

Summary
Post-translational modifications (PTMs) play key roles in regulating cell signaling and physiology both in normal and cancer cells. Advances in mass spectrometry enable high-throughput, accurate and sensitive measurement of PTM levels to better understand their role, prevalence, and crosstalk. Here, we analyze the largest collection of proteogenomics data from 1110 patients with PTM profiles across 11 cancer types (10 from the National Cancer Institute's Clinical Proteomic Tumor Analysis Consortium [CPTAC]). Our study reveals Pan-Cancer patterns of changes in protein acetylation and phosphorylation involved in hallmark cancer processes. These patterns revealed subsets of tumors, from different cancer types, including those with dysregulated DNA repair driven by phosphorylation; altered metabolic regulation associated with immune response driven by acetylation; affected kinase specificity by crosstalk between acetylation and phosphorylation; and modified histone regulation. Overall, this resource highlights the rich biology governed by PTMs and exposes potential new therapeutic avenues.
Keywords: Post-translational modifications, Pan-Cancer, Genomics, Transcriptomics, Mass Spectrometry, Proteomics, DNA Damage Response, Metabolism, CPTAC
Graphical Abstract

Introduction
Systematic genomics-based studies of tumors have revolutionized our understanding of tumor biology1 and significantly impacted patient care2. Yet, many cancers still lack effective treatments or remain poorly characterized, emphasizing their complex biology and their molecular and phenotypic heterogeneities3. Recent advances in sample processing and liquid chromatography tandem mass spectrometry (LC-MS/MS) enable quantifying protein levels and post-translational modifications (PTMs) at a large scale.
Concerted efforts by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) have generated large proteogenomic datasets for individual cancer types4–13. These studies all included PTMs and started to bridge the gap between molecular features and phenotypic consequences, identifying new cancer subtypes with potential therapeutic vulnerabilities14. Despite these advances and the critical role PTMs play in regulation and fine tuning of cellular signaling15, their shared patterns, crosstalk among PTMs (e.g., phosphorylation, acetylation, etc.), and how multiple PTMs form regulatory networks, remain poorly understood, especially across cancer types.
Previous Pan-Cancer genomic studies have demonstrated that investigating recurrent gene and pathway alterations across different cancer types can promote our understanding of the fundamental molecular events that drive cancer16,17. Here, we set out to identify shared and divergent PTM patterns across cancer types in order to investigate common post-translational regulatory mechanisms that are altered in multiple cancers in order to both expand and complement genomic studies. To this end, we generated a harmonized pan-cancer cohort using data from 11 studies, encompassing samples from 1110 treatment-naive patients, with complete genomic, transcriptomic, proteomic and PTM (phosphorylation and acetylation) data (Figure 1A). This enabled us to search for patterns that could not be identified in a single cohort due to the limited sample size of individual studies (45–140 patients). To focus on shared and tissue- independent patterns across cancer, we regressed out the tissue-specific effects in each data type as part of the harmonization process (Methods).
Figure 1 -.

Pan-Cancer dataset overview
A. Pan-Cancer analysis workflow: (left) Data harmonization of available cohorts; (middle top) Available data types and discovery of multi-omic signatures based on RNA, proteins, and phosphosites, (middle bottom) clustering of samples based on signature activities; (right top) Experimental and computational tools used to study clusters of tumors and pathways; (right bottom) Highlighted cancer pathways with altered post-translational modifications.
B. TMB across cohorts -CPTAC median: Red, TCGA median: dotted orange.
C. Upset plots showing the distribution of shared expressed genes (RNA) and the different RNA Biotypes contribution.
D. Upset plots showing the distribution of shared proteins (left) and site-level phosphorylation (middle) and acetylation (right) across the different cohorts (bars representing ~85% of the data for visibility).
We focused our analyses on (i) hallmark pathways known to be dysregulated in cancer18 that are tightly controlled by PTMs19,20, including DNA damage and repair pathways, cell immuno- metabolism, and histone-level regulation of gene expression; and (ii) potential crosstalk among different types of PTMs. PTMs have a range of potential regulatory effects – from quick, to on-going, to long term19,20. In immune and metabolic responses, the transient and reversible nature of PTMs enables the quick response needed to adapt to changes in the microenvironment21–23. PTM effects on histone modifications, on the other hand, can affect long-lasting regulation of cellular programs; indeed, in cancer, aberrant histone acetylation can inactivate tumor suppressors, or activate oncogenes24,25. In DNA repair processes, phosphorylation plays a key role in regulating the activity of DNA repair proteins26, and PTM-focused analyses may better characterize the landscape of DNA repair, particularly in DNA repair–deficient cancers. Lastly, serine/threonine phosphorylation and lysine acetylation are among the most widespread and conserved PTMs in eukaryotic organisms. While most studies to date have focused on how a single PTM type can regulate cellular processes, the recognition that proteins harbor multiple PTM types suggests that they may act together to jointly manifest complex regulatory effects, many of which remain largely unexplored.
Overall, this is the first Pan-Cancer study that details the extensive regulation of acetylation and phosphorylation and their shared patterns across cancer types. Together, our results comprise a rich resource to explore and generate hypotheses regarding PTM-governed processes in cancer that, after further experimental validation, may identify new drug targets, or suggest novel ways to affect cancer biology.
Results
Pan-Cancer dataset overview
Previous CPTAC proteogenomic studies revealed protein-based molecular tumor subtypes and identified cancer-specific pathways using PTMs. In this study, we integrate data across CPTAC cohorts to enable a Pan-Cancer analysis of gene, protein and PTM patterns shared across cancer types. To accomplish this, CPTAC Pan-Cancer working groups harmonized data from all available cohorts using standardized pipelines for assessing somatic mutations, somatic copy number alterations (SCNA), mRNA expression, protein abundance, phosphorylation, acetylation and clinical data (companion Data resource paper CANCER-CELL-D-22–00603, Figure S1). The final combined dataset comprised 1110 patients from 11 cohorts (Figure 1A). Ten tumor types were part of CPTAC, including glioblastoma (GBM)4, head and neck squamous cell carcinoma (HNSCC)5, lung adenocarcinoma (LUAD)6, lung squamous cell carcinoma (LSCC)7, breast cancer (BRCA)8, pancreatic ductal adenocarcinoma (PDAC)9, clear cell renal cell carcinoma (ccRCC)10, high-grade serous ovarian cancer (HGSC)11, uterine corpus endometrial carcinoma (UCEC)12, colorectal adenocarcinoma (COAD)13. An external Medulloblastoma (MB) dataset27 was also included, generated following the same protocols as the CPTAC datasets for all available data types, but lacking whole-exome DNA sequencing. For each patient, we identified both germline and somatic variants and quantified gene expression, protein abundance, and PTM levels (Methods). We detected a median of ~25k exonic germline variants and ~320 exonic somatic mutations per patient, with median somatic mutation burdens that matched The Cancer Genome Atlas cohorts (Figure 1B). As expected, a subset of UCEC and COAD patients showed exceptionally high tumor mutational burden (TMB), reflective of microsatellite instability (MSI) and polymerase proofreading deficiencies (POLE and POLD1 exonuclease domain mutants). In addition, we found that an average of ~24k genes, including coding and non-coding, were expressed in any cohort (TPM≥0.1 and ≥6 reads each in at least 20% of patients). We detected an average of ~10k proteins, ~22k phosphosites, and ~6k acetylation sites per patient (available for 6 cohorts) (Methods; Figure 1C–D).
Next, since we are searching for Pan-Cancer patterns, we analyzed the overlap among genes, proteins, and PTM sites. We found ~21k genes to be expressed across all cohorts (~14.5k protein coding and ~6.5k non-coding; Figure 1C); moreover, 6,333 proteins were detected across all cohorts and accounted for the majority of the data. Importantly, PTMs show a more discrete pattern in each tumor type, with relatively fewer shared across cohorts (Figure 1D, center and right panels; Table S1). This may reflect their role in fine-tuning responses at the cell- and tissue-type level beyond that of gene or protein expression alone28,29.
Pan-Cancer PTM landscape
To explore shared PTM patterns across cancer, we first integrated the data types that were available across all 11 cohorts –– specifically, gene expression, protein abundance, and phosphoprotein level data –– while regressing out tissue-specific effects to remove obvious differences among tumor types (Figure S1A–D, Methods). We applied SignatureAnalyzer, a Bayesian variant of non-negative matrix factorization30–32, across the 1110 tumors represented by a combined set of 14,057 features (Note S1, Methods) to obtain 33 Pan-Cancer multi-omic signatures. Notably, most signatures had contributions from all 3 features (Figure 2A, Methods). In addition to defining the signatures, SignatureAnalyzer estimates the activity level of each signature for each tumor. By assigning each tumor to its most active signature, we found that most signatures span multiple tumor types (Figure S2A), suggesting that, in general, the signatures reflect Pan-Cancer biological processes.
Figure 2 -.

Pan-Cancer PTM landscape
A. Hierarchical clustering of sample similarity matrices across their signature activities (middle heatmap). Tracks: (top) Cluster, and cohort annotations, (middle) whole-exome mutational signatures and (below) ESTIMATE assignments. Lower panel heatmap shows RNA, proteins and phosphosites in the top differentially expressed pathways between the left and right sides of the first split of the dendrogram.
B. Bubble plot representation of The Kinase Library enrichment based on differentially expressed substrates of each kinase between the first split of the dendrogram. Enrichment (Red), depletion (Blue).
C. CLUMPS-PTM results for the first split shows Significant 3D spatial clustering of differentially acetylated (left, triangles) or phosphorylated (right, boxes) sites. Circles represent significance based on the union of both. DDR hallmark geneset (blue). Red: Significant results - FDR <0.1, Yellow: near significance results- FDR <0.25.
D. SRSF2 phosphorylation cluster on 3D crystal structure (cyan; PDB ID: 2LEA), RRM-1 domain (amber), phosphosites (purple).
E. ARID1A acetylation cluster on 3D crystal structure (cyan,PDB ID: 6LTH), acetylsites (pink).
F. Violin plots showing protein abundances of ARID1A (left), and Glucocorticoid targets (right) between the first split of the dendrogram.
To characterize tumor subsets with both shared and divergent biology, we performed hierarchical clustering of the samples based on their activities across the 33 signatures, which more robustly reflect the Pan-Cancer biological processes active in each sample (Table S2, Methods). In addition, in order to define groups of samples that share their most prominent biology (used for certain downstream analyses), we traversed the dendrogram and defined clusters based on their most frequent dominant signature, identifying 26 non-overlapping terminal clusters (Figure S2B–C). To further explore the biology in each group, we performed pathway enrichment analyses at the RNA, protein, and PTM levels (Table S2), and applied multiple methods specifically tailored to identify PTM differences: (i) CLUMPS-PTM (Figure S2D–F), (ii) The Kinase Library33, (iii) CausalPath34, (iv) PTM Signature Enrichment Analysis (PTM-SEA)35, and (v) a method to predict differential activity of histone regulators (Note S2). Aggregating the results allowed us to comprehensively characterize differences in tumor biology across our Pan-Cancer cohort.
We started our analysis by focusing on the top split in the dendrogram comprised of a significant enrichment for DNA damage response (DDR) and proliferation pathways (MYC and E2F) on the left side of the split, and myogenesis and epithelial-mesenchymal transition (EMT) pathways on the right side (Figure 2A). We applied both a site-specific pathway enrichment analysis - PTM-SEA as well as The Kinase Library that predicts the PTM regulators based on their substrate specificity (Note S2). Both tools showed significant enrichment of cyclin-dependent kinase (CDK) activity and downregulation of p21-activated kinases (PAKs) in the left side of the first split (Figure 2B, Table S2). These results are consistent with the pathway activation differences, since CDK-mediated phosphorylation is associated with rapid cell proliferation, while PAKs are associated with the actin cytoskeletal remodeling and increased migratory phenotype that accompanies EMT36.
Using CLUMPS-PTM to identify clusters of correlated PTMs in protein 3D structures (Note S2), we found 22 proteins with significant clustering (FDR<0.1) of phosphosites that were upregulated in the first split when comparing the left vs. right side of the dendrogram (Figure 2C). One of the top hits for significant phosphorylation clustering was SRSF2 (FDR=0.044), a serine and arginine rich splicing factor. The cluster falls within the RRM-1 domain and its phosphorylated form has been shown to interact with E2F1 in order to promote transcriptional control of cell cycle target genes such as cyclin E (Figure 2D, S2F). Thereby, promoting cell proliferation in lung carcinoma cell lines37.
For acetylation sites, we found only one protein, ARID1A, with a significant clustering (FDR=0.085). ARID1A is a SWI/SNF chromatin remodeler that is commonly mutated in cancer38 and plays a complex role in tumorigenesis39. The cluster is on the C terminal tail of ARID1A, within the glucocorticoid receptor (GR) binding domain (Figure 2E, S2F). GR regulates many genes whose products increase catabolism, reduce inflammation, and increase cell survival40. Increased acetylation in this cluster can potentially block a ubiquitination site at the C-terminal of the protein41, which would reduce ARID1A degradation and potentially increase GR signaling. We indeed observe higher protein abundance of ARID1A and GR targets on the left side of the dendrogram (Figure 2F).
Mechanisms of PTM dysregulation in DNA repair-deficient tumors
Next, we leveraged our proteogenomic dataset to investigate the effects of DNA repair deficiencies that are undetectable at the genomic and transcriptomic levels. We first extracted mutational signatures across our cohort by applying SignatureAnalyzer30–32,42,43 to five partitions of our dataset by distinct environment and cell-intrinsic mutational mechanisms: POLE/POLD1-exonuclease domain mutants, mismatch repair–deficient (MMRD), smoking-related, homologous recombination deficiency (HRD)-related, and not HRD-related (Figure S3A, Methods). We extracted a total of 22 mutational signatures representing 11 distinct mutational processes, including MMRD and HRD (Figure 3A, Table S3, Methods). Using these signatures, we identified 57 MMRD and 88 HRD tumors (Figure S3B, Methods). Consistent with previous Pan-Cancer studies, most MMRD tumors were from the COAD and UCEC cohorts (21 and 28 tumors, respectively), while the HRD group encompassed 54 HGSC, 30 BRCA, and four PDAC tumors44,45.
Figure 3 -.

PTM Analysis of DNA Repair Deficiencies
A. Mutational signatures associated with each cohort. Circle size represents the proportion of tumors. Circle color indicates median mutations/Mb.
B. Volcano plot illustrating the differential phosphorylation between HRD and HRP tumors. MMEJ genes are labeled.
C. Violin plot of 1st principal component projections based on the multi-omic signature activities for HRD tumors. Points are colored by their cancer type and separated by HRD cluster.
D.Schematic diagram of the acute and chronic hypoxia HRD clusters (top). Arrow length represents duration of hypoxia. Bubble plot showing GSEA results between the acute and chronic hypoxia HRD subgroups (bottom).
E. CausalPath results of differentially expressed DDR genes between acute and chronic hypoxia HRD tumors. Acute hypoxia upregulation (red), downregulation (blue). Black dashed lines - 90th percentile scoring substrates based on The Kinase Library results.
F. Bubble plot showing GSEA results between MMRD and MMRP tumors. MMRD pathways upregulated (red), downregulated (blue).
G. Violin plots showing protein abundance (top) and RNA (bottom) levels of MRN complex proteins between MMRD and MMRP tumors (COAD [circle] and UCEC [triangle]). RAD50 microsatellite frameshift indel samples indicated in red.
H. CausalPath results of differentially expressed DDR genes between MMRD and MMRP tumors as in panel E.
HRD cancers rely on alternative repair pathways to mitigate double-strand break damage46. To investigate the PTM-directed activities of repair proteins in HRD cancers, we performed differential expression analyses (across all feature types) between HRD and homologous recombination-proficient (HRP) tumors across DNA repair genes, followed by CausalPath analysis to identify causal relationships between PTMs and their mediators (Methods, Note S2).
These comparisons revealed significant differences in the phosphorylation of 268/1596 sites residing in 112/310 measured DNA repair proteins. In particular, we found differences in 8/12 proteins representing the microhomology-mediated end-joining (MMEJ) pathway, which is the primary HRD compensatory pathway (FDR≤0.1, Figure 3B, Table S3)47,48. Notably, we found increased phosphorylation of three PARP1 phosphorylation sites, including PKA-mediated site S782 (FDR=0.05) and ATR-mediated site S179 (FDR=0.05), which are known to regulate PARP1 activity49–51. HRD tumors also exhibited significantly increased phosphorylation of POLQ on S1587 (FDR=0.05). POLQ promotes MMEJ by inhibiting RAD51-mediated HR, and its loss has been shown to elicit synthetic lethality in HRD tumors, including in cell lines resistant to PARP inhibition52,53 (Figure S3C); the functional effects of S1587 phosphorylation, however, have not been well studied. We additionally found increased phosphorylation of EXO1 S714 (FDR=0.04), an ATM-mediated site that has been proposed to attenuate EXO1 activity and hinder homologous recombination (HR) as a result54. Differential phosphorylation analysis thus revealed site-specific modifications that may regulate mechanisms that compensate for HR loss.
We observed that the 88 HRD tumors spread across the four main branches of the dendrogram (Clusters A–D, Figure 2A). We therefore explored whether this partitioning reflected different DNA repair activities, which could potentially associate with different therapeutic vulnerabilities. First, we performed principal component analysis (PCA) of the multi-omic signature weights in HRD tumors to verify that their partitioning in the Pan-Cancer dendrogram was maintained when focusing only on these tumors. We found that even the first principal component (PC1) can separate these HRD clusters (Figure 3C). To characterize the biological processes associated with each cluster, we performed pairwise multi-omic differential expression analyses between A, B, and C (excluding D due to the small sample size of n=7). Gene set enrichment analysis (GSEA) revealed that B exhibited significant upregulation of hypoxia-related proteins compared to A, and significant downregulation compared to C (FDR=0.08 and 0.03, respectively, Table S3). Previous cell line studies have described the relationship between hypoxia severity and DDR, showing that acute hypoxia with periodic reoxygenation activated DNA repair pathways to mitigate reactive oxygen species (ROS)-related DNA damage, while chronic hypoxia stalled replication and suppressed DDR55–58. We hypothesized that B represented an acute hypoxia group, and C a chronic hypoxia group. Indeed, GSEA also highlighted an upregulation of the ROS pathway at the mRNA level (FDR=0.04); DNA repair at the protein level (FDR=0.02); and DNA replication at both mRNA and protein levels in B compared to C (FDR=0.01, 0.02, respectively) (Figure 3D, Table S3). Similarly, PTM-SEA detected in the acute hypoxia HRD cluster B increased activity of the DNA damage signaling kinases ATM, CHEK1, and CHEK2 (FDR=0.09, 0.02, 0.06, respectively), as well as an enrichment of CDK1/2/4/6 activities (all FDR=0.018; Table S3), as expected55–57.
In order to identify specific differences in regulators of DDR proteins between acute and chronic hypoxia HRD tumors, we applied CausalPath on all differentially expressed features. CausalPath detected increased PARP1 and XRCC1 interaction in the acute hypoxia HRD group through increased PARP1 protein (FDR= 0.09) and decreased phosphorylation of XRCC1 sites S475, S485, and T488 (FDR= 0.102, Figure 3E)59. This interaction facilitates XRCC1 recruitment to ROS-induced base lesions and single-strand break sites, suggesting increased PARP1 activity in the base-excision repair (BER) pathway60,61, which is necessary for the PARP trapping mechanism of PARP inhibitors62. Further supporting increased PARP activity, GSEA showed protein-level enrichment of oxidative phosphorylation and downregulation of glycolysis pathways (FDR=0.0007, 0.08, respectively), consistent with a known pro-survival metabolic shift from glycolysis to oxidative phosphorylation due to PARP consumption of NAD+63. Furthermore, CausalPath highlighted CDK2 phosphorylation of WRN S1133, which is further supported by the Kinase Library (97.5th percentile of CDK2 substrates) and is a known response to collapsed replication forks48,64 (Table S3). Overall, phosphorylation-focused analysis highlighted major differences in the PTM activity of DDR proteins between the acute and chronic hypoxic HRD tumors that are indistinguishable at the mutational signature level.
As opposed to the partitioning of the HRD tumors to four groups, the MMRD tumors showed only tissue-driven separation (Figure S3D), prompting us to analyze all the MMRD tumors together. Similar to the HRD analysis, we ran differential expression analysis between MMRD and mismatch repair–proficient (MMRP) tumors across all feature types associated with DNA repair genes (Table S3). As expected, due to common MLH1 promoter hypermethylation in MMRD tumors, depletion of MLH1 RNA expression and protein abundance (FDR=1x10−33, 5x10−30, respectively) were among the most significant differences. To characterize pathway-level differences, we performed GSEA on all differentially expressed features (Table S3, Figure 3F). Intriguingly, MMRD tumors exhibited upregulation of double-strand break (DSB) repair pathways at the mRNA level and downregulation at the protein level (FDR = 0.09, 0.01, respectively; box in Figure 3F). This difference is likely due to the fact that different genes drive the mRNA vs. protein pathway activation levels (14 vs. 4 distinct leading edge genes, respectively). The 4 leading edge proteins were MRE11, RAD50, NBN (which form the double-strand break sensing and signaling MRN complex65), and ATM (FDR=2x10−17, 2x10−18, 2x10−7, 2x10−3, respectively; Figure 3G). We investigated whether truncating microsatellite mutations, which were previously found to be enriched in RAD50, MRE11, and ATM66,67,68, could explain the decreased mRNA and protein levels of these genes. We found 16 patients with truncating alterations in RAD50 (ten K722fs and four N934fs frameshift indels), all of which were in the MMRD group (16/49 vs 0/142, FDR=1x10− 10, Table S3, Methods). The analysis of MRE11 and NBN alterations was not sufficiently powered due to few truncating events (three and one, respectively). Interestingly, we found a similar decrease in protein abundance of MRE11, RAD50, and NBN in MMRD vs. MMRP cell lines, but only a marginal decrease in MRE11 mRNA, and no change (or even increase) in RAD50 and NBN, respectively (Figure S3E), raising the possibility that reducing one protein in the complex may destabilize the complex and lead to degradation of the other complex proteins. Further studies are needed to explore the mechanisms of reduced expression of the MRN complex in MMRD tumors.
Consistent with previous reports, we also found significant enrichment of ATM microsatellite indels in MMRD tumors (16/49 vs 3/142, FDR=5x10−8; Table S3). Applying CausalPath to the differential expression results revealed evidence of DSB sensing and signaling defects due to ATM loss (Figure 3H). MMRD tumors showed a decrease in ATM-mediated phosphorylation of PRKDC (DNA-PKcs) at S3205 (FDR=0.1), which induces DSB repair signaling68. We also found a decrease in ATM and PRKDC-mediated phosphorylation of PNKP at S114 and T118 (FDR=0.09, Table S3). Phosphorylation of these sites is critical for PNKP retention at DSB sites and subsequent processing of DSB ends prior to ligation in the NHEJ pathway69,70. These results highlight that proteogenomic analyses can expose effects of somatic deleterious alterations in MMRD tumors that cannot be observed at the mRNA level alone.
PTM regulation of metabolic pathways affects tumor-associated immune responses
The interplay between cell metabolism and the immune response was previously established;19,20 here, we aimed to characterize the effects of PTM regulation on this interplay across tumor types. First, we applied multiple methods that infer immune infiltration and activity: (i) ESTIMATE71, which estimates abundance of immune infiltration based on expression levels of curated gene sets; (ii) ImmuneSubtypeClassifier72, which provides granular immune phenotypes using a classifier approach; and (iii) unsupervised clustering based on enrichments of curated gene sets from CIBERSORT73. Our unsupervised clustering approach revealed four broad immune subtypes across different cancer types: immune-cold, -cool, -warm, and -hot. These subtypes aligned with the results from ESTIMATE and ImmuneSubtypeClassifier (Figure 4A, Figure S4A–B, Table S4). We observed a mixed tumor distribution in the subtypes, except for the “immune- cold” subtype, which was predominantly composed of brain tumors (93 out of 130 samples, Figure S4C), consistent with brain tumors typically being “immune-cold” due in part to the blood– brain barrier74. Next, we performed differential expression and pathway analyses in these immune subtypes using estimated tumor-intrinsic expression by removing the contribution of the immune cells (Figure S4B, Table S4, Methods). Similar to previous studies6,7, our immune-hot subtype showed an increase in immune-related pathways (Figure 4B) as well as a significant increase of immunosuppressive markers, including IDO1, CD163, ENTPD1, and PD-L1 (CD274) (immune- hot vs. immune-warm FDR<0.1, Figure S4D). The median fold change was lower than was previously reported in LUAD, potentially due to the large heterogeneity across cancer types (median FC differences between immune-hot vs. immune-warm ranges between 0.32 and 1.5 vs. >1.9 in CPTAC LUAD6). We also found significant differences in acetylation levels across multiple metabolic pathways in the immune-cool subtype, including propanoate metabolism, oxidative phosphorylation, and fatty acid metabolism (FA) pathways (Figure 4B; FDR<0.07). In these pathways, acetylation is known to play an important inhibitory role7,75,76. We observed high levels of acetylation in lipid metabolism pathways in the immune-hot group and low levels of acetylation in the immune-cool subtype (FDR<0.01), even after correction for protein abundance (denoted as acetylome_res in Figure 4B; Methods), suggesting that low acetylation levels potentially contribute to high activation of these pathways in immune-cool tumors. Indeed, FA enzymes are known to control specific gene expression77, and this effect is mainly regulated at the protein and PTM levels78. Of note, immune-cold tumors show a similar metabolic pathway activation as immune-hot tumors, perhaps due to the fact that the brain mostly relies on glucose as its energy source since it requires less oxygen for ATP generation79 (although this explanation does not explain why the few non-brain tumors clustered into the immune-cold subtype, which is yet to be determined) (Figure S4E).
Figure 4 -.
PTM regulation of immuno-metabolism across cancers
A. ssGSEA hierarchical clustering for immune-related gene sets (heatmap) showing four immune clusters: hot to cold. Tracks represent ESTIMATE and ImmuneSubtypeClassifier annotations.
B. Bubble plot representing MSigDB Hallmark and KEGG pathways enrichment among the four immune subtypes.
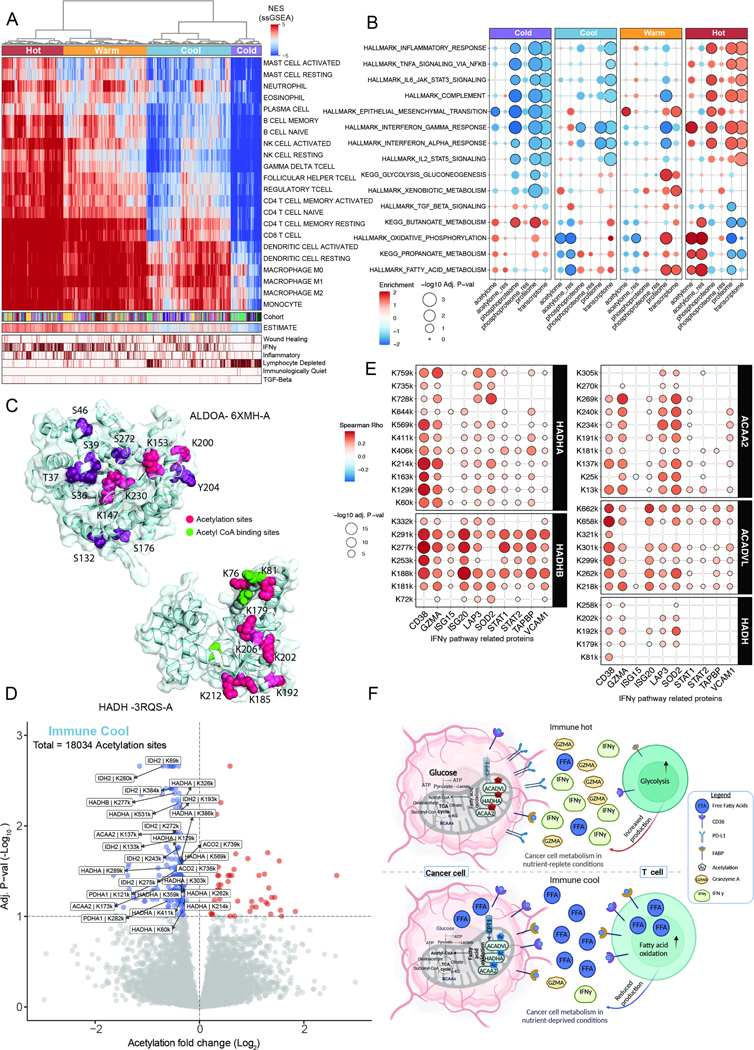
C. Significant clustering based on CLUMPS-PTM of both acetylation and phosphorylation sites on ALDOA (top panel; PDB ID: 6XMH-A) in the immune-hot group, and clustering of decreased acetylation sites on HADH in the immune-cool cluster (lower panel; PDB ID: 3RQS-A). Phosphosites (purple), acetylsites (pink), and Acetyl CoA binding sites (green).
D. Volcano plot showing differential acetylation between the immune-cool subtype and the other immune clusters. Acetylation sites on fatty acid metabolism proteins are highlighted.
E. Bubble plot representing significant correlations between fatty acid beta oxidation enzymes acetylation sites and protein levels from the IFNγ pathway.
F. Schematic representation of PTM-based metabolic changes in immune-cool vs. immune- hot tumors showing key enzymes in the glycolysis and fatty acid beta oxidation pathways and their proposed effect on T cells.
Next, we employed CLUMPS-PTM on the differentially regulated sites among these immune subtypes to identify functional regions on the 3D protein structures. The glycolytic domain of the ALDOA enzyme, which is abundant in cancer80, was found to harbor a significant cluster of 4 increased acetylated sites in the immune-hot group (K147, K153, K200, K230; FDR< 0.12, subset to glycolysis proteins), three of which are also known ubiquitination sites that can lead to protein degradation41. The same domain harbored a significant cluster of increased phosphorylated sites (FDR=0.06, subset to glycolysis proteins) in the immune-warm group (Figure 4C top panel, Table S4). In contrast, in the immune-cool group, multiple FA metabolic–related proteins (e.g., HIBCH, FASN, and HADH) display clusters of sites with decreased acetylation (FDR<0.12, subset to FA pathways). For instance, HADH has an essential role in FA beta oxidation, and the eight significantly reduced acetylation sites are clustered on the 3-hydroxyacyl-CoA dehydrogenase NAD-binding domain of the protein; this domain also contains the acetyl CoA binding sites, which would allow the binding and subsequent oxidative activity of the enzyme81,82 (Figure 4C, lower panel). Moreover, we detected a significant cluster of increased phosphorylation sites (S337, T338, and S339, FDR=0.0026) on the dehydrogenase E1 domain of BCKDH known to catalyze the overall breakdown of alpha-keto acids to acetyl-CoA83. This phosphorylation was shown to be mediated by BCKDK and inhibit BCKDH activity, further limiting the levels of acetyl-CoA and increasing FA oxidation to support the cell’s energy demand84(Figure S4F).
We then performed PTM-SEA to identify the main regulators of PTMs (e.g., kinases or phosphatases, Table S4). This analysis revealed high enrichment of (i) CDK activity in the immune-cool subtype, consistent with the high proliferation associated with this subtype (additionally supported by The Kinase Library enrichment results Figure S4G); and (ii) mTOR activity, a direct regulator of FA metabolism and oxidative phosphorylation as well as an indirect regulator of lipid homeostasis through SREBP185. Moreover, the immune-cool subtype showed an increase of FA uptake, both by transporters within the cell, such as CPT1A (FDR<7x10−7), and by cell-surface transporters including (i) FABP4, (ii) ABCA, and (iii) CD36 (FDR<1.3x10−4 immune- cool vs. immune-hot) (Figure S4H).
Recent studies have shown that lipid-enriched tumor microenvironments reduce the cytotoxicity of effector T cells21,86 since they cannot metabolize long-chain fatty acids, leading to lipotoxicity and exhaustion23. Therefore, we tested the correlation of FA acetylation levels with protein levels of immune-related effectors (Figure 4D). We observed a significant positive correlation between downregulating FA acetylation sites and immune response marker proteins in the IFNγ and cell cytotoxic pathways (Figure 4E). Some of the most significant associations were between GZMA and HADHA K214 & K759 (rho -0.3, p<1x10−13), and between CD38 and HADHB K277 & K253 (rho 0.35, p<1x10−13). Moreover, a Spearman correlation of single-sample gene set enrichment analysis (ssGSEA) for CD8+ T-cell abundance estimated from CIBERSORT showed FA as the fourth most significantly correlated association after interferon pathways (rho 0.23, FDR=2x10−13, Figure S4I). The observed associations between PTM-regulated biological processes in cancer cells and their neighboring immune cells are summarized in Figure 4F.
Alterations in histone regulation by PTMs in cancer-associated genes
Here, we leveraged the largest Pan-Cancer acetylation dataset to comprehensively study histone acetylation and phosphorylation patterns across the six cancer types with available acetylation data (Methods). To identify specific histone acetylation patterns, we classified histone-related genes into five categories: (i) the linker histone H1, (ii) the four core histones H2A (including MACROH2A1, MACROH2A2, H2A.X, H2AZ1), (iii) H2B, (iv) H3, and (v) H4 (Figure 5A). We found that histone acetylation partitioned into two structural groups, Group 1 (H3, H4, H1) and Group 2 (H2A, H2B), with significant correlations among the mean acetylation profiles of each group for pairs within the groups (all with rho>0.4 and p<2.2x10−16), and weaker to no correlation for pairs between the two groups (−0.15<rho<0.17) (Figure S5A). This is consistent with acetylation states coordination leading to enhanced nucleosome opening and subsequent gene activation87,88.
Figure 5 -.

Pan-Cancer histone regulation
A. Heatmap showing site level acetylation of various histone protein substrates across 6 cohorts. Tracks above show the cohort, cluster assignment, gender and smoking score.
B. Scatter plots showing the rankings of site-specific histone PTM levels and tobacco smoking mutational signature activities. 95% confidence intervals of the Spearman’s correlation coefficient determined by bootstrapping.
C. Bubble plot showing Pan-Cancer associations between key regulators of histone acetylation and histone acetylation sites.
D. Scatter plots showing the lasso regression associations between histone regulators and H2B acetylation levels across all tumors and in specific clusters in the dendrogram.
E. Heatmap showing the differentially acetylated histone sites in the Immune Cold subtype compared to all other immune subtypes.
F. Correlations between histone acetylation sites and close proximity phosphorylation sites.
Since tobacco smoking is known to impair HDAC activity and affect histone acetylation89, we sought to better characterize these effects by evaluating the correlation between the smoking mutational signature and histone acetylation in LUAD tumors, limiting to male patients in order to decouple effects due to strong association between gender and smoking (p=1x10−4, Figure S5B). We found two positive and two negative significant correlations (FDR≤0.1; Figure S5C)90–92. Among these correlations was the previously described dose-dependent relationship between smoking and H4–16 K9 and K13 acetylation (rho=-0.33, FDR=0.09). We also found positive correlations for H2AZ1 K8 & K12 and K12 & K14 acetylation (rho=0.34, 0.39, respectively; FDR=0.08, 0.06, respectively) (Figure S5C, Table S6). Acetylation of these sites has been shown to localize H2AZ1 to promoter regions of several cancer genes (e.g., ERBB3, CDK4, and RASEF)93,94 and to facilitate their transcription95 (Figure 5B). Next, to explore the effect of smoking on HDAC activity, we tested the correlation between smoking and phosphorylation of HDACs. We found six significant (FDR≤0.1) positive correlations on four proteins, which are all components of SIN3/HDAC complexes (Figure S5D). Among these correlations was phosphorylation of HDAC2 S422 (rho=0.36, FDR=0.04), which was shown to reduce deacetylase activity96. Moreover, phosphorylation of this site was mediated by CSNK2A1 kinase upon exposure to cigarette smoke extract96.
Following this analysis, we investigated potential transcriptional consequences of smoking-related changes in histone acetylation by correlating the smoking signature and ssGSEA pathway scores (Figure S5E). Among the top significant associations (FDR≤0.1 and rho≥0.15) was the expected upregulation of G2/M checkpoint genes, consistent with previous studies that associated cigarette smoke and increased proliferation (FDR=0.01, rho=0.26, Figure S5F, left)97,98. We also identified a significant positive correlation with mTOR signaling genes (FDR=0.01, rho=0.27, Figure S5F, right), which is consistent with previous studies and may play a role in lung tumorigenesis by altering cell proliferation and metabolism99,100.
Next, we investigated the association of key regulators (HATs, HDACs, and BRDs; Methods) with histone acetylation levels. We identified multiple positive associations between the protein abundance of histone acetyltransferases CBP/p300 and various acetyl-sites, including N-terminal H2B acetyl-sites such as K11, K15, K16, and K20 (0.2<β<0.52, Table S5), consistent with the reported substrate specificity of CBP/p300 for these sites (Figure 5C)101. The protein abundance of the histone acetyltransferase NCOA1, a known co-activator of CBP/p300, was also positively associated with H2BC9-K21 acetylation (β=0.29)101. Additionally, we identified novel associations such as the positive correlation of H3C1 at K36 acetylation with HAT1 (β=0.36). HAT1 was shown to promote acetylation of H3 at K14, and its expression was associated with poor prognosis across cancer types102,103. We also observed negative associations: HDAC5 protein abundance negatively correlates with H2BC14 K16 and K20 acetylation (β=−0.20). HDAC5, a known therapeutic target, was shown to have a role in cell differentiation, stemness, and proliferation in several cancer types104.
We also tested these associations within our 26 clusters and indeed found cluster-specific correlations (Table S5). For example, CBP showed a low correlation with H2B using all samples (β=0.25), but a much stronger correlation when evaluating tumors across cluster 22, which is enriched with brain tumors (β=0.65, Figure 5D). Furthermore, we tested the association between histone regulation and cancer hallmark pathways. We found increased acetylation levels of EP300 on known activating sites K1558 and K1560 when comparing the left vs. right side of the rightmost side of the dendrogram’s second level (C vs D, Figure 2A) (FDR=2.6x10−7)105, and a concordant increase in the acetylation of the N-terminal H2A and H2B acetylation sites regulated by CBP/p300 (H2AC21-K5K9, FDR=0.052; H2BC18-K16K20, FDR=1.3x10−4)101. Moreover, GSEA showed significant enrichment of E2F and MYC transcription targets in tumors from cluster C (both FDR=0.08). Consistently, we found a significant positive correlation between H3 acetylation and E2F, MYC, and G2/M checkpoint pathways across the six cohorts, likely reflecting increased transcription associated with cell proliferation. We also observed a significant positive correlation between the acetylation of H3 at K27 and K36 and MTORC1 signaling, consistent with previous studies106 .
We then focused on how metabolic shifts across our Pan-Cancer immune subtypes affect the regulation of histones. Our findings above indicated an increase in FA metabolism in immune-cool tumors (relative to immune-hot), and we also observe decreased acetylation at 22/61 histone acetylation sites relative to immune-hot and at 31/61 sites relative to immune-warm (FDR<0.1, Figure 5E, Table S5). As previously shown, these results reflect a possible association between histone acetylation and glycolytic flux as well as cellular acetyl-CoA abundance and availability in the different immune clusters107.
Finally, we analyzed the correlations between adjacent histone phosphorylation and acetylation sites to better understand their potential crosstalk (Methods). Out of 81 histone acetylsite and phosphosite adjacent pairs (up to five amino acids apart) tested globally, we identified 12 as significantly correlated (FDR<0.05, Table S5). For instance, H3F3A-S31 phosphorylation was strongly correlated with H3F3A-K27K36 acetylation across all samples (rho=0.38, FDR=6.2x10− 7), consistent with S31 phosphorylation stimulating H3-K2 acetylation through p300 activity108 (Figure 5F). In addition, the phosphorylation levels of H3-S28 and acetylation of H3-K27 were positively correlated (rho=0.27, FDR=3x10−5), consistent with S28 phosphorylation reducing K27 trimethylation and priming acetylation109.
Crosstalk between protein phosphorylation and acetylation in cancer
Motivated by the correlations between phosphorylation and acetylation of adjacent sites in histones, we aimed to systematically analyze this crosstalk across other proteins. Mechanistically, serine/threonine protein kinases are known for their substrate specificity based on the amino acid sequence surrounding their phosphorylation sites110–112. We therefore asked whether similarly lysine acetylation adjacent to phosphorylation sites can impact their ability to be phosphorylated.
We experimentally characterized lysine-PTM selectivity across 207 recombinant Ser/Thr kinases using degenerate peptide substrates that compared modified and unmodified lysine at the five adjacent amino acid positions in both the N- and C-terminal directions from the phosphoacceptor site110,112–115 (Figure 6A). Globally, we observed a general selection against substrates containing acetylated lysine across the kinome (Figure 6B). Nevertheless, there are some exceptions in which kinases favor acetylated lysine over unmodified lysine to carry out the phosphorylation of a nearby serine or threonine (Figure 6C). We observed a similar pattern of global selection against trimethylated lysine, but to a smaller extent than acetylation, possibly because trimethylation causes a smaller steric alteration and preserves the positive charge on lysine (Figure S6A–B). Together, our screen indicates that Ser/Thr kinases discriminate between the PTM states of lysine surrounding their phosphorylation sites and that lysine acetylation has the potential to regulate their function.
Figure 6 -.

Pan-Cancer acetylation and phosphorylation crosstalk
A. The Kinase Library overview - biochemical assay of a combinatorial peptide library with unmodified, methylated, or acetylated lysine for testing kinases affinity to peptides with modified lysins at ±5 positions relative to the Ser/Thr phospho-acceptor residue (excluding serine, threonine, and cysteine).
B. Box plot showing the average intensity for unmodified and acetylated lysine residues.
C. Heatmap showing ratio between mean intensities for acetylated and unmodified lysine residues. Kinases are colored according to their phylogenetic groups168.
D. Scatter plot showing the correlation between K23 acetylation levels and S28 phosphorylation levels on Histone H3–3A and their cohort distribution (top panel). Biochemical specificity assays showing AurB and PKACB phosphorylation between unmodified and acetylated peptides (bottom panel).
E. Volcano plot showing correlations between pairs of phosphorylation and acetylation sites. Negative correlations are highlighted.
F. Scatter plot showing the correlation between K1378 acetylation levels and S1375 phosphorylation levels on RSF1 and their cohort distribution (left panel). Biochemical specificity assays showing CDK1 phosphorylation between unmodified and acetylated peptides (right panel).
G. Inhibitory crosstalk proposed mechanism on RSF1.
Using these kinase specificity patterns, we could potentially identify the kinases involved in specific acetylation–phosphorylation crosstalks. Since different kinases may be active in different cell types and states, we first mapped the crosstalk in different dendrogram branches that may each display shared kinase activity due to their similar RNA, protein, and phosphorylation patterns. We searched for potential acetylation–phosphorylation crosstalk in adjacent pairs (up to five amino acids apart) on proteins globally as well as in the dendrogram clusters (Methods). We identified a negative correlation between H3–3A acetylation at K23 and phosphorylation at S28 in a dendrogram branch that includes terminal clusters 22 and 23 (rho=−0.58, FDR=0.04, Figures 6D, top), suggesting an inhibitory crosstalk uniquely present in these tumors compared to all tumors (rho=0.08, FDR=0.35 globally). Phosphorylation of S28 has been reported to activate transcription109, whereas acetylation at K23 inhibits transcription116. S28 on H3–3A is a reported substrate for members of the aurora kinase (AURK) and PKA families of kinases117,118. Consistent with this finding, our substrate motifs for AurB and PKACB show selection against acetylated lysine in this context (Figures 6D, bottom), indicating that K23 acetylation inhibits the ability of S28 to be phosphorylated by these upstream kinases.
We then explored the acetylation–phosphorylation crosstalk across other proteins and identified 3,952 adjacent pairs among 579 patients (Figure 6E, Methods). Among these, 74 pairs showed significant negative correlation between their levels (FDR<0.1). Among the most statistically significant examples (FDR=3x10−4) was S1375/K1378 on the centromeric protein RSF1 (Figure 6F left, Figure S6D, Table S6). RSF1 is an essential mediator of mitosis known to be overexpressed in many types of cancers119. CDK1 has been reported to phosphorylate S1375 on RSF1 during G2/M, which facilitates recruitment of the kinase PLK1 that promotes subsequent mitotic events120,121. In our peptide substrate assays, CDK1 strongly favored serines/threonines that contained lysines three positions in the C-terminal direction (+3K), matching the known motif for RSF1; moreover, the phosphorylation was almost entirely abolished when the lysine was acetylated (Figure 6F, right), consistent with CDK1’s reduced ability to phosphorylate S1375 when K1378 is acetylated, potentially explaining the observed negative correlation.
With our kinome-wide scoring system, we could also infer how phosphorylation of RSF1 facilitates the next step in this signaling cascade and the subsequent recruitment of PLK1 (Figure 6G). PLK1 binds to peptides containing phosphorylated serine or threonine that are directly preceded by an unmodified serine (S-pS/pT)122T1371 on RSF1 that matches this pattern. Therefore, when CDK1 phosphorylates S1375123,124, the phosphorylated peptide becomes a substrate for a second phosphorylation event by the ubiquitously expressed GSK3 kinases (GSK3 alpha/beta) on T1371, after which the phosphorylated peptide can be recognized by PLK1. Once lysine K1378 is acetylated, the entire cascade is inhibited. Altogether, this explains how crosstalk between S1375/K1378 on RSF1 ultimately affects the recruitment of PLK1.
Discussion
PTMs are core regulators of signal transduction, and they play major roles in protein–protein interactions, protein stability, and localization, among many other essential functions. In this study, we comprehensively investigated PTMs across 11 cancer types, and highlighted the contribution of PTMs to known cancer hallmark processes: (1) DNA repair, (2) immune response, (3) metabolism, (4) histone regulation, and (5) kinase regulation. We noted commonalities of these processes and PTM patterns across cancer types as well as important distinctions. This rich resource will enable additional investigation of PTMs across cancer types beyond the PTM work we describe here.
DNA repair deficiencies, such as HRD and MMRD, generate patterns of somatic mutations throughout tumor development, providing evidence of a given repair deficiency. Importantly, these mutational signatures do not necessarily reflect the current activities of repair pathways, which may be pertinent to understanding the variation in response to therapies that target DNA repair genes (e.g., PARP and POLQ inhibitors, etc.). In-depth analyses of DNA repair deficient cancers highlighted the ability of phosphorylation-focused analyses to reveal and characterize informative patterns that are undetectable at the genomic and transcriptomic levels. Through our analysis of HRD clusters, we found that significant differences in the phosphorylation of DNA repair proteins were strongly associated with hypoxia severity. We found decreased activity of several DNA repair proteins, including PARP1, in HRD tumors with chronic hypoxia, potentially affecting their response to PARP inhibitors. Our proteogenomic analysis of MMRD tumors linked recurrent RAD50 microsatellite indels with significant decreases in the abundance of the three proteins in the MRN complex, which is crucial for DSB sensing and signaling. Through site-specific phosphorylation analysis, we identified further evidence of DSB repair dysfunction, which may provide additional avenues for developing treatments for MMRD (i.e., MSI) cancers.
In general, immune responses are tightly regulated in order to tailor each response to a given threat encountered by the host, requiring rapid regulatory changes that can be achieved by PTMs. Similarly, cellular metabolism requires the same flexibility, and PTMs therefore play an essential role in regulating both immune and metabolic responses. As an example, these two processes can be linked with growing evidence that cancer cell regulation of lipid metabolism by PTMs on fatty acid enzymes can have an effect on the immune response21–23. In this study, we identified four immune RNA-based clusters with diverse metabolic phenotypes driven by acetylation. CLUMPS-PTM highlighted ALDOA, a glycolysis-related protein, which has both significant clusters of altered phosphorylation and acetylation sites in the immune-hot subtype that are associated with increased ALDOA activity. Inhibition of ALDOA in mice reduced lung metastases and prolonged survival125. The immune-cool subtype showed an increased activity of FA metabolism that was strongly correlated with reduced IFNγ expression, suggesting an important role of FA in immune suppression. A recent paper demonstrated that inhibition of FA oxidation in Acute myeloid leukemia (AML) can restore sensitivity to venetoclax and azacitidine in cells that became resistant126,127. These results highlight the potential of targeting lipid metabolism in cancer, as it may not only reduce the ability of tumor cells to produce higher levels of energy, but also promote a tumor microenvironment that is more conducive to immune cell infiltration and activation126,127. In addition, we identified an association between transcriptionally active metabolic pathways and reduced histone acetylation, potentially due to the reduced availability of acetyl CoA that is used by the cell for both acetylation and producing energy106; additional studies will be needed to further investigate this regulation.
Finally, we performed a comprehensive analysis of crosstalk between acetylation and phosphorylation using The Kinase Library33. This analysis revealed that most serine/threonine kinases disfavor acetylated lysine in close proximity to the phosphorylation site (shown by significantly negatively correlated pairs of neighboring acetyl/phospho sites), allowing us to then predict the responsible kinases for the crosstalk.
In summary, PTMs are an integral part of the tumor cell’s adaptation and response to intracellular and environmental changes. A deeper understanding of PTM-governed processes leading to cancer initiation and progression has the potential to uncover novel therapeutic targets, identify biomarkers of response to existing therapies, and extend our knowledge of cancer biology.
Limitations of the Study
Genomic-based Pan-Cancer studies have proved to be highly valuable resources for discovering new cancer driver genes and shared dysregulated pathways, as well as for identifying actionable therapeutic targets16–18. Our proteogenomic Pan-Cancer study was limited to 1110 samples across 11 tumor types, and we anticipate that larger-scale studies, including both more cases and more cancer types, will increase power to identify proteogenomic mechanisms underlying cancer. The analyses described in this study are all based on bulk tumor material. Similar to single-cell and spatial transcriptomic analyses128–131, technologies including single-cell proteomics and spatial proteomics or laser capture microdissection132–134 are likely to provide even more valuable insights into tumor heterogeneity and the contribution of specific cell types to cancer. While comprehensive PTM-focused research and analyses of PTM crosstalk are emerging fields, some shortcomings are worth noting: (i) current mass spectrometry analyses have relatively high false negative rates that limit our ability to perform crosstalk analyses among PTMs; (ii) to fully establish crosstalk relationship between PTMs a double MS searches for simultaneous detection of 2 or more PTMs would be needed; and (iii) while there is a growing body of phosphorylation databases and kinase prediction tools, parallel comprehensive acetylation databases and tools are currently lacking, and the functional effects of many of the acetylation sites reported in this study remain to be explored.
STAR★Methods
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead author, Gad Getz (gadgetz@broadinstitute.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Data Availability:
Raw and processed proteomics as well as open access genomic data can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov with dbGaP Study Accession: phs001287.v16.p6. Complete CPTAC pan-cancer controlled and processed data can be accessed via the Cancer Data Service (CDS). The CPTAC pan-cancer data hosted in CDS is controlled data and can be accessed through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis.
Instructions to access data:
Create an account on CGC, Seven Bridges (https://cgc-accounts.sbgenomics.com/auth/register
Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001287.v16.p6
Log into CGC to access Cancer Data Service (CDS) File Explore
Copy data into your own space and start analysis and exploration
Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data)
Code for the analysis and figures in this paper can be found at https://github.com/getzlab/CPTAC_PanCan_PTM_2023.
Software and code used in this study are referenced in their corresponding STAR Method sections and the Key Resource Table.
Key resources table
Experimental model and subject details
Human subjects and clinical data
In this study, a comprehensive dataset was assembled, comprising a total of 1110 patients from 11 different cohorts. The cohorts included 99 patients with glioblastoma(GBM)4, 110 patients with head and neck squamous cell carcinoma (HNSCC)5, 110 patients with lung adenocarcinoma (LUAD)6, 108 patients with lung squamous cell carcinoma (LSCC)7, 121 patients with breast cancer (BRCA)8, 140 patients with pancreatic ductal adenocarcinoma (PDAC)9, 110 patients with clear cell renal cell carcinoma (ccRCC)10, 82 patients with high-grade serous ovarian cancer (HGSC)11, 95 patients with uterine corpus endometrial carcinoma (UCEC)12, 96 patients with colorectal adenocarcinoma (COAD)13 and 39 patients with Medulloblastoma (MB)27.
This combined dataset comprises 531 males and 579 females, with an age range of 2–90 years (median age 62). Detailed clinical data can be found in the companion Pan-Cancer resource manuscript (CANCER-CELL-D-22–00603).
Method Details
Genomics Data processing
Harmonized genome alignment
WGS, WES, RNA-Seq sequence data were harmonized by NCI Genomic Data Commons (GDC) https://gdc.cancer.gov/about-data/gdc-data-harmonization, which included alignment to GDC’s hg38 human reference genome (GRCh38.d1.vd1) and additional quality checks. All the downstream genomic processing was based on the GDC-aligned BAMs to ensure reproducibility.
Somatic mutation detection
The Broad hg38 characterization pipeline Patient whole exome sequencing (WES) data, i.e. WES sequences of patients’ matched tumor and blood normal samples, were analyzed using the Getz Lab’s production hg38 WES characterization pipeline. While somatic whole genome sequencing data are available for 7 out of 10 cancer types, they were sequenced to an average coverage of 15X, which would limit our discovery of subclonal mutations or clonal mutations in low tumor purity and/or high ploidy samples.
The hg38 characterization pipeline runs on the Terra cloud-based analysis platform (https://terra.bio/). This pipeline is the standard computational workflow used by the Getz Lab for characterizing a tumor sample’s somatic variants through contrastive computational analysis of matched tumor-normal WES BAMs. The pipeline’s analysis steps are organized into five modules: (1) DNA Sequence Data Quality Control, (2) Somatic Copy Number Analysis, (3) Somatic Variant Discovery, which includes the discovery of SNVs and indels, (4) Post-Discovery Filtering and (5) merging of adjacent somatic SNPs into DNPs, TNPs and ONPs.
The DNA Sequence Quality Control module, at the head of the pipeline, employs (i) GATK4’s CalculateContamination (Ver GATK 4.1.4.1) tool to calculate the fraction of reads coming from cross-sample contamination and (ii) GATK4 Picard tools (ver GATK 4.0.5.1) to validate the BAM files and collect multiple classes of metrics that can be used to evaluate sequencing data quality. The pipeline’s Somatic Copy Number Analysis module runs the GATK4 Best Practices Workflow (ver GATK 4.1.4.1) for discovery of allele-specific copy-number alterations.
The Somatic Variant Discovery module employs MuTect137 for detection of somatic single nucleotide variants and Manta+Strelka v2135,136 for detecting small insertions and deletions (INDEL sizes up to 49 bases). Following this initial detection of somatic SNVs (SSNV) and INDELs DeTiN138 (v1.8.9) was run to rescue SSNVs and INDELs called by MuTect and Strelka that may have been misclassified as germline variants due to contamination of normal tissue with tumor cells. The resulting SSNV and indel VCFs are each run through the GATK4 Funcotator (ver GATK 4.1.4.1) to analyze detected variants for their function and produce annotated MAFs, which were then merged into a single MAF containing candidate SSNVs and indels.
The Post-Discovery Filtering module runs a collection of filters in parallel on the merged annotated MAF to eliminate artifacts, germline variants and common sequencing artifacts that occur in normal panels. The filtered variant calls coming out of each filter are then aggregated to create an “intersection MAF” containing only variants that pass all filters. These variants are then run through a mutation validator that validates the calls with any available orthogonal sequencing data (e.g., from WGS sequencing, targeted sequencing, low pass sequencing, RNA sequencing).
The final stage of the pipeline employs a SNVmerger subworkflow which merges SNPs to DNP/TNP/ONPs and writes the resulting oligonucleotides to a VCF, which are then re-annotated and merged into the earlier filtered/validated variant MAF.
Washington University characterization pipeline
In parallel to somatic mutation calling done by the Broad pipeline, somatic mutations and DNP calls were done by the Washington University characterization pipeline and are provided in detail in the companion Pan-Cancer Driver manuscript (CELL-D-22–01960).
Callset Harmonization
The per patient variant calls employed by the CPTAC PanCan working group were derived from the harmonization of variant calls made independently by the Broad and Washington University. ICE whole-exome capture technology was deployed by the Genomics Platform at the Broad Institute for all CPTAC projects. Therefore, as a first filtering step, we removed calls outside of the ICE capture interval list. A panel-of-normals built from an aggregation of normal blood samples from the CPTAC and TCGA cohorts, which is an integral part of the Broad’s somatic mutation calling pipeline, was used to filter recurrent artifacts arising from calls made by the Washington University pipeline. In addition, indels were left-aligned to make sure their representations were comparable.
While we got better concordance between the two pipelines, we observed 2 key differences:
Majority of divergent calls are of low AF (<0.05)
The Broad’s pipeline calls long MNP
To mitigate (1), we first removed all calls with Variant Allele Frequency (VAF) < 0.05 from both pipelines and rescued only high confident calls if either criteria is satisfied:
If a low VAF variant is called from both pipelines
If a low VAF is only called by either pipeline, but is a cancer hotspot defined in Hess 2019 150.
To mitigate (2), long MNPs were collapsed to shorter MNPs by imposing a more stringent merging criteria that requires a 2bp gap length at max.
C>A artifact in CPTAC2 cohorts
Using Asymtools242 we were able to identify a sequencing artifact affecting CPTAC2 whole exome sequencing. Asymtools2 is a framework for visualizing mutational strand asymmetries. Asymtools2 illustrated a biased enrichment of cytosine to adenine SNVs over guanine to thymine SNVs on the genomic reference strand at low allelic fractions, suggesting a previously described process driven by oxidative damage of guanine to 8-oxoguanine after bait-DNA hybridization151. We further partitioned the C>A and G>T mutations with an allele fraction of less than 0.1 into their trinucleotide contexts, establishing the G>T contexts as the null model for correction. We then corrected for the sequencing artifact by ranking each C>A mutation by its allelic fraction and removed mutations until the number of C>A mutation counts were equal to those of the G>T mutations for each context.
Functional Impact
Finally, the functional impact of harmonized calls was annotated with GATK Funcotator.
Germline SNP and short indel discovery from WES (Washington University in St Louis)
Germline variant calling was performed using the Washington University pipeline and is provided in detail in the companion Pan-Cancer Driver manuscript (CELL-D-22–01960).
RNAseq data processing and quantification
We processed the RNA-seq data from all cohorts using the GTEx/TOPMed pipeline described at https://github.com/broadinstitute/gtex-pipeline/blob/master/TOPMed_RNAseq_pipeline.md 143.The samples were aligned to the human reference genome GRCh38 with the GENCODE V34 gene annotation using STAR v2.7.5a; optical and PCR duplicates were identified with Picard 2.18.17 MarkDuplicates; quality control and gene-level quantification (in Transcripts per Million (TPM) units) were performed with RNA-SeQC 2.3.6 152, and isoform expression was quantified with RSEM 153. We defined ‘expressed’ across the combined 11 cohorts using the following criteria: both (i) ≥0.1 TPM in ≥20% of samples and (ii) ≥6 reads in ≥20% of samples.
Proteomics data processing
Proteomics LC-MS/MS data interpretation
MS/MS spectra obtained from proteins/ phosphosites and acetylation sites were interpreted by Spectrum Mill (SM) v 7.08 (proteomics.broadinstitute.org) to provide identification and relative quantitation at the protein, peptide, and post-translational modification (PTM) phospho and acetyl site levels.
Personalized sequence databases
For searching with LC-MS/MS all available datasets we generated a cohort-level personalized protein sequence database for each tumor type starting with a base human reference proteome to which we appended non-redundant somatic and germline variants and indels for each of the ~100 participants/cohort. The base proteome consisted of the human reference proteome GENCODE 34 (ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_34/) with 47,429 non-redundant protein coding transcript biotypes mapped to the human reference genome GRCh38, 602 common laboratory contaminants, 2043 curated smORFs (lncRNA and uORFs), 237,427 novel unannotated ORFs (nuORFs) supported by ribosomal profiling nuORF DB v1.0 154 for a total of 287,501 entries which yield 16,645,198 distinct 9-mers. The nuORFs alone yield 8,612,372 distinct 9-mers and thus increase the peptide search space by only a factor of ~2. The personalized protein sequence entries were prepared by processing the individual participant’s somatic and germline variant calls from whole exome sequencing data, described above, using QUILTS v3 (Ref. 155) with no further variant quality filtering using an Ensembl v100 reference proteome and reference genome for sequence identifiers consistent with the variant calling. GENCODE v34 is a contemporaneous subset of Ensembl v100 (March 2020). Using the SM Protein Database utilities, the base reference proteome and individual patient proteomes were combined and redundancy removed to produce a cohort-level protein sequence database and a variant summary table to enable subsequent mapping of sequence variants identified in TMT multiplexed LC-MS/MS datasets back to individual patients.
Spectrum quality filtering
For all datasets, similar MS/MS spectra with the same precursor m/z acquired in the same chromatographic peak were merged, the precursor MH+ inclusion range was 800–6000, and the spectral quality filter was a sequence tag length > 0 (i.e., minimum of two peaks separated by the in-chain mass of an amino acid).
MS/MS search conditions
Using the SM MS/MS search module for all datasets included the next parameters: “trypsin allow P” enzyme specificity with up to 4 missed cleavages; precursor and product mass tolerance of ± 20 ppm; 30% minimum matched peak intensityScoring parameters were ESI-QEXACTIVE- HCD- v2, for whole proteome datasets, and ESI-QEXACTIVE-HCD-v3, for phosphoproteome and acetylome. Allowed fixed modifications included carbamidomethylation of cysteine and selenocysteine. TMT labeling was required at lysine, but peptide N-termini were allowed to be either labeled or unlabeled. Allowed variable modifications for whole proteome datasets were acetylation of protein N-termini, oxidized methionine, deamidation of asparagine, hydroxylation of proline in PG motifs, pyro-glutamic acid at peptide N-terminal glutamine, and pyro-carbamidomethylation at peptide N-terminal cysteine with a precursor MH+ shift range of -18 to 97 Da. For all PTM-omes variable modifications were revised to omit hydroxylation of proline and allow deamidation only in NG motifs. The phosphoproteome was revised to allow phosphorylation of serine, threonine, and tyrosine with a precursor MH+ shift range of -18 to 272 Da. The acetylome was revised to allow acetylation of lysine with a precursor MH+ shift range of -400 to 70 Da.
PTM site localization
Using the SM Autovalidation and Protein/Peptide Summary modules for the PTM-ome datasets results were filtered and reported at the phospho and acetyl site levels. When calculating scores at the variable modification (VM) site level and reporting the identified VM sites, redundancy was addressed in SM as follows: a VM-site table was assembled with columns for individual TMT-plex experiments and rows for individual VM-sites. PSMs were combined into a single row for all non-conflicting observations of a particular VM-site (e.g., different missed cleavage forms, different precursor charges, confident and ambiguous localizations, and different sample-handling modifications). For related peptides, neither observations with a different number of VM-sites nor different confident localizations were allowed to be combined. Selecting the representative peptide for a VM-site from the combined observations was done such that once confident VM- site localization was established, higher identification scores and longer peptide lengths were preferred. While an SM PSM identification score was based on the number of matching peaks, their ion type assignment, and the relative height of unmatched peaks, the VM site localization score was the difference in identification score between the top two localizations. The score threshold for confident localization, > 1.1, essentially corresponded to at least 1 b or y ion located between two candidate sites that has a peak height > 10% of the tallest fragment ion (neutral losses of phosphate from the precursor and related ions as well as immonium and TMT reporter ions were excluded from the relative height calculation). The ion type scores for b-H3PO4, y-H3PO4, b-H2O, and y-H2O ion types were all set to 0.5. This prevented inappropriate confident localization assignment when a spectrum lacked primary b or y ions between two possible sites but contained ions that could be assigned as either phosphate-loss ions for one localization or water loss ions for another localization.
Protein grouping of PSMs, peptides and PTM sites
Using the SM Autovalidation and Protein/Peptide summary modules results were filtered and reported at the protein level. Identified proteins were combined into the same protein group if they shared a peptide with sequence length greater than 8. A protein group could be expanded into subgroups (isoforms or family members) when distinct peptides were present which uniquely represent a subset of the proteins in a group. For the proteome dataset the protein grouping method “expand subgroups, top uses shared” (SGT) was employed which allocates peptides shared by protein subgroups only to the highest scoring subgroup containing the peptide. For the PTM-ome datasets the protein grouping method “unexpand subgroups” was employed which reports a VM-site only once per protein group allocated to the highest scoring subgroup containing the representative peptide. The SM protein score is the sum of the scores of distinct peptides. A distinct peptide is the single highest scoring instance of a peptide detected through an MS/MS spectrum. MS/MS spectra for a particular peptide may have been recorded multiple times (e.g., as different precursor charge states, in adjacent bRP fractions, modified by deamidation at Asn or oxidation of Met, or with different phosphosite localization), but are still counted as a single distinct peptide.
Peptide spectrum match (PSM) filtering and false discovery rates (FDR)
Using the SM Autovalidation module peptide spectrum matches (PSMs) for individual spectra were confidently assigned by applying target-decoy based FDR estimation to achieve <1.0% FDR at the PSM, peptide, VM site and protein levels. For the whole proteome dataset thresholding was done in 3 steps: at the PSM level, the protein level for each TMT-plex, and the protein level for the cohort of 2 TMT-plexes. For the PTM omes: phosphoproteome and acetylome datasets thresholding was done in two steps: at the PSM level for each TMT-plex and at the VM site level for the cohort of 2 TMT-plexes. In step 1 for all datasets, PSM level autovalidation was done first and separately for each TMT-plex experiment using an auto-thresholds strategy with a minimum sequence length of 7; automatic variable range precursor mass filtering; with score and delta Rank1 - Rank2 score thresholds optimized to yield a PSM level FDR estimate for precursor charges 2 through 4 of < 0.8% for each precursor charge state in each LC-MS/MS run. To achieve reasonable statistics for precursor charges 5–6, thresholds were optimized to yield a PSM-level FDR estimate of < 0.4% across all runs per TMT-plex experiment (instead of per each run), since many fewer spectra are generated for the higher charge states.
In step 2 for the PTM-omes: phosphoproteome and acetylome datasets VM site polishing autovalidation was applied across both TMT plexes to retain all VM site identifications with either a minimum id score of 8.0 or observation in n TMT plexes (n=4, 3, or 2 if > 20, 7, or 1 plexes/cohort , respectively). The intention of the VM site polishing step is to control FDR by eliminating unreliable VM site level identifications, particularly low scoring VM-sites that are only detected as low scoring peptides that are also infrequently detected across both TMT plexes in the study. Using the SM Protein/Peptide Summary module to make VM-site reports the ubiquitylome and acetylome datasets are further filtered to remove peptides ending with the regular expression [^K][^K]k since trypsin and Lys-C cannot cleave at a acetylated lysine. The [^K] means retain if unmodified Lys present in one of the last two positions to allow for a missed cleavage with ambiguous PTM-site localization. C-terminally acetylated lysines are present in the acetylome dataset, but have been shown to arise from artifactual modification during TMT-labeling after trypsin digestion.
In step 2 for the whole proteome dataset, protein polishing autovalidation was applied separately to each TMT-plex experiment to further filter the PSMs using a target protein level FDR threshold of zero. The primary goal of this step was to eliminate peptides identified with low scoring PSMs that represent proteins identified by a single peptide, so-called “one-hit wonders.” After assembling protein groups from the autovalidated PSMs, protein polishing determined the maximum protein level score of a protein group that consisted entirely of distinct peptides estimated to be false-positive identifications (PSMs with negative delta forward-reverse scores). PSMs were removed from the set obtained in the initial peptide level autovalidation step if they contributed to protein groups that had protein scores below the maximum false-positive protein score. Step 3 was then applied, consisting of protein polishing autovalidation across all TMT plexes in a cohort together using the protein grouping method “expand subgroups, top uses shared” to retain protein subgroups with either a minimum protein score of 25 or observation in TMT plexes (n=4, 3, or 2 if > 20, 7, or 1 plexes/cohort, respectively). The primary goal of this step was to eliminate low scoring proteins that were infrequently detected in a cohort. As a consequence of these two proteins- polishing steps, each identified protein reported in the study comprised multiple peptides, unless a single excellent scoring peptide was the sole match and that peptide was observed in multiple TMT-plexes.
Quantitation using TMT ratios
Using the SM Protein/Peptide Summary module, a protein comparison report was generated for the proteome dataset using the protein grouping method “expand subgroups, top uses shared” (SGT). For the PTM omes: phosphoproteome and acetylome datasets Variable Modification site comparison reports limited to either phospho, or acetyl sites, respectively, was generated using the protein grouping method “unexpand subgroups.” Relative abundances of proteins and VM-sites were determined in SM using TMT reporter ion log2 intensity ratios from each PSM. TMT reporter ion intensities were corrected for isotopic impurities in the SM Protein/Peptide Summary module using the afRICA correction method, which implements determinant calculations according to Cramer’s Rule and correction factors obtained from the reagent manufacturer’s certificate of analysis for each cohort. Each protein-level or PTM site-level TMT ratio was calculated as the median of all PSM-level ratios contributing to a protein subgroup or PTM site. PSMs were excluded from the calculation if they lacked a TMT label, had a precursor ion purity < 50% (MS/MS has significant precursor isolation contamination from co-eluting peptides), or had a negative delta forward-reverse identification score (half of all false-positive identifications). Using the SM Process Report module non-quantifiable proteins and PTM sites (ex: unlabeled peptides containing an acetylated protein N-terminus and ending in arginine rather than lysine) were removed, and median/MAD normalization was performed on each TMT channel in each dataset to center and scale the aggregate distribution of protein-level or PTM site-level log-ratios around zero in order to nullify the effect of differential protein loading and/or systematic MS variation.
Normalization of phosphosites and acetylation sites
For downstream analyses with PTM data, we perform ordinary least squares fit using statsmodels.regression.linear_model.OLS for every matched value of protein and PTM site aligned by accession number (RefSeq) (Figure S1). The residuals from this are “corrected” phosphoproteome and acetylome values we term as “phosphoproteome_res” or “acetylome_res.”
Patient Signatures & Clustering
Transcriptomics
For downstream analyses, TPM values were scaled using DESeq2 size factors and then log2(x+1) transformed. Three cohorts (BRCA, HGSC, COAD, from CPTAC2) were sequenced with a polyA selection protocol, with the remainder sequenced with a total RNA protocol (rRNA-depleted using RiboZero, from CPTAC3). Medulloblastoma was also sequenced with a total RNA protocol. This protocol batch effect was regressed out from the log-transformed values using COMBAT 156. Finally, the 5,000 genes with the highest coefficient of variation were selected as inputs to clustering.
Proteomics
We first selected phospho-sites that were fully localized using mass spectrometry. Then, within each cohort, we filtered out any proteins or phosphoproteins that were identified based on SM criteria in less than ≤25% of samples (Figure S1A). After this, we further subset the feature space to include only detected proteins & phospho-sites across all 11 cohorts to avoid imputation across entire cohorts. Within each cohort, we then used K-nearest neighbors to impute missing values with sklearn.impute.KNNImputer with K=5. Finally, to correct phosphoproteome levels for protein abundance, we performed ordinary least squares fit described in the Proteomics Data Processing Section (above) with filtered and imputed data (Figure S1A).
Combining Multi-Omic Data
Across all 11 cohorts, the shared data types available for clustering were whole transcriptome RNA-seq, proteome, and phosphoproteome. To harmonize the RNA data with the normally distributed proteomic data, we applied an inverse normal transformation, median centered the data, and scaled the data using median absolute deviation. We selected samples that had matched RNA, protein, and phosphoprotein in the entire dataset and concatenated these 3 matrices. To reduce the transcriptome space to a comparable feature size to the proteome data, we selected the top 5,000 highly variable genes, ranked by coefficient of variation. The combined matrix contained gene expression for 5,000 highly variable mRNA protein coding genes, 5,716 proteins, and 3,341 phosphoproteins for 1,110 patients across 11 cancer types. Prior to any cohort-level correction, we found that cohort and tissue-specific effects were the dominant source of variation in multi-omic data (Figure S1C–D). We therefore performed cohort-level batch correction by regressing out the cohort effects (as dummy-coded covariates/ indicator variables) from the combined matrix.
Multi-Omic Signatures
We derived the expression signatures using SignatureAnalyzer (https://github.com/getzlab/SignatureAnalyzer), a Bayesian variant of non-negative matrix factorization (ARD-NMF)32,157,158. To use this tool with zero-centered, normally distributed data, SignatureAnalyzer splits the input matrix into positive and negative matrices before running the decomposition (Table S2). L2 priors were imposed on both the W and H matrices, and a Gaussian objective function was used. SignatureAnalyzer was run 100 times with random initialization, and the solution with the best objective function with the mode number of signatures (k=33) was selected (robustness analysis and additional support is provided in Supplementary note 1). To annotate these 33 signatures, we performed ranked gene-set enrichment analysis using fGSEA with factor weight as the rank.
Sample-Sample Clustering
To further cluster samples using the 33 derived expression signatures, we created a similarity matrix based on cosine similarity of the H-matrix (samples x signatures) derived from the SignatureAnalyzer algorithm. We then performed hierarchical clustering using euclidean distance as the metric and Ward linkage using SciKit-learn. For downstream analysis, we compared each pair of sample clusters at each split of the dendrogram (see Differential Expression).
Immune clustering
To estimate the abundance of immune cell-types and signatures in each sample, we ran multiple bulk RNA deconvolution approaches. We ran CIBERSORT with the LM22 signature matrix 73 using the re-processed, CPTAC transcriptomic TPM data for all 11 cohorts. We also ran ESTIMATE 71 as a separate approach for estimating tumor purity and immune infiltration using TPM data. Finally, we ran ImmuneSubtypeClassifier 72 to classify samples into different “immune types”. The methods for the immune analysis are provided at https://github.com/getzlab/CPTAC_PanCan_2021/blob/master/analysis/Fig_immuno_metabolis m/runImmuneDeconv.R. To identify broad immune clusters to probe differences in immune signals and changes in metabolic levels using matched protein and PTM data, we ran gene set enrichment analysis using the gene sets from CIBERSORT’s LM22 matrix on the z-scored transcriptomic TPM data. Next, we performed hierarchical clustering using euclidean distance and Ward-linkage with SciKit-learn 159 and selected K=4 to identify 4 broad “immune clusters” for downstream use. To perform differential expression analysis for each immune subtype, we used a ‘one vs. rest’ approach. We additionally corrected for cancer type (i.e. cohort) by modeling it as a fixed covariate along with the estimated immune abundances from CIBERSORT for “Macrophages_M0”, “Macrophages_M1”, “Macrophages_M2”, “T_cells_CD8”, and “T_cells_CD4_memory_resting” to better separate tumor intrinsic expression from contributions of the immune microenvironment.
Interpretive data analysis
Variant call tools
GISTIC
The Genomic Identification of Significant Targets in Cancer (GISTIC2.0) algorithm 141 was used to identify significantly amplified or deleted focal-level and arm-level events, with q value <0.25 considered significant. The following parameters were used: Amplification Threshold = 0.1, Deletion Threshold = 0.1, Cap Values = 1.5, Broad Length Cutoff = 0.98, Remove X-Chromosome = 0, Confidence Level = 0.99, Join Segment Size = 4, Arm Level Peel Off = 1, Maximum Sample Segments = 2000, Gene GISTIC = 1.
Each gene of every sample is assigned a thresholded copy number level that reflects the magnitude of its deletion or amplification. These are integer values ranging from -2 to 2, where 0 means no amplification or deletion of magnitude greater than the threshold parameters described above. Amplifications are represented by positive numbers: 1 means amplification above the amplification threshold; 2 means amplification larger than the arm level amplifications observed in the sample. Deletions are represented by negative numbers: -1 means deletion beyond the threshold; -2 means deletions greater than the minimum arm-level copy number observed in the sample.
MutSig2CV
The somatic variants were filtered through a panel of normals to remove potential sequencing artifacts and undetected germline variants. MutSig2CV 142 was run on these filtered results to evaluate the significance of mutated genes and estimate mutation densities of samples. These results were constrained to genes in the Cancer Gene Census 160, with false discovery rates (q values) recalculated. Genes of q value < 0.1 were declared significant.
Mutational signatures using SignatureAnalyzer
Mutational signatures were extracted for all cohorts excluding MB since WES data was unavailable. This analysis was done using SignatureAnalyzer (https://github.com/getzlab/SignatureAnalyzer), a Bayesian Non-negative matrix factorization algorithm that infers an optimal collection of signatures from the data (Additional details are provided in supplementary note 1). We performed a multi-step signature extraction workflow to mitigate noise and bias introduced by hypermutated samples with high leverage, an issue particularly exacerbated when solely using exomes.
We first focus on the 13 patients with missense mutations in the exonuclease domain of POLE or POLD1 (referred to as POLE-exo* or POLD-exo*) since they have a unique mutational signature that requires a special analysis. For these tumors, we applied SignatureAnalyzer using the 96 tri-nucleotide sequence context for single-base substitutions (SBSs) and a length-based context for insertions and deletions161. Analyzing these tumors using this particular spectra allowed us to detect 7 distinct signatures, 6 of which had strong similarity (cosine similarity > 0.75) with the previously reported signatures in POLE/POLD-exo* tumors161. In order to control for a single DNA repair deficiency in our study, we do not include POLE/POLD-exo* samples in downstream MMRD (i.e., MSI) analyses.
We then applied SignatureAnalyzer on the remaining 1056 samples using the standard composite spectra that includes the 96 tri-nucleotide sequence context for single-base substitutions (SBSs), 78 double-base substitutions (DBSs), and 83 indel features (ID), as performed in Alexandrov, Kim, Haradhvala, et. al. Nature (2020)43. Among the extracted signatures was one resembling a signature (SBS15) associated with MMRD in previous studies (cosine similarity 0.82). We inspected the distribution of this MMRD signature in COAD and UCEC samples, which are known to have higher incidences of MMRD (i.e. MSI). The minimum signature weight in these samples with evidence of mismatch repair deficiency, as evidenced by their non-trivial contributions from the signature, was 72 mutations and 19% of their mutations were attributed to this signature. Thus, we set a threshold of 72 mutations and 19% of mutations from this SBS15-like signature to identify MMRD tumors across our dataset (Figure S3B). Finally, since several known signatures associated with MMRD were merged together in the composite analysis, we re-ran SignatureAnalyzer using only the 96 SBS features on the MMRD samples and indeed recapitulated (cosine similarity > 0.85) the known MMRD-associated signatures from the COSMIC v3 catalog of signatures.
We partitioned the remaining patients into three groups by their tissue type: tobacco smoking related included LSCC, LUAD, and HNSCC, homologous recombination deficiency (HRD) related included BRCA, OV, and PDAC, and non-homologous recombination deficiency related included CCRCC, COAD, GBM, and UCEC. We used the tri-nucleotide SBS context to extract signatures from the HRD related and non-HRD related tumors, and we characterized these signatures by computing cosine similarity with the COSMIC v3 signatures.
Tobacco smoking and UV exposure (in HNSCC cancers) serve as primary mechanisms of mutagenesis in the tobacco smoking related group, and these processes have been shown to cause a wide range of single and double base substitutions as well as indels 43. Thus, we employed a composite spectra to isolate the effects of the various mutational processes in these patients, and characterized them by computing their cosine similarity to those found in the PCAWG study 43.
Identifying homologous recombination deficiency using whole exome data remains challenging due to its flat single-base substitution landscape as well as a reduction in microhomology coverage in exomes. Further complicated by the composite reference’s whole genome calibration, we opted to use the trinucleotide SBS spectra for this group of patients, as well as for the remaining COAD, UCEC, CCRCC, and GBM patients.
As previously described, whole exome sequencing provides less power for non-negative matrix factorization, such that signature bleeding can present particularly misleading attributions for low mutation burden patients. In order to more confidently classify HRD tumors, we leveraged germline exome data to identify tumors with known pathogenic variants from the ClinVar database or variants considered as “HIGH IMPACT” by ClinVar on BRCA1, BRCA2, and PALB2. We found that breast and ovarian tumors with these variants that also showed relatively high contributions from the HRD mutational signature had more than 45 mutations attributed to the signature (Figure S3B). Thus, we set our threshold for HRD classification to any tumor with greater than or equal to 45 mutations attributed to the HRD mutational signature.
HRD vs HRP Analysis
We note that classifying HRD tumors using only exome somatic and germline mutation data remains challenging, particularly due to substantially low power to detect microhomology indels, a crucial component to HRD detection 162. To prevent over-classification of HRP tumors for this analysis, we selected the bottom 20% of samples based on their HRD mutational signature contributions from both HGSC and BRCA cohorts. In order to limit repair deficiencies to HR, we excluded any MMRD or POLE exonuclease mutant tumors. In addition, due to the small number of PDAC classified as HRD (4 patients), we focused this analysis on breast and ovarian HRD tumors.
Differential Expression
RNA
Differential expression was performed using Limma-Voom163. The trimmed mean of M values (TMM) between-sample normalization164 was applied to counts using calcNormFactors, the voom transformation was applied using limma::voom, limma::lmFit for the moderated t-test, followed by empirical bayes shrinkage with limma::eBayes. The cancer type (i.e. cohort) was modeled as a fixed covariate in each analysis unless otherwise specified. FDR is computed using the Benjamini-Hochberg procedure. Versions used were edgeR_3.28.1, limma_4.32.2, and R version 3.6.1.
Protein, Phosphorylation, Acetylation
Differential expression was performed using Limma on median-MAD normalization matrices output from SpectrumMill v 7.08. limma::lmFit was used for the moderated t-test, followed by empirical bayes shrinkage with limma::eBayes. No imputation is performed prior to differential expression analyses. Proteins & PTM-sites were filtered out if they were present in <10 patients in either group being compared. The cancer type (i.e. cohort) is modeled as a fixed covariate in each analysis unless otherwise specified. Versions used limma_4.32.2 and R version 3.6.1.
fGSEA
Gene-set enrichment analysis was performed using fGSEA (http://bioconductor.org/packages/release/bioc/html/fgsea.html). To evaluate RNA- and protein-level enrichment in the context of differential expression analyses, we ranked genes by the product of log2(Fold Change) and -log10(P-value). We analyzed the enrichment of phosphorylation and acetylation of gene sets in differential expression analyses by collapsing each gene to its maximum absolute value product of log2(Fold Change) and -log10(P-value) of all corresponding peptides. To apply fGSEA to the results of SignatureAnalyzer, we performed ranked gene-set enrichment analysis by ranking the factor weights (i.e., the weights within each signature were used to rank the genes to carry out ranked gene-set enrichment analysis for each signature).
Dedicated tools for PTM analysis
Additional details and use cases for the PTM dedicated tools can be found in Supplementary Note 2.
PTM-SEA
We evaluated site-specific phosphorylation pathway enrichment using PTM-SEA (https://github.com/broadinstitute/ssGSEA2.0). Enrichment was run using the sites’ flanking amino acids, using the ptm.sig.db.all.flanking.human.v1.9.0.gmt database. We employed the heuristic method introduced by Krug et al. 35 to deconvolute multiple phosphorylated peptides to separate data points (for differential expression analyses, ranks were based on −10 * log10 (p value) * sign(log2(fold change)); for SignatureAnalyzer results, the factor weights of phosphorylation features were used at the rank).
CLUMPS-PTM
Mapping Sites
Every PTM site detected in this study was first mapped to UniprotKB to select for canonical isoforms and for ease of downstream alignment with PDB structures. blastp+ was run on every fasta sequence from the CPTAC reference (RefSeq) used for mass-spec quantification for Spectrum Mill and queried to the entire UniprotKB Sequence database. To select the appropriate hit for each RefSeq ID blasted to UniprotKB, the top hit by identity overlaps from blastp+ was selected after filtering for Uniprot IDs found in the SIFTS protein database. The SIFTS protein database was used for highly curated annotations between Uniprot protein IDs and their respective PDB structures. Next, each matched Uniprot was selected and every matching PDB in the SIFTS protein database was checked for sequence overlap. The DBREF entry in each PDB header was used to identify the offset between the Uniprot sequence and PDB sequence. Finally, the PDB entry with the most overlap between sites found in CPTAC and longest sequence length was selected as the matching PDB entry.
Algorithm
The method is based on the CLUMPS method for detecting significant clusters of mutations in 3D protein structures165. Here we search for significant clustering of differentially acetylated/phosphorylated sites. For sites mapped to an individual PDB structure, we computed an initial weighted average proximity (WAP) score based on matched PTM sites. Succinctly, WAP scores (see formula below) are a summation of all residue pairs (and), weighted by the product of the strengths of each individual residues (or respectively) and their distances (with a Gaussian decreasing weight using a scale parameter ) in the 3D protein structure. The original CLUMPS was designed to analyze mutations and the strength of each residue was the number of mutated patients at that residue. In CLUMPS-PTM, we use the differential phosphorylation/acetylation of each PTM-modified residue. The formula is:
where and are protein residues; is the Euclidean distance (in Å) between the centroids of these residues; and are the weights of each modification (i.e log2(fold-change) * -log10(FDR) in CLUMPS-PTM); is distance scale parameter (in Å). Empirical p-values are calculated by permuting the altered residues in the protein. In CLUMPS-PTM, we only test permutations of PTMs to other PTM-possible sites (e.g., only lysines are sampled for acetyl-sites). In our analysis, we wanted to average across a range of scale parameters and therefore used t=3, 4.5, 6, 8, 10, and run 10,000 permutations for each, and generate a mean empirical p-value across these parameter values. Finally, we used sites that mapped to the PDB structure and were differentially expressed (log2(fold change)>0; see Differential Expression). Two steps were taken before FDR correction of the empirical p-values. First, if the empirical p-values were zero, representing an insufficient number of permutations, we set the p-values to 0.1 over the number of permutations. Next, we reduced the number of hypotheses tested by excluding proteins from the FDR procedure if the most significant p-value that the protein and associated sites could theoretically yield is greater than 0.1. The minimal theoretical p-value is given by .
Finally, the Benjamini-Hochberg procedure was used to correct empirical p-values for multiple hypothesis testing. The code to perform all analyses is available in a python package at https://github.com/getzlab/CLUMPS-PTM.
The Kinase Library
Serine/threonine kinase substrate specificity assays: Assays, matrix processing, and scoring process were previously described at 33.
The Kinase Library enrichment analysis
The phosphorylation sites detected in this study were scored by all the characterized kinases (303 S/T kinases), and their ranks in the known phosphoproteome score distribution were determined as described above (percentile score). For every non-duplicate, singly phosphorylated site, kinases that ranked within the top-15 kinases for the S/T kinases were considered as biochemically predicted kinases for that phosphorylation site. Towards assessing a kinase motif enrichment, we compared the percentage of phosphorylation sites for which each kinase was predicted among the significantly downregulated/upregulated phosphorylation sites (i.e sites with FDR ≤ 0.1), versus the percentage of biochemically favored phosphorylation sites for that kinase within the set of unregulated (non-significant) sites in this study (sites with FDR > 0.1). Contingency tables were corrected using Haldane correction (adding 0.5 to the cases with zero in one of the counts). Statistical significance was determined using one-sided Fisher’s Exact test, and the corresponding p-values were adjusted using the Benjamini-Hochberg procedure. Kinases that were significant (FDR ≤ 0.1) for both upregulated and downregulated analysis were excluded from downstream analysis. Then, for every kinase, the most significant enrichment side (upregulated or downregulated) was selected based on the adjusted p-value and presented in the bubble plot. Bubble plots were generated with size and color strength representing the adjusted p-values and frequency factors respectively, only displaying significant kinases.
Modified lysine peptide library assays
Reagents used for the peptide library experiments include: Kinase substrate library (Anaspec). Streptavidin conjugated membranes (Promega). List of kinases information can be found in 112. To determine the substrate motifs, we performed in vitro phosphorylation assays with recombinant kinases on an oriented peptide array library of design:
in the presence of ATP[γ-32P]. Unmodified, tri-methylated, and acetylated lysines were fixed in every position across the peptide. Reactions were carried out in their designated buffers plus 20 μm ATP and 0.4 μCi of (33 nm) [γ-32P]ATP) at 30°C for 90 min. The peptides were spotted onto streptavidin-coated filter sheets (Promega SAM2 biotin capture membrane) and visualized by phosphorimaging on Typhoon FLA 7000. Detailed information on the protocol is provided elsewhere 110,115.
CausalPath
For each differential expression table that we generated in this study, we applied the CausalPath method34,166 to identify possible cause-effect relations between the detected differential values, and to understand the molecular signaling behind those changes. CausalPath uses literature-curated human mechanistic pathways to identify which protein activities have control over the observable features in the omics studies. The method uses this information to do logical reasoning over the observed differences, and generates a network of causal relations. Generated networks are tested by data label randomization to identify enrichments that indicate activation/inhibition of certain regulators.
In these CausalPath analyses, we integrated global protein, phosphoprotein, acetylprotein and mRNAseq datasets, and used a 0.1 FDR threshold. To include biologically relevant edge cases, we included RAD18 S99, XRCC1 S475/S485/T488, and CDK1 T14 (all FDR=0.102) for the acute versus chronic hypoxia HRD analysis, TOP2A S1247 (FDR=0.102) for the global HRD vs. HRP analysis, and PRKDC S3205 (FDR=0.1009) for the MMRD vs. MMRP analysis. To manage the complexity of the result networks and to focus on specific pathways, we generated sub-networks of results by taking graph neighborhoods of pre-defined gene sets encompassing DNA damage response (DDR). The DDR gene set (Supplementary Table S3) includes the union of the Reactome DNA Repair gene set and the CPTAC DNA-Damage Response (DDR) Working Group gene set (unpublished). We also included CDK1/2/4/6, AURKA, AURKB, PLK1/2/3/4/5, and WEE1 to account for cell cycle checkpoint kinases.
We also used CausalPath to investigate the downstream effects of the kinase activity predictions made by the Kinase Library method. We inserted the predictions of the Kinase Library as custom hypotheses to CausalPath to find out which differential changes are compatible/explainable by those predicted activities.
We used the Newt software 167 to visualize the CausalPath results and to generate Figs. 3E,H, and S3C.
Histone analysis
For Histone analysis we used 6 cancer types with available acetylation data (5 from the CPTAC cohorts): breast, uterine, glioblastoma, lung squamous, and lung adenocarcinoma in addition to one external dataset, medulloblastoma, that was generated and harmonized in the same manner as the CPTAC cohorts. For the global analysis, we included only acetylation sites that were detected in all cohorts. These summed up to a total of 61 histone acetylation sites: 21 sites on H1, 10 sites on H2A, 16 sites on H2B, 10 sites on H3, and 4 sites on H4.
Histone-level correlations
The imputed acetylation data was subset for histone genes by mapping RefSeq IDs to HGNC IDs, and within each group of histones (H1, H2A, H2B, H3, H4) the relative abundance of all sites within this group was averaged. Spearman correlations and p values were calculated for each of the 10 pairs.
Histone acetylation and smoking analysis
We used Spearman’s rank correlation to evaluate associations between the tobacco smoking mutational signature and histone acetylation and deacetylase phosphorylation, and we used Benjamini-Hochberg to control the FDR. We additionally calculate the 95% confidence intervals for each correlation shown in Figure 5B through bootstrapping. We sampled from the data with replacement 10,000 times and computing the Spearman’s correlation coefficient each time. Using this distribution of correlation coefficients, we determined the 95% confidence interval for the correlations between the PTM sites and tobacco smoking mutagenesis. We limited this analysis to LUAD tumors, since this cancer type presents a broad spectrum of tobacco smoking mutagenesis. Due to the strong correlation between gender and tobacco smoking mutational signature weights as well as smoking history (p=1x10−4, Figure S5B), we further limit this analysis to the 71 cancers from male patients (which are the majority of the smokers) in order to decouple these variables. Additionally, we computed the Spearman’s rank correlation between smoking signature weights and normalized enrichment scores from mRNA-based ssGSEA using the MSigDB HALLMARK gene sets. We used the 71 male LUAD and 86 male LSCC for this analysis in order to prevent tissue-driven biases at low tobacco smoking mutation levels from other cancer types that typically have relatively low levels of tobacco smoking mutagenesis.
Lasso regression analysis
Acetylation sites with detection rates of at least 80% across the entire cohort were collected, and the abundances of these sites were imputed using the “impute.knn” function from the R package “impute” using the default parameters (k = 10, rowmax = 0.5, colmax = 0.8, maxp = 1500, rng.seed = 362436069). For the protein abundance of histones and histone regulators, the abundance of proteins mapping to multiple RefSeq IDs were averaged. Within each cluster, we tested the association between the abundance of histone acetylation sites and the abundance of histone acetyltransferases (HATs), histone deacetylases (HDACs) and bromodomain proteins (BRDs) using lasso regression. In particular, each histone acetylation site was treated as the outcome variable with the HATs/HDACs/BRDs protein treated as covariates. We split the data so that 80%(training set) are randomly designated for fitting the lasso regression model and the other 20% (testing set) are used to test its performance. Lasso uses a regularization parameter, λ, to control the weight of the L1 norm in the Lasso cost function. Within the training set, we identified the site-specific optimal λ value with 10-fold cross-validation that minimizes the MSE (mean squared error) for each acetylation site in each bootstrapping. We also calculated the lasso coefficient for each outcome-covariate pairs where a non-zero lasso coefficient indicating that the covariate would have a significant effect on the outcome. We predicted the abundance of each acetylation site in the testing set using the lasso coefficient generated from the training set and calculated the test MSE between predicted value and actual value. We bootstrapped these processes for 100 replications in order to minimize any bias introduced by the random sampling. We reported average lasso coefficients (Table S5) from 100 bootstrapping of lasso regression to improve precision in variable selections. To demonstrate the reproducibility and stability of the variable selections, we computed the mean and standard error of the test MSE across all histone acetylation sites separately from 25, 50, 75 and 100 replications. The test MSE was generated from the best model selected using optimal λ in each bootstrapping of lasso regression. The mean and standard error of the MSE were averaged across all histone acetylation sites, resulting in values of 1.14±0.78, 1.14±0.79, 1.15±0.79, 1.15±0.79, across 25, 50, 75, and 100 replications, respectively. We also reported site-specific mean MSE along with their standard errors in Table S5. This demonstrates that the mean MSE is very stable across different numbers of replications. Differential acetylation of histone sites across dendro groups
The results from the global differential expression analysis (see Differential Expression) were subset to histone genes by mapping RefSeq IDs to HGNC IDs. Sites with q < 0.1 were considered significant.
Transcriptional signature analysis
Across all samples, the correlation between the ssGSEA scores of HALLMARK pathways and histone acetylation sites was assessed using the imputed PTM data. For each histone acetylsite-pathway pair, we computed the Spearman correlation and p value. P values were then adjusted to FDR values across the 50 HALLMARK pathways for each histone acetylation site tested.
Acetylation-phosphorylation crosstalk analysis
For each acetylation site detected, phosphorylation sites with the same RefSeq protein ID and within 5 amino acid residues of the acetylation site were collected. For each cluster and across all samples, Spearman correlations were then computed for each adjacent acetylation site-phosphorylation site pair and p-values were adjusted to FDR values across all site pairs tested.
Histone acetylation across immune groups
The imputed acetylation data was filtered for histone acetylation sites by mapping RefSeq IDs to HGNC IDs. The abundance of these sites was then compared between the Immune-cool samples versus all other samples using a two-sided Wilcoxon test. P values were corrected to FDR values, and sites with FDR < 0.1 were considered significant.
Quantification and statistical analysis
RNA and Protein quantification
The process of RNAseq data processing and quantification, as well as proteome quantification, has been outlined in the sections titled “RNAseq data processing and quantification” and “Proteomics Data Processing,” respectively. The statistical analysis methodology and its corresponding details can be found both within the main text and in the relevant sections of the STAR Methods.
Additional resources
Comprehensive information about the CPTAC program, including program initiatives, investigators, and datasets, are available at the CPTAC program website: https://proteomics.cancer.gov/programs/cptac.
For the Pan-Cancer proteogenomics collection papers, along with links to the data and supplementary materials associated with these publications, please visit the Proteomic Data Commons (PDC) at: https://pdc.cancer.gov/pdc/cptac-pancancer.
Note S1. SignatureAnalyzer, related to Figures 2 & 3 as well as Figure S2 & S3
Efficient algorithms have been developed by the machine learning community for matrix factorization with a strict non-negativity requisite. One of the benefits of using a non-negative factorization method is that it typically yields sparse solutions (i.e., fewer ‘signatures’ are used to represent the data of one tumor), which typically have clear interpretations. Non-negative Matrix Factorization (NMF) is an algorithm that computes a low-rank factorization of an input matrix V into two matrices W and H, such that V ~ W * H and all elements of V, W, and H are strictly non-negative. The W matrix is a N-by-k matrix, where N is the number of features and k is the number of signatures. This matrix provides the weights for each feature in each of the signatures, and thus the columns of W are called the signatures. The H matrix is a k-by-S matrix, where k, as before, is the number of signatures and S is the number of samples. This matrix provides the contribution of each signature in each sample. This matrix can be normalized for downstream analyses such that the sum of contributions for a given sample sums to 1.
NMF was originally developed for analysis of image data169 but has a long history of use in biology to deconvolute expression data170 and identify ‘meta genes,’ which we now call ‘signatures’. NMF was later used to deconvolute mutation profiles and identify mutational signatures that were then matched with different mutational processes43. The use of non-negative methods in the mutation space is intuitive since both mutation counts, and mutation rates are positive numbers, and the results have a clear interpretation (i.e., the number of mutations a particular process contributes to each tumor). In these cases, the input V is a matrix of transcriptomic or genomic features by samples, respectively. The derived W matrix represents signatures and their feature weights, while the H matrix represents signature contributions to each sample’s observed phenotype.
A common challenge with NMF is the tendency to overfit the data given an increasing input number of signatures, K. SignatureAnalyzer uses a Bayesian version of NMF, called automatic relevance determination-NMF (ARD-NMF)157, which assumes a sparse prior on both the W and H matrices and searches for a maximum posterior solution. It initializes the analysis with a large number of signatures (K0 = 50 for multi-omic NMF and K0 = # features for mutational signatures) and by virtue of the priors, signatures unnecessary to explain the data have their weights driven to zero in the respective columns/rows of the W/H matrix. We run SignatureAnalyzer multiple times (n=100) and obtain a distribution on the number of signatures (as it converges to different local maxima of the posterior distribution). We select k as the mode of the distribution and the W and H matrices that correspond to the highest posterior solution with that value of k. This allows highly interpretable and sparse representations for both signature profiles and attributions that strike a balance between data fitting and model complexity157. This method, among others, has been widely applied to analyze mutational signatures and expression signatures for molecular data (mRNA, protein, etc) 42,43,161,170–172.
In this study, we apply SignatureAnalyzer to somatic mutation data to extract mutational signatures, and to the multi-omic phenotypes to identify patterns of regulation across cancer. Mutation counts are by nature non-negative, and we can adjust the multi-omic phenotype matrix by splitting each gene, protein, and phosphorylation feature into a positive and negative row in the matrix. We negate the negative row such that all values are non-negative. This creates a less dense matrix since every feature is replaced with two values, one which is zero. The sparse matrix can be easily handled by NMF. At the end, the resulting signatures also contain rows corresponding to positive and negative values, and therefore the possibility of upregulation and downregulation is maintained. Since mutations can be represented as count data generated by independent, low frequency events (with the exception of clustered mutations such as APOBEC generated kataegis), we can approximate these mutation counts with a Poisson distribution. For multi-omic data the Poisson assumptions do not hold, and we model the likelihood of the zero-centered data using a truncated (half) Gaussian distribution (Figure S1C).
In order to reward sparsity of the resulting W and H matrices, we apply an exponential prior to all entries in the W and H matrices for mutational signatures, and a half-normal prior to all entries for the multi-omic signatures. Applying an exponential prior to the weights in the W and H matrices is equivalent to adding an L1 regularization term to the cost function being optimized. To illustrate this point, consider the pdf of the exponential distribution:
In our cases x represents an entry in the W and H matrices of the factorization. As SignatureAnalyzer follows the procedure in Tan et al.157 to minimum a cost function given by the negative log posterior, this term is incorporated as follows:
Since -log𝜆 is constant with respect to the entries of the W and H matrix, the choice of an exponential prior effectively puts an L1 penalty on the value of with regularization strength determined by 𝜆.Note that L1 penalty uses the absolute value of , ie. , but since all values in the matrices are positive, we do not need the absolute value. We can work out similar math for the half normal case to see that this is indeed equivalent to L2 regularization.
Mutational signature discovery requires deconvolution of somatic mutation counts, stratified by mutation sequence context, into a set of characteristic patterns (signatures) and inferring their activity across each of the samples in the study. The common stratification of somatic single nucleotide variants (SNVs) is based on the six base substitutions (C>A, C>G, C>T, T>A, T>C, and T>G) within the trinucleotide sequence context, which includes the bases located at positions immediately adjacent to the 5’ and 3’ directions relative to the mutated base. Thus, each of the six base substitutions have 16 possible combinations of neighboring bases, yielding 96 possible mutation types (or sequence contexts). The input matrix for mutational signature analysis is a 96 x S matrix, where S is the number of samples, and each element xij represents the number of observed mutations of type i in sample j. Using this matrix, we identified the mutational signatures and their contributions to each sample. This type of analysis has been used in numerous publications starting with Lawrence et al, 2014142 and most recently applied across cancer in the TCGA/ICGC Pan-Cancer Analysis of Whole Genomes (PCAWG) project43. We compared the signatures that we obtained to the ones reported in the COSMIC database and PCAWG paper in order to connect them to their proposed etiologies. We then used these signatures to classify tumors with mismatch repair deficiency and homologous recombination deficiency based on the activity of mutational signatures associated with these repair defects across all of our samples.
To provide additional internal validation for the robustness of the subtypes and test the effect of having fewer samples, we ran SignatureAnalyzer on downsampled cohorts of sizes N=100, 200,...1000 from our full Pan-Cancer cohort (n=1110). We performed 30 random downsamplings without replacement per value of N (“runs”), and within each run, we performed 20 independent Bayesian NMF iterations (each initialized with a unique random seed). We then matched the signatures derived in each downsampling run with the signatures obtained when analyzing the full cohort using cosine similarity with a threshold of 0.8. The percent of downsamples out of 30 runs in which a particular signature was identified is plotted in the heatmap below. As shown in the heatmap, even when downsampling to 800 samples (~72% of the dataset), 24 signatures are seen in >1/2 of the runs. Moreover, signatures S24 and S7 (which are the dominant signature in a large number of samples; 71 and 73 samples, respectively) are detected even in very small subsampled cohorts, making them more robust signatures. On the other hand, signatures S27 and S31 are less robust (i.e., “weaker”) and are only found in a minority of downsampling runs, even with large cohort sizes (800–1000).
Further exploring the effect of cohort composition on our signatures, and specifically our external MB cohort, we investigated how the inclusion of MB impacts our signatures. To this end, we reran SignatureAnalyzer excluding (i) the MB cohort, (ii) the GBM cohort, and (iii) both MB and GBM cohorts. We provide a heatmap showing the cosine similarities between the signatures derived from each of these cohort exclusion runs, and the signatures derived in the full dataset, colored by cosine similarity. The boxplot to the right indicates the number of samples assigned to each signature per decomposition (assignment based on maximally contributing signature). First, we find that Signature 31 is not derived in each of these decompositions, consistent with downsampling analyses indicating its lower robustness (‘weakness’). Next, we see that upon removal of the MB cohort, all signatures with the exception of S14 are derived with >0.85 cosine similarity to the full cohort signatures, suggesting MB itself does not yield a significant effect on the signatures. S14 is not derived when MB is excluded, but this signature is the second “weakest” signature derived from downsampling and is mixed across multiple cohorts (maximal contributions to S14 are with 47% from UCEC, 29% from CCRCC, and then 12% from MB) defined by strong transcriptomic activation of EMT (Table S2). Interestingly, we find Signatures 9 and 20 are not derived when excluding GBM and both brain cohorts (MB and GBM). S20 is a weaker signature with a 56% dominance of GBM. S9, however, is a stronger signature split between MB (34.29%) and GBM (65.71%) defined by downregulation of MYC with strong positive transcriptomic and negative phosphorylation effects. This suggests that S9 is a tissue-specific signature of biologic interest rather than a specific batch effect. S7 (immune upregulation) and S22 (myogenesis/adipogenesis in phosphoproteome) are examples of signatures without any MB samples. Notably, for these two signatures, 1 GBM patient (out of 73 in S7) maximally maps to S7 and 1 GBM patient (out of 39 in S22) maximally maps to S22 (Table S2). We believe more samples would be required to definitively suggest if these signatures are truly devoid of MB specific samples, or lack biological pathways associated with brain tissue.
Note S2. PTM dedicated tools, related to Figures 2–6, and Figures S2–S6
To explore and interpret site specific PTM data, we developed and applied PTM-tailored tools. We provide here a short description of each:
CLUMPS-PTM
PTMs play key roles in numerous cell signaling processes in the context of cancer. Previous studies have shown that close proximity of PTM sites, either in linear sequence or on the 3D protein structure, increases the chance of crosstalk among such sites and may have functional outcomes173–175. Despite advances in LC-MS increasing the throughput of PTM quantification, identifying these functionally relevant PTMs and protein domains remains a challenge. We developed CLUMPS-PTM, an algorithm for spatial clustering of PTMs, to identify clusters of correlated PTMs in protein structures that likely reflect stronger, mutually dependent signaling effects (Figure S2D). CLUMPS-PTM is built on a previous tool we developed, CLUMPS165, initially designed to identify novel driver mutations based on clustering of missense mutations in 3D protein structures. CLUMPS-PTM was developed for phosphorylation and acetylation events, but is flexibly designed to incorporate any PTM modifications (such as ubiquitination, etc.). We developed pipelines (compatible with RefSeq and GENCODE isoforms) to map over 14K phospho-sites and 13K acetyl-sites detected in the CPTAC dataset to protein data bank (PDB) files (Figure S2E). We expand this to the predicted AlphaFold proteome and recover an additional 12K phospho-sites and 10K acetyl-sites exceeding model confidence (pLDDT) of 70%. Consistent with previous research176, phosphosites are found on residues with lower AlphaFold prediction confidence than acetylsites due to their abundance in unstructured domains (p<1e-4). CLUMPS-PTM may be run on either a database of PTM sites or differential expression results. This tool has been applied to multiple CPTAC projects7 in addition to the Pan-Cancer context to propose functional domains for 1) tumor vs. normal analyses, 2) immune infiltration / subtyping, 3) phospho/acetyl co-clustering, and 4) tumor-specific subtyping. Overall, CLUMPS-PTM is an open-source tool that allows near proteome-wide spatial analysis with the growing availability of PTM data. We anticipate that it will be useful to the broader proteomic community for the discovery of novel targets and generation of insights into functional mechanisms (Methods, Figure S2F).
The Kinase Library -
The Kinase Library experimentally characterizes the substrate sequence specificity of over 300 protein Ser/Thr kinases33. This dataset is then used to computationally identify the most likely protein kinases for differentially phosphorylated sites and predict kinases that are up- or downregulated (Methods). Moreover, this library also tests the change of sequence specificity when a close proximity acetylation is added and enables the study of crosstalk and its effect on kinase regulation. In addition, this method was also applied in our companion Driver paper (CELL-D-22–01960) to characterize the activity states of kinases under different oncogenic driver events.
CausalPath
CausalPath34 aims to identify potential cause–effect relationships between the observed omic changes (proteomic and transcriptomic) using a variety of prior information, such as (i) the regulation of protein modification (phosphorylation, acetylation, and methylation), (ii) the effect of the modified protein site on the protein activity, and (iii) the regulation of gene expression. In general, the method has two essential steps. In step 1, CausalPath works on the Pathway Commons database to identify which protein activities control which measurable omic features, and in what direction. In step 2, CausalPath uses a logical equation to detect the subset of the extracted priors that can causally explain the observed coordinated changes. The logical function links omic changes to protein activities and checks if the observed downstream changes are in the expected direction. On top of these steps, CausalPath runs enrichment analysis to detect the proteins on the result network with a significant number of affected downstream targets. CausalPath was previously used in a myriad of analyses and diseases such as: platelet activation177,178, lung squamous cell carcinoma7, glioblastoma4 , acute myeloid leukemia179, pediatric high-grade glioma180, cell line drug treatments181, and breast cancer 182,183.
PTM-SEA -
PTM-SEA, similar to GSEA, is a pathway enrichment approach that identifies activated or deactivated regulators like kinases or phosphatases, using an enrichment of experimentally validated substrates 35. Most proteomic tools are performed on a gene-centric level and collapse multiple sites to their average or dominant site; this process causes loss of important information encoded at the site-specific level. The tool is based on the PTMsigDB curated database with a large advantage of site-specific annotation and directionality of the PTM regulation.
An analytical resource of post-translational modifications from over a 1000 patients across 11 cancer types reveals pan-cancer changes involved in hallmark cancer processes and reveals potential new therapeutic avenues.
Supplementary Material
Figure S1 - Pan-Cancer Data harmonization
A. Density plots showing PTM levels and their matched protein levels. Ordinary least squares (OLS) were fitted for each PTM-protein pair, and the residuals from the fit represented PTM levels corrected for protein abundance. Raw phosphoproteome abundance (left), imputed phosphoproteome abundance (used for clustering, middle), and raw acetylome abundance (right).
B. Principal component analysis (PCA) projection of transcriptome TPM across the 10 CPTAC cohorts and the medulloblastoma cohort before and after applying batch correction (using COMBAT). Top PCA plots colored by sequencing protocol (poly- adenylated mRNA enrichment [red] and total RNA [blue]), and bottom PCA plots colored by cohort.
C. Feature distribution showing the final normalized RNA data, the proteomic data, phosphorylation data corrected for protein abundance and the final multi-omic feature space following cohort correction. Distributions are colored by cohort.
D. Projection of multi-omic features input into NMF on a PCA of all 1110 samples colored by cohort; (top row) all features combined- mRNA, protein, and phosphorylation, (middle row) mRNA, (bottom row) protein and phosphorylation. (left + middle column) features before and after regressing out cohort-specific effects, (right column) features after regression and split into positive and negative values for NMF.
Figure S2 - Patient Clustering and CLUMPS-PTM method
A. Stacked barplot of patient-wise assigned maximally weighted signature colored by cohort (left). Stacked bar plot shows the breakdown, normalized by the number of patients maximally assigned to a given signature (right).
B. Multi-omic hierarchical clustering of tumors based on multi-omic NMF signature weights. Heatmap represents the pair-wise cosine similarity between tumors. Non-overlapping clusters - yellow boxes. Top track shows the cluster assignments. Dendrogram is colored based on the second level of the tree.
C. Distribution of the fraction of a sample’s NMF signature weights accounted for by the maximally contributing signature (top, left); Distribution of the number of signatures required to explain at least 90% of a sample’s signature weights (top, right); Distribution of the difference between the fraction of a sample’s NMF signature weights accounted for by the maximally contributing signature and the second strongest signature (bottom).
D. Schematic workflow of the newly developed CLUMPS-PTM algorithm.
E. Sankey plot showing the mapping between protein sequences to PDB structures. Protein sequences were aligned to the UniProtKB database (using BLASTP+ hits), then filtered to those with matching well-annotated proteins identified using the Structure Integration with Function, Taxonomy and Sequence (SIFTS) method. These were then mapped to available crystal structures with at least 3 modified PTM sites across the entire CPTAC dataset (upper left). Sankey plot of all PTM sites in the CPTAC dataset, splitting to those with accession-numbers mapped to available Uniprot input proteins, and then to proteins with available PDB structures verified in the SIFTS database (lower left). Violin plots of the BLASTP+ relative identity score to the Uniprot database for proteins and PTM sites that map or do not map to PDB structures (right).
F. Stacked bar plots summarizing CLUMPS-PTM results showing the total number of proteins tested based on group-specific differential expression test & direction (top panel). Stacks show the number of structures that are nominally significant (p<0.1; yellow) and pass multiple hypothesis testing (FDR<0.1; red) for acetylome, phosphoproteome, and combined samplers. CLUMPS-PTM phosphorylation and acetylation hits (q-value<0.1) for the left (L) vs. right (R) top split of the Pan-Cancer dendrogram (bottom, panel). The first heatmap (on the left) indicates the number of distinct peptides found in each cohort that contributed to the differential PTM cluster. The x-axis represents the cohort. The second heatmap (on the right) expands on each CLUMPS-PTM hit and shows the log-fold change between the left vs. right branches of the top split in the dendrogram for each represented peptide. Blue indicates phosphorylated clustered sites.
Figure S3 - Mutational signature extraction and analysis of DNA repair deficiency
A. Schematic of the mutational signature extraction workflow and barplot representations of the resulting mutational signatures.
B. Scatter plots illustrating the distributions for the number of mutations and the fraction of mutations attributed to the HRD-associated mutational signature (top). HRD classification threshold at 45 mutations indicated by a dotted line. Tumors with known pathogenic or high impact germline variants in BRCA1, BRCA2, or PALB2 genes are highlighted with a black outline; Scatter plots showing the distributions for the number of mutations and the fraction of mutations attributed to the mutational signature associated with MMRD in COAD and UCEC tumors (bottom). MMRD classification thresholds at 72 mutations and at 19% of mutations, respectively indicated by horizontal and vertical dotted lines. Tumors with known pathogenic or high impact variants in MSH6, MSH2, MLH1, and PMS2 are highlighted with a black outline.
C. Pathway of differentially expressed DDR genes (at the RNA [r circle], protein [box], and phosphorylation site [p circles] levels) between the HRD and homologous recombination proficient (HRP) tumors and their inferred effect on other pathway members. Upregulation in the HRD tumors in red and downregulation in blue. Dotted edges between the kinases and their substrates indicates that the interaction is within the 90th percentile of all top scoring substrates for the specific kinase (The Kinase Library).
D. PCA of multi-omic signature weights across UCEC and COAD MMRD tumors. Points are colored by tumor type (COAD - orange and UCEC - purple).
E. Distributions of MRN complex proteins (MRE11, RAD50, and NBN) relative protein abundance (top) and RNA expression in CCLE COAD and UCEC cancer cell lines from the DepMap dataset. Groups are separated and colored by mismatch repair proficiency (MMRD - purple, MMRD - yellow). Shapes indicate tumor type (UCEC - circle, COAD - triangle).
Figure S4 - Immune clustering, activity and associated metabolic markers
A. Violin plots showing the levels of different immune signatures based on ImmuneSubtypeClassifier and purity estimates from ESTIMATE for the 4 identified immune clusters across all tumors.
B. Violin plots showing the top 6 immune cell abundances from CIBERSORT that were used as covariates in differential expression to estimate tumor intrinsic gene expression in the 4 immune clusters.
C. Stacked barplot showing the distribution of 4 immune clusters within each cohort.
D. Violin plots showing significant differences between the 4 identified immune clusters for the immunosuppressive markers - PD-L1, IDO1, CD163, ENTPD1 (Pairwise comparisons with q value < 0.1 are shown).
E. Violin plots showing acetylation levels of fatty acid related proteins among different immune subtypes when immune cold is split to Brain and non-brain tumors.
F. BCKDK (PDB ID:1DTW) showing significant spatial clustering of phosphorylation sites by CLUMPS-PTM of differentially regulated sites for the immune-cool cluster. Protein 3D crystal structure space-filling model (cyan) and highlighted clumped phosphosites (purple) and thymidine-5’-triphosphate (TTP) sites are marked in green.
G. Bubble plot representing the Kinase Library enrichment score and significance, differentially expressed substrates of each kinase for the 4 immune clusters. Colors represent enrichment (Red) and depletion (Blue). Kinases are colored by to their phylogenetic groups 168
H. Violin plots showing significant differences between the 4 identified immune clusters for fatty acid transporters - CPT1A, FABP, ABCA3, CD36 (only pairwise comparisons with q value < 0.1 are shown).
I. Spearman correlation between the ssGSEA values for each sample of the MSigDB HALLMARK gene sets to the CD8+ T-cell abundance estimated from CIBERSORT.
Figure S5 - Histone analysis
A. Correlation matrix showing the correlations between mean acetylation of H1, H2A, H2B, H3 and H4. Asterisks denote correlations with p<0.05.
B. Violin plot showing the distribution of tobacco smoking mutational signature contributions (log10-scale) for males and females in LSCC and LUAD. Shapes indicate the cancer type-LUAD (circle) and LSCC (triangle). p-value (1x10−4) from Wilcoxon rank-sum test.
C. Dot plot showing significant Spearman correlations (q ≤ 0.1) across male LUAD patients (n=71) between histone acetylation levels and tobacco smoking mutational signature contributions.
D. Dot plot showing significant Spearman correlations (q ≤ 0.1) across male LUAD patients (n=71) between deacetylases phosphorylation levels and tobacco smoking mutational signature contributions.
E. Dot plot illustrating significant Spearman correlations (q ≤ 0.1, |rho|>0.15) across male LSCC and male LUAD patients (n=157) between tobacco smoking mutational signature contributions and the mRNA ssGSEA scores for the MSigDB HALLMARK gene sets.
F. Scatter plot showing the positive correlation (rho=0.332) between tobacco smoking mutational signature contributions and the ssGSEA score for the G2/M Checkpoint (left) and MTORC1 (right) Hallmark gene set in male LUAD and male LSCC tumors (n=157).
Figure S6 - The Kinase Library
A. Box plots showing the Kinase Library normalized intensity ratios of 207 Ser/Thr kinases for unmodified, trimethylated, and acetylated lysines, across 10 positions (-5 to +5) around the central phospho-acceptor site.
B. Heatmap showing the Kinase Library normalized intensity ratios between trimethylated lysine and unmodified lysine across the 207 Ser/Thr kinases. Kinases are colored according to their phylogenetic groups 168
C.Heatmap showing the expression levels of acetylated K23 and phosphorylated S28 on H3F3A across 41 patients in cluster #8. Samples are sorted based on hierarchical clustering of their correlations.
D. Heatmap showing the expression levels of acetylated K1378 and phosphorylated S1375 on RSF1 across the 89 LUAD and LUSC patients with acetylation and phosphorylation data. Samples are sorted based on hierarchical clustering of their correlations.
Table S1. Metadata of all features included in the study, related to Figure 1 and Figure S1.
A. Inclusion of Transcriptome Features
B. Inclusion of Proteome Features
C. Inclusion of Phosphoproteome Features
D. Inclusion of Acetylation Features
Table S2. Pan-Cancer Multi-omics clusterings and annotations, related to Figure 2 & 3 and Figure S2 & S3.
A. ARD NMF H-matrix
B. ARD NMF W-matrix
C. Signature Annotations
D. Dendrogram 1–4 CLUMPS-PTM
E. Dendrogram results 1–3 from The Kinase Library
Table S3. Mutational signatures and DDR analysis, related to Figure 3 and Figure S3.
A. Mutational signature W matrix (mutational spectra for each signature)
B. Mutational signature H Matrix
C. Collapsed mutational signature attributions (weights for similar signatures collapsed to a single weight)
D. Mutational signatures summary table
E. DNA repair deficiency classification
F. DNA damage response geneset
G. HRD vs HRP DDR differential expression
H. HRD acute (B) vs chronic (C) hypoxia full differential expression
I. Acute (B) vs chronic (C) hypoxia DDR differential expression
J. HRD acute (B) vs chronic (C) hypoxia GSEA
K. HRD acute (B) vs chronic (C) hypoxia PTM-SEA
L. The Kinase Library scores for CausalPath kinase-substrate pairs
M. HRD low (A) vs acute (B) hypoxia full differential expression
N. HRD low (A) vs acute (B) hypoxia GSEA
O. HRD low (A) vs chronic (C) hypoxia GSEA
P. MMRD vs MMRP full differential expression
Q. MMRD vs MMRP DDR differential expression
R. MMRD vs MMRP GSEA
S. MMRD microsatellite indel Fisher’s exact tests
Table S4. Immune classification and metabolic analysis, related to Figure 4 and Figure S4.
A. ESTIMATE scores
B. CIBERSORT scores
C. CIBERSORT ssGSEA enrichments
D. Immune Subtype Classifier
E. fGSEA Metabolic and Immune pathways per subtype
F. Immune CLUMPS-PTM results
G. Fatty acid metabolism correlations
H. Immune cold results from The Kinase Library
I. Immune cold results results from The Kinase Library
J. Immune warm results from The Kinase Library
K. Immune hot results results from The Kinase Library
Table S5. Histone analysis, related to Figure 5 and Figure S5.
A. Histone related genes
B. Histone acetylation sites and phosphorylation sites
C. Correlation across histone acetylation sites
D. Correlation between smoking signature and histone acetylation across male LUAD patients
E. Correlation between smoking signature and HDAC phosphorylation across male LUAD patients
F. Correlation between smoking signature and HALLMARK pathways across male LSCC and male LUAD patients
G. Key regulators of histone acetylation sites
H. Correlation between histone acetylation sites and Hallmark pathways
I. Differentially acetylated histone sites in the immune clusters
J. Correlation between histone acetylation and phosphorylation
K. Site specific lasso regression
Table S6. The Kinase Library crosstalk analysis, related to Figure 6 and Figure S6.
A. Unmodified lysine values
B. Unmodified lysine - normalized
C. Acetylated lysine values
D. Acetylated lysine - ratio
E. Acetylated lysine - normalized
F. Methylated lysine values
G. Methylated lysine - ratio
H. Methylated lysine - normalized
I. Acetylation-phosphorylation crosstalk correlations
Highlights.
Unsupervised clustering revealed 33 Pan-Cancer multi-omic signatures
PTM dysregulation associated with distinct DNA damage repair mechanisms
Changes in acetylation of metabolic proteins correlate with tumor immune state
Phosphorylation of Th/Sr kinases affected by proximal acetylation
Acknowledgments
This work was supported by the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC) grants U24CA210955, U24CA210985, U24CA210986, U24CA210954, U24CA210967, U24CA210972, U24CA210979, U24CA210993, U01CA214114, U01CA214116, U24 CA270823, and U01CA214125. In addition, this project has been funded in part with Federal funds from the National Cancer Institute, National Institutes of Health, under Contract No. 75N91019D00024, Task Order 75N91020F00029. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the U.S. Government. Pattern Team at the Broad Institute for figure design. Schematic figures and the graphical abstract were created using BioRender.com.
Footnotes
Consortia
The members of the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium for Pan-Cancer are François Aguet, Yo Akiyama, Eunkyung An, Shankara Anand, Meenakshi Anurag, Özgün Babur, Jasmin Bavarva, Chet Birger, Michael J. Birrer, Anna Calinawan, Lewis C. Cantley, Song Cao, Steven A. Carr, Michele Ceccarelli, Daniel W. Chan, Arul M. Chinnaiyan, Hanbyul Cho, Shrabanti Chowdhury, Marcin P. Cieslik, Karl R. Clauser, Antonio Colaprico, Daniel Cui Zhou, Felipe da Veiga Leprevost, Corbin Day, Saravana M. Dhanasekaran, Li Ding, Marcin J. Domagalski, Yongchao Dou, Brian J. Druker, Nathan Edwards, Matthew J. Ellis, Myvizhi Esai Selvan, David Fenyö, Steven M. Foltz, Alicia Francis, Yifat Geffen, Gad Getz, Michael A. Gillette, Tania J. Gonzalez Robles, Sara J. C. Gosline, Zeynep H. Gümüş, David I. Heiman, Tara Hiltke, Runyu Hong, Galen Hostetter, Yingwei Hu, Chen Huang, Emily Huntsman, Antonio Iavarone, Eric J. Jaehnig, Scott D. Jewell, Jiayi Ji, Wen Jiang, Jared L. Johnson , Lizabeth Katsnelson, Karen A. Ketchum, Iga Kolodziejczak, Karsten Krug, Chandan Kumar-Sinha, Alexander J. Lazar, Jonathan T. Lei, Yize Li, Wen-Wei Liang, Yuxing Liao, Caleb M. Lindgren, Tao Liu, Wenke Liu, Weiping Ma, D R. Mani, Fernanda Martins Rodrigues, Wilson McKerrow, Mehdi Mesri, Alexey I. Nesvizhskii, Chelsea J. Newton, Robert Oldroyd, Gilbert S. Omenn, Amanda G. Paulovich, Samuel H. Payne, Francesca Petralia, Pietro Pugliese, Boris Reva, Ana I. Robles, Karin D. Rodland, Henry Rodriguez, Kelly V. Ruggles, Dmitry Rykunov, Shankha Satpathy, Sara R. Savage, Eric E. Schadt, Michael Schnaubelt, Tobias Schraink, Stephan Schürer, Zhiao Shi, Richard D. Smith, Xiaoyu Song, Yizhe Song, Vasileios Stathias, Erik P. Storrs, Jimin Tan, Nadezhda V. Terekhanova, Ratna R. Thangudu, Mathangi Thiagarajan, Nicole Tignor, Joshua M. Wang, Liang-Bo Wang, Pei Wang, Ying Wang, Bo Wen, Maciej Wiznerowicz, Yige Wu, Matthew A. Wyczalkowski, Lijun Yao, Tomer M. Yaron, Xinpei Yi, Bing Zhang, Hui Zhang, Qing Zhang, Xu Zhang, Zhen Zhang.
Secondary author list -
Eunkyung An, Meenakshi Anurag, Jasmin Bavarva, Michael J. Birrer, Song Cao, Michele Ceccarelli, Daniel W. Chan, Arul M. Chinnaiyan, Hanbyul Cho, Shrabanti Chowdhury, Marcin P. Cieslik, Antonio Colaprico, Daniel Cui Zhou, Felipe da Veiga Leprevost, Corbin Day, Marcin J. Domagalski, Yongchao Dou, Brian J. Druker, Nathan Edwards, Matthew J. Ellis, Myvizhi Esai Selvan, David Fenyo, Steven M. Foltz, Alicia Francis, Tania J. Gonzalez Robles, Sara J.C. Gosline, Zeynep H. Gümüş, Tara Hiltke, Runyu Hong, Galen, Hostetter, Yingwei Hu, Chen Huang, Antonio Iavarone, Eric J. Jaehnig, Scott D. Jewel, Jiayi Ji, Wen Jiang, Lizabeth Katsnelson, Karen A. Ketchum, Iga Kolodziejczak, Chandan Kumar-Sinha, Jonathan T. Lei, Wen-Wei Liang, Yuxing Liao, Caleb M. Lindgren, Tao Liu, Wenke Liu, Weiping Ma, Fernanda Martins Rodrigues, Wilson McKerrow, Alexey I. Nesvizhskii, Chelsea Newton, Robert Oldroyd, Gilbert S. Omenn, Amanda G. Paulovich, Francesca Petralia, Pietro Pugliese, Boris Reva, Karin D. Rodland, Kelly V. Ruggles, Dmitry Rykunov, Sara R. Savage, Eric E. Schadt, Michael Schnaubelt, Tobias Schraink, Zhiao Shi, Richard D. Smith, Xiaoyu Song, Vasileios Stathias, Erik P. Storrs, Jimin Tan, Nadezhda V. Terekhanova, Ratna R. Thangudu,Nicole Tignor, Joshua M. Wang, Pei Wang, Ying (Cindy) Wang, Bo Wen, Maciej Wiznerowicz, Yige Wu, Matthew A. Wyczalkowski, Lijun Yao, Xinpei Yi, Bing Zhang, Hui Zhang, Xu Zhang, Zhen Zhang.
Declaration of interests
Y. G. is a consultant for Oriel Research Therapeutics. T.M.Y. is a co-founder, stockholder and on the board of directors of DESTROKE, Inc., an early-stage start-up developing mobile technology for automated clinical stroke detection. J.L.J has received consulting fees from Scorpion Therapeutics and Volastra Therapeutics. Y.E.M is a consultant for ForseeGenomics and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig and MSIDetect N.J.H. is a consultant for MorphoSys. F.A. is an inventor on a patent application related to SignatureAnalyzer-GPU, and has been an employee of Illumina, Inc., since 8 November 2021. L.C.C. is a founder and member of the board of directors of Agios Pharmaceuticals and is a founder of Petra Pharmaceuticals. L.C.C. is an inventor on patents (pending) for Combination Therapy for PI3K-associated Disease or Disorder, and The Identification of Therapeutic Interventions to Improve Response to PI3K Inhibitors for Cancer Treatment. L.C.C. is a co- founder and shareholder in Faeth Therapeutics. G.G receives research funds from IBM, Pharmacyclics and Ultima Genomics, and is also an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, MSIDetect and MinimuMM-Seq. He is also a founder, consultant, and privately held equity in Scorpion Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ding L, Bailey MH, Porta-Pardo E, Thorsson V, Colaprico A, Bertrand D, Gibbs DL, Weerasinghe A, Huang K-L, Tokheim C, et al. (2018). Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 173, 305–320.e10. 10.1016/j.cell.2018.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doroshow DB, and Doroshow JH (2020). Genomics and the History of Precision Oncology. Surg. Oncol. Clin. N. Am 29, 35–49. 10.1016/j.soc.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez H, Zenklusen JC, Staudt LM, Doroshow JH, and Lowy DR (2021). The next horizon in precision oncology: Proteogenomics to inform cancer diagnosis and treatment. Cell 184, 1661–1670. 10.1016/j.cell.2021.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L-B, Karpova A, Gritsenko MA, Kyle JE, Cao S, Li Y, Rykunov D, Colaprico A, Rothstein JH, Hong R, et al. (2021). Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 39, 509–528.e20. 10.1016/j.ccell.2021.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang C., Chen L., Savage SR., Eguez RV., Dou Y., L Y., da Veiga Leprevost F., Jaehnig EJ., Lei JT., Wen B., et al. (2021). Proteogenomic insights into the biology and treatment of HPV-negative head and neck squamous cell carcinoma. Cancer Cell 39, 361–379.e16. 10.1016/j.ccell.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gillette MA, Satpathy S, Cao S, Dhanasekaran SM, Vasaikar SV, Krug K, Petralia F, Li Y, Liang W-W, Reva B, et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. 10.1016/j.cell.2020.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Satpathy S, Krug K, Jean Beltran PM, Savage SR, Petralia F, Kumar-Sinha C, Dou Y, Reva B, Kane MH, Avanessian SC, et al. (2021). A proteogenomic portrait of lung squamous cell carcinoma. Cell 184, 4348–4371.e40. 10.1016/j.cell.2021.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krug K, Jaehnig EJ, Satpathy S, Blumenberg L, Karpova A, Anurag M, Miles G, Mertins P, Geffen Y, Tang LC, et al. (2020). Proteogenomic Landscape of Breast Cancer Tumorigenesis and Targeted Therapy. Cell 183, 1436–1456.e31. 10.1016/j.cell.2020.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao L, Huang C, Cui Zhou D, Hu Y, Lih TM, Savage SR, Krug K, Clark DJ, Schnaubelt M, Chen L, et al. (2021). Proteogenomic characterization of pancreatic ductal adenocarcinoma. Cell 184, 5031–5052.e26. 10.1016/j.cell.2021.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih T-SM, Chang H-Y, et al. (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. 10.1016/j.cell.2019.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDermott JE, Arshad OA, Petyuk VA, Fu Y, Gritsenko MA, Clauss TR, Moore RJ, Schepmoes AA, Zhao R, Monroe ME, et al. (2020). Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability. Cell Reports Medicine 1, 100004. 10.1016/j.xcrm.2020.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dou Y, Kawaler EA, Cui Zhou D, Gritsenko MA, Huang C, Blumenberg L, Karpova A, Petyuk VA, Savage SR, Satpathy S, et al. (2020). Proteogenomic Characterization of Endometrial Carcinoma. Cell 180, 729–748.e26. 10.1016/j.cell.2020.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasaikar S, Huang C, Wang X, Petyuk VA, Savage SR, Wen B, Dou Y, Zhang Y, Shi Z, Arshad OA, et al. (2019). Proteogenomic Analysis of Human Colon Cancer Reveals New Therapeutic Opportunities. Cell 177, 1035–1049.e19. 10.1016/j.cell.2019.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mani DR, Krug K, Zhang B, Satpathy S, Clauser KR, Ding L, Ellis M, Gillette MA, and Carr SA (2022). Cancer proteogenomics: current impact and future prospects. Nat. Rev. Cancer 22, 298–313. 10.1038/s41568-022-00446-5. [DOI] [PubMed] [Google Scholar]
- 15.Deribe YL, Pawson T, and Dikic I. (2010). Post-translational modifications in signal integration. Nat. Struct. Mol. Biol 17, 666–672. 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10. 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailey MH., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl MC., Kim J., Reardon B., et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 173, 371–385.e18. 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 19.Narita T, Weinert BT, and Choudhary C. (2019). Functions and mechanisms of non- histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174. 10.1038/s41580-018-0081-3. [DOI] [PubMed] [Google Scholar]
- 20.Biswas SK (2015). Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 43, 435–449. 10.1016/j.immuni.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 21.Yu W, Lei Q, Yang L, Qin G, Liu S, Wang D, Ping Y, and Zhang Y. (2021). Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J. Hematol. Oncol 14, 187. 10.1186/s13045-021-01200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rohatgi N, Ghoshdastider U, Baruah P, Kulshrestha T, and Skanderup AJ (2022). A pan-cancer metabolic atlas of the tumor microenvironment. Cell Rep. 39, 110800. 10.1016/j.celrep.2022.110800. [DOI] [PubMed] [Google Scholar]
- 23.Manzo T, Prentice BM, Anderson KG, Raman A, Schalck A, Codreanu GS, Nava Lauson CB, Tiberti S, Raimondi A, Jones MA, et al. (2020). Accumulation of long-chain fatty acids in the tumor microenvironment drives dysfunction in intrapancreatic CD8+ T cells. J. Exp. Med 217. 10.1084/jem.20191920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peleg S, Feller C, Ladurner AG, and Imhof A. (2016). The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci 41, 700–711. 10.1016/j.tibs.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Audia JE, and Campbell RM (2016). Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol 8, a019521. 10.1101/cshperspect.a019521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huen MSY, and Chen J. (2008). The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 18, 8–16. 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 27.Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK, Mahoney EL, Daniel CJ, LeNail A, Ramamoorthy D, et al. (2018). Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 34, 396–410.e8. 10.1016/j.ccell.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karabulut NP, and Frishman D. (2016). Sequence- and Structure-Based Analysis of Tissue-Specific Phosphorylation Sites. PLoS One 11, e0157896. 10.1371/journal.pone.0157896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garcia BA, Thomas CE, Kelleher NL, and Mizzen CA (2008). Tissue-specific expression and post-translational modification of histone H3 variants. J. Proteome Res. 7, 4225–4236. 10.1021/pr800044q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J., Mouw KW., Polak P., Braunstein LZ., Kamburo A., Kwiatkowski DJ., Rosenberg JE., Van Allen EM., D’Andrea A., and Getz G. (2016). Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet 48, 600–606. 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, Lawrence MS, Kiezun A, Fernandes SM, Bahl S, et al. (2015). Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun 6, 8866. 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor-Weiner A, Aguet F, Haradhvala NJ, Gosai S, Anand S, Kim J, Ardlie K, Van Allen EM, and Getz G. (2019). Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 20, 228. 10.1186/s13059-019-1836-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin T-Y, Liberatore K, Cizin DM, Cohen BM, et al. (2023). An atlas of substrate specificities for the human serine/threonine kinome. Nature 613, 759–766. 10.1038/s41586-022-05575-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babur Ö, Luna A, Korkut A, Durupinar F, Siper MC, Dogrusoz U, Vaca Jacome AS, Peckner R, Christianson KE, Jaffe JD, et al. (2021). Causal interactions from proteomic profiles: Molecular data meet pathway knowledge. Patterns (N Y) 2, 100257. 10.1016/j.patter.2021.100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krug K, Mertins P, Zhang B, Hornbeck P, Raju R, Ahmad R, Szucs M, Mundt F, Forestier D, Jane-Valbuena J, et al. (2019). A Curated Resource for Phosphosite-specific Signature Analysis. Mol. Cell. Proteomics 18, 576–593. 10.1074/mcp.TIR118.000943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong Y, Sun Y, Huang Y, Fang X, Sun P, Dwarakanath B, Kong L, and Lu JJ (2019). Depletion of MLKL inhibits invasion of radioresistant nasopharyngeal carcinoma cells by suppressing epithelial-mesenchymal transition. Ann Transl Med 7, 741. 10.21037/atm.2019.11.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edmond V, Merdzhanova G, Gout S, Brambilla E, Gazzeri S, and Eymin B. (2013). A new function of the splicing factor SRSF2 in the control of E2F1-mediated cell cycle progression in neuroendocrine lung tumors. Cell Cycle 12, 1267–1278. 10.4161/cc.24363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet 45, 592–601. 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun X, Wang SC, Wei Y, Luo X, Jia Y, Li L, Gopal P, Zhu M, Nassour I, Chuang J-C, et al. (2017). Arid1a Has Context-Dependent Oncogenic and Tumor Suppressor Functions in Liver Cancer. Cancer Cell 32, 574–589.e6. 10.1016/j.ccell.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu S, and Tang C. (2021). The Role of ARID1A in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol 11, 745187. 10.3389/fonc.2021.745187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Akimov V, Barrio-Hernandez I, Hansen SVF, Hallenborg P, Pedersen A-K, Bekker-Jensen DB, Puglia M, Christensen SDK, Vanselow JT, Nielsen MM, et al. (2018). UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol 25, 631–640. 10.1038/s41594-018-0084-y. [DOI] [PubMed] [Google Scholar]
- 42.Haradhvala NJ., Polak P., Stojanov P., Covington KR., Shinbro E., Hess JM., Rheinbay E., Kim J., Maruvka YE., Braunstein LZ., et al. (2016). Mutational Strand Asymmetries in Cancer Genomes Reveal Mechanisms of DNA Damage and Repair. Cell 164, 538–549. 10.1016/j.cell.2015.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. (2020). The repertoire of mutational signatures in human cancer. Nature 578, 94–101. 10.1038/s41586-020-1943-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kübler K, Mouw KW, et al. (2017). A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet 49, 1476–1486. 10.1038/ng.3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degasperi A, Zou X, Amarante TD, Martinez-Martinez A, Koh GCC, Dias JML, Heskin L, Chmelova L, Rinaldi G, Wang VYW, et al. (2022). Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 376. 10.1126/science.abl9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polo SE, and Jackson SP (2011). Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433. 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang H, and Xu X. (2017). Microhomology-mediated end joining: new players join the team. Cell Biosci. 7, 6. 10.1186/s13578-017-0136-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sfeir A, and Symington LS (2015). Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci 40, 701–714. 10.1016/j.tibs.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Z, Wang-Heaton H, Cartwright BM, Makinwa Y, Hilton BA, Musich PR, Shkriabai N, Kvaratskhelia M, Guan S, Chen Q, et al. (2021). ATR prevents Ca2+ overload-induced necrotic cell death through phosphorylation-mediated inactivation of PARP1 without DNA damage signaling. FASEB J. 35, e21373. 10.1096/fj.202001636RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gupte R, Liu Z, and Kraus WL (2017). PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126. 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brunyanszki A, Olah G, Coletta C, Szczesny B, and Szabo C. (2014). Regulation of mitochondrial poly(ADP-Ribose) polymerase activation by the β-adrenoceptor/cAMP/protein kinase A axis during oxidative stress. Mol. Pharmacol 86, 450–462. 10.1124/mol.114.094318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, Grande D, Grinkevich V, Heald RA, Langdon S, et al. (2021). Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun 12, 3636. 10.1038/s41467-021-23463-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MIR, O’Connor KW, Konstantinopoulos PA, Elledge SJ, Boulton SJ, et al. (2015). Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 518, 258–262. 10.1038/nature14184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bolderson E., Tomimatsu N., Richard DJ., Boucher D., Kumar R., Pandita TK., Burma S., and Khanna KK. (2010). Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 38, 1821–1831. 10.1093/nar/gkp1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bindra RS, Crosby ME, and Glazer PM (2007). Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 26, 249–260. 10.1007/s10555-007-9061-3. [DOI] [PubMed] [Google Scholar]
- 56.Ng N, Purshouse K, Foskolou IP, Olcina MM, and Hammond EM (2018). Challenges to DNA replication in hypoxic conditions. FEBS J. 285, 1563–1571. 10.1111/febs.14377. [DOI] [PubMed] [Google Scholar]
- 57.Pires IM, Bencokova Z, Milani M, Folkes LK, Li J-L, Stratford MR, Harris AL, and Hammond EM (2010). Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 70, 925–935. 10.1158/0008-5472.CAN-09-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, Belmaaza A, Wouters B, and Bristow RG (2008). Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 68, 605–614. 10.1158/0008-5472.CAN-07-5472. [DOI] [PubMed] [Google Scholar]
- 59.Kim I-K, Stegeman RA, Brosey CA, and Ellenberger T. (2015). A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem 290, 3775–3783. 10.1074/jbc.M114.624718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hegde ML, Izumi T, and Mitra S. (2012). Oxidized base damage and single-strand break repair in mammalian genomes: role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci 110, 123–153. 10.1016/B978-0-12-387665-2.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zheng F, Zhang Y, Chen S, Weng X, Rao Y, and Fang H. (2020). Mechanism and current progress of Poly ADP-ribose polymerase (PARP) inhibitors in the treatment of ovarian cancer. Biomed. Pharmacother 123, 109661. 10.1016/j.biopha.2019.109661. [DOI] [PubMed] [Google Scholar]
- 62.Rose M, Burgess JT, O’Byrne K, Richard DJ, and Bolderson E. (2020). PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol 8, 564601. 10.3389/fcell.2020.564601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murata MM, Kong X, Moncada E, Chen Y, Imamura H, Wang P, Berns MW, Yokomori K, and Digman MA (2019). NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 30, 2584–2597. 10.1091/mbc.E18-10-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Palermo V, Rinalducci S, Sanchez M, Grillini F, Sommers JA, Brosh RM Jr, Zolla L, Franchitto A, and Pichierri P. (2016). CDK1 phosphorylates WRN at collapsed replication forks. Nat. Commun 7, 12880. 10.1038/ncomms12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bian L, Meng Y, Zhang M, and Li D. (2019). MRE11-RAD50-NBS1 complex alterations and DNA damage response: implications for cancer treatment. Mol. Cancer 18, 169. 10.1186/s12943-019-1100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ikenoue T., Togo G., Nagai K., Ijichi H., Kato J., Yamaji Y., Okamoto M., Kato N., Kawabe T., Tanaka A., et al. (2001). Frameshift mutations at mononucleotide repeats in RAD50 recombinational DNA repair gene in colorectal cancers with microsatellite instability. Jpn. J. Cancer Res. 92, 587–591. 10.1111/j.1349-7006.2001.tb01134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alemayehu A, and Fridrichova I. (2007). The MRE11/RAD50/NBS1 complex destabilization in Lynch-syndrome patients. Eur. J. Hum. Genet 15, 922–929. 10.1038/sj.ejhg.5201858. [DOI] [PubMed] [Google Scholar]
- 68.Neal JA, Dunger K, Geith K, and Meek K. (2020). Deciphering the role of distinct DNA- PK phosphorylations at collapsed replication forks. DNA Repair 94, 102925. 10.1016/j.dnarep.2020.102925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zolner AE, Abdou I, Ye R, Mani RS, Fanta M, Yu Y, Douglas P, Tahbaz N, Fang S, Dobbs T, et al. (2011). Phosphorylation of polynucleotide kinase/ phosphatase by DNA-dependent protein kinase and ataxia-telangiectasia mutated regulates its association with sites of DNA damage. Nucleic Acids Res. 39, 9224–9237. 10.1093/nar/gkr647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weinfeld M, Mani RS, Abdou I, Aceytuno RD, and Glover JNM (2011). Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem. Sci 36, 262–271. 10.1016/j.tibs.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gibbs DL (2020). Robust classification of Immune Subtypes in Cancer. bioRxiv, 2020.01.17.910950. 10.1101/2020.01.17.910950. [DOI] [Google Scholar]
- 73.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, and Alizadeh AA (2015). Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457. 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sevenich L. (2019). Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front. Oncol 9, 163. 10.3389/fonc.2019.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DeBerardinis RJ, and Chandel NS (2016). Fundamentals of cancer metabolism. Sci Adv 2, e1600200. 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hitosugi T, and Chen J. (2014). Post-translational modifications and the Warburg effect.Oncogene 33, 4279–4285. 10.1038/onc.2013.406. [DOI] [PubMed] [Google Scholar]
- 77.Pégorier J-P, Le May C, and Girard J. (2004). Control of gene expression by fatty acids. J. Nutr 134, 2444S–2449S. 10.1093/jn/134.9.2444S. [DOI] [PubMed] [Google Scholar]
- 78.Nusinow DP, Szpyt J, Ghandi M, Rose CM, McDonald ER 3rd, Kalocsay M, Jané-Valbuena J, Gelfand E, Schweppe DK, Jedrychowski M, et al. (2020). Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402.e16. 10.1016/j.cell.2019.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schönfeld P., and Reiser G. (2013). Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 33, 1493–1499. 10.1038/jcbfm.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gizak A, Wiśniewski J, Heron P, Mamczur P, Sygusch J, and Rakus D. (2019). Targeting a moonlighting function of aldolase induces apoptosis in cancer cells. Cell Death Dis. 10, 712. 10.1038/s41419-019-1968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan A, Sun X-M, Huang F-Q, Liu J-F, Cai Y-Y, Wu X, Alolga RN, Li P, Liu BL, Liu Q, et al. (2022). The mitochondrial β-oxidation enzyme HADHA restrains hepatic glucagon response by promoting β-hydroxybutyrate production. Nat. Commun 13, 386. 10.1038/s41467-022-28044-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang JH, Kim NH, Yun JS, Cho ES, Cha YH, Cho SB, Lee S-H, Cha SY, Kim S-Y, Choi J, et al. (2020). Snail augments fatty acid oxidation by suppression of mitochondrial ACC2 during cancer progression. Life Sci Alliance 3. 10.26508/lsa.202000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peng H, Wang Y, and Luo W. (2020). Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 39, 6747–6756. 10.1038/s41388-020-01480-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.White PJ, McGarrah RW, Grimsrud PA, Tso S-C, Yang W-H, Haldeman JM, Grenier-Larouche T, An J, Lapworth AL, Astapova I, et al. (2018). The BCKDH Kinase and Phosphatase Integrate BCAA and Lipid Metabolism via Regulation of ATP-Citrate Lyase. Cell Metab. 27, 1281–1293.e7. 10.1016/j.cmet.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koundouros N, and Poulogiannis G. (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122, 4–22. 10.1038/s41416-019-0650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, and Hess C. (2013). Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol 14, 1064–1072. 10.1038/ni.2687. [DOI] [PubMed] [Google Scholar]
- 87.Furukawa A, Wakamori M, Arimura Y, Ohtomo H, Tsunaka Y, Kurumizaka H, Umehara T, and Nishimura Y. (2020). Acetylated histone H4 tail enhances histone H3 tail acetylation by altering their mutual dynamics in the nucleosome. Proc. Natl. Acad. Sci. U. S. A 117, 19661–19663. 10.1073/pnas.2010506117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hao F, Murphy KJ, Kujirai T, Kamo N, Kato J, Koyama M, Okamato A, Hayashi G, Kurumizaka H, and Hayes JJ (2020). Acetylation-modulated communication between the H3 N-terminal tail domain and the intrinsically disordered H1 C-terminal domain. Nucleic Acids Res. 48, 11510–11520. 10.1093/nar/gkaa949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen D, Fang L, Li H, Tang M-S, and Jin C. (2013). Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J. Biol. Chem 288, 21678–21687. 10.1074/jbc.M113.476630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sundar IK, Nevid MZ, Friedman AE, and Rahman I. (2014). Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer. J. Proteome Res. 13, 982–996. 10.1021/pr400998n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Den Broeck A., Brambilla E., Moro-Sibilot D., Lantuejoul S., Brambilla C., Eymin B., and Gazzeri S. (2008). Loss of histone H4K20 trimethylation occurs in preneoplasia and influences prognosis of non-small cell lung cancer. Clin. Cancer Res. 14, 7237–7245. 10.1158/1078-0432.CCR-08-0869. [DOI] [PubMed] [Google Scholar]
- 92.Liu F, Killian JK, Yang M, Walker RL, Hong JA, Zhang M, Davis S, Zhang Y, Hussain M, Xi S, et al. (2010). Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 29, 3650–3664. 10.1038/onc.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oshita H, Nishino R, Takano A, Fujitomo T, Aragaki M, Kato T, Akiyama H, Tsuchiya E, Kohno N, Nakamura Y, et al. (2013). RASEF is a novel diagnostic biomarker and a therapeutic target for lung cancer. Mol. Cancer Res. 11, 937–951. 10.1158/1541-7786.MCR-12-0685-T. [DOI] [PubMed] [Google Scholar]
- 94.Wu A, Wu B, Guo J, Luo W, Wu D, Yang H, Zhen Y, Yu X, Wang H, Zhou Y, et al. (2011). Elevated expression of CDK4 in lung cancer. J. Transl. Med 9, 38. 10.1186/1479-5876-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Valdés-Mora F, Song JZ, Statham AL, Strbenac D, Robinson MD, Nair SS, Patterson KI, Tremethick DJ, Stirzaker C, and Clark SJ (2012). Acetylation of H2A.Z is a key epigenetic modification associated with gene deregulation and epigenetic remodeling in cancer. Genome Res. 22, 307–321. 10.1101/gr.118919.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Adenuga D, and Rahman I. (2010). Protein kinase CK2-mediated phosphorylation of HDAC2 regulates co-repressor formation, deacetylase activity and acetylation of HDAC2 by cigarette smoke and aldehydes. Arch. Biochem. Biophys 498, 62–73. 10.1016/j.abb.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho Y-S, Chen C-H, Wang Y-J, Pestell RG, Albanese C, Chen R-J, Chang M-C, Jeng J-H, Lin S-Y, Liang Y-C, et al. (2005). Tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induces cell proliferation in normal human bronchial epithelial cells through NFkappaB activation and cyclin D1 up-regulation. Toxicol. Appl. Pharmacol 205, 133–148. 10.1016/j.taap.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 98.Schaal C, and Chellappan SP (2014). Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 12, 14–23. 10.1158/1541-7786.MCR-13-0541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Memmott RM, and Dennis PA (2010). The role of the Akt/mTOR pathway in tobacco carcinogen-induced lung tumorigenesis. Clin. Cancer Res. 16, 4–10. 10.1158/1078-0432.CCR-09-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y, Liu J, Zhou J-S, Huang H-Q, Li Z-Y, Xu X-C, Lai T-W, Hu Y, Zhou H-B, Chen H-P, et al. (2018). MTOR Suppresses Cigarette Smoke-Induced Epithelial Cell Death and Airway Inflammation in Chronic Obstructive Pulmonary Disease. J. Immunol 200, 2571–2580. 10.4049/jimmunol.1701681. [DOI] [PubMed] [Google Scholar]
- 101.Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Schölz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244.e12. 10.1016/j.cell.2018.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mishima Y., Wang C., Miyagi S., Saraya A., Hosokawa H., Mochizuki-Kashio M., Nakajima-Takagi Y., Koide S., Negishi M., Sashida G., et al. (2014). Histone acetylation mediated by Brd1 is crucial for Cd8 gene activation during early thymocyte development. Nat. Commun 5, 5872. 10.1038/ncomms6872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gruber JJ, Geller B, Lipchik AM, Chen J, Salahudeen AA, Ram AN, Ford JM, Kuo CJ, and Snyder MP (2019). HAT1 Coordinates Histone Production and Acetylation via H4 Promoter Binding. Mol. Cell 75, 711–724.e5. 10.1016/j.molcel.2019.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yang J, Gong C, Ke Q, Fang Z, Chen X, Ye M, and Xu X. (2021). Insights Into the Function and Clinical Application of HDAC5 in Cancer Management. Front. Oncol 11, 661620. 10.3389/fonc.2021.661620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, et al. (2004). Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol 11, 308–315. 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 106.Wan W, You Z, Xu Y, Zhou L, Guan Z, Peng C, Wong CCL, Su H, Zhou T, Xia H, et al. (2017). mTORC1 Phosphorylates Acetyltransferase p300 to Regulate Autophagy and Lipogenesis. Mol. Cell 68, 323–335.e6. 10.1016/j.molcel.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 107.Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, and Locasale JW (2015). The rate of glycolysis quantitatively mediates specific histone acetylation sites. Cancer Metab 3, 10. 10.1186/s40170-015-0135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng Y-C, Tastemel M, and Banaszynski LA (2019). Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat. Genet 51, 941–946. 10.1038/s41588-019-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lau PNI, and Cheung P. (2011). Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proc. Natl. Acad. Sci. U. S. A 108, 2801–2806. 10.1073/pnas.1012798108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, and Turk BE (2004). A rapid method for determining protein kinase phosphorylation specificity. Nat. Methods 1, 27–29. 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 111.Songyang Z, Carraway KL 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, and Eng C. (1995). Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature 373, 536–539. 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 112.Johnson JL, Yaron TM, Huntsman EM, Kerelsky A, Song J, Regev A, Lin T-Y, Liberatore K, Cizin DM, Cohen BM, et al. (2022). A global atlas of substrate specificities for the human serine/threonine kinome. bioRxiv, 2022.05.22.492882. 10.1101/2022.05.22.492882. [DOI] [Google Scholar]
- 113.Songyang Z, Blechner S, Hoagland N, Hoekstra MF, Piwnica-Worms H, and Cantley LC (1994). Use of an oriented peptide library to determine the optimal substrates of protein kinases. Curr. Biol 4, 973–982. 10.1016/s0960-9822(00)00221-9. [DOI] [PubMed] [Google Scholar]
- 114.Yaffe MB., and Smerdon SJ. (2004). The use of in vitro peptide-library screens in the analysis of phosphoserine/threonine-binding domain structure and function. Annu. Rev. Biophys. Biomol. Struct 33, 225–244. 10.1146/annurev.biophys.33.110502.133346. [DOI] [PubMed] [Google Scholar]
- 115.Turk BE, Huang LL, Piro ET, and Cantley LC (2001). Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nat. Biotechnol 19, 661–667. 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 116.Agricola E, Randall RA, Gaarenstroom T, Dupont S, and Hill CS (2011). Recruitment of TIF1γ to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol. Cell 43, 85–96. 10.1016/j.molcel.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 117.Yasui Y, Urano T, Kawajiri A, Nagata K-I, Tatsuka M, Saya H, Furukawa K, Takahashi T, Izawa I, and Inagaki M. (2004). Autophosphorylation of a newly identified site of Aurora-B is indispensable for cytokinesis. J. Biol. Chem 279, 12997–13003. 10.1074/jbc.M311128200. [DOI] [PubMed] [Google Scholar]
- 118.Goto H, Tomono Y, Ajiro K, Kosako H, Fujita M, Sakurai M, Okawa K, Iwamatsu A, Okigaki T, Takahashi T, et al. (1999). Identification of a novel phosphorylation site on histone H3 coupled with mitotic chromosome condensation. J. Biol. Chem 274, 25543–25549. 10.1074/jbc.274.36.25543. [DOI] [PubMed] [Google Scholar]
- 119.Cai G, Yang Q, and Sun W. (2021). RSF1 in cancer: interactions and functions. Cancer Cell Int. 21, 315. 10.1186/s12935-021-02012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee H-S, Park Y-Y, Cho M-Y, Chae S, Yoo Y-S, Kwon M-H, Lee C-W, and Cho H. (2015). The chromatin remodeller RSF1 is essential for PLK1 deposition and function at mitotic kinetochores. Nat. Commun 6, 7904. 10.1038/ncomms8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lee H-S, Lin Z, Chae S, Yoo Y-S, Kim B-G, Lee Y, Johnson JL, Kim Y-S, Cantley LC, Lee C-W, et al. (2018). The chromatin remodeler RSF1 controls centromeric histone modifications to coordinate chromosome segregation. Nat. Commun 9, 3848. 10.1038/s41467-018-06377-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Elia AEH, Cantley LC, and Yaffe MB (2003). Proteomic screen finds pSer/pThr- binding domain localizing Plk1 to mitotic substrates. Science 299, 1228–1231. 10.1126/science.1079079. [DOI] [PubMed] [Google Scholar]
- 123.Yaron TM, Heaton BE, Levy TM, Johnson JL, Jordan TX, Cohen BM, Kerelsky A, Lin T-Y, Liberatore KM, Bulaon DK, et al. (2020). The FDA-approved drug Alectinib compromises SARS-CoV-2 nucleocapsid phosphorylation and inhibits viral infection in vitro. bioRxiv. 10.1101/2020.08.14.251207. [DOI] [Google Scholar]
- 124.Zheng Y, Ramsamooj S, Li Q, Johnson JL, Yaron TM, Sharra K, and Cantley LC (2019). Regulation of folate and methionine metabolism by multisite phosphorylation of human methylenetetrahydrofolate reductase. Sci. Rep 9, 4190. 10.1038/s41598-019-40950-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chang Y-C, Chiou J, Yang Y-F, Su C-Y, Lin Y-F, Yang C-N, Lu P-J, Huang M-S, Yang C-J, and Hsiao M. (2019). Therapeutic Targeting of Aldolase A Interactions Inhibits Lung Cancer Metastasis and Prolongs Survival. Cancer Res. 79, 4754–4766. 10.1158/0008-5472.CAN-18-4080. [DOI] [PubMed] [Google Scholar]
- 126.Luby A, and Alves-Guerra M-C (2021). Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers 13. 10.3390/cancers13235912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stevens BM, Jones CL, Pollyea DA, Culp-Hill R, D’Alessandro A, Winters A, Krug A, Abbott D, Goosman M, Pei S, et al. (2020). Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat Cancer 1, 1176–1187. 10.1038/s43018-020-00126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van Galen P, Hovestadt V, Wadsworth MH Ii, Hughes TK, Griffin GK, Battaglia S, Verga JA, Stephansky J, Pastika TJ, Lombardi Story J, et al. (2019). Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 176, 1265–1281.e24. 10.1016/j.cell.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, Lieb DJ, Chen JH, Frederick DT, Barzily-Rokni M, et al. (2018). Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 175, 998–1013.e20. 10.1016/j.cell.2018.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, Bejnood A, Dionne D, Ge WH, Xu KH, et al. (2021). Spatially organized multicellular immune hubs in human colorectal cancer. Cell 184, 4734–4752.e20. 10.1016/j.cell.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstråhle J, et al. (2020). Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 182, 497–514.e22. 10.1016/j.cell.2020.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Minakshi P, Kumar R, Ghosh M, Saini HM, Ranjan K, Brar B, and Prasad G. (2019). Chapter 14 - Single-Cell Proteomics: Technology and Applications. In Single-Cell Omics, Barh Dand Azevedo V, eds. (Academic Press; ), pp. 283–318. 10.1016/B978-0-12-814919-5.00014-2. [DOI] [Google Scholar]
- 133.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. (2019). Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 9, 1102–1123. 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Le Large TY, Mantini G, Meijer LL, Pham TV, Funel N, van Grieken NC, Kok B, Knol J, van Laarhoven HW, Piersma SR, et al. (2020). Microdissected pancreatic cancer proteomes reveal tumor heterogeneity and therapeutic targets. JCI Insight 5. 10.1172/jci.insight.138290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. 10.1093/bioinformatics/btv710. [DOI] [PubMed] [Google Scholar]
- 136.Kim S., Scheffler K., Halpern AL., Bekritsky MA., No E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 137.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Taylor-Weiner A, Stewart C, Giordano T, Miller M, Rosenberg M, Macbeth A, Lennon N, Rheinbay E, Landau D-A, Wu CJ, et al. (2018). DeTiN: overcoming tumor-in-normal contamination. Nat. Methods 15, 531–534. 10.1038/s41592-018-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G. (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.The GTEx Consortium (2020). The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330. 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Johnson WE, Li C, and Rabinovic A. (2007). Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 145.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hänzelmann S, Castelo R, and Guinney J. (2013). GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinformatics 14. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aran D, Hu Z, and Butte AJ (2017). xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18, 220. 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wen B, Wang X, and Zhang B. (2019). PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations. Genome Res. 29, 485–493. 10.1101/gr.235028.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ritchie ME., Phipson B., Wu D., Hu Y., Law CW., Shi W., and Smyth GK. (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hess JM, Bernards A, Kim J, Miller M, Taylor-Weiner A, Haradhvala NJ, Lawrence MS, and Getz G. (2019). Passenger Hotspot Mutations in Cancer. Cancer Cell 36, 288–301.e14. 10.1016/j.ccell.2019.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, et al. (2013). Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67. 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Graubert A, Aguet F, Ravi A, Ardlie KG, and Getz G. (2021). RNA-SeQC 2: Efficient RNA-seq quality control and quantification for large cohorts. Bioinformatics. 10.1093/bioinformatics/btab135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Li B, Ruotti V, Stewart RM, Thomson JA, and Dewey CN (2010). RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26, 493–500. 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ouspenskaia T, Law T, Clauser KR, Klaeger S, Sarkizova S, Aguet F, Li B, Christian E, Knisbacher BA, Le PM, et al. (2022). Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat. Biotechnol 40, 209–217. 10.1038/s41587-021-01021-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ruggles KV, Tang Z, Wang X, Grover H, Askenazi M, Teubl J, Cao S, McLellan MD, Clauser KR, Tabb DL, et al. (2016). An Analysis of the Sensitivity of Proteogenomic Mapping of Somatic Mutations and Novel Splicing Events in Cancer. Mol. Cell. Proteomics 15, 1060–1071. 10.1074/mcp.M115.056226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tan VYF, and Févotte C. (2013). Automatic Relevance Determination in Nonnegative Matrix Factorization with the /spl beta/-Divergence. IEEE Trans. Pattern Anal. Mach. Intell 35, 1592–1605. 10.1109/TPAMI.2012.240. [DOI] [PubMed] [Google Scholar]
- 158.Kim J, Kwiatkowski D, McConkey DJ, Meeks JJ, Freeman SS, Bellmunt J, Getz G, and Lerner SP (2019). The Cancer Genome Atlas Expression Subtypes Stratify Response to Checkpoint Inhibition in Advanced Urothelial Cancer and Identify a Subset of Patients with High Survival Probability. European Urology 75, 961–964. 10.1016/j.eururo.2019.02.017. [DOI] [PubMed] [Google Scholar]
- 159.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine learning in Python. the Journal of machine Learning research 12, 2825–2830. [Google Scholar]
- 160.Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, and Forbes SA (2018). The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705. 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Haradhvala NJ., Kim J., Maruvka YE., Polak P., Rosebrock D., Livitz D., Hess JM., Leshchiner I., Kamburov A., Mouw KW., et al. (2018). Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun 9, 1746. 10.1038/s41467-018-04002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. (2017). HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med 23, 517–525. 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Robinson MD, and Oshlack A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, 1–9. 10.1186/gb-2010-11-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kamburov A, Lawrence MS, Polak P, Leshchiner I, Lage K, Golub TR, Lander ES, and Getz G. (2015). Comprehensive assessment of cancer missense mutation clustering in protein structures. Proc. Natl. Acad. Sci. U. S. A 112, E5486–E5495. 10.1073/pnas.1516373112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Luna A, Siper MC, Korkut A, Durupinar F, Dogrusoz U, Aslan JE, Sander C, Demir E, and Babur O. (2021). Analyzing causal relationships in proteomic profiles using CausalPath. STAR Protoc 2, 100955. 10.1016/j.xpro.2021.100955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Balci H, Siper MC, Saleh N, Safarli I, Roy L, Kilicarslan M, Ozaydin R, Mazein A, Auffray C, Babur Ö, et al. (2021). Newt: a comprehensive web-based tool for viewing, constructing and analyzing biological maps. Bioinformatics 37, 1475–1477. 10.1093/bioinformatics/btaa850. [DOI] [PubMed] [Google Scholar]
- 168.Manning G, Whyte DB, Martinez R, Hunter T, and Sudarsanam S. (2002). The protein kinase complement of the human genome. Science 298, 1912–1934. 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 169.Lee DD, and Seung HS (1999). Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791. 10.1038/44565. [DOI] [PubMed] [Google Scholar]
- 170.Brunet J-P, Tamayo P, Golub TR, and Mesirov JP (2004). Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. U. S. A 101, 4164–4169. 10.1073/pnas.0308531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Knisbacher BA, Lin Z, Hahn CK, Nadeu F, Duran-Ferrer M, Stevenson KE, Tausch E, Delgado J, Barbera-Mourelle A, Taylor-Weiner A, et al. (2022). Molecular map of chronic lymphocytic leukemia and its impact on outcome. Nat. Genet 10.1038/s41588-022-01140-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Roh W, Geffen Y, Cha H, Miller M, Anand S, Kim J, Heiman DI, Gainor JF, Laird PW, Cherniack AD, et al. (2022). High-Resolution Profiling of Lung Adenocarcinoma Identifies Expression Subtypes with Specific Biomarkers and Clinically Relevant Vulnerabilities. Cancer Res. 82, 3917–3931. 10.1158/0008-5472.CAN-22-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hunter T. (2007). The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738. 10.1016/j.molcel.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 174.Woodsmith J, Kamburov A, and Stelzl U. (2013). Dual coordination of post translational modifications in human protein networks. PLoS Comput. Biol 9, e1002933. 10.1371/journal.pcbi.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Beltrao P, Bork P, Krogan NJ, and van Noort V. (2013). Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol 9, 714. 10.1002/msb.201304521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bludau I, Willems S, Zeng W-F, Strauss MT, Hansen FM, Tanzer MC, Karayel O, Schulman BA, and Mann M. (2022). The structural context of posttranslational modifications at a proteome-wide scale. PLoS Biol. 20, e3001636. 10.1371/journal.pbio.3001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Babur Ö, Melrose AR, Cunliffe JM, Klimek J, Pang J, Sepp A-LI, Zilberman- Rudenko J, Tassi Yunga S, Zheng T, Parra-Izquierdo I, et al. (2020). Phosphoproteomic quantitation and causal analysis reveal pathways in GPVI/ITAM-mediated platelet activation programs. Blood 136, 2346–2358. 10.1182/blood.2020005496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Babur Ö, Ngo ATP, Rigg RA, Pang J, Rub ZT, Buchanan AE, Mitrugno A, David LL, McCarty OJT, Demir E, et al. (2018). Platelet procoagulant phenotype is modulated by a p38-MK2 axis that regulates RTN4/Nogo proximal to the endoplasmic reticulum: utility of pathway analysis. Am. J. Physiol. Cell Physiol. 314, C603–C615. 10.1152/ajpcell.00177.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Joshi SK, Nechiporuk T, Bottomly D, Piehowski PD, Reisz JA, Pittsenbarger J, Kaempf A, Gosline SJC, Wang Y-T, Hansen JR, et al. (2021). The AML microenvironment catalyzes a stepwise evolution to gilteritinib resistance. Cancer Cell 39,999–1014.e8. 10.1016/j.ccell.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Khadka P, Reitman ZJ, Lu S, Buchan G, Gionet G, Dubois F, Carvalho DM, Shih J, Zhang S, Greenwald NF, et al. (2022). PPM1D mutations are oncogenic drivers of de novo diffuse midline glioma formation. Nat. Commun 13, 604. 10.1038/s41467-022-28198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Keshishian H, McDonald ER 3rd, Mundt F, Melanson R, Krug K, Porter DA, Wallace L, Forestier D, Rabasha B, Marlow SE, et al. (2021). A highly multiplexed quantitative phosphosite assay for biology and preclinical studies. Mol. Syst. Biol 17, e10156. 10.15252/msb.202010156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Johnson BE, Creason AL, Stommel JM, Keck JM, Parmar S, Betts CB, Blucher A, Boniface C, Bucher E, Burlingame E, et al. (2022). An omic and multidimensional spatial atlas from serial biopsies of an evolving metastatic breast cancer. Cell Rep Med 3, 100525. 10.1016/j.xcrm.2022.100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhao M, Scott S, Evans KW, Yuca E, Saridogan T, Zheng X, Wang H, Korkut A, Cruz Pico CX, Demirhan M, et al. (2021). Combining Neratinib with CDK4/6, mTOR, and MEK Inhibitors in Models of HER2-positive Cancer. Clin. Cancer Res 27, 1681–1694. 10.1158/1078-0432.CCR-20-3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 - Pan-Cancer Data harmonization
A. Density plots showing PTM levels and their matched protein levels. Ordinary least squares (OLS) were fitted for each PTM-protein pair, and the residuals from the fit represented PTM levels corrected for protein abundance. Raw phosphoproteome abundance (left), imputed phosphoproteome abundance (used for clustering, middle), and raw acetylome abundance (right).
B. Principal component analysis (PCA) projection of transcriptome TPM across the 10 CPTAC cohorts and the medulloblastoma cohort before and after applying batch correction (using COMBAT). Top PCA plots colored by sequencing protocol (poly- adenylated mRNA enrichment [red] and total RNA [blue]), and bottom PCA plots colored by cohort.
C. Feature distribution showing the final normalized RNA data, the proteomic data, phosphorylation data corrected for protein abundance and the final multi-omic feature space following cohort correction. Distributions are colored by cohort.
D. Projection of multi-omic features input into NMF on a PCA of all 1110 samples colored by cohort; (top row) all features combined- mRNA, protein, and phosphorylation, (middle row) mRNA, (bottom row) protein and phosphorylation. (left + middle column) features before and after regressing out cohort-specific effects, (right column) features after regression and split into positive and negative values for NMF.
Figure S2 - Patient Clustering and CLUMPS-PTM method
A. Stacked barplot of patient-wise assigned maximally weighted signature colored by cohort (left). Stacked bar plot shows the breakdown, normalized by the number of patients maximally assigned to a given signature (right).
B. Multi-omic hierarchical clustering of tumors based on multi-omic NMF signature weights. Heatmap represents the pair-wise cosine similarity between tumors. Non-overlapping clusters - yellow boxes. Top track shows the cluster assignments. Dendrogram is colored based on the second level of the tree.
C. Distribution of the fraction of a sample’s NMF signature weights accounted for by the maximally contributing signature (top, left); Distribution of the number of signatures required to explain at least 90% of a sample’s signature weights (top, right); Distribution of the difference between the fraction of a sample’s NMF signature weights accounted for by the maximally contributing signature and the second strongest signature (bottom).
D. Schematic workflow of the newly developed CLUMPS-PTM algorithm.
E. Sankey plot showing the mapping between protein sequences to PDB structures. Protein sequences were aligned to the UniProtKB database (using BLASTP+ hits), then filtered to those with matching well-annotated proteins identified using the Structure Integration with Function, Taxonomy and Sequence (SIFTS) method. These were then mapped to available crystal structures with at least 3 modified PTM sites across the entire CPTAC dataset (upper left). Sankey plot of all PTM sites in the CPTAC dataset, splitting to those with accession-numbers mapped to available Uniprot input proteins, and then to proteins with available PDB structures verified in the SIFTS database (lower left). Violin plots of the BLASTP+ relative identity score to the Uniprot database for proteins and PTM sites that map or do not map to PDB structures (right).
F. Stacked bar plots summarizing CLUMPS-PTM results showing the total number of proteins tested based on group-specific differential expression test & direction (top panel). Stacks show the number of structures that are nominally significant (p<0.1; yellow) and pass multiple hypothesis testing (FDR<0.1; red) for acetylome, phosphoproteome, and combined samplers. CLUMPS-PTM phosphorylation and acetylation hits (q-value<0.1) for the left (L) vs. right (R) top split of the Pan-Cancer dendrogram (bottom, panel). The first heatmap (on the left) indicates the number of distinct peptides found in each cohort that contributed to the differential PTM cluster. The x-axis represents the cohort. The second heatmap (on the right) expands on each CLUMPS-PTM hit and shows the log-fold change between the left vs. right branches of the top split in the dendrogram for each represented peptide. Blue indicates phosphorylated clustered sites.
Figure S3 - Mutational signature extraction and analysis of DNA repair deficiency
A. Schematic of the mutational signature extraction workflow and barplot representations of the resulting mutational signatures.
B. Scatter plots illustrating the distributions for the number of mutations and the fraction of mutations attributed to the HRD-associated mutational signature (top). HRD classification threshold at 45 mutations indicated by a dotted line. Tumors with known pathogenic or high impact germline variants in BRCA1, BRCA2, or PALB2 genes are highlighted with a black outline; Scatter plots showing the distributions for the number of mutations and the fraction of mutations attributed to the mutational signature associated with MMRD in COAD and UCEC tumors (bottom). MMRD classification thresholds at 72 mutations and at 19% of mutations, respectively indicated by horizontal and vertical dotted lines. Tumors with known pathogenic or high impact variants in MSH6, MSH2, MLH1, and PMS2 are highlighted with a black outline.
C. Pathway of differentially expressed DDR genes (at the RNA [r circle], protein [box], and phosphorylation site [p circles] levels) between the HRD and homologous recombination proficient (HRP) tumors and their inferred effect on other pathway members. Upregulation in the HRD tumors in red and downregulation in blue. Dotted edges between the kinases and their substrates indicates that the interaction is within the 90th percentile of all top scoring substrates for the specific kinase (The Kinase Library).
D. PCA of multi-omic signature weights across UCEC and COAD MMRD tumors. Points are colored by tumor type (COAD - orange and UCEC - purple).
E. Distributions of MRN complex proteins (MRE11, RAD50, and NBN) relative protein abundance (top) and RNA expression in CCLE COAD and UCEC cancer cell lines from the DepMap dataset. Groups are separated and colored by mismatch repair proficiency (MMRD - purple, MMRD - yellow). Shapes indicate tumor type (UCEC - circle, COAD - triangle).
Figure S4 - Immune clustering, activity and associated metabolic markers
A. Violin plots showing the levels of different immune signatures based on ImmuneSubtypeClassifier and purity estimates from ESTIMATE for the 4 identified immune clusters across all tumors.
B. Violin plots showing the top 6 immune cell abundances from CIBERSORT that were used as covariates in differential expression to estimate tumor intrinsic gene expression in the 4 immune clusters.
C. Stacked barplot showing the distribution of 4 immune clusters within each cohort.
D. Violin plots showing significant differences between the 4 identified immune clusters for the immunosuppressive markers - PD-L1, IDO1, CD163, ENTPD1 (Pairwise comparisons with q value < 0.1 are shown).
E. Violin plots showing acetylation levels of fatty acid related proteins among different immune subtypes when immune cold is split to Brain and non-brain tumors.
F. BCKDK (PDB ID:1DTW) showing significant spatial clustering of phosphorylation sites by CLUMPS-PTM of differentially regulated sites for the immune-cool cluster. Protein 3D crystal structure space-filling model (cyan) and highlighted clumped phosphosites (purple) and thymidine-5’-triphosphate (TTP) sites are marked in green.
G. Bubble plot representing the Kinase Library enrichment score and significance, differentially expressed substrates of each kinase for the 4 immune clusters. Colors represent enrichment (Red) and depletion (Blue). Kinases are colored by to their phylogenetic groups 168
H. Violin plots showing significant differences between the 4 identified immune clusters for fatty acid transporters - CPT1A, FABP, ABCA3, CD36 (only pairwise comparisons with q value < 0.1 are shown).
I. Spearman correlation between the ssGSEA values for each sample of the MSigDB HALLMARK gene sets to the CD8+ T-cell abundance estimated from CIBERSORT.
Figure S5 - Histone analysis
A. Correlation matrix showing the correlations between mean acetylation of H1, H2A, H2B, H3 and H4. Asterisks denote correlations with p<0.05.
B. Violin plot showing the distribution of tobacco smoking mutational signature contributions (log10-scale) for males and females in LSCC and LUAD. Shapes indicate the cancer type-LUAD (circle) and LSCC (triangle). p-value (1x10−4) from Wilcoxon rank-sum test.
C. Dot plot showing significant Spearman correlations (q ≤ 0.1) across male LUAD patients (n=71) between histone acetylation levels and tobacco smoking mutational signature contributions.
D. Dot plot showing significant Spearman correlations (q ≤ 0.1) across male LUAD patients (n=71) between deacetylases phosphorylation levels and tobacco smoking mutational signature contributions.
E. Dot plot illustrating significant Spearman correlations (q ≤ 0.1, |rho|>0.15) across male LSCC and male LUAD patients (n=157) between tobacco smoking mutational signature contributions and the mRNA ssGSEA scores for the MSigDB HALLMARK gene sets.
F. Scatter plot showing the positive correlation (rho=0.332) between tobacco smoking mutational signature contributions and the ssGSEA score for the G2/M Checkpoint (left) and MTORC1 (right) Hallmark gene set in male LUAD and male LSCC tumors (n=157).
Figure S6 - The Kinase Library
A. Box plots showing the Kinase Library normalized intensity ratios of 207 Ser/Thr kinases for unmodified, trimethylated, and acetylated lysines, across 10 positions (-5 to +5) around the central phospho-acceptor site.
B. Heatmap showing the Kinase Library normalized intensity ratios between trimethylated lysine and unmodified lysine across the 207 Ser/Thr kinases. Kinases are colored according to their phylogenetic groups 168
C.Heatmap showing the expression levels of acetylated K23 and phosphorylated S28 on H3F3A across 41 patients in cluster #8. Samples are sorted based on hierarchical clustering of their correlations.
D. Heatmap showing the expression levels of acetylated K1378 and phosphorylated S1375 on RSF1 across the 89 LUAD and LUSC patients with acetylation and phosphorylation data. Samples are sorted based on hierarchical clustering of their correlations.
Table S1. Metadata of all features included in the study, related to Figure 1 and Figure S1.
A. Inclusion of Transcriptome Features
B. Inclusion of Proteome Features
C. Inclusion of Phosphoproteome Features
D. Inclusion of Acetylation Features
Table S2. Pan-Cancer Multi-omics clusterings and annotations, related to Figure 2 & 3 and Figure S2 & S3.
A. ARD NMF H-matrix
B. ARD NMF W-matrix
C. Signature Annotations
D. Dendrogram 1–4 CLUMPS-PTM
E. Dendrogram results 1–3 from The Kinase Library
Table S3. Mutational signatures and DDR analysis, related to Figure 3 and Figure S3.
A. Mutational signature W matrix (mutational spectra for each signature)
B. Mutational signature H Matrix
C. Collapsed mutational signature attributions (weights for similar signatures collapsed to a single weight)
D. Mutational signatures summary table
E. DNA repair deficiency classification
F. DNA damage response geneset
G. HRD vs HRP DDR differential expression
H. HRD acute (B) vs chronic (C) hypoxia full differential expression
I. Acute (B) vs chronic (C) hypoxia DDR differential expression
J. HRD acute (B) vs chronic (C) hypoxia GSEA
K. HRD acute (B) vs chronic (C) hypoxia PTM-SEA
L. The Kinase Library scores for CausalPath kinase-substrate pairs
M. HRD low (A) vs acute (B) hypoxia full differential expression
N. HRD low (A) vs acute (B) hypoxia GSEA
O. HRD low (A) vs chronic (C) hypoxia GSEA
P. MMRD vs MMRP full differential expression
Q. MMRD vs MMRP DDR differential expression
R. MMRD vs MMRP GSEA
S. MMRD microsatellite indel Fisher’s exact tests
Table S4. Immune classification and metabolic analysis, related to Figure 4 and Figure S4.
A. ESTIMATE scores
B. CIBERSORT scores
C. CIBERSORT ssGSEA enrichments
D. Immune Subtype Classifier
E. fGSEA Metabolic and Immune pathways per subtype
F. Immune CLUMPS-PTM results
G. Fatty acid metabolism correlations
H. Immune cold results from The Kinase Library
I. Immune cold results results from The Kinase Library
J. Immune warm results from The Kinase Library
K. Immune hot results results from The Kinase Library
Table S5. Histone analysis, related to Figure 5 and Figure S5.
A. Histone related genes
B. Histone acetylation sites and phosphorylation sites
C. Correlation across histone acetylation sites
D. Correlation between smoking signature and histone acetylation across male LUAD patients
E. Correlation between smoking signature and HDAC phosphorylation across male LUAD patients
F. Correlation between smoking signature and HALLMARK pathways across male LSCC and male LUAD patients
G. Key regulators of histone acetylation sites
H. Correlation between histone acetylation sites and Hallmark pathways
I. Differentially acetylated histone sites in the immune clusters
J. Correlation between histone acetylation and phosphorylation
K. Site specific lasso regression
Table S6. The Kinase Library crosstalk analysis, related to Figure 6 and Figure S6.
A. Unmodified lysine values
B. Unmodified lysine - normalized
C. Acetylated lysine values
D. Acetylated lysine - ratio
E. Acetylated lysine - normalized
F. Methylated lysine values
G. Methylated lysine - ratio
H. Methylated lysine - normalized
I. Acetylation-phosphorylation crosstalk correlations
Data Availability Statement
Data Availability:
Raw and processed proteomics as well as open access genomic data can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov with dbGaP Study Accession: phs001287.v16.p6. Complete CPTAC pan-cancer controlled and processed data can be accessed via the Cancer Data Service (CDS). The CPTAC pan-cancer data hosted in CDS is controlled data and can be accessed through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis.
Instructions to access data:
Create an account on CGC, Seven Bridges (https://cgc-accounts.sbgenomics.com/auth/register
Get approval from dbGaP to access the controlled study (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001287.v16.p6
Log into CGC to access Cancer Data Service (CDS) File Explore
Copy data into your own space and start analysis and exploration
Visit the CDS page on CGC to see what studies are available and instructions and guides to use the resources. (https://docs.cancergenomicscloud.org/page/cds-data)
Code for the analysis and figures in this paper can be found at https://github.com/getzlab/CPTAC_PanCan_PTM_2023.
Software and code used in this study are referenced in their corresponding STAR Method sections and the Key Resource Table.
Key resources table
Raw and processed proteomics as well as open access genomic data can be obtained via Proteomic Data Commons (PDC) at https://pdc.cancer.gov/pdc/cptac-pancancer. Raw genomic and transcriptomic data files can be accessed via the Genomic Data Commons (GDC) Data Portal at https://portal.gdc.cancer.gov with dbGaP Study Accession: phs001287.v16.p6. Complete CPTAC pan-cancer controlled and processed data can be accessed via the Cancer Data Service (CDS). The CPTAC pan-cancer data hosted in CDS is controlled data and can be accessed through the NCI DAC approved, dbGaP compiled whitelists. Users can access the data for analysis through the Seven Bridges Cancer Genomics Cloud (SB-CGC) which is one of the NCI-funded Cloud Resource/platform for compute intensive analysis.