

Abstract
To determine the potential for bacteriophage-mediated gene transfer in the marine environment, we established transduction systems by using marine phage host isolates. Plasmid pQSR50, which contains transposon Tn5 and encodes kanamycin and streptomycin resistance, was used in plasmid transduction assays. Both marine bacterial isolates and concentrated natural bacterial communities were used as recipients in transduction studies. Transductants were detected by a gene probe complementary to the neomycin phosphotransferase (nptII) gene in Tn5. The transduction frequencies ranged from 1.33 × 10−7 to 5.13 × 10−9 transductants/PFU in studies performed with the bacterial isolates. With the mixed bacterial communities, putative transductants were detected in two of the six experiments performed. These putative transductants were confirmed and separated from indigenous antibiotic-resistant bacteria by colony hybridization probed with the nptII probe and by PCR amplification performed with two sets of primers specific for pQSR50. The frequencies of plasmid transduction in the mixed bacterial communities ranged from 1.58 × 10−8 to 3.7 × 10−8 transductants/PFU. Estimates of the transduction rate obtained by using a numerical model suggested that up to 1.3 × 1014 transduction events per year could occur in the Tampa Bay Estuary. The results of this study suggest that transduction could be an important mechanism for horizontal gene transfer in the marine environment.
A virus is little more than nucleic acid encapsulated in a protein coat. The recent discovery of the abundance of viruses in the marine environment (5, 14, 42, 47, 48) has led to speculation regarding the involvement of viruses in gene transfer. In the process of viral propagation, viruses transfer nucleic acid synthesized in one bacterium to another bacterium. If a virus infecting a new host contains genetic material from the previous host rather than its own DNA, the extra genetic information may be transmitted to the new host, resulting in transduction.
Bacteriophage-mediated horizontal transduction has been known for nearly half a century (55). Transduction has been found to occur in many phage-host systems (6), and well-studied transduction systems have been routinely used as molecular cloning tools (37). However, in most transduction studies workers have focused only on the development of tools for better understanding bacterial genetics (3, 7, 9, 25, 26, 34, 41, 49). The role of phage transduction in microbial ecology was not an area of interest until very recently.
To assess the risk associated with the spread of genetically engineered microorganisms in the environment, the potential for gene transfer by transduction was studied in soil and freshwater environments. Using an Escherichia coli-phage P1 transduction system, Zeph et al. (54) studied transduction in sterile and nonsterile soils. The results of these authors demonstrated that transduction occurred in the soil and that the resulting transductants survived in soil environments for 28 days. Almost all studies of transduction in aquatic environments have been performed by Miller’s group (for a review see reference 29). These researchers demonstrated that both chromosomal and plasmid DNAs of Pseudomonas aeruginosa were transduced during in situ incubation in a freshwater lake (31, 36, 39, 40). Cell-free phage lysates, as well as temperate phages spontaneously released from lysogens, were capable of transduction (40). Also, both lysogenic and nonlysogenic bacteria can serve as recipients, but lysogenic recipients have higher transducing frequencies, possibly due to lysogenic protection from lysis (homoimmunity [29]). More recently, Ripp and Miller (35) also suggested that the presence of suspended particulates in the water column facilitates transduction by bringing the host and phage into close contact with each other.
Little is known about transduction in the marine environment. Although transducing phages have been isolated from seawater previously (16, 23, 24), the focus of these studies was to develop a gene transfer system to study the genetics of Vibrio spp. rather than to investigate the potential for gene transfer in the environment.
Viruses are abundant and active members of microbial ecosystems. The dynamic interactions of viruses with their hosts may contribute significantly to the genetic diversity and composition of microbial populations. For many years, studies of gene transfer in the environment have largely focused on the process of conjugation (2, 15, 32) and transformation (13, 33, 43). Gene transfer via transduction was considered negligible because of the lytic effect of phage infection (29). However, Zeph et al. (54) suggested that gene transduction is as important or more important than conjugation and transformation in the environment. In contrast to transforming DNA, transducing DNA is packaged inside phage capsids, which prevents nuclease degradation. Thus, viruses may serve as reservoirs for exogenous genes (44).
To estimate the potential for transduction in the marine environment, we developed marine transduction systems by using marine phage host isolates. Transduction assays were performed by using marine bacterial isolates, as well as mixed natural bacterial communities, as recipients. This work established that transduction is a mechanism for horizontal gene transfer among marine microbial communities.
MATERIALS AND METHODS
Bacteria, phages, and plasmids.
The bacteria, phages, and plasmids used in this study are listed in Table 1. Marine bacterial strains HSIC and D1B were isolated from Mamala Bay, Hawaii, on artificial seawater (ASWJP) agar plates containing 5 g of peptone per liter and 1 g of yeast extract per liter. HSIC is an unidentified gram-negative coccus, while D1B is a gram-negative slender rod which was identified as a Flavobacterium sp. by using an API-NFT test kit (BioMerieux Vitek, Inc., Hazelwood, Mo.). Phages T-φHSIC and T-φD1B were isolated by using HSIC and D1B as hosts, respectively. Both of these phages are temperate and contain double-stranded DNA. Detailed information about these phages and their hosts has been published elsewhere (17).
TABLE 1.
Bacteria, plasmids, and phages used in this study
| Strain, plasmid, or phage | Description | Reference |
|---|---|---|
| Bacterial strains | ||
| HSIC | Isolated from Mamala Bay, Hawaii (carrying plasmid upon isolation) | 17 |
| HSICrif | HSIC spontaneous mutant of rifampin-resistant strain | This study |
| D1B | Isolated from Mamala Bay, Hawaii | 17 |
| D1Brif | D1B spontaneous mutant of rifampin-resistant strain | |
| E. coli RM1259(pQSR50) | Kmr Smr | 26 |
| E. coli CA60(pNJ5000) | Tetr | 52 |
| HOPE-1 | HSICrif(pQSR50) | This study |
| HOPE-2 | D1Brif(pQSR50) | This study |
| Plasmids | ||
| pQSR50 | R1162::Tn5 Kmr Smr | 26 |
| pNJ5000 | Tetr Mob+ | 52 |
| Phages | ||
| T-φHSIC | Isolated from Mamala Bay, Hawaii, on bacterial host HSIC | 17 |
| T-φD1B | Isolated from Mamala Bay, Hawaii, on bacterial host D1B | 17 |
E. coli RM1259 contains plasmid pQSR50, which encodes resistance to kanamycin and streptomycin. The kanamycin resistance gene, nptII, is on transposon Tn5. A detailed description of this plasmid and a plasmid map have been published by Meyer et al. (27). E. coli RM1259 was used as a plasmid donor in triparental mating, and E. coli CA60 was used as a helper strain for triparental mating (52). E. coli CA60 contains a conjugative plasmid, pNJ5000, which encodes tetracycline resistance. HOPE-1 and HOPE-2 are wild-type strain HSIC and D1B derivatives containing plasmid pQSR50, respectively, which was introduced into the bacterial cells by triparental mating (see below).
Plasmid pQSR50 contains a Tn5 transposon insertion which codes for the neomycin phosphotransferase gene (nptII). Plasmid pNJ5000 has a mobilizing function and was used as a helper plasmid for mobilization of other plasmids.
Triparental mating.
Rifampin-resistant mutants of bacterial strains HSIC and D1B were selected on ASWJP nutrient plates containing 500 μg of rifampin per ml and were used as recipients for triparental mating. One milliliter of an overnight rifampin-resistant cell culture was transferred to 10 ml of ASWJP nutrient broth containing 500 μg of rifampin per ml, and the preparation was incubated with shaking until the optical density at 600 nm was 0.8. One milliliter of this culture was mixed with 1 ml of a log-phase culture of plasmid donor strain E. coli RM1259 and 1 ml of a log-phase culture of helper strain E. coli CA60 in a sterile 15-ml tube. The mixture was filtered onto a 47-mm-diameter 0.2-μm-pore-size filter under a vacuum (∼150 mm of Hg). After filtration, the filter was placed on an ASWJP nutrient plate, and incubated overnight at 28°C. The filter was then transferred to 5 ml of artificial seawater medium in a sterile tube. Cells were washed off by vortexing the filter for 30 s. The cell suspension was plated onto ASWJP nutrient plates containing 250 μg of kanamycin per ml, 250 μg of streptomycin per ml, and 500 μg of rifampin per ml. The plates were incubated at 28°C for at least 48 h. Colonies that grew on the selection plates were picked, and we confirmed that they contained pQSR50 by colony hybridization, plasmid preparation, and Southern hybridization with radiolabeled probe nptII (see below). The sensitivities of these plasmid-containing bacterial hosts to their corresponding phages were tested by phage typing. Strain HSIC containing plasmid pQSR50 was designated HOPE-1, and strain D1B containing plasmid pQSR50 was designated HOPE-2 (Table 1). Both strain HOPE-1 and strain HOPE-2 were used as donors in the transduction assays.
Transduction assays.
Strains HOPE-1 and HOPE-2 were used as plasmid donors, and transducing particles were produced by infecting these strains with the corresponding temperature phages by the soft agar overlay method. The phages were eluted from the plates after overnight incubation by using warm 0.5 M Tris-HCl (pH 8.0). A second round of phage lysate was produced with the same donor strain to ensure that the transducing particles contained DNA only from the donors (28). The transducing lysates were filtered (pore size, 0.2 μm) to remove residual donor cells. A subsample of the transducing lysate was treated with UV radiation (NIS G15T8 15-W germicidal lamp; peak wavelength, 256 nm) to reduce the phage titer to 1% of the original titer (28). UV-treated and untreated phage lysates were digested with 50 U of DNase I per ml before they were used in the transduction assays to prevent transformation from occurring.
Both bacterial hosts isolated from Mamala Bay, Hawaii, and concentrated bacterial communities from Tampa Bay, Florida, the Gulf of Mexico, and Dry Tortugas, Florida, were used as recipients for transduction. For cultured recipients, 10- to 100-ml portions of log-phase cultures were mixed with transducing phage particles at multiplicities of infection (MOI) ranging from 0.01 to 10. Each control contained an equal volume of the recipient cell culture and 1 ml of 0.5 M Tris-HCl (pH 8.0). After a 10-min adsorption period, the unabsorbed phages were removed by three rounds of centrifugation and three washes with artificial seawater. The final washed cell pellet was resuspended in 0.5 to 1.5 ml of ASWJP nutrient broth, and the cells were allowed to recover (phenotypic expression) in this nonselective medium for 10 to 20 min before they were plated onto selective seawater nutrient plates containing 250 μg of kanamycin per ml and 250 μg of streptomycin per ml. The transducing phage lysate (containing no recipient) was also plated onto selective plates as a control.
For transduction assays in which indigenous marine bacterial communities were used as recipients, natural populations (in 20 to 100 liters of water) from a variety of marine environments were concentrated to volumes of approximately 50 ml by vortex flow filtration by using a 100-kDa-cutoff filter (21). One milliliter of transducing phage lysate was added to 10 ml of a concentrated microbial population and incubated at room temperature for 10 min to allow phage adsorption. The mixture was then filtered onto a 47-mm-diameter 0.2-μm-pore-size filter. An equal volume of concentrated sample was used as a control. The filter was rinsed with sterile ASWJP to wash off the unabsorbed phages and was transferred to 2 ml of sterile ASWJP nutrient broth in a 15-ml tube. Bacteria on the filter were resuspended in medium, and the suspension was plated onto selective medium plates containing 500 μg of kanamycin per ml and 1,000 μg of streptomycin per ml. The phage lysate was also plated onto selective plates as a no-recipient control.
Purification of plasmid DNA and gene probe construction.
Plasmid DNAs from the E. coli strains and from marine bacteria were purified by the alkaline lysis miniprep method (37) or with the Promega plasmid DNA purification system (Promega, Madison, Wis.).
A BamHI- and HindIII-digested fragment of the neomycin phosphotransferase gene (nptII) of plasmid pQSR50 was cloned into Riboprobe vector pGEM 4Z (Promega) by using the manufacturer’s recommended procedure. A detailed description of cloning and the location of this fragment on the plasmid map have been published elsewhere (10). A 35S-RNA probe was prepared by transcription of the fragment with T7 polymerase by using 35S-UTP (12). This probe, designated the nptII probe on the basis of the complementary gene sequence in the plasmid (12), hybridized with the Tn5 region of the plasmid.
Fragments of T-φHSIC DNA and T-φD1B DNA were also cloned into a Riboprobe vector (Promega); 35S-labeled single-stranded RNA probes, designated the T-φHSIC probe and the T-φD1B probe, respectively, were made as previously described (17).
Colony lift, dot blot, and Southern hybridization.
Colonies grown on agar plates were lifted by using a Magna charged nylon transfer membrane (MSI, Westboro, Mass.) and were lysed by soaking them in 2× SSC (0.3 M NaCl plus 0.03 M sodium citrate, pH 7.0) containing 5% sodium dodecyl sulfate and microwaving them for 2 min on gel blot paper (Schleicher & Schuell, Keene, N.H.). The lysed colonies were then denatured on gel blot paper saturated with 1.5 M NaOH and 1.5 M NaCl, and the pH was adjusted to 8.0 with 0.5 M Tris-HCl. DNA was fixed on the membrane by baking the membrane in a vacuum oven at 80°C for 2 h.
Plasmid DNA was dotted onto charged nylon membranes (Zetaprobe; Bio-Rad, Richmond, Calif.) by using a Bio-Rad Bio-Dot microfiltration apparatus. The DNA on the membrane was denatured, neutralized, and fixed as previously described (22). Southern transfer of DNA from agarose gels to charged nylon membranes (Zetaprobe; Bio-Rad) was performed by using the method of Sambrook et al. (37).
Hybridization of DNA with the nptII probe, T-φHSIC probe, or T-φD1B probe was performed at 42°C overnight. The wash temperature was 65°C, as previously described (17).
PCR amplification.
When natural bacterial communities were used as recipients in the transduction assays, two sets of primers were employed in PCR amplification to detect the pQSR50 gene sequence and to differentiate the putative transductants from the indigenous antibiotic-resistant colonies. The primer locations and sequences are shown in Fig. 1 and Table 2, respectively. Primers JP44 and JP52 were designed to amplify the Tn5 region of the plasmid, which is the region that codes for neomycin phosphotransferase. Primers JP64 and JP65 were used to amplify the region near the EcoRI site, which is the region farthest from the Tn5 insertion site (Fig. 1). The rationale for this procedure was to differentiate transposition of Tn5 from maintenance of the rest of the plasmid.
FIG. 1.

Map of plasmid pQSR50, including the locations of PCR primers. Arrows indicate primer directions.
TABLE 2.
Oligonucleotides used as PCR primers
| Primer | Sequence (5′-3′) | Size (bp) | Location (positions) |
|---|---|---|---|
| JP44 | GGGTCGGACGACAGGATGAGGATCGTTTCG | 30 | 5473–5490 |
| JP52 | CTCGGATCCAGCGGCGATACCGTAAAG | 27 | 6214–6234 |
| JP64 | GGATGCATTGAGCCAAATGAGGCGGTCACGC | 31 | 215–238 |
| JP65 | CTCGGATCCTGACGGGTGCCGGTATCAAACGC | 32 | 14105–14128 |
For amplification, the plasmid DNA was diluted 1 to 100 times with double-distilled deionized water. The PCR mixtures (total volume, 100 μl) contained 10 mM Tris, 50 mM MgCl2, 0.01% gelatin, 0.05% Nonidet P-40, each deoxynucleoside triphosphate at a concentration of 37.5 μM, 20 pmol of each primer, approximately 1 ng of template, and 2.5 U of Taq DNA polymerase. The Taq DNA polymerase was pretreated with the TaqStart antibody (Clontech, Palo Alto, Calif.) to reduce amplification artifacts. The reaction mixture was overlaid with 3 drops of sterile mineral oil. Sterile deionized water (DI) was used as a template for a negative control. The PCR cycle included a hot start (96°C, 5 min), 40 cycles consisting of 96°C for 45 s, 58°C for 1 min, and 72°C for 1.5 min, and finally incubation at 72°C for 10 min. The amplification products were analyzed by gel electrophoresis.
RESULTS
Establishing indigenous marine phage-host transduction systems.
To study the potential for gene transduction among marine phages and bacterial hosts, we attempted to establish transducing systems by using marine phage host isolates. Four marine phage-host systems were isolated from Mamala Bay, Hawaii (17), and plasmid pQSR50 was successfully introduced into two of the bacterial hosts; the resulting strains were designated HOPE-1 and HOPE-2.
Strains HOPE-1 and HOPE-2 grew more slowly than the wild-type strains, possibly because of the burden of the new extrachromosomal element. The plaquing efficiency of phage T-φD1B on strain HOPE-2 was lower than the plaquing efficiency of this phage on its wild-type host, D1B, because the titers of phage lysates collected after infection of HOPE-2 were often 10 to 100 times lower than the titers of lysates generated from infection of strain D1B. The sensitivity of strain HOPE-1 to the corresponding phage was basically unchanged, and strain HOPE-1 yielded approximately the same number of viruses per infection cycle as when wild-type strain HSIC served as the host.
Transduction assays performed with bacterial isolates as recipients.
Both UV-treated and untreated transducing lysates were used in transduction assays. Transducing lysates of T-φHSIC were more resistant to UV radiation, requiring 1 min of UV radiation at 464 mW/cm2 for a 2-log reduction in infectivity, while the infectivities of transducing lysates of T-φD1B were reduced by 2 logs after 10 s of radiation.
Putative transductants were found in three experiments when HOPE-1 was used as the donor strain, UV-treated T-φHSIC was used as the transducing lysate, and HSIC was used as the recipient (Table 3). Only transduction assays in which a low MOI (MOI, 0.01 to 0.05) was used produced detectable transductants, even though MOIs of up to 5 were also tested (data not shown). The frequencies of transduction in lab trials ranged from 5.13 × 10−9 to 1.33 × 10−7 transductants per PFU or 4.02 × 10−10 to 6.8 × 10−10 transductants per CFU. The transduction frequencies when the untreated HOPE-1–T-φHSIC and HOPE-2–T-φD1B systems were used were below the detection limit (see below).
TABLE 3.
Marine phage transduction with bacterial isolates as recipients
| Donor | Transducing phage(s) | Recipient | MOI | Frequencies of transduction
|
|
|---|---|---|---|---|---|
| No. of transductants/PFU | No. of transductants/CFU | ||||
| HOPE-1 | UV-treated T-φHSIC lysate | HSIC (10 ml) | 0.01 | 5.13 × 10−9 | 4.02 × 10−10 |
| HSIC (100 ml) | <0.005 | <2.6 × 10−11 | |||
| 0.01 | 1.33 × 10−7 | 6.8 × 10−10 | |||
| 0.05 | 1.33 × 10−8 | 6.8 × 10−10 | |||
| 0.5 | <2.6 × 10−11 | ||||
| Untreated T-φHSIC lysate | HSIC (10 ml) | 0.01–10 | NDa | ||
| HOPE-2 | UV-treated and untreated T-φD1B lysates | D1B (10 ml) | 0.01–5 | ND | |
ND, not detected.
The putative transductants found on the selective plates coexisted with the phage. The colonies appeared to be wrinkled and to have colony morphology similar to that of the lysogen L-HSIC (17). Transductants were confirmed by probing the colony lift membranes with the nptII probe. Strong hybridization of the nptII probe was found with E. coli RM1259, donor strain HOPE-1, and the transductants (data not shown). No hybridization was observed in the wild-type recipients, which suggests that plasmid DNA was introduced into the recipients by phage transduction.
Analysis of plasmid DNA from transductants.
To ensure that the transferred plasmid DNA was maintained as a plasmid in the transductants, plasmid DNAs extracted from E. coli RM1259, donor strains, recipient strains, and transductants were dotted onto a nylon membrane and probed with the plasmid nptII probe (Fig. 2). Hybridization of the probe with DNAs from E. coli RM1259, the donor strains, and all transductants was observed in the autoradiograph. No hybridization with the plasmid DNA from recipient strain HSIC was found. This result suggested that the transferred plasmid DNA was maintained as an extrachromosomal element in the transductant’s cells.
FIG. 2.

Dot blot hybridization of plasmid DNAs from E. coli RM1259, donor strain HOPE-1, recipient strain HSIC, and transductants with the nptII probe.
The restriction patterns of the plasmid DNAs from transductants, donors, and recipients were analyzed to determine similarities and differences between the cells. Wild-type recipient strain HSIC contained a high-copy-number plasmid upon isolation, which interfered with the restriction pattern of the transduced plasmid (data not shown). Therefore, to determine the restriction patterns of the transduced plasmid, HindIII-digested and undigested plasmid DNAs from transductants, donors, recipients, and E. coli RM1259 were Southern transferred to a nylon membrane and probed with radiolabeled nptII probe. Figure 3 shows the autoradiograph from the Southern hybridization experiment. The hybridization patterns of the digested and undigested transductant plasmid DNAs were identical to those of the donor plasmid DNA but were different from those of pQSR50 DNA in E. coli RM1259. One large hybridization band was missing from undigested pQSR50 in E. coli RM1259 (Fig. 3, arrow). This change in the plasmid band pattern may have resulted from transposition of Tn5 from plasmid pQSR50 to an indigenous plasmid of the wild-type bacteria or from maintenance of the plasmid as a multimer in the new host. No hybridization with either uncut or HindIII-cut plasmid DNA of the recipient was observed.
FIG. 3.

Southern hybridization of undigested and HindIII-digested plasmid DNAs from transductants, E. coli RM1259, donor strain HOPE-1, and recipient strain HSIC, probed with the nptII probe. Lanes 1 through 20, plasmid DNAs from transductants; lanes 21 and 22, plasmid DNA from E. coli RM1259; lanes 23 and 24, plasmid DNA from donor strain HOPE-1; lanes 25 and 26, plasmid DNA from recipient strain HSIC. Odd-numbered lanes contained undigested plasmid DNA, whereas even-numbered lanes contained HindIII-digested DNA. Molecular weights (M) (in kilobases) are indicated on the left.
According to the plasmid pQSR50 gene map (Fig. 1) presented previously (10), HindIII digestion of this plasmid should generate a single 3.4-kb diagnostic band which hybridizes with the nptII probe. However, in all HindIII-digested plasmid DNAs, including the DNA of pQSR50 from parental strain RM1259, an unexpected 1.2-kb band was found to hybridize strongly with the probe. This Southern hybridization experiment was performed several times by using plasmid DNA extracted by different methods, and the presence of this 1.2-kb band was confirmed. Although the reason for this band is unknown, a similar hybridization pattern for HindIII-digested pQSR50 was also seen by other workers (51).
A replicate Southern transfer of plasmid DNAs was probed with the T-φHSIC probe, and a 9-kb HindIII fragment was found to hybridize in all digests of transductant plasmid DNA (Fig. 4), while no hybridization was found in plasmid DNA from the donor, recipient, or E. coli RM1259. This result suggests that all of the transductants were lysogenized. However, whether transduction and lysogenization occurred simultaneously, with plasmid and phage DNAs entering each cell from a single transducing phage particle, or in a sequential process involving more than one phage is not known. The sizes of the T-φHSIC genome and plasmid pQSR50 are 37 and 14.4 kb, respectively. Therefore, it is possible for a phage particle to contain both plasmid DNA and part of the phage genome. However, other possibilities are equally likely. To confirm lysogenization, all putative transductants were tested for sensitivity to T-φHSIC, and all were found to be resistant.
FIG. 4.

Southern hybridization of undigested (odd-numbered lanes) and HindIII-digested (even-numbered lanes) plasmid DNAs from transductants, E. coli RM1259, and donor and recipient strains probed with the T-ΦHSIC riboprobe. For the contents of the lanes see the legend to Figure 3.
Transduction assays performed with indigenous bacterial communities as recipients.
To understand the potential for transduction in the marine environment, concentrated bacterial communities from a variety of marine environments were used as transduction recipients. Six samples were collected from Tampa Bay, Florida, the Gulf of Mexico, and Dry Tortugas, Florida, and indigenous bacteria from four of the sample sites were resistant to kanamycin and streptomycin and hybridized with the nptII probe before transduction, which made it impossible to detect transductants in these samples by the currently used detection method. This may have been caused by Tn5-like sequences in the natural communities.
Potential transduction was found in two experiments (Table 4) performed with the bacterial recipients collected from the mouth of Tampa Bay and the deep-sea environment of the Gulf of Mexico. The indigenous kanamycin- and streptomycin-resistant bacteria from these two locations did not hybridize with the nptII probe before transducing phages were added (Fig. 5), but they were hybridization positive after transduction assays performed with the HOPE-2–T-φD1B transducing lysate and the UV-treated HOPE-2–T-φD1B transducing lysate, respectively. The frequencies of transduction in these bacterial communities ranged from 1.57 × 10−8 to 3.7 × 10−8 transductants/PFU (Table 4). No transduction was observed with T-φHSIC lysates.
TABLE 4.
Transduction performed with concentrated marine bacterial communities as recipients
| Recipient sampling site | Donor | Transducing phage(s) | Transduction frequency (no. of transductants/PFU) | Transduction detection limit (no. of transductants/PFU) |
|---|---|---|---|---|
| Mouth of Tampa Bay (27°35′, 82°43′) | HOPE-1 | T-φHSIC | NDa | 1.74 × 10−10 |
| HOPE-1 | UV-treated T-φHSIC | ND | 1.74 × 10−10 | |
| HOPE-2 | T-φD1B | 1.57 × 10−8 | 1.74 × 10−10 | |
| Gulf of Mexico (27°04’, 83°78′) | HOPE-2 | T-φD1B, UV-treated T-φD1B | —b | |
| Gulf of Mexico, Chal Max (28°38′, 84°35′) | HOPE-2 | T-φD1B, UV-treated T-φD1B | — | |
| Gulf of Mexico (depth 1,500 m) (24°55′, 85°32′) | HOPE-2 | UV-treated T-φD1B | 3.7 × 10−8 | 3.7 × 10−8 |
| African Reef, Loggerhead Key (24°39′, 82°56′) | HOPE-1 | T-φHSIC, UV-treated T-φHSIC | — | |
| HOPE-2 | T-φD1B, UV-treated T-φD1B | — | ||
| Wreck, Loggerhead Key (24°39′, 82°56′) | HOPE-2 | T-φD1B, UV-treated T-φD1B | — |
ND, not detected.
—, sequences homologous to nptII are present in the indigenous population.
FIG. 5.

Colony hybridization after a transduction assay performed with indigenous bacterial communities from Tampa Bay as recipients. (A) Control (no lysate added). (B) Transduction with HOPE-2–T-φD1b lysate. (C) Transduction with HOPE-1–T-φHSIC lysate. (D) Transduction with UV-treated HOPE-1– T-φHSIC lysate.
Plasmid miniprep analysis of these putative transductants yielded restriction patterns unlike the restriction patterns of the native pQSR50 plasmid (data not shown). Since we have observed similar phenomena after plasmid transformation of indigenous flora recently (12), we used PCR primers specific for pQSR50 to amplify the plasmid DNA sequences from the control colonies (antibiotic-resistant indigenous recipients that did not hybridize with the plasmid probe) and putative transductants. Figure 6 shows the results of amplification with primers JP44 and JP52, which specifically amplify the Tn5 region of the pQSR50 plasmid. Amplification products were observed only when the transductant plasmid and pQSR50 DNAs were used as templates (Fig. 6, lanes 1 through 7 and 13, respectively) and were hybridized with the nptII probe. Amplification products or hybridization signals were not found when the plasmid DNAs of the control colonies were used.
FIG. 6.

Autoradiograph after Southern transfer of an agarose gel containing PCR products obtained with primers JP44 and JP52. The template DNAs were plasmid DNAs of transductants (lanes 1 through 7), plasmid DNAs of indigenous antibiotic-resistant bacteria (lanes 8 through 12), and pQSR50 DNA (lane 13). Lane 14 was a negative control (no template DNA). The preparations were hybridized with the nptII probe. Lane M contained a molecular weight marker; sizes (in base pairs) are indicated on the left.
Primers JP64 and JP65, designed to specifically amplify a 635-bp region of pQSR50 containing an EcoRI site (Fig. 1), were also used to amplify the plasmid DNAs from the transductants and control colonies. Amplification products that were approximately 700 bp long were observed in the plasmid DNAs of the transductants and pQSR50 (Fig. 7). However, the transductants’ plasmid amplification products were slightly larger than the pQSR50 amplification product and seemed to have lost the EcoRI site (Fig. 7). They were resistant to EcoRI digestion, while the amplification product of pQSR50 was digested into two fragments (Fig. 7). Plasmid pQSR50 may have recombined with an indigenous extrachromosomal element(s), and the restriction site may have been lost during the process. The presence of similar gene sequences in the recipient cells may have also been a factor facilitating the transduction process (see below).
FIG. 7.

PCR amplification performed with primers JP64 and JP65 and EcoRI-digested amplification products. The template DNAs were DNAs from plasmids of transductants (lanes 1 through 10), indigenous antibiotic-resistant bacteria (lanes 11 and 12), and pQSR50 (lanes 13 and 14). Odd-numbered lanes contained uncut DNA, and even-numbered lanes contained EcoRI-cut DNA. Lane M contained a molecular weight ladder; sizes (in base pairs) are indicated on the right.
Primers JP64 and JP65 were also used to amplify plasmid DNAs from nptII hybridization-positive indigenous bacteria found in other environments. No amplification products were found in any of the samples tested (data not shown). This result suggests that the indigenous bacteria were different from the putative transductants. Only putative transductants contained gene sequences similar to the gene sequences in the region near the EcoRI restriction site of the pQSR50 plasmid.
A gene probe constructed for the T-φD1B gene was used to probe the putative transductants from the environment. No hybridization occurred, suggesting that the transductants were not lysogenized (data not shown).
DISCUSSION
The results of transduction assays performed with indigenous marine phage host isolates and mixed bacterial communities as recipients suggest that transduction can be a means of gene transfer in the marine environment. In this study, we demonstrated that a marine phage host isolate is capable of transferring an antibiotic-resistant plasmid among bacterial hosts. Plasmid transduction in this phage-host system was confirmed by selection of antibiotic-resistant colonies on nutrient plates, colony hybridization, plasmid DNA restriction analysis, and Southern hybridization. The potential for plasmid transduction in marine bacterial communities was also assayed by using concentrated indigenous bacterial populations as recipients. Putative transductants were detected in two of the six experiments performed. These putative environmental transductants were separated from indigenous antibiotic-resistant bacteria by colony hybridization and PCR amplification with two sets of primers specific for two regions of the plasmid. This was the first attempt to study the potential for transduction in mixed marine bacterial communities. Ackermann and DuBow (1) have suggested that transduction can occur in all phage-host systems because mistakes in phage replication are made in all systems. However, the frequencies of transduction are often below the detection limit, or there is not a genetic marker which can be detected.
The use of UV radiation-treated transducing lysates in this study may have increased the frequency of transduction. Many studies have shown that UV treatment of transducing lysates increases the frequency of transduction from 10- to 50-fold (28). The effect of UV radiation may be due to inactivation of virulent effects of the infectious bacteriophage particles that are present in the lysate (7, 8). Alternatively, it has been suggested that this treatment stimulates recombination within a recipient cell, which leads to increased incorporation of the transduced DNA into the recipient’s genetic elements (4). Sandri and Berger (38) showed that only about 10% of the P1 transducing DNA injected into a recipient cell was stably maintained in the cell. Recombination with the recipient native genetic elements could increase the stability of the introduced DNA. Therefore, the presence of gene sequences similar to the gene sequence of the introduced DNA in a recipient cell may increase the transduction frequency via recombination. Such a similarity may facilitate plasmid transfer to natural communities.
Potential for gene transduction in the marine environment.
Both viruses and bacteria are abundant and active in the marine environment. Recently, lysogenic bacteria have also been shown to be an important component of marine bacterial communities (18–20). It is reasonable to believe that transduction occurs in natural marine microbial populations. To predict the potential rate of transduction in the marine environment, we constructed a simple model by using the following factors: bacterial abundance, viral abundance, and the frequencies of transduction. The results of previous transduction assays (23, 30, 36, 46) and this study suggested that transduction frequency is a function of MOI and that this function is most similar to a second-order polynomial function (Y = aX2 + bX + c), with the frequency of transduction decreasing below and above the optimal MOI. Using transduction frequencies and MOI data presented in Table 4, we predicted a second-order polynomial equation by using Excel spreadsheet software (Microsoft Corp.). The relationship between transduction frequencies and MOI is expressed in a numerical model by equation 1:
 |
1 |
where Ft is the frequency of transduction and can be rewritten as the ratio of the number of transductants (T) to the number of recipient bacteria (B) and M is the MOI, which is the ratio of phage concentration (P) to recipient bacterial concentration (B). Therefore, the numerical model for transduction (equation 1) can be transformed to equations 2 and 3:
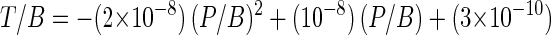 |
2 |
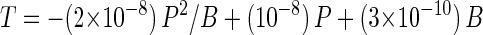 |
3 |
Assuming that there are n types of phage-host systems in the marine environment and that all natural systems fit our model of transduction based on the study of marine phage host isolates, then the total number of transductants (Tt) is sum of the number of transductants in each phage-host system (Ti) and can be written as:
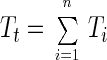 |
4 |
Therefore, the total number of transductants in an aquatic system after extrapolation from equations 3 and 4 can be modeled by equation 5:
 |
5 |
 |
where
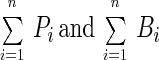 |
are the total number of viruses (Pt) and the total number of bacteria (Bt), respectively, in an environment. Therefore, equation 5 can be rewritten:
 |
6 |
In any microbial system, Pi ranges from 1 to a maximum of the total viral density. Bi ranges from the below the threshold density which supports viral replication to a maximum of the total bacterial concentration. Wiggins and Alexander (50) suggested that the threshold density for bacterial hosts which supports viral replication is about 104 cells/ml in the aquatic environment. By using bacterial and viral concentrations of 2 × 109 cells/liter and 1010 virus particles/liter, respectively, for Tampa Bay, Florida (18), the total number of transductants in 1 liter of Tampa Bay water per day was calculated from equation 6. The results ranged from negative values to 100 transductants/liter per day. Negative values resulted when the concentration of one type of phage was more than 50% of its host concentration (Pi/Bi > 0.5), which is unlikely in the environment. The maximum value resulted when the concentration of the host population was maximum and the phage concentration was the lowest concentration observed. Assuming that negative values represent zero transductants, then from 0 to 100 transductants can occur in 1 liter of water in 1 day in the Tampa Bay environment. When this transduction rate was extrapolated to the ecosystem scale of the Tampa Bay Estuary by using a bay water volume of 3.56 × 1011 liters, transduction rates of up to 1.3 × 1014 transductants per year were estimated.
This is the first attempt to quantify transduction rates in the marine environment, and our model is based on several assumptions. For example, the model assumes that all marine phages are infective. However, several researchers (45, 54) have suggested that a large number of phages in the environment are not infectious because they are inactivated by solar radiation. Furthermore, we assumed that all phage-host systems in the marine environment fit the transduction model generated with a single phage-host system, which is unlikely. Other marine phage transduction systems may have generated different numerical models. In fact, higher transduction frequencies per MOI were detected previously in a marine Vibrio-phage transduction system (16). To simplify the model, many factors which may influence the transduction rate were not considered in the model, including temperature, ionic strength, effect of predation on marine bacteria, and the nonspecific attachment of phage to other particles. To achieve a more closely fitting transduction model for the marine environment, several marine phage-host transduction systems should be used to generate polynomial empirical curves and equations, and all of the factors mentioned above should be integrated into the model. However, despite the limitations of our current transduction model, this quantitative estimation tool is important to our understanding of the potential for transduction in marine microbial systems.
In summary, this research demonstrated the potential for bacteriophage-mediated gene transfer in the marine environment. Gene transfer by transduction may be an important mechanism for gene evolution in the marine environment, and bacteriophage transduction could play an important role in contributing to the genetic diversity of marine microbial populations.
REFERENCES
- 1.Ackermann H W, DuBow M S. Viruses of prokaryotes. 1. General properties of bacteriophages. Boca Raton, Fla: CRC Press; 1987. [Google Scholar]
- 2.Bale M J, Fry J C, Day M J. Plasmid transfer between strains of Pseudomonas aeruginosa on membrane filters attached to river stones. J Gen Microbiol. 1978;133:3099–3107. doi: 10.1099/00221287-133-11-3099. [DOI] [PubMed] [Google Scholar]
- 3.Barsomina G D, Robillard N J, Throne C B. Chromosomal mapping of Bacillus thuringiensis by transduction. J Bacteriol. 1984;157:746–750. doi: 10.1128/jb.157.3.746-750.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benzinger T, Hartmen P E. Effect of ultraviolet light on transducing phage P22. Virology. 1962;18:263–268. doi: 10.1016/0042-6822(62)90064-8. [DOI] [PubMed] [Google Scholar]
- 5.Bergh O, Borsheim K Y, Bratbak G, Heldal M. High abundance of viruses found in aquatic environments. Nature. 1989;340:467–468. doi: 10.1038/340467a0. [DOI] [PubMed] [Google Scholar]
- 6.Birge E A. Bacterial and bacteriophage genetics. 3rd ed. New York, N.Y: Springer-Verlag; 1994. [Google Scholar]
- 7.Buchanan-Wollaston V. Generalized transduction in Rhizobium leguminosarum. J Gen Microbiol. 1979;112:135–142. [Google Scholar]
- 8.Ely B, Johnson R C. Generalized transduction in Caulobacter crescentus. Genetics. 1977;87:391–399. doi: 10.1093/genetics/87.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finan T M, Hartwieg E, LeMieux K, Bergman K, Walker G C, Signer E T. General transduction in Rhizobium meliloti. J Bacteriol. 1984;159:120–124. doi: 10.1128/jb.159.1.120-124.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frischer M E. Ph.D. dissertation. St. Petersburg, Fla: University of South Florida; 1994. [Google Scholar]
- 11.Frischer M E, Gregory G J, Paul J H. Plasmid transfer to indigenous marine bacterial populations by natural transformation. FEMS Microbiol Ecol. 1994;15:127–136. [Google Scholar]
- 12.Frischer M E, Thurmond J M, Paul J H. Natural plasmid transformation in a high-frequency-of-transformation marine Vibrio strain. Appl Environ Microbiol. 1990;56:3439–3444. doi: 10.1128/aem.56.11.3439-3444.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frischer M E, Thurmond J M, Paul J H. Factors affecting competence in a high frequency of transformation marine Vibrio. J Gen Microbiol. 1993;139:753–761. [Google Scholar]
- 14.Fuhrman J A, Suttle C A. Viruses in marine planktonic systems. Oceanography. 1993;6:57–63. [Google Scholar]
- 15.Gowland P C, Slater J H. Transfer and stability of drug resistance plasmids in Escherichia coli K12. Microb Ecol. 1984;10:1–13. doi: 10.1007/BF02011590. [DOI] [PubMed] [Google Scholar]
- 16.Ichige A, Matsutani S, Oishi K, Mizushima S. Establishment of gene transfer systems and construction of the genetic map of a marine Vibrio strain. J Bacteriol. 1989;171:1825–1834. doi: 10.1128/jb.171.4.1825-1834.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang S C, Kellogg C A, Paul J H. Characterization of marine temperate phage-host systems isolated from Mamala Bay, Hawaii. Appl Environ Microbiol. 1998;64:535–542. doi: 10.1128/aem.64.2.535-542.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang S C, Paul J H. Seasonal and diel abundance of viruses and occurrence of lysogeny/bacteriocinogeny in the marine environment. Mar Ecol Prog Ser. 1994;104:163–172. [Google Scholar]
- 19.Jiang S C, Paul J H. Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Mar Ecol Prog Ser. 1996;142:27–38. [Google Scholar]
- 20.Jiang S C, Paul J H. Significance of lysogeny in the marine environment: studies with isolates and an ecosystem model. Microb Ecol. 1998;35:235–243. doi: 10.1007/s002489900079. [DOI] [PubMed] [Google Scholar]
- 21.Jiang S C, Thurmond J M, Pichard S L, Paul J H. Concentration of microbial populations from aquatic environments by vortex flow filtration. Mar Ecol Prog Ser. 1992;80:101–107. [Google Scholar]
- 22.Kellogg C A, Rose J B, Jiang S C, Thurmond J M, Paul J H. Genetic diversity of related vibriophages isolated from marine environments around Florida and Hawaii. Mar Ecol Prog Ser. 1995;120:89–98. [Google Scholar]
- 23.Keynan A, Nealson K, Sideropoulos H, Hastins J W. Marine transducing bacteriophage attacking a luminous bacterium. J Virol. 1974;14:333–340. doi: 10.1128/jvi.14.2.333-340.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levisohn R, Moreland J, Nealson K H. Isolation and characterization of a generalized transducing phage for the marine luminous bacterium Vibrio fischeri MJ-1. J Gen Microbiol. 1987;133:1577–1582. [Google Scholar]
- 25.Martin M O, Long S R. Generalized transduction in Rhizobium meliloti. J Bacteriol. 1984;159:125–129. doi: 10.1128/jb.159.1.125-129.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McHenney M A, Baltz R H. Transduction of plasmid DNA in Streptomyces spp. and related genera by bacteriophage FP43. J Bacteriol. 1988;170:2276–2282. doi: 10.1128/jb.170.5.2276-2282.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer R, Laux R, Boch G, Hinds M, Bayly R, Shapiro J A. Broad-host-range IncP-4 plasmid R1162: effects of deletions and insertions on plasmid maintenance and host range. J Bacteriol. 1982;152:140–150. doi: 10.1128/jb.152.1.140-150.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller R V. Methods for evaluating transduction: an overview with environmental considerations. In: Levin M A, Seidler R J, Rogul M, editors. Microbial ecology. Principles, methods, and applications. New York, N.Y: McGraw-Hill, Inc.; 1992. [Google Scholar]
- 29.Miller R V, Ripp S, Replicon J, Ogunseitan O A, Kokjohn T A. Virus-mediated gene transfer in freshwater environments. In: Gauthier M J, editor. Gene transfers and environment. Proceedings of Third European Meeting on Bacterial Genetics and Ecology (BAGECO-3). Berlin, Germany: Springer Verlag; 1991. pp. 51–62. [Google Scholar]
- 30.Morgan A F. Transduction of Pseudomonas aeruginosa with a mutant of bacteriophage E79. J Bacteriol. 1979;139:137–140. doi: 10.1128/jb.139.1.137-140.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison W D, Miller R V, Sayler G S. Frequency of F116-mediated transduction of Pseudomonas aeruginosa in a freshwater environment. Appl Environ Microbiol. 1978;36:724–730. doi: 10.1128/aem.36.5.724-730.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Morchoe S B, Ogunseitan O, Sayler G S, Miller R V. Conjugal transfer of R68.45 and FP5 between Pseudomonas aeruginosa strains in a freshwater environment. Appl Environ Microbiol. 1988;54:1923–1929. doi: 10.1128/aem.54.8.1923-1929.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul J H, Frischer M E, Thurmond J T. Gene transfer in marine water column and sediment microcosms by natural plasmid transformation. Appl Environ Microbiol. 1991;57:718–724. doi: 10.1128/aem.57.5.1509-1515.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raya R R, Klaenhammer R. High-frequency plasmid transduction by Lactobacillus gasseri bacteriophage φadh. Appl Environ Microbiol. 1992;58:187–193. doi: 10.1128/aem.58.1.187-193.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ripp S, Miller R V. Effects of suspended particulates on the frequency of transduction among Pseudomonas aeruginosa in a freshwater environment. Appl Environ Microbiol. 1995;61:1214–1219. doi: 10.1128/aem.61.4.1214-1219.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ripp S, Ogunseitan O A, Miller R V. Transduction of a freshwater microbial community by a new Pseudomonas aeruginosa generalized transducing phage, UT1. Mol Ecol. 1994;3:121–126. doi: 10.1111/j.1365-294x.1994.tb00112.x. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 38.Sandri R M, Berger H. Bacteriophage P1-mediated generalized transduction in Escherichia coli: fate of transduced DNA in Rec+ and RecA− recipients. Virology. 1980;106:14–29. doi: 10.1016/0042-6822(80)90217-2. [DOI] [PubMed] [Google Scholar]
- 39.Saye D J, Ogunseitan O, Sayler G S, Miller R V. Transduction of linked chromosomal genes between Pseudomonas aeruginosa strains during incubation in situ in a freshwater habitat. Appl Environ Microbiol. 1990;56:140–145. doi: 10.1128/aem.56.1.140-145.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saye D J, Ogunseitan O, Sayler G S, Miller R V. Potential for transduction of plasmids in a natural freshwater environment: effect of plasmid donor concentration and a natural microbial community on transduction in Pseudomonas aeruginosa. Appl Environ Microbiol. 1987;53:987–995. doi: 10.1128/aem.53.5.987-995.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sik T, Horvath J, Chatterjee S. Generalized transduction in Rhizobium meliloti. Mol Gen Genet. 1980;178:511–516. doi: 10.1007/BF00337855. [DOI] [PubMed] [Google Scholar]
- 42.Steward G F, Smith D C, Azam F. Abundance and production of bacteria and viruses in the Bering Seas and Chukchi Seas. Mar Ecol Prog Ser. 1996;131:287–300. [Google Scholar]
- 43.Stewart G J, Sinigalliano C D. Detection of horizontal gene transfer by natural transformation in native and introduced species of bacteria in marine and synthetic environments. Appl Environ Microbiol. 1990;56:1818–1824. doi: 10.1128/aem.56.6.1818-1824.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stotzky G. Gene transfer among bacteria in soil. In: Levy S B, Miller R V, editors. Gene transfer in the environment. New York, N.Y: McGraw-Hill; 1989. pp. 165–222. [Google Scholar]
- 45.Suttle C A, Chen F. Mechanisms and rates of decay of marine viruses in seawater. Appl Environ Microbiol. 1992;58:3721–3729. doi: 10.1128/aem.58.11.3721-3729.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Svab Z, Kondorasi A, Orosz L. Specialized transduction of cysteine marker by Rhizobium meliloti phage 16-3. J Gen Microbiol. 1978;106:321–327. [Google Scholar]
- 47.Torrella F, Morita R Y. Evidence by electron micrographs for a high incidence of bacteriophage particles in the waters of Yaquina Bay, Oregon: ecological and taxonomical implications. Appl Environ Microbiol. 1979;37:774–778. doi: 10.1128/aem.37.4.774-778.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinbauer M G, Fuks D, Puskaric S, Peduzzi P. Diel, seasonal and depth related variability of viruses and dissolved DNA in the northern Adriatic Sea. Microb Ecol. 1995;30:25–41. doi: 10.1007/BF00184511. [DOI] [PubMed] [Google Scholar]
- 49.Weiss B D, Capage M A, Kessel M, Benson S A. Isolation and characterization of a generalized transducing phage for Xanthomonas campestris pv. campestris. J Bacteriol. 1994;176:3354–3359. doi: 10.1128/jb.176.11.3354-3359.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wiggins B A, Alexander M. Minimum bacterial density for bacteriophage replication: implications for significance of bacteriophages in natural ecosystems. Appl Environ Microbiol. 1985;49:19–23. doi: 10.1128/aem.49.1.19-23.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williams, H. G. Personal communication.
- 52.Winstanley C, Morgan J A W, Pickup R, Jone J G, Sauders J R. Differential regulation of lambda PL and PR promoters by a cI repressor in a broad-host-range thermoregulated plasmid marker system. Appl Environ Microbiol. 1989;55:771–777. doi: 10.1128/aem.55.4.771-777.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wommack K E, Hill R, Muller T A, Colwell R R. Effects of sunlight on bacteriophage viability and structure. Appl Environ Microbiol. 1996;62:1336–1341. doi: 10.1128/aem.62.4.1336-1341.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeph L R, Onaga M A, Stotzky G. Transduction of Escherichia coli by bacteriophage P1 in soil. Appl Environ Microbiol. 1988;54:1731–1737. doi: 10.1128/aem.54.7.1731-1737.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zinder N D, Lederberg J. Genetic exchange in Salmonella. J Bacteriol. 1952;64:679–699. doi: 10.1128/jb.64.5.679-699.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]