

Abstract
Alzheimer’s disease (AD) pathology is thought to progress from normal cognition through preclinical disease and ultimately to symptomatic AD with cognitive impairment. Recent work suggests that the gut microbiome of symptomatic patients with AD has an altered taxonomic composition compared with that of healthy, cognitively normal control individuals. However, knowledge about changes in the gut microbiome before the onset of symptomatic AD is limited. In this cross-sectional study that accounted for clinical covariates and dietary intake, we compared the taxonomic composition and gut microbial function in a cohort of 164 cognitively normal individuals, 49 of whom showed biomarker evidence of early preclinical AD. Gut microbial taxonomic profiles of individuals with preclinical AD were distinct from those of individuals without evidence of preclinical AD. The change in gut microbiome composition correlated with β-amyloid (Aβ) and tau pathological biomarkers but not with biomarkers of neurodegeneration, suggesting that the gut microbiome may change early in the disease process. We identified specific gut bacterial taxa associated with preclinical AD. Inclusion of these microbiome features improved the accuracy, sensitivity, and specificity of machine learning classifiers for predicting preclinical AD status when tested on a subset of the cohort (65 of the 164 participants). Gut microbiome correlates of preclinical AD neuropathology may improve our understanding of AD etiology and may help to identify gut-derived markers of AD risk.
INTRODUCTION
The human gut microbiome harbors a compositionally and functionally diverse community of microorganisms that influences the health and well-being of their hosts (1, 2). These communities include >1012 bacterial cells (2), representing thousands of taxa that encode a vast repertoire of pathways with diverse influences on human physiology and metabolism (1, 2). Some gut microbes train the mammalian immune system at birth, whereas others have lifelong immunomodulatory activity (3, 4). Gut microbiome dysbiosis, defined as bacterial populations correlated with disease status and typified by diminished diversity, has been associated with a number of disorders (2, 5–7). Gut dysfunction and aberrant microbial content may contribute to the pathogenesis of Alzheimer’s disease (AD) and potentially other neurodegenerative diseases (8).
AD pathobiology is thought to progress from cognitively normal with no evidence of disease to apparently cognitively normal with biomarker evidence of disease (preclinical AD) and to symptomatic AD. These transitions are based on the presence of markers identified by positron emission tomography (PET) imaging or cerebrospinal fluid (CSF) assays to detect pathogenic β-amyloid (Aβ) and tau protein, as well as markers of neurodegeneration identified by CSF assays and magnetic resonance imaging (MRI) (9). The amyloid-tau-neurodegeneration or AT(N) marker combination suggests that neuropathology occurs well before symptom onset, defined as the point when the clinical dementia rating (CDR) score becomes abnormal (10).
Several lines of evidence suggest a role for gut microbes in the evolution of AD pathogenesis. Compared with stool samples from healthy cognitively normal individuals, those with symptomatic AD have increased relative abundance of Bacteroidetes and decreased relative abundance of Firmicutes (6), an imbalance found in other chronic inflammatory conditions (11). Changes in the gut microbiome correlate with the presence of CSF markers of AD, including phosphorylated tau-181 (p-tau-181) and Aβ (measured by the Aβ42/Aβ40 ratio) (6). Variations in the bacterial composition of stool in symptomatic patients with AD were accompanied by a dys-regulated P-glycoprotein pathway in gut epithelial cells (5), an alteration that contributes to enteric inflammation and disrupted organ homeostasis (5). Symptomatic patients with AD also show increased concentrations of lipopolysaccharide in the circulation (12), presumably of gut microbial origin. In animal models of AD, manipulation of the gut microbiota decreases Aβ deposition and improves neurologic function (13, 14). Last, gut bacteria from wild-type mice diminish AD pathology in recipient mice (15).
Current AT(N) criteria do not consider gut dysbiosis. However, identification of microbial community alterations before AD symptoms ensue might enable strengthening of the AT(N) framework with microbiome-derived markers that are more accessible to assay than current markers (16–20). Early signatures of gut dysbiosis in conjunction with preclinical AD markers may inform future gut microbiome-directed therapies that could potentially slow AD progression (21, 22).
Here, we examined a Knight Alzheimer’s Disease Research Center (ADRC) cohort of cognitively normal individuals with and without preclinical AD (23–25) to determine whether cognitively normal individuals with preclinical AD may have an AD-associated dysbiotic gut microbiome, taking into account clinical covariates and dietary data. We investigated whether specific microbiome characteristics in stool samples correlated with preclinical AD status or established AD biomarkers and determined whether microbiome characteristics could improve the performance of machine learning classifiers designed to distinguish healthy individuals from those with preclinical AD.
RESULTS
Global differences in the gut microbiomes of healthy individuals and those with preclinical AD
Participants (68 to 94 years old; 45% male) recruited from existing longitudinal studies (26–28) of the Knight ADRC cohort submitted stool samples from 2019 to 2021, which were sequenced to an average depth of 21.5 million reads. Participants underwent PET imaging, MRI imaging, lumbar puncture to obtain CSF samples, stool sampling, phlebotomy, and clinical and cognitive testing, including completion of the CDR scale (10) every 3 years (<65 years old) or annually (≥65 years old) (fig. S1) (27, 28). The mean interval between stool sampling and PET imaging or lumbar puncture for quantification of Aβ and tau was 2.4 and 2.8 years, respectively, and 3.8 months from the most recent CDR assessment. All sampling intervals were summarized according to preclinical AD status (table S1). We defined preclinical AD status as CDR 0 and Aβ-positive, with Aβ plaque positivity defined as Centiloid > 16.4, corresponding to an 11C Pittsburgh compound B (PiB)–PET standardized uptake value ratio (SUVR) > 1.42 (29, 30). Eleven participants did not have PET Aβ data available, in which case Aβ positivity was defined as a CSF Aβ42/Aβ40 ratio < 0.0673 (31, 32). Similarly, healthy status was defined as CDR 0 and Centiloid ≤ 16.4 or CSF Aβ42/Aβ40 ratio ≥ 0.0673. We used these criteria to assign healthy (n = 115) or preclinical AD status (n = 49) to 164 participants. We compared clinical covariates between groups and identified differences in age, body mass index, apolipoprotein ε4 (APOE ε4) carrier status, diabetes, and hypertension, which we included as variables in our linear regression and machine learning models as well as in analyses of variance (ANOVAs) (Table 1). We also included as a variable the time interval between stool collection and PET imaging or lumbar puncture to obtain CSF samples depending on which biomarker was used to define the preclinical AD status (table S1). To account for the impact of diet on gut microbiome composition (33), where dietary behavior can rapidly (within 24 hours) (34) induce taxonomic shifts, we assessed participant nutritional profiles from stool-matched 24-hour diet logs (fig. S2). We observed no significant difference between the healthy and preclinical AD groups in the overall caloric intake, caloric source distribution, or intake of any major nutrient group (e.g., carbohydrates, fats, and total dietary fiber) or specific vitamins or minerals (fig. S2).
Table 1.
Participant demographics at the time of stool sampling stratified by AD preclinical status.
| Healthy | Preclinical AD | P | ||
|---|---|---|---|---|
| Number | 115 | 49 | ||
| Age in years, mean (SD) | 77.02 (5.80) | 78.96 (4.51) | 0.039 | |
| Sex (%) | Male | 49 (42.6) | 24 (49.0) | 0.562 |
| Years of education, mean (SD) | 16.42 (2.26) | 16.69 (2.53) | 0.49 | |
| Race(%) | Black | 18 (15.7) | 1 (2.0) | 0.034 |
| White | 96 (83.5) | 48 (98.0) | ||
| Other | 1 (0.9) | 0 (0.0) | ||
| Body mass index, mean (SD) | 28.81 (5.03) | 26.73 (5.19) | 0.018 | |
| APOE4 (%) | e4+ | 26 (22.6) | 23 (46.9) | 0.003 |
| Active depression (%) | 3 (2.6) | 1 (2.0) | 1 | |
| Alcohol abuse (%) | 7 (6.1) | 1 (2.0) | 0.481 | |
| Autoimmune disorder† (%) | 11 (9.6) | 5 (10.2) | 1 | |
| Cancer(%) | 6 (5.2) | 2 (4.1) | 1 | |
| Cardiovascular disease (%) | 14 (12.2) | 6 (12.2) | 1 | |
| Diabetes (%) | 22 (19.1) | 1 (2.0) | 0.008 | |
| Hypercholesterolemia (%) | 69 (60.0) | 25 (51.0) | 0.373 | |
| Hypertension (%) | 74 (64.3) | 23 (46.9) | 0.057 | |
| Liver disease (%) | 7 (6.1) | 3 (6.1) | 1 | |
| Thyroid disease (%) | 21 (18.3) | 10 (20.4) | 0.917 | |
| Tobacco use (past or present) (%) | 51 (44.3) | 24 (49.0) | 0.709 |
P, Student’s t test (continuous variables) or chi-square test (categorical variables).
Rheumatoid arthritis, lupus, etc.
We performed fecal metagenomic sequencing and profiled relative abundance of microbial taxa at the species level (MetaPhlAn3) (35), as well as microbial pathways (HUMAnN 3.0) (figs. S1 and S3) (35). The Firmicutes/Bacteroidetes ratio for healthy controls was 7.30, with a 95% confidence interval (CI) [4.31, 10.30], and the ratio for preclinical AD was 5.98, with 95% CI [3.90, 8.06], and there was no significant difference between these two groups (Fig. 1A and fig. S3A). Similarly, alpha (within-sample) diversities calculated on taxa or pathways (fig. S3C) were similar between groups. In contrast, principal coordinates analysis (PCoA) using between-sample UniFrac distances (36) demonstrated global differences in gut taxonomic profiles by preclinical status [P = 0.036, permutational ANOVA (PERMANOVA); P = 0.046, one-way ANOVA on PCoA1 coordinates by AD status, Benjamini-Hochberg–adjusted] (Fig. 1B and table S2). We observed a binomial structure in the ordination along PCoA2, which we found to be associated with taxonomic alpha diversity (P = 0.006, Student’s t test comparing richness for samples with PCoA2 > 0 versus PCoA2 ≤ 0); however, alpha diversity in this cohort was not associated with AD status. The PERMANOVA results prompted canonical analysis of principal coordinates (CAP) (37), in which candidate explanatory variables (“constraints”) are tested for their ability to explain variance in sample coordinates within an unconstrained ordination; preclinical AD status explained significant multivariate, i.e., taxonomic differences in the cohort (P = 0.040, PERMANOVA; P = 0.0004 and P = 0.0003, one-way ANOVA by AD status on CAP1 and CAP2 coordinates, respectively, Benjamini-Hochberg–adjusted) (Fig. 1C and table S3). Applying CAP to a PCoA ordination of microbial pathway profiles using the binary Bray-Curtis dissimilarity metric (fig. S3D), sample coordinates significantly differed by AD status along the CAP2 axis (P = 0.036, one-way ANOVA on CAP2 coordinates by AD status) (fig. S3E and tables S4 and S5). These data suggest that the human gut microbiome may change early in AD, before cognitive impairment becomes apparent (5, 6).
Fig. 1. Healthy and preclinical AD individuals have distinct gut microbiome profiles.
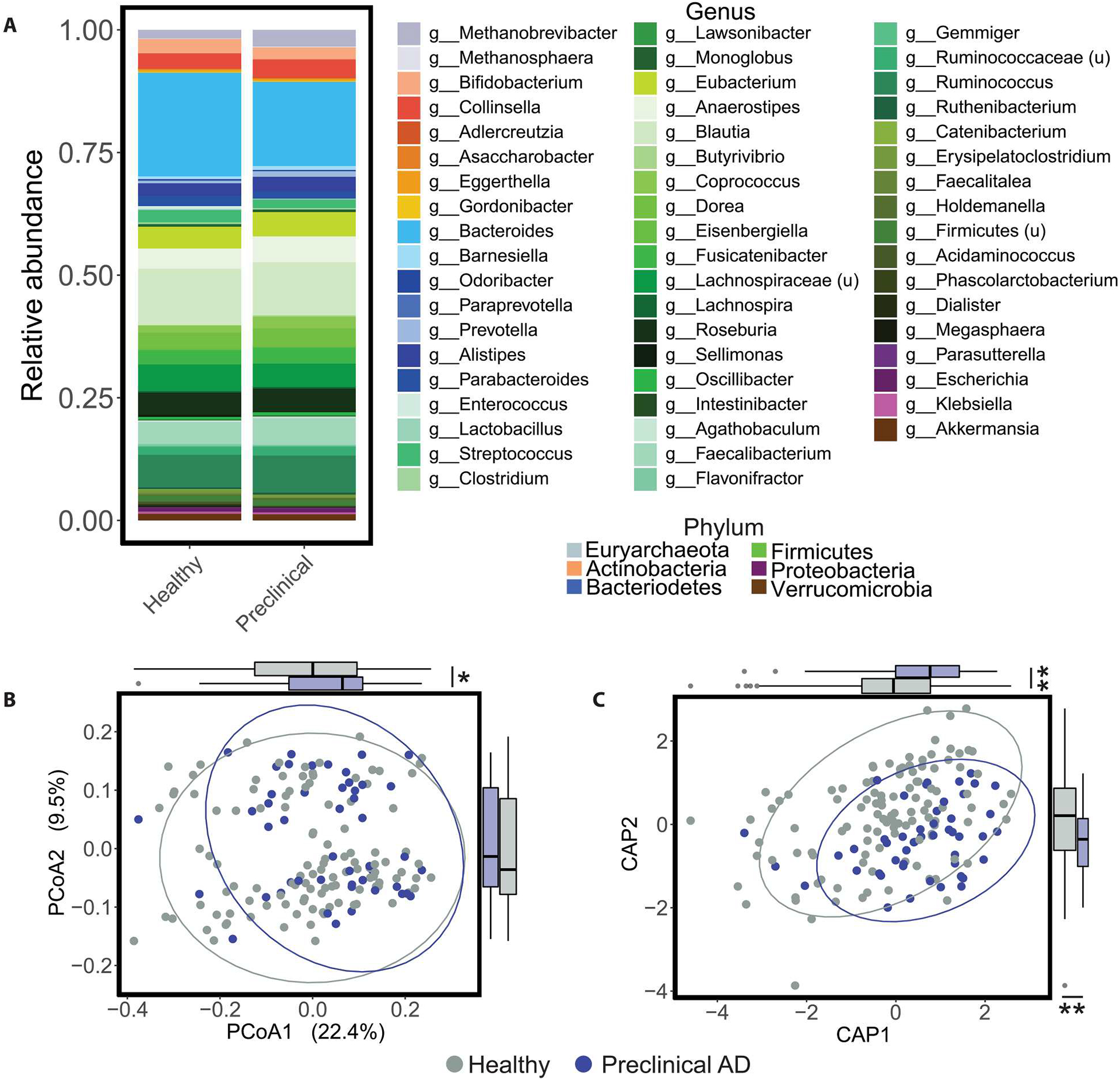
(A) Stacked taxonomic (MetaPhlAn3) bar plots at the genus level stratified by preclinical AD status are shown, with color grouping at the phylum level. u, unclassified. (B) The PCoA on unweighted UniFrac distances was calculated from the MetaPhlAn3 taxonomic profiles. Global microbiome composition was different between healthy and preclinical AD individuals after accounting for age, APOE ɛ4 carrier status, diabetes, body mass index, hypertension, and time elapsed between PET imaging or lumbar puncture for Aβ quantification and stool collection (P = 0.039, PERMANOVA; table S2). In addition, coordinates of healthy and preclinical AD samples were different along the PCoA axis 1 (P = 0.046, Student’s t test). (C) Corresponding CAP ordination on unweighted UniFrac distances was calculated from the MetaPhlAn3 taxonomic profiles using the same terms as the PERMANOVA in (B) (preclinical AD status P = 0.038, PERMANOVA; table S3). In addition, sample coordinates along the CAP1 and CAP2 axes differed by AD status (P = 0.001, Student’s t test). Ellipses represent 95% confidence bounds around group centroids. *P < 0.05 and **P < 0.01. Student’s t test; P values were adjusted using the Benjamini-Hochberg method.
Gut microbiome profiles correlate with Aβ and tau, but not neurodegeneration
We determined whether gut microbiome profiles correlated with specific characteristics of preclinical AD using pairwise Spearman correlation analyses. We compared summary measures of the gut microbiome, specifically PCoA axes 1 and 2 from ordinations of microbial taxonomic and pathway profiles, with the amount of Aβ plaques measured by PET imaging (38, 39) using the Centiloid scale (40) as well as the ratio of Aβ42/Aβ40 in CSF samples (41, 42). Gut microbiome summary measures were also compared with the amount of tau according to PET imaging (43) and the amount of p-tau-181 in CSF samples (44). They were also compared with neurodegeneration measured by total tau (t-tau) in CSF (41), a cortical signature of neurodegeneration, or hippocampus volume (45), as well as with vascular injury measured as brain white matter hyperintensities (Fig. 2A). In addition, we asked whether these gut microbiome measures were associated with genetic risk factors: APOE ε4 carrier status and polygenic risk score (46).
Fig. 2. Gut microbiome profiles correlate with Aβ and tau but not neurodegeneration.
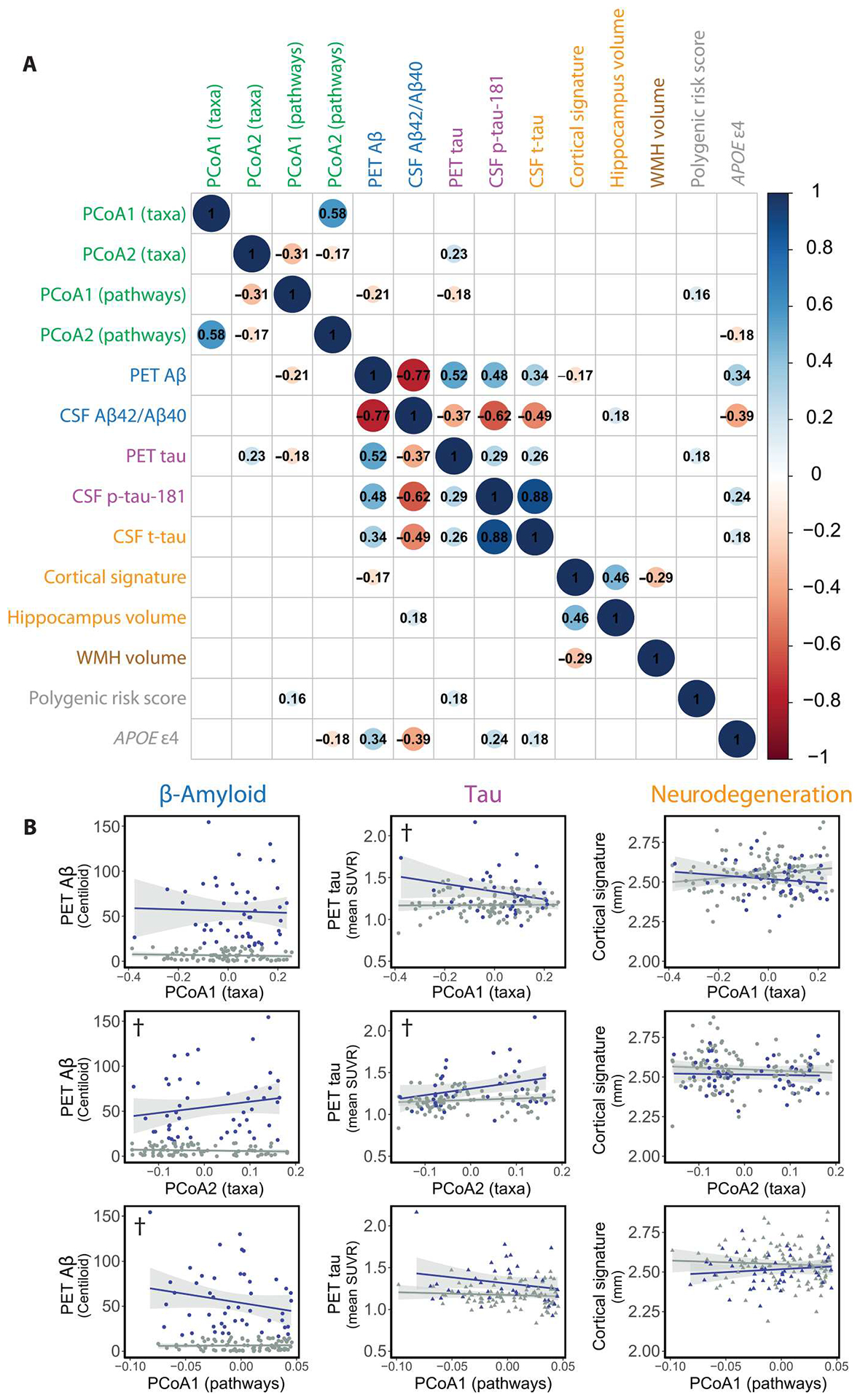
(A) Pairwise Spearman correlations between microbiome summary metrics (green) and AD biomarkers (blue, Aβ; purple, tau; orange, neurodegeneration; brown, vascular injury; gray, genetic risk factors). Significant correlations are shown (P < 0.05, Benjamini-Hochberg adjusted), with the size of the circle inversely proportional to the P value. Inset values are Spearman correlations. WMH, white matter hyperintensities. (B) Linear regressions of AD biomarkers against gut microbiome–derived axes. Specifically, regressions of PET Aβ, PET tau, or cortical thickness (a measure of neurodegeneration) against PCoA sample coordinates derived from MetaPhlan3 taxonomic profiles (top and middle rows) or HUMAnN 3.0 functional pathway profiles (bottom row). Source PCoA ordinations are from Fig. 1B and fig. S3D. †P < 0.1, ANOVAs, Benjamini-Hochberg–adjusted. ANOVAs compare models regressing biomarker ~ PCoA axis*Aβ status against null models regressing biomarker ~ Aβ status to determine significantly improved explanation of variance with addition of the gut microbiome summary feature (PCoA axis). Regression models and ANOVAs are summarized in tables S6 to S8.
PET imaging of Aβ plaques correlated with PCoA1 pathway profiles (ρ = −0.21, P = 0.015, Benjamini-Hochberg adjusted) (Fig. 2A), a result supported by linear regression models. These models additionally revealed an association of PET Aβ plaques with PCoA2 of taxonomic profiles (P = 0.080 taxa PCoA2, P = 0.052 pathways PCoA1, ANOVAs comparing models including the interaction between PCoA axis coordinates and preclinical AD status against null models with preclinical AD status as the only predictor, Benjamini-Hochberg–adjusted across all models tested) (Fig. 2B and tables S7 and S8). PET tau also correlated with PCoA2 from taxonomic profiles (ρ = 0.23, P = 0.014, Benjamini-Hochberg–adjusted) and PCoA1 from pathway profiles (ρ = −0.18, P = 0.040, Benjamini-Hochberg–adjusted) (Fig. 2A). The correlation of PET tau with PCoA axes from taxonomic profiles was supported by linear regression models (P = 0.080 taxa PCoA1, P = 0.018 taxa PCoA2, ANOVAs, Benjamini-Hochberg–adjusted) (Fig. 2B and tables S6 and S7). The proportion of Aβ-negative but tau-positive (18F-flortaucipir imaging probe > 1.22) individuals in the cohort was 16.3%, which was similar to proportions previously reported for comparable cohorts (47, 48).
No microbiome measures included in this analysis were significantly correlated with neurodegeneration markers (Fig. 2 and tables S6 to S8). Neurodegeneration is considered to be a later event than the appearance of Aβ and tau biomarkers in the AT(N) framework of preclinical AD (49). An increase in Aβ plaques as measured by PET imaging before onset of clinical symptoms of dementia is considered to be associated with a greater risk of progression to symptomatic AD (50).
Specific gut microbiome features are associated with preclinical AD status
To identify specific taxa and microbial pathways associated with preclinical AD status, we fitted negative binomial regression models to taxonomic or microbial pathway abundance data. As with the PERMANOVA and CAP analyses, in addition to preclinical AD status, we included age, APOE ε4 carrier status, body mass index, diabetes, and hypertension as model variables, as well as the time interval between stool sampling and PET imaging or lumbar puncture for CSF sampling depending on the biomarker used to define Aβ positivity (Fig. 3 and fig. S4). Species most associated with preclinical AD status by magnitude of their model coefficients included Dorea formicigenerans (coefficient = 0.661, 95% CI [0.659, 0.662], P < 0.001; all P values from regression analyses are Benjamini-Hochberg adjusted), Oscillibacter sp. 57_20 (coefficient = 0.512, 95% CI [0.510, 0.514], P < 0.001), Faecalibacterium prausnitzii (coefficient = 0.298, 95% CI [0.297, 0.298], P < 0.001), Coprococcus catus (coefficient = 0.190, 95% CI [0.187, 0.192], P < 0.001), and Anaerostipes hadrus (coefficient = 0.163, 95% CI [0.163, 0.164], P < 0.001) (Fig. 3, A and B). We highlight these taxa here because they were also identified as important features in Random Forest classifiers for preclinical AD status (Fig. 4 and fig. S3B). Ruminococcus lactaris was associated with preclinical AD status (coefficient = 0.028, 95% CI [0.026, 0.029], P < 0.001), whereas Methanosphaera stadtmanae was associated with healthy status (coefficient = −0.240, 95% CI [−0.243, −0.236], P < 0.001); both species were also identified as an important feature in Random Forest classifiers (Fig. 4 and fig. S3B) but are not included here because they did not meet filtering criteria for visualization (detected in ≥25 participants or magnitude of the coefficient ≥0.15; data file S2). Seven of 13 gut microbial species, most associated with healthy status in this cohort, belonged to the Bacteroides genus (Fig. 3).
Fig. 3. Fitting negative binomial models to gut microbiome taxonomic data identifies species associated with AD preclinical status.
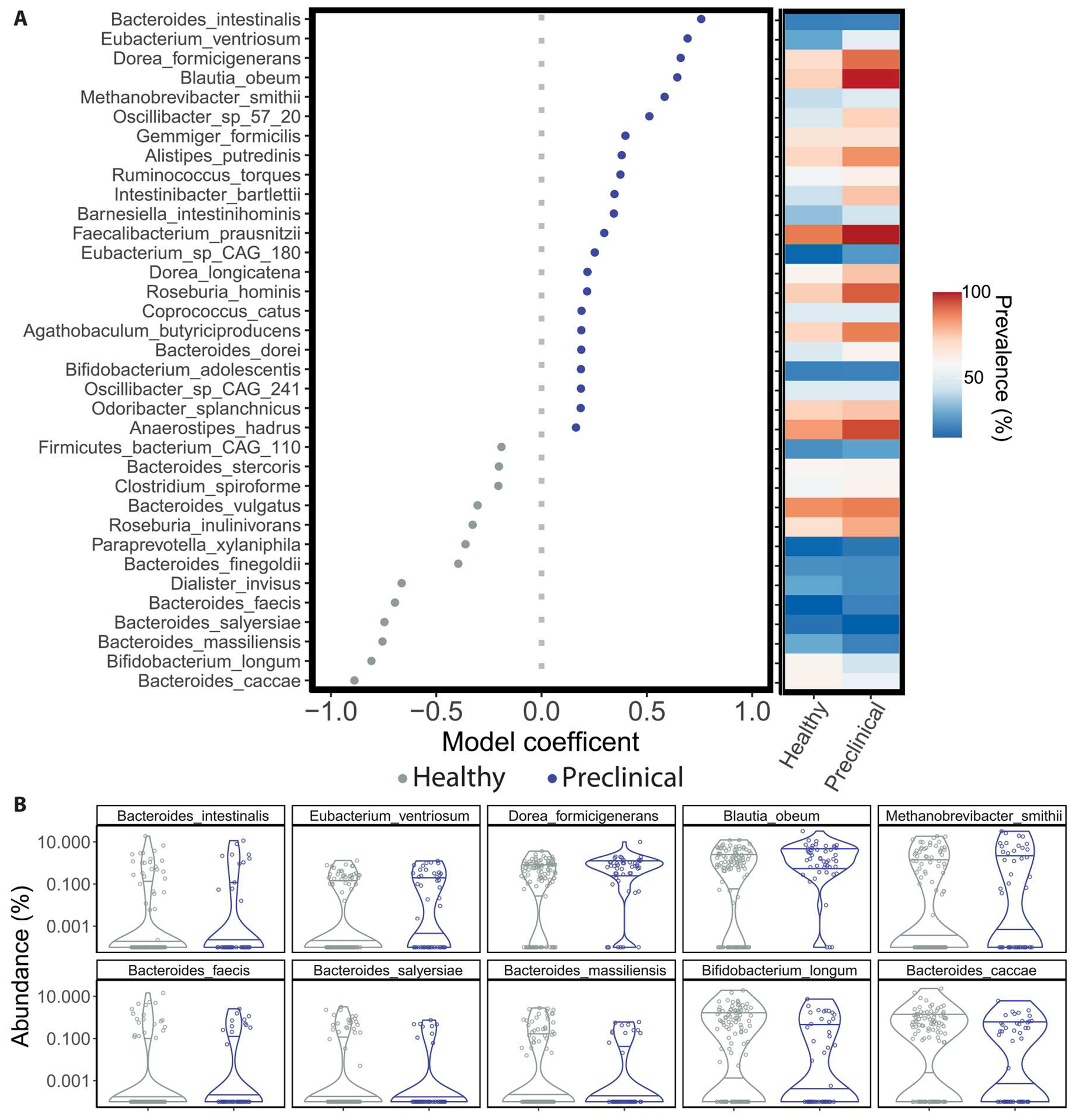
(A) Model coefficients (left) and prevalence (right) of top-ranking species significantly associated with healthy or preclinical AD status are shown. Gut microbial species detected in at least 15% of samples are shown, with Benjamini-Hochberg–adjusted P values of the coefficient < 0.05 and with the magnitude of the coefficient > 0.15. Error bars represent the SE of the coefficient and may not be visible. Taxa coefficients are from negative binomial regression models (as implemented in MaAsLin2) that additionally included participant age, APOE ɛ4 carrier status, diabetes, body mass index, hypertension, and time elapsed between PET imaging or lumbar puncture for Aβ quantification and stool collection as predictors. (B) Relative abundances of the 10 taxa most associated with preclinical AD (top row) or healthy status (bottom row) by their model coefficient. All regression model results are available in data file S2.
Fig. 4. Gut microbiome features improve the performance of Random Forest classifiers for AD status.
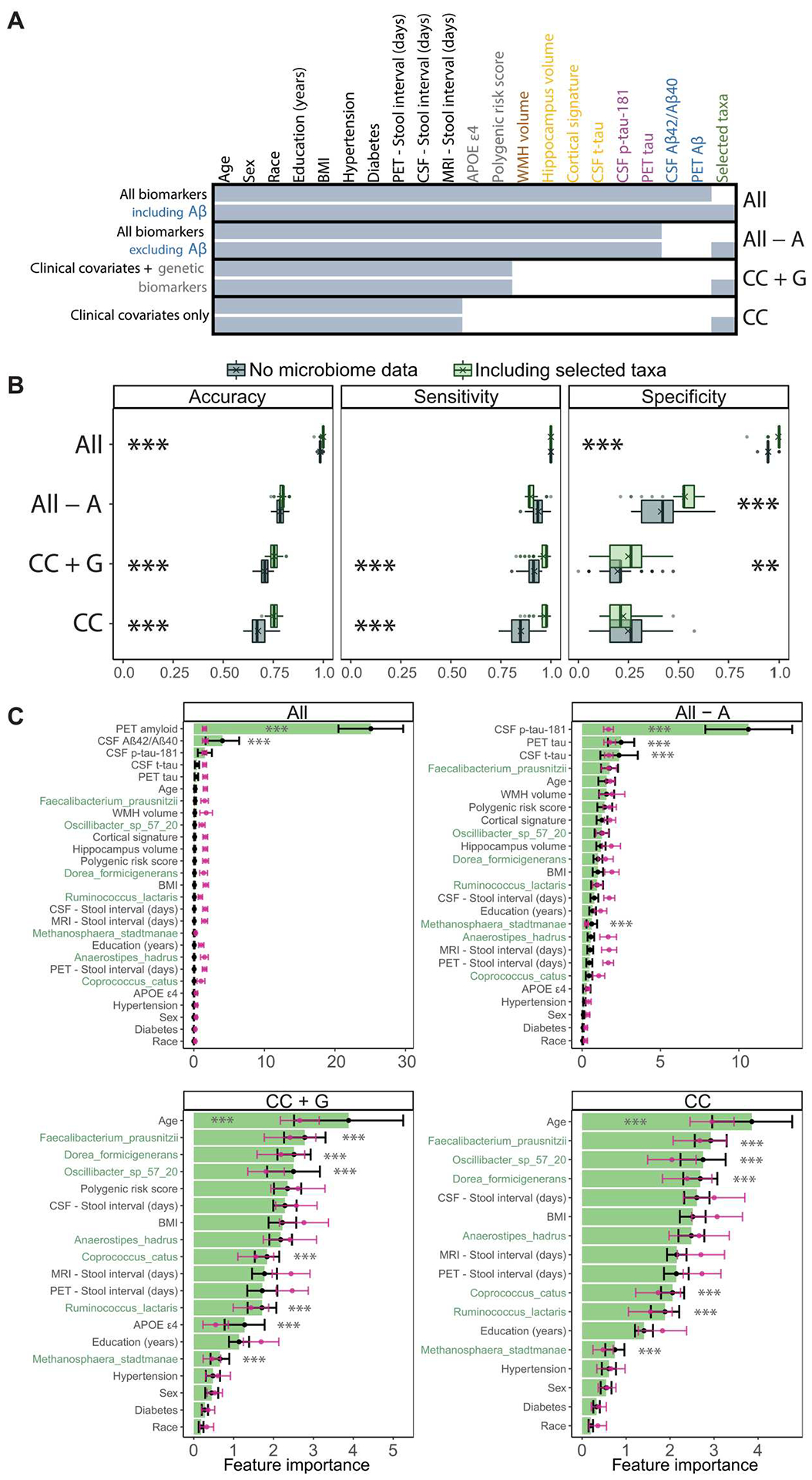
We compare the performance of Random Forest classification models with and without gut microbiome features, across combinations of AT(N) biomarkers and genetic risk factors for AD. (A) Summary of features included in each of the Random Forest models reported in (B) and (C). Feature inclusion is denoted by shaded cells. Models that include or exclude feature-selected gut taxa are compared (bottom and top of each model). Feature labels are colored by data/biomarker type (green, gut taxa; blue, Aβ; purple, tau; orange, neurodegeneration; brown, vascular injury; gray, genetic risk factors; black, clinical covariates). Except for model “All biomarkers including Aβ,” other models exclude Aβ biomarkers (PET Aβ and CSF Aβ42/Aβ40 ratio). Model shorthand names listed in the right margin: CC, clinical covariates; A, Aβ; G, genetics. Missing data were imputed before model training and are summarized in fig. S5. The feature with the most missingness was PET tau (20.7%). BMI, body mass index; WMH, white matter hyperintensities. (B) Performance metrics for Random Forest models that include or exclude feature-selected gut microbiome taxa (gray, no microbiome features; green, including relative abundances of feature-selected taxa). Boxplots summarize performance metrics on the retained validation cohort of models trained on 100 random partitions of the training cohort. Means are denoted by “X” in the boxplots. **P < 0.01 and ***P < 0.001. ANOVAs with Tukey’s post hoc test, Bonferroni-adjusted for multiple comparisons at both ANOVA and Tukey post hoc levels. (C) Importance of the features included in each model, averaged over the 100 training partitions (black), optionally with random class label shuffling at each iteration to generate null distributions (pink). Error bars represent SD. The seven taxonomic features are highlighted in green. ***P < 0.001. Student’s t test with Benjamini-Hochberg adjustment (see Table 2 and figs. S5 and S6).
The microbial pathways most strongly associated with preclinical AD status included those involved in arginine and ornithine degradation (coefficients = 0.967, 95% CI [0.954, 0.979]; 0.625, 95% CI [0.615, 0.635]; P < 0.001 in each case) (fig. S4). The pathway most associated with healthy status was glutamate degradation (coefficient = −0.992, 95% CI [−1.00, −0.98], P < 0.001) (fig. S4). Model coefficients of significant features in these regression analyses are collated in data file S2.
Gut microbiome features improve the performance of classifiers for preclinical AD status
We next sought to determine whether gut microbiome features improve the predictive performance of Random Forest classifiers for preclinical AD status, which train on basic demographics and clinical covariates, including age, APOE ε4 carrier status, body mass index, diabetes, and hypertension, as well as combinations of Aβ (PET Aβ and CSF Aβ42/Aβ40 ratio), tau (PET tau and CSF p-tau-181), neurodegeneration (CSF t-tau, cortical signature, and hippocampus volume), vascular injury (white matter hyperintensities volume), and genetics (APOE ε4 carrier status and polygenic risk score). We also tested the contribution of gut microbiome features in models that lacked canonical AD biomarkers. Aβ positivity underlies the definition of preclinical AD status; access to Aβ amounts in the brain or CSF diminished the potential relative value of putative stool markers. We rationalized that the predictive performance of gut microbiome features would be of interest in the absence of access to canonical AD biomarkers, which requires neuroimaging or lumbar puncture. To characterize the upper bound of predictive performance under this approach, we tested a model that, in addition to demographic data and clinical covariates, included all available AD biomarkers in the categories of Aβ, tau, neurodegeneration, and genetic risk (Fig. 4A). We then rationally omitted categories of AD biomarkers from these models, beginning with Aβ markers, then all AD biomarkers except for genetics, and then all biomarkers, leaving only demographics and clinical covariates (Fig. 4A). We also assessed models that included different combinations of neurodegeneration/vascular and tau biomarkers (fig. S6). We compared each model’s predictive performance without and with gut microbiome features (species relative abundances).
The cohort was randomly split into training (n = 99; 30.3% preclinical) and validation (n = 65; 29.2% preclinical) cohorts (fig. S1), reflecting the prevalence of Aβ positivity in an independent non-cognitively impaired population of the same age group (32.1%, 95% CI [27.8, 36.4]) (51). We subjected the taxonomic abundance data from the training cohort to iterative feature selection, applying the Boruta algorithm (52) 100 times using a new seed in each iteration, and identified taxa that were selected in at least 25 of 100 iterations. This identified seven candidate taxa as important features for classification of healthy versus preclinical AD samples (Fig. 4 and fig. S6), which were retained for training and testing. Missing biomarker data were imputed using the k-nearest neighbor method and are summarized in fig. S5. PET tau exhibited the most missingness (20.7%), whereas all other biomarkers had <11% missingness.
Each model (with or without the seven selected taxa) was then trained on 100 random subsets (80% or n = 79 in each iteration) of the training cohort using 10-fold cross-validation in each case. After each of these 100 iterations, the models were tested against the validation cohort. We collated accuracy, sensitivity, and specificity of predictions made on the validation cohort (Fig. 4B and fig. S6B). Unexpectedly, in the comprehensive model trained on all biomarkers, including Aβ (“All”; Fig. 4A), gut microbiome features afforded small but significant improvements in classification accuracy (difference of means = 0.014, 95% CI [0.013, 0.015], P < 0.001, ANOVAs with Tukey’s post hoc test, additionally Bonferroni-adjusted across models) and specificity (0.047, 95% CI [0.043, 0.052], P < 0.001) (Table 2). When PET Aβ and CSF Aβ42/Aβ40 were omitted (“All − A”), inclusion of the selected taxa offered significant improvements in specificity (0.119, 95% CI [0.093, 0.145], P < 0.001), but not accuracy or sensitivity (Fig. 4B and Table 2). This pattern of improvements in accuracy or specificity with the addition of taxonomic features, at the cost of sensitivity, held in other models that included various combinations of neurodegeneration/vascular, tau, and genetic biomarkers (fig. S6B and table S9). In contrast, in models that omitted all AD biomarkers except for genetics (“CC + G”; Fig. 4A), or AD biomarkers together (“CC”), inclusion of taxonomic features significantly improved accuracy of predictions (CC + G: 0.048, 95% CI [0.042, 0.055]; CC: 0.075, 95% CI [0.067, 0.083]; P < 0.001 in each case) and sensitivity of predictions (CC + G: 0.046, 95% CI [0.036, 0.056]; CC: 0.117, 95% CI [0.105, 0.128]; P < 0.001 in each case), as well as specificity of predictions (0.053, 95% CI [0.026, 0.080], P = 0.002) in the case of the model CC + G (Fig. 4B).
Table 2.
Improvements in Random Forest classifier performance after incorporating gut microbiome features.
| Model | Metric | No microbiome data | Including selected taxa | Including selected taxa – no microbiome data | |||||
|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Difference of means | CI 95% lower | CI 95% upper | P | ||
| All biomarkers including Aβ | Accuracy | 0.985 | 0.004 | 0.999 | 0.006 | 0.014 | 0.013 | 0.015 | 5.77 × 10−13 |
| Specificity | 0.948 | 0.013 | 0.996 | 0.019 | 0.047 | 0.043 | 0.052 | 5.77 × 10−13 | |
| All biomarkers excluding Aβ | Specificity | 0.413 | 0.107 | 0.532 | 0.074 | 0.119 | 0.093 | 0.145 | 8.44 × 10−13 |
| Clinical covariates + genetic biomarkers | Accuracy | 0.706 | 0.024 | 0.755 | 0.023 | 0.048 | 0.042 | 0.055 | 5.77 × 10−13 |
| Sensitivity | 0.917 | 0.036 | 0.963 | 0.036 | 0.046 | 0.036 | 0.056 | 8.38 × 10−13 | |
| Specificity | 0.196 | 0.096 | 0.249 | 0.099 | 0.053 | 0.026 | 0.080 | 0.002 | |
| Clinical covariates only | Accuracy | 0.674 | 0.036 | 0.750 | 0.019 | 0.075 | 0.067 | 0.083 | 5.77 × 10−13 |
| Sensitivity | 0.850 | 0.051 | 0.967 | 0.024 | 0.117 | 0.105 | 0.128 | 5.77 × 10−13 | |
Mean accuracy, sensitivity, and specificity for Random Forest models trained on subsets of AD biomarkers, with or without gut microbiome features (selected MetaPhlAn3 taxa), are presented. Each model was trained on 100 random subsets of the training cohort. Shown are the mean performance metrics of those 100 models on the validation cohort. Models are included if they retained significant ANOVA P values after Bonferroni adjustment across all ANOVAs [groups: no microbiome data, including selected taxa (MetaPhlAn3)]. The corresponding differences of means and 95% confidence intervals (CIs) are reported. P values: Tukey’s post hoc test after ANOVA for each model, additionally adjusted using the Bonferroni method (see Fig. 4 and table S9).
Improvements in accuracy afforded by inclusion of taxonomic features increased in magnitude as more categories of AD biomarkers (Aβ, tau, neurodegeneration/vascular, and genetics) were omitted (Spearman’s ρ = 0.975, P = 0.005), suggesting that the utility of microbial features as an indicator of preclinical AD increased with greater scarcity of available data for established AD biomarkers. When comparing the importance of specific features in these models against their importance in corresponding null models trained on class label–shuffled data (pink distributions; Fig. 4C), six of seven taxonomic features were confirmed as important, along with participants’ age (models CC + G and CC) and APOE ε4 carrier status (CC + G).
DISCUSSION
The prevalence of AD continues to grow globally as life expectancies increase (53), but therapies remain elusive (14). Considerable data suggest that an interval of at least 10 years exists between the first deposition of Aβ plaques in the brain and the first clinical signs of impairment (9). This sequence forms the basis for the concept of preclinical AD (9), during which biomarkers (e.g., Aβ plaques detected by PiB or 18F-florbetapir (AV45) radioligand during PET imaging and CSF assays of Aβ42, Aβ40, and tau) can predict disease progression (54). Early detection of molecular hallmarks of AD pathology remains critical for implementing effective treatments (55).
We asked whether distinct gut microbiome profiles are present in the preclinical stage of AD, a point in disease progression when Aβ biomarker values are abnormal but cognitive impairment has not ensued. Gut microbiome signatures of preclinical AD, readily assayed in stool, could enhance early screening measures for AD risk and improve recruitment of cohorts at this critical stage of AD progression. We found differing microbiome composition and microbial functional potential at the preclinical stage of AD. Furthermore, gut sample PCoA coordinates correlated with PET Aβ and tau biomarkers (the earliest in the biomarker cascade), but not with markers of neurodegeneration. The association of gut features with the definitive molecular hallmarks of early AD pathology strengthens their potential utility as complementary early-in-progression predictive markers.
The microbial pathways most associated with preclinical AD status in regression models (l-arginine, l-ornithine, and 4-aminobutanoate degradation) share succinate as a product. Succinate, known largely as an intermediate of the tricarboxylic acid cycle, is also a bacterial metabolite produced in the gut that has been associated with obesity (56) and inflammatory bowel disease (57) and is increasingly appreciated as immunomodulatory (58–61), where gut-driven inflammation is associated with AD pathogenesis (62). In addition, succinate is a major precursor for the short-chain fatty acid (SCFA) propionate, which has previously been found to be elevated in symptomatic individuals with AD, as well as in AD mouse models, compared with healthy controls (63). The pathway most associated with healthy individuals (l-glutamate degradation V) produces the SCFA acetate, which not only has been observed to inhibit Aβ aggregation in vitro (64) and protect against cognitive impairment in mice (65) but also has been associated with an elevated Aβ SUVR in a human cohort (66).
Of the taxa significantly associated with preclinical AD status in regression models, Alistipes, Barnesiella, and Odoribacter were previously found in symptomatic individuals with AD (5). Distinct Bacteroides species were highly associated with preclinical AD and healthy groups (Bacteroides intestinalis and Bacteroides caccae, respectively), highlighting the importance of species-level associations. We did not observe a significant difference in the overall Bacteroidetes-to-Firmicutes ratio between healthy individuals and individuals with preclinical AD, in contrast to a comparison of healthy individuals and symptomatic individuals with AD (6). Capturing the Bacteroidetes-to-Firmicutes ratio longitudinally as an individual progresses from preclinical to symptomatic AD would help to elucidate whether this metric emerges with symptomatic AD. Methanobrevibacter smithii was associated with preclinical AD and was negatively correlated with fecal concentrations of butyrate, a SCFA (67) that attenuated Aβ plaque deposition and neuroinflammation in a mouse model of AD (68).
Because preclinical AD status is defined by Aβ burden, improvements in predictive performance of Random Forest classifiers afforded by taxonomic features were small in magnitude when the more difficult-to-obtain Aβ variables were included in the model (1.4 and 5.0% improvements in mean accuracy and specificity, respectively, for model All). In contrast, in models trained just on demographics, clinical covariates, and genetics (CC + G), inclusion of taxonomic features afforded 6.8 and 27.1% improvements in mean accuracy and specificity, respectively, whereas models trained on demographics and clinical covariates only (CC) saw improvements of 11.2 and 13.7% in mean accuracy and sensitivity with inclusion of taxonomic features. Gut microbiome features could enhance early screening measures to identify candidates for follow-up CSF or PET Aβ assays to verify preclinical AD status. Of the taxonomic features included in the models after feature selection (D. formicigenerans, Oscillibacter sp. 57_20, F. prausnitzii, C. catus, A. hadrus, M. stadtmanae, and R. lactaris), all were identified as significantly associated with preclinical AD or healthy status in negative binomial regression analyses. This agreement lends support for these taxa to be considered candidate markers in preclinical AD. Of these, F. prausnitzii, Oscillibacter sp. 57_20, and D. formicigenerans ranked as the three most important taxonomic variables in the models CC + G and CC. D. formicigenerans degrades mucin and may play an inflammatory role in patients with multiple sclerosis (69). Oscillibacter spp. were correlated with decreased colonic epithelial integrity in the context of a high-fat diet in mice (70). In contrast, Oscillibacter sp. 57_20 received a beneficial score in a recent large-cohort microbiome-wide association study (PREDICT 1) (71). Similarly, F. prausnitzii is typically considered an anti-inflammatory commensal (72) and is known to be enriched in non-AD individuals compared with those with AD dementia (5). These contradictions highlight the importance of host and environmental context, disease stage, and strain-specific effects when considering potential roles of the gut microbiome in disease (73, 74). In silico analyses of F. prausnitzii genomes have revealed two phylogroups that share less than 90% average nucleotide identity (75) and a high degree of genome plasticity (76). More recently, an approach assembling genomes from metagenomes identified 22 Faecalibacterium-like species-level genome bins, suggesting that the diversity of functional potential and anti-inflammatory phenotypes within Faecalibacterium spp. is not yet fully elucidated. Strain specificity of F. prausnitzii–mediated protective effects was recently demonstrated in a mouse model of AD (77).
Overall, the associations we report in this study between the gut microbiome and preclinical AD status or AD markers support the existence of an enteric neuroimmune axis in neurodegenerative disease (8). Here, we report that such associations are established in preclinical AD, potentially positioning at least a few of these microbial species in the causal chain. However, additional investigation is needed to validate these associations in broader preclinical AD cohorts, assess causality, and determine whether these associations extend to symptomatic AD or are succeeded by other gut microbiome or immune features concomitant with disease progression. The specificity of these associations also needs to be tested in a cohort of patients with non-AD dementias. Antecedent gut microbiome signatures of AD have the potential to complement the current AT(N) framework by improving accessibility and sensitivity of early screening measures, because stool is an easily obtained analyte, and sequencing costs continue to decrease (78). Blood biomarkers such as plasma Aβ42/Aβ40, p-tau-217, and neurofilament light have emerged as predictive biomarkers of cognitive decline and subsequent AD dementia in unimpaired elderly populations (79). Use of stool analytes could similarly reduce inequities in access to imaging technologies (80), and its acquisition is much less invasive than lumbar puncture to obtain CSF. Because stool can be acquired at home, its use could increase coverage of community testing programs, especially in people with poor access to medical settings (81). Improved early detection of AD risk may also increase enrollment in research studies at a critical juncture in AD progression, before the onset of neurodegeneration and cognitive decline, and inform the development of therapies to interdict this progression. Whereas mechanisms that govern the impact of the gut on AD severity and progression have not been fully elucidated, such efforts potentially could lead to gut microbiome–directed interventions that reverse or ameliorate AD pathology (82). For example, in a recent study, specific strains of F. prausnitzii from healthy participants reduced cognitive impairment in a murine model of brain amyloidosis (77). Similarly, dietary supplementation of the 5xFAD mouse model of AD with mannan oligosaccharide–modulated SCFA production by gut microbes suppressed neuroinflammation and alleviated cognitive impairment (68). In a phase 2 randomized trial, sodium oligomannate (a marine algae–derived oligosaccharide) improved outcomes in cognitive function in AD (22).
There are important limitations to our study and clear areas where further investigation is needed. First, whereas we account for them in our analyses, the intervals of neuroimaging, serum and CSF assessments, and stool samplings raise the possibility that true biomarker concentrations in participants at the time of stool sampling differed from the biomarker values used in these analyses. Whereas the rate of AD progression in our cohort is such that biomarker values are not expected to change significantly over the course of 3 years (83) (the interval at which Knight ADRC participants undergo neuroimaging and serum and CSF assessment), this estimation is at a population level, and it is possible that a specific participant may have traversed an inflection point in biomarker values. Similarly, the composition of the adult gut microbiome is generally stable (84, 85), but it is susceptible to perturbation in response to changes in environmental factors such as activity, medication, and aging (86–88). Future studies will benefit from synchronous biomarker assessments and stool sampling, as well as from longitudinal sampling of participants to account for natural intraparticipant variation in microbiome composition. In addition, preclinical AD status does not imply that an individual will necessarily progress to symptomatic AD. Long-term longitudinal tracking of participants with time-matched neuroimaging, biological and cognitive assessments, and stool sampling will enable the identification of individuals who progress to mild cognitive impairment or symptomatic AD. Corresponding analyses will determine whether gut features predict this transition or stabilization from progression. Second, whereas stool metagenome sequencing offers abundant information about taxonomic composition and functional potential of the gut microbiome, including strain level resolution, such investigations would be bolstered by broader multiomics approaches including metatranscriptomics to assay the repertoire of microbial genes that are actively expressed at the time of sampling, as well as metabolomics to profile the bioactive small molecules present in the gut. In particular, assaying metabolites that have been implicated in neurodegenerative disease, such as SCFAs and secondary bile salts, and corresponding correlation analyses with taxonomic composition data could strengthen mechanistic hypotheses about the role of specific gut taxa in AD. Similarly, clinical measures of gut health, including markers of systemic and enteric inflammation, as well as gut permeability, will strengthen our understanding of preclinical AD-associated gut dysbiosis and dysfunction. Longitudinal measurements will enable determination of whether microbiome dysbiosis precedes host enteric dysfunction or vice versa within the context of AD.
In summary, we report global and specific differences in the gut microbiome at the preclinical stage of AD. We further demonstrate that addition of gut microbiome features improved accuracy, sensitivity, and specificity of classifiers for preclinical AD. Microbiome markers in stool might complement early screening measures for preclinical AD and generate encouraging hypotheses about potential roles of the gut in AD progression. Last, microbially at-risk populations could open new opportunities for gut-directed interventions to interdict progression to clinical AD.
MATERIALS AND METHODS
Study design
The objective of this cross-sectional, observational study was to identify differences in the gut microbiome profiles of healthy participants (n = 115) and participants with preclinical AD (n = 49), as well as any correlations of gut microbiome features with AD biomarkers. Symptomatic participants were excluded (CDR > 0) (10). The AD biomarkers included in analyses were brain Aβ plaques calculated from PET imaging (on the Centiloid scale) (40, 89) using 11C PiB or AV45 depending on data availability or, if PiB and AV45 were both available, using the most recent value. Other biomarkers included tau measured by PET imaging (43), the Aβ42/Aβ40 ratio in CSF (41, 42), p-tau-181 and t-tau in CSF (41, 44), and MRI measures of white matter hyperintensities volume, hippocampus volume, and cortical signature (45). In addition, we included the polygenic risk score (46) and APOE ε4 carrier status. Participants for this stool study were recruited from existing longitudinal cohort studies (26–28) at the Knight ADRC at Washington University School of Medicine (WUSM) in St. Louis. The Knight ADRC cohort undergoes assessments including PET, MRI, and CSF collection via lumbar puncture; blood draws every 3 years; and clinical and cognitive testing every 3 years (participants younger than 65 years old) or annually (participants 65 and older) (27, 28). Biomarker and clinical measures from the most recently available neurological and cognitive assessments for each participant were used. Time elapsed between neurological and cognitive assessments and stool collections are summarized in table S1. Processing of stool samples was done blinded to AD status. All participants underwent identical procedures, so no group randomization was carried out. Sample sizes were not determined in advance.
Participants
Participants were recruited for this study from existing longitudinal cohort studies (26–28) at the Knight ADRC at WUSM, St. Louis from July 2019 to October 2021, primarily from the Adult Children Study (ACS), which deliberately seeks to enroll individuals who may have preclinical AD (26, 27). Eligibility to the ACS includes being healthy and being between 45 and 64 years of age at the time of enrollment, as well as either (i) having two parents who were never affected by AD and lived past 70 years of age or (ii) having a parent who developed AD before the age of 80. Genetic risk data for all participants are available in Table 1 (APOE ε4 carrier status) or in data file S1. Enrollees from existing Knight ADRC cohorts were approached for enrollment in this study in a randomized fashion. Participants ranged from 68 to 94 years of age. All participants were asymptomatic, with their most recent (within 3.8 months on average) CDR equal to 0. Preclinical AD was defined as CDR = 0 and Aβ positive. Aβ-positive was defined as Centiloid > 16.4, where Centiloid was calculated from PiB SUVR or AV45 SUVR depending on availability or most recent acquisition. If PET Aβ biomarkers were not available (n = 11 participants), then Aβ positivity was defined as a CSF Aβ42/Aβ40 ratio < 0.0673 (31, 32). Healthy was defined as CDR = 0 and Aβ negative (Centiloid ≤ 16.4 or CSF Aβ42/Aβ40 ratio ≥ 0.0673). CDR rating was performed by qualified clinicians in accordance with established scoring rules (10); dementia diagnostic criteria conformed to the National Institute on Aging–Alzheimer’s Association Work Group recommendations (90). Other than CDR = 0, additional exclusion criteria were as previously described for recruitment to the Knight ADRC (28).
Study protocols were approved by the WUSM Institutional Review Board, and all participants provided written informed consent to the use of clinical and genetic information for research purposes. Demographic, biomarker, and genetic data were extracted on 22 February 2022. Clinical metadata were extracted on 3 May 2022. Data were analyzed from 1 March 2022 to 18 January 2023.
Clinical assessment
All CDR scores were obtained from assessments by experienced clinicians trained in the use of the CDR. The CDR is used to determine whether dementia is present and, if so, to stage its severity. When using global scores, a CDR of 0 indicates that the individual is “cognitively normal,” whereas a CDR of 0.5 and a CDR of 1 indicate very mild and mild dementia, respectively (91). For this study, all participants had a CDR of 0.
Other clinical metadata (demographics and health information, including body mass index, diabetes, hypertension, history of drug use, etc.) were collected according to assessment protocols from existing longitudinal cohort studies (26–28) at the WUSM Knight ADRC, under which experienced clinicians conduct interviews with participants and a collateral source to obtain, in addition to cognitive evaluations, clinical metadata according to the Uniform Data Set protocol of the National Alzheimer’s Coordination Center (92, 93).
APOE ε4 status and polygenic risk scores
DNA samples were collected at enrollment and genotyped using either an Illumina 610 or OmniExpress chip. Genotyping methods were previously published (94). To control for effects of APOE ε4 on individuals in this analysis, APOE status was converted from a genotype to a binary variable. Participants either had at least one copy of the APOE ε4 allele (“APOE ε4 positive”) or had no copies of the allele (“APOE ε4 negative”). Polygenic risk scores were calculated at a genome-wide P value threshold (5.0 × 10−8) using Kunkle stage 1 and stage 2 combined summary statistics by excluding the APOE region on chr19 (95). PRSice-2 was used to obtain the risk scores, applying the default clumping parameters (--clump-p 1; --clump-r2 0.1; --clump-kb 250) (96).
MRI acquisition
Imaging was performed using a 3.0 Tesla Trio Siemens Biograph mMR (Erlangen, Germany) or 3.0 Tesla Siemens TIM Trio (Erlangen, Germany) scanner. High-resolution three-dimensional sagittal T1 magnetization prepared–rapid gradient echo anatomical images were acquired using the Alzheimer’s Disease Neuroimaging Initiative protocol: For the Siemens Biograph mMR, scanning parameters of repetition time (TR) = 2300 ms, time to echo (TE) = 2.95 ms, flip angle = 9°, 176 slices, acquisition matrix = 240 by 256, and voxel size = 1 mm by 1 mm by 1.2 mm; for the Siemens TIM Trio, scanning parameters of TR = 2400 ms, TE = 3.16 ms, flip angle = 8°, 176 slices, acquisition matrix = 256 by 256, and voxel size = 1 mm by 1 mm by 1 mm. T1-weighted scans were segmented with FreeSurfer 5.3. Previous work has identified the temporal (inferior, middle, and superior), parietal (inferior and superior), and entorhinal cortices; precuneus; and hippocampus as the regions that are most affected by disease and change the earliest (45). We converted volumes to z scores separately in the left and right hemispheres relative to the entire cohort and averaged them to obtain an “AD signature region.” The AD signature region creates a summary metric that succinctly describes brain volume atrophy due to AD (45).
PET imaging of Aβ and tau
Imaging studies were obtained at baseline and then every 3 years thereafter by the Knight ADRC. PET images were acquired within 2 years (mean = 0.6 ± 1.2 years) of MRI using the methodology previously described (97, 98). PET data were processed using the PET Unified Pipeline (github.com/ysu001/PUP), which uses regions of interest defined using the FreeSurfer version 5.3 (Martinos Center for Biomedical Imaging, Charlestown, MA, USA) Desikan-Killiany atlas. Data were transformed into SUVRs using cerebellar gray as a reference and partial volume corrected by calculating regional spread functions as part of a geometric transfer matrix framework. PET Aβ imaging was performed using either 11C PiB or AV45. The time window for quantification was 30 to 60 min postinjection for PiB and 50 to 70 min for AV45. Centiloids were used to harmonize measures from these two different tracers (40).
PET tau imaging was performed using 18F-flortaucipir (AV1451) with SUVRs calculated for the 80- to 100-min postinjection window. A summary measure of tauopathy—previously defined as the arithmetic mean of the amygdala, entorhinal cortex, inferior temporal, and lateral occipital regions based on FreeSurfer version 5.3 segmentation—was calculated for each participant (43).
CSF collection and analysis
CSF (10 to 20 ml) was collected by lumbar puncture using a 22-gauge Sprotte spinal needle (Geisingen, Germany). Lumbar punctures were performed in the morning after overnight fasting by a trained neurologist. CSF was aliquoted (500 μl) into polypropylene tubes and was free of visible blood contamination. After collection, samples were gently inverted and frozen at −80°C. CSF Aβ42, Aβ40, t-tau, and p-tau-181 were measured as previously described using an automated chemiluminescent enzyme immunoassay (LUMIPULSE G1200, Fujirebio, Malvern, PA, USA) (32). CSF Aβ42/Aβ40 ratios were calculated for this study.
Stool collection
Stool samples were collected from 20 December 2019 to 12 October 2021 under an established community sample acquisition protocol (99, 100); samples were produced at participants’ homes with standardized instructions and collection materials and transported the same day to WUSM in insulated packages by a commercial courier, at which point samples were frozen (−80°C) and aliquoted under barcode provenance until processing for bacterial DNA extraction.
Food logs and nutritional analyses
Twenty-four-hour food log questionnaires were sent out with stool collection kits. Participants self-reported all the food, drink, and any supplements consumed in the 24 hours before stool collection. Entry fields included the time and type of meal, food item, quantity, brand or restaurant, preparation, and condiments. Food logs were processed and entered by a registered and licensed dietitian into the Food Processor Nutrition Analysis software (ESHA Inc., Salem, OR). This tool was used to match food entries to a database of >146,000 food items, which encapsulates the U.S. Department of Agriculture (USDA) Agricultural Research Service Food and Nutrient Research Database for Dietary Studies and USDA SR-Legacy nutrient databases, and to generate 24-hour stool-matched nutritional profiles for each participant. These profiles included the total caloric intake, caloric intake by the macronutrient group, and percent recommended daily (% RDV) intake of essential nutrients (e.g., carbohydrates, total dietary fiber, vitamins, and minerals). % RDV was determined in the software on the basis of participant sex and age.
Metagenomic sequencing
One hundred to 200 mg of frozen stool were used as input to the DNeasy PowerSoil Pro Kit (QIAGEN, Germantown, MD, USA) to extract genomic DNA. DNA yields were measured using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). A total of 0.5 ng of genomic DNA per sample was used to create sequencing libraries with the Nextera Kit (Illumina, San Diego, CA, USA) (101). The libraries were pooled in equimolar concentrations and sequenced on a NovaSeq 6000 (2 × 150 base pairs) to obtain an average of 21.5 million reads per sample (SD, 8.5 million; min, 7.0 million; max, 58.0 million). The reads were demultiplexed by sequencing barcode. Illumina (Nextera-PE) adaptors were removed, and reads were quality-filtered and trimmed using Trimmomatic v0.38 (102), with parameters ILLUMINACLIP: Nextera-PE.fa:2:30:10:1:TRUE, LEADING:10, TRAILING:10, SLIDINGWINDOW:4:20, and MINLEN:60. Contaminating human reads were removed using DeconSeq v0.4.3 against hsref38 (103), and paired ends were repaired using repair.sh from bbtools v38.26 (sourceforge.net/projects/bbmap/).
Processed reads were used as input to MetaPhlAn3 v3.0.7 (35) to determine per-sample taxonomic relative abundances using clade-specific marker genes, as well as HUMAnN 3.0 v3.0.0a4 (35) to functionally profile the metagenomes. For rarefaction analysis, reads were incrementally subsampled (100 to 20 million) and reanalyzed with MetaPhlAn2. The R package phyloseq v1.38.0 (104) was used to estimate sample alpha diversities (richness) on the basis of count-transformed taxonomic abundance data, followed by pairwise Wilcoxon tests between all subsampling depths, with Benjamini-Hochberg adjustment. The read threshold was defined as the lowest read depth at which there was no significant difference in sample alpha diversities compared to any higher read depth. Samples that passed the empirically determined read threshold (5 million reads) were retained in downstream analyses (n = 164).
Statistical analyses
Statistical analyses were conducted in R v4.1.3, and visualizations were generated with ggplot2 v3.3.5 unless otherwise specified. Differences in demographics or clinical covariates between healthy and preclinical AD groups were determined by Student’s t or chi-square tests. Differences in nutritional intake between groups were tested individually for each nutritional component (fig. S2) by Student’s t test, with Benjamini-Hochberg correction across all comparisons. Output from MetaPhlAn3 and HUMAnN 3.0 along with sample metadata was used to generate phyloseq objects (phyloseq v1.38.0). The phylogenetic tree for the MetaPhlAn taxonomic database was obtained from the MetaPhlAn github repository (mpa_v30_CHOCOPhlAn_201901_species_tree.nwk) and incorporated into the MetaPhlAn3 phyloseq object to calculate between-sample UniFrac distances (36). The tree was visualized using iTOL v6.5.8 (105).
The phyloseq estimate_richness function was used to calculate sample alpha diversities (richness and the Shannon diversity index) from count-transformed taxonomic and pathway abundances (MetaPhlAn3 and HUMAnN 3.0 output, respectively). Lowly abundant MetaPhlAn3 taxa (with mean relative abundance across samples ≤ 0.1%) and HUMAnN 3.0 pathways (with mean relative abundance across samples ≤ 0.01%) were filtered out before downstream analyses. Vegan v2.5.7, as called in phyloseq, was used for PCoA and CAP (37) using unweighted UniFrac distances for MetaPhlAn3 data or binary Bray-Curtis dissimilarities (106) for HUMAnN 3.0 pathway data. Significant group differences were tested by PERMANOVA using between-sample distances or dissimilarities as implemented in vegan v2.5.7 with the adonis2 function, which adds terms sequentially. Terms included in PERMANOVAs were in the following order: age, APOE ε4 carrier status, diabetes, body mass index, hypertension, interval between stool collection and Aβ biomarker acquisition, and Aβ (preclinical AD) status. Significance of the constraints (the same terms included in PERMANOVAs) in CAP analyses was tested by ANOVA as implemented in vegan v2.5.7 (anova function). Group differences in principal coordinates were assessed by one-way ANOVA, with P values adjusted using the Benjamini-Hochberg method.
Pairwise Spearman correlations of AD biomarkers and gut microbiome features were visualized using the R package corrplot v0.92 (107), filtered to show only correlations with Benjamini-Hochberg–adjusted P < 0.05. Fixed effects linear regression analyses of AD biomarkers (PET Aβ, PET tau, and cortical signature) against PCoA axes from ordinations of taxonomic (MetaPhlAn3) or pathway (HUMAnN 3.0) abundance data were carried out using the lm and aov functions (stats v4.1.3). Model summaries are provided in tables S6 to S8. Linear regression models were considered statistically significant if ANOVAs against their respective null models resulted in Benjamini-Hochberg–adjusted P < 0.1, adjusted across the models tested.
To identify specific taxa and pathways significantly associated with preclinical AD status, MaAsLin2 v1.10.0 (108) was used to fit negative binomial models to count-transformed abundance data individually for the two data types. Because relative abundances of taxa and pathway data were preserved after count transformation, the normalization parameter was set to “NONE.” The remaining MaAsLin2 execution parameters were set to the following: standardize = “TRUE,” min_abundance = 0 (because abundance data were already filtered for lowly abundant taxa or pathways), min_prevalence = 0, transform = “NONE” (because count transformation occurred outside of the MaAsLin2 model executions), analysis_method = “NEGBIN,” and max_significance = 0.05. That is, significant features were required to have Benjamini-Hochberg–adjusted P < 0.05. Fixed effects included age, APOE ε4 carrier status, diabetes, body mass index, hypertension, interval between stool collection and Aβ biomarker acquisition, and Aβ (preclinical AD) status. For visualization, significant features were additionally filtered to include only those observed in at least 25 of 164 samples (15% of the cohort) and with a magnitude of the fitted coefficient > 0.15.
Training and testing of Random Forest classifiers for AD preclinical status were implemented through caret v6.0.86. The categorical variables sex, race, hypertension, diabetes, and APOE ε4 status were numerically encoded. The data were imputed using VIM v6.1.0, using the k-nearest neighbor method with k = 5. Data missingness is summarized both by feature and by combinations of features in fig. S5. Data were then combinatorially subsetted by AT(N) or vascular injury (white matter hyperintensities volume) + genetic biomarker categories (model definitions in Fig. 4A and fig. S6A). The demographic variables age, sex, race, and education (years) as well as the clinical covariates body mass index, hypertension, and diabetes were included as predictors in every model, as were time intervals between stool collection and neurological assessments. The cohort was randomly split 60:40 into training (n = 99) and validation (n = 65) cohorts (createDataPartition function from the caret package with random seed = 42). Within the training cohort, feature selection was carried out on taxonomic abundance data by applying the Boruta (v7.0.0) (52) feature selection pipeline 100× using a new seed in each iteration (set.seed = 1:100), with maxRuns = 500 in each iteration. From this, we identified taxa that had been selected in at least 25 of 100 iterations (n = 7 taxa). Continuous variables were z-scored (“center” and “scale” methods in the caret preProcess function). Then, Random Forest models were trained either including or excluding these feature-selected taxa. Each model was trained on 100 random subsets (80%; n = 79, seed = 1100) of the training cohort using 10-fold cross-validation in each case and the “Accuracy” test metric, with default parameters ntree = 500 and search = “grid” for mtry (the number of variables to be randomly sampled at each tree node). These 100 models were applied to predict the preclinical status of the retained validation cohort. Predictive performance of models including or excluding the feature-selected taxa was compared using accuracy, sensitivity, and specificity retained as performance metrics. To identify cases in which inclusion of taxonomic features improved the performance of the trained classifier, for each model (All, All − A, CC + G, CC, CC + NT, CC + GT, CC + GN, CC + N, and CC + T) and performance metric (accuracy, sensitivity, and specificity) pair, performance was compared with or without the selected taxa using one-way ANOVA. Model-performance metric pairs with Bonferroni-adjusted P < 0.05 were considered; comparisons were retested using Tukey’s test and subjected to a second round of Bonferroni adjustment (aov and TukeyHSD functions from stats v4.1.3). To assess the importance of specific features, models were retrained with random shuffling of training cohort class labels at each iteration. The resulting null distributions for feature importance were compared against their corresponding empirical distributions with Student’s t test and Benjamini-Hochberg adjustment for multiple hypothesis testing. R code and data for statistical analyses are available at https://doi.org/10.5281/zenodo.7964088.
Supplementary Material
Acknowledgments:
We thank the participants enrolled in this study for time and contributions, as well as the staff of the Biomarker, Clinical, Genetic, and Imaging Cores of the Knight ADRC. Furthermore, we thank the staff at the Edison Family Center for Genome Science and Systems Biology at Washington University in St. Louis School of Medicine: B. Dee, K. Matheny, and K. Page for administrative support; J. Hoisington-Lopez and M. L. Crosby for technical support in high-throughput sequencing; and E. Martin and B. Koebbe for computational support. We also thank the Genome Technology Access Center at the McDonnell Genome Institute for technical support in high-throughput sequencing. We also thank members of the Dantas Lab for helpful discussions.
Funding:
This research was supported by the Infection Disease Society of America Foundation (Microbial Pathogenesis in Alzheimer’s Disease Grant 2020 to G.D.), the National Institute on Aging (P01 AG026276 to J.C.M. and R01 AG057680-01A1 to B.M.A. and S.L.S.), the Brennan Fund (to B.M.A.), the Riney Fund (to B.M.A.), and the Washington University Digestive Diseases Research Core Center (NIDDK P30 DK052574, Biobank Core, to co-director P.I.T.).
Competing interests:
G.D. is a co-founder, holder of equity in, a consultant to, and a member of the Scientific Advisory Board of Viosera Therapeutics, which is developing combination antimicrobial therapy against bacterial pathogens. G.D. is a co-inventor on a patent assigned to Viosera Therapeutics. G.D. is a consultant to and a member of the Scientific Advisory Board of Pluton Biosciences and has consulted for SNIPR Technologies Ltd. in the last 5 years. P.I.T. is a holder of equity in, a consultant to, and a member of the Scientific Advisory Board of MediBeacon Inc. and is a co-inventor on patents assigned to MediBeacon; Chair of the Scientific Advisory Board of the AGA Center for Microbiome Research and Education (a paid position); a consultant to Temple University on waterborne enteric infections; a recipient of royalties from UpToDate; and an unpaid member of the Data Safety Monitoring Board of Inmunova. D.M.H. co-founded and is on the Scientific Advisory Board of C2N Diagnostics and consults for Genentech, Denali, Cajal Neurosciences, C2N Diagnostics, and Asteroid. Washington University receives research grants to the laboratory of D.M.H. from NextCure, Eli Lilly, Novartis, Ionis, and Denali. T.L.S.B. receives research funding from Siemens as well as technical support and materials from Avid Radiopharmaceuticals, Cerveau, and Life Molecular Imaging. T.L.S.B. also receives paid and unpaid consulting and has advisory roles for Siemens, Eli Lilly, Roche, Life Molecular Imaging, Biogen, and Eisai. A.M.F. was a member of the scientific advisory boards for Roche Diagnostics, Genentech, and Diadem and also previously consulted for DiamiR and Siemens Healthcare Diagnostics Inc. S.E.S. has analyzed data provided by C2N Diagnostics to Washington University and has served on a Scientific Advisory Board for Eisai. C.C. has received research support from GlaxoSmithKline and Eisai and is a member of the advisory board of Vivid Genomics and Circular Genomics and owns stocks. J.C.M. is a member of the Barcelona Brain Research Foundation Scientific Advisory Board, the Native Alzheimer Disease-Related Resource Center in Minority Aging Research External Advisory Board, the Cure Alzheimer’s Fund Research Strategy Council, and the Longer Life Foundation Board of Governors. J.C.M. was previously a member of the Diverse VCID (Vascular Contributions to Cognitive Impairment and Dementia) Observational Study Monitoring Board.
Footnotes
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. Source data for all figures are provided in data files S1 to S4. Shotgun metagenomic reads have been deposited to NCBI SRA under BioProject ID PRJNA798058. The software packages used in the study are free and open source. R scripts and data used for analyses are available for download at https://doi.org/10.5281/zenodo.7964088. Researchers may request data and materials from the Knight ADRC at https://knightadrc.wustl.edu/data-request-form/.
REFERENCES AND NOTES
- 1.Shreiner AB, Kao JY, Young VB, The gut microbiome in health and in disease. Curr. Opin. Gastroenterol 31, 69–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valdes AM, Walter J, Segal E, Spector TD, Role of the gut microbiota in nutrition and health. BMJ 361, k2179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hooper LV, Littman DR, Macpherson AJ, Interactions between the microbiota and the immune system. Science 336, 1268–1273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruiz L, Delgado S, Ruas-Madiedo P, Sánchez B, Margolles A, Bifidobacteria and their molecular communication with the immune system. Front. Microbiol 8, 2345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haran JP, Bhattarai SK, Foley SE, Dutta P, Ward DV, Bucci V, McCormick BA, Gilbert J, Faith J, Alzheimer’s disease microbiome is associated with dysregulation of the anti-inflammatory P-glycoprotein pathway. MBio 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, Bendlin BB, Rey FE, Gut microbiome alterations in Alzheimer’s disease. Sci. Rep 7, 13537 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belizario JE, Faintuch J, Microbiome and gut dysbiosis. Exp. Suppl 109, 459–476 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Pellegrini C, Antonioli L, Colucci R, Blandizzi C, Fornai M, Interplay among gut microbiota, intestinal mucosal barrier and enteric neuro-immune system: A common path to neurodegenerative diseases? Acta Neuropathol. 136, 345–361 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Shim YS, Morris JC, Biomarkers predicting Alzheimer’s disease in cognitively normal aging. J. Clin. Neurol 7, 60–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morris JC, The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43, 2412–2414 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Rizzetto L, Fava F, Tuohy KM, Selmi C, Connecting the immune system, systemic chronic inflammation and the gut microbiome: The role of sex. J. Autoimmun 92, 12–34 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Zhang R, Miller RG, Gascon R, Champion S, Katz J, Lancero M, Narvaez A, Honrada R, Ruvalcaba D, McGrath MS, Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol 206, 121–124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez-Ramos J, Song S, Sava V, Catlow B, Lin X, Mori T, Cao C, Arendash GW, Granulocyte colony stimulating factor decreases brain amyloid burden and reverses cognitive impairment in Alzheimer’s mice. Neuroscience 163, 55–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J, The gut microbiome alterations and inflammation-driven pathogenesis of alzheimer’s disease-a critical review. Mol. Neurobiol 56, 1841–1851 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim MS, Kim Y, Choi H, Kim W, Park S, Lee D, Kim DK, Kim HJ, Choi H, Hyun DW, Lee JY, Choi EY, Lee DS, Bae JW, Mook-Jung I, Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer’s disease animal model. Gut 69, 283–294 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, Xu S, Zhang H, Cui G, Chen X, Sun R, Wen H, Lerut JP, Kan Q, Li L, Zheng S, Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut 68, 1014–1023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, Beghini F, Manara S, Karcher N, Pozzi C, Gandini S, Serrano D, Tarallo S, Francavilla A, Gallo G, Trompetto M, Ferrero G, Mizutani S, Shiroma H, Shiba S, Shibata T, Yachida S, Yamada T, Wirbel J, Schrotz-King P, Ulrich CM, Brenner H, Arumugam M, Bork P, Zeller G, Cordero F, Dias-Neto E, Setubal JC, Tett A, Pardini B, Rescigno M, Waldron L, Naccarati A, Segata N, Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med 25, 667–678 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu Q, Hou Q, Huang S, Ou Q, Huo D, Vázquez-Baeza Y, Cen C, Cantu V, Estaki M, Chang H, Belda-Ferre P, Kim HC, Chen K, Knight R, Zhang J, Compositional and genetic alterations in Graves’ disease gut microbiome reveal specific diagnostic biomarkers. ISME J. 15, 3399–3411 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masi AC, Embleton ND, Lamb CA, Young G, Granger CL, Najera J, Smith DP, Hoffman KL, Petrosino JF, Bode L, Berrington JE, Stewart CJ, Human milk oligosaccharide DSLNT and gut microbiome in preterm infants predicts necrotising enterocolitis. Gut 70, 2273–2282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B, A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puurunen MK, Vockley J, Searle SL, Sacharow SJ, Phillips III JA, Denney WS, Goodlett BD, Wagner DA, Blankstein L, Castillo MJ, Charbonneau MR, Isabella VM, Sethuraman VV, Riese RJ, Kurtz CB, Brennan AM, Safety and pharmacodynamics of an engineered E. coli Nissle for the treatment of phenylketonuria: A first-in-human phase 1/2a study. Metabolism 3, 1125–1132 (2021). [DOI] [PubMed] [Google Scholar]
- 22.Wang T, Kuang W, Chen W, Xu W, Zhang L, Li Y, Li H, Peng Y, Chen Y, Wang B, Xiao J, Li H, Yan C, du Y, Tang M, He Z, Chen H, Li W, Lin H, Shi S, Bi J, Zhou H, Cheng Y, Gao X, Guan Y, Huang Q, Chen K, Xin X, Ding J, Geng M, Xiao S, A phase II randomized trial of sodium oligomannate in Alzheimer’s dementia. Alzheimer’s Res. Ther 12, 110 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luo J, Weng H, Morris JC, Xiong C, Minimizing the sample sizes of clinical trials on preclinical and early symptomatic stage of alzheimer disease. J. Prev Alzheimers Dis 5, 110–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Babulal GM, Ghoshal N, Head D, Vernon EK, Holtzman DM, Benzinger TLS, Fagan AM, Morris JC, Roe CM, Mood changes in cognitively normal older adults are linked to alzheimer disease biomarker levels. Am. J. Geriatr. Psychiatry 24, 1095–1104 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cairns NJ, Perrin RJ, Franklin EE, Carter D, Vincent B, Xie M, Bateman RJ, Benzinger T, Friedrichsen K, Brooks WS, Halliday GM, McLean C, Ghetti B, Morris JC; the Alzheimer Disease Neuroimaging Initiative; Dominantly Inherited Alzheimer Network, Neuropathologic assessment of participants in two multi-center longitudinal observational studies: The Alzheimer Disease Neuroimaging Initiative (ADNI) and the Dominantly Inherited Alzheimer Network (DIAN). Neuropathology 35, 390–400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Day GS, Cruchaga C, Wingo T, Schindler SE, Coble D, Morris JC, Association of acquired and heritable factors with intergenerational differences in age at symptomatic onset of alzheimer disease between offspring and parents with dementia. JAMA Netw. Open 2, e1913491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bollinger RM, Keleman A, Thompson R, Westerhaus E, Fagan AM, Benzinger TLS, Schindler SE, Xiong C, Balota D, Morris JC, Ances BM, Stark SL, Falls: A marker of preclinical Alzheimer disease: A cohort study protocol. BMJ Open 11, e050820 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris JC, Schindler SE, McCue LM, Moulder KL, Benzinger TLS, Cruchaga C, Fagan AM, Grant E, Gordon BA, Holtzman DM, Xiong C, Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol. 76, 264–273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, Madison C, Ayakta N, Ghosh PM, La Joie R, Arthur-Bentil SK, Vogel JW, Marks SM, Lehmann M, Rosen HJ, Reed B, Olichney J, Boxer AL, Miller BL, Borys E, Jin LW, Huang EJ, Grinberg LT, DeCarli C, Seeley WW, Jagust W, Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: Statistical and pathological evaluation. Brain 138 (Pt. 7), 2020–2033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schindler S, Li Y, Buckles VD, Gordon BA, Benzinger TLS, Wang G, Coble D, Klunk WE, Fagan AM, Holtzman DM, Bateman RJ, Morris JC, Xiong C, Predicting symptom onset in sporadic alzheimer disease with Amyloid PET. Neurology 97, e1823–e1834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dumurgier J, Schraen S, Gabelle A, Vercruysse O, Bombois S, Laplanche JL, Peoc’h K, Sablonnière B, Kastanenka KV, Delaby C, Pasquier F, Touchon J, Hugon J, Paquet C, Lehmann S, Cerebrospinal fluid amyloid-β 42/40 ratio in clinical setting of memory centers: A multicentric study. Alzheimer’s Res. Ther 7, 30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Volluz KE, Schindler SE, Henson RL, Xiong C, Gordon BA, Benzinger TLS, Holtzman DM, Morris JC, Fagan AM, Correspondence of CSF biomarkers measured by Lumipulse assays with amyloid PET. Alzheimers Dement. 17 (S5), e051085 (2021). [Google Scholar]
- 33.Singh RK, Chang HW, Yan D, Lee KM, Ucmak D, Wong K, Abrouk M, Farahnik B, Nakamura M, Zhu TH, Bhutani T, Liao W, Influence of diet on the gut microbiome and implications for human health. J. Transl. Med 15, 73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, Turnbaugh PJ, Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, Valles-Colomer M, Weingart G, Zhang Y, Zolfo M, Huttenhower C, Franzosa EA, Segata N, Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lozupone C, Knight R, UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol 71, 8228–8235 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson MJ, Willis TJ, Canonical analysis of principal coordinates: A useful method of constrained ordination for ecology. Ecology 84, 511–525 (2003). [Google Scholar]
- 38.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B, Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann. Neurol 55, 306–319 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Johnson KA, Minoshima S, Bohnen NI, Donohoe KJ, Foster NL, Herscovitch P, Karlawish JH, Rowe CC, Carrillo MC, Hartley DM, Hedrick S, Pappas V, Thies WH; Alzheimer’s Association; Society of Nuclear Medicine and Molecular Imaging; Amyloid Imaging Taskforce, Appropriate use criteria for amyloid PET: A report of the amyloid imaging task force, the society of nuclear medicine and molecular imaging, and the alzheimer’s association. Alzheimers Dement. 9, e-1–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Su Y, Flores S, Hornbeck RC, Speidel B, Vlassenko AG, Gordon BA, Koeppe RA, Klunk WE, Xiong C, Morris JC, Benzinger TLS, Utilizing the Centiloid scale in cross-sectional and longitudinal PiB PET studies. Neuroimage Clin. 19, 406–416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Craig-Schapiro R, Fagan AM, Holtzman DM, Biomarkers of Alzheimer’s disease. Neurobiol. Dis 35, 128–140 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vlassenko AG, McCue L, Jasielec MS, Su Y, Gordon BA, Xiong C, Holtzman DM, Benzinger TLS, Morris JC, Fagan AM, Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease. Ann. Neurol 80, 379–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mishra S, Gordon BA, Su Y, Christensen J, Friedrichsen K, Jackson K, Hornbeck R, Balota DA, Cairns NJ, Morris JC, Ances BM, Benzinger TLS, AV-1451 PET imaging of tau pathology in preclinical Alzheimer disease: Defining a summary measure. Neuroimage 161, 171–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM, Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch. Neurol 64, 343–349 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Benzinger TL, Hassenstab J, Blazey T, Owen C, Liu J, Fagan AM, Morris JC, Ances BM, Spatially distinct atrophy is linked to β-amyloid and tau in preclinical Alzheimer disease. Neurology 84, 1254–1260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cruchaga C, del-Aguila JL, Saef B, Black K, Fernandez MV, Budde J, Ibanez L, Deming Y, Kapoor M, Tosto G, Mayeux RP, Holtzman DM, Fagan AM, Morris JC, Bateman RJ, Goate AM; Dominantly Inherited Alzheimer Network (DIAN); Disease Neuroimaging Initiative (ADNI); NIA-LOAD family study, Harari O, Polygenic risk score of sporadic late-onset Alzheimer’s disease reveals a shared architecture with the familial and early-onset forms. Alzheimers Dement. 14, 205–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jack CR Jr., Wiste HJ, Therneau TM, Weigand SD, Knopman DS, Mielke MM, Lowe VJ, Vemuri P, Machulda MM, Schwarz CG, Gunter JL, Senjem ML, Graff-Radford J, Jones DT, Roberts RO, Rocca WA, Petersen RC, Associations of Amyloid, Tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 321, 2316–2325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weigand AJ, Edwards L, Thomas KR, Bangen KJ, Bondi MW; for the Alzheimer’s Disease Neuroimaging Initiative, Comprehensive characterization of elevated tau PET signal in the absence of amyloid-beta. Brain Commun. 4, fcac272 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ, Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, Fagan AM, Holtzman DM, Mintun MA, Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch. Neurol 66, 1469–1475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts RO, Aakre JA, Kremers WK, Vassilaki M, Knopman DS, Mielke MM, Alhurani R, Geda YE, Machulda MM, Coloma P, Schauble B, Lowe VJ, Jack CR Jr., Petersen RC, Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population-based setting. JAMA Neurol. 75, 970–979 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kursa MB, Rudnicki WR, Feature selection with the Boruta package. J. Stat. Softw 36, 1–13 (2010). [Google Scholar]
- 53.Brown GC, Living too long: The current focus of medical research on increasing the quantity, rather than the quality, of life is damaging our health and harming the economy. EMBO Rep. 16, 137–141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, Liu E, Molinuevo JL, Montine T, Phelps C, Rankin KP, Rowe CC, Scheltens P, Siemers E, Snyder HM, Sperling R; Contributors, Elliott C, Masliah E, Ryan L, Silverberg N, NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 14, 535–562 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobs HIL, Becker JA, Kwong K, Engels-Domínguez N, Prokopiou PC, Papp KV, Properzi M, Hampton OL, d’Oleire Uquillas F, Sanchez JS, Rentz DM, el Fakhri G, Normandin MD, Price JC, Bennett DA, Sperling RA, Johnson KA, In vivo and neuropathology data support locus coeruleus integrity as indicator of Alzheimer’s disease pathology and cognitive decline. Sci. Transl. Med 13, eabj2511 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Serena C, Ceperuelo-Mallafré V, Keiran N, Queipo-Ortuño MI, Bernal R, Gomez-Huelgas R, Urpi-Sarda M, Sabater M, Pérez-Brocal V, Andrés-Lacueva C, Moya A, Tinahones FJ, Fernández-Real JM, Vendrell J, Fernández-Veledo S, Elevated circulating levels of succinate in human obesity are linked to specific gut microbiota. ISME J. 12, 1642–1657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macias-Ceja DC, Ortiz-Masiá D, Salvador P, Gisbert-Ferrándiz L, Hernández C, Hausmann M, Rogler G, Esplugues JV, Hinojosa J, Alós R, Navarro F, Cosin-Roger J, Calatayud S, Barrachina MD, Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol. 12, 178–187 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LAJ, Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fremder M, Kim SW, Khamaysi A, Shimshilashvili L, Eini-Rider H, Park IS, Hadad U, Cheon JH, Ohana E, A transepithelial pathway delivers succinate to macrophages, thus perpetuating their pro-inflammatory metabolic state. Cell Rep. 36, 109521 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Banerjee A, Herring CA, Chen B, Kim H, Simmons AJ, Southard-Smith AN, Allaman MM, White JR, Macedonia MC, Mckinley ET, Ramirez-Solano MA, Scoville EA, Liu Q, Wilson KT, Coffey RJ, Washington MK, Goettel JA, Lau KS, Succinate produced by intestinal microbes promotes specification of tuft cells to suppress ileal inflammation. Gastroenterology 159, 2101–2115.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fernández-Veledo S, Vendrell J, Gut microbiota-derived succinate: Friend or foe in human metabolic diseases? Rev. Endocr. Metab. Disord 20, 439–447 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kowalski K, Mulak A, Brain-gut-microbiota axis in Alzheimer’s disease. J. Neurogastroenterol. Motil 25, 48–60 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Killingsworth J, Sawmiller D, Shytle RD, Propionate and Alzheimer’s disease. Front. Aging Neurosci 12, 580001 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM, Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev. Neurother 18, 83–90 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng H, Xu P, Jiang Q, Xu Q, Zheng Y, Yan J, Ji H, Ning J, Zhang X, Li C, Zhang L, Li Y, Li X, Song W, Gao H, Depletion of acetate-producing bacteria from the gut microbiota facilitates cognitive impairment through the gut-brain neural mechanism in diabetic mice. Microbiome 9, 145 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marizzoni M, Cattaneo A, Mirabelli P, Festari C, Lopizzo N, Nicolosi V, Mombelli E, Mazzelli M, Luongo D, Naviglio D, Coppola L, Salvatore M, Frisoni GB, Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s disease. J. Alzheimers Dis 78, 683–697 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Abell GCJ, Conlon MA, McOrist AL, Methanogenic archaea in adult human faecal samples are inversely related to butyrate concentration. Microb. Ecol. Health Dis 18, 154–160 (2006). [Google Scholar]
- 68.Liu Q, Xi Y, Wang Q, Liu J, Li P, Meng X, Liu K, Chen W, Liu X, Liu Z, Mannan oligosaccharide attenuates cognitive and behavioral disorders in the 5xFAD Alzheimer’s disease mouse model via regulating the gut microbiota-brain axis. Brain Behav. Immun 95, 330–343 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Vacca M, Celano G, Calabrese FM, Portincasa P, Gobbetti M, de Angelis M, The controversial role of human gut lachnospiraceae. Microorganisms 8, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lam YY, Ha CWY, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, Cook DI, Hunt NH, Caterson ID, Holmes AJ, Storlien LH, Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLOS ONE 7, e34233 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Asnicar F, Berry SE, Valdes AM, Nguyen LH, Piccinno G, Drew DA, Leeming E, Gibson R, le Roy C, Khatib HA, Francis L, Mazidi M, Mompeo O, Valles-Colomer M, Tett A, Beghini F, Dubois L, Bazzani D, Thomas AM, Mirzayi C, Khleborodova A, Oh S, Hine R, Bonnett C, Capdevila J, Danzanvilliers S, Giordano F, Geistlinger L, Waldron L, Davies R, Hadjigeorgiou G, Wolf J, Ordovás JM, Gardner C, Franks PW, Chan AT, Huttenhower C, Spector TD, Segata N, Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat. Med 27, 321–332 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottière HM, Doré J, Marteau P, Seksik P, Langella P, Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U.S.A 105, 16731–16736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng D, Liwinski T, Elinav E, Interaction between microbiota and immunity in health and disease. Cell Res. 30, 492–506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yan Y, Nguyen LH, Franzosa EA, Huttenhower C, Strain-level epidemiology of microbial communities and the human microbiome. Genome Med. 12, 71 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Siles M, Duncan SH, Garcia-Gil LJ, Martinez-Medina M, Faecalibacterium prausnitzii: From microbiology to diagnostics and prognostics. ISME J. 11, 841–852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fitzgerald CB, Shkoporov AN, Sutton TDS, Chaplin AV, Velayudhan V, Ross RP, Hill C, Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genomics 19, 931 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ueda A, Shinkai S, Shiroma H, Taniguchi Y, Tsuchida S, Kariya T, Kawahara T, Kobayashi Y, Kohda N, Ushida K, Kitamura A, Yamada T, Identification of Faecalibacterium prausnitzii strains for gut microbiome-based intervention in Alzheimer’s-type dementia. Cell Rep. Med 2, 100398 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wetterstrand K, DNA sequencing costs: Data from the NHGRI Genome Sequence Program (GSP) (2020); www.genome.gov/sequencingcostsdata [accessed 17 October 2021].
- 79.Cullen NC, Leuzy A, Janelidze S, Palmqvist S, Svenningsson AL, Stomrud E, Dage JL, Mattsson-Carlgren N, Hansson O, Plasma biomarkers of Alzheimer’s disease improve prediction of cognitive decline in cognitively unimpaired elderly populations. Nat. Commun 12, 3555 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gallach M, Lette MM, Abdel-Wahab M, Giammarile F, Pellet O, Paez D, Addressing global inequities in positron emission tomography-computed tomography (PET-CT) for cancer management: A statistical model to guide strategic planning. Med. Sci. Monit 26, e926544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McDonald D, Hyde E, Debelius JW, Morton JT, Gonzalez A, Ackermann G, Aksenov AA, Behsaz B, Brennan C, Chen Y, DeRight Goldasich L, Dorrestein PC, Dunn RR, Fahimipour AK, Gaffney J, Gilbert JA, Gogul G, Green JL, Hugenholtz P, Humphrey G, Huttenhower C, Jackson MA, Janssen S, Jeste DV, Jiang L, Kelley ST, Knights D, Kosciolek T, Ladau J, Leach J, Marotz C, Meleshko D, Melnik AV, Metcalf JL, Mohimani H, Montassier E, Navas-Molina J, Nguyen TT, Peddada S, Pevzner P, Pollard KS, Rahnavard G, Robbins-Pianka A, Sangwan N, Shorenstein J, Smarr L, Song SJ, Spector T, Swafford AD, Thackray VG, Thompson LR, Tripathi A, Vázquez-Baeza Y, Vrbanac A, Wischmeyer P, Wolfe E, Zhu Q; The American Gut Consortium, Knight R, American Gut: An open platform for citizen science microbiome research. mSystems 3, e00031–18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Seo D-O, O’Donnell D, Jain N, Ulrich JD, Herz J, Li Y, Lemieux M, Cheng J, Hu H, Serrano JR, Bao X, Franke E, Karlsson M, Meier M, Deng S, Desai C, Dodiya H, Lelwala-Guruge J, Handley SA, Kipnis J, Sisodia SS, Gordon JI, Holtzman DM, ApoE isoform– and microbiota-dependent progression of neurodegeneration in a mouse model of tauopathy. Science 379, eadd1236 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hadjichrysanthou C, Evans S, Bajaj S, Siakallis LC, McRae-McKee K, de Wolf F, Anderson RM; Alzheimer’s Disease Neuroimaging Initiative, The dynamics of biomarkers across the clinical spectrum of Alzheimer’s disease. Alzheimers Res. Ther 12, 74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R, Diversity, stability and resilience of the human gut microbiota. Nature 489, 220–230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mehta RS, Abu-Ali GS, Drew DA, Lloyd-Price J, Subramanian A, Lochhead P, Joshi AD, Ivey KL, Khalili H, Brown GT, DuLong C, Song M, Nguyen LH, Mallick H, Rimm EB, Izard J, Huttenhower C, Chan AT, Stability of the human faecal microbiome in a cohort of adult men. Nat. Microbiol 3, 347–355 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Davenport ER, Mizrahi-Man O, Michelini K, Barreiro LB, Ober C, Gilad Y, Seasonal variation in human gut microbiome composition. PLOS ONE 9, e90731 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koliada A, Moseiko V, Romanenko M, Piven L, Lushchak O, Kryzhanovska N, Guryanov V, Vaiserman A, Seasonal variation in gut microbiota composition: Cross-sectional evidence from Ukrainian population. BMC Microbiol. 20, 100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fassarella M, Blaak EE, Penders J, Nauta A, Smidt H, Zoetendal EG, Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 70, 595–605 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Joshi AD, Pontecorvo MJ, Clark CM, Carpenter AP, Jennings DL, Sadowsky CH, Adler LP, Kovnat KD, Seibyl JP, Arora A, Saha K, Burns JD, Lowrey MJ, Mintun MA, Skovronsky DM; Florbetapir F 18 Study Investigators, Performance characteristics of amyloid PET with florbetapir F 18 in patients with alzheimer’s disease and cognitively normal subjects. J. Nucl. Med 53, 378–384 (2012). [DOI] [PubMed] [Google Scholar]
- 90.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr., Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH, The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 7, 263–269 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morris JC, Clinical dementia rating: A reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int. Psychogeriatr 9 (Suppl 1), 173–176(1997). [DOI] [PubMed] [Google Scholar]
- 92.Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA, The Uniform Data Set (UDS): Clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis. Assoc. Disord 20, 210–216 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Weintraub S, Salmon D, Mercaldo N, Ferris S, Graff-Radford NR, Chui H, Cummings J, DeCarli C, Foster NL, Galasko D, Peskind E, Dietrich W, Beekly DL, Kukull WA, Morris JC, The Alzheimer’s Disease Centers’ Uniform Data Set (UDS): The neuropsychologic test battery. Alzheimer Dis. Assoc. Disord 23, 91–101 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM, Benitez BA, Jeng AT, Skorupa T, Carrell D, Bertelsen S, Bailey M, McKean D, Shulman JM, de Jager PL, Chibnik L, Bennett DA, Arnold SE, Harold D, Sims R, Gerrish A, Williams J, van Deerlin V, Lee VM, Shaw LM, Trojanowski JQ, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD, Peskind ER, Galasko D, Fagan AM, Holtzman DM, Morris JC; GERAD Consortium; Alzheimer’s Disease Neuroimaging Initiative (ADNI); Alzheimer Disease Genetic Consortium (ADGC), Goate AM, GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron 78, 256–268 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier J-G, Harold D, Fitzpatrick AL, Valladares O, Moutet M-L, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Zompo MD, Fox NC, Choi S-H, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernández I, Kamboh MI, Brundin RM, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT Jr., Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Uitterlinden AGA, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, St George-Hyslop P, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu C-K, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, Guinness BM, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, Quillin AM, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, De Carli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin L-W, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, La Ferla FM, Jöckel K-H, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann H-E, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nöthen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossù P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu C-E, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues JF, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz JA, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, O’Donovan MC, De Stefano AL, Jones L, Haines JL, Deleuze J-F, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang L-S, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert J-C, Pericak-Vance MA; Alzheimer Disease Genetics Consortium (ADGC); The European Alzheimer’s Disease Initiative (EADI); Cohorts for Heart, Aging Research in Genomic Epidemiology Consortium (CHARGE); Genetic, Environmental Risk in AD/Defining Genetic, Polygenic, Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES), Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Choi SW, O’Reilly PF, PRSice-2: Polygenic Risk Score software for biobank-scale data. Gigascience 8, giz082 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, Bateman RJ, Cairns NJ, Aldea P, Cash L, Christensen JJ, Friedrichsen K, Hornbeck RC, Farrar AM, Owen CJ, Mayeux R, Brickman AM, Klunk W, Price JC, Thompson PM, Ghetti B, Saykin AJ, Sperling RA, Johnson KA, Schofield PR, Buckles V, Morris JC, Benzinger TLS; Dominantly Inherited Alzheimer Network, Partial volume correction in quantitative amyloid imaging. Neuroimage 107, 55–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, Blazey TM, Christensen JJ, Vora S, Morris JC, Mintun MA, Benzinger TLS, Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLOS ONE 8, e73377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baumann-Dudenhoeffer AM, D’Souza AW, Tarr PI, Warner BB, Dantas G, Infant diet and maternal gestational weight gain predict early metabolic maturation of gut microbiomes. Nat. Med 24, 1822–1829 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gasparrini AJ, Wang B, Sun X, Kennedy EA, Hernandez-Leyva A, Ndao IM, Tarr PI, Warner BB, Dantas G, Persistent metagenomic signatures of early-life hospitalization and antibiotic treatment in the infant gut microbiota and resistome. Nat. Microbiol 4, 2285–2297 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Baym M, Kryazhimskiy S, Lieberman TD, Chung H, Desai MM, Kishony R, Inexpensive multiplexed library preparation for megabase-sized genomes. PLOS ONE 10, e0128036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bolger AM, Lohse M, Usadel B, Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schmieder R, Edwards R, Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLOS ONE 6, e17288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McMurdie PJ, Holmes S, phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLOS ONE 8, e61217 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Letunic I, Bork P, Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bray JR, Curtis JT, An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr 27, 325–349 (1957). [Google Scholar]
- 107.Wei T, Simko V, Levy M, Xie Y, Jin Y, Zemla J, Freidank M, Cai J, Protivinsky T, R package “corrplot”: Visualization of a correlation matrix (2017). p. R package version 0.84.
- 108.Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, Chatterjee S, Thompson KN, Wilkinson JE, Subramanian A, Lu Y, Waldron L, Paulson JN, Franzosa EA, Bravo HC, Huttenhower C, Multivariable association discovery in population-scale meta-omics studies. PLOS Comput. Biol 17, e1009442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. Source data for all figures are provided in data files S1 to S4. Shotgun metagenomic reads have been deposited to NCBI SRA under BioProject ID PRJNA798058. The software packages used in the study are free and open source. R scripts and data used for analyses are available for download at https://doi.org/10.5281/zenodo.7964088. Researchers may request data and materials from the Knight ADRC at https://knightadrc.wustl.edu/data-request-form/.