

Abstract
The enantioselective amidase from Rhodococcus sp. strain R312 was produced in Escherichia coli and was purified in one chromatographic step. This enzyme was shown to catalyze the acyl transfer reaction to hydroxylamine from a wide range of amides. The optimum working pH values were 7 with neutral amides and 8 with α-aminoamides. The reaction occurred according to a Ping Pong Bi Bi mechanism. The kinetic constants demonstrated that the presence of a hydrophobic moiety in the carbon side chain considerably decreased the Kmamide values (e.g., Kmamide = 0.1 mM for butyramide, isobutyramide, valeramide, pivalamide, hexanoamide, and benzamide). Moreover, very high turnover numbers (kcat) were obtained with linear aliphatic amides (e.g., kcat = 333 s−1 with hexanoamide), whereas branched-side-chain-, aromatic cycle- or heterocycle-containing amides were sterically hindered. Carboxylic acids, α-amino acids, and methyl esters were not acyl donors or were very bad acyl donors. Only amides and hydroxamic acids, both of which contained amide bonds, were determined to be efficient acyl donors. On the other hand, the highest affinities of the acyl-enzyme complexes for hydroxylamine were obtained with short, polar or unsaturated amides as acyl donors (e.g., KmNH2OH = 20, 25, and 5 mM for acetyl-, alanyl-, and acryloyl-enzyme complexes, respectively). No acyl acceptors except water and hydroxylamine were found. Finally, the purified amidase was shown to be l-enantioselective towards α-hydroxy- and α-aminoamides.
Many bacterial amidases (EC 3.5.1.4) have been described previously because of their amide hydrolysis activities. Wide-spectrum amidases from Rhodococcus sp. strain R312 (26) and Pseudomonas aeruginosa (1), which are very similar, hydrolyze only short-chain amides. These enzymes are made up of four and six identical subunits having molecular weights of about 45,000 and 35,000, respectively. Based on the results of experiments performed with inhibitors, they have been classified as belonging to a branch of sulfhydryl enzymes (1, 26). The other amidases, the enantioselective amidases from Pseudomonas chlororaphis B23 (5), Rhodococcus erythropolis MP50 (12, 27), Rhodococcus sp. strain R312 (20), Rhodococcus sp. strain N-774 (10), Rhodococcus sp. (21), and Rhodococcus rhodochrous J1 (14), belong to a group of amidases containing a GGSS signature in the amino acid sequence (4) and are made up of two (or eight) identical subunits. The corresponding genes are located in clusters containing genes encoding the two subunits of a nitrile hydratase (EC 4.2.1.84). These amidases were also previously classified as sulfhydryl enzymes (5, 15), but no active amino acid residue was identified in any of them. Recently, Kobayashi et al. (15) showed that the real active site residues of the amidase from R. rhodochrous J1 were Asp-191 and Ser-195 rather than the generally accepted Cys-203 residue. These authors showed that aspartic acid and serine residues of this enzyme were also present in the active site sequences of aspartic proteinases and suggested that there is an evolutionary relationship between amidases and aspartic proteinases.
All of the different amidases also exhibit an acyl transfer activity in the presence of hydroxylamine: RCONH2 + NH2OH ↔ RCONHOH + NH3. This kind of reaction was previously described for the wide-spectrum amidase from Rhodococcus sp. strain R312 (6), but there has been no detailed study examining the acyl transfer reaction of amidases belonging to the GGSS signature-containing group. The final reaction products (hydroxamic acids) are known to possess high chelating properties. Some of them (particularly α-aminohydroxamic acid derivatives) are potent inhibitors of matrix metalloproteases, a family of zinc endopeptidases involved in tissue remodelling (3). Some other hydroxamic acids (α-aminohydroxamic acids, synthetic siderophores, acetohydroxamic acid, etc.) have also been investigated as anti-human immunodeficiency virus agents or antimalarial agents or have been recommended for treatment of ureaplasma infections and anemia (2, 8, 13, 28). Moreover, some fatty hydroxamic acids have been studied as inhibitors of cylooxygenase and 5-lipooxygenase with potent antiinflammatory activity (9).
Apart from these medical applications, some hydroxamic acids (particularly polymerizable unsaturated hydroxamic acids and mid-chain or long-chain hydroxamic acids) have also been extensively investigated in wastewater treatment and nuclear technology studies as a way to eliminate contaminating metal ions (11, 16, 18).
In this paper we describe the formation of a wide range of hydroxamic acids with the enantioselective amidase (a 120,000-dalton homodimer) from Rhodococcus sp. strain R312, and we provide some additional information which enhanced our comprehension of the reaction mechanism of this amidase.
MATERIALS AND METHODS
Bacterial strain and growth medium.
To produce large amounts of amidase, a construction was made in which the amdA gene was inserted under the control of the bacteriophage T7 promoter into the pET3c expression plasmid (24). First, NdeI and BglII sites were created at the ATG initiation and TGA stop codons of amdA by PCR amplification. The PCR was performed with Promega Taq polymerase, as described in the manufacturer’s recommendations, 1.5 mM MgCl2, and deoxynucleoside triphosphates at a concentration of 0.2 mM. For amplification of the 1,583-bp fragment, the following program was used: 5 min of denaturation at 95°C, 30 cycles consisting of 45 s at 95°C, 30 s at 55°C, and 60 s at 72°C, and a final step consisting of 5 min at 72°C. The following primers were used: forward primer 5′-AGCACACTTCATATGGCGACAATCCGACCTGAC-3′ and reverse primer 5′-GAAGATCTCAGGCGGGGCTGAGTTGTGGTGC-3′ (the start and stop codons are indicated by boldface type, and NdeI and BglII restriction sites are underlined). The 1,583-bp amplified fragment obtained was then digested by NdeI and BglII and ligated to pET3c which had been digested with NdeI and BamHI. The resulting plasmid (pETADI) was introduced into E. coli BL21(DE3) in which the bacteriophage T7 RNA polymerase gene, under lacUV5 promoter control, was incorporated into the chromosome (25).
Plasmid isolation from E. coli, DNA manipulations, and plasmid transformation into E. coli cells were performed as described by Sambrook et al. (22).
Recombinant cells were cultured at 29°C in shaking flasks in Luria-Bertani growth medium containing 0.5% (wt/vol) yeast extract, 1% (wt/vol) tryptone, and 1% (wt/vol) NaCl. Ampicillin (200 μg/ml) was added to maintain selection pressure. Cultures were grown without induction of enzyme biosynthesis because no increase in acyl transfer activity was observed in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG), an inducer of the lacUV5 promoter.
Preparation of amidase.
One liter of recombinant E. coli cells grown under the conditions described above was harvested during the stationary phase by centrifugation for 15 min at 10,000 × g, and the cells were resuspended in 20 mM phosphate buffer (pH 7) (NaH2PO4, Na2HPO4) in a 10-fold-smaller volume. Ten-milliliter portions of the cell suspension (150 to 200 mg, dry weight), cooled with sodium chloride-saturated ice, were subjected to 6 min of ultrasonic treatment with a Branson model 250 Sonifier (output, 50 W; duty cycle, 30%). The cell debris was removed by centrifugation for 15 min at 10,000 × g. The supernatant of the first centrifugation (S1 supernatant) was then subjected to ultracentrifugation for 90 min at 180,000 × g. The S2 supernatant was designated the crude soluble enzyme preparation.
Enzyme purification was performed with an anion-exchange carrier (Q-Sepharose Fast Flow chromatography; Pharmacia). The column (26 by 400 mm) was equilibrated with 50 mM Tris-HCl buffer (pH 7.3). Proteins were eluted with a linear 0.2 to 0.5 M sodium chloride gradient at a flow rate of 250 ml/h and were collected in 10-ml fractions. The amidase eluted as a single peak at an NaCl concentration of about 0.4 M. The active fractions were pooled, and the preparation obtained was the amidase preparation used for the kinetic studies.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis was performed by the method of Laemmli (17).
Acyl transfer activity.
Acyl transfer activity was determined spectrophotometrically by a modified version of the method of Brammar and Clarke (1). Each reaction was performed at 30°C in a glass reaction tube. Unless otherwise specified, the reaction medium contained 1 ml of a solution of amide (or amide hydrochloride adjusted to the pH required with 10 N NaOH) at a concentration that was four times the final amide concentration in the reaction medium (CA), 1 ml of a solution of hydroxylamine hydrochloride at a concentration that was four times the final hydroxylamine concentration in the reaction medium (CB) adjusted to the pH required with 10 N NaOH, 1 ml of 100 mM monosodium-disodium phosphate buffer at the pH required, and 1 ml of amidase preparation diluted in the same buffer. The hydroxylamine stock solution (whose concentration was four times the CB) was freshly prepared before the start of the assay. Aliquots (500 μl) of reaction medium were removed at regular intervals (e.g., after 1.5, 3, 4.5, and 6 min), and each aliquot was mixed with 1 ml of an acidic FeCl3 solution (356 mM FeCl3 in 0.65 M HCl). The formation of hydroxamic acids was assayed spectrophotometrically at 500 nm. The molar extinction coefficients (ɛM) of the different hydroxamic acid-Fe(III) complexes were determined previously (7). As suggested previously, for a new hydroxamic acid-Fe(III) complex the mean ɛM obtained with the corresponding class of hydroxamic acids was used. For example, the ɛM used for the butyrohydroxamic acid assay was 1,016 liters/mol/cm. This was the mean of the ɛM values for the Fe(III) complexes of acetohydroxamic acid (996 liters/mol/cm), propionohydroxamic acid (1,029 liters/mol/cm), and valerohydroxamic acid (1,023 liters/mol/cm).
Amide hydrolysis activity.
Amide hydrolysis activity was determined in the presence or absence of hydroxylamine. Each reaction was performed at 30°C in a glass reaction tube, and the reaction medium was the same as the medium used for the acyl transfer activity assay. In the absence of hydroxylamine, 1 ml of hydroxylamine hydrochloride was replaced by 1 ml of H2O. Aliquots (500 μl) of reaction medium were removed at regular intervals, and each aliquot was mixed with 500 μl of 1 M H3PO4 to stop the reaction. Formation of the corresponding carboxylic acids was determined by high-performance liquid chromatography (HPLC).
HPLC analysis.
Amides and carboxylic acids were analyzed by HPLC. The chromatographic column used was a C18 reverse-phase Spherisorb ODS-2 column (diameter of particles, 5 μm; length, 250 mm; inside diameter, 4.6 mm; Alltech Associates, Deerfield, Ill.). The aqueous solvent systems contained 25 mM H3PO4, plus 1 or 25% (vol/vol) methanol depending on the hydrophobicity of the amide, as well as the corresponding carboxylic acid assayed. Each 5-μl sample injected was eluted at a flow rate of 1.0 ml/min and was detected at 210 nm.
Protein determination.
The protein concentrations were determined spectrophotometrically by the method of Lowry et al. (19), using bovine serum albumin as the standard.
Substrate specificity for acyl transfer activity.
The experiments to determine substrate specificity for acyl transfer activity (see Table 2) were performed at room temperature. Each reaction mixture was composed of 125 μl of a solution of amide at a concentration that was four times the CA, 125 μl of 2 M hydroxylamine hydrochloride adjusted to pH 7 with 10 N NaOH, 125 μl of 100 mM phosphate buffer (pH 7), and 125 μl of amidase preparation diluted in the same buffer (6.6 μg/ml). The final amide and hydroxylamine concentrations were CA and 500 mM, respectively. The CA depended on the solubility of the amide tested. When possible, CA was 100 mM. After 30 min, 1 ml of an acidic FeCl3 solution was added to stop the reaction. The resulting stained complex was visually quantified. Reaction media lacking amidase were also included to take into account possible spontaneous chemical synthesis of hydroxamic acids.
TABLE 2.
Substrate specificity for acyl transfer activity
| Compound | Final concn in reaction medium (mM) | Structural formulaa | Response level with amidase (5.9 μg/ml) in the reaction mediumb | Response level of the spontaneous chemical reactionb |
|---|---|---|---|---|
| Urea | 100 | NH2-CONH2 | − | − |
| Formamide | 100 | H-CONH2 | +++++ | +++++ |
| Acetamide | 100 | CH3-CONH2 | +++ | + |
| Propionamide | 100 | CH3-CH2-CONH2 | +++++ | + |
| Butyramide | 100 | CH3-CH2-CH2-CONH2 | +++++ | − |
| Valeramide | 50 | CH3-CH2-CH2-CH2-CONH2 | +++++ | − |
| Hexanoamide | 25 | CH3-CH2-CH2-CH2-CH2-CONH2 | +++++ | − |
| Isobutyramide | 100 | CH3-CH(CH3)-CONH2 | ++++ | − |
| Pivalamide | 50 | (CH3)3C-CONH2 | +++ | − |
| Diacetamide | 100 | H2NCO-CONH2 | +++++ | ++++ |
| Malonamide | 100 | H2NCO-CH2-CONH2 | ++ | + |
| Succinamide | 10 | H2NCO-CH2-CH2-CONH2 | − | − |
| Adipamide | 6.25 | H2NCO-CH2-CH2-CH2-CH2-CONH2 | − | − |
| Acrylamide | 100 | CH2⩵CH-CONH2 | ++++ | ++ |
| Methacrylamide | 100 | CH2⩵C(CH3)-CONH2 | ++ | − |
| Benzamide | 7.5 |  |
++ | − |
| Nicotinamide | 100 | +++ | − | |
| Isonicotinamide | 100 | + | − | |
| Glycinamide | 100 | H3N+-CH2-CONH2 | + | + |
| dl-Lactamide | 100 | CH3-CH(OH)-CONH2 | +++++ | + |
| l-Alaninamide | 100 | CH3-CH(NH3+)-CONH2 | +++ | − |
| β-Alaninamide | 100 | H3N+-CH2-CH2-CONH2 | + | + |
| dl-α-Aminobutyramide | 100 | CH3-CH2-CH(NH3+)-CONH2 | +++ | − |
| l-Valinamide | 100 | (CH3)2CH-CH(NH3+)-CONH2 | + | − |
| l-Leucinamide | 100 | (CH3)2CH-CH2-CH(NH3+)-CONH2 | +++ | − |
| dl-Isoleucinamide | 100 | CH3-CH2-CH(CH3)-CH(NH3+)-CONH2 | + | − |
| dl-Methioninamide | 100 | CH3-S-CH2-CH2-CH(NH3+)-CONH2 | ++++ | − |
| l-Prolinamide | 100 |  |
+++ | − |
| l-Serinamide | 20 | HO-CH2-CH(NH3+)-CONH2 | − | − |
| l-Asparagine | 25 | −OOC-CH(NH3+)-CH2-CONH2 | − | − |
| l-Glutamine | 50 | −OOC-CH(NH3+)-CH2-CH2-CONH2 | − | − |
| l-Threoninamide | 100 | CH3-CH(OH)-CH(NH3+)-CONH2 | ++ | − |
| l-Tryptophanamide | 10 |  |
− | − |
| dl-Phenylalaninamide | 15 | + | − | |
| dl-Phenylglycinamide | 25 | − | − | |
α-, β-, and γ-aminoamides are shown with ionized amino and carboxy groups, as they appear at pH 7.
Response levels, based on staining intensity and ranging from − (yellow solution) to +++++ (very intense brown solution), were visually determined after 30 min of reaction.
Chemicals.
All chemicals were purchased from Sigma, Aldrich, or Fluka. Valeramide was purchased from Interchim (Montluçon, France). Benzamide was purchased from Merck (Nogent-sur-Marne, France). dl-Lactic acid, benzoic acid, and urea were purchased from Prolabo (Vaulx-en-Velin, France). l-Alaninamide, d-alaninamide, β-alaninamide, dl-methioninamide, dl-phenylalaninamide, dl-phenylglycinamide, dl-isoleucinamide, and dl-α-aminobutyramide were provided by the Laboratoire de l’Alimentation Equilibrée of Rhône-Poulenc (Commentry, France).
RESULTS
Amidase preparation.
The recombinant amidase was prepared as described in Materials and Methods. After the cell extract was prepared, amidase was purified by an anion-exchange chromatographic step. As shown in Table 1, the level of purification was only threefold, but the amidase preparation obtained was almost homogeneous. In fact, the high level of expression of the amdA gene in E. coli BL21(pETADI) (amidase accounted for about 30% of the endocellular soluble proteins) facilitated amidase purification considerably.
TABLE 1.
Purification of recombinant amidase
| Step | Total amt of protein (mg)a | Total activity (U)b | Sp act (U/mg) | Yield (%) | Purifi- cation (fold) |
|---|---|---|---|---|---|
| Crude extract (S2 supernatant) | 647 | 46,354 | 58 | 100 | 1 |
| Anion-exchange chromatography | 237 | 41,788 | 176 | 90 | 3 |
Determined as described in Materials and Methods.
Acyl transfer activity was determined at pH 7 with 50 mM valeramide and 500 mM hydroxylamine as the substrates.
Enzyme stability during storage.
Purified amidase was stored at 4°C for several months at a concentration of 4.7 mg/ml in 50 mM Tris-HCl buffer (pH 7.3) containing 0.4 M NaCl. The activity at pH 7 was regularly tested by using valeramide and hydroxylamine as substrates (CA = 50 mM and CB = 500 mM), as described in Materials and Methods. Amidase was diluted 200-fold in 100 mM phosphate buffer (pH 7) before the start of the assay. The purified enzyme was extremely stable since no loss of activity was observed for 5 months.
Substrate specificity for acyl transfer activity.
The purified amidase catalyzed the acyl transfer reaction on hydroxylamine from a broad spectrum of amides. As shown in Table 2, the best results were obtained with linear aliphatic monoamides. Only formamide gave a negative result, since no difference was observed between the level of response in the amidase-containing tube and the level of response in the very rapid spontaneous chemical reaction.
Urea and diamides were not substrates. Only the very short-chain diamides (diacetamide and malonamide) were slightly transformed enzymatically, despite of a rapid spontaneous chemical reaction.
Unsaturated aliphatic monoamides, arylamides, and heterocycle-containing amides were quite good substrates. Only acrylamide reacted spontaneously with hydroxylamine.
Of the different α-hydroxyamides and α-, β-, and γ-aminoamides tested, only polar side-chain-containing α-, β-, and γ-aminoamides (glycinamide, l-serinamide, l-asparagine, and l-glutamine) and bulky side-chain-containing α-aminoamides (l-tryptophanamide and dl-phenylglycinamide) were not amidase substrates. All of the other amides tested were quite good substrates and did not react (or reacted only very slightly) spontaneously with hydroxylamine.
Optimum working pH.
As some of the amidase substrates were ionized at pH 7, the acyl transfer reaction was performed with all amides at the following pH values: pH 4 to 9 for neutral amides and pH 6 to 10 for α-aminoamides. As shown in Fig. 1, the optimum working pH obtained with aliphatic amides, arylamides, and heterocycle-containing amides (i.e., neutral amides) was 7, whereas the optimum working pH obtained with α-aminoamides was 8. At pH 6, the acyl transfer activities obtained with α-aminoamides and neutral amides represented 10 and 80%, respectively, of the acyl transfer activities obtained at the optimum working pH. Amide electroneutrality thus seemed to be required for amidase-catalyzed formation of the corresponding hydroxamic acids. This explains the higher optimum working pH obtained with α-aminoamides, since the pKa values of such molecules were around 8 to 9.
FIG. 1.

Influence of pH on the acyl transfer activity of purified amidase with neutral amides (•) and α-aminoamides (○) as substrates. The curves are the mean plots obtained with 13 neutral amides (acetamide, propionamide, butyramide, isobutyramide, valeramide, pivalamide, hexanoamide, acrylamide, methylacrylamide, benzamide, nicotinamide, isonicotinamide, and dl-lactamide) and eight α-aminoamides (l-alaninamide, dl-α-aminobutyramide, l-leucinamide, dl-isoleucinamide, dl-methioninamide, l-prolinamide, l-threoninamide, and dl-phenylalaninamide). Reactions were performed as described in Materials and Methods. The different amide concentrations (CA) used are shown in Table 2, and the CB was 500 mM. The buffers used were 100 mM citrate-phosphate buffer for pH 4, 5, and 6, 100 mM monosodium-disodium phosphate buffer for pH 6, 7, and 8, 100 mM Tris-HCl buffer for pH 8 and 9, and 100 mM carbonate-bicarbonate buffer for pH 9 and 10.
Reaction mechanism and kinetic constants.
Nineteen acyl transfer reactions were investigated. For each of them, all combinations possible with five amide concentrations and five hydroxylamine concentrations were tested in the presence of amidase. The reaction conditions and hydroxamic acid assay used are described in Materials and Methods. The reaction pH depended on the type of amide; pH 8 was used for α-aminoamides, and pH 7 was used for the other compounds. Reciprocal plots of 1/v (where v is the initial velocity of the acyl transfer reaction) versus 1/CA with different hydroxylamine concentrations (CB) gave parallel lines for all amides. A typical graph is shown in Fig. 2A. As expected for most acyl transfer reactions or substituted enzyme reactions, the mechanism involved in the acyl transfer reactions to hydroxylamine catalyzed by the amidase from Rhodococcus sp. strain R312 was determined to be a Ping Pong Bi Bi mechanism (23). If we show the individual steps by using the Cleland notation, the reaction can be written as:
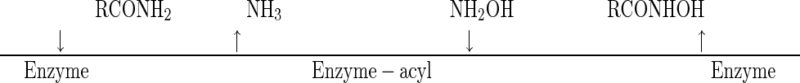 |
In the absence of NH3 and RCONHOH, the initial velocity, in reciprocal form, is given by (23):
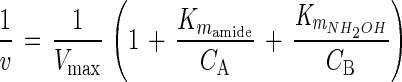 |
As shown in Fig. 2A, reciprocal plots drawn with amide as the varied substrate gave five 1/Vmaxapp values. Vmax and KmNH2OH values were then determined from the 1/v-axis intercept replot (Fig. 2B). The plots and replot obtained when hydroxylamine was the varied substrate were symmetrical with the plots and replot obtained for the various amide concentrations and led to determination of Kmamide.
FIG. 2.

Reciprocal plots obtained with propionamide as the substrate. (A) 1/v versus 1/CA at different hydroxylamine concentrations. Symbols: •, 40 mM; □, 50 mM; ▴, 66.6 mM; ○, 100 mM; ⧫, 200 mM. (B) 1/v-axis intercept replot.
All of the kinetic constants determined are described in Table 3. The Vmax values are expressed as turnover numbers (kcat) corresponding to the numbers of hydroxamic acid molecules produced per active site and per second (the amidase is made up of two identical subunits). Several types of substrates can be clearly distinguished. With respect to the Kmamide values, the substrates for which the amidase exhibited the highest affinity were the substrates containing a hydrophobic moiety lacking a polar group (e.g., all aliphatic amides with more than three carbon atoms, arylamides, heterocycle-containing amides, and hydrophobic α-aminoamides, such as l-leucinamide and l-phenylalaninamide). Substitution of an αH atom in propionamide with an OH or NH2 group, thus giving lactamide or alaninamide, considerably increased the Kmamide value.
TABLE 3.
Kinetic constants for amidase-catalyzed acyl transfer reactions
| Compound | Structural formula | kcat (s−1) | Kmamide (mM) | KmNH2OH (mM) | pH |
|---|---|---|---|---|---|
| Acetamide |  |
79 | 70 | 20 | 7 |
| Propionamide | 192 | 1 | 82 | 7 | |
| Butyramide | 229 | 0.1 | 178 | 7 | |
| Isobutyramide | 109 | 0.1 | 134 | 7 | |
| Valeramide | 313 | 0.1 | 230 | 7 | |
| Pivalamide | 15 | 0.1 | 55 | 7 | |
| Hexanoamide | 333 | 0.3 | 536 | 7 | |
| Acrylamide | 16 | 3 | 5 | 7 | |
| Methacrylamide | 8 | 0.1 | 7 | 7 | |
| Benzamide | 6 | 0.1 | 14 | 7 | |
| Nicotinamide | 8 | 0.3 | 13 | 7 | |
| Isonicotinamide | 2 | 0.6 | 62 | 7 | |
| l-Lactamide |  |
67 | 33 | 41 | 7 |
| l-Alaninamide | 66 | 12 | 25 | 8 | |
| l-Leucinamide | 63 | 1.3 | 39 | 8 | |
| l-Methioninamide | 245 | 5.4 | 36 | 8 | |
| l-Phenylalaninamide | 8.1 | 0.8 | 19 | 8 | |
| l-Prolinamide | 48 | 2 | 206 | 8 | |
| l-Threoninamide | 38 | 16 | 25 | 8 | |
On the other hand, the acyl-enzyme complex exhibited the highest affinity for hydroxylamine when the acyl group was short, polar, or unsaturated. By comparing the different KmNH2OH values obtained with acetamide, propionamide, butyramide, valeramide, and hexanoamide as substrates, we found that KmNH2OH increased considerably when the carbon chain length increased, indicating that a long acyl side chain hindered nucleophilic attack by hydroxylamine.
Finally, the highest kcat values were obtained with linear hydrophobic aliphatic amides (e.g., kcat = 333 s−1 with hexanoamide as the substrate), thus suggesting that branched-side-chain-, aryl group-, or heterocycle-containing amides were sterically hindered. By comparing the results obtained when propionamide, isobutyramide, and pivalamide were the substrates, we found that addition of one or two methyl groups at the α position considerably decreased the kcat values (192, 109, and 15 s−1, respectively), whereas addition of methyl groups at the ω position, which resulted in the linear compounds butyramide and valeramide, increased the kcat values (229 and 313 s−1, respectively). This is also why l-methioninamide turned out to be the most rapidly transformed α-aminoamide.
Undesirable reactions.
Several undesirable reactions could occur when hydroxamic acids were produced from amides (Fig. 3). At the start of the bioconversion, water and hydroxylamine were in competition with each other as acyl acceptors after enzyme acylation by amides, and carboxylate could be produced at the same time as the corresponding hydroxamic acid. As shown below, carboxylates were not amidase substrates for acyl transfer activity at pH 7 to 8. Their formation at the start of the bioconversion, due to amide hydrolysis, was thus irreversible. Moreover, we found that the amidase exhibited very high affinities for hydroxamic acids (e.g., Kmvalerohydroxamic acid = 0.2 mM and Kml-leucine hydroxamic acid = 4.9 mM). This means that the hydroxamic acids produced could also be enzymatically hydrolyzed to the corresponding carboxylates. Once again, as carboxylates were not substrates for the acyl transfer activity, their formation at the end of the bioconversion was irreversible.
FIG. 3.

Acyl transfer reaction on hydroxylamine and undesirable hydrolysis. Enz, enzyme.
The importance of these undesirable amide and hydroxamic acid hydrolysis reactions compared to the importance of the acyl transfer reaction was a function of the nature of the molecules. Linear molecules, such as valeramide and its corresponding hydroxamic acid, were slightly hydrolyzed (kcat hydrolysis = 35 and 18 s−1, respectively) compared to results of the acyl transfer reaction (kcat acyl transfer = 313 s−1). On the other hand, the kcat hydrolysis values obtained with bulky side-chain-containing amides, such as benzamide and its corresponding hydroxamic acid (kcat hydrolysis =6.2 and 4.3 s−1, respectively), were very close to the kcat acyl transfer value (kcat acyl transfer = 5.9 s−1 with benzamide). The results obtained with bulky side-chain-containing substrates could be explained by the dimensions of the molecules, which were an important limiting factor for the acyl transfer reaction.
However, as shown in Fig. 4 with benzamide as the substrate, we demonstrated that increasing the hydroxylamine concentration permitted us to considerably reduce both of the undesirable hydrolysis reactions (initial amide hydrolysis and final hydroxamic acid hydrolysis).
FIG. 4.

Influence of hydroxylamine concentration on hydrolysis of both benzamide and benzohydroxamic acid. The initial benzamide concentration and the amidase concentration in the reaction media were 5 mM and 44 μg/ml, respectively. The following four initial hydroxylamine concentrations were tested: 0 mM (A), 10 mM (B), 50 mM (C), and 500 mM (D). Symbols: ○, amount of benzoate formed; •, amount of benzohydroxamic acid.
Enantioselectivity of the purified amidase for the acyl transfer activity.
As we could not purchase all of the d-enantiomers of amides containing an asymmetrical carbon, enantioselectivity was investigated by comparing the kinetic constants obtained with the l- and d-enantiomers of lactamide and alaninamide only. Enantioselectivity towards other amides (methioninamide, leucinamide, and phenylalaninamide) was investigated by comparing the kinetic constants obtained with the l-enantiomers and the racemic mixtures. The results are shown in Table 4. With methioninamide, leucinamide, and phenylalaninamide, the amidase was found to be l-enantioselective. The kcat values obtained with the racemic mixtures were lower and the Kmamide and KmNH2OH values were higher than the values obtained with the l-enantiomers. Greater enantioselectivity was observed with leucinamide; the kcat obtained with the racemic mixture was 2.6-fold lower than that obtained with the l-enantiomer (24 versus 63 s−1), the Kmamide value was 1.8-fold higher (2.4 versus 1.3 mM), and the KmNH2OH value was 4.8-fold higher (187 versus 39 mM). We also observed that the rate of bioconversion of the racemic leucinamide mixture did not exceed 50%, whereas the rates obtained with the racemic methioninamide and phenylalaninamide mixtures did (data not shown). Finally, we showed that for leucinamide, the d-enantiomer acted as a mixed-type inhibitor after transformation of the l-enantiomer (Fig. 5), whereas this was not the case for methioninamide and phenylalaninamide.
TABLE 4.
Kinetic constants for amidase-catalyzed acyl transfer reactions with l-enantiomers, d-enantiomers, and racemic mixtures of several α-substituted amides
| Compound | Structural formula | kcat (s−1) | Kmamide (mM) | KmNH2OH (mM) | pH |
|---|---|---|---|---|---|
| l-Methioninamide |  |
245 | 5.4 | 36 | 8 |
| dl-Methioninamide | 180 | 6.4 | 75 | 8 | |
| l-Leucinamide | 63 | 1.3 | 39 | 8 | |
| dl-Leucinamide | 24 | 2.4 | 187 | 8 | |
| l-Phenylalaninamide | 8.1 | 0.8 | 19 | 8 | |
| dl-Phenylalaninamide | 6.2 | 1.1 | 19 | 8 | |
| l-Lactamide | 67 | 33 | 41 | 7 | |
| dl-Lactamide | 426 | 51 | 507 | 7 | |
| d-Lactamide | 2,504 | 270 | 2,923 | 7 | |
| l-Alaninamide | 66 | 12 | 25 | 8 | |
| d-Alaninamide | 3,353 | 379 | 1,939 | 8 |
FIG. 5.

Reciprocal plot showing the influence of d-leucinamide on the acyl transfer reaction from l-leucinamide to hydroxylamine. As we could not purchase d-leucinamide, different mixtures of l-leucinamide and dl-leucinamide allowed us to prepare solutions with different concentrations of the two enantiomers. The following five d-leucinamide concentrations were tested: 0 mM (•), 2 mM (■), 2.5 mM (▴), 3.33 mM (▾), and 5 mM (⧫). The amidase concentration in the reaction medium was 24 μg/ml.
Unexpected results were obtained with lactamide and alaninamide. The amidase was also l-enantioselective since the Kmamide values obtained with the racemic mixtures or with the d-enantiomers were higher than the values obtained with the l-enantiomers. However, the kcat values obtained with the d-enantiomers, together with the KmNH2OH values, were surprisingly very high. A possible explanation for this is discussed below.
Search for other acyl donors.
Five carboxylic acids and two methyl esters were tested as amidase substrates and compared with the corresponding amides as a function of pH. Acyl transfer activities were assayed as described in Materials and Methods. Each solution (concentration, four times the CA) was adjusted to the pH required before the start of the reaction. Standards lacking amidase were also included to take into account possible spontaneous chemical formation of hydroxamic acid. As shown in Table 5, rapid spontaneous reactions occurred with lactate and methyl esters at very basic pH values. As far as enzymatic activities are concerned, amides were undeniably the best acyl donors. Amino acids were logically not amidase substrates since they are amphoteric molecules that are electrically hindered. Similarly, carboxylic acids were very poor acyl donors. Only butyric acid and lactic acid reacted with amidase, but they reacted at pH values between the optimum working pH of the enzyme and the pKa values of the acids, as if only protonated forms of the acids could be transformed. This has also been observed for the wide-spectrum amidase from Rhodococcus sp. strain R312 (6). Both methyl esters (methyl butyrate and l-methionine methyl ester) were very slightly transformed (at pH 7 for neutral methyl butyrate and at pH 8 for l-methionine methyl ester). Under the experimental conditions used, the specific activities obtained with methyl esters represented less than 0.2% of the specific activities obtained with the corresponding amides.
TABLE 5.
Acyl transfer activities and spontaneous chemical reactions obtained with different amides, carboxylic acids, and methyl esters as acyl donorsa
| Acyl donor | Acyl transfer activity
|
Chemical reaction
|
||
|---|---|---|---|---|
| Optimum working pH | Sp act (U/mg) | pH | Activity (μmol/min/ ml) | |
| l-Alaninamide (100 mM) | 8 | 60 | 0 | |
| l-Alanine (100 mM) | 0 | 0 | ||
| l-Methioninamide (100 mM) | 8 | 160 | 0 | |
| l-Methionine (100 mM) | 0 | 0 | ||
| l-Methionine methyl ester (50 mM) | 8 | 0.25 | Very basic | 0.40 |
| Butyramide (100 mM) | 7 | 130 | 0 | |
| Butyric acid (100 mM) | 6 | 8.7 | 0 | |
| Methyl butyrate (50 mM) | 7 | 0.20 | Very basic | 0.10 |
| Nicotinamide (100 mM) | 7 | 9.9 | 0 | |
| Nicotinic acid (25 mM) | 0 | 0 | ||
| dl-Lactamide (100 mM) | 7 | 121 | 7 | 0.03 |
| dl-Lactic acid (100 mM) | 6 | 0.17 | Very basic | 0.60 |
Hydroxylamine (500 mM) was used as the acyl acceptor.
Search for other acyl acceptors.
The following five potential acyl acceptors were tested because of their polarity and were compared with water and hydroxylamine: N-methylhydroxylamine (CH3-NHOH), ethanolamine (NH2-CH2-CH2-OH), hydrazine (NH2NH2), N-methylhydrazine (CH3-NHNH2), and methanol (CH3-OH). The initial concentration of each of these compounds in the reaction medium was 500 mM. Reactions were performed at pH 7 with 100 mM butyramide as the acyl donor and with 44 μg of amidase per ml. After 40 min, 0.5 ml of 1 M H3PO4 was added to 0.5 ml of reaction medium to stop the reaction, and the mixture was analyzed by HPLC as described in Materials and Methods. Whereas 500 mM hydroxylamine totally inhibited residual butyramide hydrolysis and led to the formation of butyrohydroxamic acid with a bioconversion rate of up to 90%, the different molecules tested did not affect the hydrolysis reaction, and none of them turned out to be an acyl acceptor (data not shown). A previous study (6) showed that the wide-spectrum amidase from Rhodococcus sp. strain R312 catalyzed the acyl transfer reaction from amides to water and hydroxylamine and also to hydrazine.
DISCUSSION
The enantioselective amidase from Rhodococcus sp. strain R312 was shown to catalyze the acyl transfer reaction to hydroxylamine from a broad spectrum of amides. The reaction mechanism was determined to be a Ping Pong Bi Bi system; i.e., amides react with the enzyme to give acyl-enzyme complexes, which then transfer acyl groups to the second substrate (hydroxylamine).
Acylation was facilitated when amides contained a marked hydrophobic moiety which contrasted with the polar amide group. Diamides and amides lacking a hydrophobic area were not amidase substrates. On the other hand, the affinities of the different acyl-enzyme complexes for hydroxylamine were highest when the acyl side chain was short, polar, or unsaturated. Very low levels of affinity were obtained when long-chain aliphatic amides were the acyl donors. In these cases, attack of the carbonyl group of the acylated enzyme by hydroxylamine seemed to be hindered. With respect to the kcat values, the best results were obtained with linear molecules. Very high kcat values (up to 300 s−1) were obtained with valeramide and hexanoamide. We suppose that heptanamide, octanamide, and other linear fatty amides should be very good substrates if solubility is not a limiting factor.
The above-mentioned results suggested that amides are geometrically oriented before they enter the amidase by their polar amide groups and that the active site is located at the end of a narrow funnel. Such steric constraints could be responsible for the restricted velocities observed with amides containing branched side chains (isobutyramide, pivalamide), aryl groups (benzamide, phenylalaninamide), or heterocycles (nicotinamide, isonicotinamide). This finding differs markedly from the data obtained with the enantioselective amidase from P. chlororaphis B23, which, despite a high level of sequence homology (level of strict identity, 46%) with the enantioselective amidase from Rhodococcus sp. strain R312, exhibited the highest levels of hydrolysis activity with isobutyramide, nicotinamide, and dl-phenylalaninamide (5).
Carboxylic acids, electrically charged at pH 7, could be very slightly transformed into the corresponding hydroxamic acids when the reaction pH was between pH 7 (the optimum working pH) and the pKa of the acyl donor, but amino acids, which are amphoteric molecules, could not be transformed at all. The different kcat values obtained with α-aminoamides as substrates, also electrically charged at pH 7, were quite high at pH 8 because this optimum pH value was close both to the pKa values (about pH 8 to 9) and to pH 7.
Although methyl esters were not charged, they were found to be very bad acyl donors. The specific activities obtained with methyl butyrate and l-methionine methyl ester were less than 0.2% of the specific activities obtained with the corresponding amides, suggesting that in addition to the electrical constraints, there was an amide bond recognition site in the active site area. This would explain the importance of the hydrolysis of hydroxamic acids (which also contain amide bonds) observed at the end of the bioconversions.
So far, only water and hydroxylamine have been found to be efficient acyl acceptors. In addition to the substrate spectrum, this was an important difference from the wide-spectrum amidase family since the wide-spectrum amidase from Rhodococcus sp. strain R312 was shown to catalyze the formation of acid hydrazides from amides and hydrazine (6).
Amidases from P. chlororaphis B23, R. erythropolis MP50, Rhodococcus sp. strain R312, Rhodococcus sp., and R. rhodochrous J1 were shown to exhibit an (S)-enantioselective hydrolysis activity towards several 2-arylpropionamides (5, 12, 14, 20, 21). The amidase from Rhodococcus sp. strain R312 was also determined to be l-enantioselective [or (S)-enantioselective] towards α-substituted amides (α-hydroxyamides and α-aminoamides). This suggested that there is an additional recognition site for groups located at the α position. Surprisingly, very high kcat and KmNH2OH values were determined with the d-enantiomers of lactamide and alaninamide. A possible explanation for this is shown in Fig. 6. Because of the small size of the CH3 group at the α position, rotation of CH3, H, and OH (or NH2) around the C—CO axis of the d-enantiomer might be possible, so that the CH3 group would be positioned at the same place as the OH (or NH2) group of the l-enantiomer. In this case, the OH (or NH2) group would be positioned at a site which might hinder nucleophilic attack by hydroxylamine but which also might facilitate release of the hydroxamic acid formed. Further studies of the amidase from Rhodococcus sp. strain R312 and particularly its three-dimensional structure could provide information about the mechanism and thus confirm the geometrical organization of the active site proposed above.
FIG. 6.

Possible rearrangement of the d-enantiomer of lactamide in the active site. (A) l-Lactamide. (B) d-Lactamide.
The results described here, particularly the high kcat values obtained with aliphatic amides, α-hydroxyamides, and α-aminoamides, highlighted the interest in amidases for use in the production of a wide range of hydroxamic acids and showed that enzymatic synthesis of such molecules is an interesting alternative to chemical synthesis methods. Reactions occurred in medium lacking organic solvents and led to production of both α-aminohydroxamic acids, which are interesting because of their medical applications as enzyme inhibitors, and mid-chain or unsaturated hydroxamic acids, which are interesting because of their environmental applications as chelating agents for wastewater treatment.
REFERENCES
- 1.Brammar W J, Clarke P H. Induction and repression of Pseudomonas aeruginosa amidase. J Gen Microbiol. 1964;37:307–319. doi: 10.1099/00221287-37-3-307. [DOI] [PubMed] [Google Scholar]
- 2.Brown D A, Chidambaram M V, Clarke J J, McAleese D M. Design of iron(III) chelates in oral treatment of anemia: solution properties and absorption of iron(III) acetohydroxamate in anemic rats. Bioinorg Chem. 1978;9:255–275. doi: 10.1016/s0006-3061(00)80045-9. [DOI] [PubMed] [Google Scholar]
- 3.Cawston T E. Metalloproteinase inhibitors and the prevention of connective tissue breakdown. Pharmacol Ther. 1996;70:163–182. doi: 10.1016/0163-7258(96)00015-0. [DOI] [PubMed] [Google Scholar]
- 4.Chebrou H, Bigey F, Arnaud A, Galzy P. Study of the amidase signature group. Biochim Biophys Acta. 1996;1298:285–293. doi: 10.1016/s0167-4838(96)00145-8. [DOI] [PubMed] [Google Scholar]
- 5.Ciskanik L M, Wilczek J M, Fallon R D. Purification and characterization of an enantioselective amidase from Pseudomonas chlororaphis B23. Appl Environ Microbiol. 1995;61:998–1003. doi: 10.1128/aem.61.3.998-1003.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fournand D, Arnaud A, Galzy P. Study of the acyl transfer activity of a recombinant amidase overproduced in an E. coli strain. Application for short-chain hydroxamic acid and acid hydrazide synthesis. J Mol Catal B. 1998;4:77–90. [Google Scholar]
- 7.Fournand D, Pirat J L, Bigey F, Arnaud A, Galzy P. Spectrophotometric assay of aliphatic monohydroxamic acids and α-, β-, and γ-aminohydroxamic acids in aqueous medium. Anal Chim Acta. 1997;353:359–366. [Google Scholar]
- 8.Gao W Y, Mitsuya H, Driscoll J S, Johns D G. Enhancement by hydroxyurea of the anti-human immunodeficiency virus type 1 potency of 2′-β-fluoro-2′,3′-dideoxyadenosine in peripheral blood mononuclear cells. Biochem Pharmacol. 1995;50:274–276. doi: 10.1016/0006-2952(95)00106-a. [DOI] [PubMed] [Google Scholar]
- 9.Hamer R R L, Tegeler J J, Kurtz E S, Allen R C, Bailey S C, Elliott M E, Hellyer L, Hessley G C, Przekop P, Freed B S, White J, Martin L L. Dibenzoxepinone hydroxylamines and hydroxamic acids: dual inhibitors of cyclooxygenase and 5-lipoxygenase with potent topical antiinflammatory activity. J Med Chem. 1996;39:246–252. doi: 10.1021/jm950563z. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto Y, Nishiyama M, Ikehata O, Horinouchi S, Beppu T. Cloning and characterization of an amidase gene from Rhodococcus species N-774 and its expression in Escherichia coli. Biochim Biophys Acta. 1991;1088:225–233. doi: 10.1016/0167-4781(91)90058-t. [DOI] [PubMed] [Google Scholar]
- 11.Heitner, H. I., and R. G. Ryles. November 1992. European patent 0 514 648 B1.
- 12.Hirrlinger B, Stolz A, Knackmuss H-J. Purification and properties of an amidase from Rhodococcus erythropolis MP50 which enantioselectively hydrolyzes 2-arylpropionamides. J Bacteriol. 1996;178:3501–3507. doi: 10.1128/jb.178.12.3501-3507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holmes L B. Hydroxamic acid: a potential human teratogen that could be recommended to treat ureaplasma. Teratology. 1996;53:227–229. doi: 10.1002/(SICI)1096-9926(199604)53:4<227::AID-TERA4>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 14.Kobayashi M, Komeda H, Nagasawa T, Nishiyama M, Horinouchi S, Beppu T, Yamada H, Shimizu S. Amidase coupled with low-molecular-mass nitrile hydratase from Rhodococcus rhodochrous J1. Eur J Biochem. 1993;217:327–336. doi: 10.1111/j.1432-1033.1993.tb18250.x. [DOI] [PubMed] [Google Scholar]
- 15.Kobayashi M, Fujiwara Y, Goda M, Komeda H, Shimizu S. Identification of active sites in amidase: evolutionary relationship between amide bond- and peptide bond-cleaving enzymes. Proc Natl Acad Sci USA. 1997;94:11986–11991. doi: 10.1073/pnas.94.22.11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koide Y, Uchino M, Yamada K. Studies of collectors. IX. The flotation of a trace amount of uranium by using 2-(alkylamino)propionohydroxamic acid and cotelomer-type surfactants bearing hydroxyaminocarbonyl and pyridyl groups. Bull Chem Soc Jpn. 1987;60:3477–3483. [Google Scholar]
- 17.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature (London) 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Lewellyn, M. E. May 1996. World patent WO 96/14271.
- 19.Lowry O H, Rosebrough N J, Farr A L, Randall R J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 20.Mayaux J-F, Cerbelaud E, Soubrier F, Faucher D, Pétré D. Purification, cloning, and primary structure of an enantiomer-selective amidase from Brevibacterium sp. strain R312: structural evidence for genetic coupling with nitrile hydratase. J Bacteriol. 1990;172:6764–6773. doi: 10.1128/jb.172.12.6764-6773.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayaux J-F, Cerbelaud E, Soubrier F, Yeh P, Blanche F, Pétré D. Purification, cloning, and primary structure of a new enantiomer-selective amidase from Rhodococcus strain: structural evidence for a conserved genetic coupling with nitrile hydratase. J Bacteriol. 1991;173:6694–6704. doi: 10.1128/jb.173.21.6694-6704.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 23.Segel I H. Enzyme kinetics. New York, N.Y: John Wiley & Sons; 1975. pp. 602–625. [Google Scholar]
- 24.Studier F W, Rosenberg A H, Dunn J J, Dubendorff J W. Use of T7 polymerase to direct expression of cloned genes. Methods Enzymol. 1990;185:60–89. doi: 10.1016/0076-6879(90)85008-c. [DOI] [PubMed] [Google Scholar]
- 25.Studier F W, Moffatt B A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- 26.Thiéry A, Maestracci M, Arnaud A, Galzy P, Nicolas M. Purification and properties of an acylamide amidohydrolase (E.C. 3.5.1.4) with a wide activity spectrum from Brevibacterium sp. R312. J Basic Microbiol. 1986;26:299–311. [Google Scholar]
- 27.Trott, S. Personal communication.
- 28.Tsafack A, Golenser J, Libman J, Shanzer A, Cabantchik Z I. Mode of action of iron(III) chelators as antimalarials. III. Overadditive effects in the combined action of hydroxamate-based agents on in vitro growth of Plasmodium falciparum. Mol Pharmacol. 1995;47:403–409. [PubMed] [Google Scholar]