

Abstract
Activating transcription factor 6 (ATF6) is an endoplasmic reticulum stress responsive gene. We previously reported that conditional knockout of hepatic ATF6 exacerbated liver metabolic damage by repressing autophagy through mTOR pathway. However, the mechanism by which ATF6 influence liver metabolism has not been well established. Hydrogen sulfide (H2S) is a gaseous signaling molecule that plays an important role in regulating inflammation, and suppress nonalcoholic fatty liver in mice. Based on the previous study, we assumed that ATF6 may regulate H2S production to participate in liver metabolism. In order to clarify the mechanism by which ATF6 regulates H2S synthesis to ameliorate liver steatosis and inflammatory environment, we conducted the present study. We used the liver specific ATF6 knockout mice and fed on high-fat-diet, and found that H2S level was significantly downregulated in hepatic ATF6 knockout mice. Restoring H2S by the administration of slow H2S releasing agent GYY4137 ameliorated the hepatic steatosis and glucose tolerance. ATF6 directly binds to the promoter of cystathionine β synthetase (CBS), an important enzyme in H2S synthesis. Thus, ATF6 could upregulate H2S production through CBS. Sulfhydrated Sirtuin-1 (SIRT1) was downregulated in ATF6 knockout mice. The expression of pro-inflammatory factor IL-17A was upregulated and anti-inflammatory factor IL-10 was downregulated in ATF6 knockout mice. Our results suggest that ATF6 can transcriptionally enhance CBS expression as well as H2S synthesis. ATF6 increases SIRT1 sulfhydration and ameliorates lipogenesis and inflammation in the fatty liver. Therefore, ATF6 could be a novel therapeutic strategy for high-fat diet induced fatty liver metabolic abnormalities.
Keywords: Activating transcription factor 6 (ATF6), Cystathionine β synthetase (CBS), Sulfhydration, Non-alcoholic fatty liver disease (NAFLD), Hydrogen sulfide (H2S), Inflammation
Introduction
Endoplasmic reticulum (ER), as a vital membranous organelle, is closely involved in protein synthesis, folding and processing. However, research has verified that numerous metabolic stimuli and nutritional changes can contribute to the accumulation of unfolded and misfolded proteins, and finally result in ER stress by activating the cellular unfolded protein response (UPR) [1]. The liver, a predominantly metabolic organ, possesses abundant ER. Obviously, activation of the UPR in the liver plays a vital role in the regulation of glucose and lipid metabolism [2, 3].
Activating transcription factor 6 (ATF6), a transduction molecule in the downstream signal pathway of UPR, combines to the ER stress response element (ERSE) to regulate the homeostasis of ER and maintain cell functions. Previous studies showed that ATF6 not only inhibited gluconeogenesis, but also attenuated hepatic steatosis through interacting downstream regulators [4–6]. Our previous studies have reported that conditional knockout of hepatic ATF6 exacerbated liver metabolic damage mainly through mTOR pathway, which suggested that ATF6 may be a potential target for the remission of liver metabolic impairment [7]. However, the exact mechanism by which ATF6 affects the high-fat diet induced fatty liver metabolism is not well established.
Hydrogen sulfide (H2S), as an important gasotransmitter, is involved in various physiological and/or pathological processes including glucose metabolism and lipid synthesis ascribing to the formation of protein persulfides or protein S-sulfhydration [8–10]. The endogenous H2S is mainly generated by cystathionine β-synthase (CBS), cystathionine γ-lyase (CTH) and 3-mercaptopyruvate sulfurtransferase (MST), all of which are present in the liver [12]. Hepatic H2S metabolism dysfunction may disturb the liver metabolism and cause liver diseases [10, 11]. In the liver, a rich source of CBS involved in the H2S homeostasis [12]. It is reported that CBS is crucial to maintain the normal liver function [13]. CBS deficiency results in liver disorders by affecting the H2S function in hepatocytes under stress conditions [14]. Besides CBS, CTH together with H2S have been reported to be associated with hepatic lipid metabolism [15]. SREBPs (sterol regulatory element binding transcription factors) are master transcriptional proteins that regulate the expression of genes associated with lipid homeostasis. SREBP1 directly suppresses the expression of CTH, one of the main metabolic enzymes responsible for synthesis of H2S in the liver, and downregulates the ULK1 sulfhydration, blocks the autophagosomes with lysosomes and promotes hepatic steatosis [16]. SIRT1 is involved in the regulation of lipid and glucose metabolism in the liver, and associated with the development of NAFLD [17]. Zhou et al. reported that Resveratrol stimulated SIRT1 expression significantly, and increased deacetylation and inactivation of ATF6, and ameliorates lipid droplet accumulation in liver through SIRT1/ATF-dependent way [17]. However, the mechanism how H2S regulates liver lipid metabolism remains unclear.
Based on the previous study, we assumed that ATF6 may regulate H2S production to influence the liver metabolism, and conducted further investigation. In addition, the mechanism by which ATF6 regulates H2S synthesis to ameliorate liver steatosis, and the role of CBS and inflammatory factors in liver metabolism remains uncertain. In the present study, we demonstrated that H2S level was significantly downregulated in hepatic-specific ATF6 knockout mice, leading to hepatic steatosis and glucose tolerance. ATF6 directly binds to the promoter of CBS, and transcriptionally enhance CBS expression as well as H2S synthesis. Thus, we illustrated that ATF6 increases SIRT1 sulfhydration by H2S production, and ameliorates steatosis and inflammation in the fatty liver. Therefore, ATF6 could be a novel therapeutic strategy for high-fat diet induced fatty liver metabolic abnormalities.
Materials and methods
Animals, cell lines and plasmids
Animals
The C57BL/6J mice, B6(Cg)-Atf6tm1Hota/J mice and B6/JNjuAlbem1Cin(icre)/Nju mice were generated. The liver specific ATF6 knockout mice (hereinafter referred to as ATF6−/− mice) were generated by crossing B6(Cg) Atf6tm1Hota/J mice and B6/JNjuAlbem1Cin(icre)/Nju mice as previous reported [7]. All animals were housed and maintained on a 12 h light–dark cycle and on a regular unrestricted diet and free access to water. The male wildtype (WT) and ATF6−/− male mice were littermates and randomly selected. Since 8-week-old, the male mice were fed with high fat diet (HFD, 45% fat) for 8 weeks. At 8-week-old, there was no difference of the body weight among groups. At the beginning, the body weight in different groups were as follow, WT + HFD 25.86 ± 0.55 g, ATF6−/− + HFD 26.32 ± 0.65 g, and ATF6−/− + HFD + GYY 26.09 ± 0.84 g, (p > 0.05). The mice were administered with GYY4137 at 50 mg/kg/day by intraperitoneal injection for 4 weeks. Mice were fasted for 16 h, then scarified for serum and liver samples collection. All animal experiments were conducted under protocols approved by the Animal Research Committee of the Affiliated Hospital of Qingdao University. The agent of GYY4137 (CAS 106740-09-4) was commercial available and purchased from Santa Cruz Biotechnology (Dallas, Texas, US).
Analytical procedures
We used a glucometer (One Touch Ultra; LifeScan, Milpitas, CA, USA) to detect the blood glucose using capillary blood samples from mice tails. Serum and liver concentrations of triglyceride and cholesterol were collected after 16 h fasting, and detected by an automated Monarch device (The Affiliated Hospital of Qingdao University, Shandong, China).
Glucose and insulin tolerance test
Intraperitoneal Glucose tolerance test (ipGTT) was carried out after 16 h fasting. The mice were given glucose (1 g/kg) by intraperitoneal injection. The blood glucose was detected using glucometer at 0 min, 15 min, 30 min, 60 min, 90 min and 120 min. Insulin tolerance test (ITT) was conducted after 6 h fasting. The mice were given insulin (0.75 IU/kg) by intraperitoneal injection. Blood samples were collected and glucose levels were measured at 0 min, 15 min, 30 min, 60 min, 90 min and 120 min.
Histology
Liver tissue was extracted and fixed in 4% paraformaldehyde, then processed it for staining of paraffin-embedded sections. For histological evaluation, Hematoxylin–eosin (HE) staining was performed at the Central Research Laboratory, The Affiliated Hospital of Qingdao University following standard procedures.
Primary cell culture
Primary mouse hepatocytes were isolated from livers of male WT and ATF6−/− mice after 8 weeks HFD-feeding following the protocol. Briefly, mice were anesthetized, and livers were perfused with 0.5 mg/mL type II collagenase (Sigma–Aldrich) via the inferior vena cava to isolate hepatocytes. Primary hepatocytes were cultured in RPMI-1640 with 100 units/mL penicillin, and 0.1 mg/mL streptomycin, with serum free medium for the following investigation.
Cell lines
HepG2 cells were kind gifts from Professor Xiaopan Wu (State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, School of Basic Medicine, Peking Union Medical College, Beijing, China). HepG2 cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), and were maintained in 5% CO2 at 37 °C.
Plasmids and transfections
ATF6 overexpression plasmids and CBS promoter PGL3 plasmids were kind gifts from Professor Xiaopan Wu (State Key Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, School of Basic Medicine, Peking Union Medical College, Beijing, China). ATF6 overexpression plasmid was described as previously reported [18]. CBS promoter plasmid was constructed by Shanghai Generay Biotech Co., Ltd (Shanghai, China). The sequence of binding site of upstream promoter was predicted using Alibaba 2.1 (http://gene-regulation.com/pub/programs/alibaba2/index.html). Transfection experiments were performed using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s instructions.
Quantitative PCR
According to the manufacturer’s instruction, total RNA was extracted from the liver tissues and HepG2 cells by TRIzol reagent (Invitrogen, USA), and it was reversed transcribed into the cDNA in a 10 μL reaction volume by ReverTra Ace qPCR RT Master Mix (TOYOBO, Japan). The primers were synthesized as the report [19, 20], and the sequences of primers were listed as bellow. Mouse GAPDH-F, CAGCAACTCCCACTCTTCCA, GAPDH-R, CATGAGGTCCACCACCCTGT. Mouse ATF-6 F, GCCAGACTGTTTTGCTCTCT, ATF-6 R, CTGTCTTTCTGGTTGTCACC. CTH-F, TTGGATCGAAACACCCACAAA, CTH-R, AGCCGACTATTGAGGTCATCA. CBS-F, GCCATCAGACGAAGTCTGCAA, CBS-R, TGGTCCATCTCCAGGATGTGA. SREBP1c-F, GCGCAGATCGCGGAGCCAT, SREBP1c-R, CCCTGCCCCACTCCCAGCAT. The relative expression of targeted genes was normalized to internal control GAPDH and analyzed by the comparative Ct method.
Protein sulfhydration
The protein extracts were homogenized into RIPA lysis buffer, and 4 mL of blocking buffer was added (containing 225 mM HEPES–NaOH, pH 7.7; 0.9 mM EDTA; 0.09 mM Neocuproine; 2.5% SDS; 20 mM MMTS). Samples were kept at 50 °C for 20 min followed. Then the proteins precipitated out in 20 mL pre-ice acetone and were centrifuged for 5 min at 3100 rpm. After that, the pellets were washed with 70% acetone 2 times at room temperature, and resuspend them into HENS buffer (H250 mM EPES–NaOH, pH 7.7; 1 mM EDTA; 0.1 mM Neocuproine; 1%SDS) and 0.8 mM biotin-HPDP. The suspension liquid was rotated for 90 min at room temperature in dark, then the streptavidin resin (BBI, C006390-0005) was added into it and rotated overnight at 4 °C. The streptavidin resin was washed with HENS buffer and then centrifuged for 4 times at 4 °C for 5 min at 2500 rpm. The levels of protein in the samples were detected by western blotting.
Western blotting
The extracted proteins were separated by SDS-PAGE (1%) and transferred to a polyvinylidene fluoride membrane (Millipore, USA). After blocking with 5% non-fat milk in TBST at room temperature, the PVDF membranes were incubated with primary antibodies, anti-GAPDH (CWBIO, China), anti-ATF6 (Affinity, UK), anti-CBS (Santa Cruz, CA), anti-SIRT1 (Cusabio, China) overnight at 4 °C, followed by the incubation with the IgG (CWBIO, China) at room temperature for another 1 h. Micrographs were taken by the Tanon 5200 Multi (Shanghai, China).
Dual luciferase assay
HepG2 cells were seeded in 24-well plates and cultured for 24 h. The PGL3 plasmids without CBS promoter were used as the negative controls. Cells were co-transfected ATF6 expression vectors/empty vectors and CBS promoter PGL3 plasmids, and then harvested after 48 h. The luciferase signals were detected using Dual-Luciferase Reporter Assay System (Promega, USA) according to the manufacture’s protocol.
Electrophoretic mobility shift assay (EMSA)
According to the manufacturer’s instruction, EMSA was carried out by the Chemiluminescent EMSA Kit (Beyotime, GS009). The CBS promoter probe sequence was as below: agccttcatgatgtaactccatcc.
Chromatin immunoprecipitation (ChIP) assay
ChIP was carried out by EZ ChIPTM Chromatin Immunoprecipitation Kit (Merck, 92590) according to the manufacturer’s instruction. The cells were cross liked with formaldehyde. Subsequently the samples were subjected to sonication, and then incubated with anti-ATF6 antibody. The primers used for q-ChIP assay were as follow: sense, 5′-GACAGTCTCGCTCAGTCGCA-3′ and antisense, 5′-GAGGGGACAGGGATGGAGTT-3′.
H2S production measurement
H2S production measurement in cells
The H2S production was measured as previous reported [21]. We placed the microporous filter membrane with 1% zinc acetate on the inner side of the 12-well plates, and then washed the cells 3 times with PBS and cultured them in serum-free medium, in addition with L-cysteine (Santa Cruz Biotechnology, 168149) to normalize the concentration as 2 mM and 5-pyridoxal phosphate (BBI, PD0455) as 0.5 mM. The cells were cultured for 24 h at 37 °C with 5% CO2, followed by putting the filter in a test tube with addition of 3 mL ddH2O, 0.5 mL 0.2% N,N-dimethyl-pphenylenediamine-dihydrochloride solution (2.5 mM) and FeCl3 (3.3 mM) (Santa Cruz Biotechnology, F3629). Then the absorbance was detected at 670 nm by the microplate reader (Synergy H1; BioTek; Winooski, VT, USA). H2S measurement was calculated according to the standard curve, and presented as nM/(min 106 cells).
H2S production measurement in the liver of mice
The liver tissues (200 mg) were homogenized in the ice-cold passive lysis buffer (500 µL), and 100 µL homogenates were add into the 12-wells plates, together with 900 μL pre-cold PBS containing 5 mM L-cysteine and 2 mM pyridoxial 5-phosphate into it. The filter membrane soaked in 1% (v/v) zinc acetate solution was affixed to the inner side of the cover, and kept incubating for 24 h at 37 °C in a 5% CO2 atmosphere. The filter paper was put into centrifuge tube, and 3 mL ddH2O, 0.5 mL 0.2% N,N-dimethyl p-phenylenediamine solution, 50 µL 10% ammonium ferric sulfate were added and mixed. OD values were detected at 670 nm, and H2S measurement was calculated according to the standard curve, and presented as nM/(min 106 cells). Then the subsequent steps were performed as the procedure in cells.
Statistical analysis
The data were presented as means ± SD. The results were analyzed by Student’s t test or one-way ANOVA using SPSS 22.0. p < 0.05 was considered as statistically significant.
Results
ATF6 attenuates the HFD induced hepatic steatosis by inducing H2S accumulation
Previous studies showed that both ATF6 and H2S could moderate the fatty liver [7, 10]. In order to investigate the interaction between ATF6 and H2S in regulation of HFD induced fatty liver, the H2S level was examined in liver tissue in WT and ATF6−/− mice fed on HFD. The results showed that the H2S level was significantly decreased in ATF6−/− mice compared with WT mice (Fig. 1A), suggesting that the knockout of ATF6 reduces the H2S production. Subsequently, the same result was observed in primary mouse hepatocytes (Fig. 1B). Liver histology analysis suggested that ATF6−/− mice showed liver steatosis fed with HFD (Fig. 1C). In our previous study, we already have demonstrated that the liver specific knockout of ATF6 affects the glycolipid metabolism [7]. In current study, ATF6−/− mice showed glucose intolerance and impaired insulin sensitivity (Fig. 1D, E). To verify the effect of H2S on liver metabolism, we explore whether GYY4137, the slow-releasing H2S donor, could improve the state of glucose and lipid metabolism, and adipose accumulation on ATF6−/− deficiency mice model in HFD condition. As shown in Fig. 1D, after the treatment with GYY4137, the blood glucose level is significantly decreased and the glucose tolerance is improved. Accordingly, the serum and intracellular hepatic concentrations of triglyceride (TG) and total cholesterol (TC) are dramatically decreased compared to control mice (Fig. 1E). Taken together, these results supported the hypothesis that liver specific knockout of ATF6 exacerbates liver metabolic damage by reducing the endogenous H2S production.
Fig. 1.

ATF6 attenuates hepatic steatosis in fatty liver by inducing H2S accumulation. A H2S level was analyzed in liver tissue of WT and ATF6−/− mice fed on HFD. B H2S level was analyzed in primary mouse hepatocytes. C Histology analysis of liver in WT, ATF6−/−, and GYY4137 treated mice detected by HE stain (*200). D Glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were performed with/without GYY4137 treatment in ATF6−/− mice on HFD. E Liver and serum triglyceride and cholesterol levels in ATF6−/− mice with/without GYY4137 treatment. The asterisks (*) marked in the graph represents the significant differences (p* < 0.05; p** < 0.01; p*** < 0.001) in different groups. (n = 3–4 for each group)
ATF6 promotes CBS expression
CBS and CTH are the key H2S-metabolism enzymes in liver. First, we investigated the expression of CBS and CTH in ATF6−/− mice and WT mice on HFD, to explore the association between ATF6 and H2S-gererating enzymes. There was an obviously decreased mRNA level of CBS in liver in ATF6−/− mice compared with WT mice, while the CTH showed no significant differences between two groups (Fig. 2A). Subsequently, we also observed the similar result that CBS expression was markedly decreased in liver of ATF6−/− mice by western bolt (Fig. 2B). To further verify the effect of ATF6 on CBS, we constructed the ATF6 overexpression vector and then transfected into HepG2 cells. As expected, the mRNA levels of CBS were increased about 2.5 times along with the ATF6 overexpression. The western blot result also confirmed the same result (Fig. 2C, D). Thus, ATF6 may upregulate the expression of CBS, but the exact molecular mechanisms need further investigation.
Fig. 2.

ATF6 upregulates CBS expression. A The mRNA levels of CBS and CTH were analyzed by qPCR in liver tissues from WT and ATF6−/− mice fed with HFD. B The expression of CBS and CTH were evaluated by western bolt in liver tissues from WT and ATF6−/− mice. C The mRNA levels of CBS were analyzed by qPCR in HepG2 cells with ATF6 overexpression and control ones, respectively. D The expression of CBS was evaluated by western blot in HepG2 cells with ATF6 overexpression and control ones. The asterisks (***) marked in the graph represents the significant differences (p*** < 0.001) in two groups. (n = 5–6). WT wildtype, NC negative control
ATF6 upregulates the promoter activity and expression of the CBS gene
As a transcription factor, ATF6 can promote the transcription of downstream target genes to exert its function. Considering that ATF6 upregulates the expression of CBS, we proposed that ATF6 might affect CBS expression at transcriptional level. To examine the molecular mechanism, the CBS promoter PGL3 plasmids were constructed. HepG2 cells were co-transfected with a luciferase reporter construct harboring a human CBS gene promoter and ATF6. The results showed that overexpression of ATF6 enhance the luciferase activity of CBS promoter (Fig. 3A), indicating that ATF6 may bind directly with CBS promoter to force its expression. Furthermore, the prediction by online software AliBaba 2.1 (http://gene-regulation.com/pub/programs/alibaba2/index.html) shows that ATF6 possesses the binding motifs in the promoter region of CBS (Fig. 3B). Subsequently, the EMSA experiment was conducted to further explore whether ATF6 bind to CBS promoter. ATF6 contained in nuclear extracts specifically bound to the biotin-labeled 24 bp probe to form the protein-DNA complexes, while the excess unlabeled oligonucleotide competitor competed with the binding site (Fig. 3C). In addition, for the binding reaction with the specific antibodies against ATF6, the super shift belt was observed. Finally, the ChIP-qPCR was performed to support the results (Fig. 3D). Taken together, we conclude that ATF6 could enriched on the promoter regions of CBS. These results indicate that CBS was the downstream target gene that upregulated by ATF6.
Fig. 3.

ATF6 upregulates the promoter activity and expression of the CBS gene. A HepG2 were co-transfected with the CBS promoter luciferase plasmids and ATF6 plasmids. Luciferase assays of CBS were performed. B The binding sites of ATF6 was predicted by AliBaba 2.1. C EMSA assays was carried out to explore the Binding affinity of ATF6 to biotin-labeled probe of CBS. D The ChIP-qPCR assay was performed to detect the ATF 6 enrichment level on the CBS promoter. The asterisks (***) marked in the graph represents the significant differences (p*** < 0.001) in two groups
ATF6 promotes Sirtuin-1 (SIRT1) sulfhydration
It was reported that SIRT1, as an essential regulator in hepatic lipid metabolism, could be direct sulfhydrated by H2S [22, 23]. The sulfhydrated SIRT1 was assessed in vitro and in vivo in present study by modified biotin switch (S-sulfhydration) assay to investigate the effect of ATF6 on SIRT1 signal. As shown in Fig. 4A, the sulfhydrated SIRT1 was significantly reduced in ATF−/− mice. The upregulated sulfhydrated SIRT1 was observed in HepG2 cells with ATF6 overexpression. To explore the effect of H2S on SIRT1, the de-sulfhydration reagent, dithiothreitol (DDT) was used. The results showed that the sulfhydration was blocked after DDT pre-administration (Fig. 4B). Taken together, the results indicated that ATF6 may induce the SIRT1 sulfhydration by H2S signal.
Fig. 4.

ATF6 can promotes SIRT1 sulfhydration by H2S signal and regulate the inflammation response by mediating IL-10/IL-17A. A Effect of ATF6 on the protein expression of SIRT1 in vivo. B Effect of ATF6 on the protein expression of SIRT1 in vitro. C Effect of ATF6 on the mRNA expression of SREBP in ATF6−/− mice. D Effect of ATF6 on the mRNA expression of SREBP in HepG2 with ATF6 overexpression. E SREBP mRNA expression with DTT treatment. F The ATF6 effect on inflammatory cytokines. The asterisks (*) marked in the graph represents the significant differences (p* < 0.05; p*** < 0.001) in different groups. (n = 9 for each group). WT wildtype, NC negative control
The sterol regulatory element-binding proteins (SREBP1c), as the downstream gene of SIRT1, has been proven to be the essential target of SIRT1 in regulating lipid metabolism [23]. Thus, we further investigated whether the SREBP1c expression was changed along with ATF6 levels. The results showed that the mRNA levels of SREBP1c increased in ATF−/− mice compared with WT mice, while overexpressed ATF6 reduced SREBP1c mRNA levels (Fig. 4C, D). Subsequently, with DTT pretreatment to eliminate sulfhydration effect on SIRT1, there were no significant difference of SREBP1c mRNA levels between WT group, ATF6 overexpression group and GYY4137 co-incubation group (Fig. 4E). These data consistently showed that ATF6 could induce SIRT1 sulfhydration by H2S signal. Inflammatory cytokines play an important role in regulating liver lipid metabolism [24]. To explore the effect of ATF6 on cytokines and inflammation environment, the mRNA expression of the anti-inflammatory cytokine IL-10 (Interleukin-10) and pro-inflammatory cytokine IL-17A (Interleukin-17A) were tested in vivo subsequently. As shown in Fig. 4F, the anti-inflammatory cytokine IL-10 was downregulated, while the pro-inflammatory cytokine IL-17A was upregulated in ATF6 knockout mice, which indicated that ATF6 might regulate the inflammation response in liver (Fig. 5).
Fig. 5.
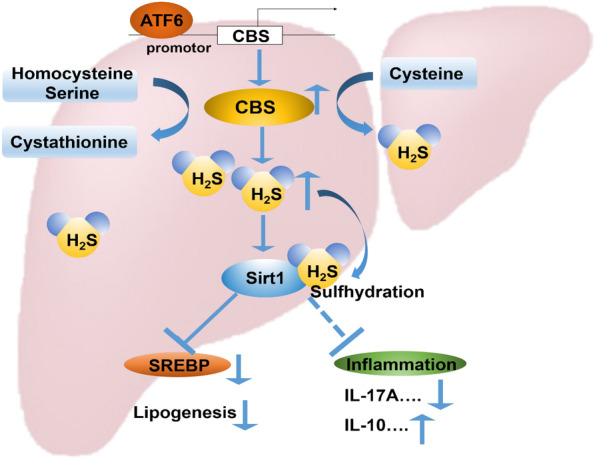
ATF6 increase Sirt1 sulfhydration by promoting H2S production through upregulating CBS expression. ATF6 binds to the promotor region of CBS, a vital H2S producing enzyme in the liver, and promote the protein transcription. The upregulation of CBS enhances H2S production. Accumulation of H2S stimulates Sirt1 sulfhydrylation, then inhibits its downstream SREBP expression, to reduce lipogenesis and fatty acid beta-oxidation. In addition, Sirt1 sulfhydrylation decrease expression of pro-inflammatory factor IL-17A, and increase anti-inflammatory factor IL-10 expression. ATF6 could mitigate the liver lipogenesis and inflammation by stimulating sulfhydrylated Sirt1 through CBS/H2S pathway
Discussion
ATF6 act as the essential ER stress responsive regulator. Three key molecules involved in the ER stress-induced adaptive response, including IRE1α (endoribonuclease inositol-requiring enzyme 1-alpha), PERK (protein kinase RNA-like endoplasmic reticulum kinase) and ATF6 [2]. ER stress is recognized by those essential regulators, and then activate the downstream signal cascade. The major ER chaperone binding-immunoglobulin protein (BIP) binds to the ER-oriented parts of PERK and IRE1α. Active PERK phosphorylates the eukaryotic translation initiation factor 2 (eIF2), resulting in the selective inhibition of translation, effectively reducing ER client protein load. IRE1α autophosphorylates and activates its endonuclease domain, resulting in the cleavage of Xbp-1 to generate a shortened Xbp-1 isoform, which drives the production of various ER chaperones to restore ER homeostasis [25]. Upon activation by dissociation of BIP, ATF6 translocates into the Golgi apparatus where it is cleaved into the functional form. ATF6 can be regulated by XBP1, suggesting interactions between the IRE1α and ATF6 branches of the UPR, then increase ER capacity, and regulate cell survival [26].
ATF6 play vital role in a variety process of physiological and pathological conditions. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling in the cardia myocytes [26]. In the cellular model of inflammatory bowel diseases, ATF6 increases expression of inflammatory cytokines CXCL1 and tumor necrosis factor (TNF) in response to ER stress [27].
ATF6 may attenuate HFD-induced fatty liver and hepatic steatosis, and it has been verified that liver specific knockout of ATF6 exacerbated HFD-induced hepatic steatosis and glucose intolerance leading to liver metabolic damage [7]. Hepatic specific deficiency of ATF6 exacerbates liver metabolic damage by repressing autophage through mTOR pathway [7]. ATF6 attenuate hepatic steatosis by increasing fatty acid oxidation through peroxisome proliferator-activated receptor α (PPAR α) [28]. Thus, investigating the mechanism of ATF6 influencing liver metabolism may be helpful for the liver disease treatment. In the present study, we concluded that ATF6 could medicate the CBS/H2S signals by sulfhydrating SIRT1 to ameliorate the liver inflammation. However, the deep molecular mechanism by which ATF6 attenuates hepatic steatosis and glucose homeostasis need further investigation in the future.
H2S has been widely recognized as a novel gasotransmitter exerting physiological, regulatory or modulatory effects in mammalian cells and tissues by sulfhydration [9, 29]. H2S plays a key role in the process of autophagy in a variety of cells and tissues. In cardiomyocytes, H2S inhibits autophagy through the PI3K/SGK1/GSK3β signaling pathway [30]. In the model of renal ischemia–reperfusion injury, H2S inhibits autophagy via mediating scavenger receptor A signaling pathway, thereby improving endothelial cell dysfunction [31]. CBS, as the vital H2S-producing enzyme, participates in balancing the production and elimination of H2S. It has been reported that H2S may exert S-sulfhydrylation on various proteins by targeting the cysteine residues [32]. Accumulating evidences identified that SIRT1, which plays a vital beneficial role in liver lipid metabolism by regulating lipogenesis and fatty acid β-oxidation, could be sulfhydrated by H2S to promote its activity [22, 23]. Here, we question whether ATF6 interacts with CBS to exert its effect on H2S production, which further affect the SIRT1 activation by sulfhydrylation. In this study, we observed that the ATF6 indeed affects the SIRT1 sulfhydrylation. Specifically, after treatment with DDT, the sulfhydrylation of SIRT1 was completely blocked. Thus, ATF6 may induce the SIRT1 sulfhydrylation by CBS/H2S pathway. In addition, SIRT1, as a vital metabolic sensor, can downregulate the expression of SREBP1, a transcription factor participating in lipid synthesis [22]. In present study, we found that ATF6 exhibited negative regulation on SREBP mRNA expression. Especially, after treatment with DTT, the effect of ATF6 on SREBP disappeared. These results suggest that ATF6 may decrease SREBP expression by SIRT1 sulfhydrylation. Previous study showed that ATF6 attenuated SREBP2-related lipogenesis. Glucose deprivation caused the proteolytic cleavage of ER stress transducer ATF6, and the cleaved ATF6 translocated into nucleus binding to SREBP2, and recruited HDAC1, thus, the SREBP2-mediated lipogenesis and gene transcription were downregulated [33]. In addition, ER Stress Induces cleavage of ATF6 by the same proteases that process SREBPs ATF6 is processed by site-1 protease (S1P) and Site-2 protease (S2P), the enzymes processing SREBPs in response to cholesterol deprivation. [34]. In our study, further investigation was needed to clarify the molecular mechanism by which ATF6 regulates SREBP expression through SIRT1 sulfhydrylation.
Liver is not only an essential metabolic organ, but also an important immunological activity site. Accumulating evidence showed that there was a crosstalk interaction between inflammatory response and ER stress in liver disease [22]. Recent study showed that inhibition of ER stress ameliorated the inflammation in liver ischemia reperfusion injury [35]. In the acute liver injury, ATF6 upregulated macrophage-derived cytokines IL-1α expression and promoted liver fibrogenesis [36]. As the ER stress responsive gene, whether ATF6 act on the inflammatory mediation was explored. Our results suggested that ATF6 deficiency downregulated the anti-inflammatory cytokine IL-10, while upregulated the pro-inflammatory cytokine IL-17A expression in mice model. Thus, ATF6 may ameliorate inflammation by regulating the inflammatory cytokines in the fatty liver. The cytokines most commonly associated with hepatic disorders are the pro-inflammatory cytokines TNF-α, IL-1, IL-6, and transforming growth factor (TGF)-β. Those cytokines are also metabolic signals trigger this transit, where it is cleaved by S1P and S2P processing enzymes [37]. In the ischemia–reperfusion injury due to the metabolic stress in liver, the production of pro-inflammatory cytokines increased, while anti-inflammatory cytokine decreased. ATF6 altered the responsiveness against Toll-like receptor (TLR) stimulation during this process, and mediates a pro-inflammatory synergy [38].
There are several limitations and shortcomings of the present study. First, in this study, we demonstrated that GYY4137, the H2S releasing agent, ameliorated the glucose intolerance and lipid metabolism abnormalities on ATF6−/− mice fed with HFD, suggesting that ATF6 attenuates hepatic steatosis in fatty liver by inducing H2S accumulation. However, we did not investigate the effect of GYY4137 on WT mice fed on normal diet or HFD. Second, our results indicated that ATF6 promote SIRT1 signal and regulate the inflammatory response in liver steatosis. Several inflammatory factors are involved in the process of inflammatory response in fatty liver. The investigation of anti-inflammatory factor IL-10 and pro-inflammatory factor IL-17A was limited. Third, we suggested that ATF6 downregulated the expression of SREBP via SIRT1. However, the molecular mechanism of regulation was unclear. Further investigation is needed to clarify those issue.
Continued on the previous study, we identified that ATF6 ameliorates liver steatosis and inflammation by upregulating CBS expression and H2S synthesis via SIRT1 sulfhydration. It would provide new insight for the development of potential therapeutic strategy for fatty liver.
Conclusion
In conclusion, ATF6 increases SIRT1 sulfhydration by promoting H2S production through transcriptionally upregulating CBS expression, meanwhile it ameliorates the inflammation by regulating the inflammatory cytokines in the liver.
Acknowledgements
We thank Prof. Xiaopan Wu (Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences and Peking Union Medical College, China) for giving ATF6 overexpression and CBS promoter PGL3 plasmids as kind gifts.
Abbreviations
- ATF6
Activating transcription factor 6
- UPR
Unfolded protein response
- GTT
Glucose tolerance test
- ITT
Insulin tolerance test
- CBS
Cystathionine β synthetase
- H2S
Hydrogen sulfide
- SIRT1
Sirtuin-1
- ER
Endoplasmic reticulum
Author contributions
DB performed the study, analyzed the data and wrote the paper. SY, CB, XY and LW provided helpful discussion and suggestions. CB participated in writing the manuscript. SX summarized all results and directed the whole study. All authors read and approved the final manuscript.
Funding
This work was supported by the grants from National Natural Science Foundation of China (Grant No. 81800769) to SX, the National Natural Science Foundation of China (Grant No. 82370890) to DB, and The Taishan Scholars Program of Shandong Province and Natural Science Foundation of Shandong Province (Grant No. ZR2023MH243) to DB.
Availability of data and materials
The datasets analyzed in the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All animal experiments were conducted under protocols approved by the Animal Research Committee of the Affiliated Hospital of Qingdao University.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 2.DeZwaan-McCabe D, Sheldon RD, Gorecki MC, et al. ER stress inhibits liver fatty acid oxidation while unmitigated stress leads to anorexia-induced lipolysis and both liver and kidney steatosis. Cell Rep. 2017;19(9):1794–1806. doi: 10.1016/j.celrep.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–2153. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Vera L, Fischer WH, et al. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460:534–537. doi: 10.1038/nature08111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Usui M, Yamaguchi S, Tanji Y, et al. Atf6a-null mice are glucose intolerant due to pancreatic b-cell failure on a high-fat diet but partially resistant to diet induced insulin resistance. Metabolism. 2012;61:1118–1128. doi: 10.1016/j.metabol.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto K, Takahara K, Oyadomari S, et al. Induction of liver steatosis and lipid droplet formation in ATF6a-knockout mice burdened with pharmaco- logical endoplasmic reticulum stress. Mol Biol Cell. 2010;21:2975–2986. doi: 10.1091/mbc.E09-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun XF, Li W, Deng YJ, et al. Hepatic conditional knockout of ATF6 exacerbates liver metabolic damage by repressing autophage through MTOR pathway. Biochem Biophys Res Commun. 2018;505:45–50. doi: 10.1016/j.bbrc.2018.09.047. [DOI] [PubMed] [Google Scholar]
- 8.Olas B. Hydrogen sulfide in signaling pathways. Clin Chim Acta. 2015;439:212–218. doi: 10.1016/j.cca.2014.10.037. [DOI] [PubMed] [Google Scholar]
- 9.Zhang DH, Macinkovic I, Devarie-Baez NO, et al. Detection of protein S-sulfhydration by a tag-switch assay. Angew Chem Int Ed Engl. 2014;53:575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mani S, Cao W, Wu L, et al. Hydrogen sulfide and the liver. Nitric Oxide. 2014;15:62–71. doi: 10.1016/j.niox.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Watanabe M, Osada J, Aratani Y, et al. Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci USA. 1995;92:1585–1589. doi: 10.1073/pnas.92.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang R. Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev. 2012;92:791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 13.Robert K, Nehme J, Bourdon E, et al. Cystathionine beta synthase deficiency promotes oxidative stress, fibrosis, and steatosis in mice liver. Gastroenterology. 2005;128:1405–1415. doi: 10.1053/j.gastro.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 14.Teng H, Wu B, Zhao K, et al. Oxygen-sensitive mitochondrial accumulation of cystathionine beta-synthase mediated by Lon protease. Proc Natl Acad Sci USA. 2013;110:12679–12684. doi: 10.1073/pnas.1308487110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ali A, Zhang YJ, Fu M, et al. Cystathionine gamma-lyase/H2S system suppresses hepatic acetyl-CoA accumulation and nonalcoholic fatty liver disease in mice. Life Sci. 2020;252:117661. doi: 10.1016/j.lfs.2020.117661. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen TTP, Kim DY, Im SS, Jeon TI. Impairment of ULK1 sulfhydration-mediated lipophagy by SREBF1/SREBP-1c in hepatic steatosis. Autophagy. 2021;17(12):4489–4490. doi: 10.1080/15548627.2021.1968608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou R, Yi L, Ye X, Zeng X, Liu K, Qin Y, Zhang Q, Mi M. Resveratrol ameliorates lipid droplet accumulation in liver through a SIRT1/ ATF6-dependent mechanism. Cell Physiol Biochem. 2018;51(5):2397–2420. doi: 10.1159/000495898. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Xin Z, Zhang W, et al. A missense polymorphism in ATF6 gene is associated with susceptibility to hepatocellular carcinoma probably by altering ATF6 level. Int J Cancer. 2014;135(1):61–68. doi: 10.1002/ijc.28649. [DOI] [PubMed] [Google Scholar]
- 19.Cui XZ, Navneet S, Wang J, et al. Analysis of MTHFR, CBS, glutathione, taurine, and hydrogen sulfide levels in retinas of hyperhomocysteinemic mic. Invest Ophthalmol Vis Sci. 2017;58:1954–1963. doi: 10.1167/iovs.16-21247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ru Y, Li HJ, Zhang RX, et al. Role of keratinocytes and immune cells in the anti-inflammatory effects of Tripterygium wilfordii Hook. F. in a murine model of psoriasis. Phytomedicine. 2020;77:153299. doi: 10.1016/j.phymed.2020.153299. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Guo J, Li W, et al. PPM1H is down-regulated by ATF6 and dephosphorylates p-RPS6KB1 to inhibit progression of hepatocellular carcinoma. Mol Ther Nucleic Acids. 2023;33:164–179. doi: 10.1016/j.omtn.2023.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding RB, Bao JL, Deng CX. Emerging roles of SIRT1 in fatty liver diseases. Int J Biol Sci. 2017;13:852–867. doi: 10.7150/ijbs.19370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du CK, Lin XJ, Xu WJ, et al. Sulfhydrated sirtuin-1 increasing its deacetylation activity is an essential epigenetics mechanism of anti-atherogenesis by hydrogen sulfide. Antioxid Redox Signal. 2019;30:184–197. doi: 10.1089/ars.2017.7195. [DOI] [PubMed] [Google Scholar]
- 24.Andus T, Bauer J, Gerok W. Effects of cytokines on the liver. Hepatology. 1991;13:364–375. doi: 10.1002/hep.1840130226. [DOI] [PubMed] [Google Scholar]
- 25.Rodvold JJ, Mahadevan NR, Zanetti M. Immune modulation by ER stress and inflammation in the tumor microenvironment. Cancer Lett. 2015 doi: 10.1016/j.canlet.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 26.Jin JK, Blackwood EA, Azizi K, et al. ATF6 decreases myocardial ischemia/reperfusion damage and links ER stress and oxidative stress signaling pathways in the heart. Circ Res. 2017;120(5):862–875. doi: 10.1161/CIRCRESAHA.116.310266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stengel ST, Fazio A, Lipinski S, et al. Activating transcription factor 6 mediates inflammatory signals in intestinal epithelial cells upon endoplasmic reticulum stress. Gastroenterology. 2020;159(4):1357–1374.e10. doi: 10.1053/j.gastro.2020.06.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen X, Zhang F, Gong Q, et al. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosisin mice through peroxisome proliferator-activated receptor α. Diabetes. 2016;65(7):1904–1915. doi: 10.2337/db15-1637. [DOI] [PubMed] [Google Scholar]
- 29.Szabo C, Papapetropoulos A. International union of basic and clinical pharma-cology. CII: pharmacological modulation of H2S levels: H2S donors and H2S bio-synthesis inhibitors. Pharmacol Rev. 2017;69:497–564. doi: 10.1124/pr.117.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H. PI3K/SGK1/GSK3beta signaling pathway is involved in inhibition of autophagy in neonatal rat cardiomyocytes exposed to hypoxia/re-oxygenation by hydrogen sulfide. Exp Cell Res. 2016;345(2):134–140. doi: 10.1016/j.yexcr.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 31.Ling Q. Roles of the exogenous H2S-mediated SR-A signaling pathway in renal ischemia/reperfusion injury in regulating endoplasmic reticulum stress-induced autophagy in a rat model. Cell Physiol Biochem. 2017;41(6):2461–2474. doi: 10.1159/000475915. [DOI] [PubMed] [Google Scholar]
- 32.Meng GL, Zhao S, Xie LP, et al. Protein S-sulfhydration by hydrogen sulfide in cardiovascular system. Br J Pharmacol. 2018;175:1146–1156. doi: 10.1111/bph.13825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng L, Lu M, Mori K, Luo S, Lee AS, Zhu Y, Shyy JY. ATF6 modulates SREBP2-mediated lipogenesis. EMBO J. 2004;23(4):950–958. doi: 10.1038/sj.emboj.7600106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ye J, Rawson RB, Komuro R, Chen X, Davé UP, Prywes R, Brown MS, Goldstein JL. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 35.Jiang Y, Huang Z, Li X, Zhou L, Zhu X, Chen F, Shi Y. Inhibition of SK2 and ER stress ameliorated inflammation and apoptosis in liver ischemia-reperfusion injury. Liver Transpl. 2023 doi: 10.1097/LVT.0000000000000210. [DOI] [PubMed] [Google Scholar]
- 36.Wang Q, Zhu X, Li Z, Feng M, Liu X. ATF6 promotes liver fibrogenesis by regulating macrophage-derived interleukin-1α expression. Cell Immunol. 2021;367:104401. doi: 10.1016/j.cellimm.2021.104401. [DOI] [PubMed] [Google Scholar]
- 37.Duvigneau JC, Luís A, Gorman AM, Samali A, Kaltenecker D, Moriggl R, Kozlov AV. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine. 2019;124:154577. doi: 10.1016/j.cyto.2018.10.018. [DOI] [PubMed] [Google Scholar]
- 38.Rao J, Yue S, Fu Y, et al. ATF6 mediates a pro-inflammatory synergy between ER stress and TLR activation in the pathogenesis of liver ischemia-reperfusion injury. Am J Transpl. 2014;14(7):1552–1561. doi: 10.1111/ajt.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analyzed in the current study are available from the corresponding author on reasonable request.