

Abstract
Numerous studies have revealed the involvement of fibrinogen in the inflammatory response. To explain the molecular mechanisms underlying fibrinogen-dependent inflammation, two bridging mechanisms have been proposed in which fibrin(ogen) bridges leukocytes to endothelial cells. The first mechanism suggests that bridging occurs via the interaction of fibrinogen with the leukocyte receptor Mac-1 and the endothelial receptor ICAM-1, which promotes leukocyte transmigration and enhances inflammation. The second mechanism includes bridging of leukocytes to the endothelium by fibrin degradation product E1 fragment through its interaction with leukocyte receptor CD11c and endothelial VE-cadherin to promote leukocyte transmigration. The role of E1 in promoting inflammation is inhibited by the fibrin-derived β15-42 fragment, and this has been suggested to result from its ability to compete for the E1-VE-cadherin interaction and to trigger signaling pathways through the Src kinase Fyn. Our recent study revealed that the β15-42 fragment is ineffective in inhibiting the E1- or fibrin-VE-cadherin interaction, leaving the proposed signaling mechanism as the only viable explanation for the inhibitory function of β15-42. We have discovered that fibrin interacts with the VLDL receptor, and this interaction triggers a signaling pathway that promotes leukocyte transmigration through inhibition of src kinase Fyn. This pathway is inhibited by another pathway induced by the interaction of β15-42 with a putative endothelial receptor. In this review, we briefly describe the previously proposed molecular mechanisms underlying fibrin-dependent inflammation and their advantages/disadvantages and summarize our recent studies of the novel VLDL receptor-dependent pathway of leukocyte transmigration which plays an important role in fibrin-dependent inflammation.
Keywords: fibrinogen, fibrin, VLDL receptor, Fibrin-VLDL receptor interaction, inflammation
Introduction
Fibrinogen is mainly known as the blood coagulation protein playing an important role in haemostasis. Activation of the blood coagulation cascade upon vascular injury or other events leads to generation of a proteolytic enzyme thrombin which converts monomeric fibrinogen into polymeric fibrin. Fibrin polymerization results in formation of blood clots that seal damaged vasculature thereby preventing the loss of blood upon vascular injury or formation of thrombi in pathological states. Besides its prominent role in haemostasis, fibrinogen participates in various physiological and pathological processes including inflammation, which represents an integral part of wound healing and is an attribute of many pathological processes. That fibrin(ogen) is a component of the inflammatory process was recognized more than 50 years ago.1,2 For example, it was shown that the severity and duration of joint inflammations relate to the amount of fibrin deposited,3 fibrin is required for efficient inflammatory responses in vivo during delayed-type hypersensitivity skin reaction,4 and fibrin deposition contributes to the pathogenesis of intraabdominal abscess formation.5 It was also shown that fibrinogen mediates acute inflammatory responses to biomaterials.6 Based on the early studies, it was proposed that fibrin(ogen) contributes to inflammation by promoting migration of leukocytes.3,4,7 Later, several studies revealed that fibrin(ogen) may modulate inflammation via increased cytokine/chemokine response and via fibrin degradation products. These studies have been reviewed by Jennewein and collaborators.8 In this regard, fibrinogen is up-regulated during inflammation and γ’ fibrinogen, a product of alternative splicing of the fibrinogen γ chain associated with inflammation,9 is disproportionally up-regulated by proinflammatory cytokine interleukin-6.10 It was also shown that the αMβ2 (Mac-1) integrin receptor interacts with fibrin through the P2 binding site of the later (γ chain residues 377-395)11 and this interaction plays an important role in driving inflammation. This inflammatory pathway and its role in inflammatory diseases and bacterial infection has been recently reviewed by M. Flick and collaborators.12 The role of this pathway in neurological diseases has been revied by K. Akassoglou and collaborators.13,14 The major objectives of the present article are to briefly describe the other previously proposed molecular mechanisms (pathways) underlying fibrin-dependent inflammation and to review our recent studies of the novel very low density lipoprotein (VLDL) receptor-dependent pathway of leukocyte transmigration which plays an important role in fibrin-dependent inflammation. Before describing these pathways, we will introduce the major fibrin-related species participating in them.
Fibrinogen, fibrin, and fibrin degradation products
The fibrinogen molecule consists of two identical subunits each of which is composed of three non-identical polypeptide chains, Aα, Bβ, and γ15 (Fig. 1A). The chains and subunits are linked together by disulfide bonds and are folded into a number of distinct domains16,17 (Fig. 1B). The N-termini of two pairs of the Aα and Bβ chains, which are located in the central part of the molecule denoted as E region,16 contain fibrinopeptides A and B (amino acid residues Aα1-16 and Bβ1-14, respectively) (Fig. 1A). Proteolytical removal of these fibrinopeptides with thrombin results in the exposure of two pairs of polymerization sites, which start with Gly17-Pro-Arg (knobs “A”) and Gly15-His-Arg (knobs “B”) sequences, and spontaneous polymerization of fibrin monomers into insoluble fibrin polymers or fibrin clots (Fig. 2A). Polymerization occurs through non-covalent interaction between the newly exposed polymerization sites “A” and “B” located in the central part of one fibrin molecule (E region) and complementary binding sites “a” and “b” located in the terminal parts (D regions) of two neighboring fibrin molecules (DD:E interaction) (Fig. 2A); non-covalent interaction between the αC-domains also contribute to this process.16 Subsequent covalent crosslinking of such fibrin polymers with activated factor XIII (factor XIIIa), which occurs between the C-terminal portions of the γ chains of fibrin (Fig. 2) and between the C-terminal portions of its α chains forming the αC-domains, reinforces fibrin structure increasing its mechanical stability.
Fig. 1.
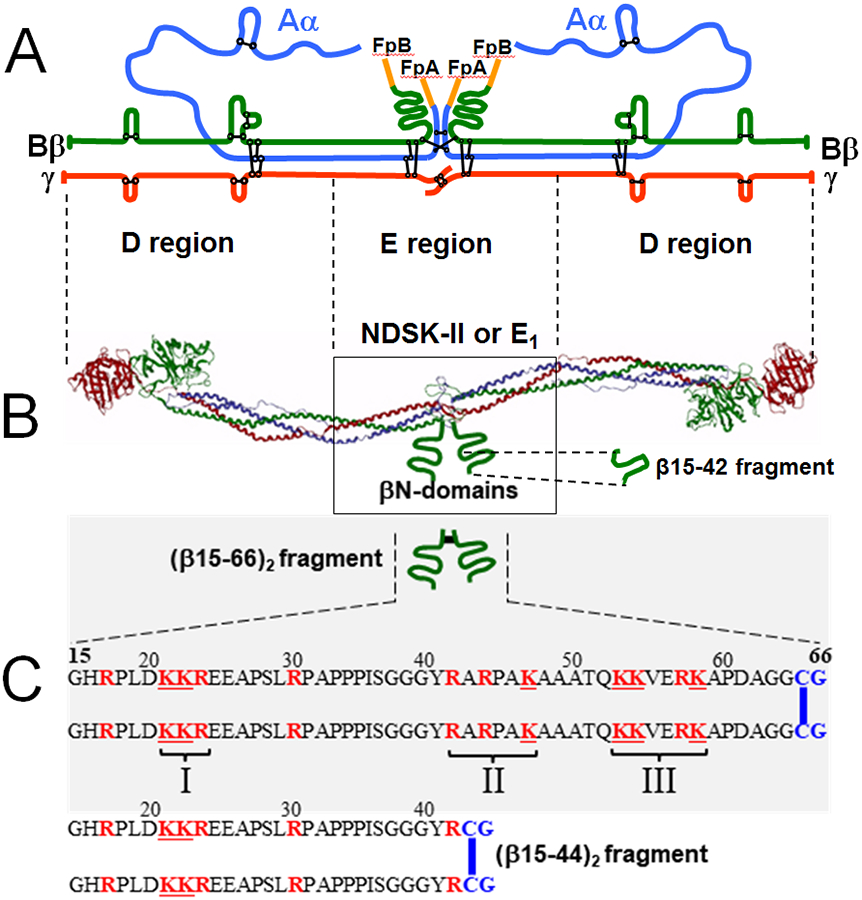
(A) Polypeptide chain composition of the fibrinogen molecule with the Aα, Bβ, and γ chains are in blue, green, and red colors, respectively, and intra- and inter-chain disulfide bonds are in black color; fibrinopeptides A and B (FpA and FpB, respectively) are in orange color. (B) Ribbon diagram of the fibrinogen molecule based on its crystal structure;17 the colors of individual polypeptide chains are the same as in panel A, the αC-domains are not shown for simplicity. The βN-domains, whose structure has not been identified, and the β15-42 fragment are shown schematically by green curved lines. The approximate boundaries between the D and E regions are shown by dashed vertical lines between panels A and B; the NDSK-II and E1 fragments derived from the E region of fibrin are shown by box. (C) Schematic representation of the recombinant disulfide-linked (β15-66)2 fragment and its amino acid sequence, as well as the sequence of the disulfide-linked (β15-44)2 fragment. The Cys-Gly (CG) amino acid residues added to dimerize both fragments are in blue color and the disulfide bonds are shown by blue vertical bars. The positively charged Lys (K) and Arg (R) residues are shown in red color. The three clusters of the positively charged residues are indicated by roman numerals.
Fig. 2.
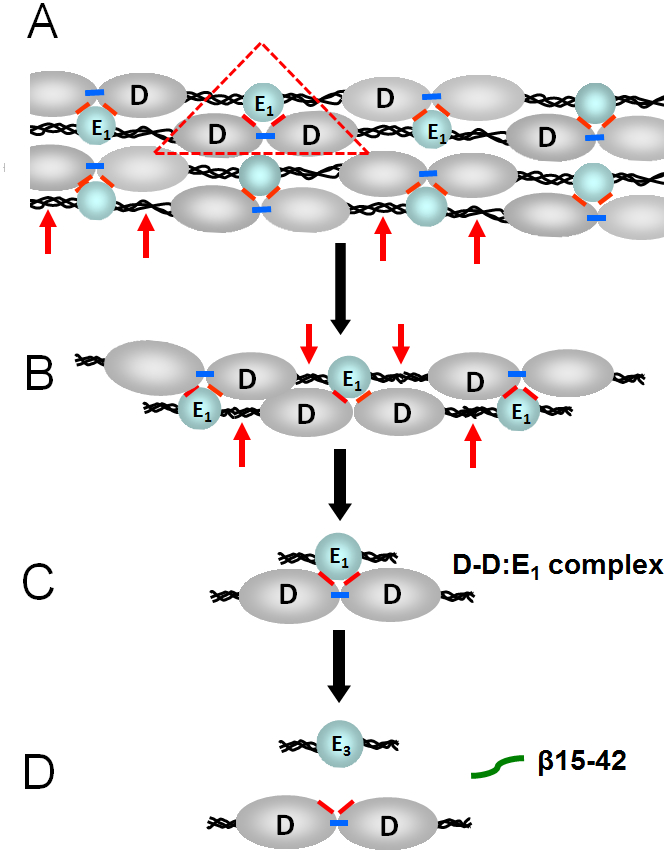
(A) Schematic representation of a fibrin polymer. D and E1 identify the D (grey color) and E (blue color) regions of individual fibrin molecules; the αC-domains are not shown for simplicity. Fibrin polymerization occurs mainly through non-covalent interactions between the complementary polymerization sites located in the D and E regions (the DD:E1 interaction shown by red triangle). Factor XIIIa crosslinks D-D regions by formation covalent bonds (shown by blue bars) between the C-terminal portions of the γ chains of these regions reinforcing fibrin polymers. Plasmin cleavage between the D and E regions of fibrin is shown by red arrows. (B and C) Cleavage of fibrin by plasmin results in high molecular mass fibrin degradation products (HMM-FDP) and the D-D:E1 complex. (D) Major final fibrin degradation products, the D-D dimer, the E3 fragment, and the β15-42 fragment.
Insoluble crosslinked fibrin is dissolved in vivo mainly by proteolytic enzyme plasmin resulting in soluble fibrin degradation products (FDP). Numerous in vitro studies revealed that the proteolytic cleavages first occur in the C-terminal portions of fibrin α chains and then in between the D and E regions (Fig. 2A-B). The resultant soluble early fibrin degradation products include some short fibrin polymers (Fig. 2B) and the D-D:E1 complex (Fig. 2C). It was previously shown that the D-D:E1 complex is the primary soluble FDP released from crosslinked fibrin upon fibrinolysis.18 This complex is maintained by non-covalent interaction between two covalently crosslinked D fragments (D-D or D dimer) and the E1 fragment, which represents the central region E of the fibrin molecule.19 Thus, the DD:E1 interactions providing fibrin polymerization, as well as the conformation of the D and E regions of fibrin, are preserved in the D-D:E1 complex. Therefore, this complex represents the simplest soluble model of fibrin polymers. The D-D:E1 complex is very stable in solution20 and its dissociation in vivo occurs only upon its further digestion with plasmin during which its E1 fragment component is converted into the E3 fragment in which α17-19 (Gly-Pro-Arg) and β15-53 sequences are proteolytically removed.18,21 The major terminal products of crosslinked fibrin are the E3 fragment, the D-dimer, and the β15-42 fragment derived from the β15-53 region of E1 (Fig. 2D). The latter, β15-42, can be purified from fibrin digest or synthesized and has been widely used in functional studies.
Fibrinogen-(ICAM-1)-dependent pathway of leukocyte transmigration
Recruitment of leukocytes from the circulation to sites of inflammation is an integral part of the inflammatory response and migration of leukocytes through the endothelium (transendothelial migration) is a key step in such recruitment.22,23 Numerous data indicate that fibrinogen plays a prominent role in leukocyte transmigration. The first mechanism to explain how fibrinogen may stimulate leukocyte transmigration and thereby promote inflammation was proposed by D. Altiery and collaborators. They identified intercellular adhesion molecule-1 (ICAM-1) as fibrinogen receptor on endothelial cells and suggested that fibrinogen mediates leukocyte adhesion to vascular endothelium through the ICAM-1-dependent pathway.24 Further, since previous studies identified αMβ2 integrin (Mac-1 or CD11b/CD18) as fibrinogen receptor on leukocytes,25-27 they proposed that fibrinogen promotes leukocyte transmigration by bridging leukocytes to the endothelium through the interaction with leukocyte receptor Mac-1 and endothelial receptor ICAM-124 (Fig. 3). This bridging mechanism, which is the essence of the proposed ICAM-1-dependent pathway of leukocyte transmigration, has been reviewed by D. Altiery.28
Fig. 3.
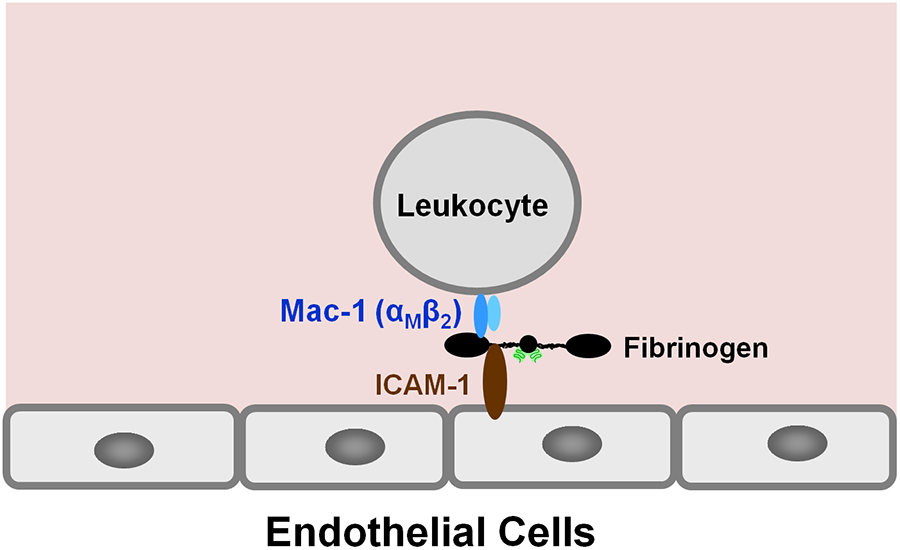
The proposed bridging mechanism24 in which fibrinogen bridges leukocytes to the endothelium through the interaction with leukocyte receptor Mac-1 and endothelial receptor ICAM-1.
It should be noted that although the fibrinogen-ICAM-1 interaction was well characterized and the proposed bridging role of fibrinogen in the (Mac-1)-fibrinogen-(ICAM-1)-dependent pathway28 is mostly accepted, subsequent studies revealed signaling role for this interaction. Specifically, using a reconstituted mammalian cell expression model it was shown that this interaction promotes leukocyte (neutrophil) transmigration, and this process was almost totally abolished by the specific anti-ICAM-1 monoclonal antibody (mAb) which blocks fibrinogen-ICAM-1 interaction.29 At the same time, anti-Mac-1 mAb OKM1, which specifically blocks fibrinogen-Mac-1 interaction, only partially inhibited leukocyte transmigration.29 Further, leukocyte transmigration, but not adhesion, was totally abolished when the ICAM-1 cytoplasmic domain was deleted, indicating that signaling inside the cell is required to mediate the fibrinogen-ICAM-1 effect on leukocyte transmigration.29 It was also shown that this signaling pathway, which promotes leukocyte transmigration, involves the GTP-binding protein Rho.29 In another study it was found that bladder high grade tumor cell lines express ICAM-1, this expression induces a fibrin-mediated migration, and this phenomenon was dependent on fibrinogen-ICAM-1 interaction as well as RhoA activity.30 These findings are in agreement with the previous observation that lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a Rho-dependent pathway.31 These findings also raise a question of whether (Mac-1)-fibrinogen interaction suggested by the bridging mechanism24,28 is required to trigger the signaling Rho-dependent pathway by the fibrinogen-ICAM-1 interaction. Further studies are required to address this question.
The fact that fibrinogen is involved in the Mac-1-(fibrinogen)-ICAM-1 bridging mechanism is surprising since this molecule is usually inert in the circulation and exhibits its binding properties only being converted into fibrin or adsorbed onto a surface. Indeed, subsequent studies from two independent laboratories including our own32,33 revealed that fibrinogen in solution does not interact with leukocyte receptor Mac-1 and it binds to this receptor only being immobilized onto a surface or converted into fibrin. Furthermore, our recent in vitro study revealed that highly purified fibrinogen has only a very small stimulating effect on leukocyte transmigration.34 These findings raise a question about the validity of the involvement of fibrinogen in the proposed bridging and signaling mechanisms. A probable explanation for such a controversy is that commercially available or in home prepared fibrinogen usually contains high molecular fraction which, from our experience, has fibrin-like activity. Thus, such fibrinogen fraction may be responsible for the observed stimulating effect in the proposed bridging and signaling mechanisms. It should be noted that in our studies mentioned above34 we used only monomeric fraction of fibrinogen (highly purified fibrinogen) which was prepared by size-exclusion chromatography and utilized immediately in our binding/stimulating experiments. Therefore, no binding to Mac-1 or very little stimulation of leukocyte transmigration were observed. Thus, the question about the involvement of fibrinogen in the fibrinogen-(ICAM-1)-dependent pathway needs further clarification. Now one can only speculate that the bridging and/or signaling in this pathway may occur through fibrin, not fibrinogen. Further studies with highly purified fibrinogen are required to test this speculation.
E1 fragment-(VE-cadherin)-dependent pathway of leukocyte transmigration
Another pathway of leukocyte transmigration, E1 fragment-(VE-cadherin)-dependent, was proposed more than a decade ago by Petzelbauer and collaborators.35,36 They found that the NDSK-II fragment,37 which derives from the central E region of fibrin and includes a pair of fibrin’s βN-domains,16,38 promotes transendothelial migration of leukocytes in a VE-cadherin-dependent manner and the β15-42 fragment, which represents a part of the βN-domain, inhibits this process.35,36 Based on this and some other observations, they proposed that the naturally occurring fibrin degradation product E1 fragment, an analogue of NDSK-II (Fig. 1B), promotes leukocyte transmigration in vivo by bridging leukocytes to the endothelium through the interaction with CD11c (integrin αX chain) on leukocytes and VE-cadherin on endothelial cells.35,36 In this bridging model, E1 binds to leukocyte integrin CD11c through its α chain Gly17-Pro-Arg sequence, which was previously shown to interact with this integrin receptor,39 and to endothelial VE-cadherin through its β15-42 sequence. The inhibitory property of the β15-42 fragment was explained by its ability to compete for the E1-VE-cadherin interaction thereby blocking this interaction and reducing leukocyte transmigration (Fig. 4A).
Fig. 4.
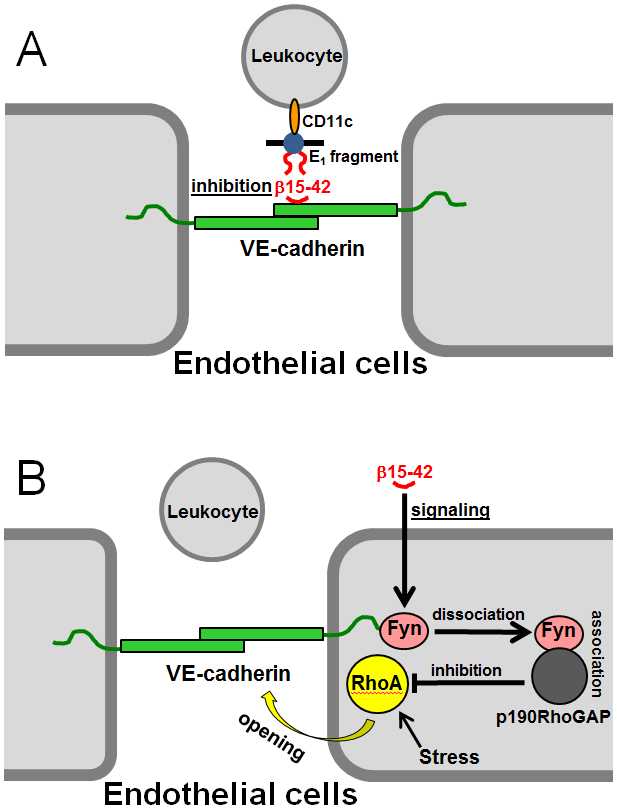
The proposed inhibitory role of the β15-42 fragment in leukocyte transmigration. (A) Fibrin degradation product, the E1 fragment, bridges leukocytes to the endothelium by interacting with endothelial VE-cadherin through its βN-domains (shown by red curved lines) and with leukocyte integrin CD11c, and β15-42 (shown by red curved line) inhibits this bridging by competing for the E1-VE-cadherin interaction.35,36 (B) The proposed additional signaling role of the β15-42 fragment in leukocyte transmigration. Treatment of endothelial cells with β15-42 results in dissociation of src kinase Fyn from VE-cadherin followed by association of Fyn with p190RhoGAP30 and this complex inhibits GTPase protein RhoA thereby preventing cell contraction and maintaining endothelial barrier function.57
Cardioprotective effect of the fibrin-derived β15-42 fragment.
As mentioned above, the β15-42 fragment is a naturally occurring fibrin degradation product released from the fibrin βN-domains upon degradation of fibrin with plasmin. It was used in several studies for testing functional properties of these domains. For example, it was shown that β15-42 inhibits fibrin-induced angiogenesis, albeit at a very high concentration;40,41 however, no subsequent studies of the anti-angiogenic properties of this fragment were performed. In contrast, the discovery of the inhibitory activity of the β15-42 fragment on leukocyte transmigration35 sparked numerous studies of its anti-inflammator6y properties using different animal models. First, Petzelbauer and collaborators35 using rat model of myocardial ischemia-reperfusion injury demonstrated that this fragment protects the myocardium against ischemia-reperfusion injury by decreasing leukocyte accumulation in the ischemic heart tissues and significantly reducing myocardial inflammation, infarct size, and scar formation. The cardioprotective effect of β15-42 was subsequently confirmed in rodent and pig models of myocardial ischemia-reperfusion injury.42,43 Because of its potent cardioprotective effect in animal models, the β15-42 fragment was under development as an anti-inflammatory drug called FX06.44 The results of Phase II trials conducted on human patients with early acute myocardial infarction revealed that administration of β15-42 (FX06) as an adjunctive agent along with reperfusion resulted in a significant reduction of the necrotic core zone and infarct size.45,46
Although the results obtained with FX06 were encouraging, this fragment is not the best inhibitor of E1 fragment-VE-cadherin interaction due to its low affinity to VE-cadherin.47,48 To address this problem, we tested VE-cadherin-binding properties of a number of βN-domain-derived recombinant fragments and synthetic peptides including the recombinant (β15-66)2 dimer mimicking the dimeric arrangement of the βN-domains in fibrin, as well as the synthetic (β15-44)2 dimer representing a pair of the N-terminal portions of these domains. The results obtained revealed that both dimeric fragments, (β15-66)2 and (β15-44)2, had much higher affinity to VE-cadherin than β15-42 and exhibited at least two-fold higher inhibitory effect on leukocyte infiltration into the peritoneum in our in vivo experiments using mouse model of peritonitis.48 Furthermore the (β15-44)2 fragment exhibited a prominent cardioprotective effect in mouse model of myocardial ischemia-reperfusion injury.48
Other anti-inflammatory properties of the β15-42 fragment.
In addition to its cardioprotective effect, the β15-42 fragment exhibited its protective effect in other inflammation-related animal models. It was subsequently shown that this fragment reduces pulmonary, myocardial, liver, and small intestine damage in a pig model of hemorrhagic shock and reperfusion,49 and exhibits organ protective effect in a pig model of hemorrhagic shock and resuscitation.50 It was also demonstrated that β15-42 significantly attenuates ischemia-reperfusion injury in cardiac and renal transplantation models,51,52 protects against kidney and liver ischemia-reperfusion injury,53-55 and exerts protective effect during acute lung injury.56 Furthermore, it was shown that β15-42 preserves endothelial barrier function in animal models for Dengue shock syndrome and LPS-induced shock,57 improves survival and neurocognitive recovery, and ameliorates vascular leakage after cardiopulmonary resuscitation.58 The β15-42 fragment was also used in combination with other procedures for successful treatment of vascular leak syndrome in the patient with Ebola virus disease.59
Such a variety of functions of the β15-42 fragment is connected not only with its inhibitory effect on E1 fragment-VE-cadherin interaction (Fig. 4A) In addition to the proposed inhibitory function of β15-42, Groger and collaborators57 suggested a signaling function for this fragment. Namely, based on their in vitro and in vivo studies they suggested that β15-42 preserves endothelial barrier function thereby reducing leukocyte transmigration by inhibiting stress-induced opening of endothelial cell adherent junctions.57 The mechanism they proposed includes dissociation of the src kinase Fyn from VE-cadherin-containing junctions upon exposure of endothelial cells to β15-42 followed by association of Fyn with p190RhoGAP30, which complex inhibits GTPase protein RhoA (Fig. 4B). Since RhoA is known to regulate actin dynamics and junction stability,60 its inactivation prevents cell contraction and maintains endothelial barrier function. Thus, the β15-42 was proposed to be an inhibitory and a signaling molecule whose dual action reduces transendothelial migration of leukocyte resulting in reduced inflammation.
Is the proposed bridging mechanism for the E1-(VE-cadherin)-dependent pathway valid?
Although the proposed bridging mechanism provides a simple and reasonable explanation for the inhibitory properties of the β15-42 fragment, some data suggested that it may not be valid. One of the problems with this mechanism is that the E1 fragment may exist in the circulation only in a complex with the D-D fragment, as mentioned above. Thus, in vivo only D-D:E1, not E1, can promote leukocyte transmigration. Since the D-D:E1 complex is maintained mainly through “A”-“a” interactions,19 the E1 fragment sequence αGly17-Pro-Arg, which actually represents a part of the polymerization site (knob “A”), is not accessible in this complex for the interaction with the leukocyte CD11c receptor. Thus, the proposed (CD11c)-E1 fragment-(VE-cadherin) bridging of leukocytes to the endothelium35,36 cannot occur in vivo through leukocyte CD11c integrin. Another problem is that the determined affinity of the β15-42 fragment to VE-cadherin (Kd = 267 μM)48 is very low in comparison with that of the E1 fragment (Kd = 69-79 nM).61 With such low affinity, efficient inhibition of E1-VE-cadherin interaction would require millimolar concentrations of β15-42. At the same time, the concentrations of this fragment that inhibited leukocyte transmigration in the in vivo experiments (−5-12 μM)35,48 was more than 20-fold lower than the Kd value for its interaction with VE-cadherin. Further, our recent experiments confirmed that even at a 10-fold higher concentration (120 μM) the β15-42 fragment was unable to inhibit binding of the E1 fragment to VE-cadherin.62 All these facts indicate that the bridging mechanism proposed to explain the inhibitory role of the β15-42 fragment35,36 is incorrect. This also leaves the proposed signaling mechanism57 (Fig. 4B) as the only viable explanation for the inhibitory effect of β15-42 on leukocyte transmigration.
Fibrin-VLDL receptor-dependent pathway of leukocyte transmigration
The VLDL receptor is involved in transendothelial migration of leukocytes.
When testing the effect of fibrin (β15-66)2 fragment, which corresponds to a pair of the fibrin βN-domains, on the morphology of endothelial cell monolayers, we noticed that treatment of the monolayers with this fragment resulted in loosening of intercellular junctions.63 This effect was inhibited by the addition of the receptor-associated protein (RAP),63 a well-known inhibitor of ligand interaction with low density lipoprotein receptor family members,64,65 suggesting the involvement of such receptor(s) in this process. Subsequent binding study revealed that one of the family members, the very low density lipoprotein receptor (VLDL receptor or VLDLR), interacts with (β15-66)2 and fibrin with a very high affinity (Kd = 5.7-9.2 nM).63 This interaction was highly specific since it was inhibited by RAP.
Our subsequent in vitro study using leukocyte transendothelial migration assay revealed that RAP inhibited NDSK-II-induced leukocyte transmigration,63 suggesting that interaction of the VLDL receptor with NDSK-II promotes leukocyte transmigration. To test this suggestion, we performed in vivo experiments with the VLDLR-deficient mice using mouse model of peritonitis. The experiments revealed that leukocyte transmigration in such mice was moderately (by ~ 22%) lower than that in wild-type mice, although the difference was not statistically significant.63 However, the most important finding was that the (β15-66)2 and (β15-44)2 fragments inhibited infiltration of leukocytes (neutrophils) into the peritoneum in wild-type mice and had no effect in VLDLR- deficient mice, providing direct evidence for the involvement of the VLDL receptor in leukocyte transmigration and thereby inflammation.63 Based on these studies, we concluded that interaction of NDSK-II and probably fibrin with the VLDL receptor promotes leukocyte transmigration.63
Fibrin-VLDL receptor interaction promotes leukocyte transmigration.
Although we confirmed that fibrin interacts with the VLDL receptor in direct binding experiments, in our in vitro transmigration study we used for stimulation of leukocyte transmigration the NDSK-II fragment as a soluble mimetic of fibrin. However, this fragment resembles more closely fibrin degradation product E1 fragment. This raised a question of whether fibrin can promote leukocyte transmigration, or this process is supported only by its degradation products, as was proposed for the E1-VE-cadherin-dependent pathway of leukocyte transmigration.35,36 To identify which fibrin species can promote leukocyte transmigration through the VLDL receptor-dependent pathway, we prepared a number of models mimicking fibrin and its degradation products. They included monomeric fibrin, fibrin polymers deposited on the endothelial monolayer, early degradation products of crosslinked fibrin including high molecular mass fibrin degradation products (HMM-FDP) and smaller D-D:E1 complex (Fig. 2B, C). Our in vitro transmigration experiments revealed that HMM-FDP and D-D:E1 both significantly stimulated leukocyte transmigration in a VLDLR-dependent manner.34 Furthermore, soluble fibrin and fibrin clots deposited on the endothelial monolayers also exhibited similar stimulating effects.34 These findings provided direct evidence for our suggestion that fibrin-VLDL receptor interaction promotes leukocyte transmigration. They also revealed that early fibrin degradation products may also be involved in this process.
Molecular mechanism of fibrin-VLDL receptor interaction.
Considering the significant role of fibrin-VLDL receptor interaction in leukocyte transmigration and thereby inflammation, we next focused on the establishment of the molecular mechanism of this interaction with the aim to identify its specific inhibitors. First, we localize d the fibrin-binding site within the VLDL receptor. This receptor consists of the extracellular portion, transmembrane domain and cytoplasmic domain. The extracellular portion includes eight cysteine-reach repeats or domains (CR domains), three EGF-like repeats (domains), and the propeller and O-linked sugar domains (Fig. 5A). Since CR domains of the LDL receptor family members are usually involved in ligand binding, we expressed the VLDLR(1-8) fragment containing all eight CR domains and the des(1-8)VLDLR fragment representing the rest of the extracellular portion and confirmed that only VLDLR(1-8) interacts with fibrin βN-domains.66 Further, we expressed a number of CR-containing fragments and localized the fibrin-binding site within CR domains 2-466 (Fig. 5B). It should be noted that the VLDLR(2-3) fragment containing CR domains 2 and 3 also exhibited high affinity to fibrin βN-domains. However, the presence of the fourth domain in the VLDLR(2-4) fragment increased its affinity to the βN-domains at least two-fold indicating that this domain is also involved in the interaction, although CR domains 2 and 3 seem to make the major contribution.66
Fig. 5.
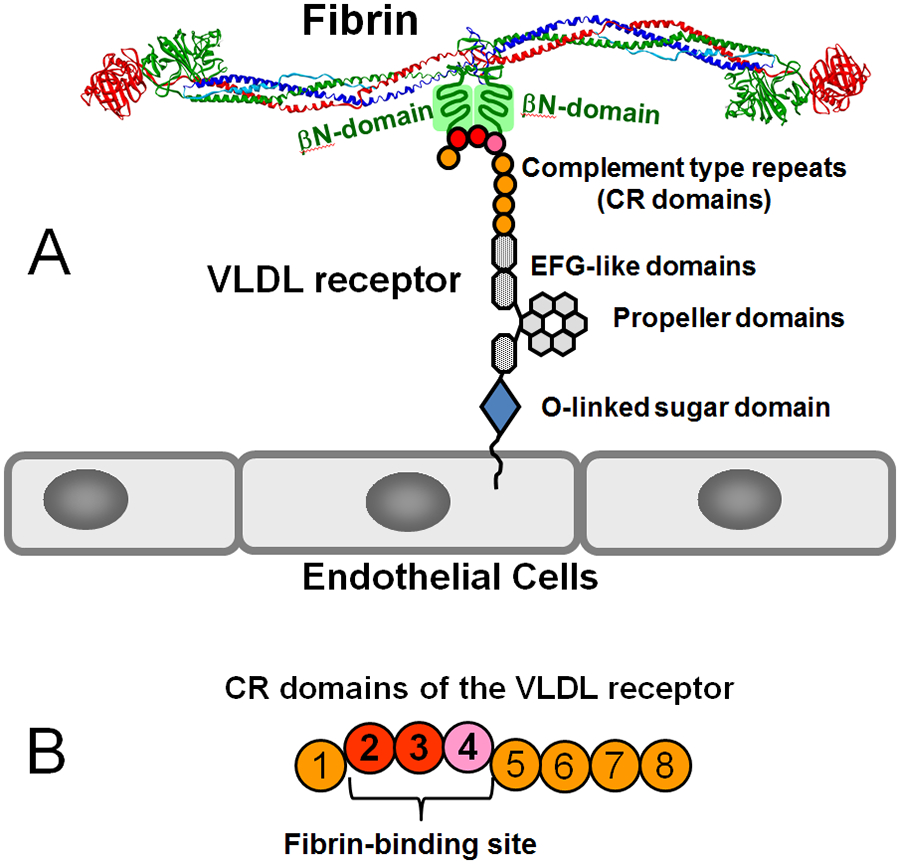
(A) Domain structure of the VLDL receptor and its interaction with the βN-domains of the fibrin molecule. (B) CR domains 2-4 of the VLDL receptor forming the fibrin-binding site.
Next, to localize the VLDLR-binding site within fibrin βN-domain, we turned to site directed mutagenesis of the VLDLR-binding (β15-66)2 fragment of fibrin. Since it was shown earlier that Lys and Arg residues play an important role in the interaction of CR domains of the LDL receptor family members with their ligands and fibrin βN-domains contain three clusters of positively charged residues (Arg and Lys) (Fig. 1C), we mutated individual Lys and Arg residues in these clusters, as well as the entire clusters.67 Subsequent binding study with these mutant fragments revealed that Arg and Lys residues of all three clusters are involved in the interaction with fibrin and cluster II and III make the major contribution to the binding.67
Finally, to establish the structural basis for fibrin-VLDL receptor interaction, we performed NMR experiments in collaboration with Nico Tjandra (NIH/NHLBI). First, we identified conditions for formation of a complex between the VLDLR(2-4) fragment of the VLDL receptor containing the fibrin-binding site and the (β15-66)2 fragment representing the fibrin βN-domains containing the VLDLR-binding site.66 Then, we established NMR solution structure of the VLDLR(2-4) fragment68 and that of the complex between this fragment and the (β15-66)2 fragment.69 The structure of this complex, which recapitulates the interaction between fibrin and VLDLR, is shown in Fig. 6. This structure revealed that the interaction occurs mainly through Lys47 and Lys53 of the second and third positively charged clusters of the βN-domain, respectively, and Lys-binding sites located in CR2 and CR3 domains. This interaction is similar to those proposed earlier for the interaction of other members of the LDL receptor family with their ligands.70 In addition, Gly15 of the βN-domain and its first positively charged cluster contributes to the high-affinity interaction with the VLDL receptor. It should be noted that it was not possible to establish exact structure of the complex due to the difficulties connected with unordered nature of the (β15-66)2 fragment and precipitation of the complex at concentrations required for standard structure determination by NMR.69 It should also be noted that although the structure of the complex shows Lys47-CR2 and Lys53-CR3 interaction, i.e. in parallel manner, the antiparallel interaction proposed earlier,67 i.e. Lys47-CR3 and Lys53-CR2, is also possible.
Fig. 6.
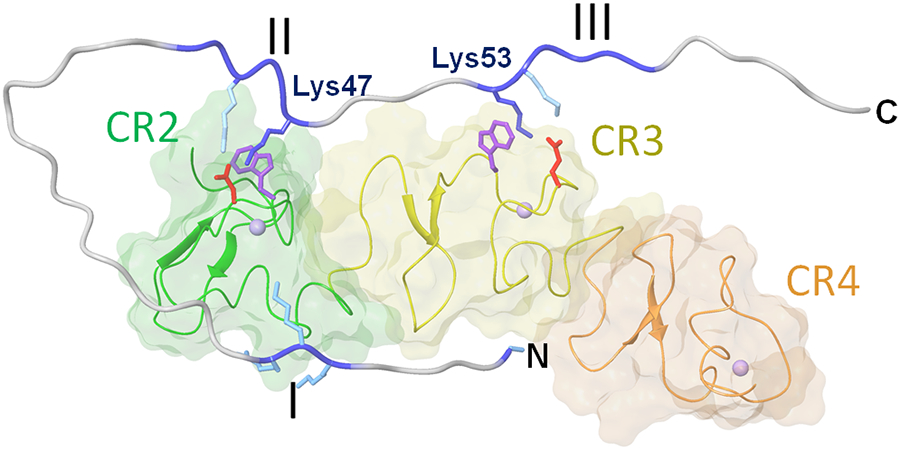
Model of the complex between the βN-domain of fibrin and the VLDLR(2-4) fragment of the VLDL receptor based on NMR study.69 The CR2, CR3 and CR4 domains of VLDLR(2-4) are shown in different colors; the three clusters of the positively charged amino acid residues of the fibrin βN-domain are colored in blue and denoted by roman numerals; Lys47 and Lys 53 interacting with the Lys-binding sites of the VLDLR(2-4) fragment are also shown. N and C indicate N- and C-terminal ends of the βN-domain.
Search for inhibitors of fibrin-VLDL receptor interaction.
Having localized the complementary binding sites in the VLDL receptor and fibrin βN-domains, we next focused on the development of inhibitors of fibrin-VLDL receptor interaction. We analyzed three monoclonal antibodies against the VLDL receptor, mAb 1H5, 1H10, and 5F3, which were previously produced by D. Strickland and collaborators,71 and found that the epitopes for two of them, mAb 1H5 and mAb 1H10, overlap with the fibrin-binding site of the VLDL receptor.72 Further experiments revealed that these two mAbs efficiently inhibited fibrin-VLDLR interaction and significantly reduced leukocyte (neutrophil) transmigration in in vitro leukocyte transendothelial migration assay.72 Furthermore, both mAbs significantly reduced infiltration of leukocytes into the peritoneum in mouse model of peritonitis and reduced infarct size in mouse model of ischemia-reperfusion injury.72 Although we tested these mAbs only in mouse model of ischemia-reperfusion injury, they may also be efficient inhibitors of leukocyte transmigration in other models of ischemia-reperfusion. Thus, they are potential candidates for the development of potent therapeutics for treatment of myocardial and other types of ischemia-reperfusion injuries.
Do the βN-domains promote or inhibit leukocyte transmigration?
It should be noted that the NDSK-II fragment containing two fibrin βN-domains was used in several studies as a potent stimulator of leukocyte transmigration.34,35,48,62,63 At the same time, in our experiments the (β15-66)2 fragment mimicking a pair of the βN-domains efficiently inhibited leukocyte transmigration.48,63 To explain this obvious paradox, we hypothesized that fibrin βN-domains have dual function in fibrin-dependent inflammation, namely, their C-terminal regions containing the VLDLR-binding sites promote leukocyte transmigration while their N-terminal regions are responsible for the inhibition of this process.73 To test this hypothesis, we prepared the dimeric (β15-44)2 and (β40-66)2 fragments corresponding to the N- and C-terminal regions of the βN-domains and studied their effect on endothelial permeability and leukocyte transmigration. The results obtained revealed that (β40-66)2 bound to the VLDLR with high affinity and promoted endothelial permeability and leukocyte transmigration while (β15-44)2 did not interact with this receptor and inhibited leukocyte transmigration,73 in agreement with our hypothesis. Further, the inhibitory properties of the βN-domains were expressed only upon their isolation from the fibrin molecule.73 The question of whether their inhibitory function may play a role in fibrin remains to be addressed.
Fibrin-VLDLR-dependent pathway promotes leukocyte transmigration by inhibiting src kinase Fyn.
Having established the molecular mechanism of fibrin-VLDL receptor interaction, we next focused on the establishment of the molecular mechanism underlying the fibrin-VLDL receptor-dependent pathway of leukocyte transmigration. Our early experiments using mouse model of peritonitis revealed that the dimeric form of the β15-42 fragment, (β15-44)2, inhibited infiltration of leukocytes into the peritoneum, as expected, but failed to inhibit this process in VLDLR-deficient mice,63 suggesting a link between the inhibitory function of β15-42 and the VLDLR-dependent pathway. Since it was proposed that the molecular key for the protective effect of β15-42 is src kinase Fyn,57 as described above, we focused on this kinase. Our in vivo and in vitro experiments revealed that interaction of fibrin with the VLDL receptor triggers signaling pathway which inhibits Fyn and that this pathway is a target for the β15-42 and (β15-44)2 fragments.62 These findings combined with the earlier proposed signaling pathway by β15-4257 allowed us to propose the molecular mechanisms underlying the fibrin-induced VLDLR-dependent pathway of leukocyte transmigration and its inhibition by β15-42 and (β15-44)2, which is schematically shown in Fig. 7. According to this mechanism, interaction of fibrin or its degradation products with the VLDL receptor triggers the VLDLR-dependent pathway which inhibits Fyn. This inhibition, according to the earlier proposed mechanism,57 prevents dissociation of Fyn from VE-cadherin and inhibition of RhoA thereby promoting leukocyte transmigration. The β15-42 and (β15-44)2 fragments, in turn, inhibit the VLDLR-dependent pathway to allow dissociation of Fyn from VE-cadherin and inhibition of RhoA thereby inhibiting leukocyte transmigration. It is unlikely that the β15-42 and (β15-44)2 fragments, which are comparatively bulky molecules, can enter endothelial cells to directly inhibit the VLDLR-dependent pathway. Therefore, we hypothesized that these fragments inhibit this pathway by interacting with a putative receptor (Fig. 7). This receptor remains to be identified. In this regard, the recent discovery that carboxypeptidase M mediates protective effect of β15-4274 may serve as a starting point for identification of this putative receptor.
Fig. 7.
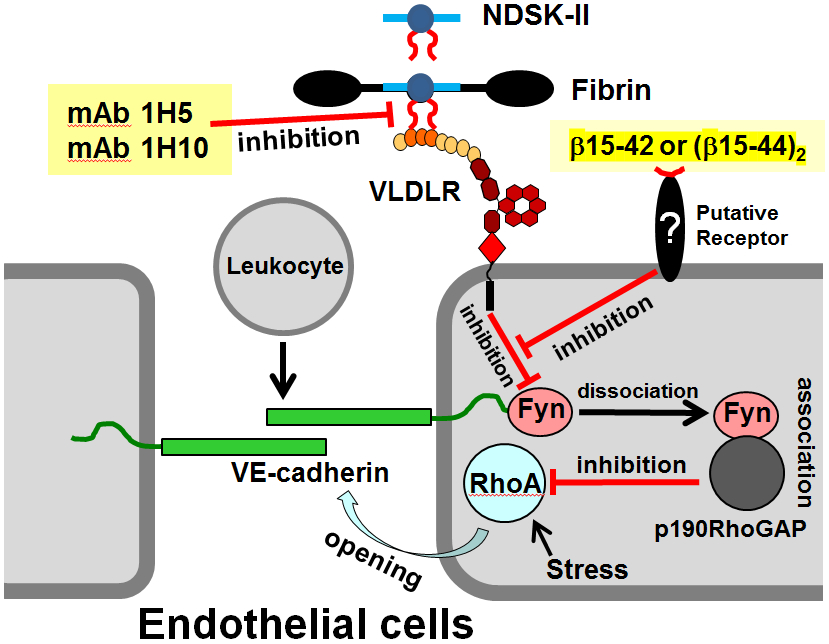
Schematic representation of the fibrin-VLDL receptor-dependent pathway of leukocyte transmigration and its inhibition by the β15-42 or (β15-44)2 fragments and by the specific anti-VLDLR monoclonal antibodies, mAb 1H5 and mAb 1H10.62,72 Interaction of fibrin or its mimetic NDSK-II with the endothelial VLDL receptor (VLDLR), which occurs through the βN-domains of the former (shown by red curved lines) and CR domains 2-4 of the later, induces intracellular pathway resulting in the inhibition of src kinase Fyn and preventing formation of its complex with p190RhoGAP. This precludes inhibition of GTPase protein RhoA, which in active state promotes stress-induced opening of endothelial adherent junctions and thereby leukocyte transmigration. The two monoclonal antibodies inhibit fibrin-VLDL receptor interaction thereby preventing activation of the fibrin-VLDL receptor-dependent pathway. Binding of the β15-42 or (β15-44)2 fragments to a putative receptor triggers a pathway that inhibits the fibrin-VLDL receptor-dependent pathway. This prevents inhibition of Fyn and results in subsequent inhibition of RhoA by the Fyn-p190RhoGAP complex thereby preventing junction opening and leukocyte transmigration.
(Patho)physiological role of the fibrin-VLDLR-dependent pathway of leukocyte transmigration.
Among fibrin species that were found to stimulate the VLDL receptor-dependent leukocyte transmigration,34 early degradation products, HMM-FDP and the D-D:E1 complex (Fig. 2), are transient. HMM-FDP are rapidly degraded by plasmin to the D-D:E1 complex in which D-D and E1 components are held together through complementary binding sites.19 This complex is also transient and dissociates upon conversion of its E1 component to the E3 fragment (Fig. 2) during which the N-terminal portions of the β chains including the βN-domains and some residues from the α chains are removed proteolytically. The final fibrin degradation products, the D-D dimer and E3 fragment do not stimulate leukocyte transmigration since they do not interact with the VLDL receptor.63 Although HMM-FDP and the D-D:E1 may contribute to stimulation of the VLDL receptor-dependent leukocyte transmigration in vivo, their contribution may be minor since they are transient products. In contrast, fibrin clots deposited on the endothelium are rather stable and, therefore, could make the major contribution to leukocyte transmigration.
Having established that endothelium-anchored fibrin clots promote transendothelial migration of leukocytes,34 one can speculate about (patho)physiological role of the fibrin-VLDLR-dependent pathway of leukocyte transmigration. Namely, during vascular injury, activation of the blood coagulation cascade results in deposition of fibrin polymers on the injured endothelium that seal vasculature to prevent blood loss. It was also shown that the procoagulant activity of inflamed endothelium due to expression of tissue factor results in activation of the coagulation cascade and deposition of polymerized fibrin (fibrin clots) on the endothelial surface.75-78 In both cases fibrin polymers come in contact with the endothelial VLDL receptor and their interaction with this receptor may promote leukocyte transmigration and thereby inflammation, which is a normal process during early stages of wound healing. Thus, the fibrin-VLDLR-dependent pathway of leukocyte transmigration may play an important role during normal wound healing by promoting inflammation. However, in pathological states, for example during myocardial ischemia-reperfusion, fibrin deposition on inflamed endothelium may promote leukocyte transmigration to the ischemic areas through this pathway thereby increasing inflammation and causing myocardial damage by ischemia-reperfusion injury.
Conclusion
Two molecular mechanisms have been proposed to explain how fibrinogen promotes leukocyte transmigration and thereby inflammation. The first one suggests that fibrinogen promotes leukocyte transmigration by bridging leukocytes to the endothelium through the interaction with leukocyte receptor Mac-1 and endothelial receptor ICAM-1.28 Subsequent studies revealed that fibrinogen-ICAM-1 interaction triggers signaling, which involves the GTP-binding protein Rho,29 resulting in enhanced leukocyte transmigration. Whether fibrinogen bridging mechanism is required for such signaling and whether fibrin, not fibrinogen, is involved in bridging and signaling remain to be established.
The second mechanism also suggests that fibrin degradation product E1 fragment promotes leukocyte transmigration by bridging leukocytes to the endothelium through the interaction with leukocyte receptor CD11c and endothelial VE-cadherin, and the β15-42 fragment inhibits E1-VE-cadherin interaction resulting in inhibition of leukocyte transmigration.35,36 The additional signaling mechanism to explain the inhibitory role of β15-42 has also been proposed.57 This mechanism includes β15-42-stimulated signaling through src kinase Fyn resulting in the inhibition of RhoA and reducing leukocyte transmigration. However, our recent study revealed that the proposed bridging mechanism and its inhibition by β15-42 is invalid62 leaving the signaling mechanism as the only viable explanation for the inhibitory function of the β15-42 fragment.
A decade ago, we have discovered that fibrin and its degradation products interact with the endothelial VLDL receptor, and this interaction promotes leukocyte transmigration.63 Our subsequent studies clarified the molecular mechanism of this interaction66,67 and established the structural basis for it.68,69 Further, we found that fibrin-VLDL receptor interaction triggers signaling pathway that inhibits src kinase Fyn thereby preventing RhoA inhibition and promoting leukocyte transmigration, and this pathway is a target for the β15-42 fragment-induced inhibitory pathway.62 We hypothesized that the β15-42 fragment induces this inhibitory pathway by interacting with a putative endothelial receptor.62 The putative receptor for β15-42 remains to be identified. We also identified two anti-VLDLR monoclonal antibodies that inhibit fibrin-VLDL receptor interaction and fibrin-induced leukocyte transmigration.72 These antibodies can be developed as potent therapeutics for treatment of fibrin-VLDLR-dependent inflammation.
Funding
This work was supported by National Institutes of Health grants R01 HL056051 to L.M. and R35 HL135743 to D.K.S.
Footnotes
Conflict of interest
Non declared.
References
- 1.Riddle JM, Barnhart ML. Ultrastructural study of fibrin dissolution via emigrated polymorphonuclear neutrophils. Am J Pathol 1964;45(5):805–823 [PMC free article] [PubMed] [Google Scholar]
- 2.Riddle JM, Barnhart ML. The eosinophil as a source for profibrinolysin in acute inflammation. Blood 1965;25(5):776–794 [PubMed] [Google Scholar]
- 3.Barnhart MI, Riddle JM, Bluhm GB, Quintana C. Fibrin promotion and lysis in arthritic joints. Ann Rheum Dis 1967;26(3):206–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colvin RB, Johnson RA, Mihm MC Jr, Dvorak HF. Role of the clotting system in cell mediated hypersensitivity. I. Fibrin deposition in delayed skin reactions in man. J Exp Med 1973;138(3):686–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McRitchie DI, Girotti MJ, Glynn MF, Goldberg JM, Rotstein OD. Effect of systemic fibrinogen depletion on intraabdominal abscess formation. J Lab Clin Med 1991;118(1):48–55 [PubMed] [Google Scholar]
- 6.Tang L, Eaton JW. Fibrin(ogen) mediates acute inflammatory responses to biomaterials. J Exp Med 1993;178(6):2147–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hurley JV. Substances promoting leukocyte emigration. Ann N Y Acad Sci 1964;116(3):918–935 [DOI] [PubMed] [Google Scholar]
- 8.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med 2011;17(5-6):568–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farrell DH. Fibrinogen as a novel marker of thrombotic disease. Clin Chem Lab Med 2012;50(11):1903–1909 [DOI] [PubMed] [Google Scholar]
- 10.Rein-Smith CM, Anderson NW, Farrell DH. Differential regulation of fibrinogen γ chain splice isoforms by interleukin-6. Thromb Res 2013;131:89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ugarova P, Solovijov DA, Zhang L, Loukinov DI, Yee VC, Medved L, et al. Identification of a novel recognition sequence for integrin αMβ2 within the γ-chain of fibrinogen. J Biol Chem 1998;273(35):22519–22527 [DOI] [PubMed] [Google Scholar]
- 12.Luyendyk JP, Schoenecker JG, Flick MJ. The multifaceted role of fibrinogen in tissue injury and inflammation. Blood 2019;133(6):511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petersen MA, Ryu JK, Akassoglou K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Nevrosci 2018;19(5):283–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akassoglou K. The immunology of blood: connecting the dots at the neurovascular interface. Nat Immunol 2020;21(7):710–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henschen A, McDonagh J. Fibrinogen, fibrin and factor XIII. In: Zwaal RFA, Hemker HC, eds. Blood Coagulation. Amsterdam: Elsevier Science Publishers; 1986:171–241 [Google Scholar]
- 16.Medved L, Weisel JW, on behalf of Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost 2009;7(2):355–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kollman JM, Pandi L, Sawaya MR, Riley M, Doolittle RF. Crystal structure of human fibrinogen. Biochemistry 2009;48(18):3877–3886 [DOI] [PubMed] [Google Scholar]
- 18.Olexa SA, Budzynski AZ. Primary soluble plasmic degradation product of human cross-linked fibrin. Isolation and stoichiometry of the (DD)E complex. Biochemistry 1979;18(6):991–995 [DOI] [PubMed] [Google Scholar]
- 19.Moskowitz KA, Budzynski AZ. The (DD)E complex is maintained by a composite fibrin polymerization site. Biochemistry 1994;33(44):12937–12944 [DOI] [PubMed] [Google Scholar]
- 20.Yakovlev S, Makogonenko E, Kurochkina N, Nieuwenhuizen W, Ingham K, Medved L. Conversion of fibrinogen to fibrin: mechanism of exposure of tPA- and plasminogen-binding sites. Biochemistry 2000;39(51):15730–15741 [DOI] [PubMed] [Google Scholar]
- 21.Olexa SA, Budzynski AZ. Binding phenomena of isolated unique plasmic degradation products of human cross-linked fibrin. J Biol Chem 1979;254(11):4925–4932 [PubMed] [Google Scholar]
- 22.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood 1994;84(7):2068–2101 [PubMed] [Google Scholar]
- 23.Springer TA. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol 1995;57:827–872 [DOI] [PubMed] [Google Scholar]
- 24.Languino LR, Plescia J, Duperray A, et al. Fibrinogen mediates leukocyte adhesion to vascular endothelium through an ICAM-1-dependent pathway. Cell 1993;73(7):1423–1434 [DOI] [PubMed] [Google Scholar]
- 25.Altieri DC, Bader R, Mannucci PM, Edgington TS. Oligospecificity of the cellular adhesion receptor Mac-1 encompasses an inducible recognition specificity for fibrinogen. J Cell Biol 1988;107(5):1893–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trezzini C, Jungi TW, Kuhnert P, Peterhans E. Fibrinogen association with human monocytes: evidence for constitutive expression of fibrinogen receptors and for involvement of Mac-1 (CD18, CR3) in the binding. Biochem Biophys Res Commun 1988;156(1):477–484 [DOI] [PubMed] [Google Scholar]
- 27.Wright SD, Weitz JI, Huang AJ, Levin SM, Silverstein SC, Loike JD. Complement receptor type three (CD11b/CD18) of human polymorphonuclear leukocytes recognizes fibrinogen. Proc Natl Acad Sci U S A 1988;85(20):7734–7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Altieri DC. Regulation of leukocyte-endothelium interaction by fibrinogen. Thromb Haemost 1999;82(2):781–786 [PubMed] [Google Scholar]
- 29.Sans E, Delachanal E, Duperray A. Analysis of the roles of ICAM-1 in neutrophil transmigration using a reconstituted mammalian cell expression model: implication of ICAM-1 cytoplasmic domain and Rho-dependent signaling pathway. J Immunol 2001;166(1):544–551 [DOI] [PubMed] [Google Scholar]
- 30.Roche Y, Pasquier D, Rambeaud JJ, Seigneurin D, Duperray A. Fibrinogen mediates bladder cancer cell migration in an ICAM-1-dependent pathway. Thromb Haemost 2003;89(6):1089–1097. [PubMed] [Google Scholar]
- 31.Adamson P, Etienne S, Couraud PO, Calder V, Greenwood J. Lymphocyte migration through brain endothelial cell monolayers involves signaling through endothelial ICAM-1 via a Rho-dependent pathway. J Immunol 1999;162(5):2964–2973 [PubMed] [Google Scholar]
- 32.Lishko VK, Kudryk B, Yakubenko VP, Yee VC, Ugarova TP. Regulated unmasking of the cryptic binding site for integrin αMβ2 in the γC-domain of fibrinogen. Biochemistry 2002;41(43):12942–12951 [DOI] [PubMed] [Google Scholar]
- 33.Yakovlev S, Zhang L, Ugarova T, Medved L. Interaction of fibrin(ogen) with leukocyte receptor αMβ2 (Mac-1): further characterization and identification of a novel binding region within the central domain of the fibrinogen γ-module. Biochemistry 2005;44(2):617–626 [DOI] [PubMed] [Google Scholar]
- 34.Yakovlev S, Medved L. Effect of fibrinogen, fibrin, and fibrin degradation products on transendothelial migration of leukocytes. Thromb Res 2018;162:93–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petzelbauer P, Zacharowski PA, Miyazaki Y, et al. The fibrin-derived peptide Bβ15-42 protects the myocardium against ischemia-reperfusion injury. Nat Med 2005;11(3):298–304 [DOI] [PubMed] [Google Scholar]
- 36.Zacharowski K, Zacharowski P, Reingruber S, Petzelbauer P. Fibrin(ogen) and its fragments in the pathophysiology and treatment of myocardial infarction. J Mol Med 2006;84(6):469–477 [DOI] [PubMed] [Google Scholar]
- 37.Blömback B, Blömback M, Henschen A, Hessel B, Iwanaga S, Woods KR. N-terminal disulphide knot of human fibrinogen. 1968;218(5137):130–134 [DOI] [PubMed] [Google Scholar]
- 38.Medved L, Yakovlev S. Structure and function of fibrinogen BβN-domains. Ukr Biochem J 2020;92(3) 22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loike JD, Sodeik B, Cao L, et al. CD11c/CD18 on neutrophils recognizes a domain at the N terminus of the Aα chain of fibrinogen. Proc Natl Acad Sci U S A 1991;88(3):1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chalupowicz DG, Chowdhury ZA, Bach TL, Barsigian C, Martinez J. Fibrin II induces endothelial cell capillary tube formation. J Cell Biol 1995;130(1):207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez J, Ferber A, Bach TL, Yaen CH. Interaction of fibrin with VE-cadherin. Ann N Y Acad Sci 2001; 936(1): 386–405 [DOI] [PubMed] [Google Scholar]
- 42.Zacharowski K, Zacharowski PA, Friedl P, et al. The effect of the fibrin-derived peptide Bβ15-42 in acute and chronic rodent models of myocardial ischemia-reperfusion. Shock 2007;27(6):631–637 [DOI] [PubMed] [Google Scholar]
- 43.Roesner JP, Petzelbauer P, Koch A, et al. The fibrin-derived peptide Bβ15-42 is cardioprotective in a pig model of myocardial ischemia-reperfusion injury. Crit Care Med 2007;35(7):1730–1735 [DOI] [PubMed] [Google Scholar]
- 44.Ahrens I, Peter K. FX-06, a fibrin-derived Bβ15-42 peptide for the potential treatment of reperfusion injury following myocardial infarction. Curr Opin Investig Drugs 2009;10(9):997–1003 [PubMed] [Google Scholar]
- 45.Atar D, Petzelbauer P, Schwitter J, et al. Effect of intravenous FX06 as an adjunct to primary percutaneous coronary intervention for acute ST-segment elevation myocardial infarction results of the F.I.R.E. (Efficacy of FX06 in the Prevention of Myocardial Reperfusion Injury) trial. J Am Coll Cardiol 2009;53(8):720–729 [DOI] [PubMed] [Google Scholar]
- 46.Hallén J, Petzelbauer P, Schwitter J, Geudelin B, Buser P, Atar D. Impact of time to therapy and presence of collaterals on the efficacy of FX06 in acute ST elevation myocardial infarction: a substudy of the F.I.R.E., the Efficacy of FX06 in the prevention of myocardial reperfusion injury trial. EuroIntervention 2010;5(8):946–952 [PubMed] [Google Scholar]
- 47.Gorlatov S, Medved L. Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: mapping of the receptor-binding site in the NH2-terminal portions of the fibrin β chains. Biochemistry 2002;41(12):4107–4116 [DOI] [PubMed] [Google Scholar]
- 48.Yakovlev S, Gao Y, Cao C, et al. Interaction of fibrin with VE-cadherin and anti-inflammatory effect of fibrin-derived fragments. J Thromb Haemost 2011;9(9):1847–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roesner JP, Petzelbauer P, Koch A, et al. Bβ15-42 (FX06) reduces pulmonary, myocardial, liver, and small intestine damage in a pig model of hemorrhagic shock and reperfusion. Crit Care Med 2009;37(2):598–605 [DOI] [PubMed] [Google Scholar]
- 50.Roesner JP, Petzelbauer P, Koch A, et al. A double blind, single centre, sub-chronic reperfusion trial evaluating FX06 following haemorrhagic shock in pigs. Resuscitation 2009;80(2):264–271 [DOI] [PubMed] [Google Scholar]
- 51.Wiedemann D, Schneeberger S, Friedl P, et al. The fibrin-derived peptide Bβ15-42 significantly attenuates ischemia-reperfusion injury in a cardiac transplant model. Transplantation 2010;89(7):824–829 [DOI] [PubMed] [Google Scholar]
- 52.Sörensen I, Rong S, Susnik N, et al. Bβ15-42 attenuates the effect of ischemia-reperfusion injury in renal transplantation. J Am Soc Nephrol 2011;22(10):1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krishnamoorthy A, Ajay AK, Hoffmann D, et al. Fibrinogen β-derived Bβ15-42 peptide protects against kidney ischemia/reperfusion injury. Blood 2011;118(7):1934–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu A, Fang H, Yang Y, et al. The fibrin-derived peptide Bβ15-42 attenuates liver damage in a rat model of liver ischemia/reperfusion injury. Shock 2013;39(4):397–403 [DOI] [PubMed] [Google Scholar]
- 55.Urbschat A, Rupprecht K, Zacharowski K, et al. Combined peri-ischemic administration of Bβ15-42 in treating ischemia reperfusion injury of the mouse kidney. Microvasc Res 2015;101:48–54 [DOI] [PubMed] [Google Scholar]
- 56.Matt U, Warszawska JM, Bauer M, et al. Bβ15-42 protects against acid-induced acute lung injury and secondary Pseudomonas pneumonia in vivo. Am J Respir Crit Care Med 2009;180(12):1208–1217 [DOI] [PubMed] [Google Scholar]
- 57.Gröger M, Pasteiner W, Ignatyev G, et al. Peptide Bβ15-42 preserves endothelial barrier function in shock. PLoS One 2009;4(4):e5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bergt S, Gruenewald M, Beltschany C, et al. The fibrin-derived peptide Bβ15-42 (FX06) ameliorates vascular leakage and improves survival and neurocognitive recovery: Implications from two animal models of cardiopulmonary resuscitation. Crit Care Med 2016;44(10):e988–995 [DOI] [PubMed] [Google Scholar]
- 59.Wolf T, Kann G, Becker S, et al. Severe Ebola virus disease with vascular leakage and multiorgan failure: treatment of a patient in intensive care. Lancet 2015;385(9976):1428–1435 [DOI] [PubMed] [Google Scholar]
- 60.Holinstat M, Knezevic N, Broman M, Samarel AM, Malik AB, Mehta D. Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J Biol Chem 2006;281(4):2296–2305 [DOI] [PubMed] [Google Scholar]
- 61.Yakovlev S, Medved L. Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: localization of the fibrin-binding site within the third extracellular VE-cadherin domain. Biochemistry 2009;48(23):5171–5179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yakovlev S, Cao C, Galisteo R, Zhang L, Strickland DK, Medved L. Fibrin-VLDL receptor-dependent pathway promotes leukocyte transmigration by inhibiting Src kinase Fyn and is a target for fibrin β15-42 peptide. Thromb Haemost 2019;119(11):1816–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yakovlev S, Mikhailenko I, Cao C, Zhang L, Strickland DK, Medved L. Identification of VLDLR as a novel endothelial cell receptor for fibrin that modulates fibrin-dependent transendothelial migration of leukocytes. Blood 2012;119(2):637–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J Biol Chem 1991;266(31):21232–21238 [PubMed] [Google Scholar]
- 65.Williams SE, Ashcom JD, Argraves WS, Strickland DK. A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J Biol Chem 1992;267(13):9035–9040 [PubMed] [Google Scholar]
- 66.Yakovlev S, Medved L. Interaction of fibrin with the very low density lipoprotein receptor: further characterization and localization of the fibrin-binding site. Biochemistry 2015;54(30):4751–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yakovlev S, Medved L. Interaction of fibrin with the very low-density lipoprotein (VLDL) receptor: further characterization and localization of the VLDL receptor-binding site in fibrin βN-domains. Biochemistry 2017;56(19):2518–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Banerjee K, Yakovlev S, Gruschus JM, Medved L, Tjandra N. Nuclear magnetic resonance solution structure of the recombinant fragment containing three fibrin-binding cysteine-rich domains of the very low density lipoprotein receptor. Biochemistry 2018;57(30):4395–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gruschus JM, Yakovlev S, Banerjee K, Medved L, Tjandra N. Structural basis for the interaction of fibrin with the very low-density lipoprotein receptor revealed by NMR and site-directed mutagenesis. Biochemistry 2021;60(33):2537–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fisher C, Beglova N, Blacklow LC. Structure of an LDL-RAP complex reveals a general mode for ligand recognition by lipoprotein receptors. Mol Cell 2006; 22(2):277–283 [DOI] [PubMed] [Google Scholar]
- 71.Ruiz J, Kouiavskaia D, Migliorini M, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res 2005;46(8):1721–1731 [DOI] [PubMed] [Google Scholar]
- 72.Yakovlev S, Belkin AM, Chen L, et al. Anti-VLDL receptor monoclonal antibodies inhibit fibrin-VLDL receptor interaction and reduce fibrin-dependent leukocyte transmigration. Thromb Haemost 2016;116(6):1122–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yakovlev S, Medved L. Dual functions of the fibrin βN-domains in the VLDL receptor-dependent pathway of transendothelial migration of leukocytes. Thromb Res 2022;214:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sörensen-Zender I, Chen R, Rong S, David S, Melk A, Haller H, Schmitt R. Binding to carboxypeptidase M mediates protective effects of fibrinopeptide Bβ15-42. Transl Res 2019;213:124–135 [DOI] [PubMed] [Google Scholar]
- 75.Kirchhofer D, Tschopp TB, Hadváry P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-α express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Invest 1994;93(5):2073–2083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kirchhofer D, Sakariassen KS, Clozel M, et al. Relationship between tissue factor expression and deposition of fibrin, platelets, and leukocytes on cultured endothelial cells under venous blood flow conditions. Blood 2003;81(8):2050–2058 [PubMed] [Google Scholar]
- 77.Campbell RA, Overmyer KA, Barnell CR, Wolberg AS. Cellular procoagulant activity dictates clot structure and stability as a function of distance from the cell surface. Arterioscler Thromb Vasc Biol 2008;28(12):2247–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Campbell RA, Overmyer KA, Selzman CH, Sheridan BC, Wolberg AS. Contributions of extravascular and intravascular cells to fibrin network formation, structure, and stability. Blood 2009;114(23):4886–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]