

Abstract
Ceramides impact a diverse array of biological functions and have been implicated in disease pathogenesis. The enzyme neutral ceramidase (nCDase) is a zinc-containing hydrolase and mediates the metabolism of ceramide to sphingosine (Sph), both in cells and in the intestinal lumen. nCDase inhibitors based on substrate mimetics, for example C6-urea ceramide, have limited potency, aqueous solubility, and micelle-free fraction. To identify non-ceramide mimetic nCDase inhibitors, hit compounds from an HTS campaign were evaluated in biochemical, cell based and in silico modeling approaches. A majority of small molecule nCDase inhibitors contained pharmacophores capable of zinc interaction but retained specificity for nCDase over zinc-containing acid and alkaline ceramidases, as well as matrix metalloprotease-3 and histone deacetylase-1. nCDase inhibitors were refined by SAR, were shown to be substrate competitive and were active in cellular assays. nCDase inhibitor compounds were modeled by in silico DOCK screening and by molecular simulation. Modeling data supports zinc interaction and a similar compound binding pose with ceramide. nCDase inhibitors were identified with notably improved activity and solubility in comparison with the reference lipid-mimetic C6-urea ceramide.
Keywords: Neutral ceramidase, colorectal cancer, neutral ceramidase inhibitor, nCDase
Introduction
Bioactive lipids impact a diverse array of biological functions and have been implicated in disease pathogenesis. Among these mediators, sphingolipids have proven to be important in cancer development and progression. Sphingolipids are a family of membrane lipids that play an important role in transducing cellular signaling processes [1–5]. Sphingosine-1-phosphate (S1P) exerts tumor-promoting functions including promoting migration, cell proliferation, and anti-apoptotic activities. S1P is generated from sphingosine by sphingosine kinases (SK). In contrast to the functions of S1P, ceramide stimulates apoptosis, induces senescence, and inhibits cell proliferation [6]. Sphingomyelin and its downstream metabolic product ceramide can be modulated through diet manipulation where excess sphingomyelin or ceramide suppresses intestinal tumor development in mouse models [7–9]. More recent human epidemiologic evidence suggests that sphingomyelin consumption is associated with reduced incidence of colorectal cancer (CRC) [10], although both mouse and human studies give little insight into the mechanisms by which exogenous or endogenous ceramide exerts its effects. Genetic deletion or inhibition of nCDase suppresses tumor development [11, 12] which is associated with increased intracellular/tumoral abundance of ceramide and promotion of cancer cell death through apoptosis, mitophagy or necroptosis [13]. Similarly, nCDase knockout mice showed a 93% reduction in carcinogen-induced colon adenocarcinomas compared with wild-type mice and a significant reduction in aberrant crypt foci (p<0.0001) [11]. Finally, colon tumors from patients have a 50% reduction in ceramide levels compared to normal mucosa [9]. The findings suggest that pharmacological inhibition of nCDase in combination with a ceramide rich diet may be useful in the treatment or prevention of colorectal cancer, though precise mechanisms are not yet defined.
nCDase is particularly enriched at the mucosal surface of the intestinal epithelium, suggesting that gut luminal levels of ceramide are susceptible to its enzymatic activity and thus its degradation [1, 3, 5, 14, 15]. Genetic ablation in mice of the nCDase/ASAH2 gene inhibits intestinal metabolism of ceramide, leading to downstream increases in luminal ceramide and ceramide within the feces. nCDase/Asah2 null feces contained 53-fold greater ceramide than wild type mice (p=0.008), which was confirmed using fluorescent NBD-ceramide [16]. Intracellular ceramides, notably in the Golgi, ER and peroxisomes, also have been implicated in inflammation and cell death processes. Finally, ceramides also have been shown to act on immune cells and the microbiota resident within intestinal and colonic mucosal microenvironments. Understanding intestinal and colonic cell responses to increased ceramide levels is an overarching aim of this study where small molecule nCDase inhibitors are needed.
Three families of ceramidases have been identified (acid, alkaline, and neutral) that are distinguished by their pH optima, subcellular localization, divergent primary structures, mechanism, and function [17]. The major CDase in intestinal tissues is neutral ceramidase (nCDase) [18], which displays little sequence or structural homology to other proteins [19, 20]. In intestinal tissues, nCDase is located at the brush-border and is tethered to the plasma membrane by a single-pass transmembrane helix and an extended highly glycosylated 60-amino acid linker. The ceramidase active site is located on the extracellular, luminal side of intestinal villi where it can hydrolyze dietary ceramides and ceramides released by sphingomyelin metabolism [16]. The enzyme also is found in intracellular compartments and can act on cellular ceramides [21]. nCDase is very specific for the endogenous stereoisomer of ceramide [22–24]. Airola et al. have overexpressed, purified, and crystallized the extracellular catalytic domain of human nCDase at 2.6 Å resolution [25], defining the active site of human neutral ceramidase as a narrow, 20 Å deep, hydrophobic pocket with a Zn2+ ion at the base.
A critical issue impeding the evaluation of ceramidase metabolism in human diseases is the lack of small molecule specific inhibitors suitable for cell-based studies. For example, the widely used nCDase inhibitor C6-urea ceramide is highly lipophilic with limited potency, aqueous solubility, and pharmaceutic properties. To identify new nCDase inhibitors we identified 42 hit compounds through HTS [26]. Here we characterized different chemical classes of active nCDase inhibitors, examining specificity, cellular activity, and use the nCDase crystal structure [25] to model the binding of inhibitors to the nCDase catalytic active site.
Materials and Methods
Chemicals and cell lines
Chemical compounds were purchased from Life Chemicals, Enamine or MedChem Express. Exact masses of active compounds were confirmed by high resolution electrospray LC-mass spectrometry, using reverse phase chromatography in-line with a Thermo Q-Exactive HF orbital trap mass spectrometer.
Recombinant nCDase
The extracellular region (residues 99–780) of human nCDase was expressed in Sf9 cells grown in Sf-900™ III SFM media (Invitrogen) in suspension at 28°C with agitation (1). Secreted nCDase protein was purified from the media through HisTrap excel column (GE Healthcare) using an AKTA FPLC system. nCDase-containing fractions were further purified by size exclusion purification on a Hi-Load 26–60 Superdex 200 column equilibrated with 100 mM NaCl, 10 mM HEPES pH 7.0. nCDase fractions were pooled, concentrated to 70 μg/mL, and flash frozen. This purification method yields approximately 1mg (~0.1 enzymatic units; μmol/min) of purified protein from a 1-liter SF9 culture [25].
Biochemical nCDase enzymatic activity
The RBM assay [27] of nCDase activity used fluorogenic substrate RBM-labeled C16-ceramide where sodium periodate allows for the release of the product umbelliferone, a hydroxycoumarin measurable by simple fluorescence detection [28]. The substrate RBM 14C16 (N-((2S,3R)-1,3-dihydroxy-5-((2-oxo-2H-chromen-7-yl)oxy) pentan-2-yl) palmitamide; Avanti Polar Lipids)) was prepared in ethanol as a stock concentration of 200 μM in glass tubes. The substrate was dried under nitrogen and re-suspended in 50 μL of 2X reaction buffer 0.6% Triton-X100, 150 mM NaCl, 25 mM sodium phosphate pH 7.4 and 20μM of substrate, sonicated for 15 min, vortexed, and sonicated for an additional five min. nCDase inhibitors were pre-incubated with the enzyme before addition of substrate. 50 μL of substrate and 50 μL of enzyme with and without inhibitor are combined in a black 96 well plate and incubated for two hours at 37°C in the dark at a final substrate concentration of 10μM. At the end of the reaction, one hundred μL/well of 2.5 mg/mL sodium periodate (NaIO4, in 10 mM glycine, pH 10.6) were added for an additional 15-min at 37°C, resulting aldehyde to undergo β-elimination, allowing release of the fluorescent product umbelliferon. Fluorescence was measured using a microplate reader and an excitation wavelength of 360 nm and emission wavelength of 460 nm. Linearity of the fluorescence output was established using a ten-point dose response for compound IC50 and percent inhibition.
The potential for chemical compound auto-fluorescence, resulting in false-positive response, was overcome by secondary assay using the synthetic fluorescence-labelled ceramide, C12-NBD Ceramide (N-[12-[(7-Nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl]-ceramide; Avanti Polar Lipids) [29] prepared in chloroform as a 1 mM stock in a glass tube. The substrate was resuspended in 50 μL of 2X reaction buffer 0.6% Triton, 150 mM NaCl, 25 mM phosphate pH 7.4 and 20μM of substrate and sonicated 15 min. One pmol of C12-NBD substrate solution was dried down under nitrogen per reaction. Purified nCDase was diluted to a volume of 50 μL, mixed with 50 μL of C12-NBD substrate solution, and incubated at 37°C for 2 hours. The reaction was extracted chloroform:methanol (1:1), the organic phase dried and lipids dissolved in 25ul chloroform/methanol (2:1, v/v), 10 μL of the reaction chloroform/methanol extract loaded on a C18:1 Luna column (2.2mm x 100mm; Phenomenex) and isocratically eluted in 100% methanol. Fluorescent NBD-fatty acid product was quantitated by peak area and compared to NBD-fatty acid standards [28].
nCDase enzyme kinetics
Functional characterization of nCDase inhibitor SB-37 by Lineweaver-Burk analysis was performed using the RBM ceramidase assay described above varying substrate and inhibitor concentrations. Purified nCDase (7ng, 0.7x10-6 U) was preincubated for 15 min with or without inhibitor at the final concentration of 0.2, 0.5, and 5μM. Substrate was then added at the final concentration of 0.75, 1.25, 2.5, 5, 10, 20, and 40μM in Triton X100 buffer (0.6% Triton-X100, 150 mM NaCl, 25 mM sodium phosphate pH 7.4). After 2 hours of reaction, sodium periodate was added to quench unmetabolized substrate and fluorescence of the reaction product was measured. Reciprocal of activity (1/fluorescence) versus the reciprocal of substrate concentration (1/substrate concentration) was plotted.
Cell-based neutral ceramidase activity
Transformed human kidney cell line 293 were transfected with full length nCDase cDNA (pCMV-nCDase) and stably transfected cells isolated by antibiotic resistance co-selection. Suspension cells were grown in Expi293™ Expression Medium (Invitrogen). A mutant S354A nCDase construct, lacking expressed catalytic activity, was used as a negative control cell line [20]. Cells were seeded at 2,000 cells per well in a 96 well plate, both cell lines were incubated with candidate inhibitors and RBM 14C16 at 20 μM for 2 hours. Media were harvested and RBM 14C16 hydrolysis measured by fluorescence using an excitation wavelength of 360 nm and emission wavelength of 460 nm. Cells expressing catalytically inactive ceramidase were used as a control and to subtract background signal. T-test was used to assess statistical significance, comparing full length wild-type nCDase treated with vehicle DMSO or nCDase inhibitor.
Measurements of acid and alkaline CDase activities
Recombinant acid ceramidase (ASAH1) was expressed in HeLa cells that lack endogenous ceramidase activity, and microsomes were isolated for biochemical experiments, [27, 29, 30]. Briefly, for acid ceramidase assay, 76 μg protein was incubated with 50 μM of C12 Ceramide with or without inhibitors (5 and 50 μM) for 30 min. Reaction buffer was 50 mM sodium phosphate, 75 mM sodium chloride, and 0.05% NP-40, pH 4.5.
For measurement of alkaline ceramidase activity [31], microsomes were prepared from RT4 cell, a human epithelial urinary bladder cell line with high expression of alkaline ceramidase (ACER1) and indetectable expression of acid or neutral ceramidase. Thirty-five μg of protein was incubated with 50 μM of C16 ceramide and inhibitor, the buffer for alkaline ceramidase assay was 25 mM Tris-HCL, 5 mM CaCl2, 0.15% Triton X-100, pH 8.8. Heat inactivated enzyme was used as a control. Conversion of ceramide to sphingosine was measured by LC-MS/MS after lipid extraction, and the results were expressed in pmol of sphingosine produced per μg of protein per min. For both acid and alkaline ceramidases t-tests were used to evaluate significance.
Protein Preparation for Docking
The protein structure used for docking was the same as reported in Airola et al [25], based on chain B from pdb code 4WGK, containing a predicted pose for the ligand ceramide. Briefly, all solvent molecules from the x-ray structure, with the exception of Ca2+ and Zn2+, were removed and apo nCDase was energy minimized in the gas-phase (distance dependent dielectric constant = 4r) with the ff99SB [32] force field using the program sander in the Amber suite of programs [33]. There are two regions in chain B with missing electron density, but these were distal from the binding site, and backbone restraints were employed during minimizations and subsequent molecular dynamics, thus they were left incomplete. Minimization was performed for 1,000 steps, with a 1,000 kcal mol-1 Å-2 restraint on all heavy atoms to optimize hydrogen bonding networks and relax any potential clashes. A four-step preparation protocol [34, 35] was then employed to prepare the minimized protein for docking: In step 1, surfaces for each protein chain were generated with the program DMS [36, 37]. In step 2, potential sphere sites for orienting ligand anchors were generated using the program sphgen [38]. In step 3, a bounding box was constructed 20 Å in all directions from the furthest spheres in each sphere set. In step 4, a 0.3 Å resolution energy grid was generated at the bounding box dimensions using the program GRID [36] which speeds up docking calculations. Grid generation employed 6-9 Lennard-Jones coefficients (van der Waals interactions) and a distance dependent dielectric constant of ε = 4.0r (Coulombic interactions).
Ligand Preparation for Docking
The ligands to be docked were downloaded in a ready-to-dock mol2 format from the ZINC15 database [39]. Compound SB-34 did not have a mol2 structure available and was constructed by modifying SB-35 using the program Chimera [40]. Partial atomic charges (AM1-BCC; [41] method) for all ligands were computed using the sqm package [42] called by the program antechamber [43] in AMBER 16 [44].
Docking Protocols for Pose Prediction
The 17 compounds were docked to the target using DOCK6 [45] using the flexible ligand (FLX) docking protocol [46] in which the rigid body and torsional degrees of freedom of the ligand are sampled and the protein is held rigid. All low-energy conformations (poses) were saved. In addition to the standard DOCK6 grid energy score, pose analysis also employed the Footprint Similarity (FPS) scoring method which enables per-residue score comparisons (the molecular footprint) between docked ligands and a reference ligand [47].
Molecular Dynamics Protocols
The stability of the docked pose for SB-26 was assessed using solvated molecular dynamics (MD) simulations performed Amber16 [44]. Briefly, the programs tleap and antechamber [48] were used to assemble each complex, assign ff14SB [49] parameters to the protein, GAFF [50] parameters to the ligand, and TIP3P [51] parameters to water. The final system comprised 673 protein residues, 22,576 water molecules, 3 ions, and 1 ligand). The solvated complex was then subjected to a multi-stage MD equilibration protocol, similar to those employed previously by our group in other studies [52–56] to relax coordinates in a systematic way prior to running production simulations: (step 1) 5,000 steps of energy minimization with 5.0 kcal mol−1 Å−2 restraints, (step 2) 50,000 steps of MD with 5.0 kcal mol−1 Å−2 restraints on the non-hydrogen atoms of the ligand and protein, (step 3) 1,000 steps of minimization with 2.0 kcal mol−1 Å−2 restraints, (step 4) 1,000 steps of minimization with 0.1 kcal mol−1 Å−2 restraints, (step 5) 1,000 steps of minimization with 0.05 kcal mol−1 Å−2 restraints, (step 6) 50,000 steps of MD with 1.0 kcal mol−1 Å−2 restraints on the non-hydrogen atoms of the ligand and protein, (step 7) 50,000 steps of MD with 0.5 kcal mol−1 Å−2 restraints on the non-hydrogen atoms of the ligand and protein, and (step 8) 100,000 steps of MD with 0.1 kcal mol−1 Å−2 restraints on just the backbone carbon and nitrogen of the protein. Production simulations (step 9) were performed at constant pressure (Berendsen barostat) at 298.15K (Langevin thermostat), with a 2 femtosecond timestep and a 0.1 kcal mol−1 Å−2 restraint on protein backbone atoms only. Final production simulations were performed for 100ns in triplicate for a total of 300ns of simulation time. Simulations employed the SHAKE [57] algorithm, and an 8.0 Å cutoff for nonbonded energies. Frames were written to file every 2500 steps, for a total of 20,000 frames per simulation. CPPTRAJ [58] was used to prepare finalized MD trajectories for each simulation and analyze both protein backbone and ligand per-frame RMSDs.
Results
nCDase inhibitory compounds were evaluated using biochemical, cell-based, and molecular modeling approaches. To identify nCDase inhibitory pharmacophores, a chemical screen of ~670K compounds was performed at the Wertheim UF Scripps Screening center [59]. The catalytically active extracellular region (residues 99–780) of human nCDase was expressed in insect cells and the RBM-14C16 assay was used in 1536 well plate format [26] . We anticipated a very low hit rate, where nCDase has a ~20 Å long hydrophobic channel with interacts with the sphingoid and fatty acid chains of ceramide, leading to an electrophilic active site which recognizes the ceramide polar head group. Forty-two hit compounds showed a response greater than fifty percent at concentrations ~17 μM (Supplementary Table 1) and were selected for validation and further characterization using biochemical, cell-based, physicochemical and in silico modeling approaches.
Zinc binding motifs and specificity in nCDase inhibitory compounds
Trichostatin A, a potent pan-histone deacetylase (HDAC) inhibitor, was shown to have nCDase inhibitory activity. HDACs also contain an active site zinc required for activity, and HDAC inhibitors frequently contain Zn-binding hydroxamic groups or other related functionality to tightly coordinate Zn2+. We considered the importance of compound interactions with the nCDase active site Zn2+ required for catalytic activity [25]. Given the affinity of Zn2+ for Asp, Glu and citrate, the finding that carboxylic acid-containing compounds valproate, anacardamate and MG149 (data not shown), exhibited nCDase inhibitory activity was not unexpected. Similarly, nCDase activity of Trichostatin A, bearing a zinc-binding hydroxamate substituent is structurally reasonable. Compounds with zinc interactions have been characterized using binding coordinates from the PDB [60] and from SAR studies of zinc chelators [61]. Review of nCDase inhibitor structures reveal multiple potential zinc binding chemotypes repeatedly observed, as shown in Table 1. Of forty-two active compounds evaluated, twenty-six compounds contained established zinc binding motifs. These data support compound interaction with the active site Zn2+ as an important factor in nCDase inhibition.
Table 1:
Zinc interaction motifs in nCDase inhibitor active compounds with >50% inhibition at 17 uM, where individual compounds are grouped by motif and designated by PubChem CID.
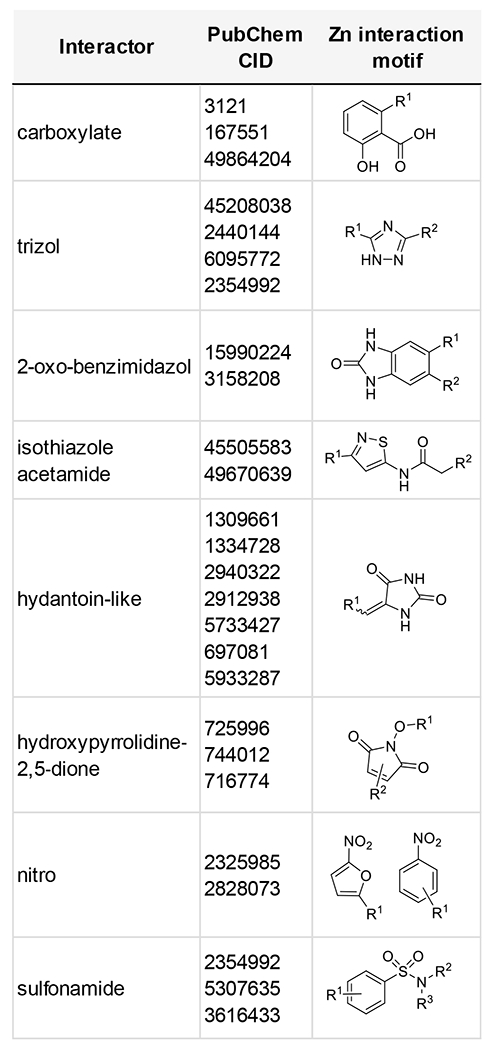
|
Isothiazole acetamide series SAR and mechanism of action
Thirteen representative nCDase pharmacophores were selected based on chemical tractability and a lack of undesired functional groups (compounds classified as for PAINS, or pan-assay interference compounds [62]). nCDase inhibitor structures, percent inhibition and physicochemical parameters are shown in Table 2. Compound activities were confirmed using distinct FRET and NBD-ceramide HPLC nCDase assays [28] (data not shown). Two compounds with acceptable IC50 values were prioritized, the isothiazole acetamides series based on SB-22 and nitrofuran carboxamide series based on SB-17. While both Trichostatin A and CPI-169 showed low uM IC50 values (not shown), both lack nCDase specificity, where Trichostatin A and CPI-169 have pan-HDAC and EZH2 family methyltransferase inhibitory activity respectively.
Table 2:
Active nCDase inhibitor chemotypes with maximal inhibition (standard deviations), molecular weight, PubChem CID, structure, polar surface area (TPSA) and cLogP.
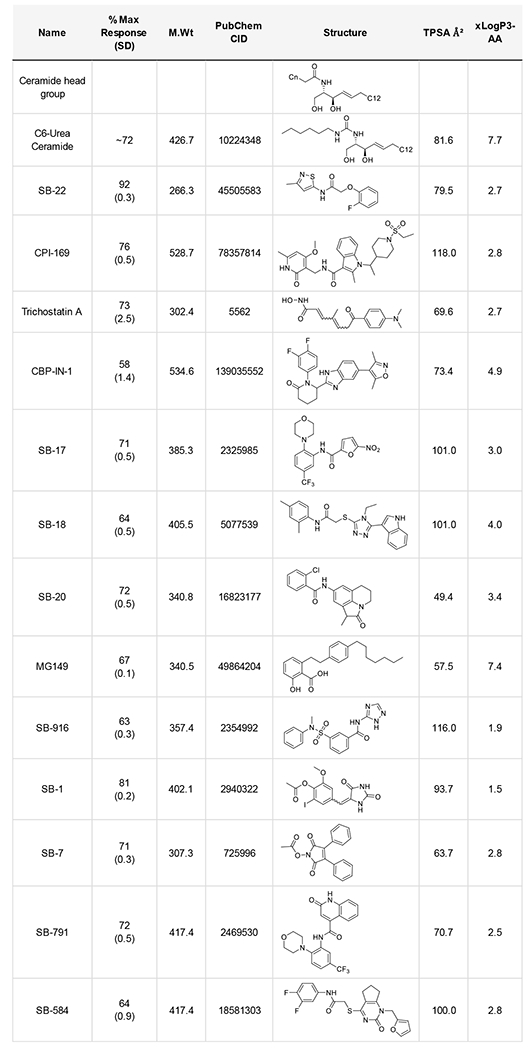
|
Structure activity relationship (SAR) studies were performed on the isothiazole acetamide series by 10-point IC50 and maximum inhibition determination in triplicate RBM assays, as shown in Table 3. All compound masses were verified by high resolution LC-MS/MS on an orbital trap instrument. We first examined SAR around the phenoxy position. Ortho substituted methyl (SB-26) or fluoro (SB-22) showed better activity than phenyl alone. Ortho substitution with bulkier groups reduced nCDase inhibitory activity (SB-28 and SB-27). Substitutions at the meta position were generally not favored and small substitutions at the para position, e.g. fluoro SB-30, were tolerated but not optimal. Bulkier substituents (methoxy, ethoxy) in the para position reduced activity (SB-32, SB-33). An ortho-para di-chloro analog (SB-23) showed reduced activity compared with the para-monochloro compound (SB-31). Increasing the amide linker lengths proved detrimental (SB-34). A heterocyclic replacement was also detrimental (SB-38). Increased potency (IC50 = 1.1 μM) and high maximal inhibition (91%) was found when a 2-chlorophenyl-substituted oxazole amide was appended to the isothiazole group (SB-37). However, preliminary SAR studies around this scaffold (not shown) suggest minimal tolerance for further changes in structure that, as even minor changes negatively impact nCDase inhibition.
Table 3:
Structure activity relationships in the isothiazole nCDase inhibitor series. Ten-point IC50 and maximum inhibition were determined by RBM biochemical assay. Compound masses were verified by high resolution orbital trap LC-MS/MS.
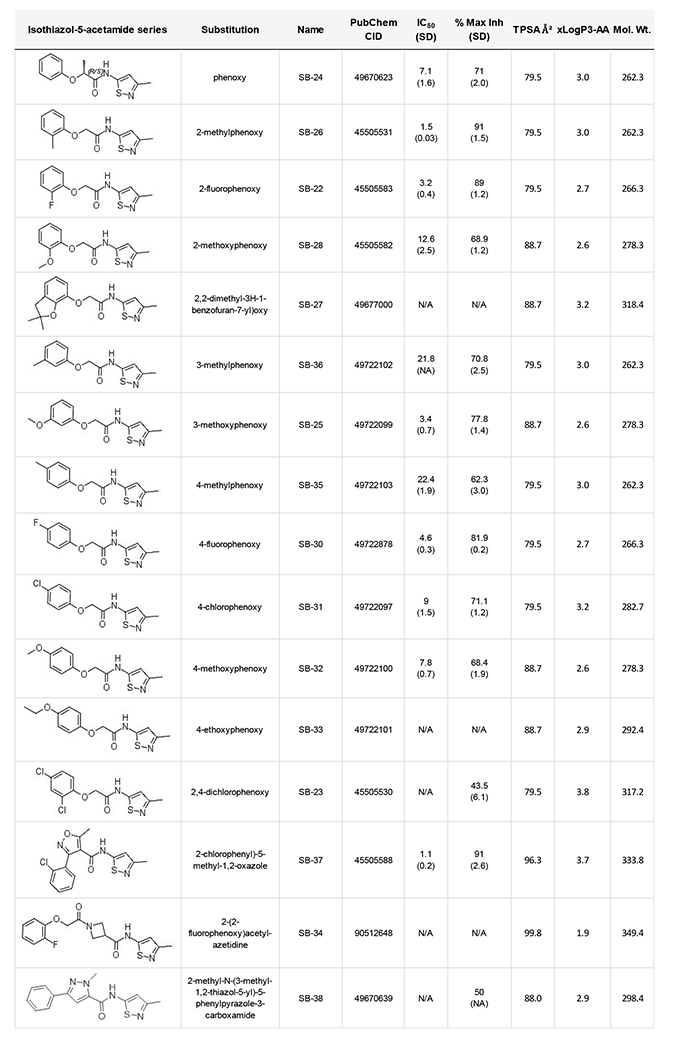
|
nCDase inhibitor specificity, mechanism of action and cell-based activity
We next asked if the most active nCDase inhibitors, isothiazole-containing SB-26 and nitro-containing SB-17, would inhibit other zinc containing enzymes. We performed activity measurements with matrix metalloproteinase 3 (MMP3) and histone deacetylase 1 (HDAC1), two zinc-containing enzymes where inhibitor interactions with zinc are well studied. We used fluorescence assays for MMP3 activity (Sigma, MAK291) and for HDAC1 (BPS Bioscience, 50061). As shown in Table 3, the nCDase inhibitors had little or no activity at a concentration of 100 μM (Table 4), while reference MMP3 compound MMP-3 inhibitor-II and HDAC inhibitor Trichostatin A (TSA) were both active.
Table 4:
nCDase inhibitors show little or no inhibitory activity toward zinc-dependent matrix metalloprotease-3 (MMP3) and histone deacetylase-1 (HDAC1). Measurement of MMP3 and HDAC1 IC50 and percent maximal inhibition activity with standard deviations (SD). IC50 values were not achieved with nCDase inhibitors SB-26 and SB-17. Little or no MMP3 or HDAC1 percent maximal inhibition was observed with 100uM SB-26 or SB-17 enzymes.
| Assay | Compound | IC50 μM (SD) |
% Max Inhibition (SD) |
|---|---|---|---|
| MMP3 inhibition | MMP-3 Inhibitor II CAS 161314-17-6 |
6.4 (0.4) |
87.1 (1.5) |
| SBU-26 | na | 2.1 (0.4) |
|
| SBU-17 | na | 2.9 (0.3) |
|
| HDAC1 inhibition | Trichostatin A | 0.9 (0.06) |
69.0 (3.0) |
| SBU-26 | na | 0.9 (5.5) |
|
| SBU-17 | na | 7.0 (1.4) |
The human ceramidase gene family is comprised of neutral ceramidase (ASAH2), acid ceramidase (ASAH1), and alkaline ceramidases 1-3 (ACER1-3) [17]. Alkaline ceramidases also harbor an active site zinc required for activity [30, 63]. We evaluated active nCDase inhibitors nitro-containing SB-17 and isothiazole-containing SB-22 and SB-26 against acid (ASAH1) or alkaline ceramidase (ACER1), measuring sphingosine production. Microsomes were isolated from HeLa cells specifically expressing acid (ASAH1) or alkaline ceramidases (ACER1) and incubated with C16 ceramide with or without nCDase inhibitor (Figure 1). Sphingosine production by ceramidases was measured by reverse phase C8 LC-MS/MS. Little or no inhibition of acid or alkaline ceramidase-1 activity by either of the two nCDase inhibitor classes was observed (p>0.2). Therefore, these two compounds demonstrate significant specificity towards nCDase among the members of the extended family of CDases.
Figure 1:
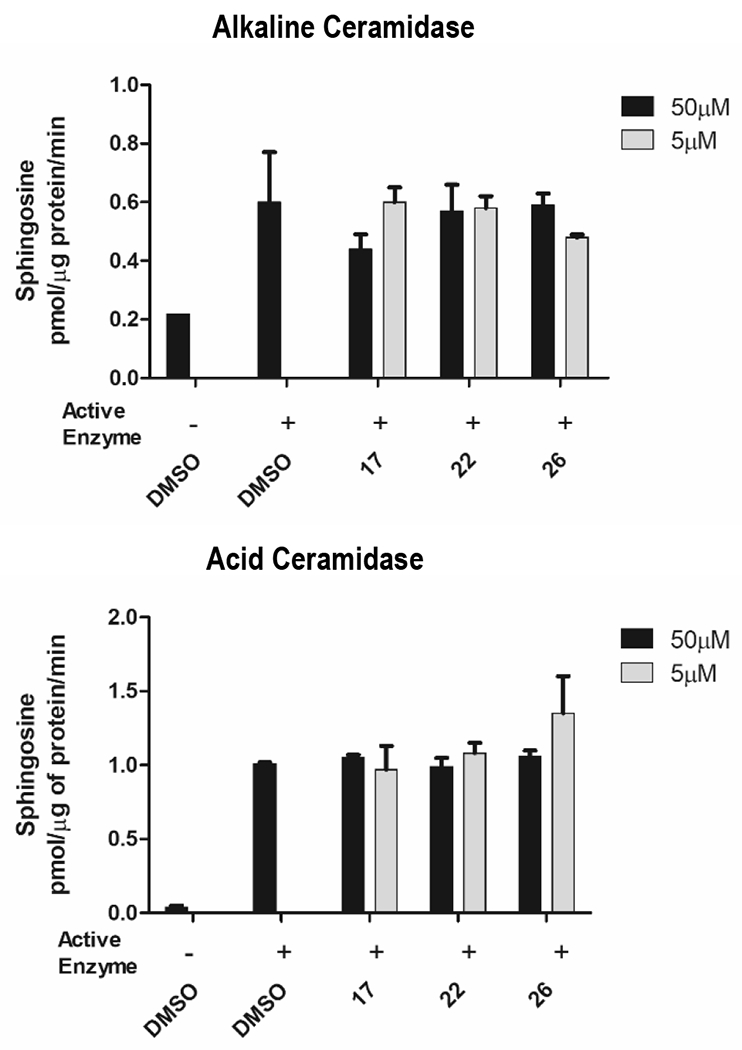
nCDase inhibitory compounds (SB-17, SB-22, SB-26) show little or no activity for activity against alkaline and acid ceramidase microsomes at concentrations of 5uM or 50uM. Hydrolysis of C16 ceramide substrate to sphingosine was measured by LC-MS/MS, as described in Materials and Methods, and expressed as pmol/g of protein/min.
We expected that nCDase inhibitors would show competitive binding with ceramide for active site occupancy. Active nCDase inhibitor SB-37 (Figure 2A) was selected for Lineweaver Burk analysis using the RBM ceramidase assay. Purified nCDase (7ng, 0.7x10-6 U) was preincubated for 15 min with or without inhibitor at the final concentration of 0.2, 0.5, and 5μM. Substrate was added at 0.75, 1.25, 2.5, 5, 10, 20, and 40μM. Reciprocal of activity (1/fluorescence) was plot against reciprocal of substrate concentration (1/substrate concentration). As shown in Figure 2B, the isothiazole acetamide SB-37 was found to be competitive with ceramide.
Figure 2.
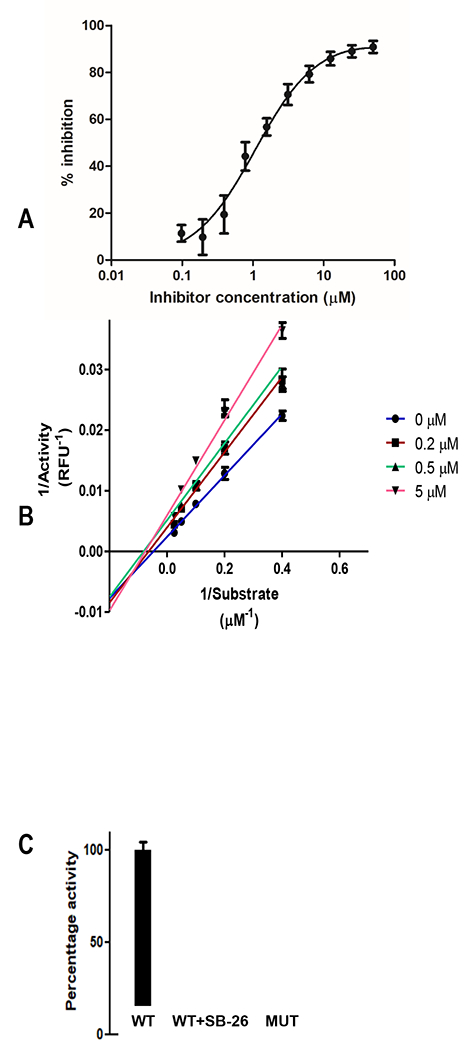
A. IC50 curve for isothiazole SB-37 by RBM assay. B. Lineweaver Burk plot of SB-37 indicating a substrate competition mode of action. Enzyme was incubated with nCDase inhibitor SB-37 (0.2, 0.5, and 5μM) and substrate added at the final concentration of 0.75, 1.25, 2.5, 5, 10, 20, and 40μM in Triton X100 buffer. C. 293 cells were transiently transfected with wild type (WT) and inactive mutant human nCDase CMV expression constructs and ceramidase activity measured by RBM assay, with and without SB-26 (30uM).
We measured nCDase inhibition in the context of full length cell membrane bound nCDase, in intact 293Expi cells, where we transiently over-expressed nCDase-WT or inactive nCDase mutant S354A [20] enzyme. Figure 2C shows SB-26 inhibition of full length nCDase (87.9% inhibition, +/−0.1 %; p=0.001) at a concentration of 30uM. The control inactive mutant nCDase displayed little or no activity. A similar analysis using a related isothiazole SB-22 showed 41.8% inhibition (+/− 3.7%; p <0.001) at a concentration of 5uM, again with no nCDase activity in the S354A nCDase mutant lysate.
Predicted binding geometries for nCDase inhibitors
Ligand binding pose prediction for this system relied on a multistage protocol based on consideration of energetics, visual information, and experimental SAR data. We reasoned that structurally related compounds would be expected to have a similar binding geometry; thus, it was desirable to generate a “consensus” pose across all ligands. The first step in the process was determining which ligand molecule(s), and associated predicted poses, could be used as the parent “reference” for re-scoring other docked molecules using footprint similarity (FPS) scoring. To achieve this, all docked conformations for all molecules were rank ordered by DOCK grid energy [49]. For each ligand, the best-ranked conformation with the most favorable electrostatic interaction (−3.5 kcal/mol or lower) was then visually examined. Based on this analysis, we ultimately chose SB-22 (the original isothiazole hit compound) as the reference since it showed better experimental activity than SB-24. The second step in the process involved computing the FPS score [52] between SB-22 and all saved conformations for the remaining 15 molecules. The objective was to identify those ligand poses with similar energetic interaction signatures as the SB-22 reference which typically correlates with similar 3D geometries.
Figure 3A highlights the consensus pose for 14 of the 17 ligands docked into nCDase using the above protocol. As expected, the procedure yielded a well clustered group of ligands that all had similar interactions in the binding site at select key positions. For example, the isothiazole rings coordinate Zn2+ (dashed cyan) and are within hydrogen bonding distance of H196 and Y579 (dashed magenta lines). An additional H-bond is predicted between the amide hydrogen and G122. For comparison, Figure 3B shows the modeled binding pose for ceramide from Airola et al. [25]. Although ceramide was not used in the present work during generation of the ligand consensus pose, there is striking similarity between the two geometries (Figure 3A versus 3B) including H-bonds with H196 and G122, completion of the octahedral coordination with zinc, and significant structural overlap with one of the fatty acid tails.
Figure 3.
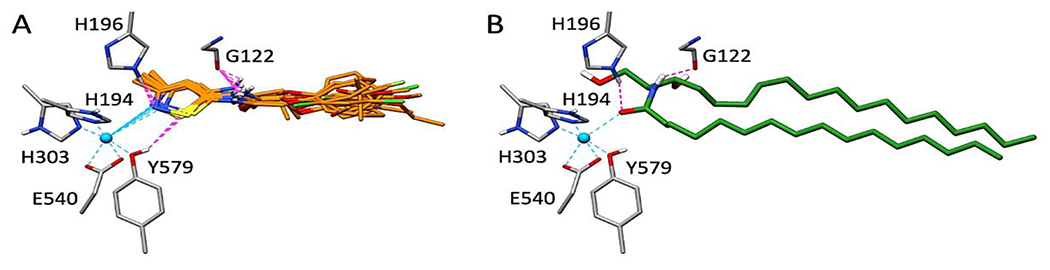
A. Predicted consensus pose for 14 out of 17 isothiazole nCDase inhibitors. B. Modeled pose for ceramide from Airola et al. (1). Proposed Zn2+ interactions in dashed cyan and hydrogen bonding in dashed magenta. Docked ligands in orange, ceramide in green, protein residues in gray.
Binding geometry comparison for ligands not adopting a consensus pose
Three of the molecules studied here (SB-34, SB-37, SB-17) did not adopt as tight an overlap with the consensus pose shown in Figure 3A although they all occupied the same binding site. Figure 4 compares poses for each of these three with compound SB-26, one of the most active ligands in the group. For ligand SB-34 (Figure 4 left) the adopted pose is flipped by about 180 degrees which yields a significantly different interaction pattern and may explain its relative lack of nCDase inhibitory activity. Interestingly, this is the only member in the class that contains a central azetidine ring and an additional carbonyl group. Ligand SB-37 also contains an added central ring, in this case a methylated isoxazole, which leads to a pose shifted downward into the site (Figure 4 middle), although the pose for SB-37 still maintains the expected interaction with zinc. Molecule SB-17, on the other hand, (Figure 4 right) has a significantly different scaffold compared to the rest of the molecules examined. This results in different interactions, though reasonable structural overlap with most of SB-26 is maintained. Zinc coordination here is achieved via SB-17’s nitro group.
Figure 4.
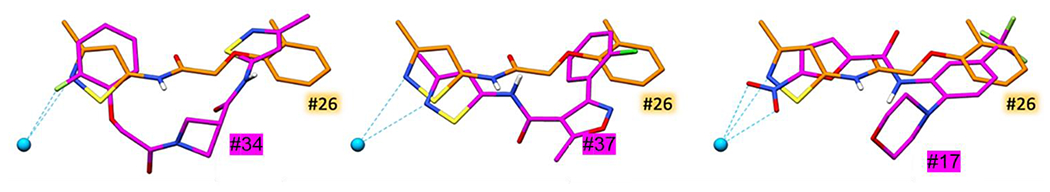
Comparison of docked poses for ligand SB-26 (#26; orange) with SB-34, SB-37, and SB-17 (#34, #37, #17; magenta).
Pose Stability with Molecular Dynamics
To further gauge the compatibility of the predicted consensus pose, we performed solvated dynamics (MD) simulations for the complex containing ligand SB-26 with nCDase. Three separate simulations were performed, with different random seeds, which yields different MD trajectories. Based on prior studies from our lab, we have shown that simulation times as short as 10-20ns are sufficient to evaluate the geometric and energetic stability of ligand poses in protein binding sites (48, 49, 58). Nevertheless, we increased the simulation time to 100ns for each replicate for a total of 300ns. Encouragingly, in most cases, the simulations yielded stable ligand RMSDs that plateaued around 2-2.5 Å (Figure 5 left) relative to the originally docked pose. Figure 5 right shows an overlaid set of frames (N=2000), from one of the three simulations of ligand SB-26, which corresponds to every 10th frame from the 100 ns trajectory. Despite the relatively large size of the binding pocket, which originally contained a much larger ceramide molecule, the significant stability observed for ligand SB-26 is indicative of an energetically and geometrically favorable pose. Taken together, these results provide strong support for the proposed consensus pose geometry for this class of inhibitors.
Figure 5.
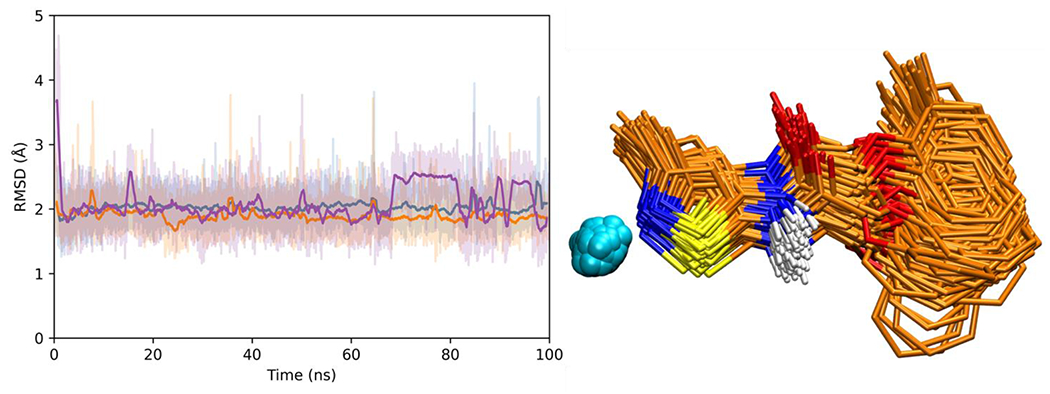
(Left) Ligand RMSD plots from three independent 100 ns MD simulations (replicates) for SB-26 docked with nCDase. Solid colored lines represent moving boxcar averages (window size = 100 frames) for each trajectory. (Right) Frame overlay plot (1000 evenly spaced frames) for SB-26 (orange) and zinc (cyan) from one MD simulation. Protein residues hidden for clarity.
DOCK nCDase structure and inhibitor action model
SB-17 and SB-37 were selected as prototypical chemical classes. Figure 6A compares the theoretical binding poses for these two ligands (magenta), relative to SB-26 (orange), and ceramide (green) [25]. Interestingly, the core scaffolds of SB-17 and SB-37 both include ring branching that allows them to engage residues across the opening of the nCDase pocket which is hypothesized to accommodate the saturated alkyl chain of ceramide (Figure 6A, left; alkyl chain in green).
Figure 6.
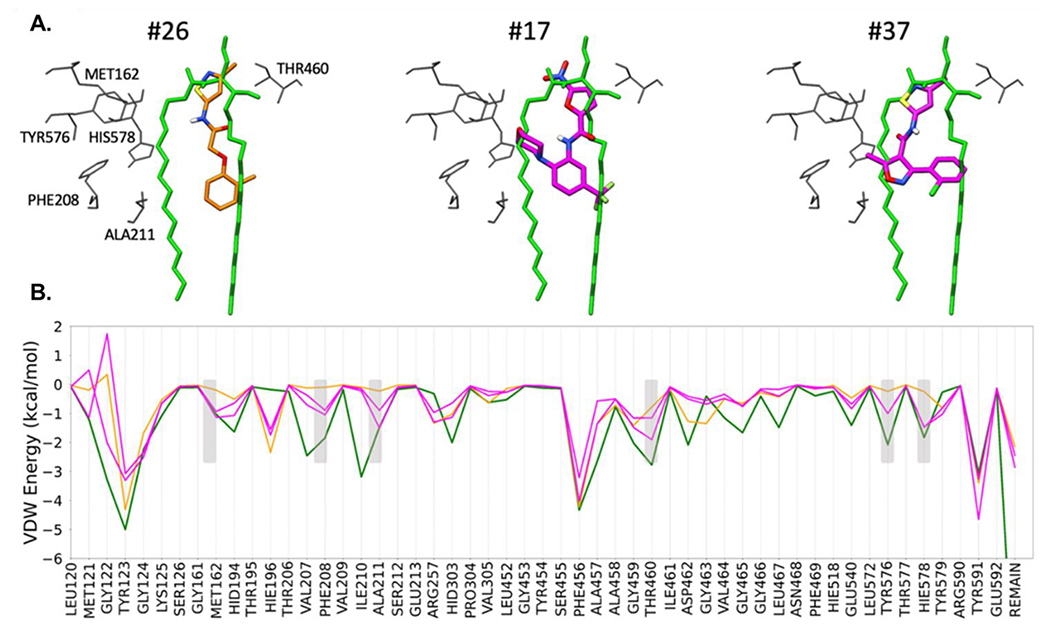
A. Proposed binding geometries for compound SB-26 (left, orange), SB-17 (middle, magenta), and SB-37 (right, magenta) with nCDase in comparison to the previously published modeled pose for ceramide (green) from Airola et al. (1). B. Protein-ligand van der Waals energies, on a per-residue basis (termed footprints) for all four species (SB-26, SB-17, SB-37, ceramide).
To help quantify these additional interactions, we used DOCK6 to decompose the protein-ligand van der Waals energies, on a per-residue basis (termed footprints) or all four inhibitors (SB-26, SB-17, SB-37, C6-urea ceramide). As shown in Figure 6B, increased favorable interactions [47] of ca −0.75 kcal or better (shaded rectangles) were observed at six positions (MET162, PHE208, ALA211, THR460, TYR576, HIS578) for inhibitor SB-17 or SB-37 (magenta lines) which more closely mimic the peaks made by ceramide (green line). In contrast, the interaction energies at these six positions for SB-26 (orange line) are less favorable. Taken together, the data suggest that the enhanced activity for SB-17 and SB-37, relative to SB-26, could be due to more complete “molecular mimicry” of the native ceramide substrate.
Discussion
Lipids impact a diverse array of biological functions and have been implicated in disease pathogenesis. Among these mediators, sphingolipids have proven to be important in cancer development and progression [2, 5]. Sphingomyelin and its downstream metabolic product ceramide can be modulated through diet manipulation in mouse models [7, 8, 11] and in human epidemiological studies [10], although both mouse and human studies give little insight into the mechanisms and cell compartments where which ceramide exerts its anti-cancer effects. In this study, chemotypes with nCDase inhibitory activity (Supplementary Table 1) were enriched in structural chemotypes capable of Zn+2 interaction (Table 1), supporting zinc interaction as a key structural element in nCDase inhibitors. After structural prioritization and a PAINS analysis, fourteen compounds were selected for further evaluation (Table 2). Low molecular weight, soluble and Lipinski guideline compliant isothiazoles SB-26 and SB-37 and nitrofuran SB-17 demonstrated IC50 values of 1.1, 1.5 and 3.0 μM respectively, and are approximately 40-50 times more potent than the current lipid-mimetic tool compound C6-urea ceramide. Isothiazoles were substrate competitive and showed cell-based activity toward full length membrane nCDase (Figure 2). The nCDase inhibitors SB-26 and SB-17 were specific for neutral ceramidase with little or no activity toward acid ceramidase (ASAH1), zinc-containing alkaline ceramidase (ACER1) (Figure 1), matrix metallopeptidase-3 (MMP3) and histone deacetylase-1 (HDAC1) (Table 4). Both neutral and alkaline ceramidases require zinc at the active site; nCDase is coordinated by histidines at positions 194 and 303, glutamic acid at 540 and tyrosine at 579, while alkaline ceramidases are coordinated by three histidines positions conserved between ACERs 1-3. The ceramidases have differing cellular localizations and functions, plasma membrane for nCDase/ASAH2, lysosomal for acid ceramidase ASAH1 and endoplasmic reticulum, Golgi and both for ACERs 1, 2 and 3 respectively. The specificity of these compounds for nCDase over alkaline or acid ceramidases is not unexpected, given the lack of sequence homology between the ceramidase family of hydrolases, even in the active sites.
SAR around isothiazoles SB-22 and SB-26 showed that even subtle changes in structure had marked impacts on nCDase inhibitory activity (Table 3). For example, ortho 2-methylphenoxy substituted SB-26 showed an IC50 of 1.5 μM, while meta 3-methylphenoxy substituted SB-36 and para 4-methylphenoxy substituted SB-35 had IC50s of 21.8 and 22.4 respectively. Most modifications of the acetamide linkage were unsuccessful (data not shown) with the exception of SB-37 where the oxazole linker insertion was favorable, with an IC50 of 1.1 μM. Overall, the SAR for all chemical series evaluated was restrictive, suggesting topological constraints on nCDase active site occupancy, which may be in keeping with the high degree of specificity of nCDase for ceramide over related sphingolipids.
Using molecular modeling, we propose a binding mode for the isothiazole nCDase inhibitor series, which align along a single chain of the native ceramide molecule. Two of the characterized compounds, SB-17 and SB-37, are branched resulting in a slightly different pose with increased interactions across the pocket (Figures 4 and 6), providing a potential physical explanation for their enhanced activity. Using SB-26, we also offer support for this pose through dynamic stability as assessed by 100 ns of backbone restrained, explicit solvent molecular dynamics (Figure 5). Several active nCDase inhibitor classes, for example containing triazoles, hydantoin-like groups, oxo-pyrimidyl, dioxo-pyrrole potential zinc-binding motifs, remain to be evaluated in future SAR studies. The identification of different classes of small molecule, soluble nCDase inhibitors, with selectivity over other ceramidases and zinc containing enzymes, will be useful in evaluating the cellular functions of nCDase.
Supplementary Material
Acknowledgements
We thank Amalia Saleh for technical assistance with nCDase enzyme assays.
Funding Sources
This work was supported by NIH grants R01CA221948 (J.D.H, L.S.), P01CA097132 (Y.A.H.) R35GM126906 (R.C.R) and R35GM128666 (M.V.A.) with student support from T32GM136572 (E.M.B) to J.D.B. Additional support was provided by the Stony Brook Cancer Center, and the Stony Brook Renaissance School of Medicine Department of Pathology. The authors would also like to thank the Stony Brook University Office of the Vice President for Research, Stony Brook Research Computing and Cyberinfrastructure, and the Institute for Advanced Computational Science at Stony Brook University for access to the high-performance LIred and SeaWulf computing systems, the latter of which was made possible by a National Science Foundation grant (#1531492).
Abbreviations:
- nCDase
neutral ceramidase
- Sph
sphingosine
- Cer
ceramide
- NBD
nitrobenzo-2-oxa-1,3-diazole
- SAR
structure activity relationship
- CRC
colorectal cancer
- S1P
sphingosine-1-phosphate
- NBD
nitrobenzo-2-oxa-1,3-diazole
- RBM14C16
N-((2S,3R)-1,3-dihydroxy-5-((2-oxo-2H-chromen-7-yl)oxy) pentan-2-yl) palmitamide
- FRET
fluorescence resonance energy transfer
- SK
sphingosine kinase
- HDAC
histone deacetylase
Footnotes
Credit author statement:
Conceptualization: YAH, JDH, RCR, LS, TS; Investigation: NC, JDB, RR, YO, Y-MC, RX, MS, CC, CM, ES, CA; Data analysis: JDH, RCR, YAH, LS, TS, TB, MVA; Writing and Editing: JDH, RCR, YAH, LS, TS, TB, MVA, PJT; Funding acquisition: JDH, LS, YAH, RCR
References
- [1].Duan RD, Nilsson A, Metabolism of sphingolipids in the gut and its relation to inflammation and cancer development, Prog Lipid Res 48(1) (2009) 62–72. [DOI] [PubMed] [Google Scholar]
- [2].Hannun YA, Obeid LM, Principles of bioactive lipid signalling: lessons from sphingolipids, Nature reviews Molecular cell biology 9(2) (2008) 139–150. [DOI] [PubMed] [Google Scholar]
- [3].Morad SA, Cabot MC, Ceramide-orchestrated signalling in cancer cells, Nature Reviews Cancer 13(1) (2012) 51–65. [DOI] [PubMed] [Google Scholar]
- [4].Ogretmen B, Hannun YA, Biologically active sphingolipids in cancer pathogenesis and treatment, Nature Reviews Cancer 4(8) (2004) 604–616. [DOI] [PubMed] [Google Scholar]
- [5].Spiegel S, Merrill AH Jr., Sphingolipid metabolism and cell growth regulation, FASEB J 10(12) (1996) 1388–97. [DOI] [PubMed] [Google Scholar]
- [6].Li F, Zhang N, Ceramide: Therapeutic Potential in Combination Therapy for Cancer Treatment, Curr Drug Metab 17(1) (2015) 37–51. [DOI] [PubMed] [Google Scholar]
- [7].Schmelz EM, Sphingolipids in the chemoprevention of colon cancer, Frontiers in bioscience: a journal and virtual library 9 (2004) 2632–2639. [DOI] [PubMed] [Google Scholar]
- [8].Schmelz EM, Dillehay DL, Webb SK, Reiter A, Adams J, Merrill AH Jr., Sphingomyelin consumption suppresses aberrant colonic crypt foci and increases the proportion of adenomas versus adenocarcinomas in CF1 mice treated with 1,2-dimethylhydrazine: implications for dietary sphingolipids and colon carcinogenesis, Cancer Res 56(21) (1996) 4936–41. [PubMed] [Google Scholar]
- [9].Zhang P, Li B, Gao S, Duan RD, Dietary sphingomyelin inhibits colonic tumorigenesis with an up-regulation of alkaline sphingomyelinase expression in ICR mice, Anticancer Res 28(6A) (2008) 3631–5. [PubMed] [Google Scholar]
- [10].Lu MS, Fang YJ, Pan ZZ, Zhong X, Zheng MC, Chen YM, Zhang CX, Choline and betaine intake and colorectal cancer risk in Chinese population: a case-control study, PLoS One 10(3) (2015) e0118661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Garcia-Barros M, Coant N, Kawamori T, Wada M, Snider AJ, Truman JP, Wu BX, Furuya H, Clarke CJ, Bialkowska AB, Ghaleb A, Yang VW, Obeid LM, Hannun YA, Role of neutral ceramidase in colon cancer, FASEB J 30(12) (2016) 4159–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schmelz EM, Roberts PC, Kustin EM, Lemonnier LA, Sullards MC, Dillehay DL, Merrill AH Jr., Modulation of intracellular beta-catenin localization and intestinal tumorigenesis in vivo and in vitro by sphingolipids, Cancer Res 61(18) (2001) 6723–9. [PubMed] [Google Scholar]
- [13].Ogretmen B, Sphingolipid metabolism in cancer signalling and therapy, Nat Rev Cancer 18(1) (2018) 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hannun YA, Obeid LM, Many ceramides, Journal of Biological Chemistry 286(32) (2011) 27855–27862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yuyama K, Mitsutake S, Igarashi Y, Pathological roles of ceramide and its metabolites in metabolic syndrome and Alzheimer’s disease, Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1841(5) (2014) 793–798. [DOI] [PubMed] [Google Scholar]
- [16].Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, Bielawski J, Bielawska A, Hannun YA, Proia RL, Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids, J Biol Chem 281(11) (2006) 7324–31. [DOI] [PubMed] [Google Scholar]
- [17].Mao C, Obeid LM, Ceramidases: regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate, Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1781(9) (2008) 424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].García-Barros M, Coant N, Truman J-P, Snider AJ, Hannun YA, Sphingolipids in colon cancer, Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].El Bawab S, Roddy P, Qian T, Bielawska A, Lemasters JJ, Hannun YA, Molecular cloning and characterization of a human mitochondrial ceramidase, Journal of Biological Chemistry 275(28) (2000) 21508–21513. [DOI] [PubMed] [Google Scholar]
- [20].Galadari S, Wu B.x.a.x., Mao C, Roddy P, El Bawab S, Hannun Y.x.a.a., Identification of a novel amidase motif in neutral ceramidase, Biochem. J 393 (2006) 687–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sakamoto W, Coant N, Canals D, Obeid LM, Hannun YA, Functions of neutral ceramidase in the Golgi apparatus, J Lipid Res 59(11) (2018) 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].El Bawab S, Bielawska A, Hannun YA, Purification and characterization of a membrane-bound nonlysosomal ceramidase from rat brain, Journal of Biological Chemistry 274(39) (1999) 27948–27955. [DOI] [PubMed] [Google Scholar]
- [23].El Bawab S, Usta J, Roddy P, Szulc ZM, Bielawska A, Hannun YA, Substrate specificity of rat brain ceramidase, Journal of lipid research 43(1) (2002) 141–148. [PubMed] [Google Scholar]
- [24].Usta J, Bawab SE, Roddy P, Szulc ZM, Hannun Y, A, A. Bielawska, Structural requirements of ceramide and sphingosine based inhibitors of mitochondrial ceramidase, Biochemistry 40(32) (2001) 9657–9668. [DOI] [PubMed] [Google Scholar]
- [25].Airola MV, Allen WJ, Pulkoski-Gross MJ, Obeid LM, Rizzo RC, Hannun YA, Structural basis for ceramide recognition and hydrolysis by human neutral ceramidase, Structure 23(8) (2015) 1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Otsuka Y, Airola MV, Choi YM, Coant N, Snider J, Cariello C, Saied EM, Arenz C, Bannister T, Rahaim R Jr., Hannun YA, Shumate J, Scampavia L, Haley JD, Spicer TP, Identification of Small-Molecule Inhibitors of Neutral Ceramidase (nCDase) via Target-Based High-Throughput Screening, SLAS Discov (2020) 2472555220945283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bedia C, Casas J, Garcia V, Levade T, Fabrias G, Synthesis of a novel ceramide analogue and its use in a high-throughput fluorogenic assay for ceramidases, Chembiochem 8(6) (2007) 642–8. [DOI] [PubMed] [Google Scholar]
- [28].Simoes M, Saleh A, Choi Y-M, Airola MV, Haley JD, Coant N, Measurement of neutral ceramidase activity in vitro and in vivo, Analytical Biochemistry (2022). [DOI] [PubMed] [Google Scholar]
- [29].Tani M, Okino N, Mitsutake S, Ito M, Specific and sensitive assay for alkaline and neutral ceramidases involving C12-NBD-ceramide, J Biochem 125(4) (1999) 746–9. [DOI] [PubMed] [Google Scholar]
- [30].Gebai A, Gorelik A, Li Z, Illes K, Nagar B, Structural basis for the activation of acid ceramidase, Nat Commun 9(1) (2018) 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sun W, Jin J, Xu R, Hu W, Szulc ZM, Bielawski J, Obeid LM, Mao C, Substrate specificity, membrane topology, and activity regulation of human alkaline ceramidase 2 (ACER2), J Biol Chem 285(12) (2010) 8995–9007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C, Comparison of multiple Amber force fields and development of improved protein backbone parameters, Proteins 65(3) (2006) 712–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ, The Amber biomolecular simulation programs, Journal of Computational Chemistry 26(16) (2005) 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Brozell SR, Mukherjee S, Balius TE, Roe DR, Case DA, Rizzo RC, Evaluation of DOCK 6 as a pose generation and database enrichment tool, Journal of computer-aided molecular design 26(6) (2012) 749–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mukherjee S, Balius TE, Rizzo RC, Docking validation resources: protein family and ligand flexibility experiments, J Chem Inf Model 50(11) (2010) 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Meng EC, Shoichet BK, Kuntz ID, Automated docking with grid-based energy evaluation, Journal of Computational Chemistry 13(4) (1992) 505–524. [Google Scholar]
- [37].Richards FM, Areas, volumes, packing and protein structure, Annu Rev Biophys Bioeng 6 (1977) 151–76. [DOI] [PubMed] [Google Scholar]
- [38].DesJarlais RL, Sheridan RP, Seibel GL, Dixon JS, Kuntz ID, Venkataraghavan R, Using shape complementarity as an initial screen in designing ligands for a receptor binding site of known three-dimensional structure, J. Med. Chem. 31(Copyright (C) 2012 American Chemical Society (ACS). All Rights Reserved.) (1988) 722–729. [DOI] [PubMed] [Google Scholar]
- [39].Sterling T, Irwin JJ, ZINC 15--Ligand Discovery for Everyone, J Chem Inf Model 55(11) (2015) 2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera-A visualization system for exploratory research and analysis, J. Comput. Chem 25 (2004) 1605–1612. [DOI] [PubMed] [Google Scholar]
- [41].Jakalian A, Jack DB, Bayly CI, Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation, J Comput Chem 23(16) (2002) 1623–41. [DOI] [PubMed] [Google Scholar]
- [42].Walker RC, Crowley MF, Case DA, The implementation of a fast and accurate QM/MM potential method in Amber, J Comput Chem 29(7) (2008) 1019–31. [DOI] [PubMed] [Google Scholar]
- [43].Wang J, Wang W, Kollman PA, Case DA, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell 25 (2006) 247–260. [DOI] [PubMed] [Google Scholar]
- [44].Case DA, Betz RM, Botello-Smith W, Cerutti DS, Cheatham III TE, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, York DM, Kollman PA, AMBER 16, University of California, San Francisco: (2016). [Google Scholar]
- [45].Allen WJ, Balius TE, Mukherjee S, Brozell SR, Moustakas DT, Lang PT, Case DA, Kuntz ID, Rizzo RC, DOCK 6: Impact of new features and current docking performance, J Comput Chem 36(15) (2015) 1132–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mukherjee S, Balius TE, Rizzo RC, Docking validation resources: protein family and ligand flexibility experiments, Journal of chemical information and modeling 50(11) (2010) 1986–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Balius TE, Mukherjee S, Rizzo RC, Implementation and evaluation of a docking-rescoring method using molecular footprint comparisons, J Comput Chem 32(10) (2011) 2273–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wang J, Wang W, Kollman PA, Case DA, Automatic atom type and bond type perception in molecular mechanical calculations, J Mol Graph Model 25(2) (2006) 247–60. [DOI] [PubMed] [Google Scholar]
- [49].Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J Chem Theory Comput 11(8) (2015) 3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field, Journal of Computational Chemistry 25(9) (2004) 1157–1174. [DOI] [PubMed] [Google Scholar]
- [51].Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML, Comparison of simple potential functions for simulating liquid water, The Journal of Chemical Physics 79(2) (1983) 926–935. [Google Scholar]
- [52].Holden PM, Allen WJ, Gochin M, Rizzo RC, Strategies for lead discovery: application of footprint similarity targeting HIVgp41, Bioorg Med Chem 22(1) (2014) 651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McGee TD Jr., Yi HA, Allen WJ, Jacobs A, Rizzo RC, Structure-based identification of inhibitors targeting obstruction of the HIVgp41 N-heptad repeat trimer, Bioorg Med Chem Lett 27(14) (2017) 3177–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Singleton CD, Humby MS, Yi HA, Rizzo RC, Jacobs A, Identification of Ebola Virus Inhibitors Targeting GP2 Using Principles of Molecular Mimicry, J Virol 93(15) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Telehany SM, Humby MS, McGee TD Jr., Riley SP, Jacobs A, Rizzo RC, Identification of Zika Virus Inhibitors Using Homology Modeling and Similarity-Based Screening to Target Glycoprotein E, Biochemistry 59(39) (2020) 3709–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhou Y, McGillick BE, Teng YG, Haranahalli K, Ojima I, Swaminathan S, Rizzo RC, Identification of small molecule inhibitors of botulinum neurotoxin serotype E via footprint similarity, Bioorg Med Chem 24(20) (2016) 4875–4889. [DOI] [PubMed] [Google Scholar]
- [57].Ryckaert J-P, Ciccotti G, Berendsen HJC, Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes, Journal of Computational Physics 23(3) (1977) 327–341. [Google Scholar]
- [58].Roe DR, Cheatham TE 3rd, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J Chem Theory Comput 9(7) (2013) 3084–95. [DOI] [PubMed] [Google Scholar]
- [59].Baillargeon P, Fernandez-Vega V, Sridharan BP, Brown S, Griffin PR, Rosen H, Cravatt B, Scampavia L, Spicer TP, The Scripps Molecular Screening Center and Translational Research Institute, SLAS Discov 24(3) (2019) 386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kawai K, Nagata N, Metal-ligand interactions: an analysis of zinc binding groups using the Protein Data Bank, Eur J Med Chem 51 (2012) 271–6. [DOI] [PubMed] [Google Scholar]
- [61].Leuci R, Brunetti L, Laghezza A, Loiodice F, Tortorella P, Piemontese L, Importance of Biometals as Targets in Medicinal Chemistry: An Overview about the Role of Zinc (II) Chelating Agents, Applied Sciences 10(12) (2020) 4118. [Google Scholar]
- [62].Baell JB, Nissink JWM, Seven Year Itch: Pan-Assay Interference Compounds (PAINS) in 2017-Utility and Limitations, ACS Chem Biol 13(1) (2018) 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vasiliauskaite-Brooks I, Healey RD, Rochaix P, Saint-Paul J, Sounier R, Grison C, Waltrich-Augusto T, Fortier M, Hoh F, Saied EM, Arenz C, Basu S, Leyrat C, Granier S, Structure of a human intramembrane ceramidase explains enzymatic dysfunction found in leukodystrophy, Nat Commun 9(1) (2018) 5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.