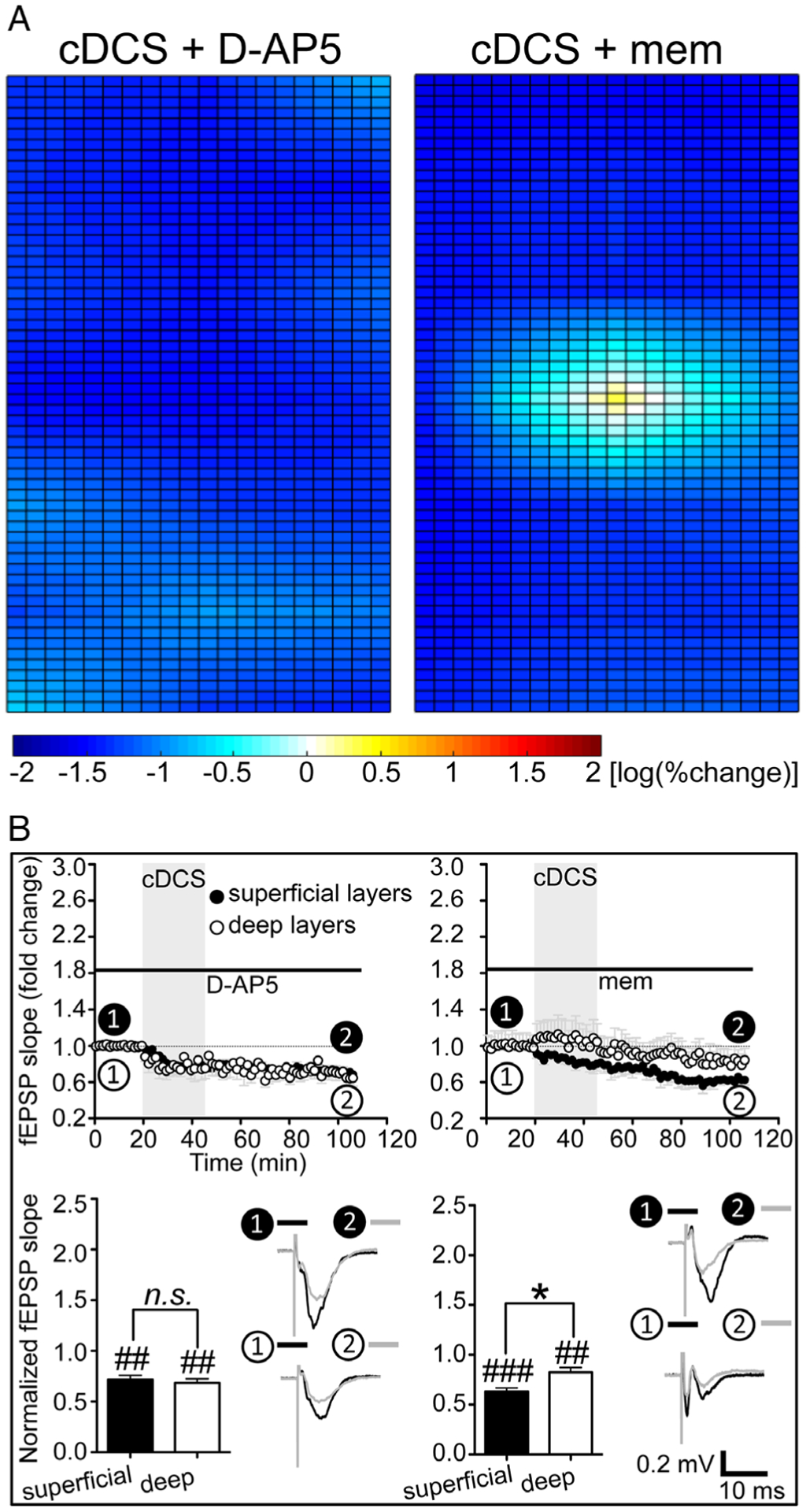
Uniform direct current stimulation (DCS)–long-term depression (LTD) can be achieved by N-methyl-D-aspartate–type glutamate receptor (NMDAR) block. (A) Application of a competitive NMDAR blocker D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5; n = 5, left) or an uncompetitive NMDAR antagonist memantine (mem; n = 7, right) could facilitate DCS-LTD in deep layers of mouse primary motor cortex slices. (B) In the presence of D-AP5 (100μM, n = 5; left column) or mem (100μM, n = 7; right column), the time courses of field excitatory postsynaptic potential (fEPSP) slope changes show that the slopes recorded from 1 channel at deep layers (open circles) are now decreased 1 hour after cathodal DCS (cDCS; 400μA/25 minutes) as compared to their individual baseline (##p < 0.01 and ###p < 0.001 indicate paired t test between the last 10 minutes of recording from each channel and their baseline; not significant [n.s.], p > 0.05 and *p < 0.05 indicate paired t test between the 2 channels).
We also tested the effect of a noncompetitive NMDAR antagonist, memantine, on the cDCS modulation of cortical excitability. Similar to D-AP5, 100μM memantine blocked the DCS-LTP in deep M1 layers and facilitated a more uniform DCS-LTD pattern across all (see Fig 4A, right column). The fEPSP slopes recorded from 2 selected channels, as above, were both decreased to 63.1 ± 3.7% (p < 0.001) and 82.3 ± 4.8% (p < 0.01; see Fig 4B, right top) of baseline, respectively, although the DCS-LTD magnitudes were different (p < 0.05; see Fig 4B, right bottom).
In Human Cortex, In Vitro, the Nonuniform cDCS Aftereffect Is Confirmed and DCS-LTD Is Facilitated by NMDAR Block
We recorded 5 human neocortical slices isolated from 5 samples obtained from 4 patients who had undergone resective epilepsy surgery. In each slice, we found that cDCS resulted in patches of either unchanged or potentiated fEPSP mixed with DCS-LTD (Fig 5B, left). The representative time course of fEPSP slope change recorded from 1 channel above the test stimulus site (reduced to 53.5% of baseline, p < 0.001) and 1 channel below the test stimulus site (101.8% of baseline, n.s.) are plotted in Figure 5D (left top). cDCS aftereffects were significantly different (p < 0.001) across these 2 channels (see Fig 5D, left bottom). The strong similarity between cDCS aftereffects in mouse and human slices suggests that cDCS aftereffect is not species-specific.
FIGURE 5:
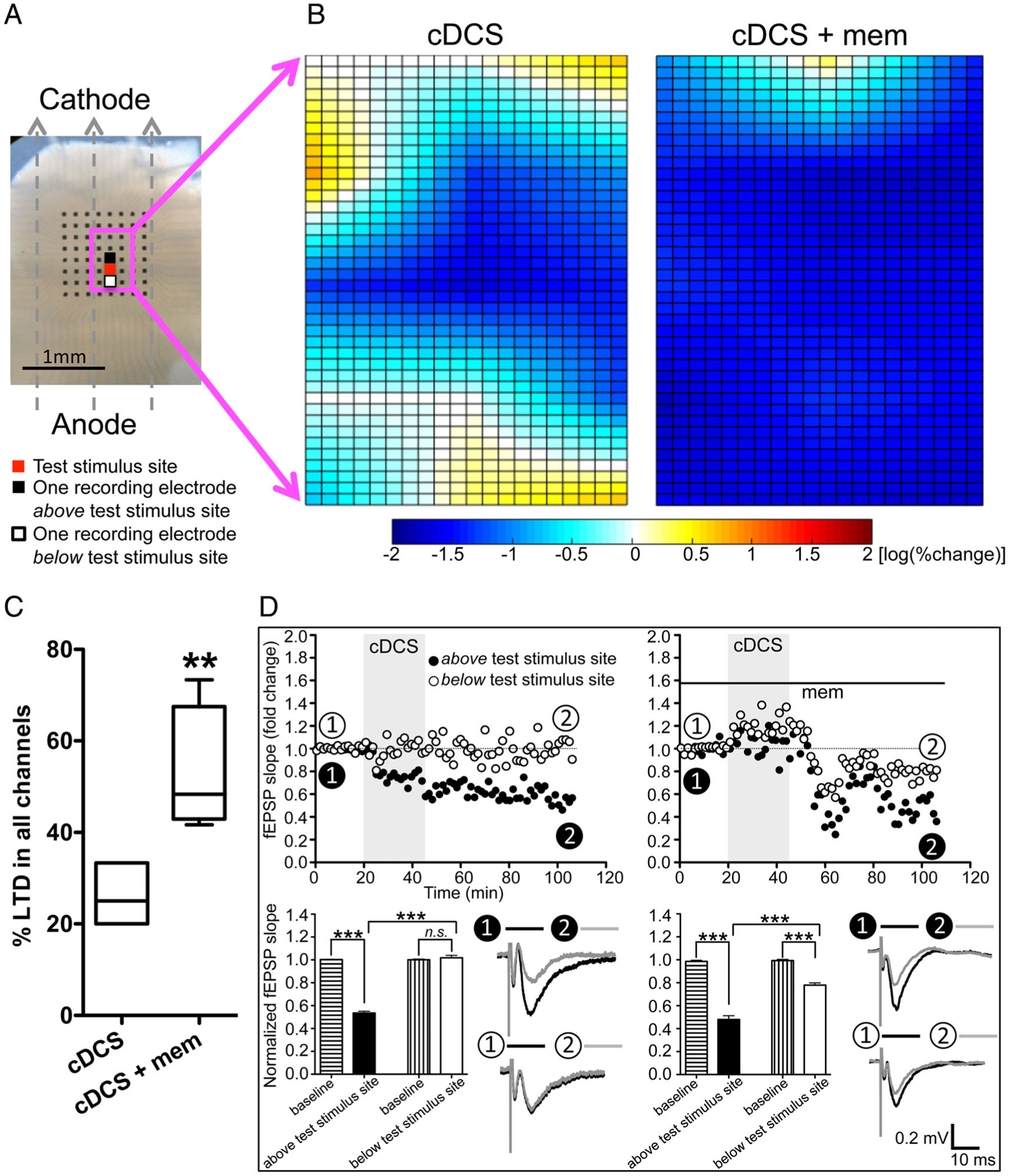
Nonuniform direct current stimulation (DCS) aftereffects were also detected in human cortical slices in vitro, and DCS–long-term depression (LTD) was facilitated by N-methyl-D-aspartate–type glutamate receptor block. (A) Cathodal DCS (cDCS) is applied over a human cortical slice. Dashed arrows indicate the electrical field orientation. The 8 × 8 electrode array is indicated as black dots. The test stimulus site is marked as red, and field excitatory postsynaptic potentials (fEPSPs) were recorded from the channels indicated by the magenta square to create color maps. Two channels selected to draw the time course of fEPSP changes are marked as black (above test stimulus site) and white (below test stimulus site). Scale bar = 1mm. (B) Due to the variable cortical layer orientation and absent control of the slice angle relative to the pial surface, instead of overlapping and computing an average of 5 human cortical slices (fEPSPs were elicited from 3 × 4 channels in 1 slice and from 3 × 5 channels in 4 slices surrounding the test stimulus site), a color map of cDCS aftereffects is obtained from a single human cortical slice (left). This representative map created from 1 human cortical slice shows that cDCS-induced depression was mixed with no change or slight potentiation of cortical excitability. One representative map created from 1 human cortical slice of 4 samples recorded (fEPSPs were elicited from 3 × 4 channels in 2 slices and from 3 × 5 channels in the other 2 slices surrounding the test stimulus site) shows the combined cDCS and memantine (mem; 100μM) effects (right). (C) The cDCS+mem-treated human cortical slices (n = 4) had an increased number of channels with DCS-LTD responses compared to the cDCS-only–treated human cortical slices (n = 5, **p < 0.01 indicates unpaired t test between the 2 treatments). (D) Time course of fEPSP slope changes in 1 cDCS-treated human cortical slice shows that DCS-LTD is only detected in the channel (closed circles) above the test stimulus site, not in the channel (open circles) below the test stimulus site (left column). In the presence of mem (right column), cDCS induces DCS-LTD in both the selected channels (***p < 0.001 and not significant [ns], p > 0.05, 1-way analysis of variance post-test comparing between the last 10 minutes of recording from each channel and their baseline or comparing between the 2 channels).
Furthermore, we tested the cDCS aftereffect coupled with memantine in 4 human cortical slices isolated from 3 individual patients with epilepsy. In all, more channels expressed DCS-LTD (52.9 ± 7.0%) in the cDCS+memantine group versus cDCS alone (26.3 ± 3.0%, p < 0.01; see Fig 5B, right and 5C). The representative time courses of fEPSP slope change 1 hour after cDCS recorded from 1 channel above the test stimulus site (48.0% of baseline, p < 0.001) and 1 channel below the test stimulus site (77.9% of baseline, p < 0.001) are plotted in Figure 5D (right top). cDCS+memantine treatment induced DCS-LTD in both of these 2 channels, although the DCS-LTD magnitudes were different from each other (p < 0.001; see Fig 5D, right bottom).
In Vivo, ctDCS Induces Nonuniform SSEP Changes in Mouse S1
To test whether the opposing plastic changes across cortical layers occurred also in vivo after ctDCS, we recorded SSEPs in S1 barrel cortex following whisker electrical stimulation, before, during, and after ctDCS at 2 depths (400–600μm and 1,000–1,500μm from brain surface) in alert mice (Fig 6). Whisker stimulation reliably evoked a short-latency negative SSEP component (N1) in both deep and superficial layers. As in vitro, ctDCS induced distinct responses in the superficial and deep cortical layers (p < 0.001). During ctDCS, superficial N1 amplitude was decreased to 40.8 ± 6.8% in the last 5 minutes of stimulation compared to baseline (p < 0.05). This superficial layer N1 reduction (resembling in vitro DCS-LTD) persisted for 60 minutes after ctDCS (77.2 ± 4.4%, p < 0.001). In contrast, whereas deep-layer N1 was unchanged during ctDCS, a sustained N1 increase to 153.2 ± 9.2% in the first 5 minutes after ctDCS was observed and persisted as an LTP-like effect for 60 minutes after stimulation (132.0 ± 6.9%, p < 0.001).
FIGURE 6:
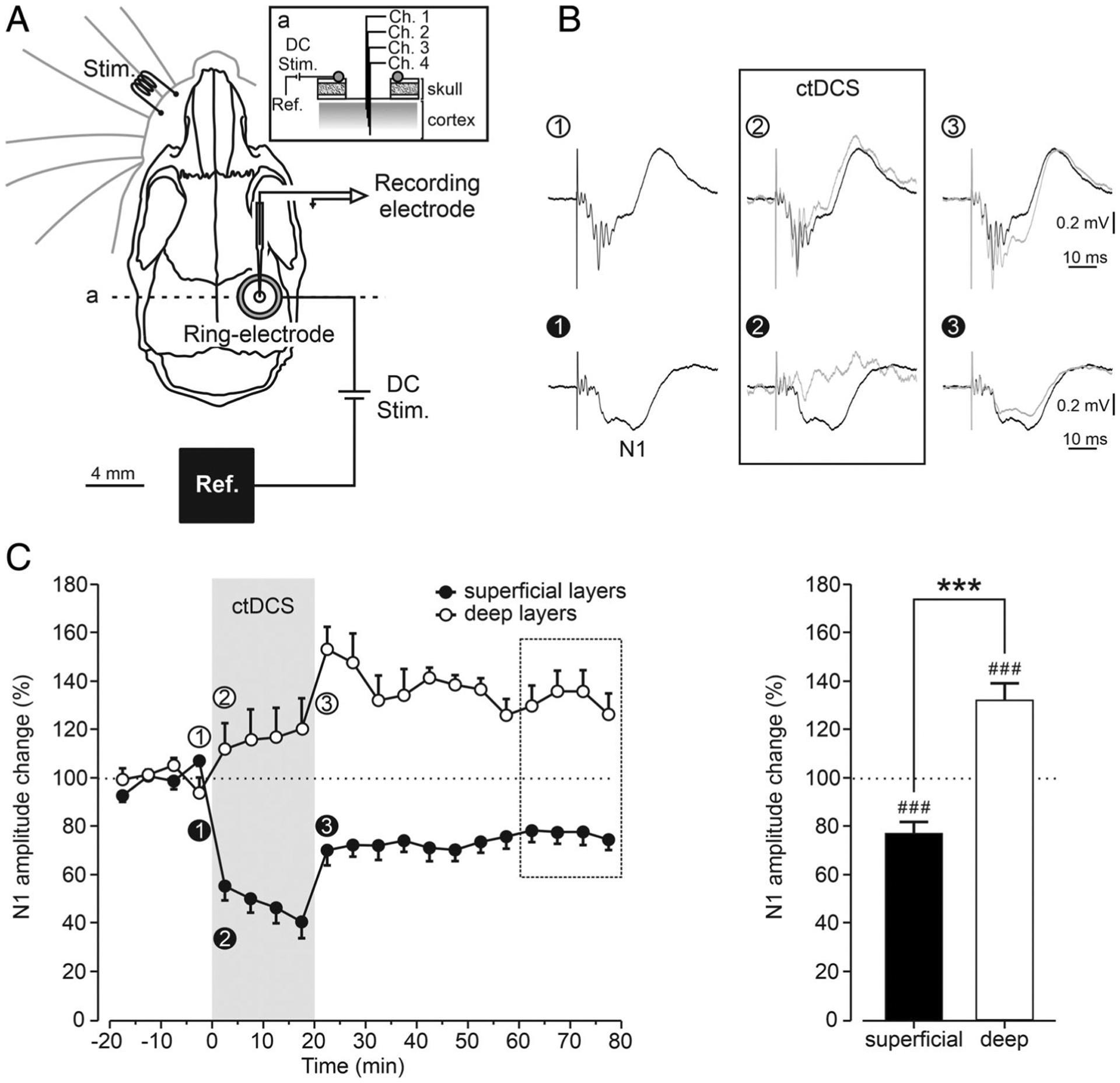
Somatosensory evoked potential (SSEP) modulation by cathodal transcranial direct current stimulation (ctDCS) recorded across the somatosensory cortex (S1) layers in vivo. (A) Illustration of the experimental design for SSEP recording and ctDCS in alert mice depicts electrode locations for ctDCS (DC Stim.), electrical whisker stimulation (Stim.), and SSEP recording from S1. ctDCS was applied between the active ring silver-chloride electrode above the right S1 and the reference (Ref.) electrode. After mapping the SSEPs by glass micropipette, a linear array of 4 recording electrodes (channels 1–4) was implanted in the appropriate S1 region corresponding to the somatosensory vibrissa representation, and the recording chamber was covered with dental cement (a: inset shows a sagittal view of the recording site). (B) Representative superficial (bottom) and deep (top) averaged SSEPs recorded from a mouse before (1), during (2), and just after (3) ctDCS are presented. To facilitate visual comparison, mean SSEPs during (gray trace) and after ctDCS (gray trace) have been superimposed with baseline SSEP (black trace) recorded just before ctDCS. (C) Time course of the effects induced by ctDCS on the normalized amplitude changes of superficial (n = 11, closed circles) and deep (n = 10, open circles) SSEPs chronically recorded across S1 (left in C). Mean normalized values were calculated for each 5-minute interval of data from 11 mice before (20 minutes), during (20 minutes), and after (60 minutes) ctDCS. ctDCS induced significant changes in the amplitude of superficial and deep SSEPs (###p < 0.001, 1-group t test for the last 20 minute of recording [dashed square box] from each superficial or deep recording and their baseline; right in C). Significant differences between superficial and deep SSEPs amplitude are also shown (***p < 0.001, unpaired t test between the 2 groups; right in C).
NMDAR Block Eliminates DCS-LTP in Deep S1 Layers In Vivo
With 300μM D-AP5, the reduction in superficial SSEPs was 58.1 ± 8.5% of baseline in the last 5 minutes of ctDCS (p < 0.05) and persisted for 60 minutes after ctDCS, averaging 81.4 ± 6.8% of baseline in the last 20 minutes of recording (p < 0.05; Fig 7A). In contrast, deep layer N1 amplitude potentiation was absent in the D-AP5 setting (n.s.). As with in vitro DCS, statistical analysis reveals a significant difference in ctDCS aftereffects between superficial and deep cortical layers (p < 0.05; see Fig 7D, left).
FIGURE 7:
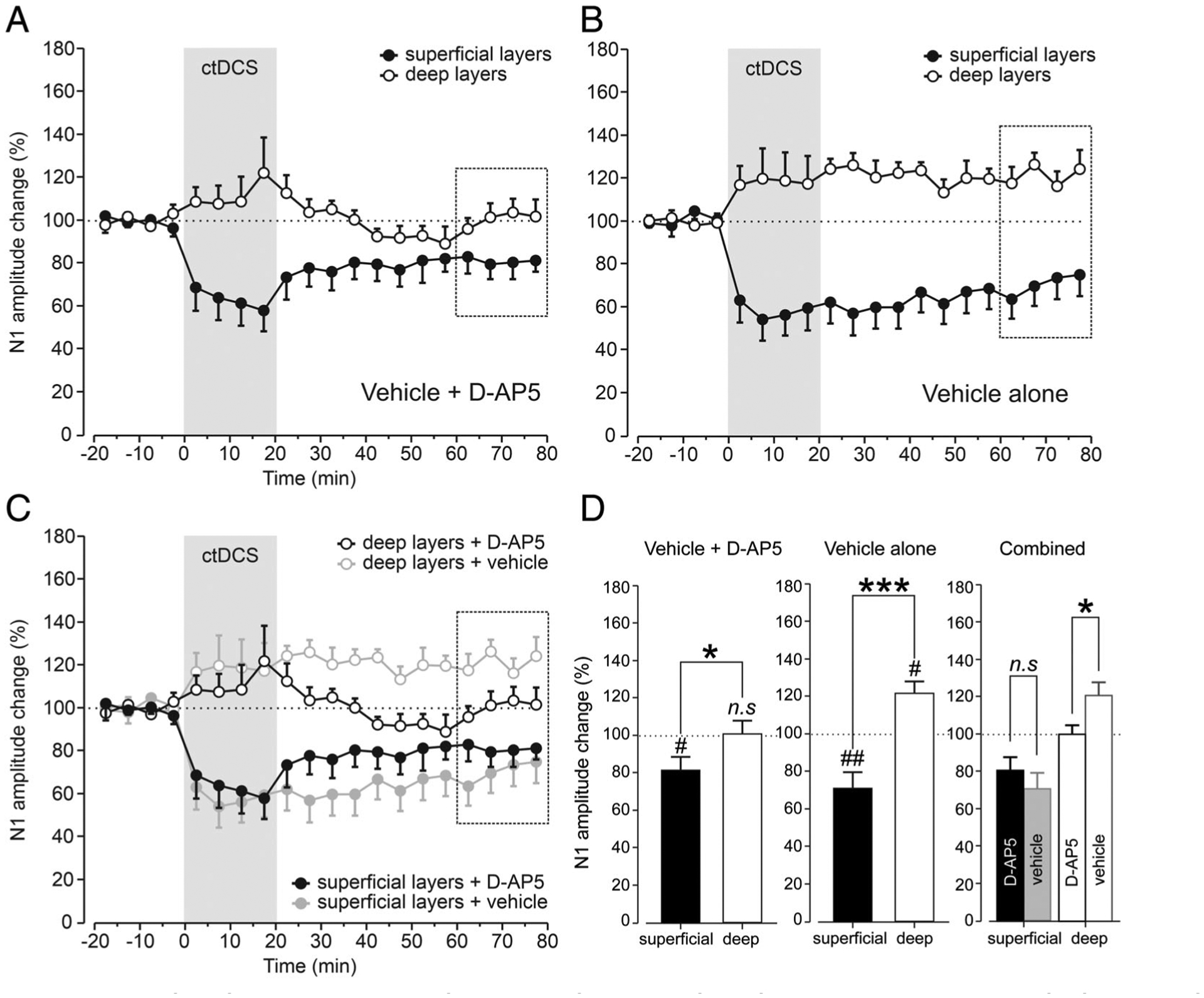
D-(−)-2-amino-5-phosphonopentanoic acid (D-AP5) eliminates deep layer somatosensory evoked potential (SSEP) enhancement by cathodal transcranial direct current stimulation (ctDCS) in vivo. Time course of ctDCS effects on superficial (closed circles) and deep (open circles) N1 amplitude in the presence of D-AP5 (A) or vehicle alone (B). Mean normalized values were calculated for each 5-minute interval of data from alert animals before (20 minutes), during (20 minutes), and after (60 minutes) ctDCS. (A) In contrast to the vehicle-only condition, ctDCS induced significant changes in the superficial (n = 9, p < 0.05, 1-group t test) but not deep N1 amplitude (n = 9, not significant [n.s.], p > 0.05, 1-group t test) in the presence of D-AP5, showing a significant difference in cDCS aftereffects in superficial somatosensory cortex (S1) layers as compared with those in deep layers (p < 0.05, unpaired t test). (B) ctDCS induced significant changes in the superficial (n = 9, p < 0.01, 1-group t test) and deep N1 amplitude (n = 9, p < 0.05, 1-group t test) in the presence of vehicle alone, showing a significant difference in ctDCS aftereffects in superficial S1 layers as compared with those in deep layers (p < 0.001, unpaired t test). (C) ctDCS aftereffects in the presence of D-AP5 and vehicle alone are superimposed to facilitate the comparison between these 2 conditions. Significant differences were found in the N1 amplitude of deep SSEPs after ctDCS between vehicle alone and D-AP5 conditions (n = 9, p < 0.05, unpaired t test), but not for superficial layers (n = 9, p > 0.05, unpaired t test). (D) Statistical analysis of vehicle+D-AP5 (left) and vehicle alone (middle) conditions is presented. A combined bar graph is also shown (right) to compare these 2 conditions; n.s. (p > 0.05), #p < 0.05, and ##p < 0.01 indicate 1-group t test for the last 20 minutes of recording (dashed square boxes) from each superficial or deep recording and their baseline; *p < 0.05, ***p < 0.001, and n.s. (p > 0.05) indicate unpaired t test between the 2 groups.
The ctDCS results in the D-AP5 setting contrasted with those of vehicle-only, replicating the above shown in vitro DCS-LTD–like (p < 0.01) and DCS-LTP–like aftereffects (p < 0.05) in superficial and deep layers, respectively (see Fig 7B and D, middle). Notably, with D-AP5 treatment, significant N1 amplitude change relative to vehicle was detected only in deep (p < 0.05) but not superficial layers (n.s.; see Fig 7C, right and D). This complements the in vitro data that indicate a more pro-found effect of NMDAR block on deep cortical layers in the DCS setting.
Antiseizure Capacity of ctDCS Coupled with NMDAR Block in Anesthetized Mice
Because NMDAR block abolished DCS-LTP and facilitated DCS-LTD across cortical layers (see Figs 4, 5, and 7), we tested whether ctDCS and drug coupling enhances the antiepileptic effect of ctDCS,12 which we hypothesized would be improved with enhanced depression of cortical excitability in the NMDAR block setting. We thus measured the antiepileptic effect of ctDCS pretreatment with or without NMDAR block by memantine.
KA injection resulted in electrographic seizures in all mice, except in only 1 in the ctDCS+memantine group. Latency to first seizure following KA injection was distinct among all groups (p < 0.01), with post hoc analysis showing significant delay by ctDCS+memantine pairing relative to sham (p < 0.01), and also relative to ctDCS alone (p < 0.01; Fig 8). Furthermore, seizure frequency was significantly different among groups (Kruskal–Wallis, p < 0.05), with post hoc analysis indicating significant reduction in the ctDCS+memantine group (median = 17 seizures/h) relative to sham (median = 121 seizures/h, p < 0.05) and ctDCS alone (median = 112 seizures/h, p < 0.01). Likewise, cumulative ictal time after KA injection varied significantly among all treatment groups (Kruskal–Wallis, p < 0.01), with post hoc comparisons indicating significant ictal time reduction in the ctDCS +memantine combination treatment group (median = 2.3 minutes/h) in comparison with sham (median = 15.5 min/h, p < 0.05) and ctDCS alone (median = 15.7 min/h, p < 0.01). Thus, ctDCS+memantine coupling not only delayed the onset of epileptic activity but also resulted in fewer seizures.
FIGURE 8:
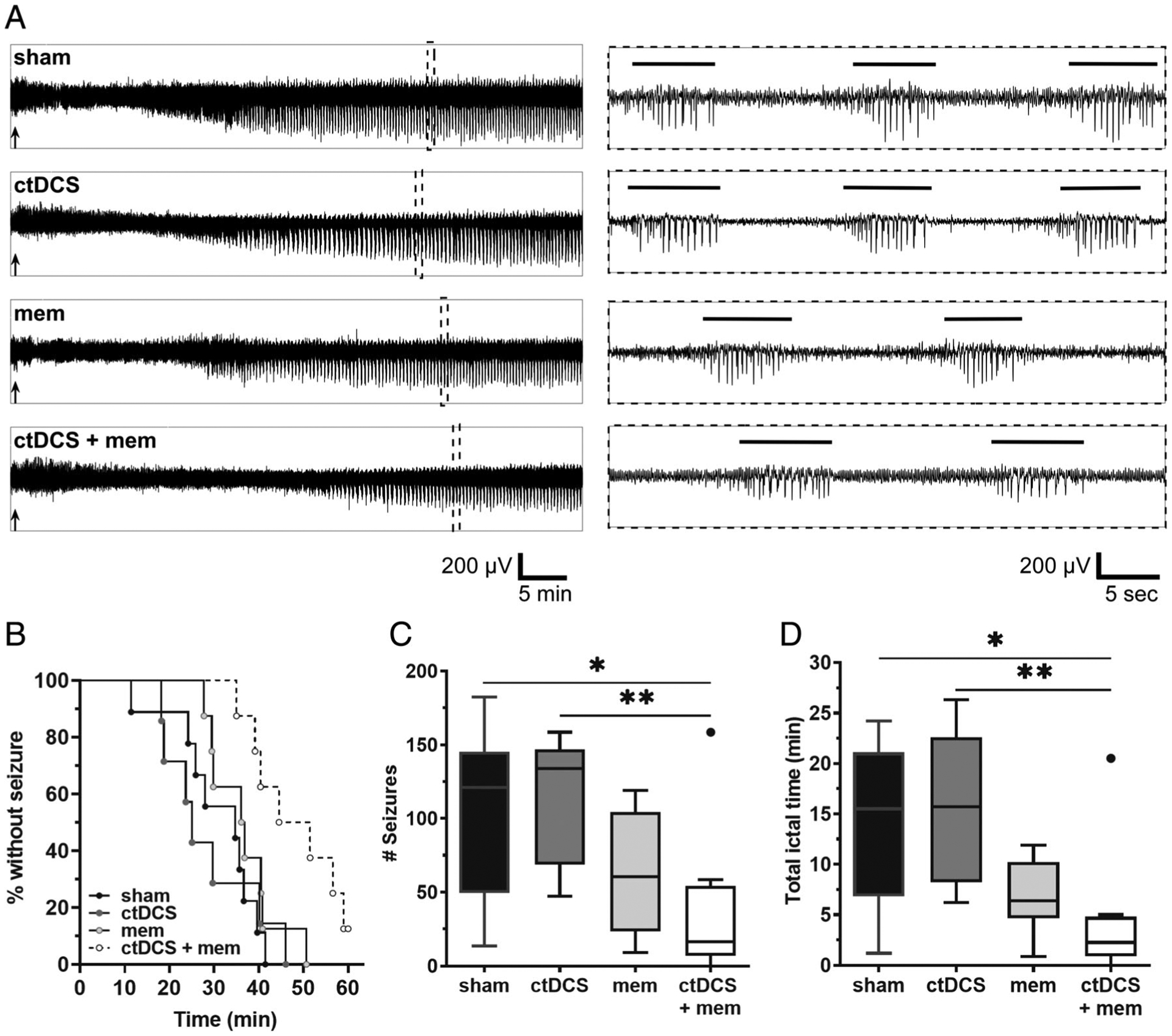
Cathodal transcranial direct current stimulation (ctDCS) and N-methyl-D-aspartate-type glutamate receptor antagonist (memantine [mem]) coupling improves kainate (KA)-induced seizure outcomes. Scalp electroencephalographic (EEG) recordings after KA (20mg/kg, intraperitoneal [IP]) administration reveal a strong effect on seizure onset among mice treated with sham (n = 9), ctDCS (n = 7), mem-only (2.5mg/kg, IP; n = 8), and ctDCS+mem (n = 8). (A) Left panel shows 60-minute representative EEG traces in each treatment group recorded after KA injection. Black arrows indicate timing of KA injection. In the right panel, dotted inserts represent 45-second traces comprising marked spike-trains (≥5 seconds) from their respective left panel traces. Horizontal bars above EEG indicate marked spike-train events that are automatically detected and manually validated. (B) Kaplan–Meier survival curve displays percent incidence of mice without seizure (y-axis) and latency to first seizure (x-axis), after KA injection. Curve comparison shows a significant difference among groups (p < 0.01, log-rank Mantel–Cox test), with clear separation of the ctDCS+mem group from others. (C) Significant reduction in total number of seizures per 60 minutes was found in the ctDCS+mem group relative to sham (*p < 0.05, Kruskal–Wallis test with Dunn post hoc) and relative to ctDCS alone (**p < 0.01). (D) Total ictal time per 60 minutes also showed significant reduction in the ctDCS+mem group compared to sham (*p < 0.05, Kruskal–Wallis test with Dunn post hoc) and compared to ctDCS alone (**p < 0.01). Boxes indicate median and first and third quartile. Tukey error bars are indicated by top and bottom whiskers. An automatically detected outlier value beyond Tukey’s error range is represented by a solid circle in C and D.
Discussion
We describe a depth-dependent c(t)DCS aftereffect pattern in vitro and in vivo, which is both cortical layer– and molecular pathway–specific. Our human tissue in vitro data also reveal strong similarities between cDCS aftereffects in mouse and human neocortex. Although the human tissues were isolated from patients with epilepsy, hence not necessarily representative of normal human cortex, and human slice orientation of cortical layering was less predictable (as tissue was often dysplastic), the findings are consistent with the animal results and provide translational insights regarding cDCS aftereffect in human cortex. Because a uniform DCS-LTD effect across cortical layers was achieved by blocking NMDARs, and this corresponded to improved seizure control in our model, we suggest the potential for NMDAR antagonism to enhance ctDCS efficiency in clinical applications.
We note that essential early DCS work identified distinct acute DCS effects as a function of depth in the cat cortex,26 but inhomogeneity of long-term modulation of cortical excitability as a function of cortical depth has not been described. Similarly, consistent with distinct DCS effects identified over a range of sulcal depths in prior work, likely reflecting distinct DC field vectors relative to the cortical surface,11,27 our data demonstrate simultaneous activation of LTD-like and LTP-like mechanisms within the same small cortical volume at a fixed stimulation intensity (in contrast to variability of the tDCS effect as a function of varying settings28,29).
Mechanisms that might explain the observed inhomogeneous c(t)DCS aftereffects on cortical excitability include a variant of the cosine hypothesis.11,27 That is, c(t)DCS effects depend on the orientation of the neuronal projections relative to the DC field vector.5,11,30 Our previous in vitro DCS studies in rat hippocampal slices showed that a constant DC field oppositely changes neuronal excitability of mossy-fiber-to-CA3 and Schaffer collaterals-to-CA1 pathways, whose axonal projections are approximately oriented in opposite directions.5 Now in the neocortex, we demonstrate that cDCS depresses excitability in the layer-5-to-layer-2/3 pathway, but potentiates excitability in the layer-5-to-layer-6 pathway. Notably, this is in contrast to completely uniform potentiation or depotentiation of the fEPSP by pharmacologic manipulation of our slice setup.
The opposing neuromodulation by cDCS may have resulted from the oppositely oriented cortical axonal pathways that were exposed to the DC field. However, investigation is needed to determine which cell type(s) and synaptic pathway(s) were stimulated, and whether distinct cellular elements might respond differently to the same c(t)DCS exposure.31 In this regard, we previously found4 that DCS-LTD persisted despite bicuculine (γ-aminobutyric acid type A receptor antagonist) administration. Furthermore, DCS-LTD was abolished with mGluR5 antagonism, and augmented with mGluR5 facilitation. Thus, we cautiously conclude that modification of excitatory synaptic strength, at the axon–dendrite interface, accounts for the majority (although likely not the entirety) of the ctDCS aftereffect.
Circuital arrangement may also explain the depth-dependent plastic effects. That is, c(t)DCS might inhibit both pyramidal and interneuron cells at superficial layers. Thus, c(t)DCS may induce a DCS-LTD of interneurons projecting to deep layers, and among these are inhibitory ionotropic serotonin receptor 3A/neuroglia form cells, vasoactive intestinal peptide cells, or parvalbumin/basket cells inhibiting deep layers via descending axons.32 Thus c(t)DCS may disinhibit, rather than potentiate, deep cortical layers. This scenario is supported in vivo by abrupt DCS-LTD observed in the superficial cortex and, in contrast, gradual DCS-LTP observed in deep layers; plausibly, this may reflect intracortical plastic changes triggered by ctDCS.
Both clinical trials33–37 and preclinical studies3,12,38 suggest prospects for ctDCS in the treatment of epilepsy, but with incomplete seizure suppression and overall inconsistent results39,40 such that participants remain with seizures (albeit in some trials reduced in frequency) after treatment.37 In addition to the role of ctDCS parameters (eg, stimulation location, intensity, duration, repetition frequency), which determine clinical efficacy,39 the inhomogeneous ctDCS aftereffects across the cortex that we demonstrate may contribute to the incomplete clinical effects in trials. Notably, incomplete depression of cortical excitability is even more likely in vivo in humans than in vitro,41 where tDCS field distribution is contingent on conductance of skin and subcutaneous or intracranial pathways, and is likely to vary in orientation relative to the cortical surface due to gyral patterns of cortical folding, all of which may result in more mixed modulatory effects by ctDCS.42
As demonstrated, a drug–device coupling approach to enhance the uniformity of ctDCS-mediated cortical suppression (and clinical efficacy) is appealing. In our prior study, the ctDCS antiepileptic effect was augmented by coadministration with lorazepam in a rat status epilepticus model.12 Our present findings show that pharmacologic NMDAR block not only facilitates DSC-LTD in human and mouse cortical slices, but also delays seizure onset when coupled with ctDCS in an in vivo seizure model. These results raise prospects for clinical use of NMDAR antagonists combined with ctDCS in treating epilepsy. For translational purposes, although specific memantine dosing schedules coupled with ctDCS will need exploration, the present dose (2.5mg/kg) is in excess of common daily dosing, but in the plausible range for acute human or animal administration.43–48
Furthermore, for translation of our results, we previously found that mGluR5-mTOR pathway activation is necessary for DCS-LTD effect, which otherwise persists despite NMDAR block.4 Separately, we and others identified that NMDAR activation is required for DCS-LTP.2 In the context of our present findings, we offer a hypothesis to explain why NMDAR block not only suppressed the DCS-LTP but also facilitates DCS-LTD in the deep layers: if neuronal orientation relative to the DC field determines the bias toward NMDAR or mGluR activation, then at any one time, both mechanisms may be competing in DCS. Thus, blockade of one (NMDAR, in our study) enables DCS-mediated activation of the other. In our setup, this would translate to unmasked mGluR5-mediated LTD in the deep cortical layers, where NMDAR block disabled DCS-LTP mechanisms. In this regard, in addition to targeting the NMDAR pathway to abolish the unwanted DCS-LTP effect, in follow-up experiments, we anticipate productive exploration of ctDCS combined with agonism or positive allosteric modulation of mGluR5-mTOR pathway to enhance the anti-epileptic effects of ctDCS.
Acknowledgment
This work was supported by grants from the Boston Children’s Hospital Translational Research Program (A.R.) and from the Spanish Ministry of Economy and Competitiveness (MINECO) and the European Fund for Regional Development (FEDER) (BFU2014-53820-P and BFU2017-89615-P) to J.M.-R. This work was also supported by the Repository Core for Neurological Disorders, Department of Neurology, Boston Children’s Hospital, and the Intellectual and Developmental Disabilities Research Center (NIH P30HD018655). A.R. has additional research support from the NIH (National Institute of Neurological Disorders and Stroke R01NS088583, and National Institute of Mental Health R01100186), the Smith Family Foundation, the Assimon Family Foundation, Sage Pharmaceuticals, Eisai Pharmaceuticals, Massachusetts Life Sciences, Neuroelectrics, and Brainsway. A.P.-L. was supported by the Sidney R. Baer Jr. Foundation, the Football Players Health Study at Harvard University, and Harvard Catalyst | Harvard Clinical and Translational Science Center (NIH National Center for Research Resources and National Center for Advancing Translational Sciences, UL1 RR025758).
We thank I. Cordones and G. Sánchez-Garrido for expert technical assistance and helpful discussions.
Footnotes
Potential Conflicts of Interest
Nothing to report.
References
- 1.Nitsche MA, Paulus W. Excitability changes induced in the human motor cortex by weak transcranial direct current stimulation. J Physiol 2000;527:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fritsch B, Reis J, Martinowich K, et al. Direct current stimulation promotes BDNF-dependent synaptic plasticity: potential implications for motor learning. Neuron 2010;66:198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang WP, Lu HC, Shyu BC. Treatment with direct-current stimulation against cingulate seizure-like activity induced by 4-aminopyridine and bicuculline in an in vitro mouse model. Exp Neurol 2015;265:180–192. [DOI] [PubMed] [Google Scholar]
- 4.Sun Y, Lipton JO, Boyle LM, et al. Direct current stimulation induces mGluR5-dependent neocortical plasticity. Ann Neurol 2016;80: 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabakov AY, Muller PA, Pascual-Leone A, et al. Contribution of axonal orientation to pathway-dependent modulation of excitatory transmission by direct current stimulation in isolated rat hippocampus. J Neurophysiol 2012;107:1881–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitsche MA, Liebetanz D, Antal A, et al. Modulation of cortical excitability by weak direct current stimulation—technical, safety and functional aspects. Suppl Clin Neurophysiol 2003;56:255–276. [DOI] [PubMed] [Google Scholar]
- 7.Nitsche MA, Paulus W. Sustained excitability elevations induced by transcranial DC motor cortex stimulation in humans. Neurology 2001;57:1899–1901. [DOI] [PubMed] [Google Scholar]
- 8.Monte-Silva K, Kuo MF, Liebetanz D, et al. Shaping the optimal repetition interval for cathodal transcranial direct current stimulation (tDCS). J Neurophysiol 2010;103:1735–1740. [DOI] [PubMed] [Google Scholar]
- 9.Marquez-Ruiz J, Leal-Campanario R, Sanchez-Campusano R, et al. Transcranial direct-current stimulation modulates synaptic mechanisms involved in associative learning in behaving rabbits. Proc Natl Acad Sci U S A 2012;109:6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefaucheur JP. A comprehensive database of published tDCS clinical trials (2005–2016). Neurophysiol Clin 2016;46:319–398. [DOI] [PubMed] [Google Scholar]
- 11.Ruffini G, Wendling F, Merlet I, et al. Transcranial current brain stimulation (tCS): models and technologies. IEEE Trans Neural Syst Rehabil Eng 2013;21:333–345. [DOI] [PubMed] [Google Scholar]
- 12.Dhamne SC, Ekstein D, Zhuo Z, et al. Acute seizure suppression by transcranial direct current stimulation in rats. Ann Clin Transl Neurol 2015;2:843–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knight ZA, Tan K, Birsoy K, et al. Molecular profiling of activated neurons by phosphorylated ribosome capture. Cell 2012;151: 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petr GT, Sun Y, Frederick NM, et al. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 2015;35: 5187–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duncan GE, Inada K, Koller BH, Moy SS. Increased sensitivity to kainic acid in a genetic model of reduced NMDA receptor function. Brain Res 2010;1307:166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gersner R, Dhamne SC, Zangen A, et al. Bursts of high-frequency repetitive transcranial magnetic stimulation (rTMS), together with lorazepam, suppress seizures in a rat kainate status epilepticus model. Epilepsy Behav 2016;62:136–139. [DOI] [PubMed] [Google Scholar]
- 17.McCord MC, Lorenzana A, Bloom CS, et al. Effect of age on kainate-induced seizure severity and cell death. Neuroscience 2008;154: 1143–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saito T, Sakamoto K, Koizumi K, Stewart M. Repeatable focal seizure suppression: a rat preparation to study consequences of seizure activity based on urethane anesthesia and reversible carotid artery occlusion. J Neurosci Methods 2006;155:241–250. [DOI] [PubMed] [Google Scholar]
- 19.Creeley C, Wozniak DF, Labruyere J, et al. Low doses of memantine disrupt memory in adult rats. J Neurosci 2006;26:3923–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhamne SC, Silverman JL, Super CE, et al. Replicable in vivo physiological and behavioral phenotypes of the Shank3B null mutant mouse model of autism. Mol Autism 2017;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL. DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology 1999;38:1577–1583. [DOI] [PubMed] [Google Scholar]
- 22.Huber KM, Roder JC, Bear MF. Chemical induction of mGluR5- and protein synthesis–dependent long-term depression in hippocampal area CA1. J Neurophysiol 2001;86:321–325. [DOI] [PubMed] [Google Scholar]
- 23.Palmer MJ, Irving AJ, Seabrook GR, et al. The group I mGlu receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 1997;36:1517–1532. [DOI] [PubMed] [Google Scholar]
- 24.Hess G Calcium-induced long-term potentiation in horizontal connections of rat motor cortex. Brain Res 2002;952:142–145. [DOI] [PubMed] [Google Scholar]
- 25.Turner RW, Baimbridge KG, Miller JJ. Calcium-induced long-term potentiation in the hippocampus. Neuroscience 1982;7:1411–1416. [DOI] [PubMed] [Google Scholar]
- 26.Creutzfeldt OD, Fromm GH, Kapp H. Influence of transcortical d-c currents on cortical neuronal activity. Exp Neurol 1962;5:436–452. [DOI] [PubMed] [Google Scholar]
- 27.Fox PT, Narayana S, Tandon N, et al. Column-based model of electric field excitation of cerebral cortex. Hum Brain Mapp 2004; 22:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Batsikadze G, Moliadze V, Paulus W, et al. Partially non-linear stimulation intensity-dependent effects of direct current stimulation on motor cortex excitability in humans. J Physiol 2013;591:1987–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosayebi Samani M, Agboada D, Jamil A, et al. Titrating the neuro-plastic effects of cathodal transcranial direct current stimulation (tDCS) over the primary motor cortex. Cortex 2019;119:350–361. [DOI] [PubMed] [Google Scholar]
- 30.Rawji V, Ciocca M, Zacharia A, et al. tDCS changes in motor excitability are specific to orientation of current flow. Brain Stimul 2018; 11:289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Molaee-Ardekani B, Marquez-Ruiz J, Merlet I, et al. Effects of transcranial direct current stimulation (tDCS) on cortical activity: a computational modeling study. Brain Stimul 2013;6:25–39. [DOI] [PubMed] [Google Scholar]
- 32.Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 2011;71:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auvichayapat N, Rotenberg A, Gersner R, et al. Transcranial direct current stimulation for treatment of refractory childhood focal epilepsy. Brain Stimul 2013;6:696–700. [DOI] [PubMed] [Google Scholar]
- 34.Fregni F, Thome-Souza S, Nitsche MA, et al. A controlled clinical trial of cathodal DC polarization in patients with refractory epilepsy. Epilepsia 2006;47:335–342. [DOI] [PubMed] [Google Scholar]
- 35.Faria P, Fregni F, Sebastiao F, et al. Feasibility of focal transcranial DC polarization with simultaneous EEG recording: preliminary assessment in healthy subjects and human epilepsy. Epilepsy Behav 2012;25:417–425. [DOI] [PubMed] [Google Scholar]
- 36.San-Juan D, Espinoza Lopez DA, Vazquez Gregorio R, et al. Transcranial direct current stimulation in mesial temporal lobe epilepsy and hippocampal sclerosis. Brain Stimul 2017;10:28–35. [DOI] [PubMed] [Google Scholar]
- 37.Yang D, Wang Q, Xu C, et al. Transcranial direct current stimulation reduces seizure frequency in patients with refractory focal epilepsy: a randomized, double-blind, sham-controlled, and three-arm parallel multicenter study. Brain Stimul 2020;13:109–116. [DOI] [PubMed] [Google Scholar]
- 38.Liebetanz D, Klinker F, Hering D, et al. Anticonvulsant effects of transcranial direct-current stimulation (tDCS) in the rat cortical ramp model of focal epilepsy. Epilepsia 2006;47:1216–1224. [DOI] [PubMed] [Google Scholar]
- 39.Nitsche MA, Paulus W. Noninvasive brain stimulation protocols in the treatment of epilepsy: current state and perspectives. Neuro-therapeutics 2009;6:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.San-Juan D, Morales-Quezada L, Orozco Garduno AJ, et al. Transcranial direct current stimulation in epilepsy. Brain Stimul 2015;8: 455–464. [DOI] [PubMed] [Google Scholar]
- 41.Bikson M, Inoue M, Akiyama H, et al. Effects of uniform extracellular DC electric fields on excitability in rat hippocampal slices in vitro. J Physiol 2004;557:175–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruffini G, Fox MD, Ripolles O, et al. Optimization of multifocal transcranial current stimulation for weighted cortical pattern targeting from realistic modeling of electric fields. Neuroimage 2014;89: 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lundbeck Canada. Product monograph: Memantine hydrochloride tablets 10 mg (N-methyl-D-aspartate (NMDA) receptor antagonist). 2016. Available at: https://www.lundbeck.com/upload/ca/en/files/pdf/pm/Ebixa.pdf. Accessed March 18, 2020.
- 44.Reisberg B, Doody R, Stoffler A, et al. Memantine in moderate-to-severe Alzheimer’s disease. N Engl J Med 2003;348:1333–1341. [DOI] [PubMed] [Google Scholar]
- 45.Stojiljkovic MP, Skrbic R, Jokanovic M, et al. Prophylactic potential of memantine against soman poisoning in rats. Toxicology 2019;416: 62–74. [DOI] [PubMed] [Google Scholar]
- 46.Tampi RR, van Dyck CH. Memantine: efficacy and safety in mild-to-severe Alzheimer’s disease. Neuropsychiatr Dis Treat 2007;3: 245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Marum RJ. Update on the use of memantine in Alzheimer’s disease. Neuropsychiatr Dis Treat 2009;5:237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winblad B, Poritis N. Memantine in severe dementia: results of the 9M-best study (benefit and efficacy in severely demented patients during treatment with memantine). Int J Geriatr Psychiatry 1999;14: 135–146. [DOI] [PubMed] [Google Scholar]