

Abstract
The cervicovaginal microbiome is highly associated with women’s health with microbial communities dominated by Lactobacillus spp. being considered optimal. Conversely, a lack of lactobacilli and a high abundance of strict and facultative anaerobes including Gardnerella vaginalis, have been associated with adverse reproductive outcomes. However, the molecular pathways modulated by microbe interactions with the cervicovaginal epithelia remain unclear. Using RNA-sequencing, we characterize the in vitro cervicovaginal epithelial transcriptional response to different vaginal bacteria and their culture supernatants. We showed that G. vaginalis upregulated genes were associated with an activated innate immune response including anti-microbial peptides and inflammasome pathways, represented by NLRP3-mediated increases in caspase-1, IL-1β and cell death. Cervicovaginal epithelial cells exposed to L. crispatus showed limited transcriptomic changes, while exposure to L. crispatus culture supernatants resulted in a shift in the epigenomic landscape of cervical epithelial cells. ATAC-sequencing confirmed epigenetic changes with reduced chromatin accessibility. This study reveals new insight into host-microbe interactions in the lower reproductive tract and suggest potential therapeutic strategies leveraging the vaginal microbiome to improve reproductive health.
Keywords: cervix, epithelial cells, lactobacillus crispatus, gardnerella vaginalis, RNA-seq, anti-microbial peptides, NLRP3 inflammasome, ATAC-seq, chromatin, women’s health
Introduction
The female lower reproductive tract is comprised of a complex ecosystem made up of host epithelial and immune cells, microorganisms including the microbiome (bacteria) 1, mycobiome (fungi) 2, virome (viruses) 3,4 and their metabolites. The cervicovaginal microbiome has become the focus of many studies due to its highly integrated and complex role in reproductive health and disease. Using high throughput 16S rRNA gene sequencing, the vaginal microbiota composition has been well characterized in both non-pregnant and pregnant individuals 5–7. The vaginal microbiome has traditionally been defined by the presence or absence of Lactobacillus spp. 5,8,9. Cervicovaginal microbial communities dominated by Lactobacillus spp. are generally considered optimal and associated with positive reproductive health outcomes, while those lacking lactobacilli and comprising a wide array of strict and facultative anaerobes are associated with a diverse spectrum of adverse gynecological and reproductive outcomes including infertility 10,11, sexually transmitted infections (STIs) (human papilloma virus (HPV) 12 and human immunodeficiency virus (HIV)) 13, and pregnancy complications (preterm birth) 14,15. Although the cervicovaginal microbiome is less taxonomically diverse than those found at other body sites, the identification of con-specific genotypes cohabitating in the vaginal microbiome adds to the complexity of these communities 16. Despite these advances in vaginal microbiome characterization and its association with clinical outcomes, there is a paucity of data regarding the precise mechanisms by which vaginal microbes modulate the functioning of host epithelia and promote adverse health outcomes.
The vaginal microbiome interacts with the epithelial barriers of the cervicovaginal space. The cervicovaginal epithelial barrier is unique as the epithelial cells that line this space have different embryological origins resulting in distinct cell-specific functions including mucus production 17–20. The cervicovaginal epithelial barrier acts as the primary site of entry for invading pathogens and disruption of the barrier is associated with adverse health outcomes including increased risk of STI (e.g., chlamydia, gonorrhea. HIV) acquisition 21,22. Undoubtedly, revealing the molecular mechanisms by which vaginal microbes modulate host responses in the lower reproductive tract is necessary to understand how vaginal microbial communities drive reproductive health and disease. Therefore, the objectives of this study were to 1) perform discovery-based RNA-sequencing to identify genes and functional pathways in cervical and vaginal epithelial cells that are altered after exposure to G. vaginalis or L. crispatus and their culture supernatants, 2) elucidate and speciate the immune pathways activated by G. vaginalis and 3) identify molecular mechanisms by which L. crispatus optimizes cervical and vaginal epithelial function.
Results
Cervicovaginal epithelial cell gene transcription and associated functional pathways are differentially modulated by L. crispatus, G. vaginalis and their culture supernatants
Exposure of cervical and vaginal epithelial cells to live L. crispatus or G. vaginalis or their bacteria-free culture supernatants resulted in significant differences in gene expression profiles. In ectocervical, endocervical and vaginal epithelial cells, PCA plots show distinctive clustering by bacterial exposure and indicates the greatest differences (Fig. 1A–C) between exposures. Exposure to L. crispatus culture supernatants and live G. vaginalis were noted to be most distant from their respective controls (NYCIII for culture supernatants and NTC for live bacteria) (Fig. 1A–C). Gene expression profiles appeared to be epithelial cell specific as exposure to G. vaginalis culture supernatant resulted in clustering that was different from the NYCIII control in endocervical and vaginal cells that was not seen in ectocervical cells (Fig. 1A–C). To avoid false discovery, we then identified differentially expressed genes with an adjusted p-value < 0.05 and a Log2 fold change of greater than 1 or less than − 1 (Supplemental Table 1). Using these criteria, in all three cell types, the number of differentially expressed genes is highest after exposure to L. crispatus culture supernatants followed by live G. vaginalis and G. vaginalis supernatants (Fig. 1D–F). In contrast, very few genes were differentially expressed in all three epithelial cell types after exposure to live L. crispatus (Fig. 1G). Figure 1G–J depicts the number of overlapping differentially expressed genes between cervicovaginal cell types for each bacterial exposure. For G. vaginalis exposures (live or supernatant), endocervical and vaginal epithelial cells have the highest number of overlapping differentially expressed genes, while exposure to L. crispatus culture supernatants illicit the most overlapping differentially expressed genes in ectocervical and endocervical epithelial cells (Supplemental Table 2). Additional comparisons were performed within each cervicovaginal epithelial cell type to identify commonly modulated genes by these different bacterial exposures (Fig. 1K–M, Supplemental Table 3). From these analyses, we were able to identify unique genes where expression was altered by both live G. vaginalis and L. crispatus culture supernatant but were differentially regulated; thus, demonstrating specific molecular effects of these bacterial exposures on regulating cervicovaginal epithelial cell transcription (Supplemental Table 4, 5, 6). In summary, genes that were upregulated by exposure to G. vaginalis and downregulated by L. crispatus point towards a pro-inflammatory response. On the other hand, those upregulated by exposure to L. crispatus and downregulated by G. vaginalis seem to be involved in transcription, epigenetic modifications, cell-cell adherence, and anti-inflammatory functions.
Figure 1.

RNA-seq identified differentially expressed genes in cervicovaginal epithelial cells after 24 hr exposure to live bacteria or culture supernatants from L. crispatus or G. vaginalis. Principle component analysis (PCA) plots showing gene expression modulation in ectocervical (A), endocervical (B) and vaginal (C) cells exposed to either live G. vaginalis (vs NTC control) or L. crispatus culture supernatants (vs NYCIII control). The total number of differentially expressed genes (adj. p<0.05, Log2FoldChange ≥ 1 and ≤ −1) in each exposure group by cell line (D-F). The number of overlapping differentially expressed genes between cervicovaginal cell types for each bacterial exposure (G-J). The number of overlapping differentially expressed genes between bacterial exposures within each cervicovaginal cell types (K-M).
Functional pathway analysis using Gene Ontology terms identified specific pathways of interest between bacterial exposures. Live G. vaginalis and G. vaginalis culture supernatant upregulated genes were mostly associated with inflammation functional pathways (Fig. 2A, 2B–D). Exposure to L. crispatus culture supernatant was associated with upregulation of transcriptional functional pathways including histone modifications, RNA polymerase II transcription and DNA binding (Fig. 2A, 2E–G). Live L. crispatus modulated the expression of too few genes to perform functional pathway analysis. Since 1) live G. vaginalis and its culture supernatant modulated the same functional pathways and 2) L. crispatus culture supernatant resulted in the highest number of differentially expressed genes while live L. crispatus had a minimal effect on epithelial cell gene expression, we chose to focus on exposures of live G. vaginalis and L. crispatus culture supernatant for further analysis.
Figure 2.

Differential clustering of significant differentially expressed genes (adj. p<0.05, Log2FoldChange ≥ 1 and ≤ −1) between exposure groups and across cervicovaginal cell lines reveal modulation of functional pathways (A). Functional pathway analysis (B-G) of RNA-seq data for ectocervical (B, E), endocervical (C, F) and vaginal (D, G) epithelial cells exposed to live G. vaginalis or L. crispatus culture supernatants.
Live G. vaginalis and its culture supernatant differentially up-regulate anti-microbial peptide gene expression and increase protein levels
RNA-seq functional pathway analysis identified “defense response to a bacterium” along with various inflammation-based pathways as Gene Ontology terms of significance after live G. vaginalis exposure (Fig. 2B–D). The specific genes associated with these pathways included the anti-microbial peptides, Chemokine Ligand 20 (CCL20), Secretory Leukocyte Peptidase Inhibitor (SLPI), Lipocalin 2 (LCN2) and S100 Calcium Binding Protein 8 (S100A8/A9, Calgranulin). Our data show that all four of these genes were significantly upregulated (adjusted p < 0.05) by live G. vaginalis exposure in ectocervical and endocervical cells with only CCL20 and SLPI being upregulated in vaginal cells (Fig. 3A, 3E, 3I). After exposure to G. vaginalis culture supernatants, CCL20 was the only transcript upregulated in all three cell lines (adjusted p < 0.05, Fig. 3B, 3F, 3J). In ectocervical cells, in addition to CCL20, S100A8 gene expression was upregulated after G. vaginalis culture supernatant exposure (adjusted p < 0.05, Fig. 3B). In endocervical cells, all four anti-microbial peptide genes were significantly upregulated by G. vaginalis culture supernatant (adjusted p < 0.05, Fig. 3F). Only expression of CCL20 was upregulated in vaginal cells after G. vaginalis culture supernatant exposure (adjusted p < 0.05, Fig. 3J). ELISAs for these antimicrobial peptides confirmed overexpression of CLL20 and S100A8 (both p < 0.05), but not SLPI and LCN2, which remained unchanged, after exposure to G. vaginalis (Fig. 3C, 3G, 3K) and G. vaginalis culture supernatants (Fig. 3D, 3H, 3L).
Figure 3.
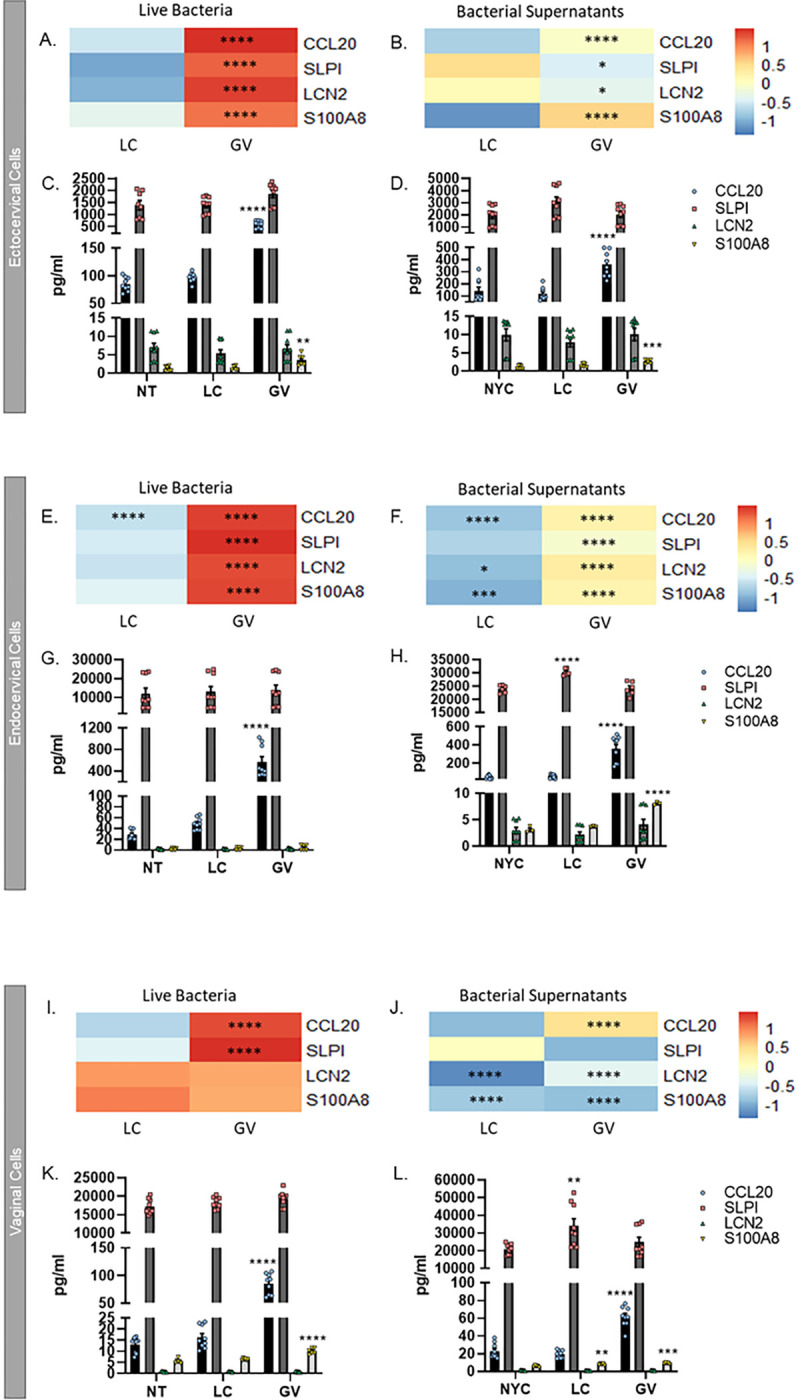
Anti-microbial peptide gene expression and proteins are significantly increased after cervicovaginal cell exposure to live bacteria or culture supernatants from G. vaginalis. RNA-seq identified CCL20, SLPI, LCN2 and S100A8 as being significantly upregulated by live G. vaginalis and its supernatants in ectocervical (A, B), endocervical (E, F) and vaginal (I, J) epithelial cells. ELISAs further identified that these anti-microbial peptides were increased after live G. vaginalis (C, G, K) or G. vaginalis culture supernatant exposure (D, H, J). Values are mean ± SEM. Asterisks over the individual bars represent comparisons to control; asterisks over solid lines represent comparisons between treatment groups. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Down-regulation of anti-microbial peptide gene expression, but not protein, by live L. crispatus and its culture supernatant
Live L. crispatus did not affect the gene expression of the four anti-microbial peptides measured (Fig. 3A, 3E, 3I), except that CCL20 mRNA was downregulated (p < 0.0001) in endocervical cells; however, the amount of CCL20 protein was not altered at this same time point (Fig. 3G). L. crispatus culture supernatants downregulated the gene expression of LCN2 (p < 0.05) and S100A8 (p < 0.001) in endocervical and vaginal cells (Fig. 3F and 3J), but not ectocervical cells (Fig. 3B). Additionally, CCL20 (p < 0.0001) gene expression was downregulated in endocervical cells (Fig. 3F). However, this decrease in anti-microbial gene mRNA expression in endocervical and vaginal cells was not associated with a change in their associated proteins (Fig. 3H and 3L).
G. vaginalis activates an inflammasome-mediated immune response in cervicovaginal epithelial cells
As noted prior, cervicovaginal cells exposed to G. vaginalis resulted in the differential expression of genes significantly associated with inflammation-related functional pathways (Fig. 2A, 2B–2D). These genes, included those coding for multiple chemokines and cytokines (e.g., IL-8, IL-6, IL-1α and TNF), have been investigated previously 23. Novel to this work, we identified several inflammasome-mediated genes that were significantly upregulated after G. vaginalis exposure including NLRP3, NLRP1, IL-1β and caspase-1 (adjusted p < 0.05) (Table 1). The gene expression changes for all detectable inflammasome-associated genes are listed in Supplemental Table 7. To determine if inflammasome activation contributes to the inflammatory response seen after G. vaginalis exposure in cervicovaginal epithelial cells, we performed a bacterial dose response experiment (1×105–1×107 CFUs/ml) for both L. crispatus and G. vaginalis. Caspase-1, IL-1β, and lactate dehydrogenase (LDH, a marker of cell death) release were significantly increased (p < 0.0001) in a dose-dependent manner from ectocervical, endocervical and vaginal cells after exposure to G. vaginalis but not L. crispatus (Supplemental Fig. 1).
Table 1:
Altered Inflammasome-related genes of interest after exposure to live L. crispatus or G. vaginalis
| Ectocervical | Endocervical | Vaginal | ||||
|---|---|---|---|---|---|---|
| Gene of Interest | LC | GV | LC | GV | LC | GV |
| NLRP3 associated genes | ||||||
| NLRP3 | 0.01 | 1.44 | 0.17 | 1.28 | 0.08 | 1.82 |
| NOD1 | 0.06 | 0.02 | 0.32 | 0.30 | −0.05 | −0.01 |
| NOD2 | −0.11 | 0.41 | 1.13 | 2.29 | −0.42 | 0.17 |
| NEK7 | −0.09 | 0.26 | −0.38 | −0.39 | 0.02 | 0.07 |
| NLRP1 associated genes | ||||||
| NLRP1 | 0.008 | 0.36 | 0.29 | 1.21 | 0.13 | 0.72 |
| Canonical Inflammasome genes | ||||||
| Caspase-1 | 0.15 | 0.29 | −0.17 | 0.79 | −0.31 | −0.59 |
| IL-1B | 0.16 | 0.96 | 0.31 | 2.06 | 0.15 | 1.37 |
| IL-18 | −0.01 | 0.41 | −0.22 | −0.46 | 0.16 | 0.10 |
| IL-1a | 0.12 | 1.29 | 0.30 | 2.07 | 0.23 | 1.12 |
Altered genes after exposure to live bacteria are shown as Log2FC from the non-treated control (NTC), gray shading denotes p≤0.01
G. vaginalis activates the NLRP3 inflammasome in THP monocytes
To determine the specificity of NLRP3 inflammasome activation in mediating G. vaginalis-induced increases in IL-1β, caspase-1 or cell death in cervicovaginal epithelial cells, we treated monocytes, THP1-Null2 cells or THP1-NLRP3-KO (knock out) cells, with LPS (positive control), live L. crispatus (control for live G. vaginalis) or live G. vaginalis. Exposure to live G. vaginalis, but not L. crispatus, resulted in a significant increase in caspase-1 (p < 0.001, Fig. 6A) and IL-1β (p < 0.0001, Fig. 6B) in THP1-Null2 cells but not in THP-NLRP3-KO cells (p < 0.001). Cell death (LDH, Fig. 6C) remained unchanged after bacterial exposure in both THP1 cell types. Additionally, we treated THP1-Null2 cells with glybenclamide (glyburide) 24, a specific inhibitor of NLRP3 (with no effects on NLRC4 or NLRP1). Glybenclamide treatment significantly reduced caspase-1 (p < 0.05) and IL-1β (p < 0.0001, Fig. 6D and 6E). As with THP1-NLRP3-KO, no reduction in G. vaginalis-mediated cell death (LDH, Fig. 6E) was observed in THP1-Null2 cells treated with the NLRP3 inhibitor.
Figure 6.

Live G. vaginalis induces inflammasome activation in THP-1 monocytes. Exposure to live G. vaginalis increases caspase-1 (A) and IL-1β (B) but not cell death (LDH) (C) in THP-null-2 cells. In THP-NLRP3 knockout cells the G. vaginalis-mediated increases in inflammasome-associated proteins are significantly reduced (A, B). LPS, included as a positive control, significantly increase IL-1β in THP-null-2 cells which was reduced in THP-NLRP3 knockout cells. Pre-treatment with glybenclamide, a specific NLRP3 inhibitor, significantly reduced the G. vaginalis-mediated increase in caspase-1 (D) and IL-1β (E). LDH was unchanged by glybenclamide (F). Values are mean ± SEM. Asterisks over the individual bars represent comparisons to control; asterisks over solid lines represent comparisons between treatment groups. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
G. vaginalis-mediated inflammasome activation in cervicovaginal epithelial cells is NLRP3 specific
Since G. vaginalis activates the NLRP3 inflammasome in THP1 monocytes, we determined if G. vaginalis could also activate an NLRP3-specific inflammasome in cervicovaginal epithelial cells. Glybenclamide treatment resulted in a significant reduction in G. vaginalis-mediated increases in caspase-1 (p < 0.01, Fig. 5A, 5D, 5G) and IL-1β (p < 0.05, Fig. 5B, 5E, 5H) in ectocervical, endocervical and vaginal cells, while G. vaginalis-induced cell death, as measured by LDH release, was unchanged (Fig. 5C, 5F, 5I).
Figure 5.

Inflammasome activation by G. vaginalis is specific to the NLRP3 inflammasome. Pre-treatment with glybenclamide, a specific NLRP3 inhibitor, was able to partially reduce the G. vaginalis-mediated increases in caspase-1 (A, D, G) and IL-1β (B, E, H) with no changes to cell death. Values are mean ± SEM. Asterisks over the individual bars represent comparisons to non-treated control; asterisks over solid lines represent comparisons between treatment groups. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
G. vaginalis activates the inflammasome-mediated immune response through caspase-1
Since inhibiting NLRP3 activation did not fully mitigate the G. vaginalis-mediated increases in IL-1β, caspase-1 or cell death in cervicovaginal epithelial cells, we sought to determine if inhibiting caspase-1 would fully block inflammasome activation. Using the irreversible caspase-1 inhibitor, Ac-YVAD-cmk 25, we determined that caspase-1 is an essential regulator of inflammasome activation by G. vaginalis. Inhibition of caspase-1 resulted in a significant reduction in the G. vaginalis-mediated increase in caspase-1 (p < 0.0001) (Fig. 4A, 4D, 4G), IL-1β (p < 0.0001) (Fig. 4B, 4E, 4H) and cell death (p < 0.0001) (LDH, Fig. 4C, 4F, 4I) in ectocervical, endocervical and vaginal cells after G. vaginalis but not L. crispatus exposure.
Figure 4.

Live G. vaginalis, but not L. crispatus, exposure initiates canonical-inflammasome activation in ectocervical (A-C), endocervical (D-F) and vaginal (G-I) epithelial cells. Exposure to live G. vaginalis for 24hrs increases caspase-1 (A, D, G), IL-1β (B, E, H) and cell death as measured by lactate dehydrogenase (LDH) (C, F, I). The G. vaginalis-mediated increases in inflammasome-associated proteins are significantly reduced after pre-treatment with YVAD, a caspase-1 inhibitor. Values are mean ± SEM. Asterisks over the individual bars represent comparisons to non-treated control; asterisks over solid lines represent comparisons between treatment groups. ****p<0.0001
L. crispatus culture supernatants alter chromatin accessibility
As most of the enriched pathways associated with differentially expressed genes in all three cervicovaginal cell types exposed to L. crispatus culture supernatants were related to alterations in histone and transcriptional regulation (Fig. 2A, 2E–G), we used assay for transposase-accessible chromatin high throughput sequencing (ATAC-seq) to ascertain whether these gene expression changes were associated with differences in chromatin accessibility 26–28. While all three cell types demonstrated substantial modulation of gene expression after exposure to L. crispatus culture supernatants, the prominence of upregulated epigenomic-associated functional pathways in ectocervical cells pointed to potential changes in chromatin accessibility in this cell type.
The number and percentage of aligned/unaligned reads were equal across all three cell types (Supplemental Fig. 2). Notably, the samples from the ectocervical cells had lower transcription start site (TSS) enrichment scores despite demonstrating similar quality control characteristics in alignment (Supplemental Fig. 3). There was a strong correlation between normalized read counts between conditions for each cell type (Supplemental Fig. 4).
We obtained a consensus peak set for each cell type (Ecto: 55,917 peaks, Endo: 46,535 peaks, VK2: 48,164 peaks). As expected, the majority of open chromatin peaks for all cell types were found in proximal promoter regions (< 1 kb) or intergenic regions (Fig. 7A). To determine if L. crispatus culture supernatant leads to cell-specific differences in chromatic organization, we compared the normalized read counts between different genomic regions. There was no difference in quantile normalized counts between the TSS and gene bodies for each cell type (Supplemental Fig. 5). However, only in the ectocervical cells, the consensus peak regions demonstrated nearly two discrete clusters (Fig. 7B). Endocervical and vaginal cells did not exhibit these two clusters of accessible sites (Fig. 7B).
Figure 7.

Chromatin accessibility was disrupted primarily in ectocervical cells exposed to L. crispatus supernatants. (A) Distribution of consensus sites by percentage. (B) Scatterplot of normalized counts between NYC and L. crispatus supernatant treatment by cell type-specific consensus peaks. (C) Distribution of differentially accessible sites in ectocervical cells across the genome. (D) Overlap of differentially accessible sites and matched number of random sites (E) all ectocervical peaks and matched number of random sites with published sites from ENCODE and multiple primary cancer specimens. (F) Motif analysis of downregulated differentially accessible sites with motif logo and graph of positional distribution based on the center of the peak of the top 5 motifs. (G) Bubble chart of DisGeNET enrichment of transcription factors identified by the motif analysis. DA: Differential Accessibility; ELS: Enhancer-like signatures; CESC: Cervical Squamous Cell Carcinoma; COAD: Colon Adenocarcinoma; BRCA: Breast Invasive Carcinoma; LUAD: Lung Adenocarcinoma; LUSC: Lung Squamous Cell Carcinoma.
We then tested whether there were regions of differential accessibility between the treatment conditions in the cell type-specific consensus peak sites. We detected 8,147 regions with differential accessibility profiles in L. crispatus supernatant-treated ectocervical cells, almost all showing reduced accessibility (8,125 with decreased accessibility, 22 with increased accessibility). There was only a fraction of sites differentially regulated in endocervical and vaginal cells (21 and 109 total sites, respectively). Notably, the distribution of differential accessibility sites was skewed towards an increase in the percentage of distal intergenic and intronic regions and a decrease in proximal promoter sites compared to the general distribution of all detected ectocervical peaks (Fig. 7C). We identified the genes neighboring the differentially regulated sites and sought to determine whether these genes overlapped with differentially expressed genes we had detected by RNA-sequencing after L. crispatus supernatant. Indeed, most genes with differential expression in ectocervical cells treated with L. crispatus supernatant had differential accessibility by ATAC-sequencing (683/762 genes)29–31.
Given the strong relationship between differentially expressed genes and differentially accessible chromatin, we sought to identify whether differentially accessible chromatin was more likely to be associated with previously identified putative tissue-specific regulatory regions or all identified human enhancer regions (enhancer-like sequences of different primary tissues described by the Encylopedia of DNA Elements (ENCODE) Project or open accessibility regions identified in different primary cancer specimens)32–34. Surprisingly, we discovered little overlap between differential accessibility in ectocervical cells and putative enhancer regions (Fig. 7D, Chi-squared test for trend p = 0.8334 in differentially accessible ectocervical peaks vs random matched control regions). However, ENCODE does not have specimens from primary cervical tissue so we could only compare the ectocervical signatures to specimens collected from the vagina and uterus. By contrast, there was a substantial overlap between all open chromatin regions identified in ectocervical cells and published likely enhancers (Fig. 7E, Chi-squared test for trend p = 0.0028 in all accessible ectocervical peaks vs random matched control regions).
Despite not being associated with known enhancer regions, motif analysis of the downregulated differentially accessible sites demonstrated enrichment for 497 transcription factor motifs with an FDR of 0.05 (Supplemental Table 8, Fig. 7F). The position of the top 5 identified motifs was at the center of the peak, consistent with the expected location of any true transcription factor binding35. Gene-disease enrichment analysis of these transcription factors demonstrated a marked enrichment of multiple pathways related to neoplasms, endometriosis and infertility, all pathologies potentially associated with Lactobacillus-dominated microbiota (Fig. 7G) 36,37. To determine if this enrichment was specific, we generated random lists of transcription factors from the same motif database and demonstrated only an overlapping enrichment in unrelated craniofacial anomalies, suggesting our findings are indeed specific (Supplemental Fig. 6).
Discussion
Host-microbe interactions have been shown to govern health and disease in multiple biological systems. With this study, we now reveal unique molecular mechanisms involved in host-microbe interactions in the cervicovaginal space, thus addressing a large knowledge gap in reproductive health. These findings confirm the induction of diverse immune pathways in response to G. vaginalis, a facultative anaerobic bacteria associated with many gynecological disorders, including STI and HPV acquisition. Importantly, we found that G. vaginalis activates the NLRP3 inflammasome in cervicovaginal epithelial and immune cells. Further advancing our understanding of host-microbe interactions in the cervicovaginal space, we report that exposure to L. crispatus culture supernatants results in epigenetic modifications in ectocervical cells. Collectively, these studies demonstrate the complexity of host-microbe interactions with divergent effects between live bacteria and their culture supernatants and epithelial cell responses to microbe-specific secreted factors.
While many studies have used high throughput sequencing technologies to identify and characterize microbial communities present in the cervicovaginal space 38–40, few have leveraged whole genome transcriptomic sequencing to investigate the genes and functional pathways altered by host-microbe interactions between different vaginal bacteria and their supernatants on all three cervicovaginal epithelial cells types. The RNA-seq results from this study reveal host epithelial cell-specific genes and functional pathways that are modulated by G. vaginalis and L. crispatus. Whole-genome transcriptomic evaluations of ectocervical, endocervical and vaginal cells exposed to G. vaginalis or L. crispatus (or their culture supernatants) revealed unexpected findings in that L. crispatus supernatant exposure modulated a high number of genes while live L. crispatus modulated very few genes. As studies have shown that L. crispatus-dominated microbiota protect the cervicovaginal epithelium from viral and bacterial infection 39,41,42, it has been hypothesized that L. crispatus secreted factors contribute to this protection. For example, a previous study identified that culture supernatant from Lactobacillus spp., including L. crispatus, is protective against Chlamydia trachomatis infection due to the protective effects of D(−) lactic acid and a reduction in vaginal epithelial cell proliferation 39. Perhaps not as surprisingly, RNA sequencing revealed that live G. vaginalis and its culture supernatant both altered the expression of a high number of genes, with live G. vaginalis modulating more than its culture supernatant. The difference in gene expression profiles induced by live bacteria or their culture supernatants by these two common bacterial species demonstrates the complexity of the host-microbial interactions in the cervicovaginal space. The induction of diverse and specific molecular pathways by vaginal bacteria serve to ascribe mechanisms by which these microbes promote reproductive health or disease.
Unique transcriptomic signatures were observed for each type of cervicovaginal epithelial cell studied. After G. vaginalis exposure, distinct sets of genes were upregulated in each epithelial cell type indicating that most altered genes appear to be cell type specific; however, many of these genes target similar innate immune pathways providing evidence of the redundancy in immune regulation. The innate immune pathways most predominantly implicated in the cervical and vaginal epithelial cell response to G. vaginalis included the upregulation of NFkB signaling, an increase in anti-microbial peptides (AMPs) and activation of the NLRP3 inflammasome. The finding that G. vaginalis initiates an NFkB-mediated inflammatory response in cervicovaginal epithelial cells is not unexpected as previous studies by our laboratory, and many others, have shown significant increases in cervical and vaginal epithelial cell cytokine levels after G. vaginalis exposure 23,43,44. Interestingly, a recent study investigated the vaginal epithelial proteome after exposure to G. vaginalis culture supernatants and identified an activation of the mTOR signaling pathway 45. As mTOR is known to regulate many immune functions including the activation of NFkB 46, it is possible that mTOR may play a role in G. vaginalis host-microbial functions, however, we did not detect a change in mTOR gene expression in vaginal cells after exposure to either live bacteria or culture supernatants in our RNA-seq analysis. Even though many studies have identified an activated immune response in Lactobacillus-deplete microbial communities, characterization of this response has been highly varied and complex putting even more emphasis on the need to clearly identify specific immune factors/pathways activated by cell and microbe specific interactions.
As a critical part of the innate immune response, AMPs, also known as host defense peptides, act to destroy invading/foreign pathogens using a variety of biological processes. The four AMPs measured in this study use overlapping but distinct mechanisms of host defense including the regulation of immune cell migration (CCL20) 47–49, acting as a chemoattractant for neutrophils (S100A8) 50,51, barrier protection against neutrophil elastase (SLPI), or sequestering iron to limit bacterial nutrients and prevent growth (LCN2). G. vaginalis-mediated increases in CCL20 and S100A8 were most consistently observed in all three cervicovaginal cells indicating that these AMPs are likely to play an essential role in the host response. Additionally, immune cell recruitment by G. vaginalis through the release of CCL20 and S100A8 is a potential plausible mechanism as to how G. vaginalis colonization results in increased risk of HIV acquisition. Overall, these data provide evidence that the host uses multiple different mechanisms, controlled by individual AMPs, to protect against pathogenic microbes and speaks to the complexity of the innate immune response and the role of unique AMPs in this response.
Interestingly, RNA-seq identified a significant upregulation of NOD-like receptor (NLR) family, pyrin domain-containing protein 3 (NLRP3) inflammasome-associated genes in cervicovaginal cells after G. vaginalis exposure. Inflammasomes are a group of proteins that act as intracellular pathogen recognition receptors (PRRs) that respond to pathogen associated molecular patterns (PAMPs) from foreign microbes. Inflammasomes are activated in a multitude of inflammatory conditions, but very little is known about microbe-mediated inflammasome activation in the cervicovaginal space. Inflammasomes have been shown to be activated in immune cells after bacterial challenge with cervicovaginal microbes including Neisseria gonorrhoeae and G. vaginalis 52,53. Specifically, G. vaginalis has been shown to activate the NLRP3 inflammasome in macrophages and THP-1 monocytes 53,54. In this study, we confirmed that G. vaginalis increases caspase-1 and IL-1β levels through NLRP3 in THP-1 monocytes, an effect that is not seen with L. crispatus.
Noting this prior work, this is the first study to demonstrate that G. vaginalis activates a canonical inflammasome pathway, not just in immune cells, but also in cervicovaginal epithelial cells. G. vaginalis activation of the inflammasome pathway seems to be mediated, at least in part, through NLRP3, as the NLRP3 specific inhibitor, glybenclamide, was able to reduce the G. vaginalis-mediated increases in caspase-1 and IL-1β. While prior reports demonstrate that NLRP3 activation is essential for the induction of the inflammasome by G. vaginalis for immune cells, we find that the induction of the inflammasome in cervicovaginal epithelial cells is more complex 53,55. RNA-seq showed significant increases in expression in several genes associated with the inflammasome pathway. As such, increased NLRP1 expression in cervicovaginal cells after exposure to G. vaginalis suggests that NLRP1 may also be contributing to the inflammasome-mediated immune activation. Future studies would be needed to further investigate the role of NLRP1, and possibly other G. vaginalis-mediated inflammasome components (Supplemental Table 7), in the regulation of host-microbial interactions with G. vaginalis. The finding that G. vaginalis can activate the inflammasome in both cervicovaginal epithelial cells and monocytes suggests that inflammasome activation may be an important and common contributor to the inflammatory response in a G. vaginalis-dominated cervicovaginal space.
While cell death (pyroptosis) is an integral part of inflammasome activation, G. vaginalis also secrets a cholesterol-dependent cytolysin, vaginolysin, that creates pores in the epithelial cell membrane causing cell death 56. Therefore, it is difficult to discern if the cell death observed after G. vaginalis exposure is due to inflammasome activation directly, an indirect result of vaginolysin or a combination of both. While our results show that cell death in cervicovaginal cells is mediated by caspase-1 activation, in agreement with studies that show caspase-1 is required for Gasdermin cleavage and activation (necessary for NLRP3-induced pyroptosis) 57, we did not see a reduction in cell death with the NLRP3 inhibitor in either cervicovaginal cells or monocytes. Further elucidation of the mechanisms leading to cell death would be important for therapeutic strategies to limit G. vaginalis-mediated epithelial dysfunction.
Demonstrating the capabilities of different vaginal bacteria species to induce unique molecular profiles, in contrast to the induction of innate inflammatory pathways by G. vaginalis, cervicovaginal cells exposed to L. crispatus culture supernatant predominantly showed alterations in RNA polymerase, histone, and transcription-related genes. These findings suggest an important role for L. crispatus culture supernatant in inducing epigenomic modifications of host epithelial cells. An abundance of Lactobacillus spp. (including L. crispatus, L. jensenii and L. gasseri) in the cervicovaginal space have been shown to be protective against microbial pathogens, vaginal infection, and cervical cancer 58. While the biological mechanisms contributing to Lactobacillus protection remain largely unknown, studies have shown that Lactobacillus spp. produce lactic acid 59,60, hydrogen peroxide 61 and bacteriocins 41 that act by creating an environment that inhibits the growth of pathogenic microbes. As evidence of a direct impact of L. crispatus on epithelial function, L. crispatus has been demonstrated to inhibit cell proliferation and migration 62.
Unique to this study, we find that L. crispatus supernatant caused substantial modulation of genes globally regulating the transcriptome and epigenome. These changes corresponded to a substantial unexpected reorganization of the epigenome as shown by ATAC-seq. L. crispatus largely reduced the number of open chromatin regions in ectocervical cells. An intriguing hypothesis is that L. crispatus may be important for increasing the resilience of cells to infection (ex. Chlamydia, HIV, HPV) by modulating the epigenetic susceptibility of the cells to a disease or pathogen 63,64. Providing evidence for the ability of L. crispatus to alter the epigenome to protect from disease, a previous study has shown that supernatant from Lactobacillus spp. can decrease C. trachomatis infection by inhibiting cell proliferation through epigenetic mechanisms. Specifically, L. crispatus culture supernatants decreased histone deacetylase 4 (HDAC4) and increased histone acetylase EP300 through mechanisms thought to involve D(−) lactic acid in vaginal cells 39. Based on this report, we can hypothesize that L. crispatus culture supernatants induce these epigenomic effects due to the presence of lactic acid isomers. However, it is also plausible that other metabolites and/or proteins within the L. crispatus culture supernatant are responsible for inducing these epigenomic changes. Importantly, understanding the ability of L. crispatus supernatants to modify cervicovaginal epithelial function may provide new therapeutic strategies to optimize reproductive health.
Intriguingly, in this study the regions of differential chromatin accessibility did not overlap with regions previously described in published putative enhancers. This discrepancy could be due to multiple reasons. First, we do not have ENCODE or ATAC profiles of healthy primary cervical tissue for comparison. In addition, the profiled cervical cancer specimens were derived from only four specimens and likely have different biological profiles than the ectocervical lines used here. Therefore, it is possible that these differentially regulated regions are cell type-specific enhancers in ectocervical cells. Second, it is notable that there is a substantial increase in the proportion of intronic sites among the ectocervical differentially accessible sites compared to all the ectocervical peaks (48.2% vs 26.7% respectively), suggesting a potential role for Lactobacillus in regulating isoform transcriptions through chromatin modulation. However, RNA-seq data in this study is limited in its ability to directly answer this question; a repeat of this study with long read RNA-seq would be more suitable65. Lastly, there is a growing literature about the roles of lactate as a precursor to acetyl-CoA needed for histone acetylation as well as a direct modifier of histones through histone lactylation66–69. Histone lactylation is generally described to lead to increased expression, putatively associated with more open chromatin. Yet, the role of lactate in modifying histones and other epigenetic regulators is a new area of research and further work would be needed to elucidate its role in decreasing chromatin accessibility in specific contexts. Nonetheless, while we cannot fully identify how these differentially regulated sites may be contributing to gene regulation, it is notable that disease gene enrichment analysis of transcription factors associated with the motifs at these sites demonstrated multiple pathologies related to women’s health, including fertility and endometriosis, posing an intriguing avenue for better understanding the molecular underpinnings of these common but poorly understood disorders.
A limitation of this study is that it relies on single strains of common vaginal bacteria. While studies investigating broader microbial communities are needed to more accurately mimic the cervicovaginal microbiota present in vivo, by focusing on single microbes of interest we were able to identify specific functional pathways that may drive adverse outcomes. An in-depth understanding of single bacteria lays the foundation for these future studies. In addition to G. vaginalis, studies investigating other high-risk anaerobic bacteria for adverse reproductive outcomes 7, including Sneathia spp, Mobiluncus spp. and Prevotella spp. would be necessary to elucidate the combinatorial effects of anaerobes more broadly on cervicovaginal function. Now that specific biological functions of both G. vaginalis and L. crispatus have been identified, these pathways can be used to develop mechanistic hypotheses to evaluate more complex microbiota or clinical populations. For the analysis of ATAC data, publicly available databases, such as the ENCODE database, do not contain comparable ATAC data in cervical tissue or vaginal tissue for comparison of normal/control human tissue32. Developing these resources should be a priority as they will be critical for further understanding of the host-microbial interactions in the reproductive tract and their role in diverse reproductive outcomes.
Overall, the results of this study identify novel transcriptomic and epigenomic pathways altered by microbes within the cervicovaginal space that are most commonly associated with reproductive health and disease. Utilizing whole genome RNA-sequencing, we identified microbe-specific functional gene pathways including activation of the innate immune response by G. vaginalis and increased RNA transcription and histone modifications by L. crispatus that are regulated by host-microbe interactions within the cervicovaginal space. These results suggest that complex interactions between host cells and live bacteria or the factors they secrete have distinct and specific functions in modifying host epithelial cell responses. Novel to this study, we identified two key areas for potential therapeutic targets: 1) activation of the inflammasome as part of the innate immune response to G. vaginalis and 2) epigenetic regulation by L. crispatus culture supernatants. Targeting specific G. vaginalis-mediated innate immune pathways may serve to modulate the inflammatory response associated with G. vaginalis, and thus, could impact STIs, BV and preterm birth. Likewise, leveraging the potential of L. crispatus to alter the epigenetic landscape may provide new opportunities to optimize the cervicovaginal epithelial barrier and prevent pathogenic (e.g. chlamydia, HPV) microbes from harming and infecting the epithelial barrier. Continued elucidation of host-microbial interactions in the female reproductive tract will undoubtedly serve to optimize reproductive health.
Materials and Methods
Cell Culture
Ectocervical (Ect/E6E7, AATC# CRL-2614) (Ecto), endocervical (End1/E6E7, AATC# CRL-2615) (Endo) and vaginal (VK2/E6E7, ATCC# CRL-2616) (VK2) human epithelial cell lines (American Type Culture Collection, Manassas, VA) were cultured in Keratinocyte-Serum Free Media (K-SFM) supplemented with 0.1 ng/mL epidermal growth factor and 50 ug/mL bovine pituitary extract (Gibco, Life Technologies), 100 U/mL penicillin, and 100 μg/mL of streptomycin at 37°C in a 5% CO2 humidified incubator. The monocyte cell lines, THP1-Null2 (Invivogen) and THP1-KO-NLRP3 (Invivogen), which have a biallelic KO of the N-terminal region of the NLRP3 gene, were cultured according to the manufacture’s protocol. Briefly, THP1-Null2 and THP1-KO-NLRP3 cells were grown in RPMI 1640 supplemented with 2 mM L-glutamine, 25 mM HEPES (Gibco, Life Technologies), 10% heat-inactivated fetal bovine serum (Gemini Bio), 100 μg/ml Normocin™, 100 U/mL penicillin, and 100 μg/mL of streptomycin. Cells were maintained in T-25 flasks at 37°C in a 5% CO2 humidified incubator.
Bacterial Cultures and Preparation of Bacteria-Free Supernatants
Bacterial strains, L. crispatus (ATCC 33197) or G. vaginalis (ATCC 14018), were obtained from the American Type Culture Collection (Manassas, VA). G. vaginalis was grown on Tryptic Soy Agar with 5% Sheep Blood plates (Hardy Diagnostics) and L. crispatus was grown on De Man, Rogosa and Sharpe agar (Fisher Scientific); both strains were grown in New York City III (NYCIII) broth. Bacteria were grown at 37°C in an anaerobic glove box (Coy Labs, Grass Lake, MI).
For each experiment the following bacterial growth protocol was followed: L. crispatus and G. vaginalis glycerol stocks were streaked on agar plates, as well as, into broth tubes and grown overnight. The broth starter cultures were diluted to an optical density of 0.2 and then used to inoculate 20ml working cultures, which were grown for 20 hours prior to use in experiments. Bacterial densities of the working cultures were estimated the day of the experiment based on optical density readings at 600 nm using an Epoch2 plate reader (Biotek, Winooski, VT), and the appropriate volume was centrifuged at 13,000 × g for 3 min. The bacterial pellets were resuspended in the appropriate cell culture media without antibiotics and added to epithelial cells at 104 −106 CFUs/well. Precise bacterial densities of the working cultures were determined by plating serial dilutions of the working cultures and counting CFUs. For all experiments, reported bacterial densities are +/− 0.5 log of the noted bacterial density (CFU/well).
To obtain bacteria-free culture supernatants, the working cultures were centrifuged at 13,000 × g for 3 min and the supernatant was filtered through a 0.22 μm filter (Fisher Scientific) to remove any remaining live bacteria. Bacteria-free culture supernatants were diluted to 1% v/v in the appropriate cell culture media without antibiotics.
In vitro Epithelial and Immune Cell - Bacteria Interactions
Ectocervical, endocervical and vaginal cells were plated at 1.5 × 105 cells/well in twenty-four well plates containing K-SFM without antibiotics. THP1-Null2 or THP1-KO-NLRP3 cells were plated at 1.5 × 105 cells/well in twenty-four well plates containing RPMI 1640 media without antibiotics. The next day, the cells were exposed to either live L. crispatus or G. vaginalis (1×105 − 1×107 CFU/well) or 1% (v/v) bacteria-free supernatants (generated from a 1×107 CFU/mL culture) for 24 hr. For cells treated with bacteria-free supernatants from L. crispatus, K-SFM media was supplemented with 50mM HEPES and sodium bicarbonate (3000 mg/L total concentration) to bring the pH of the media up to a physiological level (7.2). In additional experiments, ectocervical (n = 6/treatment), endocervical (n = 6/treatment), vaginal cells (n = 6/treatment) or THP1-Null2 (n = 6/treatment) cells were pre-treated with Glybenclamide (100 μg/ml, Invivogen), a specific NLRP3 inhibitor, for 30 mins prior to live bacteria exposure or Ac-YVAD-cmk (25 μM, Sigma-Aldrich), an irreversible caspase-1 inhibitor, for 1 hour prior to live bacteria exposure. For inflammasome experiments, LPS (10 ng/ml) exposure for 24 hr was used as a positive control of inflammasome activation. For all supernatant experiments, cells were also exposed to NYCIII bacterial growth media alone as a negative control to determine any baseline effects of the growth media on the outcomes of interest. At the end of each experiment, cell culture media was collected for cell death, ELISA assays and/or the cells were collected in Trizol (Invitrogen, Thermo-Fisher Scientific) for RNA extraction.
RNA Sequencing and Analysis
RNA was extracted from ectocervical, endocervical and vaginal cells after exposure to bacteria or culture supernatants from L. crispatus and G. vaginalis (n = 3/treatment group) collected in Trizol using the Qiagen-RNeasy Plus Mini kit by the Penn Next-Generation Sequencing Core. The resulting RNA had RIN values > 9. Illumina sequencing libraries were prepared using the Illumina TruSeq mRNA stranded library prep kit according to the manufacturer recommendations. The resulting libraries had an average molarity of 69 nM +/1 27 nM. Libraries were sequenced to a median depth of 41 million 100 bp single reads on an Illumina NovaSeq 6000. Transcript quantification from RNA-seq data was performed using Salmon and release 38 (GRCh38.p13) of the human genome 70,71. Several Bioconductor packages in R were used for subsequent steps 72,73. The output was annotated and summarized using tximeta and further annotation was completed with biomaRt 74,75. Principle Component Plots (PCA) were created using pcaExplorer 76,77. The normalizations and statistical analyses were done with DESeq2 78. The full RNA-seq dataset was submitted to Gene Expression Omnibus (accession # GSE234837).
RNA-seq Pathway Analysis
PathfindR was used for pathway enrichment analysis using Gene Ontology terms (https://github.com/egeulgen/pathfindR) 79. Upregulated and downregulated genes were grouped together for each comparison. The enrichment threshold was set at an FDR of 0.05, with a significant gene threshold of 0.02. A heatmap for enrichment scores for each comparison was created by first calculating and aggregating term scores for each sample included for each comparison and then averaging the scores across all compared samples. ComplexHeatmap package in R was then used to visualize the comparison of GO term analysis (rows) for all the comparisons (columns). Rows were clustered by the “complete” method with a kmeans = 10. A word cloud was used to represent the most significant recurring pathways in a cluster. Generic terms were excluded from word cloud (“pathway,” “cellular,” “regulation,” “positive,” “negative,” “cell,” “complex,” “process,” “factor,” “activity,” “protein,” “DNA,” “RNA,” “levels,” “binding,” “response,” “signaling,” “receptor”).
Cell Death Assay
Ectocervical, endocervical, vaginal, THP1-Null2 and THP1-KO-NLRP3 cells were grown and exposed to bacteria or bacterial culture supernatants as described above. Lactate dehydrogenase (LDH) released upon cell lysis (n = 3–9 independent experiments per cell type) was quantified using the CytoTox 96 Non-radioactive cytotoxicity assay (Promega, Madison, WI), a coupled enzymatic assay that results in the conversion of a tetrazolium salt into a red formazan product. The colorimetric output was measured using a plate reader at 490 nm and absorbance values were recorded.
ELISA
Ectocervical, endocervical, vaginal, THP1-Null2 and THP1-KO-NLRP3 cells were cultured in 24-well plates and exposed to either live bacteria or bacterial supernatants as stated above. Anti-microbial peptides, CCL20, SLPI, LCN2, S100A8/A9, or inflammasome-associated cytokines, IL-1β and caspase-1, were measured in cell culture media after 24 hr of exposure (n = 6/group). The expression of these analytes was measured by a ligand-specific commercially available ELISA kit that utilizes a quantitative sandwich enzyme immunoassay technique using reagents from R&D Systems (Minneapolis, MN).
ATAC-seq Nuclei Extraction, Tagmentation, Purification and Library Amplification
ATAC-seq was performed on ectocervical, endocervical and vaginal cells after exposure to L. crispatus bacteria-free supernatants (n = 3/treatment group). ATAC-seq libraries were generated using the ATAC-seq Kit from Diagenode (Diagenode, A Hologic Company) according to manufacturer instructions. Briefly, nuclei were extracted from 50,000 cells. Tagmentation was completed by resuspending the isolated nuclei in transposase reaction mix and the samples were purified using the kit’s provided columns. Following purification, library fragments were amplified by PCR according to the manufacturer recommendations. Unique Dual Indexes Primer Pairs were incorporated for multiplexed sequencing. To reduce amplification bias, after the first 5 cycles of the PCR reaction, qPCR was used to determine how many additional cycles were needed to produce enough library to meet the required amount for sequencing. For this, an aliquot of the PCR reaction was added to Sybr Green and amplified for 20 cycles. Libraries were amplified for a total of 11–13 cycles (with one library requiring 17 cycles for amplification). Final libraries were purified using bead purification (Beckman Coulter), then assessed for size distribution and concentration using a BioAnalyzer High Sensitivity DNA Kit (Agilent Technologies). The resulting libraries were pooled. The pool was diluted to 2 nM, denatured, and then 13 libraries were loaded onto an S1–100 (2×50) flow cell on an Illumina NovaSeq 6000 (Illumina, Inc.) according to the manufacturer’s instructions. The average read number per sample was 50M+/− 20%. De-multiplexed and adapter-trimmed sequencing reads were generated using bcl2fastq. The full ATAC-seq dataset was submitted to Gene Expression Omnibus (accession # GSE233444).
ATAC-seq Mapping and Peak Calling
ATAC-seq data analysis was adapted from a previously published approach 26. Each cervicovaginal cell type was analyzed separately. In brief, raw FASTQ files were processed and mapped to release 38 (GRCh38.p13) of the human genome using the PEPATAC pipeline 80. Reads were trimmed with Skewer and then aligned with Bowtie2 using default settings 81,82. Duplicate reads were removed using samblaster 83.
An iterative overlap peak calling strategy on fixed-sized peaks of 501 bp was used to define a set number of peaks for each cell type for downstream differential accessibility comparison 26. First, for each biological replicate, MACS2 was used to call peaks with the parameters as follows: --peak-type fixed --extend 250 84. Biological replicates of each treatment and then both treatments together from each cell type were merged using an iterative overlap approach previously described 26,34. Blacklisted regions were excluded from called peaks (accessed 4 November 2022 at https://github.com/Boyle-Lab/Blacklist) 85.
ATAC-seq Peak and Differential Accessibility Analysis
Peak location was annotated with CHIPseeker 86. Counts for peaks were calculated using Rsubread 87. We determined the differential accessibility of peaks between treatments with DESeq2 88. We compared L. crispatus culture supernatant treated to NYCIII media controls for each cell type. A Wald test was used to determine significance. A peak was defined as statistically significant in differential accessibility if |log2foldchange| > 1 and FDR < 0.05. We utilized the R package rGREAT for the nearest gene analysis to access the Genome Regions Enrichment of Annotations Tool (GREAT) web service29–31. For GREAT, we used the parameters for “the two closest genes” to a differential accessible site as it is frequently not the closest genes that is differentially regulated.
Motif analysis was performed using Simple Enrichment Analysis version 5.5 as part of the MEME Suite (https://meme-suite.org/meme/tools/sea) 89,90. CIS-BP 2.0 motifs were used for the query 91. Gene-disease enrichment was performed using disgenet2R (https://www.disgenet.org/) 92. Random gene lists for comparison were generated by sampling 497 transcription factors from the CIS-BP 2.0 database.
ATAC-seq Chromatin Accessibility Visualization
EasSeq was utilized to visualize the data 93. Biological replicates of BAM files were pooled for quantification of specific regions. Quantile normalization was used for counts per region for visualization to minimize bias from sequencing depth. Calculation of overlap performed both by any amount of overlap and the exact overlap of base pairs between all comparisons. Random regions matched of equal number comparisons to differentially accessible regions or all ectocervical open chromatin regions were generated by Regulatory Sequence Analysis Tools (random genome fragments tool; http://rsat.sb-roscoff.fr/)94. ENCODE datasets for all human enhancer-like sequences (ELS, defined as high DNAse-seq signal and high H3K37me3), or tissue-specific regulators were obtained from https://screen.encodeproject.org/ 32,33. For uterus and vaginal specimens, downloaded with “Low-DNase” were filtered out to enrich for sites that had any evidence of potential enhancer or regulator activity. However, strict enhancer-like signature criteria could not be applied because all sequencing modalities were not available for all the samples. Primary cancer cell data sets were obtained from supplemental of published ATAC profiling34.
Statistical Analysis
Statistical analyses were performed for all experiments (except for RNA or ATAC sequencing, statistical analysis is described above for each) with the GraphPad Prism Software (Version 9.0, San Diego, CA). For data that were normally distributed (as assessed by Shapiro-Wilk test), one-way analysis of variance (ANOVA) was performed. If statistical significance was reached (p < 0.05), then pair-wise comparison with a Tukey post hoc test was performed for multiple comparisons. If data were not normally distributed, then the non-parametric Kruskal-Wallis test was used and pairwise comparison was done using Dunn’s multiple comparison test. Chi test for trend was utilized to compare overlaps of indicated ectocervical peaks with the number of a random set of sites matched for size and CG content.
Acknowledgements
The authors would like to acknowledge the Microbial Culture and Metabolomics Core of the PennCHOP Microbiome Program for providing microbial culture services for these studies. Additionally, we acknowledge the Penn Genomics and Sequencing Core at the University of Pennsylvania for their help with the whole genome RNA-sequencing performed in this study, as well as RNA-sequencing bioinformatics performed by John Tobias. We would also like to thank the High Throughput Sequencing Core at the Children’s Hospital of Pennsylvania (CHOP) for completing the ATAC-sequencing. We thank Sunny Shin, Ph.D., Professor of Microbiology at the University of Pennsylvania for her scientific discussion and experimental insights surrounding microbial inflammasome activation.
Funding
Support for this research was provided by the National Institutes of Health (NIH) National Institute of Child Health and Human Development (NICHD) (R01HD102318 and R01HD098867) and March of Dimes Prematurity Research Center at the University of Pennsylvania (22-FY18-812). AGC is supported by the Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program and NIH grant K08 NS119797 (PI: AGC).
Footnotes
Declarations
Conflict of Interest
MAE receives salary support from NIH (NIAID, NINR, and NICHD). She is a consultant with equity for MIRVIE.
Contributor Information
Michal Elovitz, Icahn School of Medicine at Mount Sinai.
Lauren Anton, Perelman School of Medicine at the University of Pennsylvania.
Ana Cristancho, Children’s Hospital of Philadelphia, Perelman School of Medicine at the University of Pennsylvania.
Briana Ferguson, University of Pennsylvania.
Andrea Joseph, Ichan School of Medicine at Mount Sinai.
Jacques Ravel, University of Maryland, Baltimore.
References
- 1.Nakama C. et al. The Continuum of Microbial Ecosystems along the Female Reproductive Tract: Implications for Health and Fertility. Pathogens 11, 1244 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradford L. L. & Ravel J. The vaginal mycobiome: A contemporary perspective on fungi in women’s health and diseases. Virulence 8, 342–351 (2017). 10.1080/21505594.2016.1237332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Happel A. U., Varsani A., Balle C., Passmore J. A. & Jaspan H. The Vaginal Virome-Balancing Female Genital Tract Bacteriome, Mucosal Immunity, and Sexual and Reproductive Health Outcomes? Viruses 12 (2020). 10.3390/v12080832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madere F. S. & Monaco C. L. The female reproductive tract virome: understanding the dynamic role of viruses in gynecological health and disease. Curr Opin Virol 52, 15–23 (2022). 10.1016/j.coviro.2021.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravel J. et al. Vaginal microbiome of reproductive-age women. Proceedings of the National Academy of Sciences of the United States of America 108 Supp 1, 4680–4687 (2011). 10.1073/pnas.1002611107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium., H. M. P. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elovitz M. A. et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat Commun 10, 1305–1305 (2019). 10.1038/s41467-019-09285-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.France M. T. et al. VALENCIA: a nearest centroid classification method for vaginal microbial communities based on composition. Microbiome 8, 166 (2020). 10.1186/s40168-020-00934-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajer P. et al. Temporal Dynamics of the Human Vaginal Microbiota. Science Translational Medicine 4, 132ra152–132ra152 (2012). 10.1126/scitranslmed.3003605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campisciano G. et al. Subclinical alteration of the cervical–vaginal microbiome in women with idiopathic infertility. Journal of Cellular Physiology 232, 1681–1688 (2017). 10.1002/jcp.25806 [DOI] [PubMed] [Google Scholar]
- 11.van Oostrum N., De Sutter P., Meys J. & Verstraelen H. Risks associated with bacterial vaginosis in infertility patients: a systematic review and meta-analysis. Human Reproduction 28, 1809–1815 (2013). 10.1093/humrep/det096 [DOI] [PubMed] [Google Scholar]
- 12.Brusselaers N., Shrestha S., van de Wijgert J. & Verstraelen H. Vaginal dysbiosis and the risk of human papillomavirus and cervical cancer: systematic review and meta-analysis. Am J Obstet Gynecol 221, 9–18.e18 (2019). 10.1016/j.ajog.2018.12.011 [DOI] [PubMed] [Google Scholar]
- 13.Price J. T. et al. Highly diverse anaerobe-predominant vaginal microbiota among HIV-infected pregnant women in Zambia. PLoS One 14, e0223128 (2019). 10.1371/journal.pone.0223128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elovitz M. A. et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat Commun 10, 1305 (2019). 10.1038/s41467-019-09285-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callahan B. J. et al. Replication and refinement of a vaginal microbial signature of preterm birth in two racially distinct cohorts of US women. Proc Natl Acad Sci U S A 114, 9966–9971 (2017). 10.1073/pnas.1705899114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Potter R. F., Burnham C. D. & Dantas G. In Silico Analysis of Gardnerella Genomospecies Detected in the Setting of Bacterial Vaginosis. Clin Chem 65, 1375–1387 (2019). 10.1373/clinchem.2019.305474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsberg J.-G. Cervicovaginal epithelium: Its origin and development. American Journal of Obstetrics and Gynecology 115, 1025–1043 (1973). 10.1016/0002-9378(73)90687-X [DOI] [PubMed] [Google Scholar]
- 18.Fritsch H., Hoermann R., Bitsche M., Pechriggl E. & Reich O. Development of epithelial and mesenchymal regionalization of the human fetal utero-vaginal anlagen. Journal of Anatomy 222, 462–472 (2013). 10.1111/joa.12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurita T. Normal and abnormal epithelial differentiation in the female reproductive tract. Differentiation 82, 117–126 (2011). 10.1016/j.diff.2011.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taherali F., Varum F. & Basit A. W. A slippery slope: On the origin, role and physiology of mucus. Advanced Drug Delivery Reviews 124, 16–33 (2018). 10.1016/j.addr.2017.10.014 [DOI] [PubMed] [Google Scholar]
- 21.Nazli A. et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog 6, e1000852 (2010). 10.1371/journal.ppat.1000852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cherne M. D. et al. Matrix Metalloproteinases Expressed in Response to Bacterial Vaginosis Disrupt the Endocervical Epithelium, Increasing Transmigration of HIV. Infect Immun 88 (2020). 10.1128/iai.00041-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anton L. et al. Gardnerella vaginalis alters cervicovaginal epithelial cell function through microbe-specific immune responses. Microbiome 10, 119 (2022). 10.1186/s40168-022-01317-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamkanfi M. et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. Journal of Cell Biology 187, 61–70 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Calvo M. et al. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem 273, 32608–32613 (1998). 10.1074/jbc.273.49.32608 [DOI] [PubMed] [Google Scholar]
- 26.Grandi F. C., Modi H., Kampman L. & Corces M. R. Chromatin accessibility profiling by ATAC-seq. Nat Protoc 17, 1518–1552 (2022). 10.1038/s41596-022-00692-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajkova P. et al. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452, 877–881 (2008). 10.1038/nature06714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan F., Powell D. R., Curtis D. J. & Wong N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol 21, 22 (2020). 10.1186/s13059-020-1929-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu Z. & Hubschmann D. rGREAT: an R/bioconductor package for functional enrichment on genomic regions. Bioinformatics 39 (2023). 10.1093/bioinformatics/btac745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLean C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501 (2010). 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanigawa Y., Dyer E. S. & Bejerano G. WhichTF is functionally important in your open chromatin data? PLoS Comput Biol 18, e1010378 (2022). 10.1371/journal.pcbi.1010378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luo Y. et al. New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res 48, D882–D889 (2020). 10.1093/nar/gkz1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Consortium E. P. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corces M. R. et al. The chromatin accessibility landscape of primary human cancers. Science 362 (2018). 10.1126/science.aav1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung M. H., Guertin M. J., Baek S. & Hager G. L. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol Cell 56, 275–285 (2014). 10.1016/j.molcel.2014.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khodaverdi S. et al. Beneficial Effects of Oral Lactobacillus on Pain Severity in Women Suffering from Endometriosis: A Pilot Placebo-Controlled Randomized Clinical Trial. Int J Fertil Steril 13, 178–183 (2019). 10.22074/ijfs.2019.5584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobstyl M., Brecht P., Sobstyl A., Mertowska P. & Grywalska E. The Role of Microbiota in the Immunopathogenesis of Endometrial Cancer. Int J Mol Sci 23 (2022). 10.3390/ijms23105756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delgado-Diaz D. J. et al. Lactic acid from vaginal microbiota enhances cervicovaginal epithelial barrier integrity by promoting tight junction protein expression. Microbiome 10, 141 (2022). 10.1186/s40168-022-01337-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edwards V. L. et al. The Cervicovaginal Microbiota-Host Interaction Modulates Chlamydia trachomatis Infection. mBio 10 (2019). 10.1128/mBio.01548-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castro J., Machado D. & Cerca N. Unveiling the role of Gardnerella vaginalis in polymicrobial Bacterial Vaginosis biofilms: the impact of other vaginal pathogens living as neighbors. Isme j 13, 1306–1317 (2019). 10.1038/s41396-018-0337-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aroutcheva A. et al. Defense factors of vaginal lactobacilli. American Journal of Obstetrics and Gynecology 185, 375–379 (2001). 10.1067/mob.2001.115867 [DOI] [PubMed] [Google Scholar]
- 42.Anton L. et al. Common Cervicovaginal Microbial Supernatants Alter Cervical Epithelial Function: Mechanisms by Which Lactobacillus crispatus Contributes to Cervical Health. Frontiers in Microbiology 9 (2018). 10.3389/fmicb.2018.02181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anton L. et al. Common Cervicovaginal Microbial Supernatants Alter Cervical Epithelial Function: Mechanisms by Which Lactobacillus crispatus Contributes to Cervical Health. Front Microbiol 9, 2181 (2018). 10.3389/fmicb.2018.02181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eade C. R. et al. Identification and characterization of bacterial vaginosis-associated pathogens using a comprehensive cervical-vaginal epithelial coculture assay. PLoS One 7, e50106 (2012). 10.1371/journal.pone.0050106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berard A. R. et al. Vaginal epithelial dysfunction is mediated by the microbiome, metabolome, and mTOR signaling. Cell Reports 42, 112474 (2023). 10.1016/j.celrep.2023.112474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dan H. C. et al. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev 22, 1490–1500 (2008). 10.1101/gad.1662308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zaballos A., Varona R., Gutiérrez J., Lind P. & Márquez G. Molecular cloning and RNA expression of two new human chemokine receptor-like genes. Biochem Biophys Res Commun 227, 846–853 (1996). 10.1006/bbrc.1996.1595 [DOI] [PubMed] [Google Scholar]
- 48.Hieshima K. et al. Molecular cloning of a novel human CC chemokine liver and activation-regulated chemokine (LARC) expressed in liver. Chemotactic activity for lymphocytes and gene localization on chromosome 2. J Biol Chem 272, 5846–5853 (1997). 10.1074/jbc.272.9.5846 [DOI] [PubMed] [Google Scholar]
- 49.Liao F. et al. STRL22 Is a Receptor for the CC Chemokine MIP-3α. Biochemical and Biophysical Research Communications 236, 212–217 (1997). 10.1006/bbrc.1997.6936 [DOI] [PubMed] [Google Scholar]
- 50.Yano J. et al. Vaginal Epithelial Cell-Derived S100 Alarmins Induced by Candida albicans via Pattern Recognition Receptor Interactions Are Sufficient but Not Necessary for the Acute Neutrophil Response during Experimental Vaginal Candidiasis. Infection and Immunity 82, 783–792 (2014). 10.1128/IAI.00861-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yano J., Lilly E., Barousse M. & Fidel P. L. Jr. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun 78, 5126–5137 (2010). 10.1128/iai.00388-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duncan J. A. et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol 182, 6460–6469 (2009). 10.4049/jimmunol.0802696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiang N., Yin T. & Chen T. Gardnerella vaginalis induces NLRP3 inflammasome-mediated pyroptosis in macrophages and THP-1 monocytes. Exp Ther Med 22, 1174 (2021). 10.3892/etm.2021.10609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vick E. J. et al. Gardnerella vaginalis triggers NLRP3 inflammasome recruitment in THP-1 monocytes. Journal of Reproductive Immunology 106, 67–75 (2014). 10.1016/j.jri.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 55.Vick E. J. et al. Gardnerella vaginalis triggers NLRP3 inflammasome recruitment in THP-1 monocytes. J Reprod Immunol 106, 67–75 (2014). 10.1016/j.jri.2014.08.005 [DOI] [PubMed] [Google Scholar]
- 56.Gelber S. E., Aguilar J. L., Lewis K. L. & Ratner A. J. Functional and phylogenetic characterization of Vaginolysin, the human-specific cytolysin from Gardnerella vaginalis. J Bacteriol 190, 3896–3903 (2008). 10.1128/jb.01965-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He W. t. et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Research 25, 1285–1298 (2015). 10.1038/cr.2015.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y. et al. Vaginal Microbiome Dysbiosis is Associated with the Different Cervical Disease Status. J Microbiol 61, 423–432 (2023). 10.1007/s12275-023-00039-3 [DOI] [PubMed] [Google Scholar]
- 59.Donders G. G. et al. Pathogenesis of abnormal vaginal bacterial flora. Am J Obstet Gynecol 182, 872–878 (2000). 10.1016/s0002-9378(00)70338-3 [DOI] [PubMed] [Google Scholar]
- 60.Aroutcheva A. et al. Defense factors of vaginal lactobacilli. Am J Obstet Gynecol 185, 375–379 (2001). 10.1067/mob.2001.115867 [DOI] [PubMed] [Google Scholar]
- 61.Tachedjian G., O’Hanlon D. E. & Ravel J. The implausible “in vivo” role of hydrogen peroxide as an antimicrobial factor produced by vaginal microbiota. Microbiome 6, 29 (2018). 10.1186/s40168-018-0418-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wan B., Wei L. J., Tan T. M., Qin L. & Wang H. Inhibitory effect and mechanism of Lactobacillus crispatus on cervical precancerous cells Ect1/E6E7 and screening of early warning factors. Infect Agent Cancer 18, 5 (2023). 10.1186/s13027-023-00483-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Crimi E., Benincasa G., Figueroa-Marrero N., Galdiero M. & Napoli C. Epigenetic susceptibility to severe respiratory viral infections and its therapeutic implications: a narrative review. Br J Anaesth 125, 1002–1017 (2020). 10.1016/j.bja.2020.06.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vicente-Duenas C., Hauer J., Cobaleda C., Borkhardt A. & Sanchez-Garcia I. Epigenetic Priming in Cancer Initiation. Trends Cancer 4, 408–417 (2018). 10.1016/j.trecan.2018.04.007 [DOI] [PubMed] [Google Scholar]
- 65.Stark R., Grzelak M. & Hadfield J. RNA sequencing: the teenage years. Nat Rev Genet 20, 631–656 (2019). 10.1038/s41576-019-0150-2 [DOI] [PubMed] [Google Scholar]
- 66.Torrini C. et al. Lactate is an epigenetic metabolite that drives survival in model systems of glioblastoma. Mol Cell 82, 3061–3076 e3066 (2022). 10.1016/j.molcel.2022.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dai X., Lv X., Thompson E. W. & Ostrikov K. K. Histone lactylation: epigenetic mark of glycolytic switch. Trends Genet 38, 124–127 (2022). 10.1016/j.tig.2021.09.009 [DOI] [PubMed] [Google Scholar]
- 68.Hagihara H. et al. Protein lactylation induced by neural excitation. Cell Rep 37, 109820 (2021). 10.1016/j.celrep.2021.109820 [DOI] [PubMed] [Google Scholar]
- 69.Zhang D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). 10.1038/s41586-019-1678-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patro R., Duggal G., Love M. I., Irizarry R. A. & Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419 (2017). 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frankish A. et al. GENCODE 2021. Nucleic Acids Res 49, D916–d923 (2021). 10.1093/nar/gkaa1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gentleman R. C. et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biology 5, R80 (2004). 10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.R: A language and environment for statistical computing. R Foundation for Statistical Computing; (2021). [Google Scholar]
- 74.Love M. I. et al. Tximeta: Reference sequence checksums for provenance identification in RNA-seq. PLOS Computational Biology 16, e1007664 (2020). 10.1371/journal.pcbi.1007664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Durinck S., Spellman P. T., Birney E. & Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191 (2009). 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marini F. & Binder H. pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinformatics 20, 331 (2019). 10.1186/s12859-019-2879-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ludt A., Ustjanzew A., Binder H., Strauch K. & Marini F. Interactive and Reproducible Workflows for Exploring and Modeling RNA-seq Data with pcaExplorer, Ideal, and GeneTonic. Current Protocols 2, e411 (2022). 10.1002/cpz1.411 [DOI] [PubMed] [Google Scholar]
- 78.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550 (2014). 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ulgen E., Ozisik O. & Sezerman O. U. pathfindR: An R Package for Comprehensive Identification of Enriched Pathways in Omics Data Through Active Subnetworks. Front Genet 10, 858 (2019). 10.3389/fgene.2019.00858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith J. P. et al. PEPATAC: an optimized pipeline for ATAC-seq data analysis with serial alignments. NAR Genom Bioinform 3, lqab101 (2021). 10.1093/nargab/lqab101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jiang H., Lei R., Ding S. W. & Zhu S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014). 10.1186/1471-2105-15-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Faust G. G. & Hall I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014). 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Amemiya H. M., Kundaje A. & Boyle A. P. The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354 (2019). 10.1038/s41598-019-45839-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu G., Wang L. G. & He Q. Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015). 10.1093/bioinformatics/btv145 [DOI] [PubMed] [Google Scholar]
- 87.Liao Y., Smyth G. K. & Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47, e47 (2019). 10.1093/nar/gkz114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bailey T. L. & Grant C. E. SEA: Simple Enrichment Analysis of motifs. bioRxiv, 2021.2008.2023.457422 (2021). 10.1101/2021.08.23.457422 [DOI] [Google Scholar]
- 90.Bailey T. L., Johnson J., Grant C. E. & Noble W. S. The MEME Suite. Nucleic Acids Res 43, W39–49 (2015). 10.1093/nar/gkv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weirauch M. T. et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). 10.1016/j.cell.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pinero J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res 48, D845–D855 (2020). 10.1093/nar/gkz1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lerdrup M., Johansen J. V., Agrawal-Singh S. & Hansen K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat Struct Mol Biol 23, 349–357 (2016). 10.1038/nsmb.3180 [DOI] [PubMed] [Google Scholar]
- 94.Nguyen N. T. T. et al. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res 46, W209–W214 (2018). 10.1093/nar/gky317 [DOI] [PMC free article] [PubMed] [Google Scholar]