

Abstract
Glycogen, a branched glucose polymer, helps regulate glucose homeostasis through immediate storage and release of glucose. Reprogramming of glycogen metabolism has recently been suggested to play an emerging role in cancer progression and tumorigenesis. However, regulation of metabolic rewiring for glycogen synthesis and breakdown in cancer cells remains less understood. Despite the availability of various glycogen detection methods, selective visualization of glycogen in living cells with high spatial resolution has proven to be highly challenging. Here, we present an optical imaging strategy to visualize glycogen in live cancer cells with minimal perturbation by combining stimulated Raman scattering microscopy with metabolic incorporation of deuterium-labeled glucose. We revealed the subcellular enrichment of glycogen in live cancer cells and achieved specific glycogen mapping through distinct spectral identification. Using this method, different glycogen metabolic phenotypes were characterized in a series of patient-derived BRAF-mutant melanoma cell-lines. Our results indicate that cell-lines manifesting high glycogen storage level showed increased tolerance to glucose deficiency among the studied melanoma phenotypes. This method opens up the possibility for non-invasive study of complex glycogen metabolism at subcellular resolution and may help reveal new features of glycogen regulation in cancer systems.
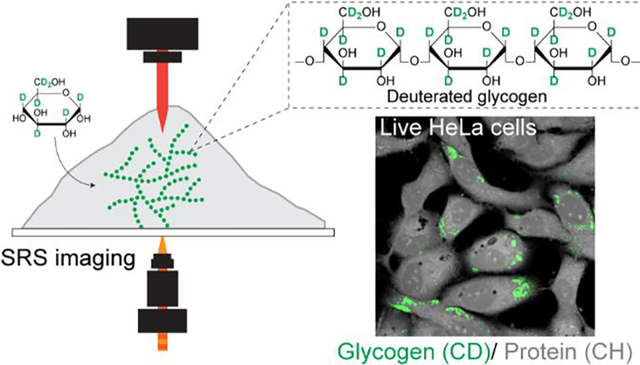
Graphical Abstract:
INTRODUCTION
Cancer cells undergo significant metabolic reprogramming to meet their high energy and anabolic demands.1 Numerous studies have revealed that metabolic pathways that branch from glycolysis, including glycogenesis, tricarboxylic acid (TCA) cycle, pentose phosphate pathway (PPP), serine biosynthesis, and fatty acid synthesis, can be altered to promote tumorigenesis and cancer cell survival.2–4 Although in-depth understandings have been achieved for many of these metabolic pathways, valuable insights have only started to emerge on the role of rewired glycogen metabolism in cancer-cell pathophysiology.5,6 Glycogen, in the form of branched polysaccharides of glucose residues, has mostly been regarded as the glucose storage site in mammalian tissues (e.g. liver, muscle, kidney, brain, and lungs).6 Yet more recently, it has been suggested that glycogen metabolism is upregulated in cancer cells in response to hypoxia as a sign of metabolic adaptation.7 In addition, the shuttling of glycogen was found in metastasizing ovarian cancer cells co-cultured with cancer-associated fibroblasts.8 With increasing evidence demonstrating the importance of cell-type and organelle specific glycogen metabolism in cancer systems,8,9 a live-cell imaging platform for investigating the glycogen activities and tracking the mobile glycogen dynamics10–12 with good selectivity, subcellular resolution, and fast temporal acquisition is in high demand.
However, subcellular interrogation of glycogen in live cancer cells remains challenging due to limitations of existing methods.6 Commonly used glycogen biochemical assays require extensive post-processing and lack spatial information.13 Magnetic resonance imaging (MRI) with isotope-labeled (i.e. 13C) glucose and positron emission tomography (PET) with radiotracer are powerful for in vivo glycogen imaging but are limited to millimeter resolution.14,15 Fourier transform infrared (FTIR) has recently been adopted for label-free imaging of glycogen in brain tissues but lacks subcellular resolution.16 Chemical staining and imaging (e.g. periodic acid-Schiff) provides subcellular information but requires sample fixation, which is known to lose more than 70% of glycogen.6 Mass spectrometry-based methods such as nano secondary ion mass spectrometry (nano SIMS) or transmission electron microscopy (TEM) are sample-destructive and not compatible for live-cell studies. Fluorescence imaging of 2-NBDG, a popular fluorescent glucose analog, offers subcellular and live-cell visualization17,18 but has a few limitations from the fluorophore labeling. First, 2-NBDG is significantly more hydrophobic compared to regular glucose,19 which can yield an affinity to lipids that decreases the specificity of 2-NBDG labeling of glycogen.20,21 Second, 2-NBDG is non-metabolizable after phosphorylation and may not provide downstream metabolic information.22
In this study, we report a strategy of stimulated Raman scattering (SRS) imaging with deuterated glucose (d7-glucose) labeling for visualizing subcellular glycogen in living cancer cells. We also demonstrate simultaneous metabolic phenotyping of glycogen together with glucose-derived proteins and lipids. By incubating cells in d7-glucose containing medium and targeting carbon-deuterium (C-D) vibrations using SRS, we visualized the incorporation of C-D bonds into subcellular glycogen. Taking advantage of the characteristic C-D Raman peaks of various types of macromolecules,23 we then constructed concentration maps of C-D labeled glycogen, lipids, and proteins using a robust linear combination algorithm in live HeLa and U87 cells. After validating the detection specificity of glycogen, we applied our technique to quantitatively study the subcellular accumulation of glycogen in a series of melanoma cancer cells that share the same BRAF-mutation but have different phenotypes. Our glycogen phenotyping results show consistent trend with gene expression profiles of glycogen metabolic enzymes. Guided by our observations, we further uncovered a previously unknown metabolic feature of increased resistance for glucose deficiency for one melanoma subtype. This imaging strategy sheds light on the role of glycogen and furthers our understanding of complex metabolic dynamics in cancer cells.
MATERIALS AND METHODS
Cell-lines and cell culture.
U87 and HeLa cells were cultured in DMEM (Gibco), supplemented with 10% fetal bovine serum (Corning, 35–015-CV), and 1% penicillin-streptomycin (Sigma-Aldrich). Patient-derived melanoma cell lines used in this study were generated under UCLA IRB approval # 11–003254. Melanoma cells were cultured in RPMI 1640 (Gibco, 11875119), supplemented with 10% fetal bovine serum (Corning, 35–015-CV), and 0.2% MycoZap Plus-CL antibiotics (Lonza, VZA-2011). Cultures were incubated in a water-saturated incubator at 37°C with 5% CO2. Cells were passaged 3–5 days once confluence reached 80%.
Spontaneous Raman spectra.
Fixed cell pellets were washed two times with pure water, and then resuspended into water to be cell solution to avoid influence from salt crystals after drying. The cell solution containing 5k cells was added dropwise on a glass slide. After air dry, glass slides with cells were then used to take Raman spectra. Spontaneous Raman spectra were acquired using an upright confocal Raman spectrometer (Horiba Raman microscope; Xplora plus). A 532 nm YAG laser is used to illuminate the sample with a power of 12 mW on sample through a 100 x, N.A. 0.9 objective (MPLAN N; Olympus) with slit 100 μm and hole 500 μm. Spectro/Raman shift center was set to be 2000.04 cm−1. With a 1200 grating (750 nm), Raman shift ranges from 690.81 cm−1 to 3141.49 cm−1 was acquired to cover whole cellular Raman peaks. Acquisition time for one spectrum was set to be 5 s, and then averaged for 5 times. The target cell was chosen randomly and spectra of five points (center, top, bottom, left, right) on the cell were acquired. The acquired spectra were processed by the LabSpec 6 software for baseline correction.
Stimulated Raman scattering microscopy.
Fig. 1B illustrates the SRS microscopy system used in this study. A picoEMERALD (Applied Physics and Electronics) system provided the pump (770–990 nm wavelength, 2 ps pulse width, 80 MHz repetition rate) and Stokes (1032 nm wavelength, 2 ps pulse width, 80 MHz repetition rate). Stokes beam was modulated by a built-in electro-optic modulator (EOM) at 20 MHz. The pump and Stokes beams were spatially and temporally overlapped within picoEMERALD. To fill the back aperture of a 25X water objective (XLPLN25XWMP, 1.05 N.A., Olympus), the two beams were expanded using a telescope and were guided into an inverted multiphoton laser scanning microscopy (FV3000, Olympus). The beams were aligned to maximize SRS signal of deuterium oxide (99.9 at% D, 151882 ALDRICH). The beams transmitted through the sample were collected using a high-NA condenser (oil immersion, 1.4 N.A., Olympus). Before detection of stimulated Raman loss using a large area (10×10 mm) Si photodiode (S3590–09, Hamamatsu), the Stokes beam was blocked out using a bandpass filter (893/209 BrightLine, 25 mm, AVR Optics) to filter out the Stokes beam. 64 V DC voltage was used on the photodiode to increase saturation threshold and reduce response time. The output current is terminated by a 50 Ω terminator and pre-filtered by a 19.2–23.6-MHz band-pass filter (BBP-21.4+, Mini-Circuits) to reduce laser and scanning noise. A lock-in amplifier (SR844, Stanford Research Systems) demodulated the signal at shot-noise-limited detection sensitivity. The in-phase X output of the lock-in amplifier was sent to analog channel of the FV1200 software (FV30-ANALOG). Image acquisition speed is limited by 30 μs time constant set for the lock-in amplifier. Correspondingly, we use 80 μs pixel dwell time, which gives a speed of 8.5 s/frame for a 320-by-320-pixel field of view. For unmixing, 3 images at channels 2123 cm−1, 2151 cm−1 and 2192 cm−1 are required, which leads to total acquisition time of approximately 1 min. The total acquisition to record an SRS spectral data cube (37 images) was approximately 9 minutes including image acquisition and wavelength tuning. For 2151 cm−1 (C-D, d7-glucose), 2845 cm−1 (CH2) and 2940 cm−1 (CH3), the wavelengths of Pump laser are 844, 797.3, and 791.3 nm, respectively. Laser powers on sample are measured to be 30 mW for modulated Stokes beam and 160 mW for Pump beam. Laser powers are monitored through image acquisition by an internal power meter and power fluctuation are controlled within 5% by the laser system. 16-bit grey scale images are acquired by Fluoview software. SRS spectra were acquired by fixing the Stokes beam at 1031.2 nm and scanning the pump beam through the designated wavelength range point by point.
Fig 1. Glycogen mapping by SRS microscopy with d7-glucose labeling.

(A) Metabolic incorporation of d7-glucose into glycogen through glycogenolysis pathway. G6P: glucose-6-phosphate, G1P: glucose-1-phosphate, UDP: uridine diphosphate. (B) Experimental scheme of SRS microscopy. EOM: electro-optic modulator. REF: reference. (C) Spontaneous Raman spectra of HeLa cells after culturing in regular glucose (black) and d7-glucose media (red) for 3 days. Zoomed-in spectra of the boxed region show C-D Raman peaks in the cell-silent window.
Coating of imaging dish.
In experiments where melanoma cell lines were used, imaging dish (MatTEK, P35G-1.5–14-C) was coated with a 2% sterile gelatin solution (Sigma, G1393) for 30 minutes, then the coating solution was removed and the dish was left for air dry for another 30 minutes before using. For U87 and HeLa cell lines, cells were directedly seeded on to the imaging dish without any coating.
Metabolic deuterium labeling.
Deuterated glucose RPMI 1640 medium was made by supplying 11.1 mM d7-glucose (Cambridge Isotope Laboratories, DLM-2062–1) into glucose deficient RPMI 1640 medium (Gibco, 11879020). The solution was then added 10% FBS and 0.2% MycoZap Plus-CL antibiotics. Cultured melanoma cells were seeded onto an imaging dish to optimal confluency. Deuterated glucose DMEM was made by supplying 25 mM d7-glucose (Cambridge Isotope Laboratories, DLM-2062–1) into glucose deficient DMEM (Gibco). The solution was then added 10% FBS and 1% penicillin-streptomycin.
GPI and cell viability assay.
Glycogen phosphorylase inhibitor (Sigma, 361515–1MG) was dissolved in DMSO (ATCC, 4-X) to a concentration of 5 mM before diluting at designated concentrations into cell culture media. To conduct cell viability assay, 3k to 5k cells were seeded into six well dishes (Corning, 3516). After culturing for 2 days, growth medium was replaced with fresh medium containing drugs with indicated concentration, and the incubation continued for another 3 days. For glucose deficiency experiment, the growth medium was replaced with glucose-deficient medium. Cell viability was measured by counting cell numbers of each well. Cell number was normalized by number of cells in normal glucose-containing medium to compute the relative viability.
Ratio image processing and data analysis.
Images were analyzed and assigned color by ImageJ. For CDP/CDL ratio imaging, a threshold (mask) image was first generated by adjusting threshold using Huang method, then non-zero values were normalized to one. CDP images were then divided by the same set of CDL, and the resulting ratio image multiplied with mask image to create the final CDP/CDL ratio image.
Linear combination algorithm for spectral unmixing.
To create the linear combination matrix in Fig. 2D, SRS hyperspectral images were acquired for U87 cells. Then spectra of intracellular glycogen, nucleoli, and lipid droplets were averaged. The SRS intensities at 2192 cm−1, 2151 cm−1, and 2123 cm−1 were extracted from the spectra and were used as coefficient after normalization to create the 3×3 matrix.23 For CH channel unmixing, lipid-channel (CHL) image was processed from a linear combination algorithm of 5· [CH2] – 0.4·[CH3] from the CH2 and CH3 images.24 Protein-channel (CHP) image was processed from a linear combination algorithm of [CH3] – [CH2] from the CH2 and CH3 images.24
Fig. 2. Identification of subcellular glycogen by SRS imaging of d7-glucose labeling.
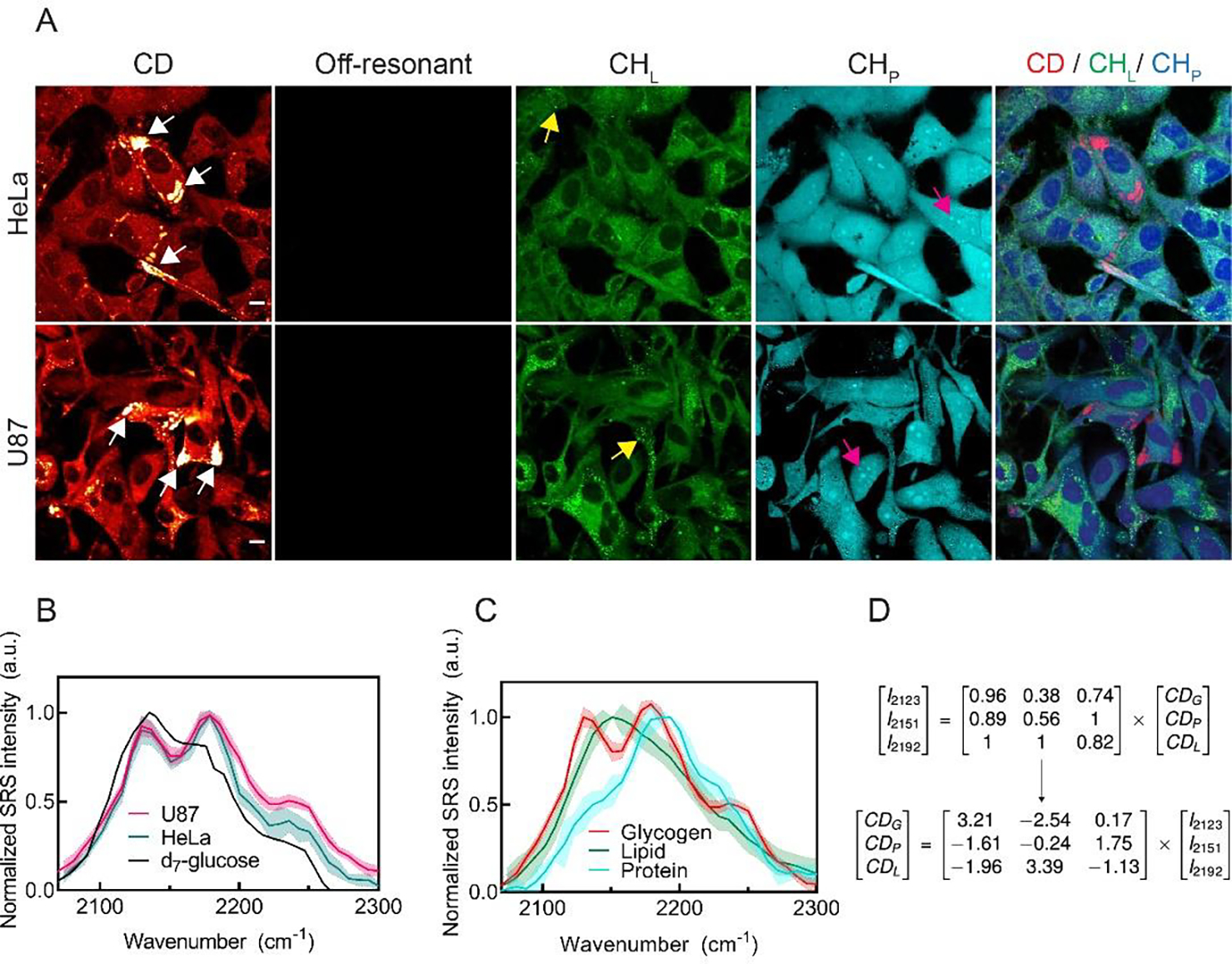
(A) Live-cell SRS images of HeLa and U87 cells after incubating in d7-glucose media for 3 days. CD (2151 cm−1) channel shows highly enriched subcellular signals, indicated by white arrows. Off-resonant image (1836 cm−1) of the same cells is background free. CHL and CHP images for endogenous lipids and proteins are unmixed from CH images acquired at 2845 cm−1 & 2940 cm−1 channels using a spectral linear combination algorithm (see Methods for details). Yellow and magenta arrows indicate lipid droplets and nucleoli at which SRS spectra were acquired, respectively. (B) Normalized SRS spectra from the subcellular C-D enriched spots (arrowed in (A)) in U87 (shaded purple), HeLa (shaded pink) cells and 1.1 M d7-glucose solution (red). (C) Representative glycogen (red), lipid (green), and protein (blue) SRS spectra generated from the subcellular C-D enriched spots (white arrows), lipid droplets (yellow arrows), and nucleoli (magenta arrows) in cells. (D) Linear combination matrix with coefficients derived from (C). Error bar: SD. Scale bars, 10 μm.
Triton and perchloric acid wash.
PBS solution containing 0.5% Triton X-100 (Sigma, T8787), PBS-T solution, was used to wash cells in imaging dish. 1 ml PBS-T detergent solution was gently added into imaging dish and the dish was placed in 4 °C for 10 minutes. Then the PBS-T washing solution was removed and the samples were washed with PBS for two times before imaging. Glycogen was removed by incubation in 10% perchloric acid for 5 minutes and washing with PBS.
RNA extraction, library construction, and sequencing.
Total RNA was extracted from frozen cells pellets (~ 1 million cells) using the RNeasy Micro Kit (Qiagen, 74004) according to the manufacturer’s protocol. Then the RNA sequencing (RNA-seq) was performed using BGISEQ-500 platform at BGI Genomics (Wuhan, China). The library preparation was followed by BGI’s standard procedure.
2-NBDG fluorescence and SRS concurrent imaging.
HeLa cells were co-incubated with 25mM d7-glucose for 60 hours and 100μM of 2-NBDG for the last 14 hours. Then fluorescence imaging was conducted with 488 nm excitation laser. Fluorescence was detected at wavelength range 500–600 nm. On the same set of cells, SRS images were taken at pump wavelengths 841 nm (2192 cm−1), 844 nm (2151 cm−1), and 846 nm (2123 cm−1).
Periodic acid acid-Schiff (PAS) staining.7
HeLa cells incubated with d7-glucose for three days were fixed using 4% PFA for 15 minutes. After several washing steps using PBS buffer, the cells were then incubated in 1% periodic acid (Sigma, 3951) for 30 minutes at room temperature. After several washing steps using deionized water, the cells were then incubated with Schiff’s reagent (Sigma, 3952016) for 30 minutes. The cells were washed multiple times with deionized water before they were imaged under an Olympus microscope (FV3000) installed with a color CMOS camera (Thorlabs, CS165CU).
Immuno-labeling using anti-glycogen antibodies.17
Fixed cells incubated with d7-glucose were permeabilized with 2% BSA-0.2% Triton X-100 PBS for 5 minutes on ice. Then non-specific bindings were blocked with 2% BSA-PBS for 1 h at room temperature. The cells were incubated in monoclonal mouse anti-glycogen primary antibody, IV58B6, diluted 1:60 in 2% BSA-PBS at 4 °C overnight. After incubating the cells for 30 minutes in BSA-PBS, the cells were incubated with Alexa Fluor 594-conjugated goat anti-mouse IgM (Invitrogen, A-21044) diluted 1:100 in 2% BSA-PBS for 4 hours at room temperature. The cells were incubated in BSA-PBS for 30 minutes and were washed multiple times in PBS before imaging. Fluorescence imaging was conducted with 561 nm excitation laser. Fluorescence was detected at wavelength range 570–670 nm.
Glycogen extraction using KOH/ethanol.8
Cells incubated with d7-glucose were fixed after 3 days. Cell suspension was centrifuged for 5 minutes at 2000 rpm. After removing the supernatant, the cells were incubated in 30% KOH for 30 minutes at 95 °C. After centrifugation and removing the supernatant, pure ethanol was added to precipitate the glycogen. After another round of centrifugation and supernatant removal, white glycogen powder was collected. The powder was then dried on a microscope glass slide before spontaneous spectrum acquisition.
RESULTS
Identifying Subcellular Glycogen by SRS Microscopy of D7-glucose Labeling.
The coupling of d7-glucose labeling with SRS imaging is ideal for subcellular visualization of glycogen in cancer cells. Isotope labeling with deuterium is a minimum labeling strategy. D7-glucose shares almost identical physicochemical properties to glucose and therefore can be metabolized into downstream metabolites. We expected that d7-glucose, after being transported into cells, will be phosphorylated to d7-glucose-6-phosphate (G6P) and then be converted to d7-glucose-1-phosphate (G1P). G1P then reacts with uridine triphosphate (UTP) to synthesize d7-uridine diphosphate (UDP) glucose (Fig. 1A). This activated glucose metabolite is incorporated into the growing chain of glycogen by glycogen synthase, the key regulatory enzyme in glycogen synthesis. Consequently, all newly synthesized glycogen is composed of repeating units of d7-glucose monomers (Fig. 1A). Specifically targeting the vibrational frequency of the C-D bonds from glycogen, SRS microscopy could, in principle, map out the intracellular enrichment of glycogen with fine spatial resolution (~ 450 nm) without interference from endogenous cellular background in live cells (Fig. 1B & Fig. 1C). In addition to minimum perturbation, d7-glucose labeling offers high imaging sensitivity for glycogen. Our detection sensitivity for d7-glucose solution is determined to be 4 mM.23,25 Although this number is less satisfying for imaging glucose uptake since the intracellular glucose concentration is about 3–5 mM,19 it is ideal for glycogen imaging with a detection limit as sensitive as 33 nM, since glycogen is a polymer composed of up to 120,000 repeating glucose residues.26 In contrast, a recently reported alkyne-labeled Raman glucose analogue, 3-OPG, offered a higher detection sensitivity (1.4 mM) for glucose imaging but could not be metabolized via the glycolysis pathway.19,27
Besides being incorporated into glycogen, d7-glucose can be converted into deuterated pyruvate, which subsequently enters TCA cycles to produce anabolic precursors including citrate, oxaloacetate, aspartate, and glutamate (Fig. S1). Consequently, macromolecules (e.g. lipids and proteins) synthesized from deuterated precursors are sparsely labelled with deuterium (Fig. S1). The deuterated G6P can also be metabolized through the pentose phosphate pathway for generating sparsely deuterated DNA/RNA (Fig. S1). The labeling patterns for different chemical structures of the macromolecules (i.e. proteins, lipids, DNA/RNA and glycogen) result in varied Raman C-D peak shapes23. Therefore, we reasoned that labeled glycogen could be identified by its unique C-D spectral shape.
Driven by our rationale above, we sought to validate the use of d7-glucose to label glycogen in both HeLa (cervical cancer) and U87 (glioblastoma) cells, two widely adopted model cancer cell-lines. We cultured both cells in d7-glucose medium for three days and imaged the cells by targeting the C-D peak at 2151 cm−1 by SRS (Fig. 2A, CD, white-arrowed; Fig. S2).23 Interestingly, we observed highly enriched subcellular C-D signals in the cytoplasm of a subset of cells (Fig. 2A, CD at 2151 cm−1 vs off-resonance at 1836 cm−1, arrowed). Such enrichment is distinct from previous reported distributions of lipids, including lipid droplets, and proteins, which yield more diffusive cytosolic patterns present in all the cells cultured in d7-glucose medium.28 Since this enrichment pattern resembles the intracellular glycogen pools reported previously by TEM,7 we hypothesized that it originated from glycogen accumulation. To test this, we first compared the CD image with label-free CHL and CHP images, representing pre-existing lipids and proteins, respectively (Fig. 2A). As expected, the enriched C-D pattern (red) does not co-localize with either lipid (green) or protein (blue) distributions in the CHL and CHP images from the same cells (Fig. 2A CD/CHL/CHP overlay). This suggests that the macromolecules in the accumulated spots are indeed not lipids or proteins but may be glycogen.
Next, we adopted a hyperspectral SRS (hSRS) strategy by acquiring SRS spectra specifically from these bright spots in both HeLa and U87 cells (Fig. 2A, arrowed) to spectrally identify the accumulated molecules. The obtained C-D spectra are mountain-like with three characteristic peaks at 2123, 2151, and 2192 cm−1 (Fig. 2B, pink and green). The overall spectral shape resembles that directly generated from d7-glucose solution (Fig. 2B, black), which suggests that the accumulated molecules preserve a similar chemical environment for C-D bond to that of glucose. To confirm whether the bright spots are indeed glycogen, we purified the deuterated glycogen from cells cultured in d7-glucose using the standard KOH/ethanol glycogen extraction protocol8. The spontaneous Raman spectrum from extracted deuterated glycogen closely resembles that of the in situ SRS spectrum (Fig. S3). Such high resemblance indicates that the bright glycogen spots, which can be as large as a few μm (Fig. S2), were indeed composed of pure glycogen. A similar spectral shape was previously reported from glycogen extracted from the liver of mice fed with d7-glucose.23 In contrast, the C-D spectra (Fig. 2C, red) are very different from those generated from lipid droplets (Fig. 2C, green) and nucleoli (Fig. 2C, blue). The in-situ SRS spectra from deuterated lipid droplets and nucleoli (Fig. 2A, blue and orange arrowed) also agree well with that previous reported from purified lipids and proteins in cells cultured in d7-glucose,28,29 indicating that the lipid droplets and nucleoli contained close-to-pure newly-synthesized lipids and proteins, respectively. Moreover, this accumulation pattern is retained in paraformaldehyde (PFA) fixed cells, ruling out the possibility of direct clustering of d7-glucose. Finally, we compared SRS images of d7-glucose cultured cells with that of the established glycogen staining methods, the periodic acid-Schiff (PAS) staining and the immuno-labeling using anti-glycogen antibody. We confirmed that SRS images present excellent correlation with brightfield images after PAS staining (Fig. S4) as well as immunofluorescence images (Fig. S5). We note that PAS also stains glycoproteins and glycolipids which may result in signals in the cytosols (Fig. S4). Together, these data provide direct evidence that the identity of the accumulated molecules is glycogen.
Specific Imaging of Glycogen with a Linear Combination Algorithm.
After confirming our imaging capability for intracellular glycogen through d7-glucose labeling, we then aimed to distinguish deuterated glycogen with other d7-glucose derived macromolecules to achieve high imaging specificity. Because of the unique C-D spectral shapes of the macromolecules (Fig. 2C), we could generate a linear combination algorithm to specifically retrieve the distribution maps for each C-D labeled metabolic end products. We derived a robust linear-combination algorithm (Fig. 2D), in which SRS signals are the products of normalized cross-section matrix, generated from Fig. 2C, and macromolecule concentrations. A similar strategy was recently adopted for multi-component imaging in mouse liver after starvation.23 We included glycogen, lipids, and proteins but excluded DNA due to an almost invisible C-D nucleus signal, which suggested minimum incorporation of C-D bonds to DNA/RNA in cancer cells. With this matrix, we can construct concentration maps for glycogen, lipids and proteins metabolically derived from d7-glucose by acquiring SRS images at three designated frequencies (Fig. 2D, i.e. 2123, 2151, and 2192 cm−1) without the need of hyperspectral images, which require much longer acquisition time and could lead to motion artifacts for live cells.
We applied the algorithm (Fig. 2D) to SRS images acquired from live HeLa and U87 cells cultured in d7-glucose medium. The same algorithm was also validated on fixed cells to avoid any potential issues from movement of live cells. Our unmixed results successfully distinguished deuterated glycogen, lipids, and proteins (Fig. 3 & Fig. S6, before vs after unmixing). First, CDG images clearly retrieved the bright subcellular spots. Second, CDL and CDP images for newly synthesized lipids and proteins showed high resemblance to those in the CHL and CHP channels (Fig. 3 & Fig. S6, after unmixing, see methods). In particular, lipid droplets were shown in both CDL and CHL channels; and nucleoli with fast protein turnover are highlighted in both CDP and CHP channels. Third, we performed a molecular-specific washing experiment with either triton or perchloric acid to eliminate lipids and glycogen, respectively and then compared the unmixed images before and after wash from the same set of cells. Correspondingly, the resulting images clearly showed the selective removal of lipids in the CDL channel or glycogen in the CDG channel (Figs. S7 and S8). This validates our algorithm for unequivocal SRS imaging of glycogen, together with lipids and proteins metabolized from d7-glucose in both live and fixed cells. We note that, unlike CDL and CDP signals that are mostly homogeneous across the cells, CDG signals are highly localized intracellularly and only appear in a subset of cells (~10–20%). Such enrichment signals would have been averaged out in bulk measurement, losing valuable cell-to-cell and subcellular heterogeneity information. This underscores the importance of subcellular imaging in studying glycogen.
Fig. 3. 5-Channel SRS imaging of newly synthesized glycogen, lipids, proteins with pre-existing lipids, and proteins in live (top) and fixed (bottom) HeLa cells.
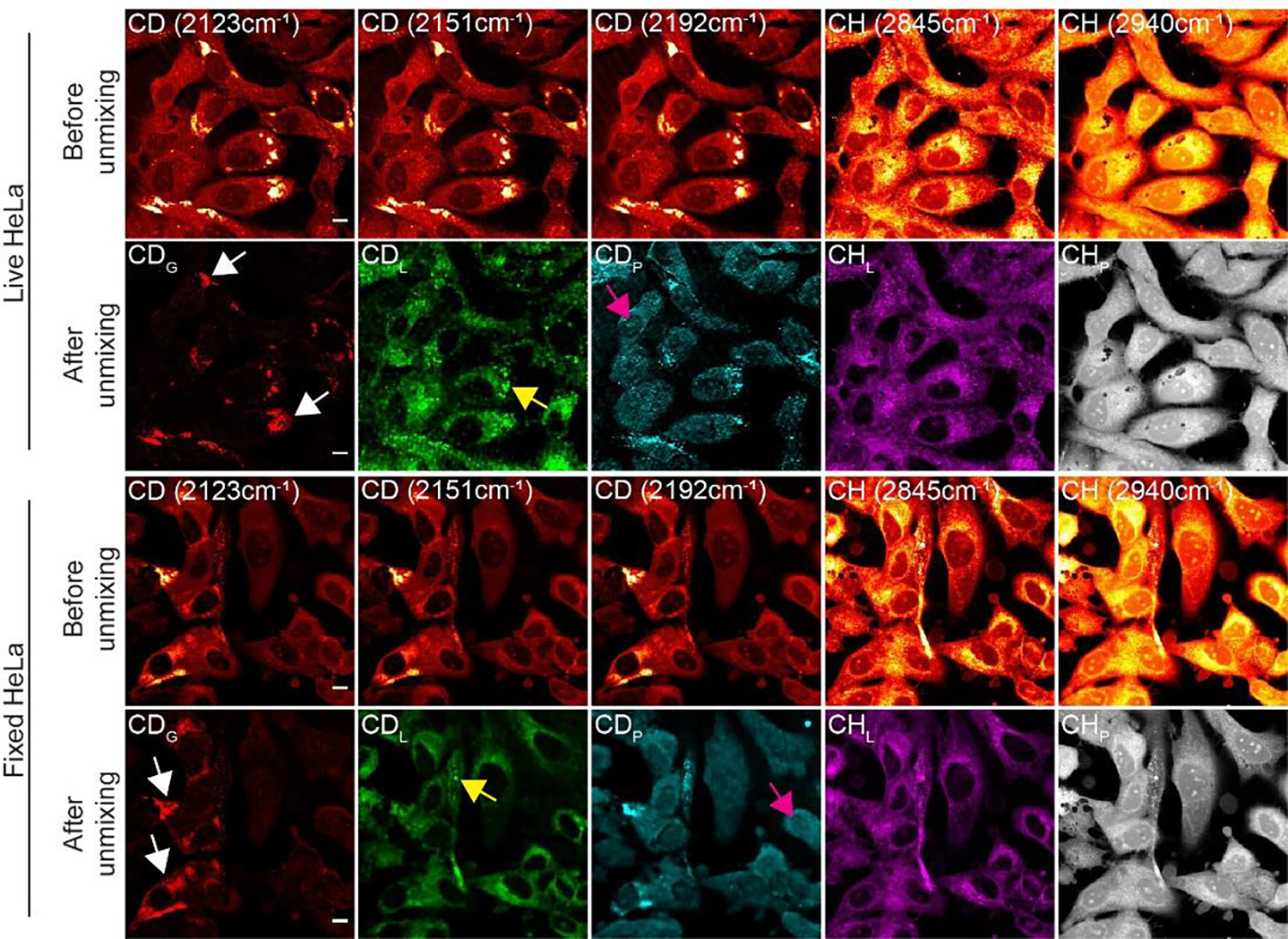
For both image sets of fixed and live HeLa cells after culturing in d7-glucose medium: top row (Before unmixing) shows SRS images acquired at C-D channels of 2123, 2151, 2192 cm−1 and C-H channels of 2845, 2940 cm−1 from the same set of cells; bottom row (After unmixing) shows concentration maps for d7-glucose-derived glycogen (CDG, red), lipids (CDL, green), and proteins (CDH, blue) unmixed from C-D channel SRS images and concentration maps for pre-existing lipids (CHL, purple) and proteins (CHP, gray) unmixed from C-H channel SRS images. White, yellow, and magenta arrows indicate glycogen, lipid droplets, and nucleoli, respectively. Scale bars, 10 μm.
After validating high imaging sensitivity and specificity together with metabolic phenotyping using this SRS strategy of d7-glucose labeling, we compared our results to those from fluorescence imaging of glycogen using 2-NBDG in live cells. 2-NBDG has been reported to be capable of being incorporated into glycogen through glycogenesis.17,18 We co-incubated HeLa cells with 100 μM of 2-NBDG and 25 mM d7-glucose and performed correlative fluorescence and SRS imaging. Surprisingly, almost no fluorescence signal from 2-NBDG colocalized with signals from the CDG channel (Fig. 4A, Fluorescence (blue) vs CDG (red), white arrowed). Instead, the fluorescence image correlated well with the unmixed lipid image (Fig. 4A, Fluorescence (blue) vs CDL (green), yellow arrowed). Three-channel correlative imaging with SRS imaging of C-D bonds, brightfield imaging of PAS staining and fluorescence imaging from 2-NBDG on the same set of cells was further performed (Fig. S9), as well as two-channel correlative PAS and 2-NBDG on starved cells following a previous reported incubation condition (Fig. S10)17. Both experiments indicated the raised specificity issue from 2-NBDG. This decreased specificity of 2-NBDG labeling for glycogen may likely due to the increased hydrophobicity of 2-NBDG compared to regular D-glucose from the substitution of the 2-hydroxyl group with large 7-nitobenzofurazan.20,21
Fig. 4. Correlative and dynamic imaging of glycogen.

(A) Colocalization of the fluorescence (cyan) image of 2-NBDG and unmixed SRS images of glycogen (CDG, red), lipids (CDL, green), and proteins (CDP, blue) in HeLa cells co-incubated with 25 mM d7-glucose and 100 μM of 2-NBDG. White arrows indicate location of glycogen. Yellow arrows indicate enriched lipid region. (B) Pulse-chase imaging of glycogen synthesis and degradation dynamics in live HeLa cells incubated in d7-glucose medium in the pulse period and replaced with regular glucose medium in the chase period. Arrows indicate the glycogen pools. (C) Normalized SRS CD signals from subcellular enriched glycogen and cytoplasm in (B) (N=20 cells for each time point). Data shown as mean ± SD. Scale bars, 10 μm.
Investigating the Metabolic Dynamics of Glycogen.
Leveraging the fine specificity and biocompatibility of SRS and d7-glucose labeling, we then monitored the glycogenesis and glycogenolysis in live cells with a pulse-chase experiment to characterize their turnover dynamics (Fig. 4B). We first pulse-incubated HeLa cells with d7-glucose medium for 24, 48, and 72 hours to probe the glycogenesis (Fig. 4B). After 24 hours, glycogen with high C-D enrichment started to emerge in the cytoplasm (Fig. 4B arrowed, 24 h). Longer incubation led to an increase in both C-D signals and intracellular numbers of glycogen (Fig. 4B arrowed, 48 h and 72 h). The C-D intensity gradually reached a plateau at 72 h (Fig. 4C, red). We then chased the cells by replacing d7-glucose medium back to regular medium after 72 h of pulse incubation to interrogate the glycogenolysis (Fig. 4B). C-D signals from glycogen were lowered to about 50% after 24 h of chase (Fig. 4B, 96 h, arrowed & Fig. 4C, red). Quantification on signal increase and decrease for both C-D enriched bright spots of glycogen and cytosolic C-D labeled lipids and proteins (Fig. 4B) revealed similar turnover dynamics (Fig. 4C). These results suggest a dynamic role of glycogen going beyond static storage sites in cancer cells even under nutrient abundant conditions, which is possibly related to glycogen shunt pathway to maintain metabolic and ATP homeostasis.30
Metabolic Phenotyping of Glycogen, Lipids, and Proteins in Metastatic Melanoma.
We aimed to demonstrate the utility of this technique and the importance of glycogen in a more clinically relevant model. To this end, we investigated a series of patient-derived BRAF mutant melanoma cell-lines that have been extensively characterized at both the genomic and transcriptome levels.31–33 Melanoma is the most deadly form of skin cancer due to its high metastatic potential.34 Recently, it has been found that these derived cell-lines exhibit different transcriptional programs and phenotypic behaviors that are associated with degrees of cellular de-differentiation.31–33 The different melanoma cancer cell phenotypes would likely have varied metabolic programs to meet phenotype-specific energy and growth demands. Thus, we hypothesized that the different cell-lines would exhibit unique accumulation rates and spatial patterns of d7-glucose derived glycogen, lipids, and proteins.
We selected five melanoma cancer cell-lines representing different de-differentiation status—melanocytic/differentiated (M262), transitory (M397, M229), neural-crest (M409), and undifferentiated (M381)—and cultured them in d7-glucose medium. We acquired SRS images at the three designated channels (Fig. S11) and adopted the linear combination algorithm to construct the concentration maps of C-D labeled lipids, proteins, and glycogen in the corresponding cell-lines (Fig. 5A). To obtain good unmixing results without potential motion artifacts, we started from fixed cells. Interestingly, we found that M397 and M262 cells showed relatively high accumulation of glycogen, whereas the three other cell-lines exhibited almost no evidence of glycogen formation (Fig. 5A, CDG, arrowed). Quantitatively, the normalized C-D intensity from the glycogen (CDG) channel of M397 cells is about twice of that in M262 cells, and orders of magnitudes higher compared to the other three cell-lines (Fig. 5B). This suggests that M397 cells have a higher dependence on glycogen-accumulation-associated metabolism. We asked whether this revealed metabolic phenotype is consistent with transcriptomics data. We computed the ratios of expression levels from RNA-sequencing between glycogen synthase (GYS1) to glycogen phosphorylase (PGYL), rate-limiting enzymes for glycogenesis and glycogenolysis, respectively (Fig. 5C). The overall trend of ratios is indeed consistent with our measured CDG intensities across cell-lines (Fig. 5B vs 5C). The lower magnitude difference in the transcriptomics data likely arises because only a subset of cells presents significantly enhanced glycogen accumulation, and so the effect is smoothed out in bulk measurements. This again highlights the importance of subcellular imaging in uncovering cell-to-cell and subcellular heterogeneities. In addition to single-channel imaging, ratiometric images between different metabolic channels also serve as phenotype indicators. We found that CDP/CDL ratios follow a similar trend to that of CDG signals across cell-lines (Fig. 5D).
Fig. 5. Metabolic phenotyping of metastatic melanoma.

(A) Representative unmixed SRS image sets for glycogen (CDG, red), lipid (CDL, green), and protein (CDP, blue) with ratiometric CDP/CDL images across selected melanoma cell-lines (M397 to M381). Each column indicates the representative image set on the same set of cells for one cell-line. Scale bar, 10 μm. (B) Normalized CDG intensity across melanoma cell-lines. (C) Ratios of gene expression levels between glycogen synthase (GYS1) to glycogen phosphorylase (PGYL). (D) Normalized CDP/CDL ratios across cell-lines. (E) Relative viability of melanoma cells cultured in glucose-deficient medium for up to 5 days. (F) Quantification of relative viability across melanoma cell-lines with 3-day glucose deficiency in (E). (G-H) Relative cell viability for M397 cells cultured in glucose-deficient medium (CT) and M397 cells cultured in glucose-deficient medium with 1 μM glycogen phosphorylase inhibitor (GPI) after 3 days (G) and 4 days (H). Statistical significance was determined by unpaired two-tailed t test. ns, not significant (p>0.05), *p < 0.05, **p < 0.01, ****p < 0.0001. Data shown as mean ± SD. Scale bars, 10 μm
Since M397 cells show the most active glycogen accumulation, we examined the spatial pattern of glycogen distribution in M397 cells. We found extensive formation of large (larger than 1 μm) glycogen-enriched extracellular vesicles (EVs) that were only observable in PFA-fixed M397 cells, but not in live cells (Fig. S12 & S13). We performed time-lapse imaging on the same set of M397 cells immediately after PFA addition (Fig. S14A & S14B). We found that the intensity increase of the EVs is accompanied by the intensity decrease of intracellular glycogen, suggesting transportation of intracellular glycogen (up to 80%) to secreted EVs under PFA fixation (Fig. S14B & S14C). This result again highlights the importance of live-cell imaging for glycogen metabolism to rule out potential complications due to PFA fixation.
Observation of Increased Tolerance on Glucose Deficiency for M397 Cells.
Since M397 cells present a much-pronounced subcellular glycogen accumulation, we next sought to understand the functional role of glycogen in this cancer cell phenotype. Elevated level of glycogen storage suggests a possible higher tolerance of glucose deficiency. Indeed, many studies report that increased accumulation of glycogen in cancer cells support cancer cell survival in nutrient and oxygen deficient conditions.7,35,36 To test this potential role of glycogen storage, we cultured the five cell-lines in glucose-deficient medium and performed time-dependent cell viability assays. Surprisingly, M397 cells showed minimally decreased proliferation even after culturing in glucose-deficient medium for three days (Fig. 5E & 5F). In contrast, the other cell-lines all showed significantly reduced growth (Fig. 5E & 5F). To test whether glycogen is involved in the extended survival, we applied glycogen phosphorylase inhibitor (GPI), which inhibits glycogenolysis to UDP-glucose, to the culture medium during glucose deficiency treatment. We first showed that GPI is non-toxic to cells in normal glucose medium for at least 5 days (Fig. S15). In glucose-deficient condition, the added GPI lowered the viability of M397 after day 3, at which point the glycogenolysis activities may be the most intensive (Fig. 5G & 5H). Our demonstration here hence emphasizes the critical role of glycogen metabolism in specific cancer cell types and may help advance our fundamental biological understanding in elucidating glycogen-related metabolism pathways with further explorations.
DISCUSSION
The reprogramming of glycogen metabolism in cancer cells is not well understood, and existing probes of glycogen metabolism have a number of limitations.6,13 We proposed SRS imaging with d7-glucose labeling as a sub-cellular, minimally perturbative probe for glycogen metabolism in live cancer cells. First, we demonstrated incorporation of d7-glucose into de novo synthesized glycogen, validating the use of d7-glucose as a probe to label glycogen in cancer cells. Second, highly specific and sensitive imaging of glycogen was shown in live cancer cells using a linear-unmixing algorithm. Third, we surveyed the subcellular glycogen, lipid, and protein metabolism in a series of metastatic melanoma cells and revealed a high glycogen-accumulation phenotype (i.e. M397 cells). This cell-line further showed a high resistance to glucose deficiency. As several recent studies have suggested the critical roles of glycogen metabolism in cancer systems,7–9 this method can help shed light on glycogen function, which is a necessary step towards targeting glycogen metabolism for therapeutic purposes.
Although the above proof-of-concept study already demonstrates the potential of our strategy, further technical improvements may lead to better imaging performance. The speed of SRS imaging can be improved by using techniques such as spectral focusing,37 which can enable much improved speed for hyperspectral imaging. Spectral analysis for hyperspectral images, such as multivariate curve resolution,38 may further reduce mapping errors and allow more sensitive detection of metabolites, such as d7-glucose derived DNA/RNA. Going beyond cellular studies, this method may also be applicable for in vivo research due to the biocompatibility of SRS imaging and d7-glucose labeling. We envision this technique as a valuable tool in investigating glycogen metabolism and for metabolic phenotyping in both cancer cells and in vivo.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Antoni Ribas for sharing the melanoma cell-lines. We also thank Dr. Otto Baba and Dr. Morita for sharing the anti-glycogen antibodies. We also would like to thank Dr. C. Qian, K. Miao, X. Bi, L. Lin and Dr. L. Voong for helpful discussions. We acknowledge the following agencies and foundations for support: NIH Grant U01 CA217655 (to J.R.H.) and the WA State Andy Hill CARE Foundation (to J.R.H.), and an ISB Innovator Award (Y.S.). L.W acknowledges the support for start-up funds from California Institute of Technology.
REFERENCES
- (1).DeBerardinis RJ; Lum JJ; Hatzivassiliou G; Thompson CB The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008, 7 (1), 11–20. [DOI] [PubMed] [Google Scholar]
- (2).Lunt SY; Vander Heiden MG Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27 (1), 441–464. [DOI] [PubMed] [Google Scholar]
- (3).Yan L; Raj P; Yao W; Ying H Glucose Metabolism in Pancreatic Cancer. Cancers 2019, 11 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Heiden MGV; Cantley LC; Thompson CB Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324 (5930), 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Dauer P; Lengyel E New Roles for Glycogen in Tumor Progression. Trends Cancer 2019, 5 (7), 396–399. [DOI] [PubMed] [Google Scholar]
- (6).Zois CE; Harris AL Glycogen Metabolism Has a Key Role in the Cancer Microenvironment and Provides New Targets for Cancer Therapy. J. Mol. Med. 2016, 94 (2), 137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Favaro E; Bensaad K; Chong MG; Tennant DA; Ferguson DJP; Snell C; Steers G; Turley H; Li J-L; Günther UL; Buffa FM; McIntyre A; Harris AL Glucose Utilization via Glycogen Phosphorylase Sustains Proliferation and Prevents Premature Senescence in Cancer Cells. Cell Metabolism 2012, 16 (6), 751–764. [DOI] [PubMed] [Google Scholar]
- (8).Curtis M; Kenny HA; Ashcroft B; Mukherjee A; Johnson A; Zhang Y; Helou Y; Batlle R; Liu X; Gutierrez N; Gao X; Yamada SD; Lastra R; Montag A; Ahsan N; Locasale JW; Salomon AR; Nebreda AR; Lengyel E Fibroblasts Mobilize Tumor Cell Glycogen to Promote Proliferation and Metastasis. Cell Metabolism 2019, 29 (1), 141–155.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sun RC; Dukhande VV; Zhou Z; Young LEA; Emanuelle S; Brainson CF; Gentry MS Nuclear Glycogenolysis Modulates Histone Acetylation in Human Non-Small Cell Lung Cancers. Cell Metab. 2019, 30 (5), 903–916.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Prats C; Graham TE; Shearer J The Dynamic Life of the Glycogen Granule. J. Biol. Chem. 2018, 293 (19), 7089–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bak LK; Walls AB; Schousboe A; Waagepetersen HS Astrocytic Glycogen Metabolism in the Healthy and Diseased Brain. J. Biol. Chem. 2018, 293 (19), 7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shulman RG; Hyder F; Rothman DL Cerebral Energetics and the Glycogen Shunt: Neurochemical Basis of Functional Imaging. Proc. Natl. Acad. Sci. 2001, 98 (11), 6417–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Passonneau JV; Lauderdale VR A Comparison of Three Methods of Glycogen Measurement in Tissues. Anal. Biochem. 1974, 60 (2), 405–412. [DOI] [PubMed] [Google Scholar]
- (14).Zhou Y; van Zijl PCM; Xu X; Xu J; Li Y; Chen L; Yadav NN Magnetic Resonance Imaging of Glycogen Using Its Magnetic Coupling with Water. Proc. Natl. Acad. Sci. 2020, 117 (6), 3144–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Witney TH; Carroll L; Alam IS; Chandrashekran A; Nguyen Q-D; Sala R; Harris R; DeBerardinis RJ; Agarwal R; Aboagye EO A Novel Radiotracer to Image Glycogen Metabolism in Tumors by Positron Emission Tomography. Cancer Res. 2014, 74 (5), 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hackett MJ; Sylvain NJ; Hou H; Caine S; Alaverdashvili M; Pushie MJ; Kelly ME Concurrent Glycogen and Lactate Imaging with FTIR Spectroscopy To Spatially Localize Metabolic Parameters of the Glial Response Following Brain Ischemia. Anal. Chem. 2016, 88 (22), 10949–10956. [DOI] [PubMed] [Google Scholar]
- (17).Louzao MC; Espiña B; Vieytes MR; Vega FV; Rubiolo JA; Baba O; Terashima T; Botana LM “Fluorescent Glycogen” Formation with Sensibility for in Vivo and in Vitro Detection. Glycoconj. J. 2007, 25 (6), 503. [DOI] [PubMed] [Google Scholar]
- (18).Zhu Y; Fan Z; Wang R; Xie R; Guo H; Zhang M; Guo B; Sun T; Zhang H; Zhuo L; Li Y; Wu S Single-Cell Analysis for Glycogen Localization and Metabolism in Cultured Astrocytes. Cell. Mol. Neurobiol. 2020, 40 (5), 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hu F; Chen Z; Zhang L; Shen Y; Wei L; Min W Vibrational Imaging of Glucose Uptake Activity in Live Cells and Tissues by Stimulated Raman Scattering. Angew. Chem. Int. Ed. 2015, 54 (34), 9821–9825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hughes LD; Rawle RJ; Boxer SG Choose Your Label Wisely: Water-Soluble Fluorophores Often Interact with Lipid Bilayers. PLOS ONE 2014, 9 (2), e87649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zanetti-Domingues LC; Tynan CJ; Rolfe DJ; Clarke DT; Martin-Fernandez M Hydrophobic Fluorescent Probes Introduce Artifacts into Single Molecule Tracking Experiments Due to Non-Specific Binding. PLOS ONE 2013, 8 (9), e74200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yamada K; Saito M; Matsuoka H; Inagaki N A Real-Time Method of Imaging Glucose Uptake in Single, Living Mammalian Cells. Nat. Protoc. 2007, 2 (3), 753–762. [DOI] [PubMed] [Google Scholar]
- (23).Zhang L; Shi L; Shen Y; Miao Y; Wei M; Qian N; Liu Y; Min W Spectral Tracing of Deuterium for Imaging Glucose Metabolism. Nat. Biomed. Eng. 2019, 3 (5), 402–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Shen Y; Zhao Z; Zhang L; Shi L; Shahriar S; Chan RB; Di Paolo G; Min W Metabolic Activity Induces Membrane Phase Separation in Endoplasmic Reticulum. Proc Natl Acad Sci USA 2017, 114 (51), 13394–13399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Miao K; Wei L Live-Cell Imaging and Quantification of PolyQ Aggregates by Stimulated Raman Scattering of Selective Deuterium Labeling. ACS Cent. Sci. 2020, 6 (4), 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Voet D; Voet JG; Pratt CW Fundamentals of Biochemistry, 1st ed.; J. Wiley: New York, 1999. [Google Scholar]
- (27).Long R; Zhang L; Shi L; Shen Y; Hu F; Zeng C; Min W Two-Color Vibrational Imaging of Glucose Metabolism Using Stimulated Raman Scattering. Chem. Commun. 2018, 54 (2), 152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li J; Cheng J-X Direct Visualization of De Novo Lipogenesis in Single Living Cells. Sci. Rep. 2015, 4 (1), 6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Wei L; Hu F; Shen Y; Chen Z; Yu Y; Lin C-C; Wang MC; Min W Live-Cell Imaging of Alkyne-Tagged Small Biomolecules by Stimulated Raman Scattering. Nat. Methods 2014, 11 (4), 410–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shulman RG; Rothman DL Homeostasis and the Glycogen Shunt Explains Aerobic Ethanol Production in Yeast. Proc. Natl. Acad. Sci. 2015, 112 (35), 10902–10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Su Y; Wei W; Robert L; Xue M; Tsoi J; Garcia-Diaz A; Homet Moreno B; Kim J; Ng RH; Lee JW; Koya RC; Comin-Anduix B; Graeber TG; Ribas A; Heath JR Single-Cell Analysis Resolves the Cell State Transition and Signaling Dynamics Associated with Melanoma Drug-Induced Resistance. Proc Natl Acad Sci USA 2017, 114 (52), 13679–13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Berger MF; Levin JZ; Vijayendran K; Sivachenko A; Adiconis X; Maguire J; Johnson LA; Robinson J; Verhaak RG; Sougnez C; Onofrio RC; Ziaugra L; Cibulskis K; Laine E; Barretina J; Winckler W; Fisher DE; Getz G; Meyerson M; Jaffe DB; Gabriel SB; Lander ES; Dummer R; Gnirke A; Nusbaum C; Garraway LA Integrative Analysis of the Melanoma Transcriptome. Genome Res. 2010, 20 (4), 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tsoi J; Robert L; Paraiso K; Galvan C; Sheu KM; Lay J; Wong DJL; Atefi M; Shirazi R; Wang X; Braas D; Grasso CS; Palaskas N; Ribas A; Graeber TG Multi-Stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33 (5), 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Davies H; Bignell GR; Cox C; Stephens P; Edkins S; Clegg S; Teague J; Woffendin H; Garnett MJ; Bottomley W; Davis N; Dicks E; Ewing R; Floyd Y; Gray K; Hall S; Hawes R; Hughes J; Kosmidou V; Menzies A; Mould C; Parker A; Stevens C; Watt S; Hooper S; Wilson R; Jayatilake H; Gusterson BA; Cooper C; Shipley J; Hargrave D; Pritchard-Jones K; Maitland N; Chenevix-Trench G; Riggins GJ; Bigner DD; Palmieri G; Cossu A; Flanagan A; Nicholson A; Ho JWC; Leung SY; Yuen ST; Weber BL; Seigler HF; Darrow TL; Paterson H; Marais R; Marshall CJ; Wooster R; Stratton MR; Futreal PA Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417 (6892), 949–954. [DOI] [PubMed] [Google Scholar]
- (35).Chen J; Lee H-J; Wu X; Huo L; Kim S-J; Xu L; Wang Y; He J; Bollu LR; Gao G; Su F; Briggs J; Liu X; Melman T; Asara JM; Fidler IJ; Cantley LC; Locasale JW; Weihua Z Gain of Glucose-Independent Growth upon Metastasis of Breast Cancer Cells to the Brain. Cancer Res 2015, 75 (3), 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu J; Gao L; Zhang H; Wang D; Wang M; Zhu J; Pang C; Wang C Succinate Dehydrogenase 5 (SDH5) Regulates Glycogen Synthase Kinase 3β-β-Catenin-Mediated Lung Cancer Metastasis. J. Biol. Chem. 2013, 288 (41), 29965–29973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fu D; Holtom G; Freudiger C; Zhang X; Xie XS Hyperspectral Imaging with Stimulated Raman Scattering by Chirped Femtosecond Lasers. J. Phys. Chem B. 2013, 117 (16), 4634–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhang D; Wang P; Slipchenko MN; Ben-Amotz D; Weiner AM; Cheng J-X Quantitative Vibrational Imaging by Hyperspectral Stimulated Raman Scattering Microscopy and Multivariate Curve Resolution Analysis. Anal. Chem. 2013, 85 (1), 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.