

Summary
Down syndrome (DS) results from trisomy of human chromosome 21 (HSA21), and DS research has been conducted by the use of mouse models. We previously generated a humanized mouse model of DS, TcMAC21, which carries the long arm of HSA21. These mice exhibit learning and memory deficits, and may reproduce neurodevelopmental alterations observed in humans with DS. Here, we performed histologic studies of the TcMAC21 forebrain from embryonic to adult stages. The TcMAC21 neocortex showed reduced proliferation of neural progenitors and delayed neurogenesis. These abnormalities were associated with a smaller number of projection neurons and interneurons. Further, (phospho-)proteomic analysis of adult TcMAC21 cortex revealed alterations in the phosphorylation levels of a series of synaptic proteins. The TcMAC21 mouse model shows similar brain development abnormalities as DS, and will be a valuable model to investigate prenatal and postnatal causes of intellectual disability in humans with DS.
Subject areas: Neuroscience, Cell biology, Proteomics
Graphical abstract
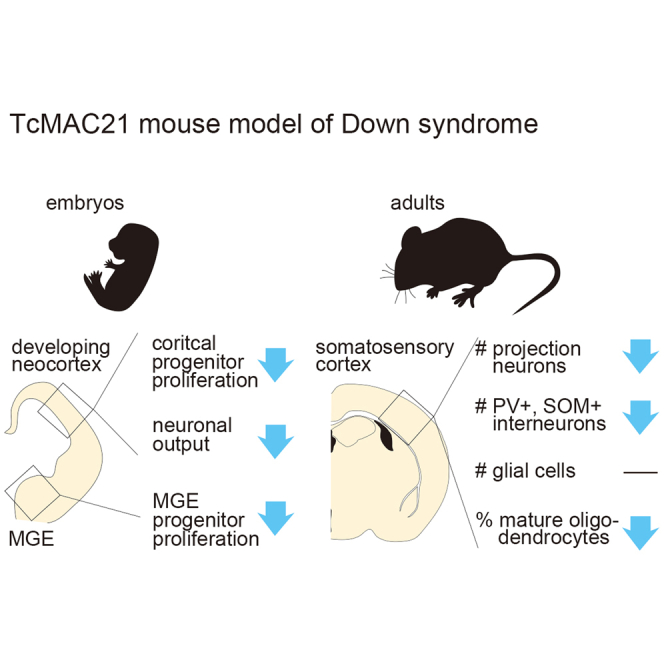
Highlights
-
•
TcMAC21 developing neocortices show reduced proliferation of progenitors
-
•
TcMAC21 developing neocortices show impaired neurogenesis
-
•
TcMAC21 adult cortices show reduced densities in projection neurons and interneurons
-
•
TcMAC21 adult cortices reveal changes in phosphorylation levels of synaptic proteins
Neuroscience; Cell biology; Proteomics
Introduction
Down syndrome (DS), which has an incidence of 1 in 700–800 live births,1 is caused by trisomy of human chromosome 21 (HSA21). DS is a common genetic cause of intellectual disability, which can range from mild to severe,2 and is associated with prenatal and postnatal abnormalities in brain development.3 For example, DS brains are smaller than normal adult brains,4,5 and histologic analyses of the cerebral cortex reveal a reduced number and density of neurons in adult and fetal DS brains.6,7,8 In addition, cortical neurons in DS fetuses exhibit primitive dendritic arborization with fewer synapses.9,10
To study the pathogenic mechanisms underlying DS phenotypes, several mouse models have been generated based on the syntenic regions within the human and mouse genomes. HSA21 genes are conserved in orthologous regions of mouse chromosomes (Mmu) 10, 16, and 17, the largest of which is on Mmu16. Therefore, several mouse models that are trisomic for large segments of Mmu16 have been created and used for molecular, cellular, and behavioral studies.11 The Ts65Dn, Ts1Cje and Dp(16)Yey/+ (Dp16) strains are frequently used models that recapitulate some aspects of brain development and behavioral phenotypes observed in people with DS.12,13,14,15,16,17 Among these mouse models, the Ts65Dn strain is unique in that it carries a freely segregating extra chromosome, making this model an aneuploid model of DS. This aneuploidy may produce additional phenotypes compared with models having intrachromosomic duplications. The triplicated segment in Ts65Dn mice, however, has ∼44 Mmu17 genes that are not triplicated in DS.18,19 More recently, the Ts66Yah mouse model of DS, which derived from the Ts65Dn lineage but not carrying the extra Mmu17 genes, has been developed and characterized.20,21 This model remains a partial DS model because the triplicated region encompasses 102/187 of the Hsa21 orthologous protein coding genes,20,21 thus, raising the need for mouse models with a better genetic representation of trisomy 21.
We previously generated a humanized mouse model of DS, TcMAC21, which carries a freely segregating mouse artificial chromosome containing a copy of HSA21q.22 It is the most complete genetic model of DS generated to date with 93% of HSA21q protein-coding genes, and notably, expression of these human genes is regulated similarly to their mouse orthologs.22 Importantly, this mouse model recapitulates several features of DS, including learning and memory deficits.22 Prenatal and postnatal brain development, however, has not yet been assessed in TcMAC21, preventing a comprehensive comparison of TcMAC21 with other DS mouse models and human patients with DS. Here, we report first phenotypic characterization of developing TcMAC21 brain.
Results
Impaired proliferative and neurogenic potential of progenitors in the TcMAC21 embryonic neocortex
During development of the mammalian brain, neural progenitor cells in the dorsal telencephalon give rise to projection neurons. Previous studies using various mouse models such as Ts65Dn and Ts1Cje demonstrated alterations in the number of apical radial glial cells (aRGCs) and intermediate progenitor cells (IPCs).14,16 We therefore began our study by measuring these progenitor cells following immunohistostaining for SOX2 and TBR2, markers for aRGCs and IPCs, respectively. The number of SOX2-positive aRGCs was similar between TcMAC21 and euploid controls at embryonic day (E) 14.5 and E16.5 (Figures 1A and 1C). On the other hand, the number of TBR2-positive IPCs in the TcMAC21 neocortex was modestly decreased at E14.5 and E16.5 (Figures 1B and 1C). Of note, we did not observe significant alterations in cleaved caspase 3-positive cells (Figure S1) in the TcMAC21 neocortex, indicating that apoptosis does not account for the phenotypes observed.
Figure 1.

Reduced proliferative and neurogenic potential of progenitors in the TcMAC21 neocortex
(A–C) The E14.5 and E16.5 brain sections of euploid and TcMAC21 were immunostained with antibodies against SOX2 (A), and TBR2 (B). Images of the entire cerebral wall are shown. Dashed line outlines the ventricular and pial surface. Scale bars, 50 μm. The number of SOX2-and TBR2-positive cells were counted and plotted in (C) by setting the mean of euploid to 100. Data are presented as means ± SEM (for E14.5, n = 6–7 (euploid) and n = 6 (TcMAC21) from 3 dams, for E16.5, n = 7 (euploid) and n = 6 (TcMAC21) from 3 dams.) ∗p < 0.05; ∗∗p < 0.01; multiple t-tests with Holm-Sidak correction for multiple comparisons.
(D) The E14.5 and E16.5 brain sections of euploid and TcMAC21 were immunostained with antibodies against P-H3. Images of the entire cerebral wall are shown. Dashed line outlines the ventricular and pial surface. Scale bars, 50 μm.
(E) The number of apical and basal P-H3-positive cells were counted and plotted by setting the mean of euploid to 100. Data are presented as means ± SEM (for E14.5, n = 7 (euploid) and n = 6 (TcMAC21) from 3 dams, for E16.5, n = 7 (euploid) and n = 6 (TcMAC21) from 3 dams.) ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; multiple t-tests with Holm-Sidak correction for multiple comparisons.
(F) Representative images of mitotic P-H3+ cells in the MGE of E14.5 euploid and TcMAC21 animals are shown, with the boxed regions magnified to the right. Scale bars, 50 μm.
(G) Quantification of the number of P-H3+ cells at the VZ surface and away from the VZ surface (Extra-VZ surface) in the MGE and the CGE at E14.5. Data are presented as means ± SEM (n = 6 for euploid and TcMAC21 from 3 dams.). ∗∗p < 0.01; ∗∗∗p < 0.001; multiple t-tests with Holm-Sidak correction for multiple comparisons.
(H) E13.5 and E15.5 timed pregnant TcMAC21 females were injected with EdU, and embryos were harvested 24 h later. The E14.5 and E16.5 brain sections were immunostained with antibodies against SOX2. Representative images of EdU+ cells throughout the entire cerebral wall are shown, with the boxed regions magnified to the bottom. Scale bars, 50 μm for entire cerebral wall; 25 μm in magnified views.
(I) The percentage of EdU-labeled cells in the VZ, SVZ, IZ and CP and (J) the fraction of SOX2+ cells in EdU+ cells was calculated and is plotted as means ± SEM (for E14.5, n = 6 for euploid and TcMAC21 from 3 dams, for E16.5, n = 5 (euploid) and n = 6 (TcMAC21) from 3 dams.) ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; multiple t-tests with Holm-Sidak correction for multiple comparisons.
The accumulated findings suggest that impaired progenitor proliferation is a common phenotype in humans with DS and in mouse models of DS.14,16,23,24 Thus, we next assessed the proliferation of progenitor cells in the neocortex by labeling dividing cells with the M-phase marker phospho-histone H3 (P-H3). The numbers of both apical (ventricular) and basal (abventricular) p-H3-positive mitotic cells were reduced in the TcMAC21 neocortex at E14.5 and E16.5 (Figures 1D and 1E). Given the similar number of aRGCs and the slight reduction in IPCs (Figures 1A–1C), these results suggest the reduced proliferation of aRGCs and IPCs in the TcMAC21 neocortex. We also quantified the number of P-H3-positive cells in the medial ganglionic eminence (MGE) and caudal ganglionic eminence (CGE) areas of the ventral telencephalon, the major source of cortical inhibitory neurons.25,26,27,28 The number of P-H3-positive cells at the ventricular (VZ) surface and outside of the ventricular zone surface (Extra-VZ surface) in the MGE of the TcMAC21 mice was significantly reduced compared with that of the euploid control mice at E14.5 (Figures 1F and 1G). In contrast, no significant alterations in the number of P-H3-positive cells were observed in the CGE (Figure 1G). These results suggest that the proliferative potential of MGE progenitors as well as cortical progenitors is reduced in the developing TcMAC21 forebrain.
Neurogenic output is also altered in several DS mouse models.13,14,16,29 To assess this in TcMAC21, pregnant TcMAC21 females were injected with the cell proliferation marker 5-ethynyl-2′-deoxyuridine (EdU) and embryos were collected 24 h later. EdU-labeled cells were counted in the VZ, subventricular zone (SVZ), intermediate zone (IZ), and cortical plate (CP) and the fraction of cells outside of the VZ/SVZ was used as an estimation of neuronal output in the neocortex during the 24-h window. We found a smaller fraction of EdU+ cells in the TcMAC21 IZ/CP at E14.5 and E16.5 (Figures 1H and 1I). In contrast, fraction of EdU+ cells in the TcMAC21 VZ was larger than that of euploid (Figures 1H and 1I). Further, we performed immunostaining of EdU+ cells with SOX2 antibody to see whether EdU+ cells remain as aRGCs (SOX2+) or differentiated from aRGCs (SOX2-). We found that the fraction of SOX2+ cells in EdU+ cells was larger in the TcMAC21 cortex (Figures 1H and 1J). Together, these results suggest continued impairment in neurogenesis from TcMAC21 cortical progenitors, although we could not rule out the possibility that the difference in the number of cells initially labeled with EdU affects the fraction of SOX2+/BrdU+ cells.
Reduced neuronal densities in the TcMAC21 neocortex
Previous studies of DS human brains and DS mouse models revealed alterations in the number and density of cortical neurons.6,7,8,12,13,14,15 Therefore, we evaluated changes in the neuronal number/density in the developing TcMAC21 cortex following immunohistostaining for various neuronal markers. The density of neurons positive for TBR1, a marker for postmitotic projection neurons and highly expressed in deep layer neurons,30 was reduced across the E14.5 to P0.5 time window in the TcMAC21 neocortex (Figures 2A–2C and 2E). We used CTIP2 as another marker for deep layer neurons,31 and found a smaller number of CTIP2-positive neurons in the TcMAC21 brain (Figures 2B, 2C, and 2E). A similar reduction was observed in the density of SATB2-positive intra-telencephalic projection neurons in TcMAC21 (Figures 2D and 2E). Together, these results suggest an overall reduction in the number of projection neurons in the TcMAC21 neocortex.
Figure 2.

Reduced number of projection neurons in the developing neocortex of TcMAC21
(A–D) The E14.5 (A), E16.5 (B), and P0.5 (C and D) brain sections of euploid and TcMAC21 were immunostained with antibodies indicated. Representative images of the entire cerebral wall are shown, with the boxed regions magnified to the right. Scale bars: 50 μm for entire cerebral wall; 25 μm in magnified views.
(E) The number of TBR1-, CTIP2- and SATB2-positive cells were counted and plotted by setting the mean of euploid to 100. Data are presented as means ± SEM (for E14.5, n = 7 (euploid) and n = 6 (TcMAC21) from 3 dams, for E16.5, n = 7 (euploid) and n = 6 (TcMAC21) from 3 dams, for P0.5, n = 4 (euploid) and n = 5 (TcMAC21) from 3 dams.) ∗∗p < 0.01; ∗∗∗p < 0.001; multiple t-tests with Holm-Sidak correction for multiple comparisons.
To gain further insight into the histologic abnormalities of the TcMAC21 neocortex, we extended our immunohistochemical analysis from prenatal and perinatal stages to the adult stage. Immunohistostaining for projection neuron markers revealed a reduced density of TBR1-, CTIP2- and SATB2-positive neurons in the somatosensory cortex of 3-month-old TcMAC21 mice (Figures 3A and 3D), as expected from the results above (Figure 2). In addition to this, we used several markers such as parvalbumin (PV), somatostatin (SOM), vasoactive intestinal peptide (VIP), and calretinin (CR) to assess the density of cortical interneurons. Fewer PV- and SOM-positive cells, which are derived from MGE progenitors, were observed in the TcMAC21 cortex (Figures 3B and 3D). In contrast, the density of VIP- and CR-positive cells, most of which are derived from CGE, was unchanged (Figures 3B and 3D). Therefore, TcMAC21 mice exhibit reductions in subsets of cortical cell populations in parallel with reduced cell proliferation of progenitors in the neocortex and MGE (Figure 1).
Figure 3.

Neuronal and glial population abnormalities in adult TcMAC21 somatosensory cortex
(A–D) The 3-month brain sections of euploid and TcMAC21 were immunostained with markers for projection neurons (A), inhibitory neurons (B), and glial cells (C) indicated. Representative images of the entire cerebral wall are shown, with the boxed regions magnified to the right. Scale bars: 100 μm for entire cerebral wall; 50 μm in magnified views. The number of positive cells for indicated markers in (A–C) were counted and plotted in (D) by setting the mean of euploid to 100. Data are presented as means ± SEM (n = 7 for euploid and n = 5 for TcMAC21 from 5 dams.). ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; multiple t-tests with Holm-Sidak correction for multiple comparisons.
In addition to the altered number of neurons, previous studies reported mis-regulation of production and differentiation of glial cells in DS. For example, spatiotemporal transcriptome differences suggest impaired oligodendrocyte maturation in DS.32 Therefore, we investigated whether similar cellular changes were observed in the TcMAC21 cortex. The density of cells positive for ALDH1L1 (astrocytes), OLIG2 (oligodendrocytes and their progenitors), and IBA1 (microglia) was similar between TcMAC21 and euploid controls (Figures 3C and 3D). The GSTπ-positive fraction of OLIG2-positive cells, reflecting matured oligodendrocytes, was reduced in TcMAC21 (Figures 3C and 3D). These results suggest that TcMAC21 has similar abnormalities during brain development as DS6,7,8,32,33 (also see discussion).
Proteomic analysis suggests an effect of HSA21q gene dosage on synaptic regulation
We previously demonstrated that TcMAC21 exhibits significant learning and memory deficits.22 To unravel the potential molecular mechanisms underlying the cognitive phenotypes associated with the neocortical abnormalities in TcMAC21, we performed (phospho-) proteomic profiling of the somatosensory cortex of 3-month-old TcMAC21 mouse. In our dataset, when focused on mouse proteins, 3859 proteins and 6107 phospho-peptides from 1852 proteins were detected (Tables S1 and S2). In addition, 14 HSA21 proteins (SYNJ1, GATD3A, CBR1, CCT8, APP, ATP5O, SOD1, CLIC6, PDKX, ATP5PF, CSTB, ITGB2, HSPA13, BACH1) were detected in the TcMAC21 cortex, validating our proteomic analysis (Table S3). Note that all of these HSA21genes have mouse orthologs (i.e., no human-specific proteins were detected in the analysis). However, because mass spectrometry-based proteomic analysis cannot be used to compare the expression levels of human proteins to their mouse orthologs, we excluded these human proteins and peptides from the following analysis. With regard to protein abundance, we found no apparent difference between TcMAC21 and control, nor any statistically significant differences (q < 0.2) (Figure 4A and Table S4). In the phospho-proteomic dataset, we found a large alteration in the TcMAC21 cortex, though statistically significant differences were detected for only a few phospho-peptides (Figure 4C and Table S5). To obtain a better description of the 2 datasets, we performed a principal component analysis (PCA). As expected, proteomic profiles of the TcMAC21 samples and control samples do not segregate in the principal component (PC) space (Figure 4B). On the other hand, phospho-proteomic profiles from the 2 strains are plotted in distinct regions of the PC space (Figure 4D). These results suggest that perturbations in phosphorylation levels can account for functional alterations in the TcMAC21 brain.
Figure 4.

The (phospho-)proteomic alterations in the TcMAC21 neocortex
(A) Log2 fold change of the WT/TcMAC21 protein levels and significance (-log2 of the q-value) of all evaluated proteins. Q-vale, or false discovery ratio (FDR) correcting for multiple testing were calculated using the HB (Benjamini & Hochberg) method correction.34
(B) PCA result using the proteome data from cortex in euploid control (black) and TcMAC21 (red). Each symbol represents one cortex.
(C) Log2 fold change of the WT/TcMAC21 phospho-peptide levels and significance (-log2 of the q-value) of all evaluated phospho-peptides. Significant peptides are indicated in magenta. Q-vale, or false discovery ratio (FDR) are calculated as described above.
(D) PCA result using the phospho-proteome data from cortex in euploid control (black) and TcMAC21 (red). Each symbol represents one cortex.
(E and F) GO terms representing the significantly enriched biological processes for “PC2-positive” (E) and “PC2-negative” (F) proteins. Enrichment analysis was performed using Metascape.35
To estimate the kinase whose activity is mis-regulated in TcMAC21 cortex, we performed kinase substrate enrichment analysis (KSEA) on the phospho-proteome dataset.36 This analysis suggests that the activity of PKA is up-regulated in TcMAC21 cortex (enrichment score 1.02, p = 0.025). Although the detailed mechanism is unclear, we can speculate that expression of HSA21 proteins up-regulates PKA activity, thereby results in an altered phosphorylation status in the TcMAC21 cortex.
Next, we examined the factors characterizing TcMAC21 phospho-proteomic profiles. Because the second principal component (PC2) appears to well explain the difference between TcMAC21 and control, we performed gene ontology (GO) analysis of the top 100 phosphorylated peptides that contributed positively or negatively to the PC2 (Table S6). According to the functional annotations, many “PC2-positive” proteins were related to “actin filament-based processes” or “microtubule cytoskeleton organization” (Figure 4E), as exemplified by Ank2 (ankyrin-2) and Map (microtubule associated proteins) (Figures S2A and S2B). Therefore, the difference in the phosphorylation state between euploid and TcMAC21 appears to result in misregulation of the cytoskeleton. On the other hand, many “PC2-negative” proteins are related to “neuron projection development”, “cell junction organization”, or “modulation of chemical synaptic transmission” (Figure 4F). Examples of these proteins include Farp1 (FERM, Rho/ArhGEF, and Pleckstrin domain protein 1) and the DLGAP (discs-large-associated protein) protein family (Figures S2C–S2E). These findings suggest that alterations in the development of neurons and synapses contribute to the phenotypic expression in TcMAC21.
Discussion
The neuropathologic consequences of DS begin during fetal development with decreased proliferation and neuronal output of cortical progenitors.7,23,37,38,39,40 Because these alterations can influence synaptic development and circuit formations, thereby affecting cognitive function, it is essential to identify early brain abnormalities in DS. We recently generated the TcMAC21 mouse, the most complete genetic model of DS created to date with 93% of the HSA21q protein-coding genes.22 We demonstrated that the expression of most HSA21q genes is regulated similarly to their mouse orthologs and cause perturbations in gene expression throughout the genome.22 Similar to other mouse models of DS, TcMAC21 mice exhibit significant learning and memory deficits,22 but the mechanisms underlying the alterations in brain development have not yet been examined. Characterizing embryonic brain development in this mouse model will advance our understanding of the cognitive deficits observed in DS. In this study, we revealed progenitor proliferation abnormalities in the embryonic TcMAC21 forebrain in association with reduced neuronal production. Further, in the postnatal period, neuronal density was reduced in the TcMAC21 neocortex. These findings suggest that prenatal changes underlie the abnormal brain development observed in TcMAC21 and demonstrate the usefulness of this mouse model for studying the contribution of aberrations in prenatal and postnatal development to the cognitive deficits observed in DS.
In addition, our findings revealed impaired proliferation and neuronal output of cortical progenitors in the developing neocortex of TcMAC21 (Figure 1). Similar phenotypes are observed in several DS mouse models and fetal DS human brain,7,14,16,23,37,38,39,40,41 suggesting that mis-regulation of cortical progenitors is a common phenotype, although the Dp16 mouse model shows normal progenitor proliferation and neurogenesis.15 Whether or not the progenitors of cortical interneurons are mis-regulated in DS brains, however, remains an unanswered question due to discrepancies across mouse models. Our study suggests the impaired proliferation of progenitors in the MGE in TcMAC21 (Figures 1F and 1G). No MGE progenitor abnormalities have been found in Dp16 and Ts1Cje.12,15 In contrast, increased proliferation of MGE progenitors is observed in Ts65Dn.12,13 Importantly, a comparison of behavioral phenotypes between Ts65Dn and Ts66Yah suggests that triplication of the non-Hsa21 orthologous Mmu17 genes significantly contributes to the behavioral phenotype of Ts65Dn.20,21 Thus, it is possible that increased proliferation of MGE progenitors in Ts65Dn is associated with the Mmu17 non-Hsa21 orthologous region. Future investigation of the MGE progenitor phenotype in developing Ts66Yah brains would help to clarify this issue. Together, these findings highlight the possibility that each mouse model, including TcMAC21, reproduces different aspects of brain development in DS, and the need to carefully evaluate the inconsistencies between models and their relevance to the human phenotype.
Accumulating evidence from studies of humans with DS and DS mouse models has led to a consensus that DS brains have a lower density and number of glutamatergic projection neurons than normal brains.12,13,14,15,16,42 Consistent with this, we observed a reduction in the density of multiple subtypes of projection neurons in the TcMAC21 cortex. Whether the number of inhibitory interneurons is increased or decreased in DS brains, however, remains unclear due to the sparseness of information and the discrepancies across studies. Nonetheless, recent single nucleus RNA-seq analysis using Brodmann areas 8/9 of DS cortex revealed increased inhibitory:excitatory neuron ratios and increased CGE-derived, but not MGE-derived, inhibitory neuron ratios.33 Though we focused on the somatosensory cortex in this study, we observed similar changes in the TcMAC21 cortex; the density of excitatory neurons and PV- and SOM-positive interneurons was reduced to a similar extent (i.e., no apparent alterations in MGE-derived inhibitory neuron ratios), but that of VIP- and CR-positive cells was unchanged (i.e., increase in CGE-derived inhibitory neuron ratios) (Figure 3). Although the number of cortical interneurons has been assessed in several mouse models, the results have not been consistent. For example, an increase in the number of PV- and SOM-positive inhibitory neurons was observed in the Ts65Dn neocortex.13 In contrast, Dp16 mice exhibit a reduced number of these interneuron subtypes.12,15 Ts65Dn mice have either a similar or larger number of CR-positive neurons, while no such alteration is observed in the Dp16 cortex.13,15,43 In terms of alterations in the number of cortical neurons, the phenotype observed in TcMAC21 is similar to that in Dp16. The underlying mechanism is likely different, however, because Dp16 shows normal progenitor proliferation and neurogenesis.15 As for the number of glial cells, previous studies reported that the numbers of microglia and astrocytes in the young adult DS cortex were similar to those in the control cortex.44,45 These findings are consistent with our observations that the density of ALDH1L1-positive astrocytes and IBA1-positive microglia in the somatosensory cortex is similar between 3-month-old euploid mice and TcMAC21. Also, the modest reduction of the GSTπ-positive fraction of OLIG2-positive cells, reflecting matured oligodendrocytes, observed in TcMAC21 is consistent with impaired oligodendrocyte maturation in DS.32 Together, these findings highlight a unique neurodevelopmental phenotype in TcMAC21 and its similarity with the human DS phenotype. We expect that further studies using this mouse model will produce more relevant results, supporting a wide range of basic and preclinical research.
In this study, we used the classical cell counting method employed in previous studies of different DS mouse models,12,13,14,15,16 allowing us to compare the current work to previously published literature. Of note, cell counting methods in neuroscience have been subject to extensive debates regarding validity and reliability, as reviewed in.46 In this context, stereological counts using modern 3-dimensional probes and systematic random sampling of sections through the tissue of interest may be more accurate for cell number estimation. Indeed, this method has been implemented in the study of the brains of DS patients and DS mouse models.6,47 Future investigation of cell numbers in TcMAC21 brains with modern stereological cell count and comparing the results with those in other DS mouse models would validate our conclusion and advance our understanding of the brain structural abnormalities in DS.
In addition to a reduced number of neurons in the cortex, mis-regulation of synapse structure and function could lead to cognitive impairment in DS. Our phospho-proteomic analysis of the TcMAC21 cortex revealed alterations in the phosphorylation levels of a series of synaptic proteins (Table S5). For example, DLGAPs (discs-large-associated proteins) are important scaffold proteins in the post-synaptic density48; CaMKII, PKCγ, Synapsin-1, Syntaxin-1b, and Rims1 contribute to neurotransmitter secretion49,50,51; and β-adductin, bassoon, neuroligin2, and FARP1 are required for synapse assembly.52,53,54 The phosphorylation levels of these proteins are downregulated in the TcMAC21 cortex and contributed negatively to PC2 in our PCA (Figures 4, S2, and Table S6). The role of phosphorylation has not been examined in most proteins, but functional changes in multiple synaptic proteins due to altered phosphorylation levels are likely to contribute to the abnormal brain function observed in TcMAC21 mice. Interestingly, similar mis-regulation (i.e., altered phosphorylation levels of synaptic protein) is also observed in other mouse models of DS, such as Ts65Dn and Dp16, and is counteracted by environmental enrichment and/or pharmacologic treatment, which improve cognition in the mouse models.55,56,57 Together with these findings, our phospho-proteomic analysis of the TcMAC21 cortex provides support for the idea that phosphorylation signaling in synapses is a promising target for enhancing cognition in DS.
Through phospho-proteomic analysis, we found that several proteins involved in neuron projection, cytoskeleton arrangements or modulation of chemical synaptic transmission are differentially phosphorylated in the TcMAC21 cortex (Figures 4 and S2). Unfortunately, we could not validate these results with immunoblotting, because phospho-specific antibodies were not commercially available when we focused on the top 100 phosphorylated peptides that contributed positively or negatively to the PC2 in our PCA (Table S6). The lack of phospho-specific antibodies highlights the advantage of mass spectrometry-based phospho-proteome analysis that can evaluate phosphorylation levels when phosphorylation sites are novel and phospho-specific antibodies are not available. The development of phospho-specific antibodies against mis-regulated proteins in TcMAC21 would not only help validate our phospho-proteomic analysis, but also advance our understanding of how neuronal function is perturbed in the TcMAC21 cortex.
A number of symptoms observed in patients with DS are thought to result from dysfunction in several cortical areas (reviewed in Dierssen et al.58). For example, a large proportion of patients with DS have difficulties with sensory integration of the tactile system,59 which is likely mediated by deficits in the somatosensory cortex. In this study, we revealed alterations in the development and phosphorylation levels of a series of proteins in the somatosensory cortex of the TcMAC21 mouse model. These observations help to elucidate the mechanisms underlying functional abnormalities in the somatosensory cortex in DS. Importantly, in addition to the somatosensory cortex, other cortical regions are thought to exhibit functional alterations in DS. Thus, the development of cortical regions other than the somatosensory cortex may be disturbed in TcMAC21 as well, and should be examined further in future studies.
The findings of the present study provide insights into the neurodevelopmental defects of TcMAC21 mice at the cellular and molecular levels. Given that TcMAC21 contains a larger number of HSA21q protein-coding genes compared with other mouse models, this mouse model will be valuable for studying the contributions of prenatal changes in brain development to cognition in DS. Further analysis investigating the effect of pharmacologic treatment on brain development and cognitive function in TcMAC21 will advance preclinical studies aimed at the prevention and treatment of DS.
Limitation of the study
In human pregnancies, fetuses with trisomy 21 develop in a normal intrauterine environment of euploid mothers. In the case of TcMAC21, at least in the genetic background used in this study, trisomic males are sterile and thus transmission of the trisomic chromosome is only possible through an aneuploid dam. Recently, Sarver et al. demonstrated that TcMAC21 mice show hypermetabolism.60 This could affect embryonic development, but the impact of an abnormal intrauterine environment on the development of TcMAC21 embryos and their euploid littermates is unknown. The development of fertile TcMAC21 male mice in the future will help to clarify this point.
Conclusion
Here, we examined the embryonic brain development of TcMAC21 and found reduced proliferation of progenitors and delayed neurogenesis in the TcMAC21 forebrain. We extended our findings to early postnatal and adult stages, and found a reduced density of both projection neurons and a subset of interneurons in the TcMAC21 neocortex. Further, a comprehensive (phospho-)proteome analysis of TcMAC21 cortex revealed alterations in the phosphorylation levels of a series of synaptic proteins. In summary, TcMAC21 shows similar brain development abnormalities as DS and may be a good model for investigating the prenatal and postnatal causes of intellectual disability in patients with DS.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-SOX2 | Cell Signaling Technology | 23064; RRID:AB_2714146 |
| rat anti-TBR2 | Invitrogen | 14-4875-82; RRID:AB_11042577; |
| rabbit anti-P-H3 | millipore | 06-570; RRID:AB_310177 |
| rabbit anti-TBR1 | Abcam | ab183032; RRID:AB_2936859 |
| rat anti-CTIP2 | Abcam | ab18465; RRID:AB_2064130 |
| rabbit anti-SATB2 | Abcam | ab92446; RRID:AB_10563678 |
| mouse anti-PV | Merck | MAB1572; RRID:AB_2174013 |
| rat anti-SOM | Merck | MAB354; RRID:AB_2255365 |
| rabbit anti-CR | Swent | 7697; RRID:AB_2721226 |
| rabbit anti-VIP | Immunostar | 20077; RRID:AB_572270 |
| rabbit anti-ALDH1L1 | Abcam | ab87117; RRID:AB_10712968 |
| mouse anti-OLIG2 | Merck | MABN50; RRID:AB_10807410 |
| rabbit anti-GST-π | MBL | 312; RRID:AB_591792 |
| goat anti-IBA1 | FUJIFILM | 011-27991; RRID:AB_2935833 |
| rabbit anti-cleaved caspase 3 | Cell Signaling Technology | 9664; RRID:AB_2070042 |
| Cy™3 AffiniPure Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | 711-165-152; RRID:AB_2307443 |
| Cy™3 AffiniPure Donkey Anti-Mouse IgG (H + L) | Jackson ImmunoResearch Labs | 715-165-151, RRID:AB_2315777 |
| Cy™3 AffiniPure Donkey Anti-Rat IgG (H + L) | Jackson ImmunoResearch Labs | 712-165-153, RRID:AB_2340667 |
| Cy™3 AffiniPure Donkey Anti-goat IgG (H + L) | Jackson ImmunoResearch Labs | 705-165-147, RRID:AB_2307351 |
| Cy™5 AffiniPure Donkey Anti-Rabbit IgG (H + L) | Jackson ImmunoResearch Labs | 711-175-152, RRID:AB_2340607 |
| Cy™5 AffiniPure Donkey Anti-Mouse IgG (H + L) | Jackson ImmunoResearch Labs | 715-175-151, RRID:AB_2340820 |
| Cy™5 AffiniPure Donkey Anti-Rat IgG (H + L) | Jackson ImmunoResearch Labs | 712-175-153, RRID:AB_2340672 |
| Chemicals, peptides, and recombinant proteins | ||
| EdU (5-ethynyl-2′-deoxyuridine) | Thermofisher Scientific | A10044 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) | Thermofisher Scientific | D1306 |
| Critical commercial assays | ||
| Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 555 dye | Thermofisher Scientific | C10338 |
| Deposited data | ||
| Dataset of proteomic and phospho-proteomic analysis of TcMAC21 | This paper | PXD044343 |
| Experimental models: Organisms/strains | ||
| Mouse: TcMAC21 (Tc (HSA21, CAG-EGFP)1Yakaz) | Kazuki et al.,22 2020 | RRID:IMSR_JAX:035561 |
| Software and algorithms | ||
| GraphPad Prism | Dotmatics | RRID:SCR_002798 |
| Adobe Photoshop | Adobe | RRID:SCR_014199 |
| Adobe Illustrator | Adobe | RRID:SCR_010279 |
| Fiji | NIH | RRID:SCR_002285 |
| Proteome discoverer | Thermofisher Scientific | RRID:SCR_014477 |
| Zeiss Zen Lite | Zeiss | RRID:SCR_023747 |
| Python Programming Language | Python Software Foundation | RRID:SCR_008394 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Keizo Takao (takao@cts.u-toyama.ac.jp).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
All data reported in this paper will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Animals
TcMAC21 (Tc (HSA21, CAG-EGFP)1Yakaz) mice were maintained on B6D2F1 (C57BL/6J (B6) x DBA/2J (D2)) background by crossing B6D2F1 males with carrier females. A genotyping of TcMAC21 mouse was performed by polymerase chain reaction (PCR) as previously described.22 All mice were housed under 12-h light-12-h dark cycle and ad libitum access to food and water. For timed pregnancies, the date of vaginal plug was designated as E0.5 and the date of birth was defined as P0.5. The sex of the embryos used was not examined. Only male mice were used in postnatal analyses. Embryos at stages E14.5 and E16.5 were used for embryonic analysis. P0.5- and 3-month-old mice were used for postnatal analysis. None of the animals had been involved in previous experiments. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) protocols of Tokyo Metropolitan Institute of Medical Science, Tottori University, and University of Toyama.
Method details
Antibodies
The following antibodies were used for immunostaining. Rabbit anti-SOX2 (1:1,000; 23064, Cell Signaling Technology), rat anti-TBR2 (1:2,000; 14–4875, eBioscience), rabbit anti-P-H3 (1:1,000; 06–570, Upstate), rabbit anti-TBR1 (1:2,000; ab183032, Abcam), rat anti-CTIP2 (1:500; ab18465, Abcam), anti-SATB2 (1:2,000; ab92446, Abcam), mouse anti-PV (1:1,000; MAB1572, Merck), rat anti-SOM (1:200; MAB354, Merck), rabbit anti-CR (1:1,000; 7697, Swent), rabbit anti-VIP (1:1,000; 20077, Immunostar), rabbit anti-ALDH1L1 (1:500; ab87117, Abcam), mouse anti-OLIG2 (1:500; MABN50, Merck), rabbit anti-GST-π (1:1,000; 312, MBL), goat anti-IBA1 (1:1,000; 011–27991, FUJIFILM), rabbit anti-cleaved caspase 3 (1:1,000; 9664, Cell Signaling Technology).
Immunohistochemistry
Prenatal and perinatal brains were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (for E14.5 and E16.5 brains) or 2 h (for P0.5 brains) and cryoprotected in 30% sucrose in PBS overnight at 4°C. Thereafter, the brains were embedded in a solution of a 2:1 mixture of 30% sucrose/PBS and OCT compound (Sakura), frozen by liquid nitrogen, and stored at −80°C until use. Thick cryosections (20 μm) were made. For adult brain, mice were perfused with saline and then 4% PFA in PBS. Brains were excised and fixed in 4% PFA in PBS overnight at 4°C. Sixty μm coronal sections were prepared using an automated vibratome (Leica VT1200S). Brain sections were washed with PBS, incubated with blocking solution (3% [w/v] BSA, 5% [v/v] FBS, and 0.2% [w/v] Triton X-100 in PBS) and then incubated with primary antibodies overnight at 4°C. The sections were then incubated with Cy3/Cy5-conjugated secondary antibodies overnight at 4°C and mounted in a DAPI-Fluoromount-G mounting solution (SouthernBiotech). For staining with SOX2, TBR2, CTIP2, ALDH1L1, and GST-π, brain sections were pretreated with HistoVT One solution (Nacalai Tesque) for 15 min at 70°C. Images were obtained with a 20× or 63× objective on Zeiss LSM780 or LSM980 confocal microscope.
Cell counting
For cell counting of immunopositive cells, the classical method employed in previous studies of different DS mouse models was used.12,13,14,15,16 Coronal sections for cell counting were carefully selected using specific anatomical landmarks and slides that are at the similar antero-posterior level were used. Labeled cells in the lateral cortical wall (the future somatosensory cortex) in embryonic tissue of E14.5, E16.5 and P0.5 embryos were counted in a 200 μm-wide column, with the exception of P-H3+ and cleaved caspase-3+ cells that were counted in a 400 μm-wide and 600 μm-wide columns, respectively. In E14.5 and E16.5 MGE/CGE samples, the entire surface of the MGE/CGE was measured; the number of P-H3+ cells were quantified within the measured area and normalized. The position of MGE and CGE was determined by cell packing density revealed by DAPI staining. For cell counting of the adult somatosensory cortex, sections around bregma −1.5 mm (Paxinos and Franklin’s THE MOUSE BRAIN, fifth edition) were selected. Labeled cells in adult sections were counted in a wide column of 500 μm. All cell counts were blinded and performed on a minimum of 2 sections. It should be noted that cell count was done manually using ImageJ and all immunopositive cells could be unambiguously identified. In addition, cell counts were not acquired using randomly sampled unbiased stereological standards and therefore cannot necessarily be extrapolated to the entire cortex.
EdU injection and cell counting
Pregnant females (E13.5 or E15.5) were injected with EdU (5 mg/kg body weight; Thermofisher Scientific), and sacrificed 24 h later. Embryonic brains were fixed, cryoprotected and cryosectioned as described before. EdU was detected in coronal sections using the Click-iT EdU Alexa Fluor 555 kit (Thermofisher Scientific) according to the manufacturer’s instructions. Images were obtained with a 63× objective on a Zeiss LSM980 confocal microscope, and EdU+ cell counts were performed in a 200 μm-wide field of the dorsolateral wall in a minimum of 2 sections per embryo. It should be noted that all EdU+ cells could be unambiguously identified, and all cells in which EdU-signal was detected, regardless of signal intensity or localization, were counted as positive cells.
Preparation of phosphopeptides
Preparation of phospho-peptides was performed as previously described with minor modifications.61 The somatosensory cortices of 3-month-old mice were dissected, frozen by liquid nitrogen, and stored at −80°C until use (n = 4 for euploid and n = 3 for TcMAC21 from 3 dams.). Frozen cortices were sonicated in 220 μL of PTS buffer (100 mM NH4HCO3, 12 mM sodium deoxycholate, and 12 mM sodium N-lauroylsarcosinate) containing Phosphatase Inhibitor Cocktails 2 and 3 (Sigma-Aldrich) using an ultrasonic disruptor (UR-21P; TOMY; output power 2 for 30–45 s) and centrifuged at 21,600 × g for 20 min at 4°C. The supernatants were reduced with 10 mM DTT at 60°C for 30 min and then alkylated by incubation with 22 mM iodoacetamide at 37°C for 30 min in the dark. The resultant protein samples were diluted with 100 mM NH4HCO3 solution up to 1 mL and digested with trypsin (Sigma-Aldrich) at 1:100 (w/w) by incubation at 37°C for 18 h in the dark. After the digestion, an equal volume of ethyl acetate was added to the samples, and the mixtures were acidified with 0.5% TFA and then well mixed to transfer the detergents into the organic phase. After the samples were centrifuged at 15,700 × g for 2 min at room temperature, the aqueous phase containing peptides was collected. The samples were concentrated by a centrifugal evaporator (EYELA) and desalted using a MonoSpin C18 column (GL Sciences). After 10% of the eluates were dried by the evaporator, they were analyzed by LC-MS/MS analysis as samples labeled “total peptides.” The remaining eluates were dried and applied to a highselect Fe-NTA phosphopeptide enrichment kit (Thermo Fisher Scientific). The enriched samples, labeled “phosphopeptides,” were dried before LC-MS/MS analysis. LC-MS/MS-Based Proteomic Analysis.
Proteomic analysis was performed as previously described62 with minor modifications. The dried and desalted peptides were dissolved in distilled water containing 2% acetonitrile and 0.1% TFA. The LC-MS/MS analyses were performed using a mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific) equipped with a nano ultra-HPLC system (Dionex Ultimate 3000; Thermo Fisher Scientific). The peptides were loaded to the LCMS/MS system with a trap column (0.3 × 5 mm L-column ODS; Chemicals Evaluation and Research Institute) and a capillary column (0.1 × 150 mm L-column ODS; Chemicals Evaluation and Research Institute) at a flow late of 20 μL/min. The loaded peptides were separated by a gradient using mobile phases A (1% formic acid in distilled water) and B (1% formic acid in acetonitrile) at a flow late of 300 nL/min (0% B for 5 min, 0–30% B for 150 min, 30–50% B for 10 min, 50–95% B for 0.1 min, 95% B for 9.8 min, 95–0% B for 0.1 min, and 0% B for 5 min). The eluted peptides were electrosprayed (2.0 kV) and introduced into the MS equipment (positive ion mode, data-dependent MS/MS). Each of the most intense precursor ions (up to the top 10) was isolated and fragmented by higher collision energy dissociation (HCD) with the normalized collision energy (27%). For full MS scans, the scan range was set to 350–1,500 m/z at a resolution of 70,000, and the automatic gain control (AGC) target was set to 3e6 with a maximum injection time of 60 ms. For MS/MS scans, the precursor isolation window was set to 1.6 m/z at a resolution of 17,500, and the AGC target was set to 5e5 with a maximum injection time of 100 ms. The Orbitrap mass analyzer was operated with the “lock mass” option to perform shotgun detection with high accuracy. The raw spectra were extracted using Proteome Discoverer 2.2 (Thermo Fisher Scientific) and searched against the mouse SwissProt database (TaxID 10,090 and subtaxonomies, v2017-10-25) and human protein list of the long arm of HSA21 (Supplementary file 1) with following settings. The parameter of the cleavage was set to trypsin, and the missed cleavage was allowed up to 2. The mass tolerances were set to 10 ppm for the precursor ion and 0.02 Da for the fragment ion. As for protein modifications, we set carbamidomethylation (+57.021 Da) at Cys as static (fixed) modifications for peptide, oxidation (+15.995 Da) at Met, phosphorylation (+79.966 Da) at Ser and Thr as dynamic (nonfixed) modifications for peptide and acetylation (+42.011 Da) at the amino-terminus as a dynamic modification for protein terminus. The amount of each peptide was semiquantified using peak area with Precursor Ions Quantifier in Proteome Discoverer 2.2. For downstream analysis, pseudovalue 2 was added to avoid zero value. The kinase substrate enrichment analysis (KSEA) was performed using kinact package36 based on dataset from PhosphoSitePlus.63
Quantification and statistical analysis
Statistical analysis
For histological studies, all statistical analyses were performed using Prism 8/9 software. Comparisons of mean differences between groups were based on a Welch’s two-tailed t-test. When evaluating differences of the density of immunopositive cells in the histological analysis, Holm-Sidak corrections64 were applied to avoid false discoveries because of multiple comparisons. The significance level was set at p < 0.05 for all tests. For proteomic experiments, statistical analysis was performed by Python 3.10. For a list of statistical tests and p values, see Table S7.
Acknowledgments
We thank A. Kurabayashi for technical assistance with proteomic analysis. The work was supported in part by JSPS KAKENHI Grant Number 18K06458, 18KT0026, research grants from the Takeda Science Foundation, The Uehara Memorial Foundation, Kato Memorial Bioscience Foundation (N.K.), JSPS KAKENHI Grant Number 21H05130, JST FOREST Program (JPMJFR2150) (H.Y.), Research Support Project for Life Science and Drug Discovery (BINDS) from AMED (JP23ama121046), Joint Research of the Exploratory Research Center on Life and Living Systems (ExCELLS) (ExCELLS program No, 21–101), and JST, CREST Grant Number JPMJCR18S4 (Y.K.), JSPS KAKENHI Grant Number 19H03538 (K.T.).
Author contributions
Conceptualization, N.K.; Methodology, N.K.; Formal Analysis, N.K., Y.O., S.H., M.H., and H.Y.; Investigation, N.K., K.F., Y.O., S.H., and M.H.; Resources, Y.K.; Writing—Original Draft, N.K.; Writing—Review and Editing, K.F., Y.O., S.H., M.H., H.Y., Y.K., and K.T.; Supervision, K.T.; Funding acquisition, N.K., H.Y., Y.K., and K.T.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 2, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108379.
Contributor Information
Nobuhiro Kurabayashi, Email: kurabayashi-nb@igakuken.or.jp.
Keizo Takao, Email: takao@cts.u-toyama.ac.jp.
Supplemental information
References
- 1.Canfield M.A., Honein M.A., Yuskiv N., Xing J., Mai C.T., Collins J.S., Devine O., Petrini J., Ramadhani T.A., Hobbs C.A., Kirby R.S. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999-2001. Birth Defects Res. A Clin. Mol. Teratol. 2006;76:747–756. doi: 10.1002/bdra.20294. [DOI] [PubMed] [Google Scholar]
- 2.Chapman R.S., Hesketh L.J. Behavioral phenotype of individuals with Down syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2000;6:84–95. doi: 10.1002/1098-2779(2000)6:2<84::AID-MRDD2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 3.Haydar T.F., Reeves R.H. Trisomy 21 and early brain development. Trends Neurosci. 2012;35:81–91. doi: 10.1016/j.tins.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinter J.D., Eliez S., Schmitt J.E., Capone G.T., Reiss A.L. Neuroanatomy of Down’s Syndrome: A High-Resolution MRI Study. Aust. J. Pharm. 2001;158:1659–1665. doi: 10.1176/appi.ajp.158.10.1659. [DOI] [PubMed] [Google Scholar]
- 5.Schmidt-Sidor B., Wisniewski K.E., Shepard T.H., Sersen E.A. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin. Neuropathol. 1990;9:181–190. [PubMed] [Google Scholar]
- 6.Golden J.A., Hyman B.T. Development of the superior temporal neocortex is anomalous in trisomy 21. J. Neuropathol. Exp. Neurol. 1994;53:513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 7.Larsen K.B., Laursen H., Graem N., Samuelsen G.B., Bogdanovic N., Pakkenberg B. Reduced cell number in the neocortical part of the human fetal brain in Down syndrome. Ann. Anat. 2008;190:421–427. doi: 10.1016/j.aanat.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 8.Wisniewski K.E. Down syndrome children often have brain with maturation delay, retardation of growth, and cortical dysgenesis. Am. J. Med. Genet. Suppl. 1990;7:274–281. doi: 10.1002/ajmg.1320370755. [DOI] [PubMed] [Google Scholar]
- 9.Petit T.L., LeBoutillier J.C., Alfano D.P., Becker L.E. Synaptic development in the human fetus: a morphometric analysis of normal and Down’s syndrome neocortex. Exp. Neurol. 1984;83:13–23. doi: 10.1016/0014-4886(84)90041-4. [DOI] [PubMed] [Google Scholar]
- 10.Takashima S., Becker L.E., Armstrong D.L., Chan F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down’s syndrome. A quantitative and qualitative golgi study. Brain Res. 1981;225:1–21. doi: 10.1016/0006-8993(81)90314-0. [DOI] [PubMed] [Google Scholar]
- 11.Das I., Reeves R.H. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis. Model. Mech. 2011;4:596–606. doi: 10.1242/dmm.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aziz N.M., Guedj F., Pennings J.L.A., Olmos-Serrano J.L., Siegel A., Haydar T.F., Bianchi D.W. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Dis. Model. Mech. 2018;11 doi: 10.1242/dmm.031013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakrabarti L., Best T.K., Cramer N.P., Carney R.S.E., Isaac J.T.R., Galdzicki Z., Haydar T.F. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 2010;13:927–934. doi: 10.1038/nn.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakrabarti L., Galdzicki Z., Haydar T.F. Defects in Embryonic Neurogenesis and Initial Synapse Formation in the Forebrain of the Ts65Dn Mouse Model of Down Syndrome. J. Neurosci. 2007;27:11483–11495. doi: 10.1523/JNEUROSCI.3406-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goodliffe J.W., Olmos-Serrano J.L., Aziz N.M., Pennings J.L.A., Guedj F., Bianchi D.W., Haydar T.F. Absence of Prenatal Forebrain Defects in the Dp(16)1Yey/+ Mouse Model of Down Syndrome. J. Neurosci. 2016;36:2926–2944. doi: 10.1523/JNEUROSCI.2513-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishihara K., Amano K., Takaki E., Shimohata A., Sago H., Epstein C.J., Yamakawa K. Enlarged Brain Ventricles and Impaired Neurogenesis in the Ts1Cje and Ts2Cje Mouse Models of Down Syndrome. Cereb. Cortex. 2010;20:1131–1143. doi: 10.1093/cercor/bhp176. [DOI] [PubMed] [Google Scholar]
- 17.Sago H., Carlson E.J., Smith D.J., Kilbridge J., Rubin E.M., Mobley W.C., Epstein C.J., Huang T.-T. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. USA. 1998;95:6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duchon A., Raveau M., Chevalier C., Nalesso V., Sharp A.J., Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling down syndrome. Mamm. Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta M., Dhanasekaran A.R., Gardiner K.J. Mouse models of Down syndrome: gene content and consequences. Mamm. Genome. 2016;27:538–555. doi: 10.1007/s00335-016-9661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duchon A., Del Mar Muñiz Moreno M., Chevalier C., Nalesso V., Andre P., Fructuoso-Castellar M., Mondino M., Po C., Noblet V., Birling M.-C., et al. Ts66Yah, a mouse model of Down syndrome with improved construct and face validity. Dis. Model. Mech. 2022;15 doi: 10.1242/dmm.049721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guedj F., Kane E., Bishop L.A., Pennings J.L.A., Herault Y., Bianchi D.W. The Impact of Mmu17 Non-Hsa21 Orthologous Genes in the Ts65Dn Mouse Model of Down Syndrome: The Gold Standard Refuted. Biol. Psychiatry. 2023;94:84–97. doi: 10.1016/j.biopsych.2023.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazuki Y., Gao F.J., Li Y., Moyer A.J., Devenney B., Hiramatsu K., Miyagawa-Tomita S., Abe S., Kazuki K., Kajitani N., et al. A non-mosaic transchromosomic mouse model of Down syndrome carrying the long arm of human chromosome 21. Elife. 2020;9 doi: 10.7554/eLife.56223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Contestabile A., Fila T., Ceccarelli C., Bonasoni P., Bonapace L., Santini D., Bartesaghi R., Ciani E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. doi: 10.1002/hipo.20308. [DOI] [PubMed] [Google Scholar]
- 24.Lu J., Lian G., Zhou H., Esposito G., Steardo L., Delli-Bovi L.C., Hecht J.L., Lu Q.R., Sheen V. OLIG2 over-expression impairs proliferation of human Down syndrome neural progenitors. Hum. Mol. Genet. 2012;21:2330–2340. doi: 10.1093/hmg/dds052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butt S.J.B., Fuccillo M., Nery S., Noctor S., Kriegstein A., Corbin J.G., Fishell G. The Temporal and Spatial Origins of Cortical Interneurons Predict Their Physiological Subtype. Neuron. 2005;48:591–604. doi: 10.1016/j.neuron.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 26.Miyoshi G., Hjerling-Leffler J., Karayannis T., Sousa V.H., Butt S.J.B., Battiste J., Johnson J.E., Machold R.P., Fishell G. Genetic Fate Mapping Reveals That the Caudal Ganglionic Eminence Produces a Large and Diverse Population of Superficial Cortical Interneurons. J. Neurosci. 2010;30:1582–1594. doi: 10.1523/JNEUROSCI.4515-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pleasure S.J., Anderson S., Hevner R., Bagri A., Marin O., Lowenstein D.H., Rubenstein J.L. Cell Migration from the Ganglionic Eminences Is Required for the Development of Hippocampal GABAergic Interneurons. Neuron. 2000;28:727–740. doi: 10.1016/S0896-6273(00)00149-5. [DOI] [PubMed] [Google Scholar]
- 28.Wonders C.P., Anderson S.A. The origin and specification of cortical interneurons. Nat. Rev. Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- 29.Kurabayashi N., Sanada K. Increased dosage of DYRK1A and DSCR1 delays neuronal differentiation in neocortical progenitor cells. Genes Dev. 2013;27:2708–2721. doi: 10.1101/gad.226381.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bulfone A., Smiga S.M., Shimamura K., Peterson A., Puelles L., Rubenstein J.L. T-Brain-1: A homolog of Brachyury whose expression defines molecularly distinct domains within the cerebral cortex. Neuron. 1995;15:63–78. doi: 10.1016/0896-6273(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 31.Arlotta P., Molyneaux B.J., Chen J., Inoue J., Kominami R., Macklis J.D. Neuronal Subtype-Specific Genes that Control Corticospinal Motor Neuron Development In Vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 32.Olmos-Serrano J.L., Kang H.J., Tyler W.A., Silbereis J.C., Cheng F., Zhu Y., Pletikos M., Jankovic-Rapan L., Cramer N.P., Galdzicki Z., et al. Down Syndrome Developmental Brain Transcriptome Reveals Defective Oligodendrocyte Differentiation and Myelination. Neuron. 2016;89:1208–1222. doi: 10.1016/j.neuron.2016.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palmer C.R., Liu C.S., Romanow W.J., Lee M.-H., Chun J. Altered cell and RNA isoform diversity in aging Down syndrome brains. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2114326118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 35.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A.H., Tanaseichuk O., Benner C., Chanda S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019;10:1523. doi: 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wirbel J., Cutillas P., Saez-Rodriguez J. In: Cancer Systems Biology Methods in Molecular Biology. Von Stechow L., editor. Springer New York; 2018. Phosphoproteomics-Based Profiling of Kinase Activities in Cancer Cells; pp. 103–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guihard-Costa A.-M., Khung S., Delbecque K., Ménez F., Delezoide A.-L. Biometry of Face and Brain in Fetuses with Trisomy 21. Pediatr. Res. 2006;59:33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- 38.Rotmensch S., Liberati M., Bronshtein M., Schoenfeld-Dimaio M., Shalev J., Ben-Rafael Z., Copel J.A. Prenatal sonographic findings in 187 fetuses with Down syndrome. Prenat. Diagn. 1997;17:1001–1009. doi: 10.1002/(sici)1097-0223(199711)17:11<1001::aid-pd186>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 39.Weitzdoerfer R., Dierssen M., Fountoulakis M., Lubec G. Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J. Neural. Transm. Suppl. 2001:59–70. doi: 10.1007/978-3-7091-6262-0_5. [DOI] [PubMed] [Google Scholar]
- 40.Wisniewski K.E., Laure-Kamionowska M., Wisniewski H.M. Evidence of arrest of neurogenesis and synaptogenesis in brains of patients with Down’s syndrome. N. Engl. J. Med. 1984;311:1187–1188. doi: 10.1056/NEJM198411013111818. [DOI] [PubMed] [Google Scholar]
- 41.Tyler W.A., Haydar T.F. Multiplex Genetic Fate Mapping Reveals a Novel Route of Neocortical Neurogenesis, Which Is Altered in the Ts65Dn Mouse Model of Down Syndrome. J. Neurosci. 2013;33:5106–5119. doi: 10.1523/JNEUROSCI.5380-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross M.H., Galaburda A.M., Kemper T.L. Down’s syndrome: is there a decreased population of neurons? Neurology. 1984;34:909–916. doi: 10.1212/wnl.34.7.909. [DOI] [PubMed] [Google Scholar]
- 43.Pérez-Cremades D., Hernández S., Blasco-Ibáñez J.M., Crespo C., Nacher J., Varea E. Alteration of inhibitory circuits in the somatosensory cortex of Ts65Dn mice, a model for Down’s syndrome. J. Neural. Transm. 2010;117:445–455. doi: 10.1007/s00702-010-0376-9. [DOI] [PubMed] [Google Scholar]
- 44.Flores-Aguilar L., Iulita M.F., Kovecses O., Torres M.D., Levi S.M., Zhang Y., Askenazi M., Wisniewski T., Busciglio J., Cuello A.C. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain. 2020;143:3653–3671. doi: 10.1093/brain/awaa326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mito T., Becker L.E. Developmental changes of S-100 protein and glial fibrillary acidic protein in the brain in Down syndrome. Exp. Neurol. 1993;120:170–176. doi: 10.1006/exnr.1993.1052. [DOI] [PubMed] [Google Scholar]
- 46.Bjerke I.E., Yates S.C., Carey H., Bjaalie J.G., Leergaard T.B. Scaling up cell-counting efforts in neuroscience through semi-automated methods. iScience. 2023;26 doi: 10.1016/j.isci.2023.107562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lorenzi H.A., Reeves R.H. Hippocampal hypocellularity in the Ts65Dn mouse originates early in development. Brain Res. 2006;1104:153–159. doi: 10.1016/j.brainres.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 48.Rasmussen A.H., Rasmussen H.B., Silahtaroglu A. The DLGAP family: neuronal expression, function and role in brain disorders. Mol. Brain. 2017;10:43. doi: 10.1186/s13041-017-0324-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bednarek E., Caroni P. β-Adducin Is Required for Stable Assembly of New Synapses and Improved Memory upon Environmental Enrichment. Neuron. 2011;69:1132–1146. doi: 10.1016/j.neuron.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 50.Cheadle L., Biederer T. The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J. Cell Biol. 2012;199:985–1001. doi: 10.1083/jcb.201205041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ziv N.E., Garner C.C. Cellular and molecular mechanisms of presynaptic assembly. Nat. Rev. Neurosci. 2004;5:385–399. doi: 10.1038/nrn1370. [DOI] [PubMed] [Google Scholar]
- 52.Südhof T.C., Rizo J. Synaptic Vesicle Exocytosis. Cold Spring Harb. Perspect. Biol. 2011;3:a005637. doi: 10.1101/cshperspect.a005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun M.-K., Alkon D.L. In: Progress in Molecular Biology and Translational Science. Teplow D.B., editor. Elsevier; 2014. The “Memory Kinases.”; pp. 31–59. [DOI] [PubMed] [Google Scholar]
- 54.Yasuda R., Hayashi Y., Hell J.W. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat. Rev. Neurosci. 2022;23:666–682. doi: 10.1038/s41583-022-00624-2. [DOI] [PubMed] [Google Scholar]
- 55.Brault V., Nguyen T.L., Flores-Gutiérrez J., Iacono G., Birling M.-C., Lalanne V., Meziane H., Manousopoulou A., Pavlovic G., Lindner L., et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021;17 doi: 10.1371/journal.pgen.1009777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen T.L., Duchon A., Manousopoulou A., Loaëc N., Villiers B., Pani G., Karatas M., Mechling A.E., Harsan L.-A., Limanton E., et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis. Model. Mech. 2018;11 doi: 10.1242/dmm.035634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Toma I.D. Re-establishment of the epigenetic state and rescue of kinome deregulation in Ts65Dn mice upon treatment with green tea extract and environmental enrichment. Sci. Rep. 2020;10:16023. doi: 10.1038/s41598-020-72625-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dierssen M., Herault Y., Estivill X. Aneuploidy: From a Physiological Mechanism of Variance to Down Syndrome. Physiol. Rev. 2009;89:887–920. doi: 10.1152/physrev.00032.2007. [DOI] [PubMed] [Google Scholar]
- 59.Pueschel S.M., Gallagher P.L., Zartler A.S., Pezzullo J.C. Cognitive and learning processes in children with Down syndrome. Res. Dev. Disabil. 1987;8:21–37. doi: 10.1016/0891-4222(87)90038-2. [DOI] [PubMed] [Google Scholar]
- 60.Sarver D.C., Xu C., Rodriguez S., Aja S., Jaffe A.E., Gao F.J., Delannoy M., Periasamy M., Kazuki Y., Oshimura M., et al. Hypermetabolism in mice carrying a near complete human chromosome 21 (Physiology) bioRxiv. 2023 doi: 10.1101/2023.01.30.526183. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imamura K., Yoshitane H., Hattori K., Yamaguchi M., Yoshida K., Okubo T., Naguro I., Ichijo H., Fukada Y. ASK family kinases mediate cellular stress and redox signaling to circadian clock. Proc. Natl. Acad. Sci. USA. 2018;115:3646–3651. doi: 10.1073/pnas.1719298115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abe Y.O., Yoshitane H., Kim D.W., Kawakami S., Koebis M., Nakao K., Aiba A., Kim J.K., Fukada Y. Rhythmic transcription of Bmal1 stabilizes the circadian timekeeping system in mammals. Nat. Commun. 2022;13:4652. doi: 10.1038/s41467-022-32326-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aickin M., Gensler H. Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. Am. J. Public Health. 1996;86:726–728. doi: 10.2105/AJPH.86.5.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are publicly available as of the date of publication. DOIs are listed in the key resources table.
This paper does not report original code.
All data reported in this paper will be shared by the lead contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.