

CONSPECTUS:
The development of palladium-catalyzed cross-coupling methods for the activation of C(sp2)–Br bonds facilitated access to arene-rich molecules, enabling a concomitant increase in the prevalence of this structural motif in drug molecules in recent decades. Today, there is a growing appreciation of the value of incorporating saturated C(sp3)-rich scaffolds into pharmaceutically active molecules as a means to achieve improved solubility and physiological stability, providing the impetus to develop new coupling strategies to access these challenging motifs in the most straightforward way possible. As an alternative to classical two-electron chemistry, redox chemistry can enable access to elusive transformations, most recently, by interfacing abundant first-row transition-metal catalysis with photoredox catalysis. As such, the functionalization of ubiquitous and versatile functional handles such as (aliphatic) carboxylic acids via metallaphotoredox catalysis has emerged as a valuable field of research over the past eight years.
In this Account, we will outline recent progress in the development of methodologies that employ aliphatic and (hetero)aromatic carboxylic acids as adaptive functional groups. Whereas recent decarboxylative functionalization methodologies often necessitate preactivated aliphatic carboxylic acids in the form of redox-active esters or as ligands for hypervalent iodine reagents, methods that enable the direct use of the native carboxylic acid functionality are highly desired and have been accomplished through metallaphotoredox protocols. As such, we found that bench-stable aliphatic carboxylic acids can undergo diverse transformations, such as alkylation, arylation, amination, and trifluoromethylation, by leveraging metallaphotoredox catalysis with prevalent first-row transition metals such as nickel and copper. Likewise, abundant aryl carboxylic acids are now able to undergo halogenation and borylation, enabling new entry points for traditional, primarily palladium- or copper-catalyzed cross-coupling strategies. Given the breadth of the functional group tolerance of the employed reaction conditions, the late-stage functionalization of abundant carboxylic acids toward desired targets has become a standard tool in reaction design, enabling the synthesis of various diversified drug molecules. The rapid rise of this field has positively inspired pharmaceutical discovery and will be further accelerated by novel reaction development. The achievement of generality through reaction optimization campaigns allows for future breakthroughs that can render protocols more reliable and applicable for industry. This article is intended to highlight, in particular, (i) the employment of aliphatic and (hetero)aryl carboxylic acids as powerful late-stage adaptive functional handles in drug discovery and (ii) the need for the further development of still-elusive and selective transformations.
We strongly believe that access to native functionalities such as carboxylic acids as adaptive handles will further inspire researchers across the world to investigate new methodologies for complex molecular targets.
Graphical Abstract
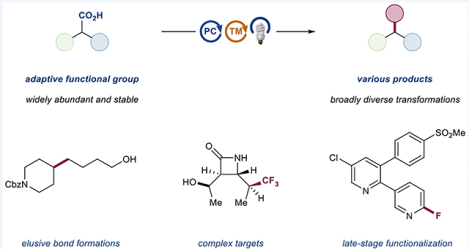
INTRODUCTION
The controlled release of CO2 from organic molecules, known as decarboxylation, is a fundamental chemical transformation that is critical to both chemical biology and organic synthesis. In nature, the enzyme-mediated regioselective decarboxylation of amino acids facilitates critical biological processes, such as the conversion of glutamic acid to the neurotransmitter γ-aminobutyric acid (GABA), a crucial pathway for mammalian central nervous system signaling.5 Taking a page from nature’s toolbox, organic chemists have leveraged such decarboxylation strategies toward regioselective activation and the functionalization of carboxylic acids, with fundamental developments achieved in the past one and a half centuries.
While thermal decarboxylation at elevated temperatures has long been explored, such two-electron strategies require forcing conditions and are inherently energy-intensive.6,7 Alternatively, a common strategy is the oxidative generation of a carboxyl radical that then rapidly extrudes CO2 to generate an aliphatic carbon-centered radical. As early as 1848, Kolbe employed electrochemistry to facilitate oxidative decarboxylation utilizing stoichiometric metal oxidants such as silver, mercury, and lead to generate the key carboxyl radical intermediate.8
Over the past decade, photoredox catalysis has emerged as a privileged technology for the generation of these high-energy species under mild conditions, mediating both oxidation and reduction events by a single catalyst in one reaction vessel.9,10 Our laboratory pioneered the merger of photoredox and transition-metal catalysis, termed metallaphotoredox catalysis, to develop a new mechanistic manifold that has helped to reconfigure synthetic logic and had enabled diverse options toward the assembly of complex molecular scaffolds.11
In this vein, we conceived of a general metallaphotoredox-catalyzed decarboxylation platform that would enable the use of bench-stable aliphatic carboxylic acids as adaptive functional groups (Figure 1). Carboxylic acids, as native functional groups, are widely available from both commercial and natural sources. The ability to harness these ubiquitous motifs as open-shell C(sp3)–C(sp3) cross-coupling partners would be of enormous benefit to practitioners of medicinal and process chemistry.12 We also sought to design a mild protocol for the activation of aromatic carboxylic acids, which have historically been resistant to standard decarboxylation chemistry. To this end, we reasoned that a light-mediated strategy, combined with copper catalysis, could utilize ligand-to-metal charge transfer (LMCT) to overcome the thermodynamic and kinetic obstacles, thus providing access to open-shell aryl radicals in functionalization reactions.
Figure 1.
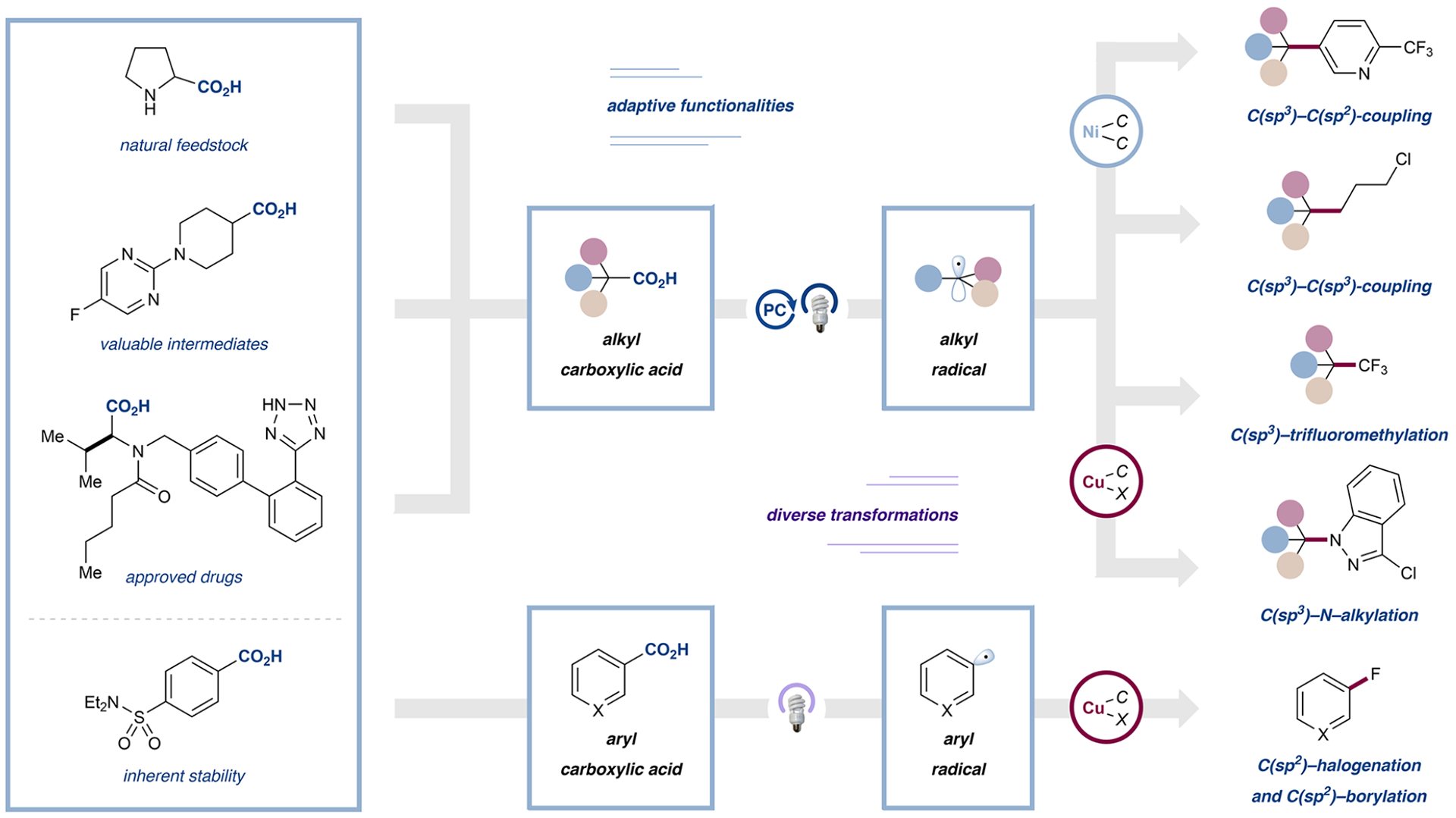
Development of various decarboxylative transformations under metallaphotoredox conditions, enabling arylation, alkylation, amination, and trifluoromethylation from aliphatic carboxylic acids. Inherently stable aromatic carboxylic acids were converted to electrophiles and nucleophiles.
Below, we present an Account of our laboratory’s research on metallaphotoredox-catalyzed decarboxylative functionalization, from its inception as a new activation platform for alkyl carboxylic acids to our most recent developments in leveraging a new mode of metallaphotocatalysis as a strategy for the decarboxylative functionalization of aryl carboxylic acids. The conceptual developments from seminal discoveries to rational reaction design and reaction generality will be outlined. We are aware of a vast variety of potent metallaphotoredox-catalyzed decarboxylative transformations, which are beyond the scope of this Account.13,14,11 We will focus on key relevant developments, which often follow our seminal works.
DECARBOXYLATIVE DUAL NICKEL PHOTOREDOX CATALYSIS
Our laboratory has had a longstanding interest in using open-shell chemistry to forge new C–C bonds. Initially, we found success by coupling persistent radicals with transient radicals, but we quickly realized that the scope of this strategy was intrinsically limited due to the relatively few types of organic radicals that exhibit persistency. Preactivation with organocatalysts15 or the electronic differentiation of α-amine16 and allylic positions17 gave access to a new but restricted chemical space. We recognized that in order to develop reactions with the most potential impact, we would need to draw from more diverse precursors, such as aryl or alkyl halides. For decades, transition-metal catalysis has been used to engage these electrophiles in productive cross-coupling chemistry, and we wondered if merging this traditional platform with photoredox catalysis could enable previously unknown reactivity. The bond-breaking and bond-forming capacities of nickel spurred our interest in this area. By interfacing nickel catalysis with the photocatalytic cycle in a dual catalytic platform, it could be possible to employ abundant, bench-stable, and readily accessible alkyl carboxylic acids in place of conventional nucleophilic organometallic coupling partners as a means to rapidly forge new C(sp2)–C(sp3) bonds. Importantly, the implementation of this general concept has enabled the use of nickel catalysis in a wide variety of C–C cross-coupling methodologies (vide infra).
C(sp3)–C(sp2) Cross-Coupling with Nickel-Based Radical Trapping after Initial Decarboxylation
In 2014, our laboratory disclosed an early example of decarboxylative cross-coupling. Enabled by the merger of photoredox and nickel catalysis, this method was among the first examples of a powerful C(sp2)–C(sp3) cross-coupling reaction (Figure 2).18 We envisioned that a plausible mechanism could begin with light excitation of an iridium photocatalyst to a long-lived triplet state (τ = 2.3 μs) capable of undergoing single-electron oxidation of a carboxylate anion ({Ir[dF(CF3)ppy]2(dtbbpy)}PF6 (PC-1), E1/2red[*Ir(III)/Ir-(II)] = +1.21 V vs SCE in MeCN and BocProOCs, and E1/2red[Pro−/Pro•] = +0.95 V vs SCE in MeCN). The resulting carboxy radical would rapidly extrude CO2 (k ≈ 109 s−1)19,20 to generate carbon-centered radical 1. At the same time, low-valent Ni(0) (formed in situ from Ni(II)) could readily undergo oxidative addition with an aryl halide to access a Ni(II)–Ar intermediate.21 Upon trapping of the alkyl radical, the resulting Ni(III) species could then undergo facile reductive elimination, forging the desired C(sp3)–C(sp2) bond. The resulting Ni(I) species could then be turned over by reduced Ir(II), closing both catalytic cycles simultaneously.
Figure 2.
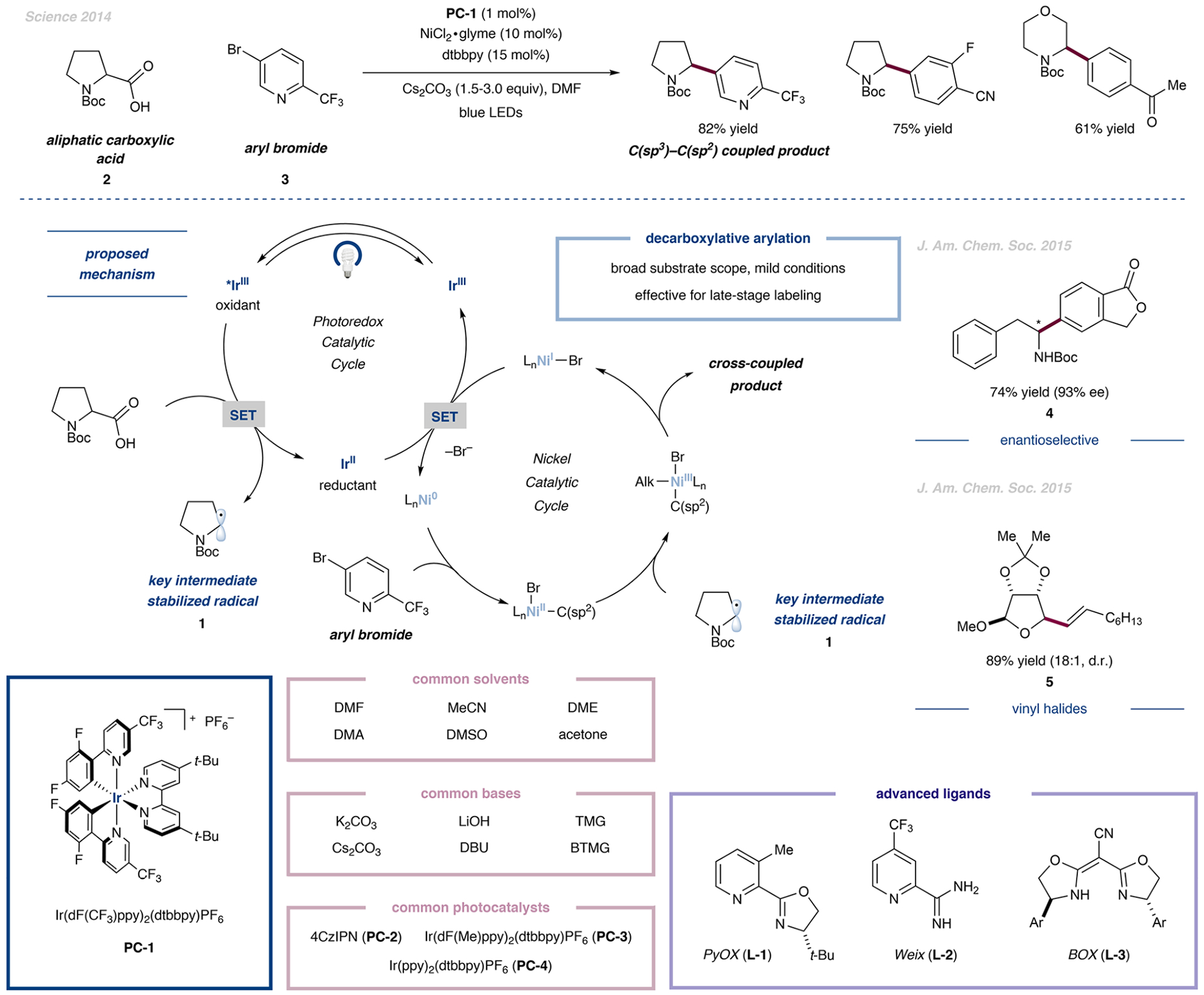
Decarboxylative arylations, pioneered by MacMillan et al., were adapted by many groups and led to diverse conditions toward smallmolecule drugs and biomolecules. Further developments led to enantioselective and vinyl halide transformations. Typical photocatalysts, bases, and solvents are displayed. The choice of ligands has a distinct effect on the nickel catalysis, and the proper ligands are highlighted. Data are from refs 18, 32, and 33.
This preliminary mechanistic proposal18 was further validated by mechanistic studies disclosed by Johannes and co-workers in 2015.22 This powerful transformation was capable of effecting C(sp2)–C(sp3) cross-coupling between a wide variety of aliphatic acids 2 and aryl halides 3. This seminal work opened the door to the use of a number of groups as precursors, and a variety of arylation strategies have been described for a broad range of small molecules.23–26 For example, the mild nature of decarboxylative arylation in aqueous environments has allowed facile access to DNA-encoded libraries.27,28 Ligand development (e.g., L-1)29 as well as large scale synthesis under constant flow conditions expanded the general utility of decarboxylative arylations.30,31
In collaboration with the Fu group, we were able to achieve the first asymmetric variant of this decarboxylative arylation toward the synthesis of enantioenriched α-amino arenes (Figure 2, middle right).32 The key to successful stereoinduction was the use of commercially available bis-oxazoline ligands (BOX, L-3) bearing bulky aromatic substituents. The chiral products 4 were obtained in generally higher that 90% ee and good to excellent yields.
Beyond our initial conversion of aromatic bromides, we wondered whether vinyl halides could serve as viable coupling partners because they present a powerful functional handle but often undergo unproductive radical addition reactions. The first decarboxylative olefination was described by our laboratory in 2015 via dual photoredox- and nickel-catalyzed cross-coupling of aliphatic carboxylic acids with vinyl iodides or bromides (Figure 2, middle right).33 Similar to our initial study, we leveraged the facile oxidative addition step of in situ-generated Ni(0) into a C(sp2)–X bond. The iridium photocatalyst {Ir[dF(Me)ppy]2(dtbbpy)}PF6 (PC-3) was employed to increase the stability toward radical-mediated decomposition at higher concentrations, affording the respective alkenylated products 5 in high yields with the retention of olefin stereochemistry. With respect to the carboxylic acid coupling partner, a ribose-derived cyclic α-oxy acid was coupled in excellent yield and diastereoselectivity, demonstrating the utility of this protocol for orthogonal nucleoside functionalization to access antiviral agents in drug discovery. The utility of this cross-coupling manifold in late-stage functionalization was demonstrated through a short synthesis of trans-rose oxide, a natural product and widely used fragrance.
A modular example of leveraging nickel photoredox decarboxylative functionalization for the synthesis of ketones was disclosed by our laboratory in 2015. In this transformation, an α-keto acid was converted to the corresponding acyl radical species 6,34 which could then engage a nickel catalyst and R–X electrophile in cross coupling (Figure 3).35 Optimization studies revealed that the deprotonation of aliphatic or aromatic α-keto acids appeared to be most efficient in the presence of lithium carbonate as the base. Under these optimized conditions, a range of aliphatic and aromatic α-keto acids were readily coupled to 4-iodo-toluene in moderate to high yields (cf. 7).
Figure 3.
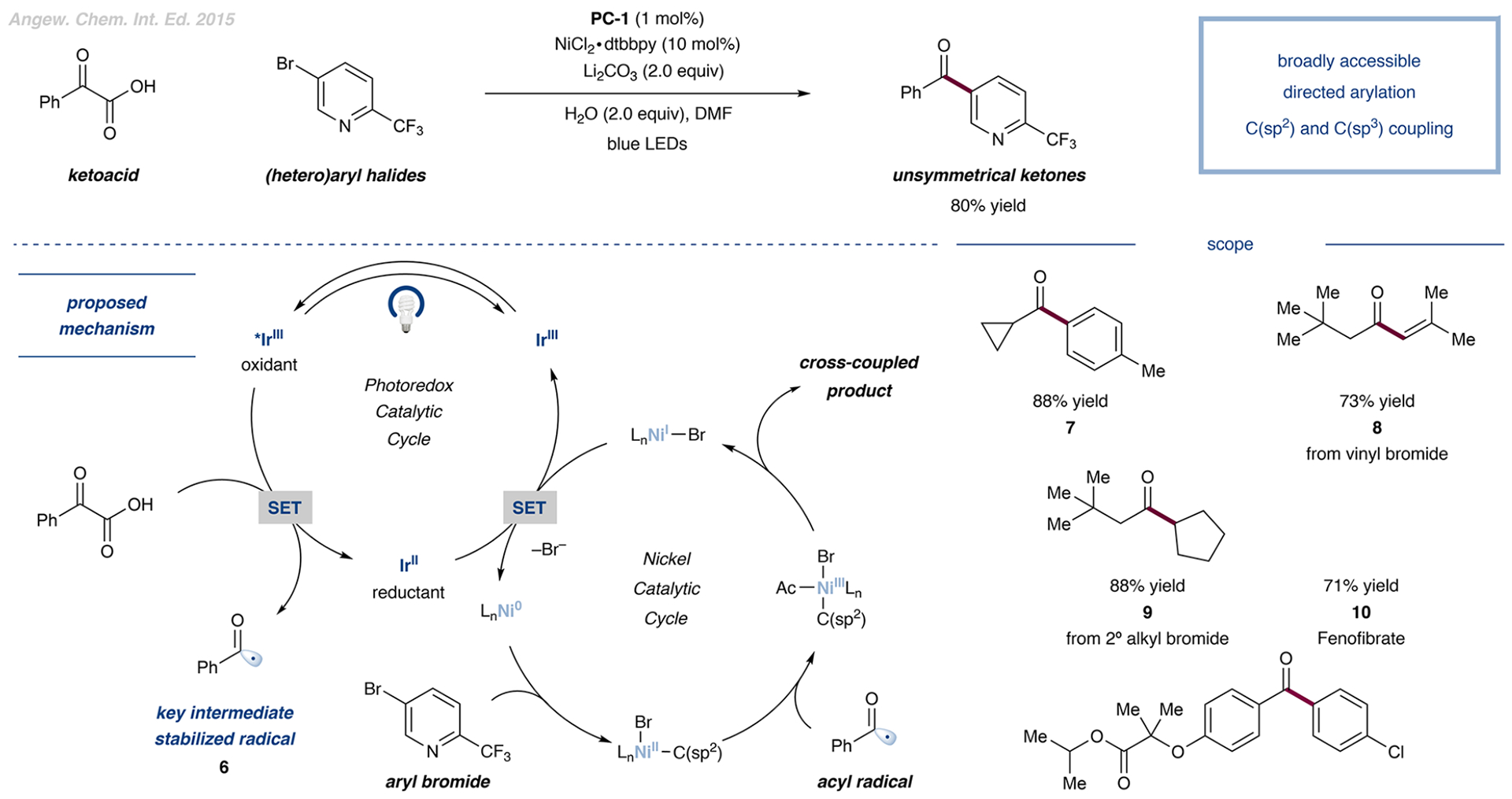
Decarboxylative formation of unsymmetrical ketones from α-keto carboxylic acids via a versatile acyl radical intermediate 6. Data are from ref 35.
Different phenyl iodides and (hetero)aryl bromides also served as viable coupling partners, reacting with 2-phenyl-α-keto acid to deliver products in yields of up to 90%. Furthermore, the coupling of alkyl bromides with aliphatic α-keto acids was successful in forming unsymmetrical dialkyl ketones (cf. 9), which are challenging synthetic targets that are difficult to access under mild conditions. Finally, the practical utility of the procedure was demonstrated through the efficient total synthesis of fenofibrate 10 from commercial precursors, featuring a 71% yield of the final metallaphotoredox-mediated step.
Our strategy of using α-keto acids as precursors in the decarboxylative synthesis of ketones has since been adapted by multiple groups, who have employed modified conditions to ketones.36–38
CO2 Extrusion Approach from In Situ-Generated Anhydrides
In 2015, we reported a new, mechanistically distinct synthesis of unsymmetrical ketones starting from commercially available carboxylic acids and acid chlorides (Figure 4).39 These two components were premixed to form an unsymmetric anhydride, with DBU as a soluble base, without further purification. This anhydride was susceptible to the oxidative addition of a Ni(0) catalyst to the C–O bond to form a complex, 11, which was further oxidized by photoexcited Ir(III). The subsequent loss of CO2 and radical rebound afforded an acyl–Ni(III)–alkyl intermediate, 12. Reductive elimination from this Ni(III) species generated the unsymmetrical ketone product and the resulting Ni(I) complex, which was reduced to Ni(0) by Ir(II), closing both catalytic cycles. The cross-coupling reaction was high-yielding for a range of α-amino acid derivatives and a menu of aliphatic or aromatic acyl chlorides (up to 86%, cf. 13). This cross-coupling method was also used to synthesize intermediate 14 en route to edivoxetine. Importantly, the use of the electron-rich 4,4′-dimethoxy-2,2′-bipyridine (dOMebpy) ligand for the nickel catalyst was required to suppress symmetrical ketone byproduct formation.
Figure 4.
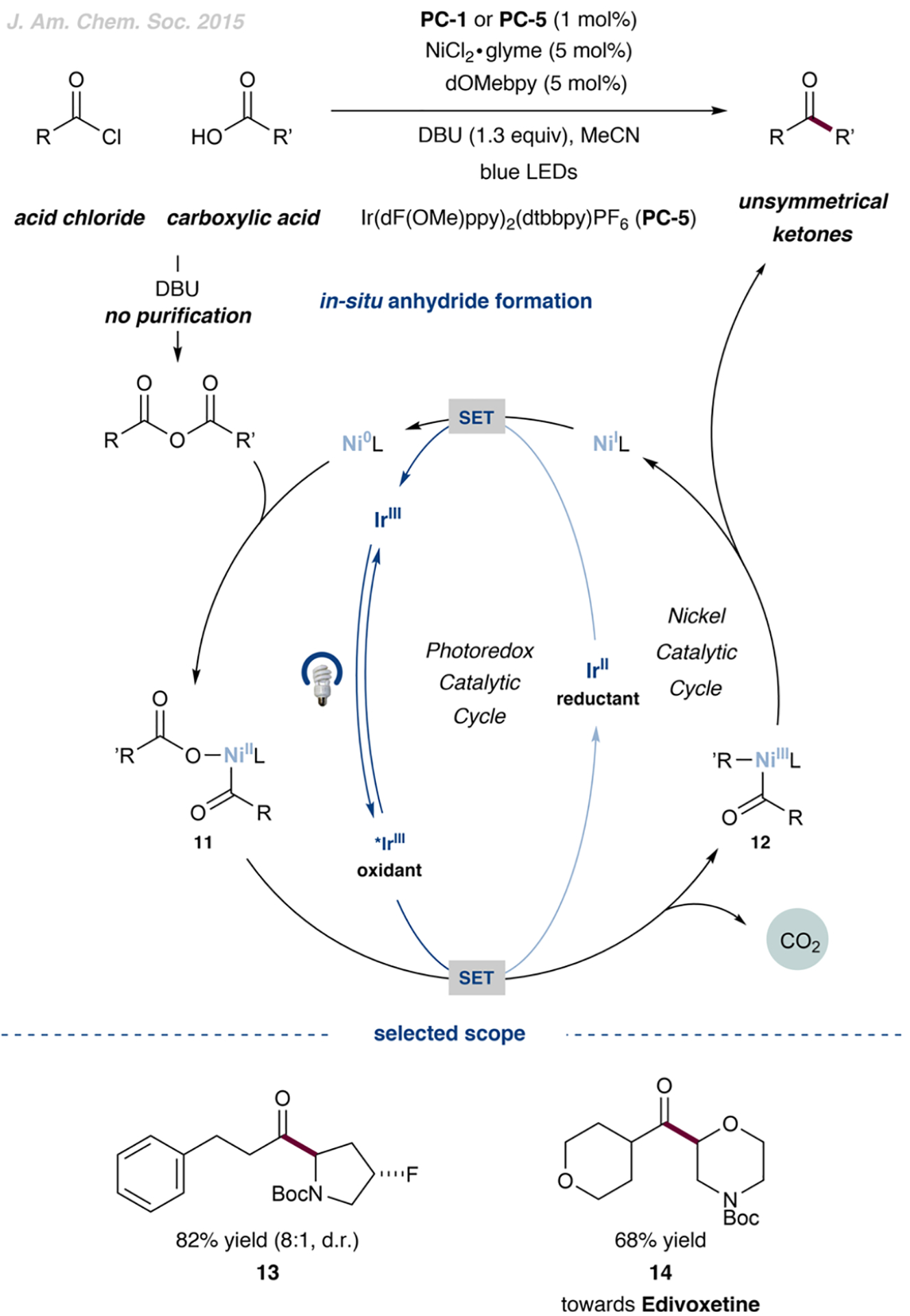
Unsymmetrical dialkyl ketones obtained from the CO2 extrusion of anhydrides. Data are from ref 39.
C(sp3)–C(sp2) Cross-Coupling with Nickel-Mediated Migratory Insertion
Radical addition to unactivated alkynes to obtain olefinic products remains a significant challenge in the field of open-shell chemistry. A polarity mismatch between nucleophilic carbon-centered radicals and neutral alkynes results in sluggish rates of radical addition and poor reaction efficiency. Furthermore, high-energy vinyl radicals resulting from such an addition are configurationally unstable.40 However, vinyl C(sp2)–nickel bonds are configurationally stable and can be stereospecifically protodemetalated to afford stereodefined olefins. Nickel is also known to capture alkyl radicals at rates approaching diffusion to afford NiII–alkyl species which can undergo stereo- and regioselective migratory insertion.41 As such, we set out to achieve a catalyst-controlled stereo- and regioselective hydroalkylation of unactivated alkynes, which would provide a complementary approach to cross-coupling with vinyl bromides.
Gratifyingly, subjecting an aliphatic carboxylic acid and an unactivated alkyne to blue-light irradiation in the presence of an iridium photocatalyst and a nickel cocatalyst provided excellent yields of a variety of vinylated products, 15 to 18 (Figure 5).42 In the case of electronically differentiated and sterically similar acetylide substituents, the migratory insertion steps are (i) controlled by a preference for alkyl insertion at the site of lowest electron density and (ii) dominated by steric factors, placing the alkyl substituent at the least sterically demanding position.
Figure 5.
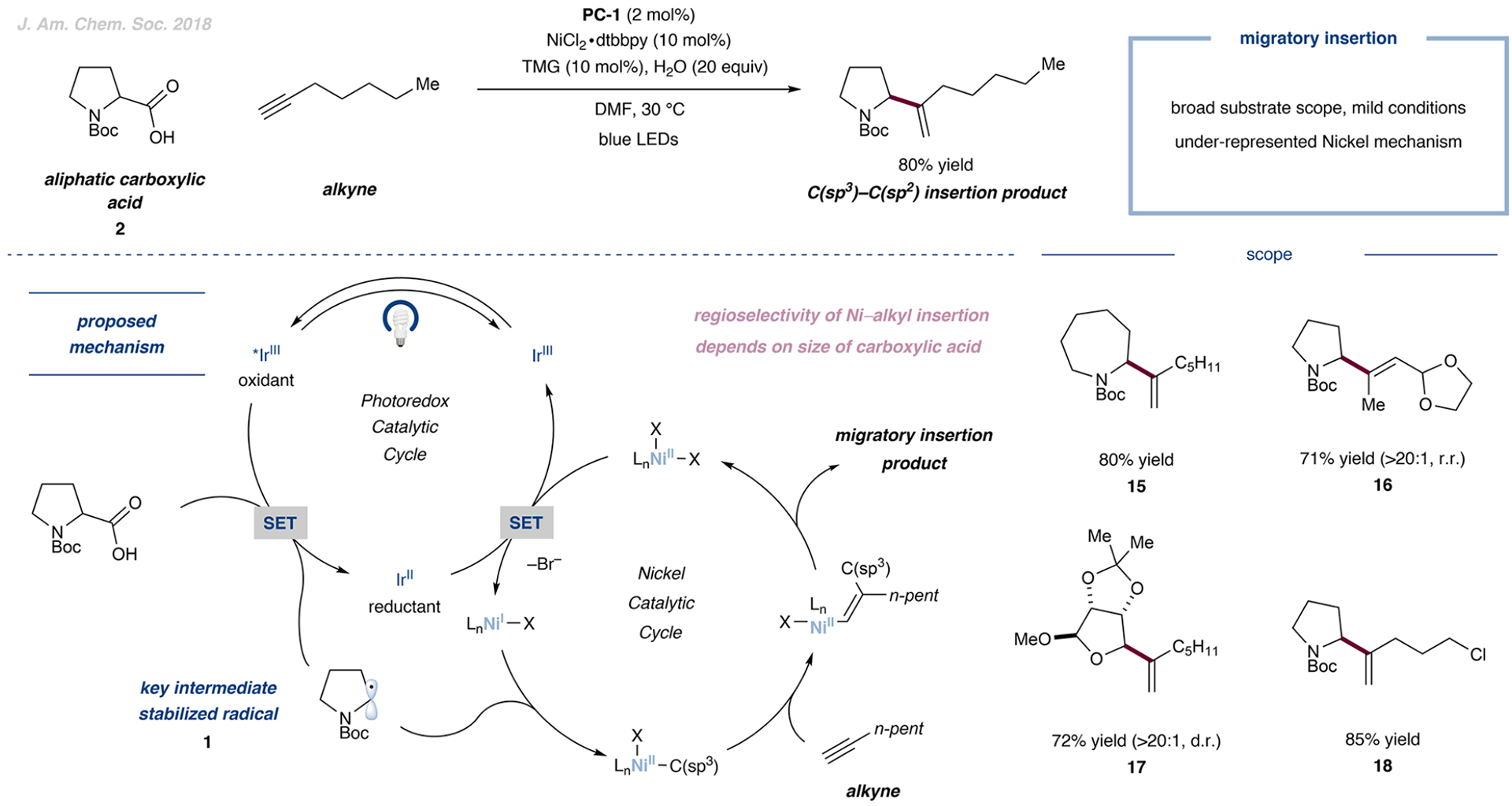
Decarboxylative mechanism merged with the migratory insertion of nickel into alkynes to obtain substituted olefins. Data are from ref 42.
Following our initial efforts, decarboxylative alkynylations were reported by copper or nickel metallaphotoredox catalysis to yield (E)- or (Z)-olefins, respectively, by the Pericas and Rueping groups.43,44
Decarboxylative C(sp3)–C(sp3) Coupling
One of the most challenging transformations in organic synthesis is the formation of a C(sp3)–C(sp3) bond from abundant and bench-stable precursors. Typically, this bond formation requires highly reactive precursors, such as organometallic reagents. The first example of a metallaphotoredox-catalyzed coupling of aliphatic carboxylic acids with alkyl halides was described by our laboratory in 2016 (Figure 6).1 We leveraged open-shell reactivity to bypass deleterious side reactions, such as β-hydride elimination in palladium catalysis or the challenging oxidative addition of alkyl halides that hampers many C(sp3)–C(sp3) bond-forming protocols. Additionally, by replacing pregenerated organometallic nucleophiles with alkyl carboxylic acids, we succeeded in efficiently accessing a broad scope of aliphatic cross-coupled products. The reaction proceeds by first generating a carbon-centered radical via decarboxylation, which can then be trapped by a Ni(0) species.21 The resulting Ni(I)-alkyl complex undergoes oxidative addition into an alkyl bromide bond and, following-reductive elimination from the Ni(III) catalyst, liberates the final C(sp3)–C(sp3) cross-coupled product 19.
Figure 6.
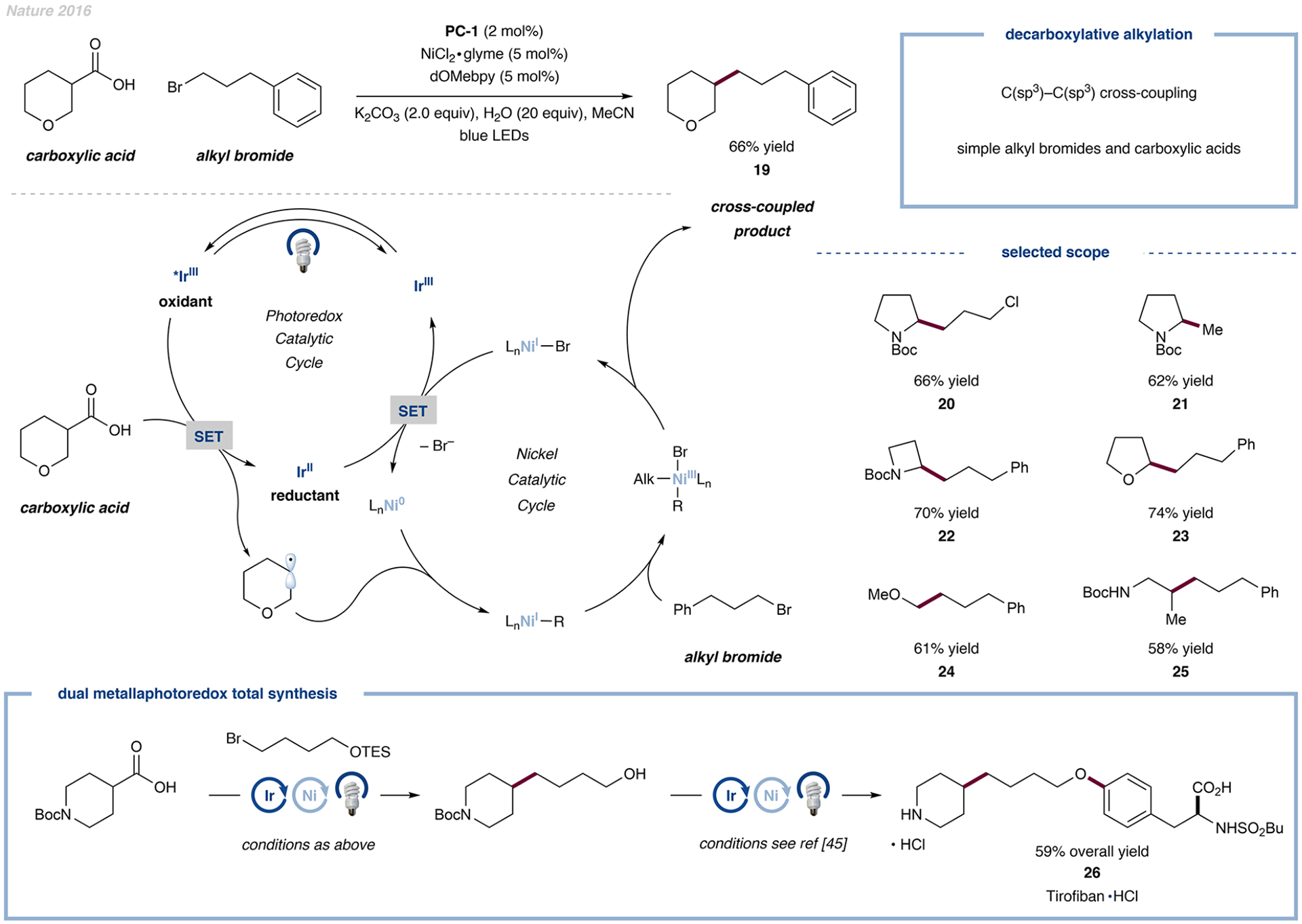
Decarboxylative cross-electrophile coupling between aliphatic carboxylic acids and alkyl bromides to yield the respective C(sp3)–C(sp3) coupled product. Data are from ref 1.
We achieved optimal reaction efficiency with acetonitrile as the solvent, K2CO3 as the base, a more electron-rich ligand (i.e., dOMebpy), and the addition of water. Water was found to be critical in suppressing undesired carboxylic acid alkylation, presumably by tempering the nucleophilicity of the carboxylate via hydrogen bonding. A variety of aliphatic carboxylic acids, including primary acids and acids with or without a stabilizing adjacent α-heteroatom, could be employed as substrates. Additionally, primary and secondary aliphatic bromides were good substrates, including (notably) bromomethane, to afford products 20 to 25 in good yields. The synthetic utility of this transformation was highlighted by a three-step synthesis of the antiplatelet drug tirofiban 26 from commercial substrates through two consecutive metallaphotoredox-mediated bond formations.45
General Reactivity in Decarboxylative C(sp3)–C(sp2) Cross-Coupling
Although thousands of novel chemical reactions are reported each year, few find longstanding and broad adoption by the synthetic community. Those that do share one common feature: they are general. General reactions are high-yielding in a wide variety of contexts and are relatively insensitive to substrate complexity. Historically, this level of success is achieved only after years of meticulous optimization enabled by deep mechanistic understanding.
The decarboxylative arylation reaction developed by our group and the Doyle group in 2014 (vida supra) performed well across a number of substrate classes but struggled in three key areas: (1) coordinating substrates, (2) electron-rich aryl halides, and (3) unactivated carboxylic acids (i.e., acids that result in unstabilized radicals upon the loss of CO2). Traditional optimization campaigns by our group and others yielded little improvement in substrate scope and failed to provide a deep mechanistic understanding of these shortcomings. We began to consider an alternative approach that would accelerate the generalization process while at the same time providing mechanistic insight.
To this end, we were inspired by phenotypic screening campaigns in drug discovery, which find solutions to complex biological problems but are initially agnostic to the underlying mechanism. Phenotypic screening involves systematically perturbing a complex system and studying how a single end point, drug efficacy, responds to that initial perturbation. We wondered whether we could apply a phenotypic strategy to problems in synthetic organic chemistry by perturbing a chemical system with a variety of additives and studying how a single end point, reaction yield, responded.
In collaboration with Merck & Co. (MSD), we initiated an unbiased screen of over 700 unconventional additives to investigate reaction robustness. To our delight, we identified phthalimide as a simple, bench-stable, nontoxic additive that broadly improved the reaction scope, decreased the number of reaction poisons, and increased the yield of each previously challenging substrate class by 2- to 3-fold (Figure 7).4 In the presence of 1 equiv of this additive, the average yield of the nickel-catalyzed decarboxylative arylation of the Merck Aryl Halide Informer Library, a benchmark set of the most challenging aryl halides for metal-catalyzed cross coupling, increased by over 3-fold (7.7 to 29.4%) for 11 out of 18 complex molecules.
Figure 7.
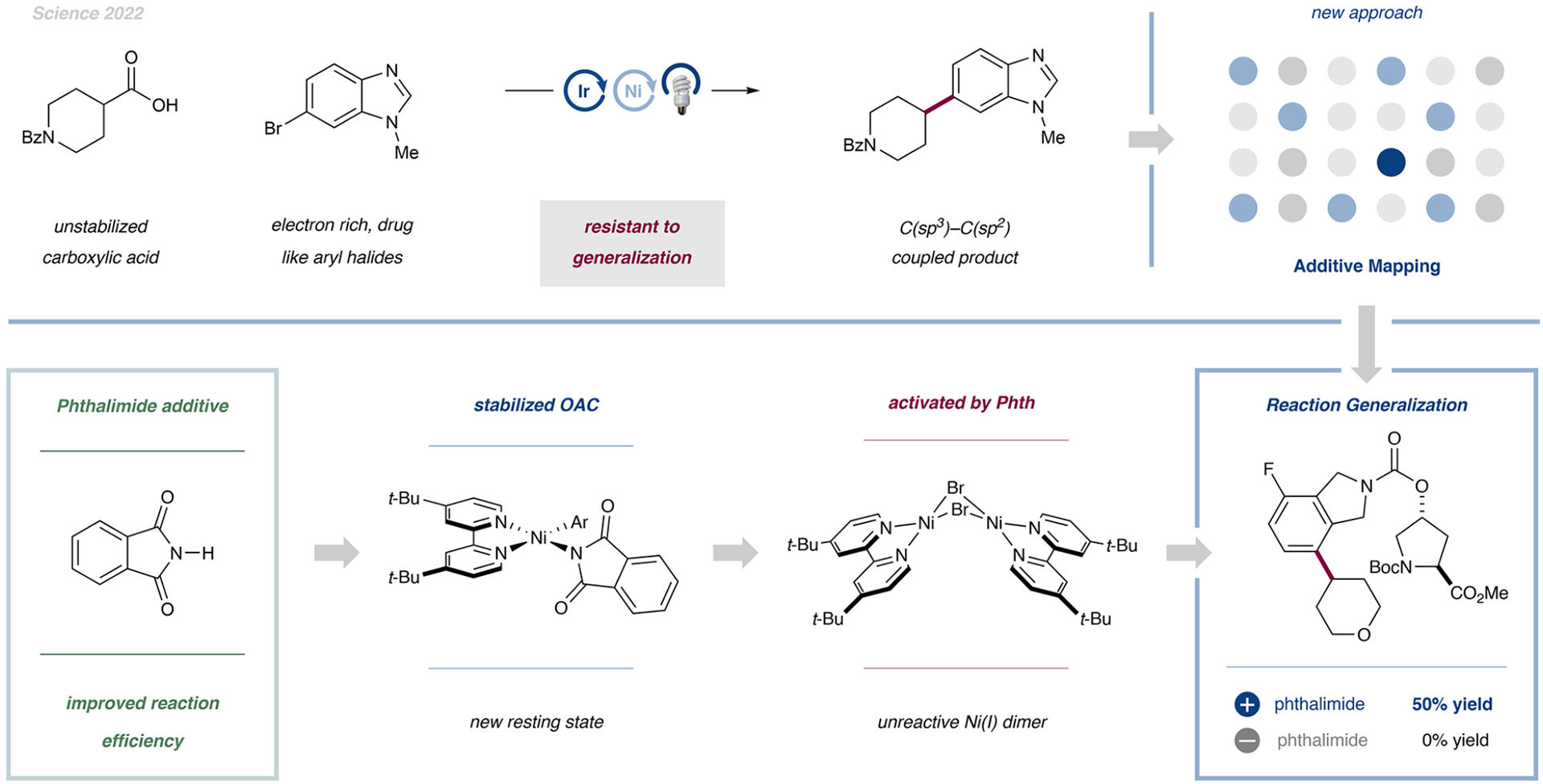
Decarboxylative arylations were adapted by many groups, but no reaction generality was observed. Phenotypic screening of hundreds of additives resulted in phthalimide as a powerful and reliable reagent to improve the conversions generally. OAC: Oxidative addition complex. Data are from ref 4.
Having identified phthalimide as uniquely capable of improving reaction yields and generality, we sought to understand how this additive was imparting its beneficial effects. While we believe the “phthalimide effect” is multifactorial, we are confident that it has two key functions: (i) stabilizing nickel oxidative addition complexes leading to suppressed protodehalogenation and facilitating radical capture by nonactivated carboxylic acids and (ii) activating unreactive, low-valent nickel species, thereby switching on catalytic activity from nickel species that would otherwise be inactive. We expect that the discovery of this improved and more general catalytic system will not only accelerate its adoption by the synthetic community but will also demonstrate how mechanistic insights can result from mechanistically agnostic optimization campaigns. We anticipate that additional transformations beyond C(sp3)–C(sp2) bond formations will benefit from future phenotypic additive screening efforts.
DECARBOXYLATIVE DUAL COPPER PHOTOREDOX CATALYSIS
Nickel has provided extensive access to C(sp2)–C(sp3) bonds formed by decarboxylative dual catalysis with a photoredox catalyst. Key to this advance is the exploitation of the facile oxidative addition of in situ-generated Ni(0) into C(sp2)–X or C(sp3)–X bonds (X = halide) as well as facile reductive elimination from Ni(III) intermediates to form C(sp2)– C(sp3) bonds. Beyond C–C bond-forming reactions, our group became increasingly interested in the coupling of carbon centers with heteroatoms and heteroatom-containing groups. However, reductive elimination from nickel to form heteroatom-containing fragments was inefficient. By contrast, we found that copper catalysts could be utilized in radical trapping events, resulting in highly reactive Cu(III) complexes. High-valent copper species undergo facile reductive elimination and thus can be used in transformations beyond the scope of their nickel counterparts.
Decarboxylative C(sp3)–CF3 Bond Formation Reactions
Given the importance of the trifluoromethyl group in drug discovery and the lack of methods for the synthesis of alkyl– CF3 motifs, our laboratory became interested in developing a general strategy for the trifluoromethylation of alkyl carboxylic acids. The merger of copper and photoredox catalysis, in the presence of an electrophilic trifluoromethylation reagent, enabled the copper-catalyzed decarboxylative trifluoromethylation of alkyl carboxylic acids, which we reported in 2018 (Figure 8).46
Figure 8.
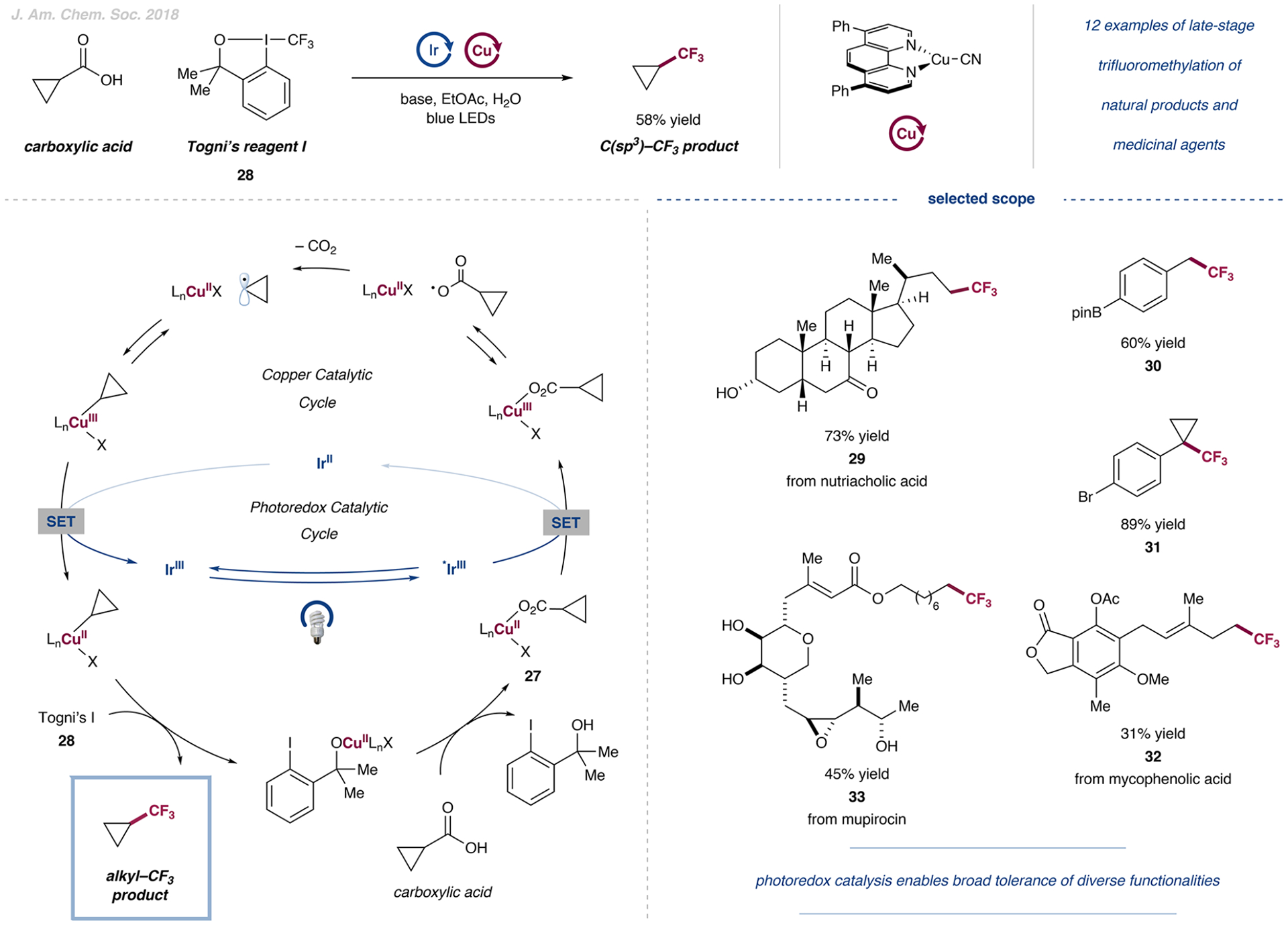
Decarboxylative copper-catalyzed trifluoromethylation using Togni’s reagent I as an electrophilic CF3 source. Data are from ref 46.
In the proposed mechanism, a carboxylate anion, derived from its corresponding carboxylic acid, would ligate Cu(II) to form 27. Simultaneously, photoexcitation of the Ir(III) photocatalyst with visible light would generate a highly oxidizing *Ir(III) catalyst, which then would deliver Cu(III) carboxylate or the corresponding dissociated carboxyl radical and Cu(II) pair. The loss of CO2 would generate an alkyl radical, and an alkyl–Cu(III) species would be formed upon radical rebound and recombination. The single-electron reduction of this species by Ir(II) would regenerate Ir(III) and deliver an alkyl–Cu(II) complex. This complex would then deliver an alkyl–CF3 product upon engaging with Togni’s reagent I (28). While control reactions indicate that copper is responsible for the construction of the key C(sp3)–CF3 bond, which likely proceeds through reduction elimination from a transient Cu(III) intermediate, the precise mechanism of this bond formation has not been fully elucidated. Finally, ligand exchange with a carboxylic acid on copper would complete both catalytic cycles.
A survey of the substrate scope revealed that the trifluoromethylation of benzylic and nonbenzylic primary carboxylic acids as well as secondary and tertiary carboxylic acids (including cyclic strained three- and four-membered rings) was achieved in good to excellent yields (29 to 32). Additionally, bicyclic and spirocyclic compounds that serve as aryl ring bioisosteres can be readily employed in this reaction. In particular, mupirocin, which is a natural product containing an epoxide, diol, and Michael acceptor, was functionalized in 45% yield (33), highlighting the remarkable breadth of functional group tolerance.
Decarboxylative C(sp3)–N Bond Formation Reactions
Traditionally, the alkylation of N-heterocycles has been accomplished via SN1 or SN2 reactions with a corresponding alkyl halide. However, some strained alkyl halides suffer from prohibitively slow reaction rates, precluding their synthesis through these classical methods. Furthermore, differentiation between nucleophilic sites in nitrogen-rich heterocycles remains a significant challenge, often leading to intractable mixtures of regioisomers. Facilitating drug discovery programs through novel reaction development has long been a central tenant of our laboratory’s research program, so we became interested in pursuing a general and mechanistically distinct C(sp3)–N bond-forming reaction.
To circumvent high kinetic barriers associated with some closed-shell SN1 and SN2 alkylations, we chose to pursue an open-shell strategy for C–N bond formation. To this end, we recognized that the propensity of Cu(II) to capture both alkyl radicals and iodine(III) carboxylate reagents would provide a convenient method for the reductive decarboxylation of alkyl carboxylic acids, rendering the overall transformation redox-neutral and obviating the need for exogenous oxidants.
As shown in Figure 9, premixing a carboxylic acid with hypervalent iodomesityl diacetate (34) leads to ligand exchange at iodine(III); we found that a variety of aliphatic carboxylic acids could be activated in this way as electrophilic iodonium coupling partners. This strategy allows the in situ activation of the carboxylic acids without cumbersome purification,47,48 thus enabling in situ activation,49,50 as was originally described by Minisci in 1989.51 Upon accepting an electron from a reduced Ir(II) photocatalyst, the iodonium reagent undergoes fragmentation with the rapid loss of CO2 to generate the desired alkyl radical, which can be captured by preformed Cu(II) amido species 35. Facile C–N reductive elimination from the resulting metastable Cu(III) species 36 effects the desired bond formation. Finally, the resulting Cu(I) intermediate is oxidized by the excited state of the iridium photocatalyst, simultaneously closing both catalytic cycles.
Figure 9.
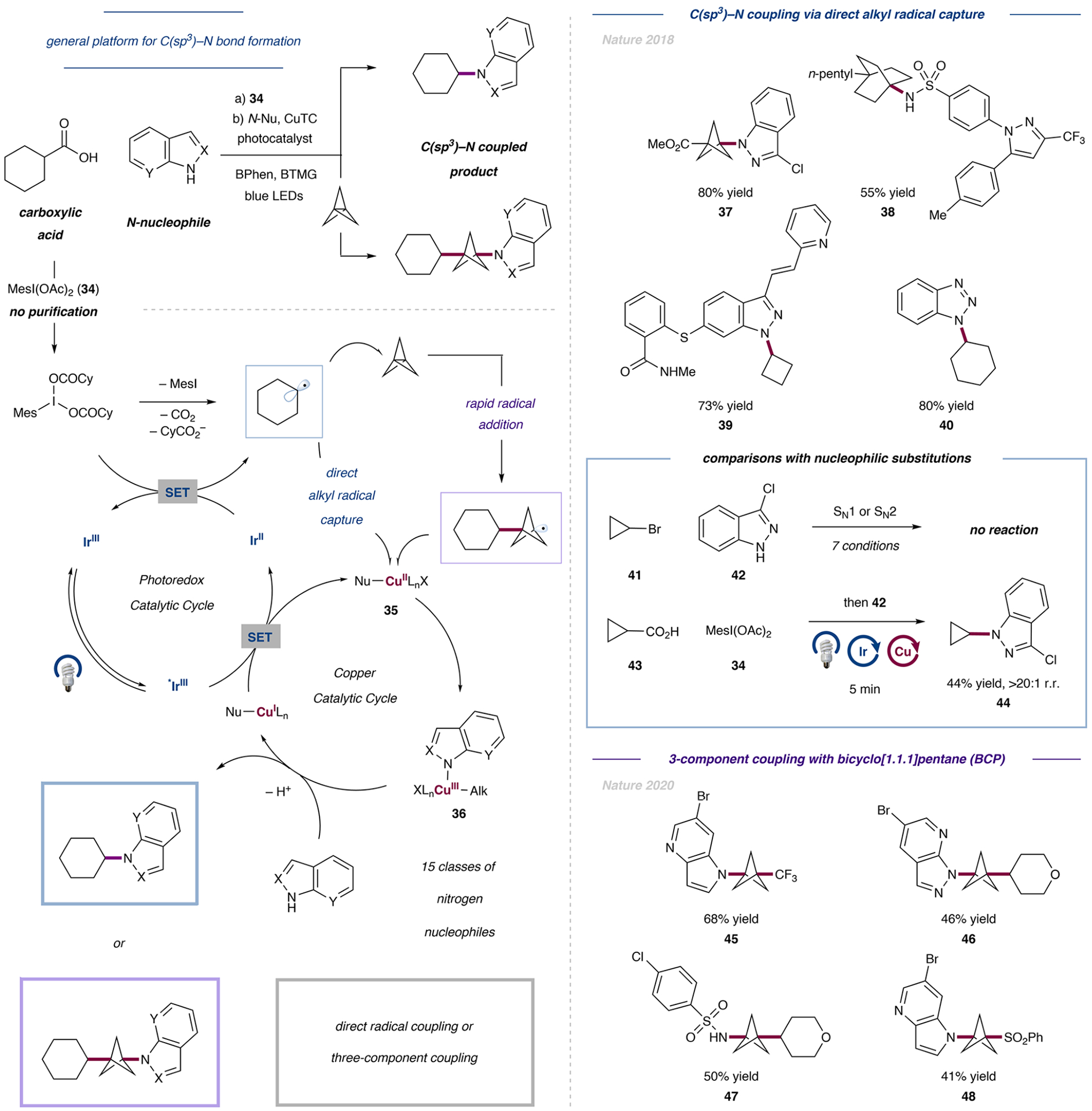
Decarboxylative N-alkylation of various heterocycles. Coupling proceeded either directly with the N-heterocycles or by previous interception with propellane to obtain the corresponding 1,3-disubstituted BCP derivatives. Data are from refs 2 and 3.
Gratifyingly, the described transformation could be applied to an exceptionally wide variety of N-heterocycles as well as a breadth of aliphatic carboxylic acids (e.g., 37 to 40). Of particular interest to us were substrates, such as 41, that were unreactive under any SN1 or SN2 conditions yet readily amenable to coupling in synthetically useful yields using our open-shell protocol (e.g., 43 to 44; Figure 9).2 Furthermore, N-nucleophiles that contain multiple nucleophilic sites could be alkylated with complete regiocontrol as a function of preferential coordination by the most aromatic heterocycle tautomer.52
Given the growing interest in 1,3-disubstituted bicyclopentane (BCP) moieties in drug discovery53 and the propensity for the highly strained ring system [1.1.1]propellane to react with open-shell intermediates, we wondered if we could adapt our decarboxylative C–N coupling strategy to a three-component coupling reaction. If the rate of carbon-centered radical addition across the strained central bond of [1.1.1]-propellane could outcompete radical capture by copper, then we should be able to achieve “interrupted” open-shell C–N coupling in which a BCP motif would be inserted between an N-nucleophile and a carbon-centered radical. Indeed, subjecting a preactivated carboxylic acid, an N-heterocycle, and [1.1.1]propellane to blue-light irradiation in the presence of an iridium photocatalyst alongside a copper cocatalyst provided high yields of previously inaccessible bicyclopentane products.3
Key to the success of this transformation was the utilization of diketonate ligands. We postulate that these X-type ligands increase the electron density on the copper center, creating a polarity mismatch with nucleophilic carbon-centered radicals and reducing the rate of radical capture by the metal relative to the rate of radical capture by [1.1.1]propellane.
This efficient procedure was capable of furnishing 1,3-disubstituted BCP derivatives with a variety of primary, secondary, and tertiary carboxylic acid coupling partners, all obtained from either acyclic or cyclic derivatives. In addition, with our strategy in hand, alkylation with alkyl bromides and trifluoromethylation (45) was achieved in good yields. A scope of 13 different N-heterocycle classes (e.g., 46 or 47) as well as sulfur- (48) and phosphorus-based nucleophiles could be successfully coupled under this protocol. Efficient late-stage functionalization of drug candidates and the synthesis of bioisosteres led to the discovery of a leflunomide derivative with significantly improved metabolic stability.
Decarboxylative C(sp2)–X Bond-Formation Reactions
Aryl carboxylic acids are attractive precursors for functionalization because they are abundant, structurally diverse, and readily accessible from feedstock chemicals such as tolyl groups and esters. In fact, over the past century, these motifs have been harnessed to achieve the site-specific installation of various functionalities via decarboxylative functionalization, often through thermal activation or the use of metal mediators.54 However, the generality of such strategies is limited by difficulties associated with aryl carboxylic acid decarboxylation and the need for reagents that often inhibit efficient functionalization due to their high reactivity. Furthermore, aryl carboxylic acids are less susceptible than their alkyl counterparts to decarboxylative functionalization through photocatalytic manifolds that proceed through intermolecular quenching. Several factors contribute to this challenge: (1) rapid back-electron transfer of the aroyloxy radical can occur with a photocatalyst and (2) undesired reactions (such as hydrogen-atom transfer) can occur at faster rates than decarboxylation (k ≈ 104 to 106 M−1 s−1).
In light of these challenges, we recognized that a new manifold would be needed to advance this arena of research. In contrast to photocatalytic methods that activate carboxylate substrates through bimolecular collisional quenching, ligandto-metal charge transfer (LMCT) can enable the formation of open-shell species via direct light-induced excitation of a metal–substrate complex. We envisioned that an aryl carboxylate–metal complex (50) could, upon irradiation and LMCT, undergo single electron transfer from the aryl carboxylate to the metal center to form an open-shell O-centered carboxyl radical (51), which would subsequently lose CO2 to generate an aryl radical intermediate (49). Indeed, following an evaluation of potential first-row transition metals for LMCT reactivity with aryl carboxylates, we found that the decarboxylative functionalization of aryl carboxylic acids could be achieved using a combination of catalytic Cu(I) and an oxidant in acetonitrile, forming the parent arene via hydrodecarboxylation upon irradiation with 365 nm light. Furthermore, utilizing deuterated acetonitrile as a solvent resulted in deuterium atom transfer when the reaction was conducted in the absence of a halogenation reagent. These initial results not only provided support for ligand-to-copper charge transfer (LCCT)-induced decarboxylation but also supported the generation of a key aryl radical intermediate that could be intercepted using various strategies. We anticipated that subsequent functionalization could occur via one of two pathways: (1) atom or group transfer via radical trapping reagents or (2) aryl radical capture by copper followed by copper-mediated bond formation.
With these results in hand, we leveraged this design strategy to access a unified general platform for the decarboxylative functionalization of (hetero)aryl carboxylic acids, including decarboxylative halogenations to access any desired aryl halide as well as borylation, C(sp2)–O bond formation, and hydrodecarboxylation (Figure 10). Adoption of this overarching strategy enabled us to develop a robust, general approach to decarboxylative functionalization that is unified by a common set of general conditions (copper source, oxidant, solvent, light source, etc.) yet capable of engaging a key (hetero)aryl radical intermediate via diverse functionalization pathways.
Figure 10.
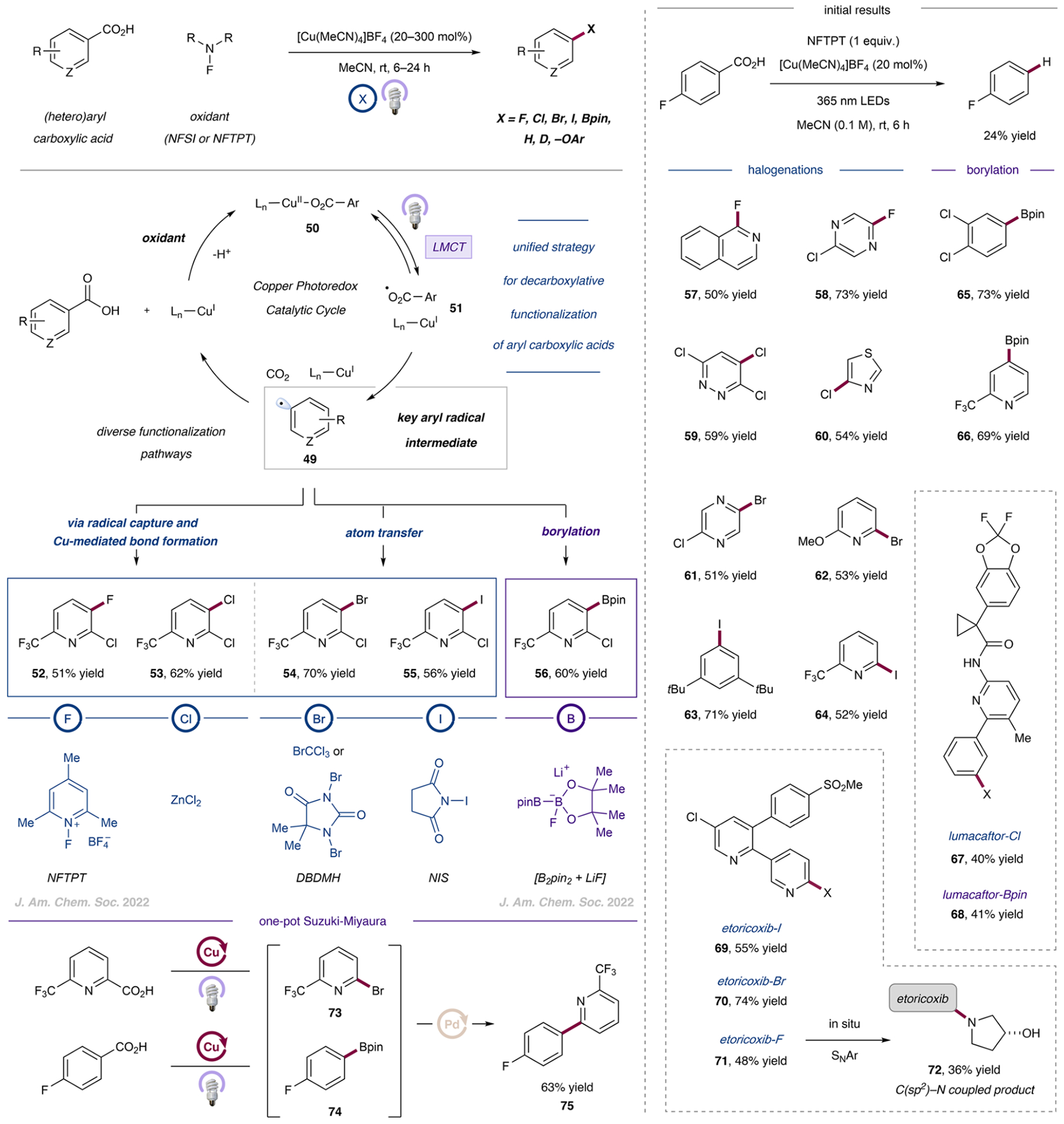
General platform for the decarboxylative halogenation of aryl carboxylic acids via copper-to-ligand charge transfer. Data are from refs 55 and 56.
Indeed, both electrophilic halogenation/atom-transfer reagents (cf. 54 to 55)55 and the group transfer reagent, B2pin2 (cf. 56),56 could be employed to access electrophilic aryl halide and nucleophilic aryl boronic ester coupling partners. Using electrophilic halogenation reagents (i.e., 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) or N-iodosuccinimide (NIS)), we achieved the decarboxylative bromination and iodination of electronically and substitutionally diverse (hetero)aryl substrates (e.g., 61 to 64). Similarly, the decarboxylative borylation protocol delivered aryl boronic esters in good yields (e.g., 65 to 66), using B2pin2 as the borylation reagent with N-fluorobenzenesulfonimide (NFSI) as the optimal oxidant; in situ activation of the diboronate was achieved with LiF (itself generated from NaF and LiClO4). In contrast, for (hetero)aryl halides with stronger C(sp2)–X bonds, such as chloro- or fluoroarenes, atom transfer using the respective electrophilic halogenation reagents was insufficiently rapid to prevent the competitive formation of side products. In these more challenging cases, we achieved the desired decarboxylative halogenations via LCCT-induced decarboxylation, followed by aryl radical capture via copper and subsequent copper-mediated C(sp2)–Cl or C(sp2)–F bond formation. Fluorodecarboxylation was achieved for diverse (hetero)aryl substrates (e.g., 57 to 58) under ambient conditions using the combination of stoichiometric copper and 1-fluoro-2,4,6-trimethylpyridinium tetrafluoroborate (NFTPT) as both the oxidant and fluoride source. Chlorodecarboxylation was demonstrated with a broad (hetero)aryl chloride scope (e.g., 59 to 60) using ZnCl2 as the chloride source. The formation of the chloroarene products putatively proceeds through ligand exchange on copper and subsequent copper-facilitated C(sp2)–Cl bond formation from the in situ-generated copper-(aryl)(chloride) complex.
To demonstrate the broad functional group tolerance and generality of this platform, we employed aryl carboxylic acids as modular functional handles in complex bioactive molecules. The decarboxylative halogenation and borylation of lumacaftor (67 and 68, respectively) were achieved in useful yields. Furthermore, we envisioned that a formal two-step demethy-lative functionalization could be achieved through sequential tolyl group oxidation and decarboxylative functionalization via Cu–LMCT. Indeed, we converted etoricoxib to its respective halogenated analogues—iodo-, bromo-, and fluoro-etoricoxib (69 to 71)—in moderate to good yields. The fluorinated analogue could be further reacted under in situ SNAr conditions to obtained complex heteroatom-coupled (e.g., C(sp2)–N, C(sp2)–O, or C(sp2)–S) products, such as 72.
Finally, we demonstrated that a direct one-pot Suzuki-Miyaura cross-coupling could be achieved using two different (hetero)aryl carboxylic acids that were first converted to the respective aryl bromide (73) and aryl boronate (74). Combining the resulting crude reaction mixtures via a telescoping procedure and utilizing palladium cross-coupling catalysis conditions afforded the one-pot Suzuki-Miyaura cross-coupling product 75 in 63% isolated yield.
Recent reports by the Rovis,57 Yoon,58 and Ritter groups have also provided support that ligand-to-metal charge-transfer activation is a fruitful approach to generating reactive open-shell intermediates. Ritter and co-workers have described protocols for the fluorodecarboxylation of aryl carboxylic acids via copper(II)–LMCT as well as the formation of the homocoupled benzoic ester product to access phenols after in situ hydrolysis.59,60
In summary, our novel approach to photocatalytic copper-catalyzed decarboxylative C(sp2)–X bond formation allows facile access to a diversity of fundamentally useful arene products and has been efficiently applied in the setting of complex, pharmaceutically relevant molecules. We envision that this unified strategy will provide a valuable synthetic toolbox for academic and industrial chemists alike, and we are working to expand this platform to include additional useful transformations.
CONCLUDING REMARKS
The research highlighted in this Accounts was conducted within the last eight years and represents a paradigm shift in modern retrosynthetic analysis. The merger of photoredox-mediated decarboxylative radical formation and metal-mediated bond formation has provided a platform upon which a variety of novel bond formations have been achieved from widely available starting materials. We hope that practitioners of synthetic organic chemistry can now view aliphatic carboxylic acids as modular and enabling functional handles, producing fragments rich in C(sp3)-hybridized carbon centers. In addition, aryl carboxylic acids, widely available and readily obtainable from feedstock chemicals, such as those including tolyl or ester moieties, can now be harnessed through a ligand-to-copper charge-transfer manifold to generate a spectrum of functional handles of broad utility and differing polarity. We expect that the continued adoption of these methods will streamline innovative applications in chemical biology and drug discovery.
We aim to inspire and motivate synthetic organic chemists to tackle underexplored reactions, such as the (i) fluorination, (ii) methylation, and (iii) hydroxylation of aliphatic carboxylic acids as well as the decarboxylative C(sp2)–N or C(sp2)– C(sp3) cross-coupling of aryl carboxylic acids, and to expand the platform of metallaphotoredox-catalyzed decarboxylation.
Finally, we anticipate that the approach outlined in our initial study of reaction generality in decarboxylative arylations will be adapted for other transformations, thus enabling more reliable and broadly applicable synthetic procedures to become accessible in the future.
ACKNOWLEDGMENTS
The authors are grateful for financial support provided by the National Institute of General Medical Sciences (NIGMS), the NIH (under award R35GM134897), the Princeton Catalysis Initiative, and kind gifts from Merck, Janssen, BMS, Genentech, Celgene, and Pfizer. S.B.B. acknowledges the Leopoldina National Academy of Sciences for a postdoctoral fellowship. N.E.I. thanks Princeton University, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. S.B.B. is grateful to Prof. Dr. Max von Delius for generously providing laboratory space during the Covid-19 pandemic.
Biography
Sebastian B. Beil was born and raised in Kiel, Germany and obtained his M.Sc. in chemistry from the Christian Albrechts University of Kiel in 2015. As a Ph.D. student, he worked in the laboratories of Siegfried Waldvogel in Mainz and Phil Baran at Scripps Research in La Jolla. His research focus was new avenues in organic electro-oxidations and -reductions. After graduation in 2019, he was a postdoctoral fellow with Max von Delius in Ulm and joined the MacMillan laboratory as a postdoctoral fellow supported by the Leopoldina National Academy of Sciences in 2020, where he explored new avenues in decarboxylative transformations. His independent career started at the Stratingh Institute for Chemistry at the University of Groningen in 2021.
Tiffany Q. Chen grew up in southern California and obtained an A.B. in anthropology from Harvard University. In 2013, she joined the laboratory of K. N. Houk at UCLA to conduct research on transannular and [6 + 4] cycloadditions. She obtained a B.S. in chemistry from UC Berkeley in 2016, where she worked on trifluoromethyl gold complexes in the group of F. Dean Toste. She began doctoral studies in 2016 at Princeton with David MacMillan, where she developed metallaphotocatalytic methods for cross-coupling and aromatic decarboxylative functionalization. After completing her Ph.D., she joined the laboratory of Timothy Swager at MIT in 2021 as a postdoctoral fellow to explore the interface of chemistry and materials science.
Nicholas E. Intermaggio is originally from New Haven, CT but moved to the suburbs of Philadelphia early in high school. He completed his undergraduate degree at Boston University in 2016, graduating with a B.A. in chemistry. While at BU, Nick conducted undergraduate research in John A. Porco Jr.’s laboratory, where he worked on the total synthesis of polycyclic xanthone natural products. After graduation, he joined the research and development department at E Ink Corporation in Billerica, MA. In 2017, Nick joined the medicinal chemistry department at Amgen Inc. in Cambridge, MA. After 2 years at Amgen, Nick joined the MacMillan group at Princeton University to conduct research in novel reaction development.
David W. C. MacMillan - was born in Bellshill, Scotland and received his undergraduate degree in chemistry at the University of Glasgow, where he worked with Dr. Ernie Colvin. In 1990, he began his doctoral studies under the direction of Professor Larry Overman at the University of California, Irvine, before undertaking a postdoctoral position with Professor Dave Evans at Harvard University (1996). He began his independent career at the University of California, Berkeley, in July of 1998 before moving to Caltech in June of 2000 (Earle C. Anthony Chair of Organic Chemistry). In 2006, Dave moved to the East Coast of the U.S. to take up the position of the James S. McDonnell Distinguished University Professor at Princeton University, and he served as department chair from 2010 to 2015. Dave’s research interests encompass a wide range of organic chemistry, including the development of new areas in organocatalysis and photoredox catalysis.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.2c00607
The authors declare no competing financial interest.
Contributor Information
Sebastian B. Beil, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States; Present Address: Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands;
Tiffany Q. Chen, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States; Present Address: Department of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Boston, Massachusetts 02139, United States.
Nicholas E. Intermaggio, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States;
REFERENCES
- (1).Johnston CP; Smith RT; Allmendinger S; MacMillan DWC Metallaphotoredox-Catalysed Sp3–Sp3 Cross-Coupling of Carboxylic Acids with Alkyl Halides. Nature 2016, 536, 322–325. [DOI] [PMC free article] [PubMed] [Google Scholar]; The transition-metal-catalyzed bond formation of two C(sp3) centers was previously unknown and was achieved by the independent activation of carboxylic acids and alkyl halides.
- (2).Liang Y; Zhang X; MacMillan DWC Decarboxylative Sp3 C-N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]; N-Alkylation usually proceeds from the alkyl bromide and requires harsh reaction conditions. Even more challenging is the introduction of secondary and tertiary alkyl derivatives, which became possible by the combination of copper catalysis and a straightforward decarboxylation strategy.
- (3).Zhang X; Smith RT; Le C; McCarver SJ; Shireman BT; Carruthers NI; MacMillan DWC Copper-Mediated Synthesis of Drug-like Bicyclopentanes. Nature 2020, 580, 220–226. [DOI] [PMC free article] [PubMed] [Google Scholar]; The pharmacologic relevance of BCP derivatives is well-appreciated, yet efficient and modular syntheses are scarce. This work describes the difunctionalization of BCP derivatives with diverse substituents.
- (4).Prieto Kullmer CN; Kautzky JA; Krska SW; Nowak T; Dreher SD; MacMillan DWC Accelerating Reaction Generality and Mechanistic Insight through Additive Mapping. Science 2022, 376, 532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]; A broader mechanistic understanding of the seminal metallaphotoredox decarboxylative arylation from 2014 led to the development of a broadly applicable procedure with improved performance. The identification of suitable additives, such as phthalimide, was the key to this generality.
- (5).Li T; Huo L; Pulley C; Liu A Decarboxylation Mechanisms in Biological System. Bioorganic Chem 2012, 43, 2–14. [DOI] [PubMed] [Google Scholar]
- (6).Rodríguez N; Goossen LJ Decarboxylative Coupling Reactions: A Modern Strategy for C-C-Bond Formation. Chem. Soc. Rev 2011, 40, 5030–5048. [DOI] [PubMed] [Google Scholar]
- (7).Wei Y; Hu P; Zhang M; Su W Metal-Catalyzed Decarboxylative C-H Functionalization. Chem. Rev 2017, 117, 8864–8907. [DOI] [PubMed] [Google Scholar]
- (8).Weaver JD; Recio A; Grenning AJ; Tunge JA Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem. Rev 2011, 111, 1846–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Romero NA; Nicewicz DA Organic Photoredox Catalysis. Chem. Rev 2016, 116, 10075–10166. [DOI] [PubMed] [Google Scholar]
- (10).Tucker JW; Stephenson CRJ Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. J. Org. Chem 2012, 77, 1617–1622. [DOI] [PubMed] [Google Scholar]
- (11).Chan AY; Perry IB; Bissonnette NB; Buksh BF; Edwards GA; Frye LI; Garry OL; Lavagnino MN; Li BX; Liang Y; Mao E; Millet A; Oakley JV; Reed NL; Sakai HA; Seath CP; MacMillan DWC Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev 2022, 122, 1485–1542. [DOI] [PubMed] [Google Scholar]
- (12).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (13).Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Twilton J; Le C; Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nat. Rev. Chem 2017, 1, 0052. [Google Scholar]
- (15).Nicewicz DA; MacMillan DWC Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science 2008, 322, 77–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McNally A; Prier CK; MacMillan DWC Discovery of an α-Amino C-H Arylation Reaction Using the Strategy of Accelerated Serendipity. Science 2011, 334, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pirnot MT; Rankic DA; Martin DBC; MacMillan DWC Photoredox Activation for the Direct -Arylation of Ketones and Aldehydes. Science 2013, 339, 1593–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl Sp3-Carbons with Aryl Halides. Science 2014, 345, 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hilborn JW; Pincock JA Rates of Decarboxylation of Acyloxy Radicals Formed in the Photocleavage of Substituted 1-Naphthylmethyl Alkanoates. J. Am. Chem. Soc 1991, 113, 2683–2686. [Google Scholar]
- (20).Snider MJ; Wolfenden R The Rate of Spontaneous Decarboxylation of Amino Acids. J. Am. Chem. Soc 2000, 122, 11507–11508. [Google Scholar]
- (21).Diccianni JB; Diao T Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem 2019, 1, 830–844. [Google Scholar]
- (22).Oderinde MS; Varela-Alvarez A; Aquila B; Robbins DW; Johannes JW Effects of Molecular Oxygen, Solvent, and Light on Iridium-Photoredox/Nickel Dual-Catalyzed Cross-Coupling Reactions. J. Org. Chem 2015, 80, 7642–7651. [DOI] [PubMed] [Google Scholar]
- (23).Kolahdouzan K; Khalaf R; Grandner JM; Chen Y; Terrett JA; Huestis MP Dual Photoredox/Nickel-Catalyzed Conversion of Aryl Halides to Aryl Aminooxetanes: Computational Evidence for a Substrate-Dependent Switch in Mechanism. ACS Catal 2020, 10, 405–411. [Google Scholar]
- (24).Kanishchev OS; Dolbier WR Jr. Ni/Ir-Catalyzed Photoredox Decarboxylative Coupling of S-Substituted Thiolactic Acids with Heteroaryl Bromides: Short Synthesis of Sulfoxaflor and Its SF5 Analog. Chem.—Eur. J 2017, 23, 7677–7681. [DOI] [PubMed] [Google Scholar]
- (25).Ma Y; Liu S; Xi Y; Li H; Yang K; Cheng Z; Wang W; Zhang Y Highly Stereoselective Synthesis of Aryl/Heteroaryl- C -Nucleosides via the Merger of Photoredox and Nickel Catalysis. Chem. Commun 2019, 55, 14657–14660. [DOI] [PubMed] [Google Scholar]
- (26).Wang H; Liu C-F; Song Z; Yuan M; Ho YA; Gutierrez O; Koh MJ Engaging α;-Fluorocarboxylic Acids Directly in Decarboxylative C-C Bond Formation. ACS Catal. 2020, 10, 4451–4459. [Google Scholar]
- (27).Phelan JP; Lang SB; Sim J; Berritt S; Peat AJ; Billings K; Fan L; Molander GA Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc 2019, 141, 3723–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kölmel DK; Meng J; Tsai M-H; Que J; Loach RP; Knauber T; Wan J; Flanagan ME On-DNA Decarboxylative Arylation: Merging Photoredox with Nickel Catalysis in Water. ACS Comb. Sci 2019, 21, 588–597. [DOI] [PubMed] [Google Scholar]
- (29).Pezzetta C; Bonifazi D; Davidson RWM Enantioselective Synthesis of N -Benzylic Heterocycles: A Nickel and Photoredox Dual Catalysis Approach. Org. Lett 2019, 21, 8957–8961. [DOI] [PubMed] [Google Scholar]
- (30).Abdiaj I; Alcázar J Improving the Throughput of Batch Photochemical Reactions Using Flow: Dual Photoredox and Nickel Catalysis in Flow for C(Sp2) C(Sp3) Cross-Coupling. Bioorg. Med. Chem 2017, 25, 6190–6196. [DOI] [PubMed] [Google Scholar]
- (31).Hsieh H-W; Coley CW; Baumgartner LM; Jensen KF; Robinson RI Photoredox Iridium-Nickel Dual-Catalyzed Decarboxylative Arylation Cross-Coupling: From Batch to Continuous Flow via Self-Optimizing Segmented Flow Reactor. Org. Process Res. Dev 2018, 22, 542–550. [Google Scholar]
- (32).Zuo Z; Cong H; Li W; Choi J; Fu GC; MacMillan DWC Enantioselective Decarboxylative Arylation of α-Amino Acids via the Merger of Photoredox and Nickel Catalysis. J. Am. Chem. Soc 2016, 138, 1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Noble A; McCarver SJ; MacMillan DWC Merging Photoredox and Nickel Catalysis: Decarboxylative Cross-Coupling of Carboxylic Acids with Vinyl Halides. J. Am. Chem. Soc 2015, 137, 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Chatgilialoglu C; Crich D; Komatsu M; Ryu I Chemistry of Acyl Radicals. Chem. Rev 1999, 99, 1991–2070. [DOI] [PubMed] [Google Scholar]
- (35).Chu L; Lipshultz JM; MacMillan DWC Merging Photoredox and Nickel Catalysis: The Direct Synthesis of Ketones by the Decarboxylative Arylation of α-Oxo Acids. Angew. Chem., Int. Ed 2015, 54, 7929–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Cheng W-M; Shang R; Yu H-Z; Fu Y Room-Temperature Decarboxylative Couplings of α-Oxocarboxylates with Aryl Halides by Merging Photoredox with Palladium Catalysis. Chem.—Eur. J 2015, 21, 13191–13195. [DOI] [PubMed] [Google Scholar]
- (37).Zhou C; Li P; Zhu X; Wang L Merging Photoredox with Palladium Catalysis: Decarboxylative Ortho-Acylation of Acetanilides with α-Oxocarboxylic Acids under Mild Reaction Conditions. Org. Lett 2015, 17, 6198–6201. [DOI] [PubMed] [Google Scholar]
- (38).Xu N; Li P; Xie Z; Wang L Merging Visible-Light Photocatalysis and Palladium Catalysis for C-H Acylation of Azo- and Azoxybenzenes with α-Keto Acids. Chem.—Eur. J 2016, 22, 2236–2242. [DOI] [PubMed] [Google Scholar]
- (39).Le C; MacMillan DWC Fragment Couplings via CO2 Extrusion-Recombination: Expansion of a Classic Bond-Forming Strategy via Metallaphotoredox. J. Am. Chem. Soc 2015, 137, 11938–11941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Fessenden RW; Schuler RH Electron Spin Resonance Studies of Transient Alkyl Radicals. J. Chem. Phys 1963, 39, 2147–2195. [Google Scholar]
- (41).Huggins JM; Bergman RG Mechanism, Regiochemistry, and Stereochemistry of the Insertion Reaction of Alkynes with Methyl(2,4-Pentanedionato)(Triphenylphosphine)Nickel. A Cis Insertion That Leads to Trans Kinetic Products. J. Am. Chem. Soc 1981, 103, 3002–3011. [Google Scholar]
- (42).Till NA; Smith RT; MacMillan DWC Decarboxylative Hydroalkylation of Alkynes. J. Am. Chem. Soc 2018, 140, 5701–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Mastandrea MM; Cañellas S; Caldentey X; Pericás MA Decarboxylative Hydroalkylation of Alkynes via Dual Copper-Photoredox Catalysis. ACS Catal 2020, 10, 6402–6408. [Google Scholar]
- (44).Yue H; Zhu C; Kancherla R; Liu F; Rueping M Regioselective Hydroalkylation and Arylalkylation of Alkynes by Photoredox/Nickel Dual Catalysis: Application and Mechanism. Angew. Chem., Int. Ed 2020, 59, 5738–5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Terrett JA; Cuthbertson JD; Shurtleff VW; MacMillan DWC Switching on Elusive Organometallic Mechanisms with Photoredox Catalysis. Nature 2015, 524, 330–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kautzky JA; Wang T; Evans RW; MacMillan DWC Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 6522–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47). While redox-active esters are an orthogonal mode to reductively activate aliphatic carboxylic acid, they are beyond the scope of this Accounts and have been reviewed. See ref 48.
- (48).Murarka S N-(Acyloxy)Phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- (49).He Z; Bae M; Wu J; Jamison TF Synthesis of Highly Functionalized Polycyclic Quinoxaline Derivatives Using Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2014, 53, 14451–14455. [DOI] [PubMed] [Google Scholar]
- (50).Genovino J; Lian Y; Zhang Y; Hope TO; Juneau A; Gagné Y; Ingle G; Frenette M Metal-Free-Visible Light C-H Alkylation of Heteroaromatics via Hypervalent Iodine-Promoted Decarboxylation. Org. Lett 2018, 20, 3229–3232. [DOI] [PubMed] [Google Scholar]
- (51).Minisci F; Vismara E; Fontana F; Claudia Nogueira Barbosa M A New General Method of Homolytic Alkylation of Protonated Heteroaromatic Bases by Carboxylic Acids and Iodosobenzene Diacetate. Tetrahedron Lett 1989, 30, 4569–4572. [Google Scholar]
- (52).Zhao X; Liu Y; Zhu R; Liu C; Zhang D Mechanistic Study on the Decarboxylative Sp3 C-N Cross-Coupling between Alkyl Carboxylic Acids and Nitrogen Nucleophiles via Dual Copper and Photoredox Catalysis. Inorg. Chem 2019, 58, 12669–12677. [DOI] [PubMed] [Google Scholar]
- (53).Mykhailiuk PK Saturated Bioisosteres of Benzene: Where to Go Next? Org. Biomol. Chem 2019, 17, 2839–2849. [DOI] [PubMed] [Google Scholar]
- (54).Goossen LJ; Deng G; Levy LM Synthesis of Biaryls via Catalytic Decarboxylative Coupling. Science 2006, 313, 662–664. [DOI] [PubMed] [Google Scholar]
- (55).Chen TQ; Pedersen PS; Dow NW; Fayad R; Hauke CE; Rosko MC; Danilov EO; Blakemore DC; Dechert-Schmitt A-M; Knauber T; Castellano FN; MacMillan DWC A Unified Approach to Decarboxylative Halogenation of (Hetero)Aryl Carboxylic Acids. J. Am. Chem. Soc 2022, 144, 8296–8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Dow NW; Pedersen PS; Chen TQ; Blakemore DC; Dechert-Schmitt A-M; Knauber T; MacMillan DWC Decarboxylative Borylation and Cross-Coupling of (Hetero)Aryl Acids Enabled by Copper Charge Transfer Catalysis. J. Am. Chem. Soc 2022, 144, 6163–6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Treacy SM; Rovis T Copper Catalyzed C(Sp 3)-H Bond Alkylation via Photoinduced Ligand-to-Metal Charge Transfer. J. Am. Chem. Soc 2021, 143, 2729–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Li QY; Gockel SN; Lutovsky GA; DeGlopper KS; Baldwin NJ; Bundesmann MW; Tucker JW; Bagley SW; Yoon TP Decarboxylative Cross-Nucleophile Coupling via Ligandto-Metal Charge Transfer Photoexcitation of Cu(Ii) Carboxylates. Nat. Chem 2022, 14, 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Xu P; López-Rojas P; Ritter T Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination. J. Am. Chem. Soc 2021, 143, 5349–5354. [DOI] [PubMed] [Google Scholar]
- (60).Su W; Xu P; Ritter T Decarboxylative Hydroxylation of Benzoic Acids. Angew. Chem., Int. Ed 2021, 60, 24012–24017. [DOI] [PMC free article] [PubMed] [Google Scholar]