

Abstract
The replacement of aryl rings with saturated carbocyclic structures has garnered significant interest in drug discovery due to the potential for improved pharmacokinetic properties upon substitution. In particular, 1,3-difunctionalized bicyclo[1.1.1]pentanes (BCPs) have been widely adopted as bioisosteres for parasubstituted arene rings, appearing in a number of lead pharmaceutical candidates. However, despite the pharmaceutical value of 2-substituted BCPs as replacements for ortho- or meta-substituted arene rings, general and rapid syntheses of these scaffolds remain elusive. Current approaches to 2-substituted BCPs rely on installation of the bridge substituent prior to BCP core construction, leading to lengthy step counts and often nonmodular sequences. While challenging, direct functionalization of the strong bridge BCP C–H bonds would offer a more streamlined pathway to diverse 2-substituted BCPs. Here, we report a generalizable synthetic linchpin strategy for bridge functionalization via radical C–H abstraction of the BCP core. Through mild generation of a strong hydrogen atom abstractor, we rapidly synthesize novel 2-substituted BCP synthetic linchpins in one pot. These synthetic linchpins then serve as common precursors to complex 2-substituted BCPs, allowing one-step access to a number of previously inaccessible electrophile and nucleophile fragments at the 2-position via two new metallaphotoredox protocols. Altogether, this platform enables the expedient synthesis of four pharmaceutical analogues, all of which show similar or improved properties compared to their aryl-containing equivalents, demonstrating the potential of these 2-substituted BCPs in drug development.
Graphical Abstract
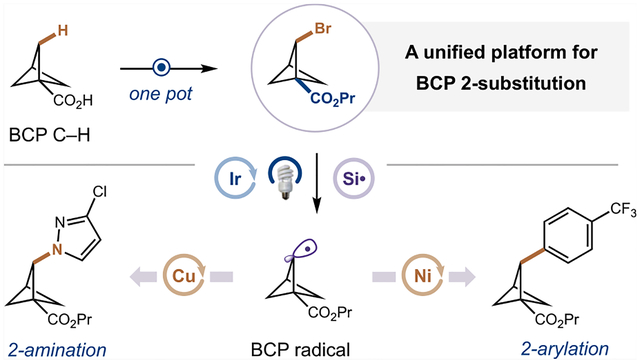
INTRODUCTION
The substitution of arene rings with BCPs in pharmaceutical candidates often leads to improved properties, including increased solubility, greater metabolic stability, and decreased nonspecific binding.1–6 Given these advantages and the synthetic accessibility of bridgehead-substituted BCP cores (i.e., BCPs substituted at the 1(,3)-position) from [1.1.1]propellane,7–11 1,3-disubstituted BCPs have been investigated in a number of lead candidates as replacements for para-substituted aryl rings, appearing in over 700 patents to date (Figure 1A).12,13 However, despite the wide adoption of 1(,3)-substituted BCPs in medicinal chemistry programs, the corresponding 2-substituted BCPs are virtually nonexistent in the patent literature. While there is substantial interest in synthesizing these bridge-substituted BCPs (i.e., BCPs substituted at the 2-position) as a means to access novel chemical space and replace ortho- or metasubstituted arene rings in bioactive molecules,5,14,15 no methods exist that allows the direct preparation of complex 2-substituted BCP scaffolds. Although current approaches have enabled the introduction of some drug-like substituents at the bridge position, they suffer from long step counts and often lack modularity as they require preinstallation of the desired 2-substituents prior to BCP core construction.16–19 Conversely, direct functionalization of the bridge BCP C–H bonds would dramatically streamline the synthesis of these 2-substituted BCP scaffolds, enabling rapid library generation of these sought-after structures for drug development. While appealing, BCP C–H activation represents a significant synthetic challenge due to the high BDEs (bond dissociation energies) of the methylene C–H bonds (estimated BDE ~106 kcal mol−1),20 meaning no general methods exist to convert these strong bonds to useful functional handles in good efficiency.21,22 Furthermore, selectivity for the monofunctionalized bridge BCP is difficult, given that past BCP C–H functionalization strategies all favor the difunctionalization or bridgehead-substituted product over the desired bridge-functionalized BCP.22–25 A third challenge to modular bridge functionalization is the lack of cross-coupling procedures that can elaborate 2-substituted BCPs, which are hindered by the high s character of the bridge C–X bonds, and corresponding intermediates, compared to typical alkyl systems (hybridization of bridge C–H ~ sp2.5).26 To overcome these long-standing issues, we hypothesized that a programmable platform for 2-substituted BCP synthesis could be achieved through radical-mediated C–H functionalization and metallaphotoredox cross coupling (Figure 1B). First, we would directly access linchpin 2-monosubstituted BCPs by selective radical abstraction of the strong bridge BCP C–H bonds under mild visible-light conditions to minimize multifunctionalization and/or ring-opened products. Importantly, these intermediates would have orthogonal functional handles at the bridgehead and bridge positions, enabling modular elaboration. Following the synthesis of these linchpin BCPs, we hypothesized that new metallaphotoredox cross-coupling procedures could transform our common intermediates to an unprecedented array of drug-like 2-substituted BCP scaffolds in only one step (Figure 1C). Overall, this strategy would considerably expand the number of obtainable bridge-substituted BCPs, allowing their wide application in the pharmaceutical sector across various therapeutic areas.
Figure 1.
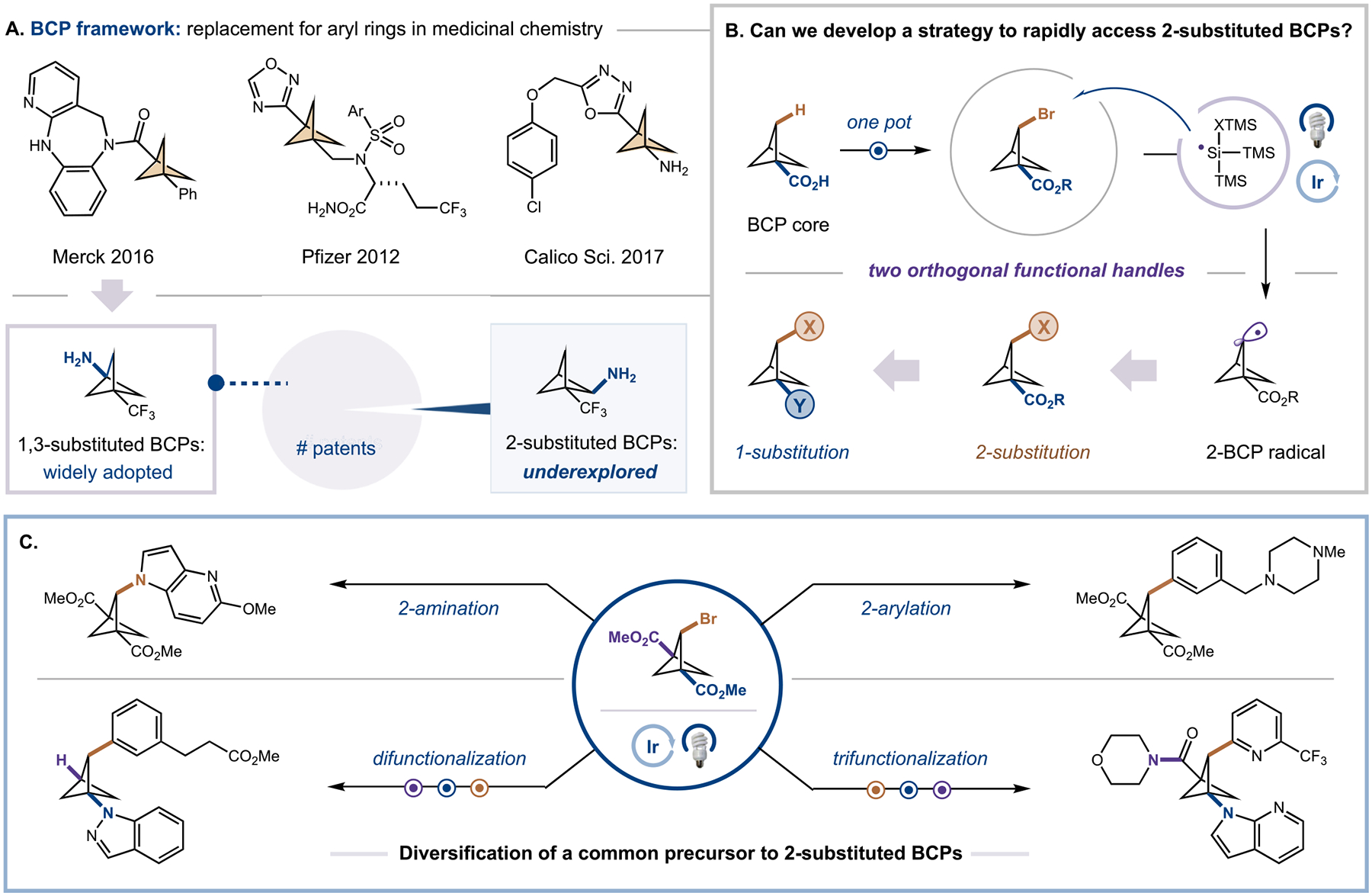
Rapid synthesis of 2-substituted bicyclo[1.1.1]pentanes. (A) Examples of the BCP framework in pharmaceutical compounds. Relative number of patents containing 1,3- and 1,2-disubstituted BCPs. (B) One-pot C–H functionalization to access a key disubstituted BCP intermediate with orthogonal functional handles. Functionalization of this intermediate is done via photoredox catalysis to access diverse 1,2-disubstituted BCPs. (C) Photoredox catalysis enables diversification of a common 1,2,3-trisubstituted BCP intermediate to 2-aminated, 2-arylated, difunctionalized, and trifunctionalized BCPs.
RESULTS AND DISCUSSION
To enable installation of diverse functionality at the bridge and bridgehead positions of the BCP scaffold, we targeted a previously unreported 1(,3)-carboxylated, 2-brominated BCP. To access this intermediate, we hypothesized that a hydrogen atom transfer (HAT) mechanism could selectively activate the strong bridge C–H bonds of the commercially available mono- and di-substituted carboxylated BCPs, leading to brominated products in one step (Figure 2A). Within the field of radical-mediated HAT, a range of regioselective strong C–H bond functionalizations have been developed using polarity-matched hydrogen-atom abstraction by electrophilic radicals, such as quinuclidinium or halogen radicals, followed by radical trapping by an appropriate radical acceptor or metal catalyst.27 Considering the thermodynamic dependence of HAT rates and the high BDE of H–Cl (103 kcal mol−1),28 we postulated that chlorine radicals could be suitable for accomplishing this difficult BCP bridge C–H abstraction. In particular, we were inspired by studies demonstrating that chlorine gas could activate the strong methylene C–H bonds of BCPs to generate di- and trisubstituted chlorinated products in moderate yields under photochemical conditions.23–25 In our proposed monobromination protocol, we would generate high-energy radicals by photolysis of N-chlorosuccinimide (NCS) under visible-light conditions, which would then abstract the BCP bridge C–H bonds of 1 leading to a BCP bridge radical 2. This radical would then abstract a bromine atom from a brominating agent to give our desired 2-brominated BCP 3. Overall, we speculated that these milder conditions would favor bridge monofunctionalization over multifunctionalization and/or ring-opened products which are traditionally favored in BCP C–H bridge functionalization protocols.14
Figure 2.
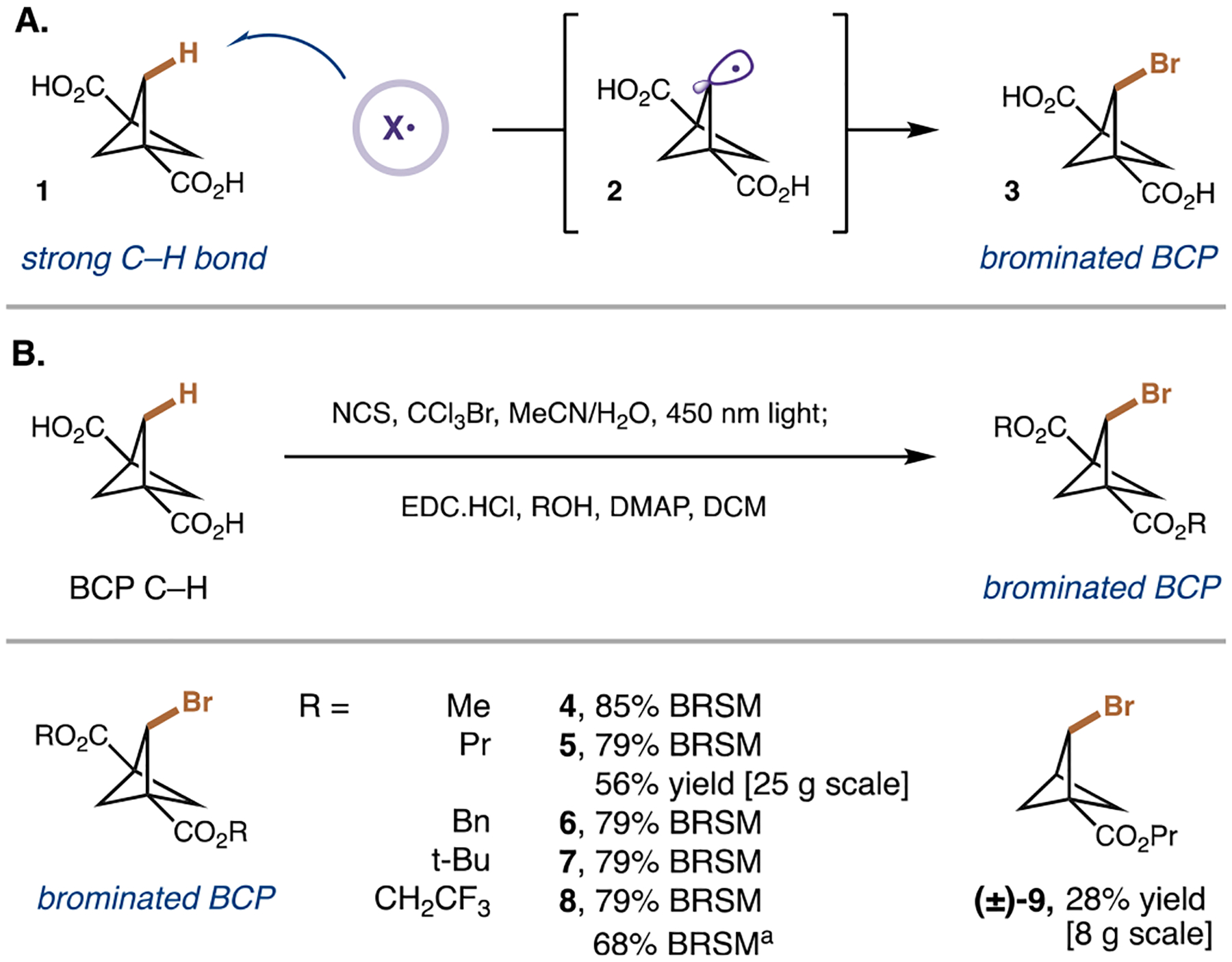
Synthesis of C–H bridge-brominated BCP intermediates. (A) Proposed mechanism for bromination. (B) A range of 2-brominated BCPs can be synthesized. All yields are isolated. Standard conditions: BCP acid (1 equiv.) NCS (1.1 equiv), CCl3Br (5.0 equiv), MeCN/H2O; EDC·HCl (6 equiv), ROH (10 equiv), DMAP (4 equiv), DCM; NaOH, THF/H2O. See the Supporting Information for full experimental details. BRSM, based on recovered starting material; DMAP, 4-dimethylaminopyridine; EDC, N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide; NCS, N-chlorosuccinimide.
We first examined this bromination protocol with the commercially available 1,3-dicarboxylic acid BCP 1. Pleasingly, exposure of 1,3-dicarboxylic acid BCP and NCS to visible-light (450 nm) conditions, in the presence of brominating agent bromotrichloromethane, led to the formation of 2-brominated-1,3-dicarboxylic acid BCP 3 in moderate yields (Tables S1–S7). Control reactions omitting NCS, light, or brominating agent proved that each component was necessary for efficient bromination (Table S5). Noting our desired use of these brominated BCPs in cross-coupling reactions and sequential functionalization sequences, we next targeted a one-pot bromination/esterification sequence to place the linchpin functionality at the bridgehead positions that would be tolerated in further metal-catalyzed procedures (Figure 2B). The desired esterification could be achieved following bromination through a solvent swap with the desired alcohol and addition of EDC and DMAP as the activator and the base, respectively, to yield a range of BCP esters in synthetically useful yields (4–8). Flow conditions enabled excellent yield of the brominated BCP 4 on a 25 g scale, meaning this synthetic linchpin can be rapidly accessed for library generation. To avoid volatility, the monosubstituted carboxylic acid BCP was esterified with propanol, yielding building block 9 in a decent yield on an 8 g scale.
Given the ubiquity of aryl groups and the amine functionality in pharmaceuticals, we next targeted 2-amination and 2-arylation reactions on our newly synthesized brominated BCP intermediates. To the best of our knowledge, there are presently no cross-coupling methods to aminate the bridge position of BCPs; consequently, very few 2-heteroamine BCPs have been synthesized to date. Furthermore, current cross-coupling methods to arylate the 2-position of BCPs are limited in arene scope and do not couple nitrogen-containing heterocycles,16 which are present in the majority of approved drug molecules.
To activate the 2-bromine handle of our BCPs, we aimed to use silyl-radical chemistry developed by our group over the last several years (see Figure 1B).29–32 In 2016, our laboratory established that alkyl halides could be activated by photo-catalytically generated silyl radicals to produce the corresponding alkyl radical under visible-light conditions.29 These open shell intermediates have been engaged in a number of copper and nickel cross-coupling pathways, yielding a number of valuable transformations, such as C(sp3)-arylation, -alkylation, -trifluoromethylation, and -amination.29–32 Using these silyl-radical-mediated technologies, we anticipated that under photocatalytic conditions we could form BCP radicals at the bridge position from our 2-brominated BCPs, which could engage in subsequent cross-coupling with an aryl bromide or amine via a nickel or copper metal catalyst, respectively.
From the outset, we recognized that a fundamental challenge to high product yield would be the high s character of the proposed BCP radical intermediate (see Figure 1B).20,26 In contrast to typical alkyl radicals, we anticipated that the more electrophilic character of this BCP radical and the high BDE of the corresponding bridge C–H bonds would likely favor the protodehalogenation product over the desired cross-coupling product. Indeed, initial attempts at silyl-radical-mediated C(sp3) arylation29 and amination32 using our previously published conditions gave only a minimal yield (<10%) of the desired cross-coupling product, instead leading to solvent-coupled products, dimer formation, and/or protodehalogenated BCP.26 To prevent this byproduct formation, we found that solvents lacking weaker, hydridic C–H bonds, combined with rigorous control of the reaction temperature and slow addition of aryl bromide enabled efficient formation of the arylated product (see the Supporting Information for more details). Furthermore, use of a different silane source, 1-adamantyl aminosilane,33 also an efficient quencher of photocatalyst Ir(dF(CF3)ppy)2(dtbpy)PF6,32 improved the product yield. Conversely, for the amination reaction, use of more electron-rich bipyridine (bipy) ligands, a copper(I) source, and organic bases, such as DBN, led to high yields of 2-aminated BCP in preference to protodehalogenated BCP and detrimental BCP hydrolysis side products (see the Supporting Information for more details).
Following optimization, we turned to evaluate the scope of the arylation reaction for a range of BCP bromides and aryl bromide coupling partners (Figure 3). Pleasingly, both disubstituted and monosubstituted BCP bromides could be successfully coupled (10–15). A wide range of aryl bromides were also found to serve as productive coupling partners: para-substituted, electron-deficient, and electron-rich substituents could be coupled in excellent yields (16–21). Furthermore, ortho-, meta-, and parasubstituents on the aryl ring were amenable to our reaction conditions (16, 22, and 23). The conditions were applicable to a range of potentially sensitive functional groups and useful synthetic handles, such as tertiary amines (24), carbamates (20 and 25), nitriles (26), and aryl chlorides (19 and 27). Notably, acidic functionalities such as NH bonds, which are commonly problematic in transition-metal-catalyzed reactions,15 could also be accommodated in this reaction (25).
Figure 3.
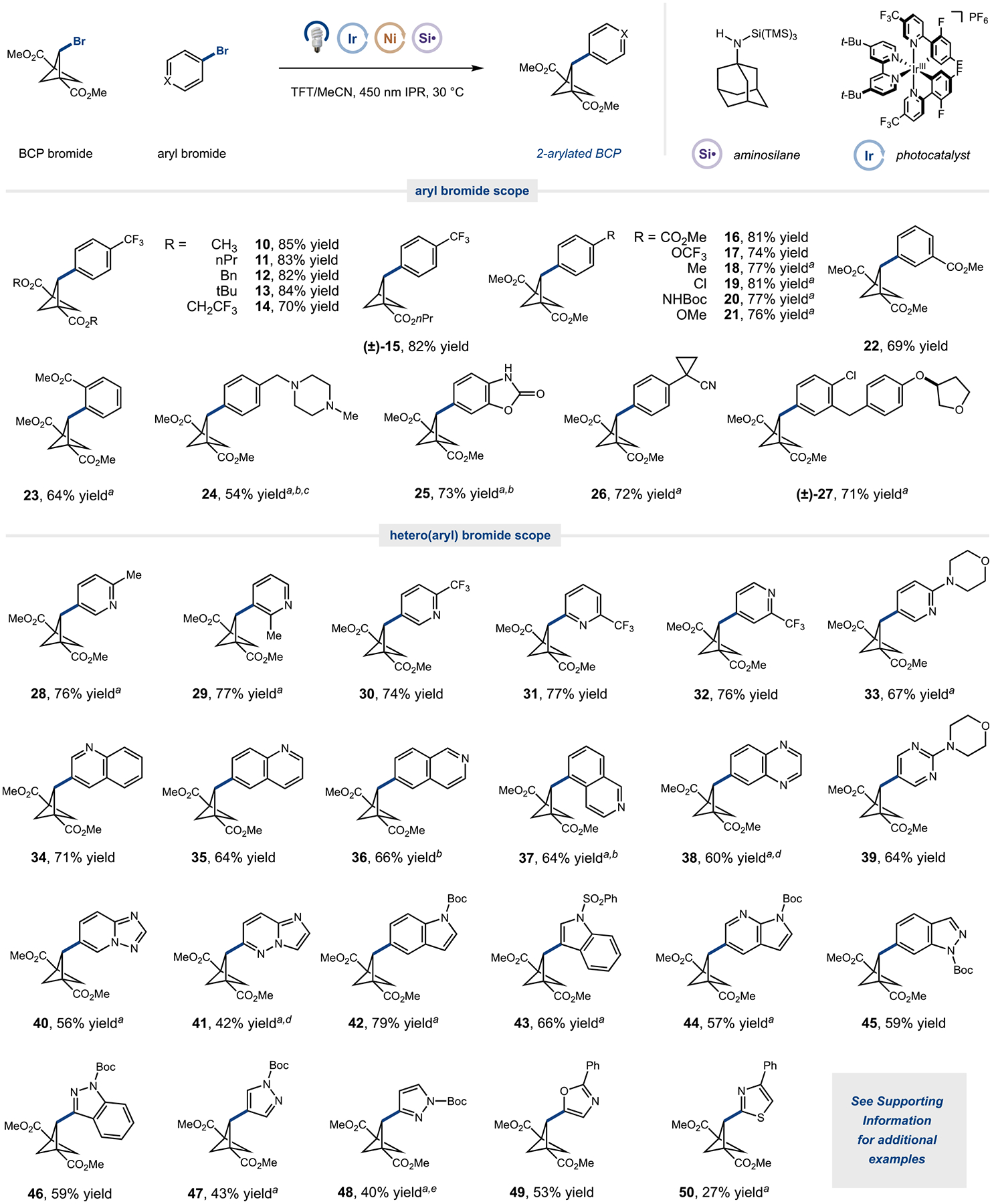
Scope for silyl-radical-mediated BCP 2-arylation. A range of hetero(aryl) bromides can be used as aryl coupling partners. All yields are isolated. Standard conditions: aryl bromide (0.5 mmol, 1 equiv), BCP bromide (2 equiv), aminosilane (1.6 equiv), Ir(dF(CF3)ppy)2(dtbbpy)PF6 (2 mol %), Ni(dtbbpy)Br2 (5 mol %), Cs2CO3 (2 equiv), TFT (0.05 M), IPR 450 nm (100% intensity) for 2 h. See the Supporting Information for full experimental details. a10 mol % Ni(dtbbpy)Br2. bReaction performed in TFT/tBuOH (v/v = 3/1). cIsolated as the dihydrochloride. dReaction performed in TFT/tBuOH (v/v = 1/1). eIrradiation for 240 min. Boc, tert-butyloxycarbonyl; Bn, benzyl; IPR, integrated photoreactor; TFT, trifluorotoluene; TMS, trimethylsilane.
Given the prevalence of heteroaryl moieties in pharmaceutical molecules, we were pleased to find that a number of heteroaryl bromides could also be successfully coupled in this reaction. A range of 2-, 3-, and 4-pyridyl bromides bearing electronically diverse substituents all served as competent coupling partners (28–33). Heteroaryl bromides with extended pi systems, such as quinolines and quinoxalines, performed well under these reaction conditions (34–38). Pleasingly, excellent yields were still observed for heterocyclic systems containing multiple nitrogen atoms, including pyrazines (39), triazolopyridines (40), and imidazopyridazines (41). Furthermore, traditional heterocycles such as indoles (42 and 43), azaindoles (44), and indazoles (45 and 46) were all well tolerated in this reaction. Notably, a number of five-membered ring heteroaryl bromides, compounds that are notoriously challenging for transition-metal cross-coupling procedures, were also effective substrates albeit in modest yields (47–50).
Our optimized amination conditions also proved highly general: over 10 classes of amines were capable nucleophiles in our procedure (Figure 4). These results are particularly notable, given that these bridge-substituted heteroamines have been scarcely synthesized. A variety of indazoles (51–53), pyrazoles (54–56), and azaindazoles (57 and 58) were coupled in excellent yields and regioselectivities, with reaction only at the designated nitrogen site. Amines with only one reactive nitrogen site, such as azaindoles (59–61), benzophenone imine (62), carbazoles (63–65), azacarbazole (66), pyrroles (67 and 68), and indoles (69 and 70) were all competent reaction partners. Notably, a number of substrates containing heteroaryl chloride substituents, which can be easily elaborated in further transition-metal-catalyzed protocols, were successful partners in this reaction (51, 55, 59, 60, 63, and 70). Furthermore, heteroamines that are traditionally challenging in these silyl-radical-mediated copper-catalyzed aliphatic aminations, such as imidazoles (71) and triazoles (72 and 73), could be coupled in synthetically useful yields. Both trisubstituted and disubstituted BCPs (55, 60, and 75) were amenable to these reaction conditions. Finally, to determine the necessity of employing metal cross-coupling procedures for installation of nucleophiles at the bridge position, we evaluated basic and/or thermal SN2 conditions for a variety of amines and iodide sources—no reaction was observed under these alternate conditions (Table S8 and Figure S2, see the Supporting Information for more details).
Figure 4.
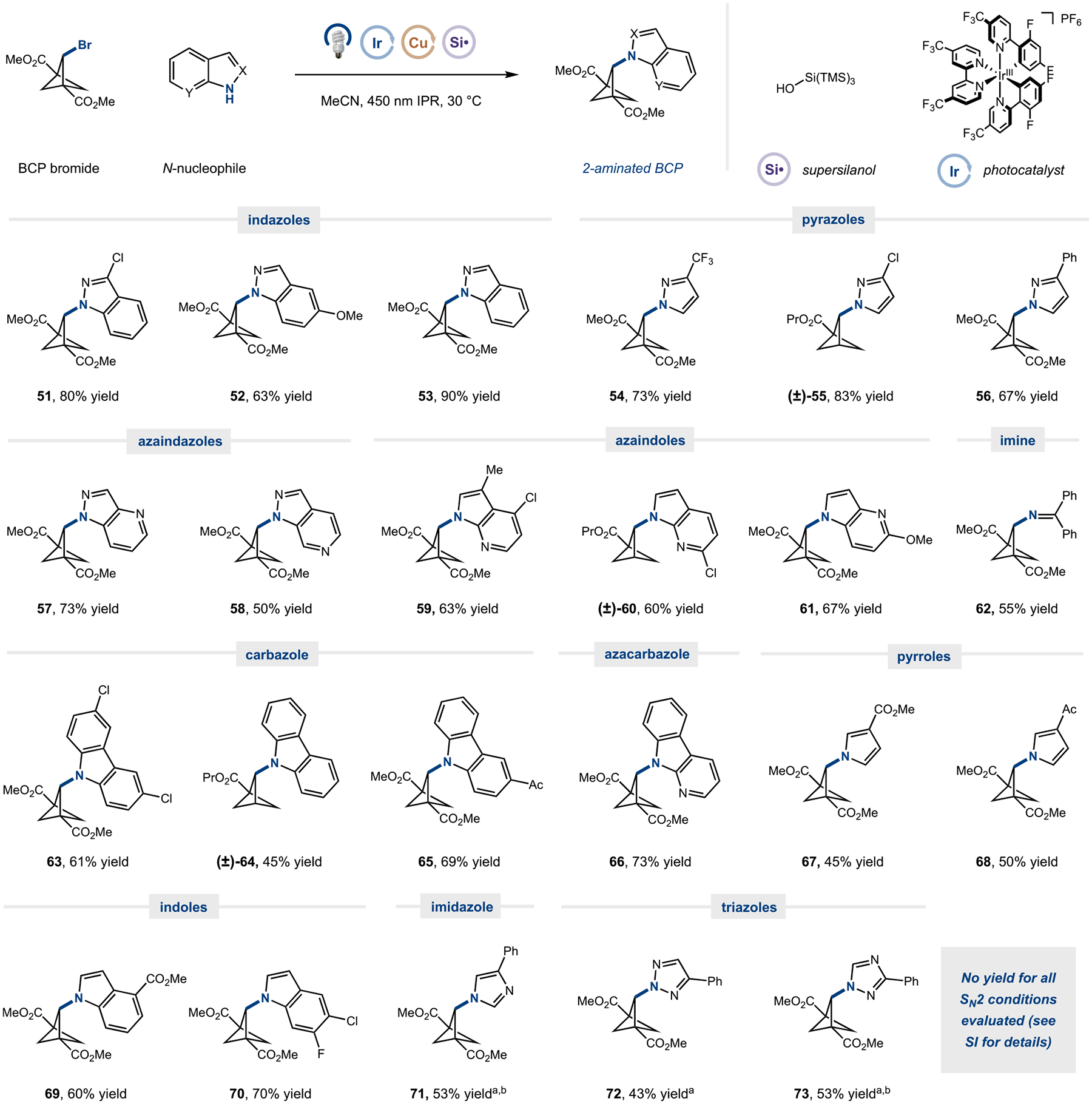
Scope for silyl-radical-mediated BCP 2-amination. Numerous (hetero)amine classes can be used in this transformation. All yields are isolated. Standard conditions: amine (0.25 mmol, 1 equiv), BCP bromide (2 equiv), silanol (2.5 equiv), Ir(dF(CF3)ppy)2(4,4′-dCF3bpy)PF6 (0.8–1.6 mol %), CuTC (50 mol %), 4,4′-dOMebpy (50 mol %), DBN (2 equiv), MeCN, H2O (25 equiv), IPR 450 nm (25–30% intensity) for 12 h. See the Supporting Information for full experimental details. a5 mol % 4CzIPN. b5 × 0.5 mmol. Ac, acetyl; CuTC, copper(I)-thiophene-2-carboxylate; DBN, 1,5-diazabicyclo(4.3.0)non-5-ene.
With optimized protocols in hand, we next sought to apply these photoredox transformations in sequential functionalization reactions (Figure 5A). In these sequences, we hoped to employ our previously published decarboxylative amination chemistry at the bridgehead carboxylic acid,34 followed by a metallaphotoredox silyl-radical cross-electrophile coupling reaction to yield difunctionalized 1-amine, 2-aryl BCP cores. Pleasingly, using the aforementioned amination conditions, we were able to couple indazoles (S11, 84% yield), azaindoles (S14, 73% yield), and azaoxindoles (S15, 46% yield) at the bridgehead position of our brominated BCPs in excellent yields. Furthermore, the subsequent silyl-radical-mediated arylation could tolerate all of these amine functionalities (74–76) and utilize diverse hetero(aryl) bromides, including pyridines (74), quinolines (77), indoles (78), and unprotected carbamates (79) in high yields. To showcase our platform for the synthesis of densely functionalized BCP cores with three exit vectors, we took BCP 74, and, following hydrolysis, installed a morpholine substituent through an amide coupling (S13, 45% overall yield over five steps). Alternatively, the third bridgehead substituent can be decarboxylated through a photoredox decarboxylation procedure from the redox active ester to yield 1,2-difunctionalized BCP scaffolds (see the Supporting Information for details).
Figure 5.
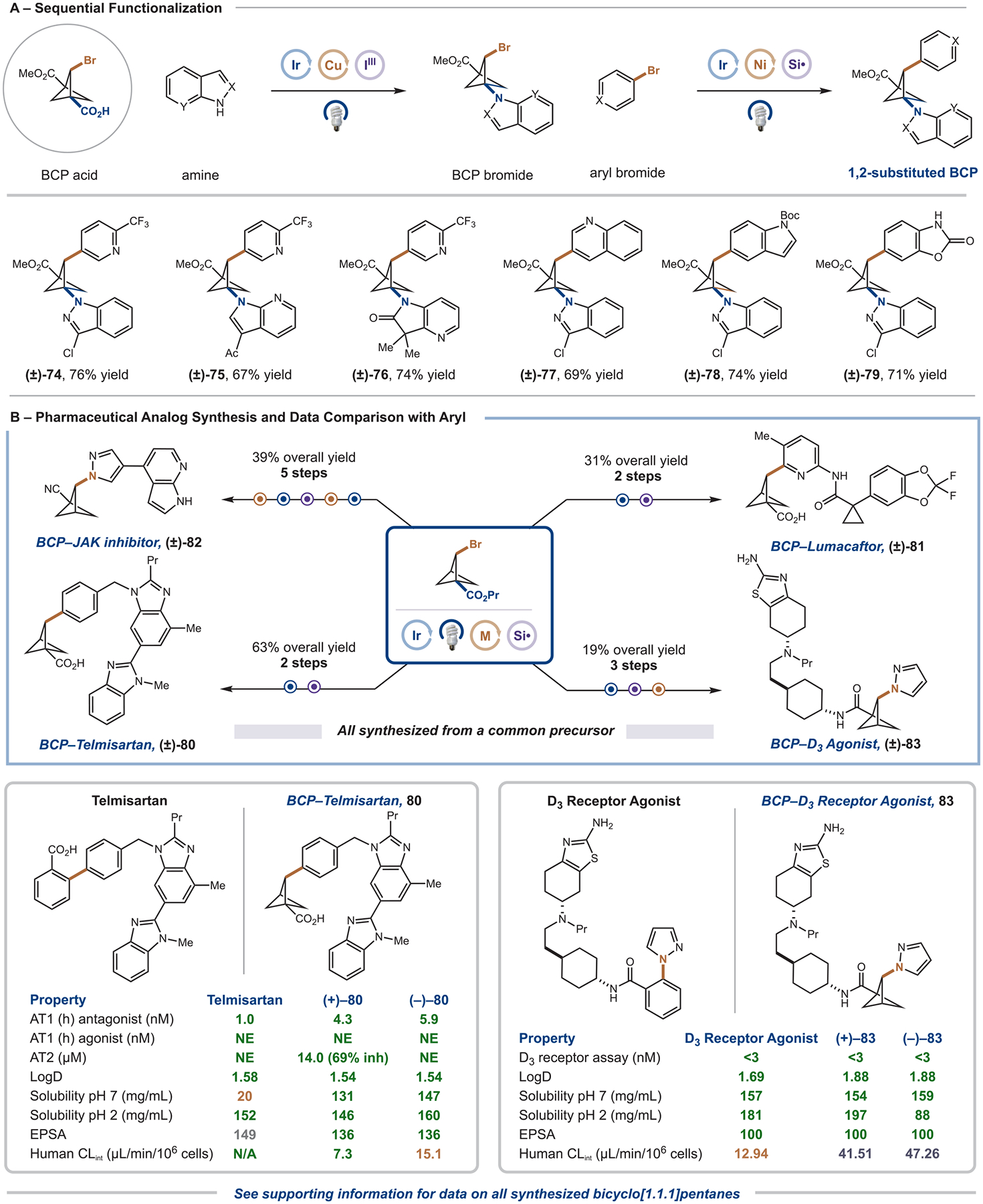
Sequential functionalization and drug analogues. (A) A 2-brominated, 1-carboxylic acid BCP can be leveraged in sequential photoredox-catalyzed decarboxylative coupling, followed by silyl-radical-mediated 2-arylation. Reported yield is for the silyl-radical-mediated 2-arylation. All yields are isolated. Standard conditions: aryl bromide (0.3 mmol, 1 equiv), BCP bromide (2 equiv), aminosilane (1.6 equiv), Ir(dF(CF3)ppy)2(dtbbpy)PF6 (2 mol %), Ni(dtbbpy)Br2 (5 mol %), Cs2CO3 (2 equiv), TFT (0.05 M), IPR 450 nm (100% intensity) for 2 h. See the Supporting Information for full experimental details. (B) These silyl-radical-mediated 2-arylation and 2-amination procedures can be applied to the rapid preparation of pharmaceutical analogues and bioactive molecules. Data is compared with the aryl compound. All yields are isolated. See the Supporting Information for full experimental details. inh, Inhibition; NE, no significant effect.
To demonstrate the potential of these 1,2-difunctionalized BCP cores as ortho- or meta-disubstituted aryl bioisosteres and the applicability of our procedures to drug discovery settings, we next applied our new metallaphotoredox coupling reactions to the synthesis of four pharmaceutical analogues (Figure 5B). Toward this end, BCP–telmisartan 80 was synthesized in just two steps and 63% overall yield from our common BCP precursor and the corresponding aryl bromide (previous best: 10 steps and 10% overall yield).16 This approach was also applied to the synthesis of BCP–lumacaftor 81 from the corresponding aryl bromide and the BCP intermediate in two steps and 31% overall yield. To test our amination chemistry in pharmaceutically relevant contexts, we targeted the BCP analogues of a JAK inhibitor (82) and a D3-dopamine agonist (83). Excitingly, we could synthesize the BCP–JAK inhibitor analogue 82 in five steps and 39% overall yield, through coupling of the appropriate protected azaindole–pyrazole, followed by amidation of the bridgehead ester, and subsequent dehydration to the nitrile. Furthermore, BCP–dopamine D3 agonist 83 could be prepared by coupling of pyrazole, followed by hydrolysis, and amide coupling of a complex amine (three steps and 19% overall yield).
We next tested these BCP pharmaceutical analogues in comparison to their aryl-containing counterparts (Figures 5B and S4). Most notably, three out of four of the BCP compounds retained potency when moving from the arene ring to the BCP scaffold. In addition, all BCP compounds tested showed improved or similar lipophilicity, solubility, and EPSA over the corresponding 2-arylated and 2-aminated BCP drug molecule, with most retaining or improving the metabolic profile. Altogether, these results demonstrate the high potential of 2-substituted BCPs for improving the pharmacological properties of drug candidates containing ortho- or meta-substituted aryl rings.
CONCLUSIONS
In summary, we have established a unified platform for 2-arylation and 2-amination of bicyclo[1.1.1]pentanes through HAT and photoredox catalysis mechanisms. Rapid synthesis of a diversifiable BCP synthetic linchpin is achieved via development of a novel C–H bromination of the strong BCP bridge C–H bond. Our two new silyl-radical-mediated protocols for BCP 2-arylation and 2-amination enable more modular and expedient access to 2-substituted BCPs than previous methodology. We demonstrate that these methods can be applied to the synthesis of complex drug substrates, showcasing the applicability of these reactions to drug discovery. Through biological testing of 2-substituted BCP analogues of known bioactive compounds, we demonstrate that 2-substituted BCPs are valuable to the pharmaceutical industry as replacements for ortho- or meta-substituted aryl rings.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the NIH National Institute of General Medical Sciences (NIGMS), the NIH (R35GM134897-03), the Princeton Catalysis Initiative, and kind gifts from Merck, Bristol-Myers Squibb (BMS), Celgene, Genentech. Janssen Research and Development LLC and Pfizer. O.L.G. thanks BMS for a BMS Graduate Fellowship. J.C. and O.L.G. thank Princeton University, E. Taylor, and the Taylor family for an Edward C. Taylor Fellowship. M.H. thanks the Swiss National Science Foundation for an Early Postdoc. Mobility fellowship (grant number P2BSP2_195373). The authors thank Feng Peng (Merck & Co., Inc) for help with external chemistry; Jennifer Piesvaux, John P Imredy, Richard L Kraus and Brian Lacey (Merck & Co., Inc) for help with biological profiling; Adam Beard, Miroslawa Darlak, Spencer McMinn, Lisa Nogle, Mark Pietrafitta, David Smith, and Yingchun Ye for help with reverse phase chromatography and SFC chiral separation.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.2c12163
The authors declare the following competing financial interest(s): D.W.C.M. declares a competing financial interest with respect to the Integrated Photoreactor. A provisional U.S. patent has been filed by D.W.C.M., O.G., Y.L. and M.H. based in part on this work, 63/151,974. International Ap-plication No. PCT/US2022/070770. The remaining authors declare no competing interests.
Contributor Information
Olivia L. Garry, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Michael Heilmann, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Jingjia Chen, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Yufan Liang, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Xiaheng Zhang, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States.
Xiaoshen Ma, Department of Discovery Chemistry, Merck & Co., Inc., Boston, Massachusetts 02115, United States.
Charles S. Yeung, Department of Discovery Chemistry, Merck & Co., Inc., Boston, Massachusetts 02115, United States
David Jonathan Bennett, Department of Discovery Chemistry, Merck & Co., Inc., Boston, Massachusetts 02115, United States.
David W. C. MacMillan, Merck Center for Catalysis at Princeton University, Princeton, New Jersey 08544, United States
REFERENCES
- (1).Pu Q; Zhang H; Guo L; Cheng M; Doty AC; Ferguson H; Fradera X; Lesburg CA; McGowan MA; Miller JR; Geda P; Song X; Otte K; Sciammetta N; Solban N; Yu W; Sloman DL; Zhou H; Lammens A; Neumann L; Bennett DJ; Pasternak A; Han Y Discovery of Potent and Orally Available Bicyclo[1.1.1]-Pentane-Derived Indoleamine-2,3-Dioxygenase 1 (IDO1) Inhibitors. ACS Med. Chem. Lett 2020, 11, 1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Stepan AF; Subramanyam C; Efremov IV; Dutra JK; O’Sullivan TJ; DiRico KJ; McDonald WS; Won A; Dorff PH; Nolan CE; Becker SL; Pustilnik LR; Riddell DR; Kauffman GW; Kormos BL; Zhang L; Lu Y; Capetta SH; Green ME; Karki K; Sibley E; Atchison KP; Hallgren AJ; Oborski CE; Robshaw AE; Sneed B; O’Donnell CJ Application of the Bicyclo[1.1.1]Pentane Motif as a Nonclassical Phenyl Ring Bioisostere in the Design of a Potent and Orally Active γ-Secretase Inhibitor. J. Med. Chem 2012, 55, 3414–3424. [DOI] [PubMed] [Google Scholar]
- (3).Measom ND; Down KD; Hirst DJ; Jamieson C; Manas ES; Patel VK; Somers DO Investigation of a Bicyclo[1.1.1]Pentane as a Phenyl Replacement within an LpPLA2 Inhibitor. ACS Med. Chem. Lett 2017, 8, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Auberson YP; Brocklehurst C; Furegati M; Fessard TC; Koch G; Decker A; LaVecchia L; Briard E Improving Nonspecific Binding and Solubility: Bicycloalkyl Groups and Cubanes as Para-Phenyl Bioisosteres. ChemMedChem 2017, 12, 590–598. [DOI] [PubMed] [Google Scholar]
- (5).Mykhailiuk PK Saturated Bioisosteres of Benzene: Where to Go Next? Org. Biomol. Chem 2019, 17, 2839–2849. [DOI] [PubMed] [Google Scholar]
- (6).Sodano TM; Combee LA; Stephenson CRJ Recent Advances and Outlook for the Isosteric Replacement of Anilines. ACS Med. Chem. Lett 2020, 11, 1785–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kanazawa J; Uchiyama M Recent Advances in the Synthetic Chemistry of Bicyclo[1.1.1]Pentane. Synlett 2018, 30, 1. [Google Scholar]
- (8).Ma X; Nhat Pham LN Selected Topics in the Syntheses of Bicyclo[1.1.1]Pentane (BCP) Analogues. Asian J. Org. Chem 2020, 9, 8–22. [Google Scholar]
- (9).Grygorenko OO; Volochnyuk DM; Vashchenko BV Emerging Building Blocks for Medicinal Chemistry: Recent Synthetic Advances. Eur. J. Org. Chem 2021, 2021, 6478–6510. [Google Scholar]
- (10).Locke GM; Bernhard SSR; Senge MO Nonconjugated Hydrocarbons as Rigid-Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry. Chem.—Eur. J 2019, 25, 4590–4647. [DOI] [PubMed] [Google Scholar]
- (11).Levin MD; Kaszynski P; Michl J Bicyclo[1.1.1]Pentanes, [n]Staffanes, [1.1.1]Propellanes, and Tricyclo[2.1.0.02,5]Pentanes. Chem. Rev 2000, 100, 169–234. [DOI] [PubMed] [Google Scholar]
- (12).Simov V; Kaplan WP; Acton JJ III; Ardolino MJ; Chen JL; Fuller PH; Gunaydin H; Li D; Liu P; Logan KM; Methot J; Morriello Gregori J; Neelamkavil SF; Torres L; Yan X; Zhou H N-Heteroaryl Indazole Derivatives as LRRK2 Inhibitors, Pharmaceutical Compositions, and Uses Thereof. WO 2020/092136 Al International Patent., May 7, 2020.
- (13).Hewings DS; Jaeschke G; Kuhn B; Nagel YA; Ricci A EGFR Inhibitors. WO 2021/123087 Al, June 24, 2021. [Google Scholar]
- (14).Anderson JM; Measom ND; Murphy JA; Poole DL Bridge Functionalisation of Bicyclo[1.1.1]Pentane Derivatives. Angew. Chem., Int. Ed 2021, 60, 24754–24769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (16).Zhao J-X; Chang Y-X; He C; Burke BJ; Collins MR; Del Bel M; Elleraas J; Gallego GM; Montgomery TP; Mousseau JJ; Nair SK; Perry MA; Spangler JE; Vantourout JC; Baran PS 1,2-Difunctionalized Bicyclo[1.1.1]Pentanes: Long–Sought-after Mimetics for Ortho/Meta-Substituted Arenes. Proc. Natl. Acad. Sci. U.S.A 2021, 118, No. e2108881118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Klopsch R; Schlüter A-DA [1.1.1]Propellane with an Unprotected Hydroxy Group in the Side Chain. Tetrahedron 1995, 51, 10491–10496. [Google Scholar]
- (18).Ma X; Han Y; Bennett DJ Selective Synthesis of 1-Dialkylamino-2-Alkylbicyclo-[1.1.1]Pentanes. Org. Lett 2020, 22, 9133–9138. [DOI] [PubMed] [Google Scholar]
- (19).Yang Y; Tsien J; Hughes JME; Peters BK; Merchant RR; Qin T An Intramolecular Coupling Approach to Alkyl Bioisosteres for the Synthesis of Multisubstituted Bicycloalkyl Boronates. Nat. Chem 2021, 13, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Feng Y; Liu L; Wang J-T; Zhao S-W; Guo Q-X Homolytic C–H and N–H Bond Dissociation Energies of Strained Organic Compounds. J. Org. Chem 2004, 69, 3129–3138. [DOI] [PubMed] [Google Scholar]
- (21).Wiberg KB; Connor DS Bicyclo[1.1.1]Pentane1. J. Am. Chem. Soc 1966, 88, 4437–4441. [Google Scholar]
- (22).Wiberg KB; Williams VZ Bicyclo[1.1.1]Pentane Derivatives. J. Org. Chem 1970, 35, 369–373. [Google Scholar]
- (23).Kaleta J; Rončević I; Císařová I; Dračínský M;Šolínová V; Kašička V; Michl J Bridge-Chlorinated Bicyclo[1.1.1]Pentane-1,3-Dicarboxylic Acids. J. Org. Chem 2019, 84, 2448–2461. [DOI] [PubMed] [Google Scholar]
- (24).Kaszynski P; Michl J A Practical Photochemical Synthesis of Bicyclo[1.1.1]Pentane-1,3-Dicarboxylic Acid. J. Org. Chem 1988, 53, 4593–4594. [Google Scholar]
- (25).Le TP; Rončević I; Dračínsky M; Císařová I;Šolínová V; Kašička V; Kaleta J Polyhalogenated Bicyclo[1.1.1]Pentane-1,3-Dicarboxylic Acids. J. Org. Chem 2021, 86, 10303–10319. [DOI] [PubMed] [Google Scholar]
- (26).Jarret RM; Cusumano L 13C-13C Coupling in [1.1.1]Propellane. Tetrahedron Lett. 1990, 31, 171–174. [Google Scholar]
- (27).Galeotti M; Salamone M; Bietti M Electronic Control over Site-Selectivity in Hydrogen Atom Transfer (HAT) Based C(Sp3)–H Functionalization Promoted by Electrophilic Reagents. Chem. Soc. Rev. 2022, 51, 2171–2223. [DOI] [PubMed] [Google Scholar]
- (28).Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- (29).Zhang P; Le C; MacMillan DWC Silyl Radical Activation of Alkyl Halides in Metallaphotoredox Catalysis: A Unique Pathway for Cross-Electrophile Coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kornfilt DJP; MacMillan DWC Copper-Catalyzed Trifluoromethylation of Alkyl Bromides. J. Am. Chem. Soc 2019, 141, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Smith RT; Zhang X; Rincón JA; Agejas J; Mateos C; Barberis M; García-Cerrada S; de Frutos O; MacMillan DWC Metallaphotoredox-Catalyzed Cross-Electrophile Csp3–Csp3 Coupling of Aliphatic Bromides. J. Am. Chem. Soc 2018, 140, 17433–17438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dow NW; Cabré A; MacMillan DWC A General NAlkylation Platform via Copper Metallaphotoredox and Silyl Radical Activation of Alkyl Halides. Chem 2021, 7, 1827–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sakai HA; Liu W; Le C; MacMillan DWC Cross-Electrophile Coupling of Unactivated Alkyl Chlorides. J. Am. Chem. Soc 2020, 142, 11691–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liang Y; Zhang X; MacMillan DWC Decarboxylative Sp3 C–N Coupling via Dual Copper and Photoredox Catalysis. Nature 2018, 559, 83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.