

Abstract
The present United States opioid crisis requires urgent and innovative scientific intervention. This perspective highlights a role for the chemical sciences by expounding upon three key research areas identified as priorities by the National Institute on Drug Abuse (NIDA). Specifically, important advances in chemical interventions for overdose reversal, strategies for opioid use disorder (OUD) treatment, including immunopharmacotherapies, and next-generation alternatives for pain management will be discussed. Ultimately, progress made will be presented in light of remaining challenges for the field.
1. INTRODUCTION
The United States is in the midst of an opioid crisis, with more than 3.6 million current (past-month), nonmedical opioid users; 2.1 million meet the diagnostic criteria for opioid use disorder (OUD).1 From 2004 to 2011, opioid-associated emergency department (ED) visits tripled;2 similarly, from 1999 to 2014, opioid overdose mortalities tripled, and the increasing trends continue.3 The Centers for Disease Control and Prevention (CDC) sponsored Enhanced State Opioid Overdose Surveillance (ESOOS) Program reports an alarming 30% increase in ED-observed overdose fatalities from July 2016 to September 2017 alone.4 In North America, overdose now exhibits mortality rates higher than motor vehicle accidents and human immunodeficiency virus (HIV-1) combined (16.3 deaths per 100 000 people vs 10.9 and 2.0 deaths per 100 000 in 2015, respectively).3,5–8 In addition to mortality, illicit opioid use has grave health and psychosocial consequences, such as higher incidence of infectious disease comorbidity and criminal behavior, and, frequently, reduced educational achievement and gainful employment. One estimate of total lost productivity costs now exceeds one trillion dollars.9 Clearly, innovative solutions to combat the opioid crisis are in dire need, presenting an opportunity for the chemical sciences to develop new pharmacotherapeutic interventions. This perspective will discuss what pharmaceuticals are available and highlight some of the most promising recent advances in therapeutic interventions. We will also provide a brief commentary on the challenges that remain for improving treatment and a general synopsis for the interplay of chemistry and the opioid crisis.
2. THE μ-OPIOID RECECPTOR AND CURRENTLY AVAILABLE THERAPEUTICS
The opioid crisis is fueled in part by the misuse of prescription opioids and ineffective management of chronic pain. Opioids elicit analgesic effects by binding μ-opioid receptors (MORs), a subtype of G protein-coupled receptor (GPCR), in the central nervous system (CNS). Agonist-occupied MORs signal analgesic responses through heterotrimeric G protein-coupled nociceptive ion channel inhibition.10 Analgesia, however, is complicated by on-target effects including respiratory depression, gastrointestinal complications (nausea, vomiting, and constipation), drug tolerance, abuse, and dependence, which are equally engendered upon opioid binding.11 To date, pharmacotherapeutic discovery for overdose reversal and OUD has primarily focused on agonists and antagonists of the MOR and has produced four Federal Drug Administration (FDA)-approved treatment options: methadone, buprenorphine, naltrexone, and naloxone. While these agents have demonstrated some success, they are limited by considerable side effects, most notably their nonbiased activation or inactivation of all MOR pharmacology.
2.1. Opioid Overdose Reversal Agent: Naloxone.
Opioid overdose is the result of cardiorespiratory arrest caused by opioid binding at brainstem respiratory center MORs. The single therapeutic available for opioid overdose reversal is naloxone, a nonselective, competitive μ, δ, and κ opioid receptor antagonist (Figure 1). Naloxone is not only fast-acting (2–5 min), but also rapidly metabolized with a half-life (t1/2) of 30 to 80 min.12 Accordingly, repeat dosing of naloxone is sometimes required to out-compete longer-acting opioid agonists to prevent renarcotization. When administered correctly, naloxone exhibits a nearly 95% success rate in rescuing opioid overdose, and laypersons alone are estimated to have reversed over 26 000 opioid overdoses since 1996.13
Figure 1.
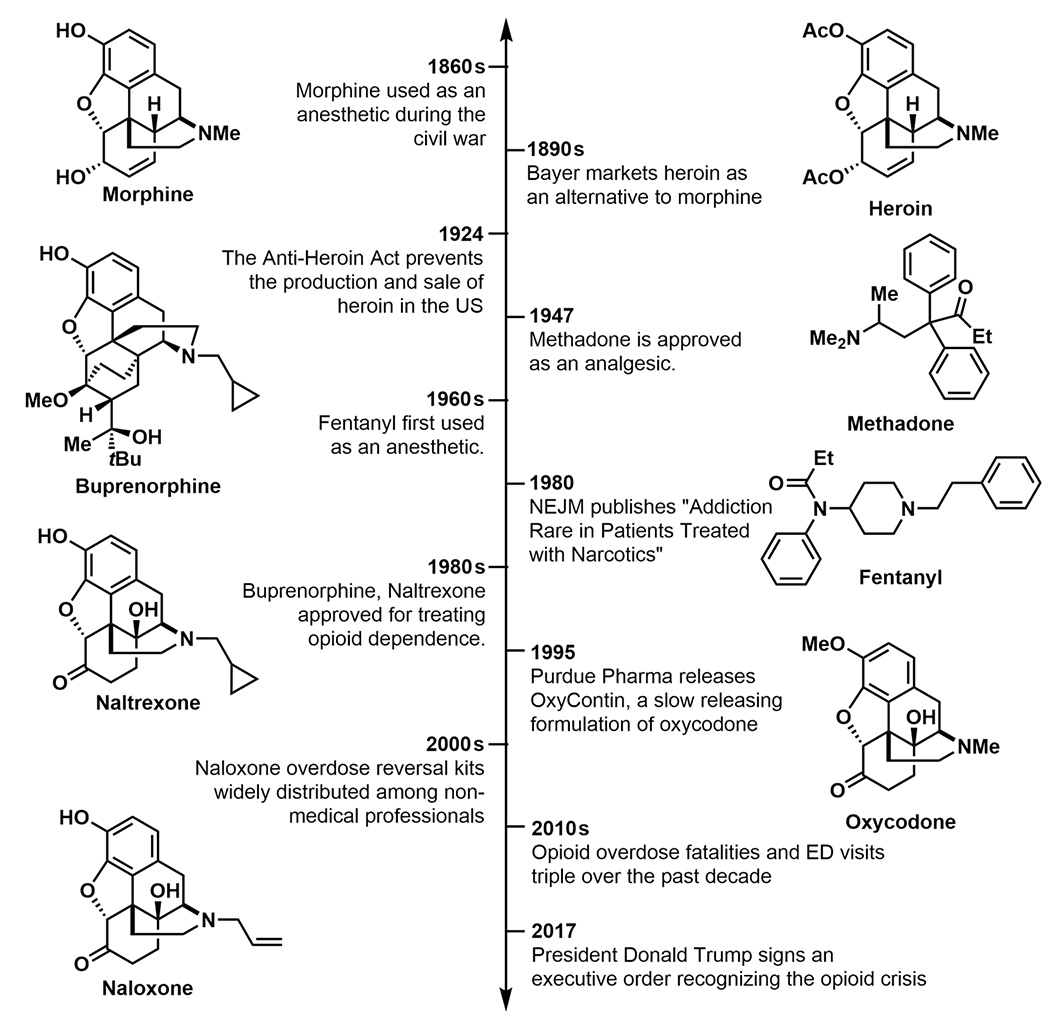
Timeline of defining events in the opioid crisis.
2.2. Approved Treatments for Opioid Use Disorder (OUD).
Because of the rewarding and incentivizing properties of opioids, illicit use can escalate into OUD, a chronic illness defined by compulsive use, withdrawal, and relapse. Understanding the currently available treatment options for OUD requires comprehension of the phase of intervention, specifically whether patients are in the detoxification or maintenance phases.14 Detoxification is a required step in transitioning patients from illicit drug use to maintenance, and often maintenance to abstinence. During the detoxification phase, pharmacotherapies are required to manage withdrawal symptoms for optimal treatment retention and relapse prevention, for which the MOR agonists methadone and buprenorphine are available (Figure 1). Methadone is an orally available, full agonist of the MOR, characterized by delayed action onset, long half-life (t1/2 = 24 to 36 h), and ability to block opioid-induced euphoria.15 For successful withdrawal management, methadone doses are tapered over days to months, with longer, smaller tapers typically yielding better results. Buprenorphine is a high-affinity, partial agonist of the MOR with an improved safety profile and reduced potential for misuse, especially when prescribed as the abuse deterrent formulation Suboxone (4:1 buprenorphine/naloxone).14 Methadone and buprenorphine, in addition to naltrexone, are also used for opioid maintenance therapy (OMT). For methadone and buprenorphine, OMT functions by maintaining MOR agonism for significant time periods (years to life) to control drug cravings and block relapse-induced euphorigenic effects. Naltrexone is the only FDA-approved MOR antagonist for OMT, which functions to block the pharmacological effects of competing opioid in relapse situations. Oral naltrexone use, however, is marked by poor treatment retention and relapse, likely as a result of the drug’s lack of intrinsic reward.14 Recently, naltrexone delivery, including extended release formulations (XR-NTX), implants, and multidrug formulations, has seen significant improvements in naltrexone-based OMT.14
A treatment plan of methadone, buprenorphine, or naltrexone combined with psychosocial therapy aims to alleviate opioid withdrawal symptoms and maintain abstinence; however, the current opioid crisis highlights the shortcomings of these therapeutics. In addition to the abuse liabilities inherent to these treatments, nonpharmacological limitations include the regulation of methadone and buprenorphine at federally controlled clinics. Promising steps toward better alternatives have been taken with Suboxone, given its improved access at office-based prescribers and abuse deterrent formulation, but non-MOR or biased MOR modulating therapeutics are also needed. The National Institute on Drug Abuse (NIDA) has recently identified three priority areas for chemical development including: (1) improved overdose-reversal and prevention therapeutics, (2) new, innovative treatments for opioid addiction, and (3) safe, effective, and nonaddictive pharmacotherapies for managing chronic pain.16 The remaining sections of this perspective will highlight recent accomplishments in each of these priority areas.
3. NEXT-GENERATION INTERVENTIONS FOR OVERDOSE REVERSAL
Despite naloxone’s success, overdose fatalities are becoming more prevalent in part because of synthetic opioids, including fentanyl and carfentanil, which are increasingly laced in heroin. Fentanyl and its analogues are produced for legitimate medical use; however, hundreds of designer drugs have been introduced into the illicit market over the past decade. Minor changes in fentanyl’s structure often translate to dramatic changes in the drug’s potency. For example, 3-methylfentanyl is 4–60-times more potent and carfentanil is 100–1000-times more potent than fentanyl. The significant potency of fentanyl and its analogues, being 50- to to 10 000-times stronger than morphine, may complicate naloxone’s effectiveness due to its short half-life.17 Overdose deaths resulting from fentanyl misuse increased by 80% from 2013 to 2014, a likely underrepresentation as an estimated 41% of heroin-attributed deaths resulted from fentanyl adulteration.18 Fentanyl and its analogues also pose a risk to the general public as chemical warfare agents (CWAs). In 2002, Russian Special Forces deployed carfentanil and remifentanil aerosols into the Melnikov Street Theater in Moscow, which resulted in 125 deaths that included civilians.19 To address the increasing incidence of overdose and the threat of malicious fentanyl use, more interventions than naloxone are needed, especially to counteract the increased potency of synthetic opioids.
3.1. AMPA Receptor Positive Allosteric Modulators.
Promising non-MOR strategies to allay opioid-induced respiratory depression (OIRD), and thus overdose, include targeting glutaminergic and serotonergic neurotransmission in the brainstem. AMPAkines, a diverse class of CNS-acting, positive allosteric modulators of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors (AMPARs), effect respiratory rhythmogenesis by regulating the duration of glutamate-induced inward currents.20,21 AMPAkine binding modulates glutamate channel closing to promote inspiration and expiration through the strength and timing of respiratory muscle contractions.22 The AMPAkine class is a structurally diverse group of small molecules suitable for oral administration, with the most well-studied, CX717, exhibiting metabolic stability and safety for human use (Figure 2).23 In preclinical studies, administration of CX717 prior to fentanyl markedly attenuated OIRD in rats, and postadministration successfully rescued rats from lethal overdose.24 Importantly, AMPAkines maintain the analgesic block while reversing OIRD in contrast to naloxone, which nullifies all MOR pharmacology. This characteristic has dramatic implications for improving pain management in surgical settings as AMPAkines offer a strategy for separating the analgesic power of opioids from their tendency to suppress respiratory drive. Moreover, AMPAkines could be employed to reverse OIRD without inducing the fast-onset withdrawal symptoms associated with naloxone. CX717 completed a double-blind, placebo-controlled human clinical trial measuring effective antagonism of OIRD without impacting analgesia.23 Pretreatment of 16 healthy men with CX717 prior to alfentanil dosing demonstrated significantly higher respiratory frequency compared to placebo. An alfentanil model was selected because of the opioid’s fast equilibration between plasma and effect site, which enabled short experimental duration and quick patient recovery. CX717 had no effect on alfentanil-induced analgesia as measured through electrical and heat-based pain models. Recently, CX1739, a proprietary CX717 analogue (RespireRx Pharmaceuticals, Inc.), underwent a Phase II clinical trial; however, mixed results were obtained. CX1739 only antagonized OIRD brought on by slow, lower dose (300 mg) infusions of remifentanil versus single bolus, higher doses (600 mg).25
Figure 2.
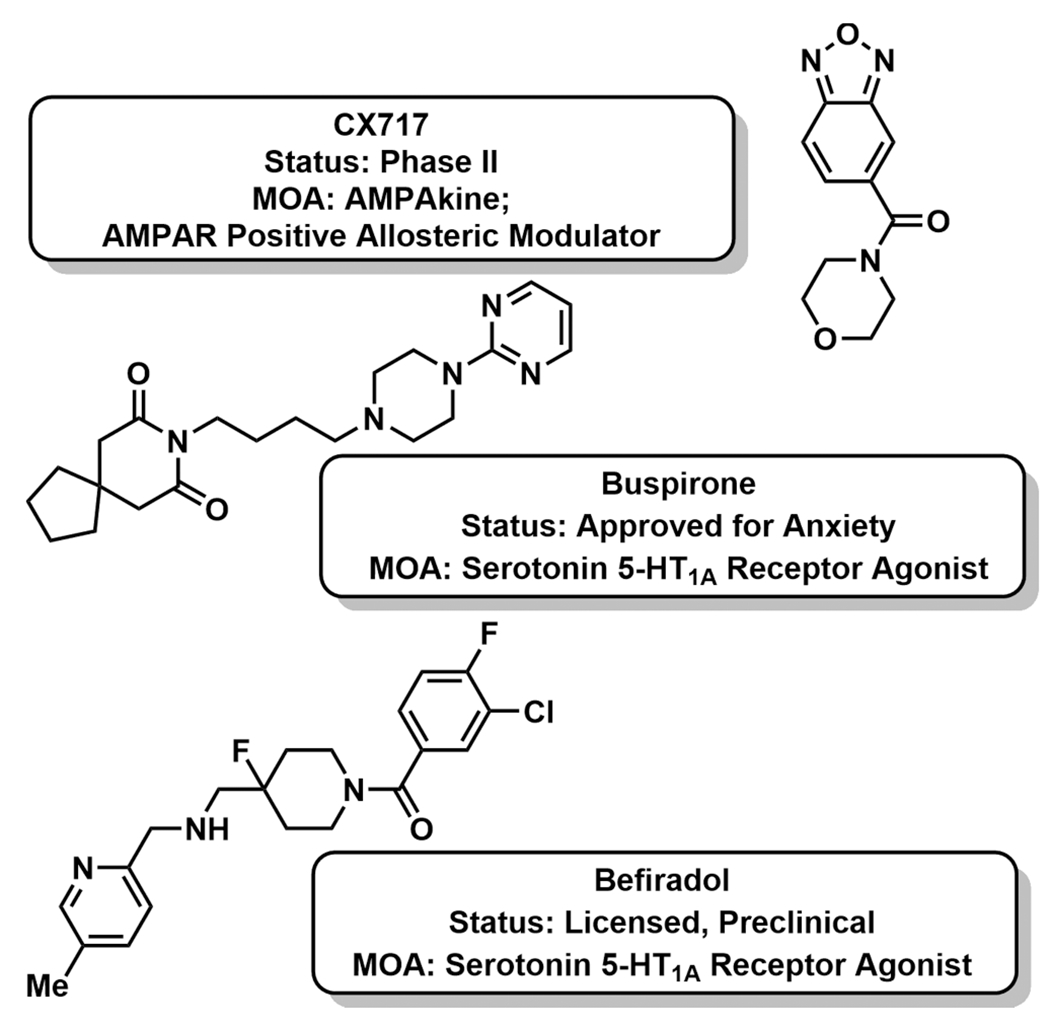
Investigatory AMPAkine and 5-HT1A modulators for opioid-induced respiratory depression (OIRD) reversal.
3.2. 5-HT1A Serotonergic Receptor Agonists.
Agonism of 5-HT1A serotonergic receptors (5-HT1AR) offers another opportunity to increase respiratory activity during periods of OIRD without negating analgesia. Serotonin (5-HT), similar to glutamate, excites respiratory motoneurons in the brainstem.26 Several 5-HT1AR agonists, including buspirone, repinotan, and befiradol, have been investigated for this indication in rodents and humans; however, the findings have been convoluted (Figure 2).27,28 For example, befiradol, a highly selective 5-HT1A agonist, potently reversed fentanyl-induced respiratory depression in rats, but also negated analgesia.26 Moreover, befiradol manifested abnormal behavior, including hyperalgesia, hyperventilation, and “behavioral syndrome”. In vitro experiments with befiradol suggested that the in vivo alleviation of fentanyl-induced respiratory depression may be via a mechanism unique to brainstem 5-HT1AR activation. Similarly, despite early results suggesting that buspirone reversed OIRD in mice, a human trial failed to demonstrate efficacy.27 Contrary to the AMPAkine family, which has demonstrated a favorable safety and efficacy profile in human clinical trials and concrete mechanism of action studies, the field of 5-HT1A requires considerably more target validation to demonstrate that 5-HT1A agonism is an effective strategy for human OIRD reversal.
4. STRATEGIES FOR TREATING OUD
Despite the approval of methadone, buprenorphine, and naltrexone, OUD diagnoses remain at staggering levels. Moreover, because methadone and buprenorphine prescriptions are limited by federal regulations and physician licensing requirements, alternative non-MOR binding treatments for opioid withdrawal and maintenance therapy are urgently needed. This section will discuss three strategies for treating OUD, two pharmacotherapeutic and one immunopharmacotherapeutic.
4.1. α2-Adrenergic Receptor Agonists.
Agonists of the α2-adrenergic receptor, particularly clonidine, have been used off-label for withdrawal alleviation since the 1980s (Figure 3).29 α2-Adrenergic receptor agonists function by dampening norepinephrine release; norepinephrine’s effects on the autonomous nervous system produce the anxiety, agitation, insomnia, muscle pain, sweating, nausea, vomiting, and diarrhea associated with withdrawal. As a result of two randomized, double-blind, placebo-controlled clinical trials, lofexidine was approved by the US FDA in May 2018 for mitigation of opioid withdrawal symptoms after abrupt drug discontinuation (Figure 3).30–32 Lofexidine had been approved for opioid detoxification in the United Kingdom since 1992.31 In the first clinical trial, opioid dependent participants stabilized on morphine were abruptly withdrawn from opioid, then given lofexidine (0.8 mg QID, p.o.) for 5 days.32 Lofexidine treatment yielded significantly reduced self-reported scores on the Modified Himmelsbach Opiate Withdrawal Scale (MHOWS) and fewer study dropouts compared to placebo. In the second Phase III, multicenter study for FDA registration, lofexidine similarly reduced SOWS-Gossop (Short Opioid Withdrawal Scale) scores and demonstrated increased study retention compared to placebo.31 FDA approval of lofexidine marks the first nonopioid treatment for withdrawal; however, there is still room for chemical optimization. Lofexidine is not a long-term solution for OUD, as it treats the symptoms and not the underlying cause of the addiction. Accordingly, lofexidine is only prescribed for 14 days immediately following abrupt discontinuation of chronic opioid use. The FDA is requiring postmarketing studies to evaluate longer-term use of lofexidine, particularly in indications where pain patients undergo slow opioid tapers.30 Moreover, lofexidine has notable side effects including, and most importantly, hypotension that can spike to hypertension upon therapy completion.
Figure 3.
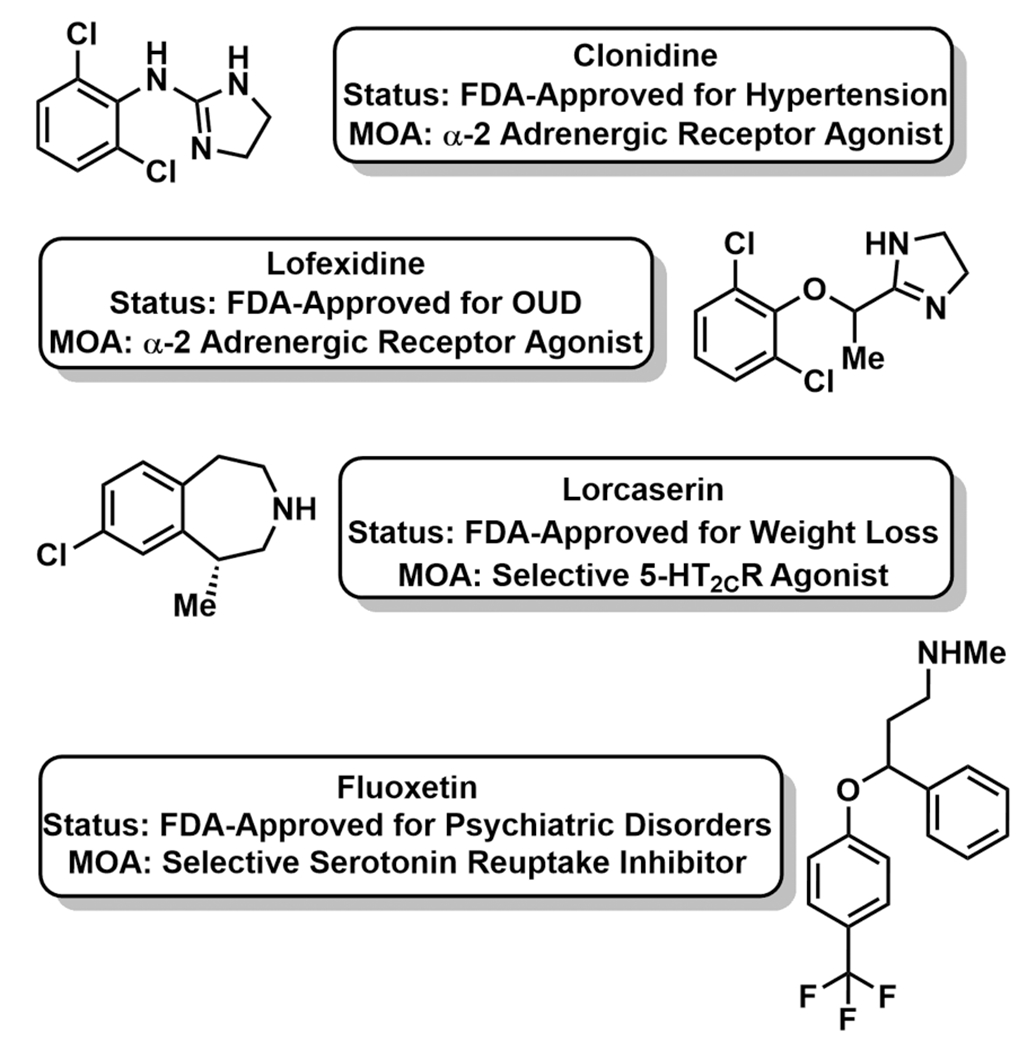
Next-generation, non-MOR modulating treatments for OUD.
4.2. 5-HT2C Receptor Agonists.
In a complementary approach, modulators of serotonin (5-HT) neurotransmission, particularly 5-HT2C receptor agonists, are being investigated to curb opioid-associated reward and cue reactivity.33 Cue reactivity is defined by a conditioned response to environmental or material stimuli, such as drug paraphernalia, that induces drug cravings, drug-seeking behaviors, and relapse.34 In a study with heroin-abstinent users post detoxification, heightened cue reactivity, as measured by elevated heart rate and self-reported cravings, accurately correlated to relapse.35 Accordingly, pharmacotherapies to control cue reactivity may enable more effective management of OUD. Lorcaserin is an FDA-approved, antiobesity medication that functions as a high-affinity, full agonist of the 5-HT2C receptor (Figure 3).
Lorcaserin and second-generation 5-HT2CR agonists have been shown to suppress oxycodone self-administration and cue reactivity in preclinical rodent models.33 Similarly, fluoxetine, a selective serotonin reuptake inhibitor (SSRI), alleviates symptoms of naloxone-induced withdrawal when dosed to rodents.36 Dexfenfluramine and fenfluramine, compounds that stimulate nonreceptor specific 5-HT release, suppress heroin self-administration in rats and reduce cue reactivities in nonhuman primates.37,38 Continued investigation into next-generation fenfluramine analogues will be required to overcome the significant cardiovascular side effects of these agents, which ultimately led to their withdrawal from the market. Clearly, chemical modulation of serotonin neurotransmission elicits profound effects on opioid-associated behaviors, with possible implications for managing OUD. Accordingly, three Phase I–II clinical trials are recruiting participants to study lorcaserin’s effects on opioid relapse prevention (clinicaltrials.gov).
4.3. Immunopharmacotherapies for Opioid Use Disorder.
For OMT, immunopharmacotherapy has emerged as an alternative approach, encompassing both active immunization with opioid-specific vaccines and passive immunization with preformed antibodies. Mechanistically inherent to both strategies, circulating drug is sequestered in the periphery by high-affinity antiopioid antibodies, the resulting antibody–drug complex being too large to cross the blood–brain barrier (BBB) to reach CNS MORs. Compared to pharmacotherapeutic antagonists that compete directly for receptor binding sites within the CNS, antiopioid antibodies act as immunoantagonists by targeting and sequestering drug in circulation.
Active immunization uses a series of vaccinations to train the immune system to mount an antiopioid antibody response. The efficacy of an antiopioid vaccine hinges on its ability to achieve large concentrations of highly specific IgG antibodies in serum (antibody titer). Opioid conjugate vaccines are composed of three components, a hapten, immunogenic carrier, and adjuvant, that must be optimized to enhance and fine-tune the elicited immune response.39 Haptens are small molecule drug mimics that exhibit no inherent immunogenicity but become immunogenic when covalently conjugated to a larger carrier protein. Moreover, addition of an adjuvant further stimulates the immune response, increases antibody titers, and provides long-lasting protection. The oft-used, FDA-approved adjuvant colloidal aluminum salt (alum) functions as a depot by lumping immunoconjugates into larger size particles near the injection site, enabling uptake by antigen-presenting cells for successful downstream immune activation. Other adjuvants that directly stimulate Toll-like receptors (TLRs) of the innate immune system are now commonly used in combination with alum to enhance the immune response.
The first heroin vaccine was reported in the early 1970s; 6-hemisuccinylmorphine-bovine serum albumin (BSA) successfully affected heroin self-administration in a rhesus monkey (Figure 4A).40 Importantly, heroin itself is a prodrug that undergoes rapid deacetylation at the 6-position to form 6-acetylmorphine (6AM), and subsequently at the 3-position, to form morphine, both of which are MOR agonists (Figure 4B). 6AM, not heroin, is the major mediator of psychoactivity. Accordingly, hapten design must consider both the structures of heroin and its metabolites to maximize vaccine efficacy, which is accomplished by maintaining the highest structural congruence between the hapten and the psychoactive opioids. In the initial design, the heroin hapten incorporated a 6-position succinyl linker for bioconjugation to BSA; however, this strategy fails to present each of the dynamic haptens (heroin, 6AM, morphine) to the immune system. To demonstrate this concept, our laboratory hypothesized that a single, appropriately designed heroin hapten could yield multiple drug-like antigens in vivo.41 To access such an immunoconjugate, nitrogen was chosen for linker attachment and a heroin-like hapten containing labile acetyl esters at the 3 and 6 positions was synthesized (Her-KLH, Figure 5). We envisioned that by adsorbing Her-KLH onto alum, the depot matrix would minimize rapid hydrolysis of the labile esters. Advantageously, slow desorption of the immunoconjugate from the adjuvant would provide a steady and chemically dynamic source of multiple drug-like antigens for presentation to the immune system. In subsequent studies, the metabolically dynamic nature of this immunoconjugate was demonstrated to be critical for antiheroin potency.42,43 In rats, a high titer immune response was observed in Her-KLH vaccinated animals and was characterized by antibodies with high affinity for 6AM, heroin, and morphine. The antinociceptive effects and the acquisition of heroin self-administration were blocked in vaccinated rats. For comparison, a static morphine vaccine (Mor-KLH, Figure 5), deacetylated at the 3 and 6 positions, produced antibodies with high affinity for morphine only, with reduced affinity to heroin and no affinity for 6AM. The shift in antibody response toward morphine was reflected in Mor-KLH’s inability to prevent heroin self-administration in rats, presumably due to a lack of 6AM-antibody binding in the periphery. Following an intravenous injection of heroin, Her-KLH vaccinated rats showed significantly greater retention of 6AM and heroin in the blood, compared to Mor-KLH vaccinated rats. The dynamic heroin vaccine was demonstrated to be efficacious not only in a prophylactic scenario with drug naïve animals, but also in the treatment of drug-dependent animals.43 Following a period of opioid abstinence, dependent, Her-KLH immunized rats exhibited extinguished heroin self-administration. The latter paradigm represents a relevant clinical situation encompassing individuals who have achieved abstinence as part of a relapse prevention program.
Figure 4.
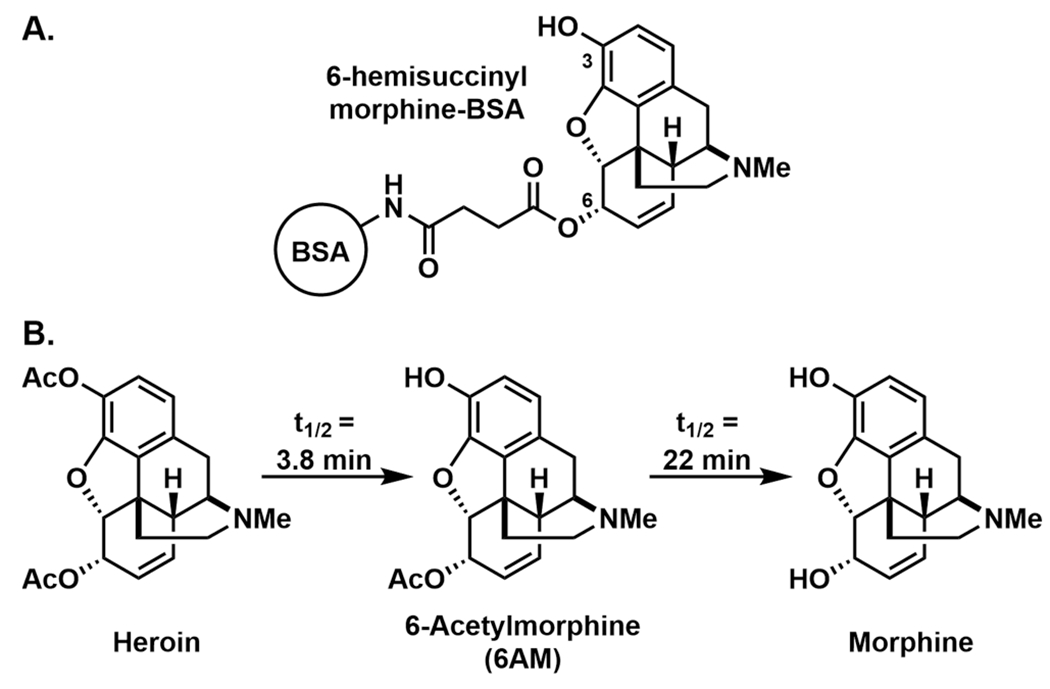
(A) Structure of the first opioid immunopharmacotherapy, 6-hemisuccinylmorphine-BSA; (B) metabolic pathway of heroin, including half-lives in humans.
Figure 5.
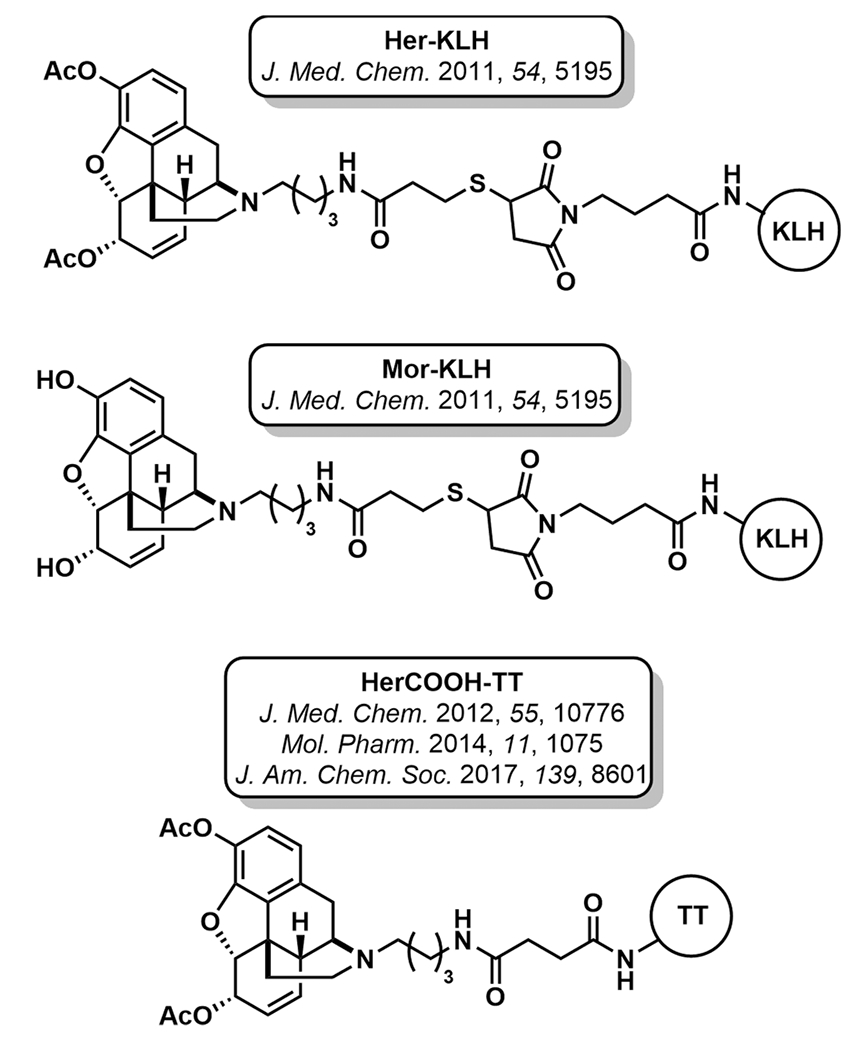
Structures of heroin immunopharmacotherapies.
For the aforementioned reasons, we have chosen to highlight nitrogen-linked opioid haptens for the remainder of this perspective. While several laboratories have continued investigating 3 and 6-linked haptens, the vaccines exhibit a notable lack of specificity, including an inability to differentiate between heroin and the commonly prescribed pain relievers oxycodone and hydrocodone.44,45 While a broad-spectrum opioid vaccine may find utility in certain indications, nitrogen-linked haptens allow for specific tunability, which could be invaluable in managing chronic pain in individuals with OUD.
Though hapten design is a critical element for antiopioid vaccine development imparting both antibody affinity and specificity, carrier proteins and adjuvants also play an important role. An optimal combination of all three components can greatly enhance a vaccine’s efficacy. A series of systematic studies with the dynamic Her-KLH vaccine uncovered a superior formulation, HerCOOH-tetanus toxoid (TT) + alum and CpG ODN 1826 adjuvants. HerCOOH is conjugated to the carrier protein TT through an amide linkage, thus imparting greater stability and coupling efficiency (Figure 5).42,46,47 The optimized vaccine formulation reduced heroin potency by >15-fold in rodents and the effects persisted for over eight months.46
The clinical potential of this lead heroin vaccine was evaluated in nonhuman primates (NHPs).46 Immunized rhesus monkeys produced high antibody titers with submicromolar and low nanomolar binding affinities for heroin and 6AM, respectively. Importantly, minimal cross-reactivity with other clinically used, medical opioids (oxycodone, hydrocodone) was observed. In a subsequent study, HerCOOH-TT vaccinated monkeys showed long-lasting IgG titers in both previously vaccinated as well as vaccine naïve monkeys. Interestingly, previously vaccinated monkeys produced significantly higher antibody titers and improved binding affinities compared to previously nonvaccinated NHPs. The previously nonvaccinated monkeys did show a gradual increase in binding affinity for 6AM over the course of the study. Therefore, the vaccine was able to boost immunity in immunized monkeys, an important feature of any vaccine.
Heroin pharmacokinetic results paralleled findings in the prior versus nonprior vaccinated groups. Both groups produced high concentrations of antibodies capable of binding 6AM in the blood; however, the revaccinated group showed a much larger AUC shift (193-fold) compared to the previously nonvaccinated group (19-fold). AUC is indicative of the time of actual drug exposure. Heroin potency was diminished in all vaccinated monkeys based on behavioral studies and the vaccine produced antiopioid effects equivalent to naltrexone.
Because oxycodone and hydrocodone are among the most commonly misused prescription opioids, vaccine development to manage their abuse has also been pursued. Investigation of nitrogen-linked oxy- and hydrocodone haptens demonstrated significant right shifting (5- to 10-fold) of opioid dose–effect curves in antinociceptive testing between vaccinated and nonvaccinated mice (Figure 6).48 Moreover, the vaccinated mice demonstrated resistance to lethal opioid doses and reduced opioid biodistribution to the CNS. Isolated serum IgG antibodies exhibited subnanomolar affinity for both oxycodone and hydrocodone with minimal cross-reactivity to other opioids.
Figure 6.
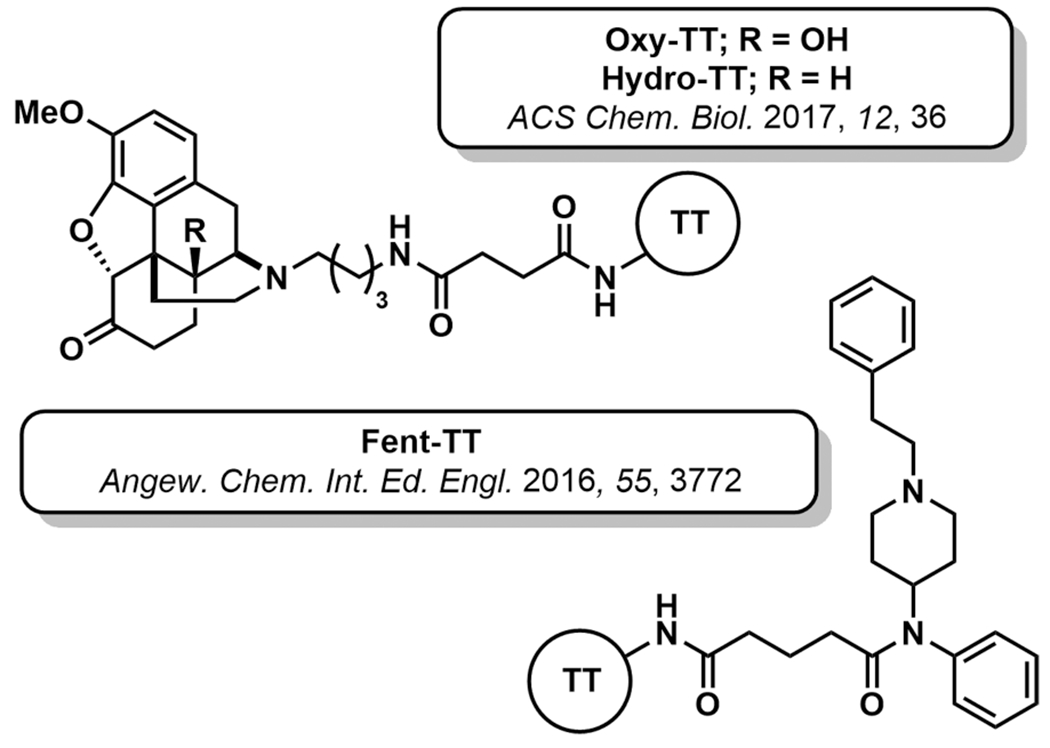
Structures of non-heroin opioid immunopharmacotherapies.
Recently, immunopharmacotherapies for fentanyl and its structural analogues have been investigated. Hapten design and linker positioning are crucial to accomplish a vaccine that elicits a broad antibody response capable of neutralizing many fentanyl derivatives. We designed the immunoconjugate Fen-TT consisting of the fentanyl core, N-(1-phenethylpiperidin-4-yl)-N-phenylacetamide, conjugated through its propanoyl group (Figure 6).49 Mice vaccinated with Fen-TT + alum and CpG ODN 1826 produced exceptionally high antifentanyl antibody titers with low nanomolar binding affinities. Biodistribution studies illustrated the peripheral binding capacity of the raised serum antibodies, as 45-times less fentanyl crossed the BBB compared to nonvaccinated mice. This blood partitioning correlated to a >30-fold decrease in fentanyl potency in behavioral studies and significant protection from lethal doses of fentanyl. Importantly, the Fen-TT vaccine displayed broad specificity to fentanyl class drugs and minimal cross-reactivity to the clinically used opioids methadone and oxycodone.
With the recent trend of fentanyl-laced heroin translating to increased opioid-associated mortality rates, a heroin and fentanyl admixture vaccine was investigated.50 Similar studies had been reported for the structurally congruent opioids morphine and oxycodone with promising results; however, no studies had been performed on structurally unique opioids like heroin and fentanyl.51 Mice vaccinated with a combination of Her-KLH46 and Fen-KLH49 showed preferential opioid partitioning to the blood and reduced opioid concentrations in the brain when challenged with a mixture of heroin and fentanyl (10% w/w). Antiserum displayed nanomolar binding affinity for fentanyl and cross-reactivity to a panel of six fentanyl derivatives. This proof-of-concept study demonstrated the potential for dual vaccine strategies.
Passive vaccination involves the immunization of antibodies preselected against a specified target. The goal of passive immunization, analogous to active immunization, is to bind and sequester opioid in the peripheral circulation, thereby reducing the drug’s pharmacodynamic effects. Passive antibody administration (immediate immunity) has many positive attributes including rapid onset of therapeutic benefit, preclusion of immune response differences among individuals, and ease of antibody manipulation through protein engineering. Additionally, the antibody dosage can be accurately tailored to each therapeutic context. Passive immunization has the benefit of providing long-lasting protection as fully human IgG antibodies have a long in vivo half-life (~23 days), limited tissue distribution, and low risk of immune response liability. Because of this sustained protection, passive immunization exhibits significant potential for overdose indications. While the short half-life of naloxone enables possible renarcotization, passive immunization offers an opportunity for improved MOR immunoantagonism.
The ability of monoclonal antibodies (mAbs) to confer passive immunity has shown promise for stimulant drugs, such as cocaine and methamphetamine, in preclinical and clinical settings. Notably, a human anticocaine mAb, IgG GNCgzk, with subnanomolar affinity for cocaine (Kd = 0.18 nM) provided full protection against an LD50 dose when administered prophylactically and reduced lethality to 20% when administered after cocaine challenge.52 A humanized IgG mAb for 6AM has been reported.53,54 Prophylactic administration of this anti-6AM mAb to mice produced a significant decrease in CNS 6AM levels and translated to diminished 6AM- and heroin-induced behavior. However, mAb pretreatment was more effective against a 6AM challenge compared to heroin.
Antiopioid vaccines and mAbs present viable therapeutic options for treating opioid addiction, morbidity, and mortality, though translation to humans has yet to be realized. Many questions still need to be answered before vaccines reach their full potential as clinically sound alternatives. Concrete benchmarks must be established to quantify the concentration of antiopioid antibody required to outcompete high dose and repeat opioid challenge. Moreover, boosting regimes need to be determined so that effective levels of antiopioid protection are maintained uninterrupted. Future work for antiopioid vaccines will investigate combination strategies of vaccine-induced immunoantagonism with pharmacotherapeutic agents for withdrawal or pain management. To successfully function as OMT, vaccines will be required to function in the presence of other analgesics and adjunctive therapies.
5. ALTERNATIVES FOR PAIN TREATMENT
The opioid crisis is in part fueled by the overprescription of opioid pain medication. In 2010, chronic pain affected an estimated 100 million Americans, corresponding to an economic burden of approximately $600 billion per year: greater than annual cost of cancer, heart disease, and diabetes combined.55 Even with significant advances in treatment options for overdose and OUD, the crisis will not be adverted without alternative options for addressing chronic pain. Accordingly, safer and nonaddictive analgesics are urgently needed.
5.1. G-Protein Pathway Selective MOR Agonists.
Classically, GPCRs, including the MOR, were considered binary “ON–OFF” sensors, with agonist bound receptors triggering G-protein coupled responses. Analgesic drug discovery thus developed agonists and antagonists for the MOR, which activated or blocked entire polypharmacological signaling paradigms. Advances in the structural understanding of the MOR have now illuminated the ability to design biased ligands that impart selective activation of individual signaling pathways.56,57 In the case of the MOR, this strategy enables activation of G-protein coupled analgesia without β-arrestin recruitment. The gastrointestinal and respiratory adverse, on-target processes associated with opioids are mediated by β-arrestin. Trevena, Inc. has developed oliceridine, an opioid specifically biased for Gi protein-coupled MOR signaling (Figure 7).58 Originally identified from a high-throughput screen of a diversity library, oliceridine functions as a highly selective agonist of MOR coupled G-protein activity with efficacy comparable to morphine and superior to buprenorphine. Because of nearly undetectable levels of β-arrestin recruitment, oliceridine demonstrates potent analgesia in multiple rodent pain models, with significantly reduced gastrointestinal complications and respiratory depression. Oliceridine has passed both Phase I and Phase II clinical trials with a positive safety profile and dose-dependent μ-opioid activity, as determined by pupil constriction.59 It is now posed for a head-to-head comparison with approved, unbiased opioid treatments. If successful, oliceridine would offer the first opioid analgesic with reduced overdose and side effect risk, though the molecule’s effects on abuse and reward remain unknown.
Figure 7.
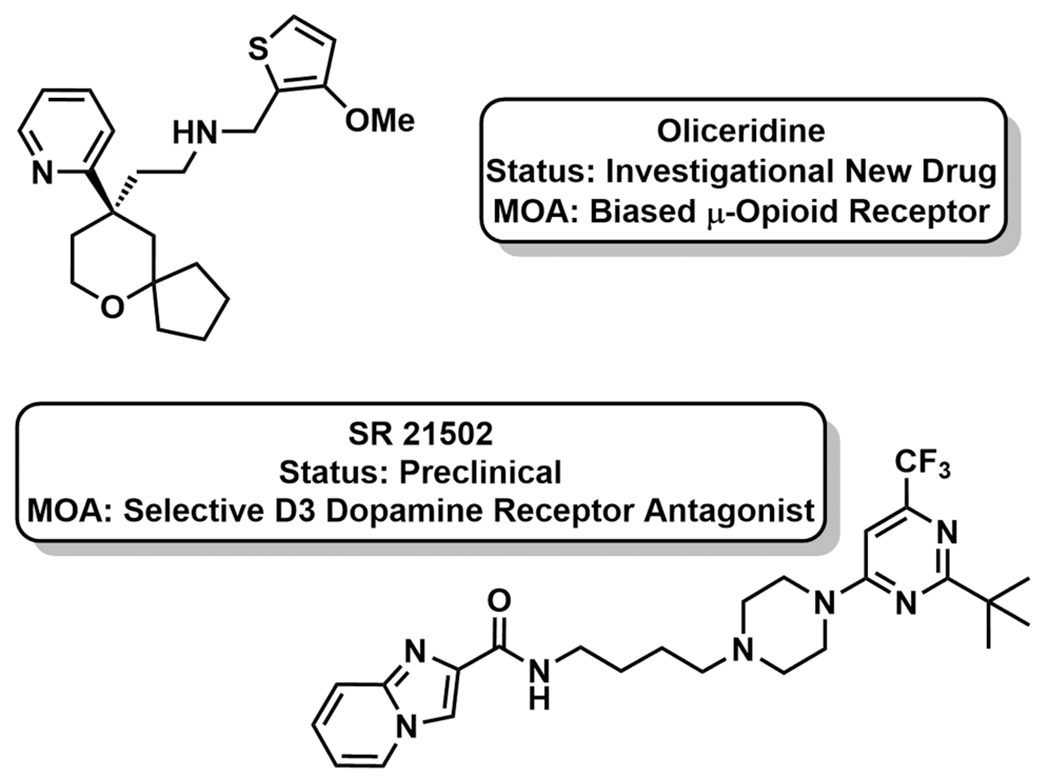
New pharmacotherapeutic strategies in pain management.
Dopamine (DA) D3 receptor antagonism is also being investigated as a strategy to improve the safety profile of currently available opioids.60 DA D3 receptor activation contributes to the addictive and rewarding feedback of both stimulant and opioids drugs.61 Opiate cues induce DA release into the mesocorticolimbic system, and repeated exposure to drug upregulates DA D3 receptor expression, inducing opioid tolerance. DA D3 receptor antagonism has been shown to reduce cue-induced drug reinstatement (relapse) and conditioned place preference in rodents for various opioids. The selective DA D3 receptor antagonist SR 21502 was evaluated for its ability to reduce morphine tolerance in the presence of analgesia (Figure 7).60,61 Ultimately, SR 21502 reduced morphine tolerance two-fold, as evidenced by a less rightward shift in the dose–response curve. In a model of naloxone-induced withdrawal/chronic dependence, SR 21505 reduced vertical jumping and urine and feces output over the observation window. Together, these results demonstrate that codosing SR 21505 with morphine exhibits fewer negative side effects (tolerance, gastrointestinal complications, addictive tendencies) than opioid alone. SR 21505 remains in preclinical development.
6. REMAINING CHALLENGES AND PHARMACOTHERAPEUTIC OUTLOOK
One of the greatest challenges remaining for next-generation overdose, OUD, and pain treatment is the accomplishment of therapeutics with low abuse liabilities. While significant progress has been made to overcome many opioid-associated polypharmacological effects, abuse and reward remain adverse on-target outcomes. This goal in pharmacotherapeutic design is complicated by the complexity of neurotransmission mechanisms of the brain. Going forward, the chemical field will be required to rely heavily on fundamental neuroscience discoveries for insights into unique opportunities for chemical modulation. From a realistic viewpoint, successful treatment of OUD and overdose prevention will require a detailed treatment plan including therapeutics from each of the priority areas described in this perspective in addition to psychosocial support including counseling, behavior modification, and management of other underlying psychiatric disorders. Yet, there is significant opportunity for the chemical sciences to design next-generation overdose reversal agents, OUD treatments, and analgesics to combat the opioid crisis.
ACKNOWLEDGMENTS
Research was supported by the National Institutes of Health grants F32 DA044692 (to M.E.O.) and UH3 DA041146 (to K.D.J.).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Substance Abuse and Mental Health Services Administration. Results from the 2016 National Survey on Drug Use and Health: Detailed Tables; Center for Behavioral Health Statistics and Quality: Rockville, MD, 2017. [Google Scholar]
- (2).Drug Abuse Warning Network. National estimates of drug-related emergency department visits, 2004–2011 - illicits (excluding alcohol); SAMHSA, 2011. https://www.samhsa.gov/data/report/national-estimates-drug-related-emergency-department-visits-2004-2011-illicits-excluding (accessed Aug 26, 2018). [Google Scholar]
- (3).Rudd RA; Seth P; David F; Scholl L Increases in Drug and Opioid-Involved Overdose Deaths - United States, 2010–2015. MMWR Morb. Mortal. Wkly. Rep 2016, 65, 1445–1452. [DOI] [PubMed] [Google Scholar]
- (4).Vivolo-Kantor AM; Seth P; Gladden RM; Mattson CL; Baldwin GT; Kite-Powell A; Coletta MA Vital Signs: Trends in Emergency Department Visits for Suspected Opioid Overdoses — United States, July 2016–September 2017. Morb. Mortal. Wkly. Rep 2018, 67, 279–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).King NB; Fraser V; Boikos C; Richardson R; Harper S Determinants of Increased Opioid-Related Mortality in the United States and Canada, 1990–2013: A Systematic Review. Am. J. Public Health 2014, 104 (8), e32–e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).National Center for Health Statistics. Drug Overdose Mortality by State; Centers for Disease Control and Prevention, 2018. https://www.cdc.gov/nchs/pressroom/sosmap/drug_poisoning_mortality/drug_poisoning.htm (accessed Nov 13, 2018). [Google Scholar]
- (7).General Statistics; Insurance Institute for Highway Safety Highway Loss Data Institute, 2016. https://www.iihs.org/iihs/topics/t/general-statistics/fatalityfacts/state-by-state-overview/2015 (accessed Nov 13, 2018). [Google Scholar]
- (8).National Center for Health Statistics. AIDS and HIV; Centers for Disease Control and Prevention, 2017. https://www.cdc.gov/nchs/fastats/aids-hiv.htm (accessed Nov 13, 2018). [Google Scholar]
- (9).Altarum. Economic Toll of Opioid Crisis in U.S. Exceeded $1 Trillion Since 2001; Altarum, 2018. https://altarum.org/news/economic-toll-opioid-crisis-us-exceeded-1-trillion-2001 (accessed Aug 27, 2018). [Google Scholar]
- (10).Ikeda K; Kobayashi T; Kumanishi T; Yano R; Sora I; Niki H Molecular mechanisms of analgesia induced by opioids and ethanol: is the GIRK channel one of the keys? Neurosci. Res 2002, 44 (2), 121–131. [DOI] [PubMed] [Google Scholar]
- (11).Kieffer BL Opioids: first lessons from knockout mice. Trends Pharmacol. Sci 1999, 20 (1), 19–26. [DOI] [PubMed] [Google Scholar]
- (12).Ngai SH; Berkowitz BA; Yang JC; Hempstead J; Spector S Pharmacokinetics of naloxone in rats and in man: basis for its potency and short duration of action. Anesthesiology 1976, 44 (5), 398–401. [DOI] [PubMed] [Google Scholar]
- (13).Wheeler E; Stephen Jones T; K Gilbert M; J Davidson P Opioid Overdose Prevention Programs Providing Naloxone to Laypersons - United States, 2014. Morb. Mortal. Wkly. Rep 2015, 64, 631–5. [PMC free article] [PubMed] [Google Scholar]
- (14).Ayanga D; Shorter D; Kosten TR Update on pharmacotherapy for treatment of opioid use disorder. Expert Opin. Pharmacother 2016, 17 (17), 2307–2318. [DOI] [PubMed] [Google Scholar]
- (15).Ward J; Bell J; Mattick RP; Hall W Methadone Maintenance Therapy for Opioid Dependence. CNS Drugs 1996, 6 (6) , 440–449. [Google Scholar]
- (16).Volkow ND; Collins FS The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med 2017, 377 (4), 391–394. [DOI] [PubMed] [Google Scholar]
- (17).Vardanyan RS; Hruby VJ Fentanyl-related compounds and derivatives: current status and future prospects for pharmaceutical applications. Future Med. Chem 2014, 6 (4), 385–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Frank RG; Pollack HA Addressing the Fentanyl Threat to Public Health. N. Engl. J. Med 2017, 376 (7), 605–607. [DOI] [PubMed] [Google Scholar]
- (19).Riches JR; Read RW; Black RM; Cooper NJ; Timperley CM Analysis of clothing and urine from Moscow theatre siege casualties reveals carfentanil and remifentanil use. J. Anal. Toxicol 2012, 36 (9), 647–56. [DOI] [PubMed] [Google Scholar]
- (20).Greer JJ; Ren J Ampakine therapy to counter fentanyl-induced respiratory depression. Respir. Physiol. Neurobiol 2009, 168 (1–2), 153–7. [DOI] [PubMed] [Google Scholar]
- (21).Arai AC; Xia YF; Suzuki E Modulation of AMPA receptor kinetics differentially influences synaptic plasticity in the hippocampus. Neuroscience 2004, 123 (4), 1011–1024. [DOI] [PubMed] [Google Scholar]
- (22).Lynch G. Glutamate-based therapeutic approaches: ampakines. Curr. Opin. Pharmacol 2006, 6 (1), 82–8. [DOI] [PubMed] [Google Scholar]
- (23).Oertel BG; Felden L; Tran PV; Bradshaw MH; Angst MS; Schmidt H; Johnson S; Greer JJ; Geisslinger G; Varney MA; Lütsch J Selective Antagonism of Opioid-Induced Ventilatory Depression by an Ampakine Molecule in Humans Without Loss of Opioid Analgesia. Clin. Pharmacol. Ther 2010, 87 (2), 204–211. [DOI] [PubMed] [Google Scholar]
- (24).Ren J; Ding X; Funk GD; Greer JJ Ampakine CX717 protects against fentanyl-induced respiratory depression and lethal apnea in rats. Anesthesiology 2009, 110 (6), 1364–70. [DOI] [PubMed] [Google Scholar]
- (25).RespireRx Pharmaceuticals Inc. Announces Data for CX1739 Clinical Study in Opioid Induced Respiratory Depression; RespireRx Pharmaceuticals Inc., 2016. https://www.sec.gov/Archives/edgar/data/849636/000149315216015894/ex99-1.htm (accessed Aug 29, 2018). [Google Scholar]
- (26).Ren J; Ding X; Greer JJ 5-HT1A receptor agonist Befiradol reduces fentanyl-induced respiratory depression, analgesia, and sedation in rats. Anesthesiology 2015, 122 (2), 424–34. [DOI] [PubMed] [Google Scholar]
- (27).Oertel BG; Schneider A; Rohrbacher M; Schmidt H; Tegeder I; Geisslinger G; Lotsch J The partial 5-hydroxytryptamine1A receptor agonist buspirone does not antagonize morphine-induced respiratory depression in humans. Clin. Pharmacol. Ther 2007, 81 (1), 59–68. [DOI] [PubMed] [Google Scholar]
- (28).Guenther U; Theuerkauf NU; Huse D; Boettcher MF; Wensing G; Putensen C; Hoeft A Selective 5-HT(1A)-R-agonist repinotan prevents remifentanil-induced ventilatory depression and prolongs antinociception. Anesthesiology 2012, 116 (1), 56–64. [DOI] [PubMed] [Google Scholar]
- (29).Kleber HD; Riordan CE; Rounsaville B; et al. Clonidine in outpatient detoxification from methadone maintenance. Arch. Gen. Psychiatry 1985, 42 (4), 391–394. [DOI] [PubMed] [Google Scholar]
- (30).U.S. Food & Drug Administration. FDA Approves the First Nonopioid Treatment for Management of Opioid Withdrawal Symptoms in Adults; U.S. FDA: Silver Spring, MD, 2018. [Google Scholar]
- (31).Gorodetzky CW; Walsh SL; Martin PR; Saxon AJ; Gullo KL; Biswas K A phase III, randomized, multi-center, double blind, placebo controlled study of safety and efficacy of lofexidine for relief of symptoms in individuals undergoing inpatient opioid withdrawal. Drug Alcohol Depend. 2017, 176, 79–88. [DOI] [PubMed] [Google Scholar]
- (32).Yu E; Miotto K; Akerele E; Montgomery A; Elkashef A; Walsh R; Montoya I; Fischman MW; Collins J; McSherry F; Boardman K; Davies DK; O’Brien CP; Ling W; Kleber H; Herman BH A Phase 3 placebo-controlled, double-blind, multi-site trial of the alpha-2-adrenergic agonist, lofexidine, for opioid withdrawal. Drug Alcohol Depend. 2008, 97 (1), 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Neelakantan H; Holliday ED; Fox RG; Stutz SJ; Comer SD; Haney M; Anastasio NC; Moeller FG; Cunningham KA Lorcaserin Suppresses Oxycodone Self-Administration and Relapse Vulnerability in Rats. ACS Chem. Neurosci 2017, 8 (5), 1065–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Carter BL; Tiffany ST Meta-analysis of cue-reactivity in addiction research. Addiction 1999, 94 (3), 327–40. [PubMed] [Google Scholar]
- (35).Marissen MA; Franken IH; Waters AJ; Blanken P; van den Brink W; Hendriks VM Attentional bias predicts heroin relapse following treatment. Addiction 2006, 101 (9), 1306–12. [DOI] [PubMed] [Google Scholar]
- (36).Singh VP; Jain NK; Kulkarni SK Fluoxetine suppresses morphine tolerance and dependence: modulation of NO-cGMP/DA/serotoninergic pathways. Methods Find. Exp. Clin. Pharmacol 2003, 25 (4), 273–80. [DOI] [PubMed] [Google Scholar]
- (37).Higgins GA; Wang Y; Corrigall WA; Sellers EM Influence of 5-HT3 receptor antagonists and the indirect 5-HT agonist, dexfenfluramine, on heroin self-administration in rats. Psychopharmacology 1994, 114 (4), 611–9. [DOI] [PubMed] [Google Scholar]
- (38).Li JX; Koek W; Rice KC; France CP Effects of direct- and indirect-acting serotonin receptor agonists on the antinociceptive and discriminative stimulus effects of morphine in rhesus monkeys. Neuropsychopharmacology 2011, 36 (5), 940–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bremer PT; Janda KD Conjugate Vaccine Immunotherapy for Substance Use Disorder. Pharmacol. Rev 2017, 69 (3), 298–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Bonese KF; Wainer BH; Fitch FW; Rothberg RM; Schuster CR Changes in heroin self-administration by a rhesus monkey after morphine immunisation. Nature 1974, 252 (5485), 708–10. [DOI] [PubMed] [Google Scholar]
- (41).Stowe GN; Vendruscolo LF; Edwards S; Schlosburg JE; Misra KK; Schulteis G; Mayorov AV; Zakhari JS; Koob GF; Janda KD A vaccine strategy that induces protective immunity against heroin. J. Med. Chem 2011, 54 (14), 5195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bremer PT; Janda KD Investigating the effects of a hydrolytically stable hapten and a Th1 adjuvant on heroin vaccine performance. J. Med. Chem 2012, 55 (23), 10776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Schlosburg JE; Vendruscolo LF; Bremer PT; Lockner JW; Wade CL; Nunes AA; Stowe GN; Edwards S; Janda KD; Koob GF Dynamic vaccine blocks relapse to compulsive intake of heroin. Proc. Natl. Acad. Sci U. S. A 2013, 110 (22), 9036–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sulima A; Jalah R; Antoline JFG; Torres OB; Imler GH; Deschamps JR; Beck Z; Alving CR; Jacobson AE; Rice KC; Matyas GR A Stable Heroin Analogue That Can Serve as a Vaccine Hapten to Induce Antibodies That Block the Effects of Heroin and Its Metabolites in Rodents and That Cross-React Immunologically with Related Drugs of Abuse. J. Med. Chem 2018, 61 (1), 329–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pravetoni M; Le Naour M; Tucker AM; Harmon TM; Hawley TM; Portoghese PS; Pentel PR Reduced antinociception of opioids in rats and mice by vaccination with immunogens containing oxycodone and hydrocodone haptens. J. Med. Chem 2013, 56 (3), 915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Bremer PT; Schlosburg JE; Banks ML; Steele FF; Zhou B; Poklis JL; Janda KD Development of a Clinically Viable Heroin Vaccine. J. Am. Chem. Soc 2017, 139 (25), 8601–8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bremer PT; Schlosburg JE; Lively JM; Janda KD Injection route and TLR9 agonist addition significantly impact heroin vaccine efficacy. Mol. Pharmaceutics 2014, 11 (3), 1075–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Kimishima A; Wenthur CJ; Zhou B; Janda KD An Advance in Prescription Opioid Vaccines: Overdose Mortality Reduction and Extraordinary Alteration of Drug Half-Life. ACS Chem. Biol 2017, 12 (1), 36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bremer PT; Kimishima A; Schlosburg JE; Zhou B; Collins KC; Janda KD Combatting Synthetic Designer Opioids: A Conjugate Vaccine Ablates Lethal Doses of Fentanyl Class Drugs. Angew. Chem., Int. Ed 2016, 55 (11), 3772–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Hwang CS; Smith LC; Natori Y; Ellis B; Zhou B; Janda KD Efficacious Vaccine against Heroin Contaminated with Fentanyl. ACS Chem. Neurosci 2018, 9 (6), 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pravetoni M; Raleigh MD; Le Naour M; Tucker AM; Harmon TM; Jones JM; Birnbaum AK; Portoghese PS; Pentel PR Co-administration of morphine and oxycodone vaccines reduces the distribution of 6-monoacetylmorphine and oxycodone to brain in rats. Vaccine 2012, 30 (31), 4617–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Treweek JB; Janda KD An antidote for acute cocaine toxicity. Mol. Pharmaceutics 2012, 9 (4), 969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Bogen IL; Boix F; Nerem E; Morland J; Andersen JM A monoclonal antibody specific for 6-monoacetylmorphine reduces acute heroin effects in mice. J. Pharmacol. Exp. Ther 2014, 349 (3), 568–76. [DOI] [PubMed] [Google Scholar]
- (54).Kvello AM; Andersen JM; Oiestad EL; Morland J; Bogen IL Pharmacological Effects of a Monoclonal Antibody against 6-Monoacetylmorphine upon Heroin-Induced Locomotor Activity and Pharmacokinetics in Mice. J. Pharmacol. Exp. Ther 2016, 358 (2), 181–9. [DOI] [PubMed] [Google Scholar]
- (55).Gaskin DJ; Richard P Appendix C: The Economic Costs of Pain in the United States. In Relieving Pain in America: A Blueprint for Transforming Prevention, Care, Education, and Research; National Academies Press: Washington, DC, 2011. [PubMed] [Google Scholar]
- (56).Huang W; Manglik A; Venkatakrishnan AJ; Laeremans T; Feinberg EN; Sanborn AL; Kato HE; Livingston KE; Thorsen TS; Kling RC; Granier S; Gmeiner P; Husbands SM; Traynor JR; Weis WI; Steyaert J; Dror RO; Kobilka BK Structural insights into μ-opioid receptor activation. Nature 2015, 524, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Violin JD; Crombie AL; Soergel DG; Lark MW Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci 2014, 35 (7), 308–316. [DOI] [PubMed] [Google Scholar]
- (58).Chen XT; Pitis P; Liu G; Yuan C; Gotchev D; Cowan CL; Rominger DH; Koblish M; Dewire SM; Crombie AL; Violin JD; Yamashita DS Structure-activity relationships and discovery of a G protein biased mu opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem 2013, 56 (20), 8019–31. [DOI] [PubMed] [Google Scholar]
- (59).Singla N; Minkowitz HS; Soergel DG; Burt DA; Subach RA; Salamea MY; Fossler MJ; Skobieranda F A randomized, Phase IIb study investigating oliceridine (TRV130), a novel μ-receptor G-protein pathway selective (μ-GPS) modulator, for the management of moderate to severe acute pain following abdominoplasty. J. Pain Res 2017, 10, 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Eon V; Giuvelis D; Ananthan S; Bilsky E Efficacy of Dopamine D3 Receptor Antagonist SR 21502 in Reducing Opioid Tolerance and Dependence. FASEB J. 2015, 29 (1_supplement), 614.5. [Google Scholar]
- (61).Galaj E; Manuszak M; Babic S; Ananthan S; Ranaldi R The selective dopamine D3 receptor antagonist, SR 21502, reduces cue-induced reinstatement of heroin seeking and heroin conditioned place preference in rats. Drug Alcohol Depend. 2015, 156, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]