

Abstract
Background & Aims:
The consumption of sugar and a high-fat diet (HFD) promotes the development of obesity and metabolic dysfunction. Despite their well-known synergy, the mechanisms by which sugar worsens the outcomes associated with a HFD are largely elusive.
Methods:
Six-week-old, male, C57Bl/6 J mice were fed either chow or a HFD and were provided with regular, fructose- or glucose-sweetened water. Moreover, cultured AML12 hepatocytes were engineered to overexpress ketohexokinase-C (KHK–C) using a lentivirus vector, while CRISPR-Cas9 was used to knockdown CPT1α. The cell culture experiments were complemented with in vivo studies using mice with hepatic overexpression of KHK–C and in mice with liver-specific CPT1α knockout. We used comprehensive metabolomics, electron microscopy, mitochondrial substrate phenotyping, proteomics and acetylome analysis to investigate underlying mechanisms.
Results:
Fructose supplementation in mice fed normal chow and fructose or glucose supplementation in mice fed a HFD increase KHK–C, an enzyme that catalyzes the first step of fructolysis. Elevated KHK–C is associated with an increase in lipogenic proteins, such as ACLY, without affecting their mRNA expression. An increase in KHK–C also correlates with acetylation of CPT1α at K508, and lower CPT1α protein in vivo. In vitro, KHK–C overexpression lowers CPT1α and increases triglyceride accumulation. The effects of KHK–C are, in part, replicated by a knockdown of CPT1α. An increase in KHK–C correlates negatively with CPT1α protein levels in mice fed sugar and a HFD, but also in genetically obese db/db and lipodystrophic FIRKO mice. Mechanistically, overexpression of KHK–C in vitro increases global protein acetylation and decreases levels of the major cytoplasmic deacetylase, SIRT2.
Conclusions:
KHK–C-induced acetylation is a novel mechanism by which dietary fructose augments lipogenesis and decreases fatty acid oxidation to promote the development of metabolic complications.
Keywords: Fructose, Ketohexokinase, Nonalcoholic fatty liver disease, fatty acid oxidation, carnitine palmitoyltransferase 1a, SIRT2, mass spectrometry
Graphical Abstract
Introduction
Obesity-associated fatty liver disease1 affects approximately 1 billion individuals worldwide and is associated with cardiometabolic risk factors, such as dyslipidemia and type 2 diabetes.2 The consumption of sugar-sweetened beverages and a high-fat diet (HFD) are major risk factors for development of these metabolic disturbances.3,4 Sugar-sweetened beverages most commonly contain high-fructose corn syrup (55% fructose and 45% glucose monosaccharides) or table sugar (sucrose, a disaccharide of 50% glucose and 50% fructose). Despite their structural similarities, and the fact that glucose can be endogenously converted to fructose,5 glucose and fructose metabolism are different. For instance, the catabolism of fructose occurs primarily in the liver, intestine, and kidney, mediated by the rate-limiting enzyme ketohexokinase-C (KHK–C).5 Fructose phosphorylation by KHK–C is 10x faster than phosphorylation of glucose by glucokinase and is not regulated by insulin or the end products of its metabolism, citrate or ATP. Additionally, glucose is used by almost every cell type and glycolysis is a highly regulated process. Thus, fructose, but not glucose, metabolism has been proposed to drive the development of metabolic complications.6,7
The major effects of fructose on metabolic dysfunction are thought to be secondary to its lipogenic properties. Indeed, KHK–C expression is controlled by the lipogenic transcription factor, carbohydrate response element-binding protein (ChREBP),8 and fructose strongly supports lipogenesis in mice on a normal chow diet.9 However, consumption of fructose in mice on a standard chow diet for 10 weeks does not lead to development of metabolic complications.6 Similarly, consumption of fructose from fruits and vegetables, as part of a well-balanced low-fat diet, does not promote development of metabolic complications in humans.10,11 On the other hand, consumption of fructose on HFD worsens metabolic dysfunction in rodents.12,13 Moreover, the most commonly used obesogenic diets utilize a combination of fructose and HFD.14 Despite its widespread use, the mechanism by which a combined intake of fructose and HFD accelerates the development of obesity and metabolic dysfunction is largely unknown.
Several groups have demonstrated that dietary fructose, but not glucose supplementation, impairs fatty acid oxidation (FAO) across many species.15-21 Long-chain FAO is mediated through a series of steps which rely on carnitine shuttle and fatty acid transport for proper oxidation.22 The rate-limiting enzyme of fatty acid oxidation, carnitine palmitoyltransferase 1a (CPT1α), resides on the outer mitochondrial membrane where it utilizes carnitine and acyl-CoAs to produce acylcarnitines, enabling fatty acid entry into the mitochondria. Deficiency of CPT1α is an inborn error of metabolism characterized by hepatic steatosis, increased risk of liver failure, and systemic inflammation.23-26 Malonyl-CoA, an intermediate in fatty acid synthesis, is an endogenous inhibitor of CPT1α. We have observed that chronic fructose supplementation of a HFD does not alter malonyl-CoA compared to the intake of HFD alone. However, chronic fructose feeding has been reported to decrease CPT1α,22,27 and we have shown that knocking down total KHK via small-interfering RNA (siRNA) increases CPT1α protein and acylcarnitine levels in the liver.20 Despite these observations, the mechanisms by which fructose catabolism and a concomitant increase in KHK–C influence CPT1α-mediated FAO have not been fully elucidated. Moreover, it remains to be determined whether upregulation of KHK–C can independently promote metabolic derangements or if the described effect on FAO is purely mediated by the lipogenic effects of fructose. Herein, we used cell culture and mouse models to elucidate the mechanisms underlying these previous observations.
Materials and methods
Experimental model and subject details
All animal protocols were in accordance with NIH guidelines and approved by the IACUC of the Joslin Diabetes Center and the University of Kentucky, as previously described.20 Mice were housed at 20-22 °C on a 12 h light/dark cycle with ad libitum access to food and water. Male C57BL/6 J mice at 6-weeks of age were purchased from Jackson Laboratory and were fed either chow (Mouse Diet 9 F, PharmaServ) or 60% HFD (Research Diets, D12492) for 2 weeks. Mice were provided either tap water or 30% (weight/volume) fructose or glucose-sweetened water. The mice were sacrificed from 8-11 AM, with the first mouse from each dietary group sacrificed prior to moving to the second mouse. This was repeated until all four mice per cage were sacrificed. For insulin injections, food was removed from the mouse cages from 7-9 AM, then mice were injected with saline or insulin (1 U/mouse) via the inferior vena cava. After 10 min, the mice were sacrificed and tissues were collected. For IV glucose tolerance test, mice were fasted overnight and 10 μl per gram of body weight of a 20% glucose solution was injected into the peritoneal cavity. Serum glucose levels were measured using the INFINITY glucometer and glucose strips (ADW). Serum insulin was quantified using the ultra-sensitive mouse insulin ELISA kit (Crystal Chem), according to manufacturer’s instructions.
For in vivo KHK–C overexpression (OE) experiments, 6–8-week-old male C57Bl/6 J mice were injected via the tail vein with adeno-associated virus carrying GFP or KHK–C plasmid. For in vivo KHK knockdown, C57Bl/6 J or FIRKO mice were injected subcutaneously every 2 weeks with luciferase control or 20 mg/kg GalNAc siRNA targeting total KHK. The KHK siRNA achieves liver-specific knockdown, as previously shown.6 For CPT1α KO experiments, CPT1α-floxed mice were obtained from Dr. Peter Carmeliet28 and bred with the Albumin-Cre transgenic mice to achieve liver-specific knockout (LKO). Six- to 8-week-old littermate controls (floxed & LKO) were fed a semipurified LFD (D12450 K) for 15 weeks prior to necropsy after a 16-hour fast.
A detailed description of materials and methods can be found in the supplementary information.
Results
Short-term sugar supplementation of HFD worsens hepatic steatosis
We have previously shown that dietary sugar supplementation for 10 weeks in mice on a chow diet or a HFD modifies the development of obesity, insulin resistance and non-alcoholic fatty liver disease (NAFLD).20 To examine the short-term effects of dietary sugar supplementation prior to the onset of obesity, we fed 6-week-old male C57BL/6 J mice a chow diet (21.6% calories from fat) or HFD (60.0% calories from fat) and provided regular water or water-containing 30% (w/v) fructose or 30% glucose for 2 weeks (Fig. S1A). Indeed, body weights were similar in sugar-supplemented groups on chow or HFD, and all HFD-fed mice had slightly higher body weight than the chow groups (Fig. S1B). Chow-fed mice supplemented with fructose (Chow+Fru) or glucose (Chow+Glu) consumed ~40% of calories from sugar water, but the total caloric intake was not different from chow-fed control mice (Chow+H2O) (Fig. S1C). As expected, caloric intake was greater in the HFD-fed mice (HFD+H2O) than in the Chow+H2O group. On HFD, mice supplemented with fructose (HFD+Fru) consumed 17% of their calories from sugar water, while mice supplemented with glucose (HFD+Glu) consumed ~30% of their calories from the liquid. The HFD+Fru group had similar caloric intake, but HFD+Glu-fed animals consumed 26% less total calories, compared to the HFD+H2O group (Fig. S1C). Metabolic phenotyping revealed that HFD+Fru-fed mice had impaired glucose tolerance (Fig. S1D,E). On the other hand, serum insulin was elevated in all three HFD-fed groups compared to chow-fed groups, and there was no difference with sugar supplementation of either chow or HFD (Fig. S1F). In spite of elevated insulin, hepatic insulin signaling in terms of insulin receptor β, protein kinase B, and extracellular-signal-regulated kinase phosphorylation was intact in all groups of mice at this early time point (Fig. S1G).
Hepatic lipid accumulation is a hallmark of NAFLD; hence, we aimed to understand the early pathophysiology that underpins the development of fatty liver disease. Hepatic steatosis was assessed by Oil red-O staining, which showed slightly higher neutral lipid accumulation in Chow+Fru mice and a greater increase in Chow+Glu compared to Chow+H2O mice (Fig. 1A). All mice fed a HFD had increased lipid accumulation. The HFD+Fru group had modestly increased Oil red-O staining, but a stronger induction was observed in the HFD+Glu group (Fig. 1A). Liver triglycerides were increased in all mice fed a HFD and glucose supplementation of either chow or HFD further increased hepatic triglycerides (Fig. 1B). Triglycerides are considered inert lipids, and we and others have shown that hepatic triglyceride accumulation is not associated with development of metabolic complications.6,29,30 Hepatic cholesterol was increased in Chow+Fru and Chow+Glu groups compared to the Chow+H2O group. Mice on HFD+H2O had even higher cholesterol, but there was no further elevation with sugar supplementation (Fig. 1B). Collectively, hepatic lipid accumulation was primarily increased by HFD feeding and sugar supplementation worsened steatosis in mice on a HFD. Interestingly, only the HFD+Fru group developed glucose intolerance, whereas glucose supplementation of chow and HFD augmented hepatic triglycerides without inducing glucose intolerance.
Fig. 1. Sugar supplementation of a HFD worsens hepatic steatosis.

(A) Representative Oil red-O staining to assess neutral lipid accumulation across all groups. Scale bar = 20 μm. (B) Hepatic triglyceride and cholesterol levels were quantified enzymatically (n = 5-6). (C) Long-chain acyl-CoA levels quantified by mass spectrometry (n = 6). (D) Representative cytoplasmic and nuclear protein was blotted for ChREBP, SREBP1, GAPDH (cytoplasmic control), and Lamin A/C (nuclear control). (E) Liver mRNA levels of Acly, Acc1, Fasn, and Scd1 were quantified by qPCR (n = 7-8). (F, G) Protein levels of ACLY, ACC1, FASN, SCD1, KHK–C, and ACSS2 were measured by western blot (F) and quantified using densitometry (G; n = 4). (H) KHK–C-overexpressing hepG2 hepatocytes were treated with 25 μg/ml of cycloheximide (CHX) or CHX+25 mM fructose (C+Fru) for 4 h. After 4-hour incubation, protein levels of ACLY, ACC1, FASN, ACSS2, and ACTIN (loading control) were measured using western blot. Baseline (BL) was compared to CHX and C+Fru treatment. (I) Densitometry quantification of ACLY protein in Fig. 1H and Fig. S2E. Significance was determined by two-way ANOVA with Sidak’s post hoc analysis of sugar supplementation (H2O, Fru, Glu) and solid diet (chow and HFD). Significance denoted with ($) compares sugar-supplemented groups to their chow+H2O control (*) comparing HFD+H2O to chow+H2O group or the difference between adjacent groups using a post hoc Student’s t test, and lastly (#) denotes significance of sugar-supplemented groups compared to their HFD+H2O control. #p<0.05; ##p<0.01; ###p<0.001; ####p<0.0001.
To understand the early processes that mediate hepatic lipid accumulation we first assessed de novo lipogenesis (DNL). Acyl-CoAs, the intermediates of fatty acid synthesis, were quantified by mass spectrometry.6,31 Chow+Fru and Chow+Glu groups did not exhibit a significant increase in the most abundant endogenously produced acyl-CoAs, including palmitoyl- (C16), stearoyl- (C18:0), or oleoyl-CoAs (C18:1), compared to Chow+H2O group (Fig. 1C). HFD-fed mice exhibited a 2-4-fold increase in these acyl-CoAs, while mice fed HFD+Fru and HFD+Glu actually exhibited a significant decrease in palmitoyl- and oleoyl-CoA compared to HFD+H2O (Fig. 1C). An increase in hepatic acyl-CoAs in the HFD+H2O group can be attributed to a higher intake of dietary free fatty acids (FFAs), as illustrated by a ~2.5-fold increase in essential fatty acyl-CoAs, linoleoyl- and linolenoyl-CoA (Fig. S2A).
Next, we assessed protein levels of the major lipogenic transcription factors, ChREBP and sterol regulatory element-binding protein 1 (SREBP1). Nuclear translocation of ChREBP and SREBP1 was not significantly affected by fructose or glucose supplementation of chow or HFD at this early time point (Figs. 1D and Fig. S2B,C). ChREBP can transcriptionally activate itself as part of a feed-forward mechanism.32 We measured Chrebpβ mRNA as a readout of ChREBP activation and observed no significant changes across all groups (Fig. S2D). ChREBP and SREBP1c promote DNL by inducing mRNA expression of a series of enzymes, including ATP citrate lyase (Acly), acetyl-CoA carboxylase 1 (Acc1), fatty acid synthase (Fasn), and stearoyl-CoA desaturase 1 (Scd1). Mice fed Chow+Fru exhibited only a slight increase in Acc1 mRNA and a significant 6-fold increase in Scd1 mRNA, compared to the Chow+H2O group (Fig. 1E). The expression of other genes or in other groups was not affected, consistent with a lack of nuclear translocation of ChREBP and SREBP1. Despite relatively minor changes at the mRNA level, the Chow+Fru group had markedly induced ACLY (7.0-fold), ACC1 (4.5-fold), FASN (1.8-fold), and SCD-1 (6.9-fold) protein levels, compared to Chow+H2O-fed animals (Fig. 1F, G). On HFD, ACLY (~3.5-fold) and ACC1 (~3-fold) proteins were significantly upregulated in HFD+Fru and HFD+Glu mice, whereas FASN (1.8-fold) and SCD1 (1.5-fold) tended to only be higher in HFD+Fru mice (Fig. 1F, G). An increase in DNL proteins in Chow+Fru, HFD+Fru and HFD+Glu groups correlated with increased KHK–C, the rate-limiting enzyme of fructolysis (Fig. 1F). The increase in KHK–C in the HFD+Glu compared to the HFD group is likely mediated by endogenous fructose production, which is further explored in Fig. 3. Acetyl-CoA is a substrate for esterification of FFAs required for DNL, so we also examined protein levels of acyl-CoA synthetase short-chain family member 2 (ACSS2), an enzyme involved in acetyl-CoA metabolism. Protein (Fig. 1F) levels of ACSS2 tended to follow the pattern for DNL enzymes, again without a change in its mRNA levels (Fig. S2D). Collectively, short-term sugar supplementation did not alter nuclear translocation of lipogenic transcription factors or mRNA expression of their DNL targets, but it robustly increased KHK–C and protein levels of enzymes mediating DNL.
Fig. 3. KHK–C is inversely associated with CPT1α protein and long-chain acylcarnitine levels.

(A-B) Protein levels measured by western blot (A) of fructolysis (KHK–C, ALDOB, TKFC), polyol (AR, SDH), and fat oxidation proteins (CPT1α, ACADS), and quantified by densitometry (B) normalized to vinculin (VINC) loading control (n = 4). (C) Hepatic long-chain acylcarnitine levels measured by GC-MS (n = 5-6). (D) Liver CPT1α mRNA expression across the six groups (n = 8). (E) Hepatic malonyl-CoA levels quantified by mass spectrometry (n = 8). (F) Linear regression analysis using KHK–C and CPT1α protein normalized to vinculin from panel A (n = 24) and from db/db mice (Fig. S5D; n = 8 for a total of 32 samples included in the linear regression). Significance was determined by two-way ANOVA with Sidak’s post hoc analysis of sugar supplementation (H2O, Fru, Glu) and solid diet (Chow and HFD). Significance denoted with ($) compares sugar-supplemented groups to their chow+H2O control (*) comparing HFD+H2O to chow+H2O group or the difference between adjacent groups using a post hoc Student’s t test, and lastly (#) denotes significance of sugar-supplemented groups compared to their HFD+H2O control. #p<0.05; ##p<0.01; ###p<0.001; ####p<0.0001.
The selective increase in DNL proteins, without much effect on their mRNA in Chow+Fru, HFD+Fru and HFD+Glu groups, prompted us to consider that sugar-induced upregulation of KHK–C may affect post-translational stability of these enzymes. Indeed, ACLY has been reported to undergo lysine acetylation, which renders the protein stable against ubiquitin-mediated degradation.33 To test if fructose promotes post-translational stability of these lipogenic enzymes via KHK–C, we generated stable human HepG2 hepatocytes overexpressing KHK–C.34 Cultured hepatocytes express little to no KHK–C, as compared to its expression in the liver.20 KHK–C-overexpressing hepG2 cells cultured in low glucose media were treated with 25 μg/ml of cycloheximide (CHX), a protein synthesis inhibitor, or with CHX plus 25 mM fructose (C+Fru). Compared to baseline, 4-hour CHX treatment reduced ACLY protein levels by 30%, which was improved to a 20% reduction with fructose treatment (Fig. 1H, I). These changes in ACLY were not observed in wild-type (WT) HepG2 cells treated with fructose (Fig. S2E). The protein levels of ACC1, FASN, and ACSS2 were minimally responsive to CHX and fructose treatment (Fig. 1H). These results suggest that fructose metabolism via KHK–C promotes post-translational stability of lipogenic enzymes, especially ACLY. In spite of an increase in protein levels of DNL enzymes in Chow+Fru, HFD+Fru and HFD+Glu-fed mice, acyl-CoA intermediates of DNL were surprisingly reduced with sugar supplementation of HFD. Thus, DNL does not explain the increase in steatosis observed in HFD-fed mice supplemented with sugar at this early time point.
Upregulation of KHK–C correlates with mitochondrial histology, but minimally affects TCA cycle and amino acid metabolism
Next, we sought to determine if tricarboxylic acid (TCA) cycle (Fig. 2A) metabolism was altered with short-term sugar supplementation. Pyruvate, the end product of fructolysis, may be converted to lactate or acetyl-CoA to feed into the TCA cycle. Hepatic pyruvate was unaltered, while lactate was decreased in all mice on a HFD, and acetyl-CoA was significantly increased only in the Chow+Glu group (Fig. 2B). Downstream TCA cycle intermediates were largely unaltered, except succinyl-CoA, which was decreased in the HFD+H2O compared to the Chow+H2O group (Fig. S3A). Uric acid, a byproduct of fructose metabolism, has been shown to inhibit aconitase to increase citrate levels.35 We observed a trend for short-term fructose supplementation of chow or HFD to increase citrate levels (Fig. S3A). Overall, sugar and HFD feeding for 2 weeks was not long enough to induce a difference in TCA cycle metabolism.
Fig. 2. Upregulation of KHK–C correlates with mitochondrial histology, but minimally affects TCA cycle and amino acid metabolism.

(A) Graphical representation of the TCA cycle. (B-D) Hepatic TCA cycle intermediates (B), branched chain (C) and essential amino acids (D) measured by mass spectrometry (n = 6). (E-G) Representative transmission electron microscopy (EM) 5,000x images. (E) and pie-chart representation (F) of mitochondrial length shown as a percent of four equal length subgroups. (n = 3 mice, EM images contained 26-78 mitochondria per slide). (G) Percent of small mitochondria from 0-500 nm in length (n = 3 mice per group). Significance was determined by two-way ANOVA with Sidak’s post hoc analysis of sugar supplementation (H2O, Fru, Glu) and solid diet (Chow and HFD). Significance denoted with ($) compares sugar-supplemented groups to their chow+H2O control (*) comparing HFD+H2O to chow+H2O group or the difference between adjacent groups using a post hoc Student’s t test, and lastly (#) denotes significance of sugar-supplemented groups compared to their HFD+H2O controls. #p<0.05; ##p<0.01; ###p<0.001; ####p<0.0001.
Impaired amino acid metabolism contributes to the progression of NAFLD.36,37 Perhaps, the most widely studied are branched-chain amino acids (BCAA, leucine/isoleucine and valine) whose serum levels correlate with insulin resistance.38 However, in patients with cirrhosis, BCAA supplementation reduces oxidative stress39,40 and decreases hepatic fat accumulation.41 We found decreased hepatic leucine/isoleucine levels with sugar supplementation of both chow and HFD, as well as reduced valine with sugar supplementation of HFD (Fig. 2C). Essential amino acids (methionine and phenylalanine, but not histidine) were decreased in the Chow+Fru and Chow+Glu groups compared to the Chow+H2O group (Fig. 2D). Tyrosine levels were decreased with Chow+Fru, Chow+Glu and in all mice on a HFD (Fig S3B), consistent with a decrease in its precursor phenylalanine (Fig. 2D). Citrulline, which is predominantly synthesized by the mitochondria as part of the urea cycle, was increased in the Chow+Fru (p = 0.0025) and Chow+Glu (p = 0.0371) groups compared to the Chow+H2O group (Fig S3C). Arginine and ornithine are the main precursors for citrulline in mice.42 Arginine was decreased with Chow+Glu (p = 0.0162) and HFD+H2O (p = 0.0007) compared to Chow+H2O, while no changes were observed with ornithine (Fig S3C). Other amino acids were not significantly affected by the pelleted diet intake or sugar supplementation (Fig S3D). In conclusion, amino acid metabolism is partially affected by sugar supplementation, but these effects are unlikely to explain the changes in hepatic lipid accumulation.
We previously reported that chronic sugar supplementation alters mitochondrial morphology and size.20 Using electron microscopy, we observed a decrease in mitochondrial length with fructose supplementation of chow or with addition of either sugar on HFD (Fig. 2E,F). The largest percent of small mitochondria (black part of the pie chart) was observed in Chow+Fru, HFD+Fru and HFD+Glu groups (Fig 2G), in agreement with increased KHK–C in these groups. Collectively, an increase in hepatic lipid accumulation with sugar supplementation of HFD cannot be explained by the minimal differences in amino acid or TCA cycle metabolism, while mitochondrial length was decreased most profoundly in mice supplemented with fructose on a HFD.
Activation of fructolysis is inversely associated with CPT1α protein and long-chain acylcarnitine levels
Given the association between mitochondrial length and KHK–C protein, we hypothesized that increased fructolysis may lower mitochondrial FAO. We quantified the first, second, and third steps of fructolysis mediated by KHK–C, Aldolase B (ALDOB), and triokinase/FMN cyclase (TKFC), respectively. Fructose supplementation of both chow and HFD increased KHK–C, ALDOB, and TKFC proteins >2 fold (Fig. 3A, B), while mRNA expression was only increased 1.7-fold for Khk-c, but was unchanged for AldoB and Tkfc (Fig. S4A). Interestingly, glucose supplementation of HFD, but not chow, increased KHK–C, ALDOB, and TKFC protein (Fig. 3A, B) and mRNA levels (Fig. S4A). Hyperglycemia may activate the polyol pathway,43 where excess glucose is endogenously converted into sorbitol via aldose reductase (AR), and then to fructose via sorbitol dehydrogenase (SDH).5 Indeed, HFD+Glu-fed mice had significantly higher AR and SDH protein levels (Fig. 3A,B), consistent with increased endogenous fructose production. Once again, an increase in AR and SDH proteins was observed without a change in their mRNA expression (Fig. S4B). Collectively, fructose supplementation induced KHK–C protein and mRNA in mice fed chow or a HFD, whereas glucose supplementation only induced the fructolysis pathway in mice fed a HFD, likely due to endogenously produced fructose via the polyol pathway. Fructose-induced KHK–C elevation correlated with increased ALDOB and TKFC proteins, without a change in their mRNA.
To determine if increased KHK–C is associated with reduced FAO, we measured CPT1α, the rate-limiting enzyme in mitochondrial FAO. CPT1α protein tended (p = 0.065) to be reduced in Chow+Fru-compared to Chow+Glu-fed mice, but it was significantly reduced in the HFD+Fru- (p = 0.018) and HFD+Glu (p = 0.002) groups compared to the HFD+H2O group (Fig. 3A,B). Given these results, we quantified other proteins mediating carnitine transport (Fig. S4C). OCTN2, CACT and CPT2 proteins were not affected by sugar supplementation of chow or HFD (Fig. S4D,E). Similarly, mRNA expression of the carnitine transporters Slc22a5 (encodes OCTN2), Slc25a20 (encodes CACT), and Crat were not affected by fructose, but were increased with glucose supplementation of both chow and HFD (Fig. S5A). Enzymes mediating mitochondrial beta oxidation, such as very long-chain acyl-CoA dehydrogenase (ACADVL) and long-chain acyl-CoA dehydrogenase (ACADL), were largely not affected by fructose supplementation (Fig. S4D). However, short-chain acyl-CoA dehydrogenase (ACADS), which mediates downstream oxidation of short-chain fatty acids, was significantly reduced with fructose supplementation of chow and HFD (Fig. 3A, B). The expression of genes regulating FAO, Acadvl, Acadl, and Acadm were largely not affected by fructose, but were predominantly increased with glucose supplementation of both chow and HFD (Fig. S5A). Thus, after 2 weeks of sugar feeding an increase in KHK–C protein was associated with a corresponding decrease in CPT1α protein, without significantly affecting other proteins involved in long-chain FAO and carnitine transport. Moreover, protein levels of FAO and carnitine transport enzymes largely do not correlate with their mRNA expression.
CPT1α catalyzes the production of long-chain acylcarnitines, such as C16 palmitoylcarnitine, from acyl-CoAs and carnitine. Given the inverse correlation between KHK–C and CPT1α, we next quantified hepatic long-chain acylcarnitine products of CPT1α activity. Consistent with CPT1α protein, saturated long-chain acylcarnitines (C16, C18 and C20) tended to be minimally decreased in the Chow+Fru group compared to the Chow+Glu group (Fig. 3C). Mice fed HFD+H2O had significantly elevated long-chain acylcarnitines compared to Chow+H2O mice (Fig. 3C); however, both HFD+Fru (p = 0.0002 for C16) and HFD+Glu (p = 0.0001 for C16) groups had significantly lower saturated long-chain acylcarnitines. Medium- and short-chain acylcarnitines (C12, C8, C6, and C4/Ci4) are not produced by CPT1α, so their levels were not affected by sugar supplementation, while C6 and C4/Ci4 were actually decreased in the HFD+H2O group compared to the Chow+H2O group (Fig. 3C). Overall, fructose supplementation of chow had minimal effect on long-chain acyl-carnitines, while both fructose and glucose supplementation of HFD significantly decreased CPT1α protein and acylcarnitine products of its enzymatic activity.
The CPT1α enzyme is regulated transcriptionally44 but also post-translationally via allosteric inhibition by the fatty acyl-CoA intermediate, malonyl-CoA.45 Despite altered protein levels, the mRNA levels of CPT1α were not different with pelleted diet or sugar supplementation (Fig. 3D). Hepatic malonyl-CoA levels were significantly decreased in the HFD+H2O group compared to the chow+H2O group (Fig. 3E), consistent with a corresponding increase in long-chain acylcarnitines. However, malonyl-CoA was not affected by sugar supplementation of HFD (Fig. 3E), despite decreased hepatic acylcarnitines in these groups (Fig. 3C). Given that CPT1α protein does not correlate with its mRNA expression or malonyl-CoA levels, we asked if KHK–C protein correlates negatively with CPT1α in additional in vivo models of NAFLD. We overexpressed KHK–C using adeno-associated virus and knocked down KHK using siRNA in mice fed a HFD. We observed that OE of KHK–C decreased CPT1α, while KHK knockdown (KD) restored CPT1α protein levels (Fig. S5B). Similarly, KHK–C was elevated in lipodystrophic (fat-specific insulin receptor knockout [FIRKO]) mice46 consuming a standard chow diet and this elevation correlated with lower CPT1α protein (Fig. S5C). Conversely, KHK KD in FIRKO mice restored CPT1α protein to the levels observed in their littermate controls. Lastly, we found a negative correlation between KHK–C and CPT1α proteins in obese leptin receptor deficient mice (db/db) compared to heterozygote controls on standard chow diet (Fig. S5D). We then performed a linear regression analysis between KHK–C and CPT1α protein across the six dietary groups and in db/db mice. KHK–C protein correlated negatively with CPT1α protein (Fig. 3F; R = −0.487; p = 0.005). These data suggest that KHK–C may negatively regulate CPT1α protein, independent of transcriptional control and malonyl-CoA inhibition, explaining the decrease in acylcarnitines following sugar supplementation of HFD. Interestingly, OE of KHK–C did not lower CPT1α in mice fed chow (Fig. S5E), indicating that a HFD or liver fat accumulation observed in db/db and FIRKO mice is required to observe this effect.
Overexpression of ketohexokinase-C impairs fatty acid oxidation in mouse hepatocytes
We previously reported that cultured mouse AML12 hepatocytes do not express KHK–C protein.20 Similarly, ALDOB, TKFC, AR, and SDH proteins were not detected in AML12 hepatocytes (Fig. S6A). To further study the inverse relationship between KHK–C and CPT1α, we overexpressed GFP-tagged mouse KHK–C in AML12 hepatocytes using a lentivirus. There was a ~35-fold increase in Khk-c mRNA, with no changes in Khk-a, in KHK–C-overexpressing cells compared to WT hepatocytes (Fig. 4A). Similarly, KHK–C protein was significantly higher (Fig. 4B), as was its enzymatic activity when exposed to 5 mM fructose (Fig. S6B). Thus, we generated a cell culture model of selective OE of KHK–C in the absence of other fructolytic enzymes.
Fig. 4. Ketohexokinase-C overexpression impairs long-chain fatty acid oxidation in mouse hepatocytes.

(A) qPCR was used to measure Khk-c and Khk-a mRNA levels in WT and KHK–C-overexpressing AML12 hepatocytes (n = 6) (B–C) KHK–C and carnitine shuttle/FAO proteins (CPT1α, OCTN2, CACT, CPT2) assessed by western blot (B) and quantified by densitometry using actin as a loading control (C; n = 3) (D-E) Seahorse fatty acid oxidation assay (D) recording oxygen consumption rate (OCR) in WT and KHK–C-overexpressing AML12 hepatocytes upon stimulation with free fatty acids (FFAs) and treated with the CPT1α inhibitor etomoxir (E; n = 10-11) (F) Acetyl-CoA levels were quantified in WT and KHK–C-overexpressing AML12 hepatocytes (n = 5). (G-H) Mitosubstrate assay from BioLog quantifying palmitoyl-carnitine (G) and octanoyl-carnitine (H) utilization in WT and KHK–C-overexpressing AML12 cells (n = 5). The panels to the right quantify the rate of substrate utilization over 45 min. (I) Triglycerides were quantified enzymatically from WT and KHK–C-overexpressing cells treated with BSA or 0.4 mM FFA for 24 h (n = 6). (J) Seahorse glycolysis assay recording extracellular acidification rate (ECAR) in WT and KHK–C-overexpressing cells in response to glucose, oligomycin (Oligo), and 2-deoxyglucose (2DG). Glycolysis is quantified in the panel on the right as area under curve. Significance was determined using unpaired, Student’s t test. For panel I, significance was determined by two-way ANOVA with Sidak’s post hoc analysis comparing OE vs. WT groups. Significance is defined as *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Next, we quantified proteins involved in carnitine shuttle and FAO pathways. Hepatocytes with OE of KHK–C exhibited a 69% reduction in CPT1α, but also a 45-70% decrease in OCTN2, CACT, and CPT2 proteins (Fig. 4B,C). Proteins mediating FAO, such as ACADVL, ACADL, or ACADS were not affected (Fig. S6C,D). We then quantified FAO using a Seahorse XFE96 analyzer.20 WT hepatocytes treated with a 2:1 oleate to palmitate FFA mixture exhibited a 4-fold increase in oxygen consumption rate (OCR) compared to the basal state (Fig. 4D,E). Treatment with etomoxir, a CPT1α inhibitor, decreased OCR in WT hepatocytes, in agreement with reduced FAO. KHK–C-overexpressing cells had lower basal OCR (Fig. 4D, Fig. S6E), and a blunted response to both FFA (Fig. 4D,E) and etomoxir treatment, consistent with lower CPT1α activity and impaired FAO. An end product of mitochondrial FAO, acetyl-CoA, was also decreased in KHK–C-overexpressing hepatocytes, consistent with impaired FAO (Fig. 4F). Next, we employed a MitoPlates assay to examine the ability of KHK–C OE and WT cells to oxidize 30 pre-set substrates by utilizing tetrazolium redox dye, an electron acceptor, as a readout of mitochondrial activity.47 Hepatocytes with KHK–C OE had an impaired ability to oxidize long-chain palmitoylcarnitine, compared to WT cells (Fig. 4G). Notably, this was one of the most significantly altered metabolites in this assay (Fig. S7A-D). Despite decreased palmitoylcarnitine utilization, medium-chain octanoylcarnitine was metabolized at the same rate in both WT and KHK–C-overexpressing hepatocytes (Fig. 4H). We then sought to determine the impact of FFAs on triglyceride accumulation in WT and KHK–C-overexpressing hepatocytes. FFA treatment significantly increased triglyceride accumulation in KHK–C-overexpressing hepatocytes compared to WT cells (Fig. 4I). Collectively, we show that KHK–C OE selectively reduced CPT1α, OCTN2, CACT and CPT2 proteins leading to impaired long-chain FAO resulting in increased FFA-induced triglyceride accumulation.
FAO is intricately linked to glycolysis to balance cellular energy production. Extracellular acidification rates (ECAR), as measured by Seahorse, reflect glycolysis. Addition of 25 mM glucose to sugar-free media increased ECAR in WT hepatocytes (Fig. 4J). Glycolysis was further stimulated by oligomycin, an ATP synthase inhibitor, but was completely abrogated by 2-deoxyglucose, a non-metabolizable analogue of glucose. The KHK–C-overexpressing hepatocytes exhibited markedly greater increases in ECAR in response to glucose and oligomycin (Fig. 4J), compared to WT hepatocytes. Consistent with elevated glycolysis, utilizing MitoPlates, KHK–C-overexpressing hepatocytes showed increased capacity to utilize the end metabolite of glycolysis, pyruvate (p = 0.004; Fig. S7B), and the first metabolite in the TCA cycle, citrate (p = 0.042; Fig. S7C). Collectively, OE of KHK–C shifted global cellular metabolism towards decreased long-chain FAO with compensatory increases in glycolysis.
Reduced levels of serine and glycine have been reported in patients with NAFLD.48,49 Mechanistically, both serine and glycine deprivation leads to impaired oxidative metabolism,50,51 while supplementation of these amino acids promotes FAO and lowers hepatic steatosis in mice.51-53 We could not measure glycine oxidation using the MitoPlates assay, but KHK–C-overexpressing hepatocytes showed an impaired capacity to oxidize serine, compared to WT hepatocytes (p = 0.012; Fig. S7D). This prompted us to measure hepatic serine and glycine levels by mass spectrometry. Hepatic serine was not different (Fig. S7E), while glycine was decreased in all groups with elevated KHK–C, including Chow+Fru, HFD+Fru, and HFD+Glu. Collectively, KHK–C OE impairs serine oxidation in mouse AML12 hepatocytes, while KHK–C protein is inversely associated with hepatic glycine levels.
Knockdown of CPT1α, in part, mirrors KHK–C overexpression
We next asked if the KHK–C-induced decrease of CPT1α could explain the impaired FAO observed in these cells and used CRISPR/Cas9N to knockdown endogenous CPT1α in mouse AML12 hepatocytes. This modified CRISPR technique increases specificity at a given locus by utilizing two guide RNAs and a Cas9 nickase54 (Fig. S8A). Following puromycin selection, we quantified CPT1α protein for each clone and observed that clones 3, 4, and 5 had significantly reduced CPT1α (Fig. S8B). Next, we quantified proteins involved in carnitine shuttle and FAO pathways in clones 4 and 5. Relative to WT control, clones 4 and 5 had significantly reduced (by 50-70%) CPT1α and CACT proteins, while there was a compensatory increase in CPT2 and ACADL proteins (Fig. 5A,B and Fig. S8C). Given that both clones exhibited similar downstream effects, we used clone 4 for subsequent experiments (KD-4). Similar to KHK–C-overexpressing hepatocytes (Fig. 4D), CPT1α KD-4 hepatocytes exhibited reduced basal OCR (Fig. 5C) and a blunted response to both FFA and etomoxir treatment (Fig. 5C,D), compared to WT hepatocytes. Likewise, when treated with FFAs, CPT1α KD-4 hepatocytes accumulated more triglycerides (65.6 ± 5.26 μg/mg protein) than WT hepatocytes (27.9 ± 7.6 μg/mg protein) (Fig. 5E). This was confirmed with Nile Red staining for neutral lipids (Fig. S8D). To confirm this effect in vivo, we knocked out CPT1α specifically in the liver using the Albumin-Cre/CPT1α-floxed method (LKO). Compared to littermate controls, male LKO mice showed reduced CPT1α and OCTN2 proteins, while they had a compensatory increase in CPT2, ACADL and ACADS (Fig 5F, G). Moreover, LKO mice accumulated more triglyceride in the liver (Fig 5H) and developed mild hepatic steatosis (Fig 5I). Collectively, KD of CPT1α (Fig. 5) in part replicates the phenotype observed in KHK–C-overexpressing hepatocytes (Fig. 4) in terms of reduced carnitine transport, impaired long-chain FAO, and increased triglyceride accumulation.
Fig. 5. CPT1a knockdown, in part, mirrors KHK–C overexpression in mouse hepatocytes.

(A-C) Carnitine shuttle/FAO protein levels (CPT1α, CACT, OCTN2, CPT2, ACADVL, ACADL, ACADS) were measured by western blot in WT and CPT1α knockdown clones 4 (KD-4) and 5 (KD-5) (A), and were further quantified by densitometry (B, KD-4 and S8C, KD-5) using actin as a loading control (n = 3). (C-D) Seahorse fatty acid oxidation assay (C) recording oxygen consumption rate (OCR) in WT and CPT1α KD-4 AML12 hepatocytes upon stimulation with free fatty acid (FFA) (D; n = 15-18 wells per group). (E) Triglycerides were quantified enzymatically from WT and KD-4 cells treated with BSA or 0.4 mM FFA for 24 h (n = 6). (F, G) Carnitine shuttle and FAO protein levels (CPT1α, CACT, OCTN2, CPT2, ACADVL, ACADL, ACADS) were measured by western blot (F) and were further quantified by densitometry (G) from CPT1a LKO and littermate control male mice. (H, I) Hepatic triglycerides were quantified (H) and livers were stained with H&E (I). 40x magnification and scale bar = 100 μm. Significance was determined using unpaired, Student’s t test. Significance is defined as *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. CV, central vein; PV, portal vein.
Acetylation at lysine 508 decreases CPT1α protein
Our group has previously shown that dietary sugars profoundly alter acetylation of the mitochondrial proteome after 2- and 10-weeks supplementation in mice fed either chow or a HFD.20,55 We re-analyzed our 2-week acetylation data55 focusing on CPT1α acetylation. We identified three new CPT1α acetylated lysine (K) residues 195, 508, and 634 using mass spectrometry. Chow+Fru-fed mice exhibited a significant increase in CPT1α acetylation of K508 (false discovery rate [FDR] = 3.83x10−8; Fig. 6A) compared to Chow+Glu mice. An increase in K508 acetylation in these mice was associated with lower CPT1α protein (Fig. 6A). Conversely, the Chow+Glu group had a significant decrease in K508 (FDR = 1.45x10−6) and K195 (FDR =0.006) acetylation (Fig. S9A), compared to the Chow+H2O group and this was associated with a concomitant increase in CPT1α protein (Fig. S9A). There was no difference in K195, K508, and K634 acetylation of CPT1α in the Chow+Fru compared to the Chow+H2O group (Fig. S9B) and similarly CPT1α protein was unchanged between these two groups (Fig. S9B). Compared to the Chow+H2O group, the HFD+H2O group had markedly lower acetylation at K508 (FDR <4.44x10−17), K634 (FDR = 9.81x10−8), and K195 (FDR = 3.16x10−7; Fig. S9C), consistent with higher CPT1α protein (Fig. S9C). The HFD+Fru group exhibited an increase in acetylation at K508 (FDR = 7.90x10−8; Fig. 6B) and had lower total CPT1α protein compared to the HFD+H2O group (Fig. 6B). Similarly, the HFD+Glu group had increased K508 acetylation (FDR = 7.49x10−16; Fig. 6C), and a decrease in CPT1α protein, compared to the HFD+H2O group (Fig. 6C). Lastly, HFD+Fru and HFD+Glu-fed mice exhibited no difference in CPT1α acetylation or CPT1α protein levels (Fig. S9D). Collectively, acetylation of CPT1α at K508 was increased in all groups with elevated KHK–C, i.e. Chow+Fru, HFD+Fru and HFD+Glu, and this was inversely associated with CPT1α protein levels.
Fig. 6. Acetylation at lysine 508 decreases CPT1α protein levels.

(A-C) Volcano plots (A, B, C) highlighting all acetylation sites that are different between two dietary groups. CPT1α acetylation sites are highlighted in red. The horizontal black bar denotes the significance cut-off of FDR = 0.01. The vertical black bars denote a minimal threshold for effect size of 1.5 (Log2 fold-change = ±0.58). Quantification of total CPT1α protein levels (A, B, C; right) normalized to vinculin (loading control) across the same two groups. Protein levels of CPT1α were measured by western blot (D) in COS-7 cells transfected with plasmids encoding β-galactosidase control (β-gal), wild-type (WT) CPT1α, and mutant K195Q, K508Q, and K634Q CPT1α. (E) Densitometry quantification of the data in panel D. (F) Western blot for c-MYC tag in cells transfected with β-galactosidase control (Cont) plasmid, wild-type (WT) CPT1α c-MYC tagged plasmid and mutant CPT1α K508Q c-MYC tagged plasmid. Significance was determined by one-way ANOVA with Dunnet’s post hoc analysis comparing Cont to WT and mutant CPT1α proteins. Significance is defined as *p<0.05; **p<0.01; ****p<0.0001.
To mechanistically determine the impact of lysine acetylation on protein stability, we used site-directed mutagenesis to induce a point mutation in CPT1α, from lysine to glutamine (K→Q), which mimics acetylated lysine.56 Importantly, each mutant was sequenced to ensure no random mutations were integrated during site-directed mutagenesis (data not shown). We transfected WT, K195Q, K508Q, and K634Q plasmids into African green monkey-derived COS kidney cells, which have low endogenous CPT1α protein.57,58 Transfection of these plasmids induced a significant increase in mouse CPT1α mRNA compared to the β-galactosidase transfection control (Fig. S9E). Transfection efficacy of WT CPT1α was similar to CPT1α K195 and K508 mutants, while the K634Q mutant had greater CPT1α mRNA levels (Fig. S9E). As expected, CPT1α protein was increased following transfection of WT CPT1α (Fig. 6D). Despite similar transfection efficiency among the WT, K195Q and K508Q CPT1α mutants, these mutants had significantly lower CPT1α protein than WT CPT1α-transfected cells (Fig. 6D, E). Since COS cells express some CPT1α, we tagged CPT1α with c-MYC and overexpressed WT and K508Q CPT1α mutant. Focusing only on exogenous c-MYC-tagged protein, we confirmed that acetylation mimicking the K508Q mutation lowered CPT1α protein levels by 30%, compared to WT CPT1α (Fig.6F, Fig. S9F). These in vitro data show that acetylation mimicking the K508Q mutation of CPT1α led to decreased CPT1α protein levels, explaining the inverse relationship between KHK–C-induced K508 acetylation and total CPT1α protein in vivo.
KHK–C OE promotes global protein acetylation and alters sirtuin levels
We identified that an increase in KHK–C was associated with increased acetylation of CPT1α at K508, which led to reduced CPT1α protein in vivo. Next, we asked if simple KHK–C OE affects protein acetylation in cultured hepatocytes. WT and KHK–C-overexpressing AML12 cells were incubated in high glucose media containing deacetylase inhibitors, trichostatin A and nicotinamide, for 24 h. Using a pan-acetylation antibody, KHK–C-overexpressing hepatocytes exhibited elevated acetylation compared to WT control hepatocytes (Fig. S10A). To more precisely quantify protein acetylation induced by KHK–C OE, we performed global untargeted mass spectrometry analysis of the total proteome, as well as mass spectrometry on the anti-acetyl lysine immunoenriched, trypsin-digested proteins from WT and KHK–C-overexpressing cells59 using data-independent acquisition.60-62 Partial least squares-discriminant analysis showed that the proteome in KHK–C-overexpressing cells was profoundly different from that of WT cells, accounting for up to 54% of the effect on principal component (PC)1 and up to 31% on PC2 (Fig. 7A). Some of the most significantly downregulated proteins in cells with KHK–C OE are trans-2,3-enoyl-CoA reductase and alkylglycerone phosphate synthase, which are involved in lipid metabolism (Fig. S10B), while one of the most significantly upregulated proteins was acyl-CoA thioesterase 1, which is involved in acyl-CoA metabolism. Indeed, gene ontology-enrichment analysis revealed that 5 out of the top 10 most downregulated pathways in KHK–C-overexpressing cells were involved in acyl-CoA, sterol and fatty acid metabolism (Fig. 7B). Collectively, KHK–C OE drastically changed the proteome, with alterations in proteins involved in acyl-CoA metabolism and FAO.
Fig. 7. KHK–C OE drives global protein acetylation in AML12 hepatocytes and alters sirtuin levels.

(A) Partial least squares-discriminant analysis comparing WT and KHK–C OE total protein levels. (B) Gene ontology analysis highlighting the top 10 most downregulated biological processes in KHK–C-overexpressing hepatocytes. (C) Partial least squares-discriminant analysis comparing acetylated peptides in WT and KHK–C-overexpressing cells. (D) Volcano plot analysis highlighting acetylated peptides that are increased (in red) or decreased (in purple) with KHK–C OE. The horizontal black bar denotes the significance cut-off of p value = 0.05. The vertical black bars denote a minimal threshold for effect size of 1.5 (Log2 normalized fold-change = ±0.58) (E, F) Sirtuin protein levels (SIRTS2-5) were measured by western blot in WT and KHK–C-overexpressing (E) cells and were quantified by densitometry using Vinculin (VINC) as a loading control. Sirt3 full-length (3-FL) and Sirt3 cleaved (3-CL) proteins (F; n = 3). Significance was determined using unpaired, Student’s t test, and is defined as *p<0.05; **p<0.01; ***p<0.001.
Next, we determined the impact KHK–C OE on acetylated proteins. We identified 2,684 acetylated sites on 2,457 proteins (Table S1). Only proteins with two or more unique peptides were quantified in order to increase specificity (Table S2). After normalization for the total amount of a specific protein, 318 acetylated sites met pre-determined statistical (p value ≤0.05) and effect size cut-offs (Log2 normalized fold-change [FC] of ±0.58). Consistent with our pan-acetylation western blot, KHK–C-overexpressing cells had 64.2% increase in acetylation (204 of 318 total significantly altered sites), compared to WT cells. Partial least squares-discriminant analysis showed the acetyl-proteome of KHK–C-overexpressing cells was profoundly different from the WT group, accounting for up to 41% of the effect on PC1 and up to 24% on PC2 (Fig. 7C). Volcano plot analysis identified that the key enzymes involved in acyl-CoA, citrate, and FAO metabolism had altered acetylation in WT and KHK–C-overexpressing hepatocytes (Fig. 7D). For example, an enzyme involved in acetyl-CoA production, ACLY, had higher acetylation at K544 (Log2 FC = 14.73) in KHK–C-overexpressing cells compared to control cells. Similarly, acyl-CoA binding protein was acetylated at K51 (Log2 FC = 2.61) in KHK–C-overexpressing cells. TCA cycle enzyme aconitase (ACON), which converts citrate to isocitrate, was hyper-acetylated at K523 (Log2 FC = 14.27) with KHK–C OE. In addition, the mitochondrial isocitrate dehydrogenase enzymes, IDHP (NADP+ isocitrate dehydrogenase 2) and IDHG1 (NAD+ isocitrate dehydrogenase 3, subunit gamma 1) were hyper-acetylated at K400 (Log2 FC = 9.94) and K206 (Log2 FC = 12.11), respectively, in KHK–C-overexpressing hepatocytes. We also identified ETFA (electron transfer flavoprotein a), an enzyme required for transport of electrons from FAO to the electron transport chain, as being highly acetylated (Log2 FC = 3.07) in KHK–C-overexpressing cells. On the contrary, proteins hyper-acetylated in WT control cells include IDH3A (NAD+ isocitrate dehydrogenase 3, subunit alpha) at K214 (Log2 FC = −0.87), and the carnitine shuttle/FAO proteins ACADVL at K299 (Log2 FC = −1.35) and CACP (carnitine O-acetyltransferase; encoded by Crat) at K315 (Log2 FC = −1.45). Unfortunately, we did not detect CPT1α K508 acetylation in either WT or KHK–C-overexpressing cells. Collectively, KHK–C OE largely altered lysine acetylation of proteins involved in acyl-CoA, citrate/isocitrate, and FAO metabolism.
The sirtuin (SIRT) family of enzymes (SIRT 1-7) share conserved NAD+-dependent deacetylase activity, and regulate cellular protein acetylation.63 Given the strong differences in acetylation of isocitrate dehydrogenase enzymes, which regulate NAD+ metabolism, we measured protein levels of cytoplasmic (SIRT2) mitochondrial (SIRT3, 4, 5) and nuclear (SIRT1, 6, 7) sirtuins. The protein level of SIRT2 was significantly reduced by 85% in KHK–C-overexpressing hepatocytes (Fig. 7E,F) while its mRNA level was only reduced by ~15% (Fig. S10E). SIRT3 encodes a full-length 44-kDa protein that upon cellular stress64 undergoes proteolytic processing to an active 28-kDa protein, localized on mitochondria.65 We observed a significant decrease in full-length SIRT3 with a concomitant increase in cleaved SIRT3 in KHK–C-overexpressing hepatocytes (Fig. 7E,F). Furthermore, we also observed a significant decrease in SIRT4 and increase in SIRT5 with KHK–C OE (Fig. 7E, F). SIRT1/6/7 were not affected by KHK–C OE (Fig. S10C, D). Importantly, mRNA levels of SIRT3-7 were unchanged between WT and KHK–C-overexpressing hepatocytes (Fig. S10E). Given a strong decrease in SIRT2 with KHK–C OE, we then quantified SIRT2, as well as SIRT1, in two cohorts of mice fed a HFD supplemented with sugars and treated with KHK siRNA (Fig S10F-H). SIRT1 was not affected by sugar supplementation or KD of KHK. However, SIRT2 tended to be reduced in the livers of mice fed HFD+Fru diet and it was increased following KD of KHK. An increase in SIRT2 in these mice was associated with an increase in CPT1α (Fig S10F-H). Collectively, KHK–C OE promotes acetylation of enzymes regulating NAD+ metabolism and thus, influences protein levels of Sirtuin family members, most notably SIRT2.
Discussion
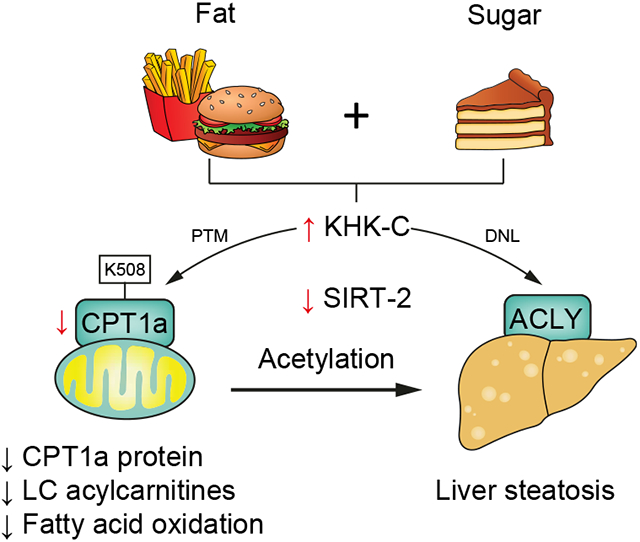
In the present study, we have investigated the mechanisms by which short-term sugar supplementation, fructose or glucose, of either chow or a HFD, contributes to the early pathophysiology of fatty liver disease prior to the onset of severe obesity and insulin resistance. We demonstrate that 2-week fructose supplementation of chow, and fructose or glucose supplementation of HFD increases KHK–C and protein levels of lipogenic enzymes, independent of their mRNA expression and nuclear translocation of lipogenic transcription factors ChREBP and SREBP1. Sugar-induced increases in lipogenic proteins are the highest on a chow diet, while marked steatosis develops with sugar supplementation of a HFD, pointing to the intricate relationship between sugar and HFD metabolism. Increased KHK–C correlates with small mitochondrial size and there is a negative relationship between KHK–C and CPT1α proteins in mouse liver. This relationship is preserved in cultured hepatocytes where a simple overexpression of KHK–C reduces the levels of proteins involved in carnitine shuttle, impairs long-chain FAO, and promotes FFA-induced triglyceride accumulation. These in vitro effects of KHK–C can be, in large part, replicated by a knockdown of CPT1α. Mechanistically, we show that increased KHK–C correlates with acetylation of CPT1α at lysine 508 in vivo, which is associated with lower CPT1α protein. OE of mutant CPT1α that mimics hyper-acetylated lysine 508 leads to a lower CPT1α protein level in vitro. Interestingly, upregulation of KHK–C has a profound effect on global protein acetylation and modifies the key proteins involved in fatty acid metabolism and acyl-CoA production. In summary, we describe a novel pathway by which fructolysis through KHK–C lowers CPT1α via acetylation at K508 to promote ectopic lipid accumulation in the liver.
Herein, we report a novel role of KHK–C in the control of global protein acetylation, which may stabilize proteins involved in DNL. First, we observed that increased KHK–C protein in vivo correlates with increased levels of lipogenic enzymes ACLY, ACC1 and FASN without significantly affecting their mRNA expression. Second, fructose treatment of KHK–C-overexpressing hepatocytes stabilized ACLY protein. Third, simple overexpression of KHK–C in cultured hepatocytes increased ACLY acetylation at K544 more than 13-fold. Acetylation of ACLY at K540, 546, 554 in human cells stabilizes its protein by blocking ubiquitin-mediated degradation.33,66 In mice, ACLY is post-translationally stabilized by acetylation at 544, K530, and 546, which are homologous to the three ubiquitination sites listed above for human ACLY.33,66,67 Conversely, the protein deacetylase SIRT2 deacetylates and destabilizes ACLY.33 Acetylated ACLY has been found to promote progression of NAFLD.68 Thus, KHK–C-mediated acetylation of proteins that mediate DNL, such as ACLY, may contribute to the lipogenic effects of fructose and its association with advanced forms of NAFLD.
Additionally, we find that an increase in KHK–C is associated with acetylation of CPT1α at K508 in vivo, whereas acetylation of CPT1α at K195 and K634 is not affected in a KHK–C-dependent manner. Acetylation of CPT1α has previously been mentioned in large proteomic data sets,69 but the functional significance of this post-translational protein modification is unknown. We show that overexpression of mutant CPT1α, with K508 substituted with glutamine to mimic the acetylated state, decreases CPT1α protein levels compared to WT CPT1α. Indeed, OE of KHK–C induces acetylation of several proteins mediating FAO and the FAO pathway is one of the most downregulated processes in cells with KHK–C OE. In vitro, overexpression of KHK–C phenocopies a knockdown of CPT1α in terms of carnitine shuttle, fat oxidation and increased triglyceride accumulation. Indeed, KHK–C OE in vitro decreases oxidation of long-chain fatty acids, such as palmitoyl-carnitine, but not medium-chain octanoyl-carnitine, which does not rely on CPT1α for mitochondrial transport. Moreover, KHK–C OE decreased CPT1α, but also OCTN2, CACT and CPT2 carnitine transporters. Similarly, a knockdown of CPT1α is sufficient to perturb the carnitine shuttle by inducing a decrease in OCTN2 and CACT. Taken together, we report a novel relationship between KHK–C in regulation of CPT1α and FAO to explain how the combined intake of fructose and a HFD accelerates the development of metabolic complications. This relationship offers a mechanistic explanation for studies in mice, rats and humans reporting that dietary fructose decreases FAO.15-19,21 Previously it was thought that sugar-induced malonyl-CoA decreases CPT1α activity and FAO.45 This mechanism was discovered in muscle tissue, but liver CPT1α is 10x less sensitive than muscle CPT1β to the inhibitory effects of malonyl-CoA.70,71 Additionally, we did not find that hepatic malonyl-CoA is increased with fructose supplementation of a HFD. Thus, KHK–C-induced acetylation of CPT1α is the likely explanation of how sugar intake decreases fat oxidation in the liver.
Our data indicate that several aspects of fructose metabolism can be explained by KHK–C-mediated effects on protein acetylation. Clearly, the most important finding here is that simple overexpression of KHK–C profoundly affects global protein acetylation. This results in even greater perturbation of the total proteome, leading to both increased and decreased abundance of proteins detected in their non-modified forms. As expected, the most downregulated pathways in hepatocytes with KHK–C OE include acyl-CoA metabolism and FAO. The observed effects of KHK–C on protein acetylation are analogous to the historic reports that fructose profoundly induces protein fructosylation,72 a post-translational modification of lysine residues.73 A profound effect of KHK–C on global protein acetylation can be, in part, explained by a decrease in a major cytoplasmic deacetylase SIRT2. SIRT2 is expressed in highly metabolically active tissues such as the liver.74 A reduction in SIRT2 has been observed in humans and mice with NAFLD, while restoration of SIRT2 alleviates insulin resistance, and hepatic steatosis.75 Further, SIRT2 affects mitochondrial biogenesis by deacetylating PGC-1α,76 possibly accounting for the increased number of small and fragmented mitochondria in the groups with increased KHK–C. Moreover, SIRT2 deacetylates several isocitrate dehydrogenase isoforms involved in oxidation of isocitrate to generate α-ketoglutarate and NADPH. It has been reported that acetylation of isocitrate dehydrogenase 2 at K413 decreases its enzymatic activity, leading to a shift from oxidative to glycolytic metabolism.77 While we did not identify K413 acetylation, acetylation at a nearby K400 was ~10-fold higher in cells with KHK–C OE. Other isocitrate dehydrogenase isoforms (IDHG1 at K206, IDH3A at K214), as well as the upstream aconitase (ACON K523, ACON K401) are also differentially acetylated following KHK–C OE. Mitochondrial sirtuins, SIRT3 and SIRT5, possess deacetylase activity, while SIRT4 has an ADP-ribosyl transferase function.74 An increase in SIRT3&5 in cells with KHK–C OE is likely a compensatory mechanism for increased mitochondrial stress and hyperacetylation induced by lowering of SIRT2. Indeed cleaved SIRT3 translocates to the mitochondria in response to cellular stress.78 An increase in KHK–C-induced acetylation agrees with our previous work showing that sugar supplementation of both chow and a HFD markedly alters mitochondrial protein acetylation.20,55 Taken together, these data indicate that KHK–C OE profoundly affects global protein acetylation, which is associated with a decrease in SIRT2, uncovering a new mechanism by which sugar metabolism through KHK–C decreases FAO and alters global metabolism.
A KHK–C-mediated decrease in fat oxidation may explain, in part, why our “Western Diet” which is high in both fructose and fat leads to development of obesity and its complications. A recent systemic review of 3,920 rodent models of NAFLD found that a high-fat, high-fructose diet most closely resembles the phenotype of advanced human NAFLD.14 Indeed, our data shows that only a combination of fructose and a HFD induces glucose intolerance. Fructose supplementation of chow robustly increases KHK–C, but these mice did not consume sufficient fat to induce metabolic complications. Likewise, consumption of fructose from fruits and vegetables, as a part of well-balanced, low-fat diet, does not lead to metabolic complications. Moreover, KHK–C is increased in the HFD+Glu-fed group, but this group had a significant reduction in HFD intake compared to the HFD and HFD+Fru groups, and did not develop glucose intolerance. The upregulation of KHK–C in HFD+Glu is mediated by endogenous fructose production. This pathway contributes a small portion of fructose to the hepatic pool, so that over time its effects pale in comparison to dietary fructose intake. Indeed, in our longer 10-week study we did not observe an increase in KHK–C in mice fed HFD+Glu compared to HFD+Fru diet and these mice remained glucose tolerant.6 The interaction of KHK–C and fat metabolism is most evident when we overexpress KHK–C in mice on a HFD. Moreover, the interaction between KHK–C and CPT1a is evident in genetically obese db/db and lipodystrophic FIRKO mice on a chow diet, since these mice accumulate a large amount of lipids in their livers.
KO79-81 and KD6,20 studies in mice show that KHK is required to observe the detrimental effects of fructose. Moreover, increased KHK has been reported in patients with NAFLD.6,82 Based on these findings, small molecule inhibitors of KHK kinase activity have been developed and studied for the treatment of NAFLD and insulin resistance.83-85 They have shown a benefit primarily in terms of decreasing hepatic DNL.86 However, KHK inhibitors appear to be less effective than genetic KO of KHK, as inhibitors have only documented a benefit in rats,87 which have much slower metabolism than mice, and in perfused human livers.88 Johnson & Johnson and more recently Pfizer89 have discontinued the clinical development of KHK inhibitors. Since KHK inhibitors are designed to decrease kinase function of the enzyme, they are likely only inhibiting DNL. Our data provide an explanation for why genetic KO of KHK may show a stronger effect than KHK inhibition, since KO abrogates both kinase and non-kinase functions of KHK. The effects of KHK–C on CPT1α and FAO appear to be mediated via its non-kinase effect, e.g. through acetylation. This is supported by decreased CPT1α and increased acetylation in cells with KHK–C OE in fructose-free media. Additionally, these cells do not express downstream ALDOB and TKFC enzymes needed for breakdown of fructose into lipogenic substrates, so increased acetylation is not dependent on fructose metabolism. In summary, KHK–C controls several important metabolic pathways that regulate the development of hepatic steatosis. A complete knockdown of KHK via siRNA may be a more effective therapy for metabolic dysfunction since it abrogates both kinase-dependent and -independent functions of KHK.
In this study, we show that the combined effects of sugar and a HFD have additive detrimental effects in terms of metabolic dysfunction. These effects are dependent on KHK–C whose newly recognized function includes a control of global protein acetylation. KHK–C-induced acetylation of ACLY may support lipogenesis, while its acetylation of CPT1α is associated with lower CPT1α protein levels and decreased FAO. Future studies are needed to explore the kinase-independent function of KHK–C in order to design more efficient drugs for management of NAFLD and insulin resistance.
Supplementary Material
Highlights.
Fructose increases de novo lipogenic proteins without altering their mRNA expression.
There is an inverse relationship between KHK–C and CPT1a proteins.
KHK–C induced acetylation of CPT1a at K508 decreases its protein levels.
Overexpression of KHK–C induces global protein acetylation and impairs FAO.
Impact and implications.
Fructose is a highly lipogenic nutrient whose negative consequences have been largely attributed to increased de novo lipogenesis. Herein, we show that fructose upregulates ketohexokinase, which in turn modifies global protein acetylation, including acetylation of CPT1a, to decrease fatty acid oxidation. Our findings broaden the impact of dietary sugar beyond its lipogenic role and have implications on drug development aimed at reducing the harmful effects attributed to sugar metabolism.
Acknowledgements
The authors would like to thank Christopher B. Newgard for performing metabolomics analysis from liver tissue.
Financial support
This work was supported in part by NASPGHAN Foundation Young Investigator Award, Pediatric Scientist Development Program Award (HD000850) and COCVD Pilot and Feasibility Grant (GM127211)awarded to SS, and the National Institutes of Health grants K01DK128022 and NIH National Center for Advancing Translational Sciences through grant number UL1TR001998 awarded to RNH. We acknowledge the support of instrumentation for the Orbitrap Eclipse Tribrid from the NCRR shared instrumentation grant 1S10 OD028654 (PI: Birgit Schilling).
Abbreviations
- ACADL
long-chain acyl-CoA dehydrogenase
- ACADS
short-chain acyl-CoA dehydrogenase
- ACADVL
very long-chain acyl-CoA dehydrogenase
- ACLY
ATP citrate lyase
- ACON
aconitase
- ALDOB
aldolase B
- AR
aldose reductase
- BCAAs
branched-chain amino acids
- ChREBP
carbohydrate response element-binding protein
- CHX
cycloheximide
- CPT1α
carnitine palmitoyltransferase 1a
- DNL
de novo lipogenesis
- ECAR
extracellular acidification rates
- FAO
fatty acid oxidation
- FC
fold-change
- FDR
false discovery rate
- FFA
free fatty acid
- FIRKO
fat-specific insulin receptor knockout
- HFD
high-fat diet
- KD
knockdown
- KHK–C
ketohexokinase-C
- KO
knockout
- LKO
liver-specific knockout
- NAFLD
nonalcoholic fatty liver disease
- OCR
oxygen consumption rate
- OE
overexpression
- siRNA
small-interfering RNA
- SREBP1
sterol regulatory element-binding protein 1
- TCA
tricarboxylic acid
- TKFC
triokinase/FMN cyclase
- WT
wild-type
Footnotes
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2023.02.010.
Conflict of interest
SS receives grant funding from Alnylam Pharmaceuticals, Inc., to study KHK as a target for management of NAFLD. No other conflicts of interest are reported.
Please refer to the accompanying ICMJE disclosure forms for further details.
Data availability statement
Raw data and complete MS data sets have been uploaded to the Center for Computational Mass Spectrometry, to the MassIVE repository at UCSD, and can be downloaded using the following link: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=b9ba416b2a0b44c8b34a29e3228e6eb3 (MassIVE ID number: MSV000089882; ProteomeXchange ID: PXD035316).
[Note: To access the data repository MassIVE (UCSD) for MS data, please use: Username: MSV000089882_reviewer; Password: winter].
References
- [1].Softic S, Kahn CR. Fatty liver disease: is it nonalcoholic fatty liver disease or obesity-associated fatty liver disease? Eur J Gastroenterol Hepatol 2019;31:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Perumpail BJ, Khan MA, Yoo ER, Cholankeril G, Kim D, Ahmed A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol 2017;23:8263–8276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Malik VS, Hu FB. Sweeteners and risk of obesity and type 2 diabetes: the role of sugar-sweetened beverages. Curr Diab Rep 2012;12:195–203. [DOI] [PubMed] [Google Scholar]
- [4].Malik VS, Hu FB. Sugar-sweetened beverages and cardiometabolic health: an update of the evidence. Nutrients 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Helsley RN, Moreau F, Gupta MK, Radulescu A, DeBosch B, Softic S. Tissue-specific fructose metabolism in obesity and diabetes. Curr Diab Rep 2020;20:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest 2017;127:4059–4074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One 2012;7:e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci 2016;61:1282–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kuzma JN, Schmidt KA, Kratz M. Prevention of metabolic diseases: fruits (including fruit sugars) vs. vegetables. Curr Opin Clin Nutr Metab Care 2017;20:286–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Semnani-Azad Z, Khan TA, Blanco Mejia S, de Souza RJ, Leiter LA, Kendall CWC, et al. Association of major food sources of fructose-containing sugars with incident metabolic syndrome: a systematic review and meta-analysis. JAMA Netw Open 2020;3:e209993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dowman JK, Hopkins LJ, Reynolds GM, Nikolaou N, Armstrong MJ, Shaw JC, et al. Development of hepatocellular carcinoma in a murine model of nonalcoholic steatohepatitis induced by use of a high-fat/fructose diet and sedentary lifestyle. Am J Pathol 2014;184:1550–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Radhakrishnan S, Yeung SF, Ke JY, Antunes MM, Pellizzon MA. Considerations when choosing high-fat, high-fructose, and high-cholesterol diets to induce experimental nonalcoholic fatty liver disease in laboratory animal models. Curr Dev Nutr 2021;5:nzab138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Im YR, Hunter H, de Gracia Hahn D, Duret A, Cheah Q, Dong J, et al. A systematic review of animal models of NAFLD finds high-fat, high-fructose diets most closely resemble human NAFLD. Hepatology 2021;74:1884–1901. [DOI] [PubMed] [Google Scholar]
- [15].Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr 2007;85:1511–1520. [DOI] [PubMed] [Google Scholar]
- [16].Couchepin C, Le KA, Bortolotti M, da Encarnacao JA, Oboni JB, Tran C, et al. Markedly blunted metabolic effects of fructose in healthy young female subjects compared with male subjects. Diabetes Care 2008;31:1254–1256. [DOI] [PubMed] [Google Scholar]
- [17].Cox CL, Stanhope KL, Schwarz JM, Graham JL, Hatcher B, Griffen SC, et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur J Clin Nutr 2012;66:201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rebollo A, Roglans N, Baena M, Padrosa A, Sanchez RM, Merlos M, et al. Liquid fructose down-regulates liver insulin receptor substrate 2 and gluconeogenic enzymes by modifying nutrient sensing factors in rats. J Nutr Biochem 2014;25:250–258. [DOI] [PubMed] [Google Scholar]
- [19].Roglans N, Sanguino E, Peris C, Alegret M, Vazquez M, Adzet T, et al. Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J Pharmacol Exp Ther 2002;302:232–239. [DOI] [PubMed] [Google Scholar]
- [20].Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab 2019;30:735–753 e734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Topping DL, Mayes PA. The immediate effects of insulin and fructose on the metabolism of the perfused liver. Changes in lipoprotein secretion, fatty acid oxidation and esterification, lipogenesis and carbohydrate metabolism. Biochem J 1972;126:295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Inci MK, Park SH, Helsley RN, Attia SL, Softic S. Fructose impairs fat oxidation: implications for the mechanism of western diet-induced NAFLD. J Nutr Biochem 2022:109224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bennett MJ, Santani AB. Carnitine palmitoyltransferase 1A deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al. , editors. GeneReviews((R)); 1993. Seattle (WA). [Google Scholar]
- [24].Choi JS, Yoo HW, Lee KJ, Ko JM, Moon JS, Ko JS. Novel mutations in the CPT1A gene identified in the patient presenting jaundice as the first manifestation of carnitine palmitoyltransferase 1A deficiency. Pediatr Gastroenterol Hepatol Nutr 2016;19:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Phowthongkum P, Suphapeetiporn K, Shotelersuk V. Carnitine palmitoyl transferase 1A deficiency in an adult with recurrent severe steato hepatitis aggravated by high pathologic or physiologic demands: a roller-coaster for internists. Clin Mol Hepatol 2019;25:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sinclair G, Collins S, Arbour L, Vallance H. The p.P479L variant in CPT1A is associated with infectious disease in a BC First Nation. Paediatr Child Health 2019;24:e111–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ohashi K, Munetsuna E, Yamada H, Ando Y, Yamazaki M, Taromaru N, et al. High fructose consumption induces DNA methylation at PPARalpha and CPT1A promoter regions in the rat liver. Biochem Biophys Res Commun 2015;468:185–189. [DOI] [PubMed] [Google Scholar]
- [28].Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature 2015;520:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Monetti M, Levin MC, Watt MJ, Sajan MP, Marmor S, Hubbard BK, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab 2007;6:69–78. [DOI] [PubMed] [Google Scholar]
- [30].Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest 2012;122:2176–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, et al. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med 2004;10:268–274. [DOI] [PubMed] [Google Scholar]
- [32].Herman MA, Peroni OD, Villoria J, Schon MR, Abumrad NA, Bluher M, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature 2012;484:333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol Cell 2013;51:506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Park SH, Helsley RN, Noetzli L, Tu HC, Wallenius K, O’Mahony G, et al. A luminescence-based protocol for assessing fructose metabolism via quantification of ketohexokinase enzymatic activity in mouse or human hepatocytes. STAR Protoc 2021;2:100731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lanaspa MA, Sanchez-Lozada LG, Choi YJ, Cicerchi C, Kanbay M, Roncal-Jimenez CA, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gaggini M, Carli F, Rosso C, Buzzigoli E, Marietti M, Della Latta V, et al. Altered amino acid concentrations in NAFLD: impact of obesity and insulin resistance. Hepatology 2018;67:145–158. [DOI] [PubMed] [Google Scholar]
- [37].Jin R, Banton S, Tran VT, Konomi JV, Li S, Jones DP, et al. Amino acid metabolism is altered in adolescents with nonalcoholic fatty liver disease-an untargeted, high resolution metabolomics study. J Pediatr 2016;172:14–19 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 2012;15:606–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Marchesini G, Bianchi G, Merli M, Amodio P, Panella C, Loguercio C, et al. Nutritional supplementation with branched-chain amino acids in advanced cirrhosis: a double-blind, randomized trial. Gastroenterology 2003;124:1792–1801. [DOI] [PubMed] [Google Scholar]
- [40].Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A, et al. Overweight and obesity increase the risk for liver cancer in patients with liver cirrhosis and long-term oral supplementation with branched-chain amino acid granules inhibits liver carcinogenesis in heavier patients with liver cirrhosis. Hepatol Res 2006;35:204–214. [DOI] [PubMed] [Google Scholar]
- [41].Arakawa M, Masaki T, Nishimura J, Seike M, Yoshimatsu H. The effects of branched-chain amino acid granules on the accumulation of tissue triglycerides and uncoupling proteins in diet-induced obese mice. Endocr J 2011;58:161–170. [DOI] [PubMed] [Google Scholar]
- [42].Marini JC. Arginine and ornithine are the main precursors for citrulline synthesis in mice. J Nutr 2012;142:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Cheng HM, Gonzalez RG. The effect of high glucose and oxidative stress on lens metabolism, aldose reductase, and senile cataractogenesis. Metabolism 1986;35:10–14. [DOI] [PubMed] [Google Scholar]
- [44].Song S, Attia RR, Connaughton S, Niesen MI, Ness GC, Elam MB, et al. Peroxisome proliferator activated receptor alpha (PPARalpha) and PPAR gamma coactivator (PGC-1alpha) induce carnitine palmitoyltransferase IA (CPT-1A) via independent gene elements. Mol Cell Endocrinol 2010;325:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest 1977;60:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Softic S, Boucher J, Solheim MH, Fujisaka S, Haering MF, Homan EP, et al. Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes 2016;65:2187–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lei XH, Bochner BR. Optimization of cell permeabilization in electron flow based mitochondrial function assays. Free Radic Biol Med 2021;177:48–57. [DOI] [PubMed] [Google Scholar]
- [48].Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, Nielsen J. Genome-scale metabolic modelling of hepatocytes reveals serine deficiency in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:3083. [DOI] [PubMed] [Google Scholar]
- [49].Yamakado M, Tanaka T, Nagao K, Imaizumi A, Komatsu M, Daimon T, et al. Plasma amino acid profile associated with fatty liver disease and co-occurrence of metabolic risk factors. Sci Rep 2017;7:14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gao X, Lee K, Reid MA, Sanderson SM, Qiu C, Li S, et al. Serine availability influences mitochondrial dynamics and function through lipid metabolism. Cell Rep 2018;22:3507–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Rom O, Liu Y, Liu Z, Zhao Y, Wu J, Ghrayeb A, et al. Glycine-based treatment ameliorates NAFLD by modulating fatty acid oxidation, glutathione synthesis, and the gut microbiome. Sci Transl Med 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sim WC, Kim DG, Lee W, Sim H, Choi YJ, Lee BH. Activation of SIRT1 by l-serine increases fatty acid oxidation and reverses insulin resistance in C2C12 myotubes (l-serine activates SIRT1 in C2C12 myotubes). Cell Biol Toxicol 2019;35:457–470. [DOI] [PubMed] [Google Scholar]
- [53].Zhou XH, He LQ, Zuo SN, Zhang YM, Wan D, Long CM, et al. Serine prevented high-fat diet-induced oxidative stress by activating AMPK and epigenetically modulating the expression of glutathione synthesis-related genes. Bba-mol Basis Dis 2018;1864:488–498. [DOI] [PubMed] [Google Scholar]
- [54].Trevino AE, Zhang F. Genome editing using Cas9 nickases. Method Enzymol 2014;546:161–174. [DOI] [PubMed] [Google Scholar]
- [55].Meyer JG, Softic S, Basisty N, Rardin MJ, Verdin E, Gibson BW, et al. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One 2018;13:e0208973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang X, Hayes JJ. Acetylation mimics within individual core histone tail domains indicate distinct roles in regulating the stability of higher-order chromatin structure. Mol Cell Biol 2008;28:227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Brown NF, Mullur RS, Subramanian I, Esser V, Bennett MJ, Saudubray JM, et al. Molecular characterization of L-CPT I deficiency in six patients: insights into function of the native enzyme. J Lipid Res 2001;42:1134–1142. [PubMed] [Google Scholar]
- [58].Hada T, Kato Y, Obana E, Yamamoto A, Yamazaki N, Hashimoto M, et al. Comparison of two expression systems using COS7 cells and yeast cells for expression of heart/muscle-type carnitine palmitoyltransferase 1. Protein Expr Purif 2012;82:192–196. [DOI] [PubMed] [Google Scholar]
- [59].Schilling B, Meyer JG, Wei L, Ott M, Verdin E. High-resolution mass spectrometry to identify and quantify acetylation protein targets. Methods Mol Biol 2019;1983:3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bruderer R, Bernhardt OM, Gandhi T, Xuan Y, Sondermann J, Schmidt M, et al. Optimization of experimental parameters in data-independent mass spectrometry significantly increases depth and reproducibility of results. Mol Cell Proteomics 2017;16:2296–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Collins BC, Hunter CL, Liu Y, Schilling B, Rosenberger G, Bader SL, et al. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat Commun 2017;8:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, et al. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 2012;11:016717. 0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zhou S, Tang X, Chen HZ. Sirtuins and insulin resistance. Front Endocrinol (Lausanne) 2018;9:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Iwahara T, Bonasio R, Narendra V, Reinberg D. SIRT3 functions in the nucleus in the control of stress-related gene expression. Mol Cell Biol 2012;32:5022–5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Marcus JM, Andrabi SA. SIRT3 regulation under cellular stress: making sense of the ups and downs. Front Neurosci 2018;12:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang C, Liu J, Huang G, Zhao Y, Yue X, Wu H, et al. Cullin3-KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression. Genes Dev 2016;30:1956–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Tian M, Hao F, Jin X, Sun X, Jiang Y, Wang Y, et al. ACLY ubiquitination by CUL3-KLHL25 induces the reprogramming of fatty acid metabolism to facilitate iTreg differentiation. Elife 2021;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Guo L, Guo YY, Li BY, Peng WQ, Chang XX, Gao X, et al. Enhanced acetylation of ATP-citrate lyase promotes the progression of nonalcoholic fatty liver disease. J Biol Chem 2019;294:11805–11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 2015;43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Jackson VN, Cameron JM, Fraser F, Zammit VA, Price NT. Use of six chimeric proteins to investigate the role of intramolecular interactions in determining the kinetics of carnitine palmitoyltransferase I isoforms. J Biol Chem 2000;275:19560–19566. [DOI] [PubMed] [Google Scholar]
- [71].Saggerson ED. Carnitine acyltransferase activities in rat liver and heart measured with palmitoyl-CoA and octanoyl-CoA. Latency, effects of K+, bivalent metal ions and malonyl-CoA. Biochem J 1982;202:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].McPherson JD, Shilton BH, Walton DJ. Role of fructose in glycation and cross-linking of proteins. Biochemistry 1988;27:1901–1907. [DOI] [PubMed] [Google Scholar]
- [73].Dills WL Jr. Protein fructosylation: fructose and the Maillard reaction. Am J Clin Nutr 1993;58:779S–787S. [DOI] [PubMed] [Google Scholar]
- [74].Yamamoto H, Schoonjans K, Auwerx J. Sirtuin functions in health and disease. Mol Endocrinol 2007;21:1745–1755. [DOI] [PubMed] [Google Scholar]
- [75].Ren H, Hu F, Wang D, Kang X, Feng X, Zhang L, et al. Sirtuin 2 prevents liver steatosis and metabolic disorders by deacetylation of hepatocyte nuclear factor 4alpha. Hepatology 2021;74:723–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, et al. Dietary obesity-associated Hif1alpha activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev 2012;26:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zou X, Zhu Y, Park SH, Liu G, O’Brien J, Jiang H, et al. SIRT3-Mediated dimerization of IDH2 directs cancer cell metabolism and tumor growth. Cancer Res 2017;77:3990–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Scher MB, Vaquero A, Reinberg D. SirT3 is a nuclear NAD+-dependent histone deacetylase that translocates to the mitochondria upon cellular stress. Genes Dev 2007;21:920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Miller CO, Yang X, Lu K, Cao J, Herath K, Rosahl TW, et al. Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am J Physiol Endocrinol Metab 2018;315:E386–E393. [DOI] [PubMed] [Google Scholar]
- [80].Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A 2012;109:4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013;58:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Maryanoff BE, O’Neill JC, McComsey DF, Yabut SC, Luci DK, Jordan AD Jr, et al. Inhibitors of ketohexokinase: discovery of pyrimidinopyrimidines with specific substitution that complements the ATP-binding site. ACS Med Chem Lett 2011;2:538–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Huard K, Ahn K, Amor P, Beebe DA, Borzilleri KA, Chrunyk BA, et al. Discovery of fragment-derived small molecules for in vivo inhibition of ketohexokinase (KHK). J Med Chem 2017;60:7835–7849. [DOI] [PubMed] [Google Scholar]
- [85].Futatsugi K, Smith AC, Tu M, Raymer B, Ahn K, Coffey SB, et al. Discovery of PF-06835919: a potent inhibitor of ketohexokinase (KHK) for the treatment of metabolic disorders driven by the overconsumption of fructose. J Med Chem 2020;63:13546–13560. [DOI] [PubMed] [Google Scholar]
- [86].Roberto Calle AB, Somayaji Veena, Kristin Chidsey, David Kazierad. PS-110-Ketohexokinase inhibitor PF-06835919 administered for 6 weeks reduces whole liver fat as measured by magnetic resonance imaging-proton density fat fraction in subjects with non-alcoholic fatty liver disease. J Hepatol 2019;70:e69–e70. [Google Scholar]
- [87].Gutierrez JA, Liu W, Perez S, Xing G, Sonnenberg G, Kou K, et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol Metab 2021;48:101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Shepherd EL, Saborano R, Northall E, Matsuda K, Ogino H, Yashiro H, et al. Ketohexokinase inhibition improves NASH by reducing fructose-induced steatosis and fibrogenesis. JHEP Rep 2021;3:100217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Taylor NP. Pfizer dumps midphase NASH prospect, slew of early efforts in Q2 clear-out. 2021. [cited 2022 April 1]; Available from: https://www.fiercebiotech.com/biotech/pfizer-dumps-midphase-nash-prospect-quarterly-pipeline-cull. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data and complete MS data sets have been uploaded to the Center for Computational Mass Spectrometry, to the MassIVE repository at UCSD, and can be downloaded using the following link: https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=b9ba416b2a0b44c8b34a29e3228e6eb3 (MassIVE ID number: MSV000089882; ProteomeXchange ID: PXD035316).
[Note: To access the data repository MassIVE (UCSD) for MS data, please use: Username: MSV000089882_reviewer; Password: winter].