

Abstract
Objective:
GNAO1-related disorders (OMIM #615473 and #617493), caused by variants in the GNAO1 gene, are characterized by developmental delay or intellectual disability, hypotonia, movement disorders, and epilepsy. Neither a genotype–phenotype correlation nor a clear severity score have been established for this disorder. The objective of this prospective and retrospective observational study was to develop a severity score for GNAO1-related disorders and to delineate the correlation between the underlying molecular mechanisms and clinical severity.
Methods:
Sixteen individuals with GNAO1-related disorders harboring 12 distinct missense variants, including four novel variants (p.K46R, p.T48I, p.R209P, and p.L235P) were examined with repeated clinical assessments, video-EEG monitoring, and brain MRI. The molecular pathology of each variant was delineated using a molecular deconvoluting platform.
Results:
The patients displayed a wide variability in the severity of their symptoms. This heterogeneity was well represented in the GNAO1-related disorders severity score, with a broad range of results. Patients with the same variant had comparable severity scores, indicating that differences in disease profiles are not due to inter-patient variability but rather to unique disease mechanisms. Moreover, we found a significant correlation between clinical severity scores and molecular mechanisms.
Interpretation:
The clinical score proposed here provides further insight into the correlation between pathophysiology and phenotypic severity in GNAO1-related disorders. We found that each variant has a unique profile of clinical phenotypes and pathological molecular mechanisms. These findings will contribute to better understanding GNAO1-related disorders. Additionally, the severity score will facilitate standardization of patients categorization and assessment of response to therapies in development.
Keywords: GNAO1, developmental and epileptic encephalopathy, movement disorders, epilepsy, missense
Introduction
Developmental and epileptic encephalopathies (DEEs) are a heterogeneous group of rare neurological disorders defined by early-onset refractory epilepsy, specific EEG abnormalities, developmental delay or regression, and intellectual disability1,2. With the advancement of genetic testing, a total of 110 genes associated with DEEs have been identified to date (https://www.omim.org/phenotypicSeries/PS308350). In addition to genetic heterogeneity, there is also a great deal of clinical variability. This is not only because the signs and symptoms are caused by a specific genetic variant but also because brain physiology is influenced by chronic seizures and prolonged drug exposure3. The accurate and timely identification of the genetic etiology of DEEs has the potential to enhance our understanding of the pathophysiology of these disorders and promote the development of targeted therapies 4.
DEE 17 (OMIM #615473), caused by variants in GNAO1, was initially identified by Nakamura et al. in 20135. Since then, our awareness of the condition has been improved by the extensive description of clinical cases 6–10 and a greater understanding of its pathogenesis11–13. This has led to the description of two recognized OMIM phenotypes: 1) Developmental and epileptic encephalopathy 17 (OMIM#615473)5–10 and 2) Neurodevelopmental disorder with involuntary movements (OMIM#617493)8,14. However, growing evidence suggests the severity of the disease lies on a spectrum with intermediate clinical phenotypes15.
While a few studies have examined genotype-phenotype correlation in GNAO1-related disorders 16–18, natural history data or severity scores have not been developed, nor have prognostic factors been identified. Defining subgroups of patients with GNAO1-related disorders by severity is a prerequisite for designing more precise natural history studies to identify domains and windows for potential therapeutic interventions and to plan clinical trials4.
The GNAO1 gene encodes the G protein Gαo subunit. Gαo plays an important role in the control of nervous system function. Among many things, it is involved in cytoskeletal remodeling and firing of developing neurons, regulation of synaptic function, and neuronal excitability11–13,19. Mechanistically, Gαo modulates both inhibitory and stimulatory neuromodulatory signaling to cAMP, a major determinant in the pathophysiology of movement disorders 18,20. From a molecular perspective, Gαo serves as a transducer of G protein Coupled Receptor (GPCR) signals as a part of the heterotrimeric complex with Gβγ subunits 21,22. Understanding how specific variants affect Gαo ability to transduce GPCR signals will be key to the development of interventional strategies. Recent progress in this area suggests that GNAO1-associated variants perturb Gαo function by different mechanisms, including loss-of-function, dominant-negative, and debatable gain-of-function effects, which ultimately lead to GNAO1-related disorders 11,15,18,20,23–26. Yet, our understanding of the molecular pathology of GNAO1 and its relationship with the disease symptomatology is far from complete, and many disease-causing variants remain to be characterized.
In this study, we investigated 16 individuals carrying de novo missense GNAO1 variants and deeply analyzed their phenotype, culminating in the development of an all-encompassing disease severity score. We further delineated the molecular pathology of these variants using a molecular deconvoluting platform and mapped these results onto a disease severity score to facilitate prognosis and the development of precision interventions.
Material and methods
Standard protocol approvals, registrations, and patient consents.
This study was approved by the Ethics Committee of the Institut de Recerca Sant Joan de Déu, Barcelona, Spain (PIC-77–21). Written informed consent was obtained from all guardians of participants according to the Declaration of Helsinki.
Patient ascertainment.
Sixteen individuals with missense variants in GNAO1 were identified as part of the ongoing study “Prospective and retrospective study of phenotypic and genotypic characterization of patients affected by GNAO1-related disorders” at the Hospital Sant Joan de Déu, Barcelona, Spain. This is a prospective three-year study that annually evaluates children with GNAO1-related disorders with a standardized protocol, in addition to retrospectively analyzing their evolution prior to inclusion in the study. In this article, partial data from the first and second years of assessment are utilized. A case report on one patient (P6) was previously published 27 and data from some of the cohort members were included in the caregiver survey by Axeen et al. 7
Movement disorders.
Fourteen of the 16 individuals were characterized by direct clinical examination, and in all 16, video recordings were independently reviewed by two movement disorder specialists, reaching a consensus agreement. We assessed the baseline movement disorders both at rest and when performing voluntary movements. During these assessments, the patients were taking their usual medications for epilepsy and movement disorders. Observed movements were classified according to established criteria and rated using specific and validated severity scales: the Burke-Fahn-Marsden Dystonia Rating Scale (BFMDRS) for dystonia and the Abnormal Involuntary Movement Scale (AIMS) for chorea or stereotypies. We defined a dyskinetic crises as a sudden and marked exacerbation of abnormal involuntary movements (dyskinesias), which are distinct in onset and duration from the baseline dyskinetic movements of the patient, such as dystonia, chorea or athetosis. During a dyskinetic crises, alterations in facial expression may manifest, which are different from epileptic seizures as there is no loss of awareness or disconnection from the surrounding environment. Additionally, post-episode somnolence is not typically observed, unlike in seizures. These events are typically identified by parents or regular caregivers. Video 1 displays examples of patients with GNAO1-related disorders experiencing dyskinetic crises. As the physical examination of children with GNAO1-related disorders can be highly variable, we scored each item according to the highest severity observed.
Epilepsy.
Seizures were classified according to the International League Against Epilepsy (ILAE) criteria. Ten of 16 patients underwent 24-hour video-EEG monitoring in an epilepsy unit during this study. Retrospective data on epilepsy history, seizures, previous video-EEG characteristics, antiseizure medication or other treatments used, and their responses were also collected.
Neurodevelopmental assessment.
Motor and language development were methodically assessed through the Vineland Adaptive Behavior Scales, Second Edition (Vineland-II), the Bayley Scales of Infant and Toddler Development, Third Edition (Bayley-III), and the Gross Motor Function Measure (GMFM-88).
For each patient, a comprehensive retrospective review of medical records and clinical investigations was also conducted.
MRI.
Brain MRI data were available for all patients. Ten patients underwent brain MRIs during this study. In addition, all previous MRIs available for 14 out of 16 patients were reviewed.
Lumbar puncture.
Available neurotransmitter data from lumbar punctures were collected in 5 out of 16 patients.
Severity of GNAO1-related disorders.
To develop the GNAO1-related disorders severity score, a team of expert pediatric neurologists specializing in movement disorders and epilepsy discussed the items for inclusion in the scale. Several iterations of the scale were constructed and tested, with input from other experts in the field, until the final version was obtained. The process involved a thorough review and analysis of other scales utilized in the assessment of neurodevelopmental disorders and epilepsy, with particular attention given to the unique characteristics and quality of life concerns of individuals with GNAO1-related disorders. The final scale reflects the severity of our patient population and was designed to be reliable and valid for clinical use. We have included the following items: 1) epilepsy; 2) movement disorders; 3) gross motor development; 4) language development; and 5) feeding. For items 1) epilepsy and 2) movement disorders, the average of four subitems was taken into account: a) frequency, b) intensity and duration, c) falls or injuries, and d) medication or therapy. Unless otherwise specified, the condition of the patient for the last year was used when calculating the score. Regarding item 3) gross motor development. Infants older than 3 months could be evaluated; otherwise, we computed 0 points. The same applies to item 4) language development. Infants older than 6 months could be evaluated; otherwise, we computed 0 points. The GNAO1-related disorders severity score is presented in Table 1. The scores range from 0 to 13. Finally, we categorize the phenotype as mild (total severity score ranging from 0 to 3.9 points), moderate (4 to 7.9 points), or severe (> 8 points).
Table 1.
GNAO1-related disorders severity score
| GNAO1-related disorders severity score | |
|---|---|
| 1. Epilepsy (clinical situation for the past year unless specified) | 2. Movement disorders (clinical situation for the past year unless specified) |
|
A. Frequency of
seizures 0: less than one seizure per year 1: more than one seizure per year, less than one per month 2: more than one seizure per month, less than one per week 3: more than one seizure per week or daily B. Intensity and duration of seizures 0: no seizures in the previous year 1: <30 seconds or without impairment of awareness (low risk of apnea or bronchial aspiration) 2: 30 seconds to 3 minutes (risk of apnea or bronchial aspiration) 3: >3 minutes or rescue medication needed C. Falls or injuries during seizures 0: no seizures in the previous year 1: no falls or injuries 2: sleep-disrupting seizures or causing falls or injuries 3: seizures resulting in apnea or bronchial aspiration D. Amount of antiseizure medications (ASM) / therapy for epilepsy: 0: no ASM 1: 1 ASM / therapy 2: 2 ASM / therapy 3: >3 ASM / therapies or rescue medication required in the last 3 months or vagal nerve stimulation implanted or epilepsy surgery performed |
A. Frequency of movement
disorders 0: no movement disorders in the previous year 1: stable or persistent choreoathetosis and dystonia, stereotypies or other movement disorders 2: rare dyskinetic crisis (one episode per month or less) 3: frequent dyskinetic crisis (weekly or daily episodes) B. Intensity and duration of dyskinetic crisis 0: no dyskinetic crisis in the previous year 1: <1 minute 2: 1 to 3 minutes 3: >3 minutes or rescue medication or hospitalization required C. Falls or injuries during dyskinetic crisis or as a result of any movement disorders 0: absence of both movement disorders or dyskinetic crisis in the previous year 1: no falls or injuries (daily short dyskinetic crisis or persistent movement disorders) 2: sleep-disrupting dyskinetic crisis or causing falls or injuries (joint dislocations, bites, etc) or pain 3: movement disorders resulting in rhabdomyolysis, renal or respiratory failure or other life threating conditions D. Amount of medications / therapy for movement disorders: 0: no medication needed 1: 1 drug 2: 2 drugs 3: >3 drugs, rescue medication required in the last three months or deep brain stimulation implanted |
| Total item1: (A+B+C+D)/4 = | Total item2: (A+B+C+D)/4 = |
| 3. Gross motor development (this item can be assessed on infants older than 3 months; otherwise, compute 0 points) | 4. Language development (this item can be assessed on infants older than 6 months; otherwise, compute 0 points) |
| 0: can walk alone or rarely need
help 1: can walk with support 2: can sit without support 3: had acquired head control 4: absence of gross motor development (not even head control) |
0: normal or can speak at least
two-word phrases 1: can speak single words or babbling 2: absence of expressive language |
| 5. Feeding | |
| 0: orally fed 1: fed by a gastrostomy or any other type of enteral feeding tube |
|
| TOTAL: (items 1+2+3+4+5)= | |
Genetic analysis.
All patients were diagnosed with GNAO1 missense variants as part of their routine clinical care using available testing, whether through 1) targeted GNAO1 gene sequencing, 2) a multiple gene panel for DEEs, or 3) whole exome secuencing (WES).
Functional studies.
Cell culture and transfection—
HEK293FT cells were grown in DMEM supplemented with 10% FBS, minimum Eagle’s medium non-essential amino acids, 1mM sodium pyruvate at 37°C in a humidified incubator containing 5% CO2. At the time of transfection, cells were supplemented with 0.1% Matrigel (Corning).
For bioluminescence resonance transfer (BRET) assays, cells were seeded in 96-well flatbottomed white microplates (Greiner Bio-One) at a density of 5 × 104 cells/well. At the same time the cells were plated, they were transfected with expression constructs (total 0.09 μg/well), PLUS reagent (0.1 μL/well) and Lipofectamine LTX (0.5 μL/well). The expression constructs transfected were as follows (number in parentheses indicates the relative amount of DNA, where 1 = 0.015 μg): Flag-D2R (1), GαoA (2), Venus 156–239-Gβ1 (1), Venus 1–155Gγ2 (1), and masGRK3ct-Nluc-HA (1).
For NanoBiT assays, cells were seeded into 6-cm dishes at a density of 4 × 106 cells/dish. After 4 hr., expression constructs (total 2.1 μg/dish) were transfected into the cells using PLUS reagent (7.5 μL/dish) and Lipofectamine LTX (12 μL/dish) reagents. The expression constructs transfected were as follows (number in parentheses indicates the relative amount of DNA, where 1 = 0.42 μg): D2R-mycSmBiT (1), GαoA (0.1), LgBiT-Gβ1 (1), and Gγ2 (1). A calibration curve of wildtype GαoA cDNA was run and the amount of GαoA cDNA used in the assay was chosen so that the assay was not saturated.
BRET assay—
BRET between Venus-Gβ1γ2 and masGRK3ctNluc-HA was used to measure trimer formation, agonist-induced G protein activation, and dominant-negative activity of Gαo mutants in living cells28,29. 16 to 24 hr. post-transfection, cells were washed with once BRET buffer (Phosphate-Buffered Saline (PBS) containing 0.5mM MgCl2 and 0.1% glucose. Cells were harvested with centrifugation at 500 g for 5 min and resuspended in BRET buffer. The substrate for Nano luciferase (Nluc), furimazine (Promega), was diluted in BRET buffer according to the manufacturer’s instructions and added to the cells. BRET measurements were made every 100 ms over a course of 1 minute using a microplate reader (PHERAstar FSX; BMG Labtech) equipped with two emission photomultiplier tubes. 100 μM dopamine (Sigma) was added after obtaining a baseline BRET value. All measurements were performed at 37 °C. The BRET signal was calculated as the ratio of the light emitted by the Venus-Gβ1γ2 (535 nm ± 30 nm) over the light emitted by the masGRK3ct-Nluc-HA (475 nm ± 30 nm). The average baseline BRET value recorded prior to dopamine stimulation was subtracted from the experimental BRET signal values.
NanoBiT assay—
Measurement of bioluminescence caused by complementation between D2R-SmBiT and LgBiT-Gβ1 was performed to examine the interaction between G proteins and the receptor. 16 to 24 hr. post-transfection, HEK293FT cells were washed once with BRET buffer and detached by gentle pipetting. Approximately 50,000 to 100,000 cells/well were transferred to a 96-well flatbottomed white microplate (Greiner Bio-One). The substrate for Nano luciferase (Nluc), furimazine (Promega), was diluted in BRET buffer according to the manufacturer’s instructions and added to the cells. Luminescence measurements were made every 0.74 s for 172.66 s using a microplate reader (PHERAstar FSX; BMG Labtech) equipped with two emission photomultiplier tubes. 100 μM dopamine (Sigma) was added after obtaining a baseline luminescence value after 55s. All measurements were performed at 37 °C.
Quantification and statistical analysis.
For clinical studies, we used Spearman’s rank correlation to establish the correlation between the GNAO1-related disorders severity score and Vineland-II, Bayley-III (cognitive, receptive language and expressive language sub scores), GMFM-88, the Burke-Fahn-Marsden Dystonia Rating Scale, and the Abnormal Involuntary Movement Scale. For functional studies, samples with only pcDNA3.1+ transfected in place of Gαo were used as baseline measurements. The max amplitude of these measurements was subtracted from the max amplitude of all other measurements. Statistical analysis was performed using GraphPad Prism 9.4. All data are represented as mean ± SEM. Comparisons were computed using one-way ANOVA. Asterisks indicate statistical significance (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001).
Results
Genetics.
We detected many different GNAO1 variants, all of them de novo and classified as pathogenic according to the ACMG criteria. While several variants were previously reported in the literature, we found four novel variants: p.K46R, p.T48I, p.R209P, and p.L235P (Table 2). The variants p.T182I (P4 and P5), p.G203R (P11, P12, and P13), and p.R209C (P14 and P15) were identified in multiple patients in this study.
Table 2. Genetics variants included in this study.
VOUS: Variant of unknown significance, LP: Likely pathogenic, B: benign, P: Pathogenic
| Patient | Genomic DNA Variant (chr16; GRCh37) | cDNA Change HGVS NM_138736.3 | Protein Change | Previously reported patients (PMID) | Inheritance | ACMG Classification | CADD Score | Mutation Taster | PROVEAN | SIFT | gnomAD Minor Allele Frequency |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | chr16-56226265-G-C | c.118G>C | G40R | 28357411 and 26485252 | De novo | P (PM2, PM5, PP3, PS1, PM1, PP2, PP5, PS2) | 33 | Disease causing (1) | Damaging (−6.37, −6.38) | Damaging (0) | 0% |
| P2 | chr16-5622650-A-G | c.137A>G | K46R | No | De novo | P (PM1, PM2, PM5, PP2, PP3, PS2) | 1.083 | Disease causing (1) | Neutral (−2.41) | Damaging (0) | 0% |
| P3 | chr16-56226510-C-T | c.143C>T | T48I | No | De novo | P (PM1, PM2, PM5, PP2, PP3, PS2) | 27 | Disease causing (1) | Damaging (−4.8, −4.81) | Damaging (0) | 0% |
| P4, P5 | chr16-56368721-C-T | c.545C>T | T182I | 32581362 | De novo | P (PM2, PM1, PP2, PP3, PP5, PS2) | 28,8 | Disease causing (1) | Damaging (−5.44, −5.54) | Damaging (0) | 0% |
| P6 | chr16-56370645-T-C | c.596T>C | L199P | 27072799 (this patient) | De novo | P (PM1, PM2, PP2, PP3, PP5, PS2) | 28,5 | Disease causing (1) | Damaging (−4.67, −4.68) | Damaging (0) | 0% |
| P7 | chr16-56370675-G-C | c.626G>C | R209P | No | De novo | P (PM1, PM2, PM5, PP2, PP3, PS2) | 32 | Disease causing (1) | Damaging (−6.59) | Damaging(0.001) | 0% |
| P8 | chr16-56370741-A-G | c.692A>G | Y231C | 30682224, 28503590, and 27072799 | De novo | P (PM1, PM2, PP3, PP5, PP2, PS2) | 29,1 | Disease causing (1) | Damaging (−8.27, −8.24) | Damaging (0) | 0% |
| P9 | chr16-56370753-T-C | c.704T>C | L235P | No | De novo | P (PM2, PM1, PP2, PP3, PS2) | 32 | Disease causing (1) | Damaging (−6.56, −6.58) | Damaging (0) | 0% |
| P10 | chr16-56374893-T-A | c.871T>A | Y291R | 30682224, 34139551 (this patient) | De novo | P (PM2, PM1, PP2, PP3, PS2) | 26 | Disease causing (1) | Damaging (−8.3) | Damaging (0) | 0% |
| P11, P12, P13 | chr16-56370656-G-C | c.607G>C | G203R | 27864847, 27476654, 26485252, 25966631 and 23993195 | De novo | P (PM1, PM2, PS1, PM5, PP5, PP2, PP3, PS2) | 32 | Disease causing (1) | Damaging (−7.56) | Damaging (0) | 0% |
| P14, P15 | chr16-56370674-C-T | c.625C>T | R209C | 30103967, 33358199, 32581362, 28688840, 28357411, 27916449,27864847, 27625011, 27068059, 27476654, 26485252, 26060304,25966631 and 23993195, | De novo | P (PP5, PM1, PM5, PM2, PP2, PP3, PS2) | 32 | Disease causing (1) | Damaging (−7.55) | Damaging (0) | 0% |
| P16 | chr16-56370758-G-A | c.709G>A | E237K | 30103967, 29935962 and 32581362 | De novo | P (PP5, PM2, PM1, PP2, PP3) | 32 | Disease causing (1) | Damaging (−3.77, −3.75) | Damaging (0) | 0% |
General clinical, radiologic, and biochemical features.
Patient ages ranged from 11 months to 15 years and 11 months (median 8.4 years, mean 7.3 years) within the cohort. Fifty-six percent of individuals were female. We collected retrospective data on P5, who died at the age of 3 years and 2 months due to status epilepticus. Additionally, P6 died during our study at the age of 7 years and 7 months in the context of respiratory failure resulting from pneumonia and a bronchospasm crisis. First symptoms were identified at an age ranging from the prenatal period (P4 and P5 mother described, subjectively, increased fetal movements) to 1.5 years, with hypotonia or motor developmental delay (6 patients, 37.5%), epilepsy (5 patients, 31%), and movement disorder (3 patients, 18.7%) being the most frequently reported (Table 3).
Table 3. Summary of Clinical Features in GNAO1-Related Disorders patients.
F = female; M = male; y = years; NP = neonatal period; PDD = psychomotor developmental delay; MD = movement disorder; m = months; SZ = seizure; freq = frequency; LEV = levetiracetam; OXC = oxcarbazepine; LTG = lamotrigine; PB = phenobarbital; VPA = valproate; VGB = vigabatrin; TPM = topiramate; pyr = pyridoxine; LCM = lacosamide; CLN = clonazepam; RUF = rufinamide; CBZ = carbamazepine; ZNS = zonisamide; CLB = clobazam; KD = ketogenic diet; PER = perampanel; BRV = brivaracetam; Choreodystonia = choreoathetosis and dystonia; DBS = deep brain stimulation; NA = not available; TP = transpyloric.
| Patient | Sex | Age at last assessment | First symptoms / age at onset | Epilepsy / SZ freq (higher SZ freq / longer SZ free period) | Epilepsy
treatments (number tried before / current) |
Type of MD / age at onset / Dyskinetic crisis | Movement disorder treatment | Gross motor Development | GMFM-88 (%) | Language Development | Gastrostomy |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | F | 8,6 y | Epilepsy / NP | Yes / Daily / 5 y |
3 / LEV + OXC | Choreodystonia + stereotypies / 6 m / No | No | Sits without support | 26.23 | Absent | No |
| P2 | F | 4,7 y | PDD / 18 m | No / - | - | Dystonia / 2y / No | No | Walks without support | 94.89 | Absent | No |
| P3 | F | 0,9 y | Epilepsy and irritability / NP | Yes / Daily | 5 / LEV + VGB + TPM + pyr | Choreodystonia + stereotypies / NP / No | No | No head control | NA | Absent | No |
| P4 | F | 12,2 y | Increased fetal movements / Prenatal | Yes / Four seizures over a period of 5 y | 2 / LEV + LCM | Choreodystonia / Prenatal / Yes | DBS, baclofen, chloral hydrate, clonidine, tetrabenazine | No head control | 0 | Absent | Yes |
| P5 | F | 3 y | Increased fetal movements / Prenatal | Yes / Daily | 4 | Choreodystonia / Prenatal / Yes | Baclofen, CBZ | No head control | NA | Absent | Yes |
| P6 | F | 7,6 y | Epilepsy / NP | Yes /Daily (every 15 minutes) / 11 m |
12 | Choreodystonia / 3 y / Yes | DBS, baclofen, chloral hydrate, trihexyphenidyl, tizanidine, | No head control | 0 | Absent | Yes |
| P7 | M | 8,1 y | Dystonia and PDD / 19 m | No / - | - | Choreodystonia / 17 m /Yes | CBZ, tetrabenazine | Walks without support | 76.29 | Single Words | No |
| P8 | M | 12,9 y | PDD / 5 m | Yes / Weekly / 8 m | 5 / VPA + OXC | Dystonia / Yes | Baclofen | No head control | 7.67 | Absent | No |
| P9 | M | 3,6 y | Hypotonia and PDD / 12 m | No / - | - | Choreodystonia / 12 m / No | No | Walks without support | NA | Babbling | No |
| P10 | M | 15,9 y | Episodes of facial redness during feeding / 5 m | No / - | - | Choreodystonia + stereotypies / 12 m / Yes | Baclofen | Walks without support | 35.67 | Absent | Yes |
| P11 | F | 3,9 y | Neonatal seizures / NP | Yes / No seizures since NP | LEV | Choreodystonia / 3 m / Yes | Tetrabenazine, CLZ | No head control | 6.91 | Absent | Yes |
| P12 | F | 0,9 y | Epilepsy / NP | Yes / Daily / 2 m | 5 / LEV + OXC + CLN | Choreodystonia / 6 m / No | No | No head control | 6.82 | Babbling | No |
| P13 | M | 1,25 y | MD / 2 m | No / - | No | Choreodystonia / NP / Yes (daily) | CZP | No head control | NA | Guttural sounds | TP tube |
| P14 | M | 9 y | Dystonia / 9 m | No / - | - | Dystonia / 12 m / Yes | L-dopa, 5-OH-thryptohan | Walks without support | 69 | Dysarthric speech | No |
| P15 | M | 11.5 y | PDD / 12 m | No / - | - | Choreodystonia / 9 m / No | DBS | Walks without support | NA | Dysarthric speech | No |
| P16 | F | 8,8 y | Irritability / 3 m | No / - | - | Choreodystonia / 5 m / Yes | CZP, Baclofen | Head control acquired | 8.67 | 4-5 single words | No |
Movement disorders.
Abnormal involuntary movements were present in all patients, in the majority of cases by 12 months of age (14 patients, 87.5%). Generalized choreoathetosis and dystonia (13 patients, 81.3%) and stereotypies (3 patients, 18.8%) were the most frequent movement disorders (Video1, in Supplementary material). Ten patients (62.5%) presented with dyskinetic crises. Six patients (37.5%) were hospitalized specifically for management of their movement disorder. A number of medications were tried in our cohort, with no obvious benefit in many cases (Table 3). Four patients underwent globus pallidus deep brain stimulation that was partially beneficial: Dyskinetic crises in P4 were less frequent and severe, avoiding the need for further hospitalizations; generalized dystonia in P14 showed only a slight improvement; P6 demonstrated a significant but transient improvement in her dyskinetic crises; whereas P7 has a brief follow-up period after the DBS to draw a conclusion. The outcomes of the assessment of dystonia and chorea utilizing the BFMDRS and AIMS rating scales are presented in Table S1.
Epilepsy.
Eight patients (50%) were diagnosed with epilepsy, with several seizure types identified and heterogeneous video-EEG findings (Table 3). Of note, the patients with the highest scores on the GNAO1-related disorders severity score had abnormal background activity, with diffuse slowing during wakefulness (5 patients, 31.3%) and absence of physiological sleep elements and periods of diffuse low voltage activity during sleep (3 patients, 18.7%). Variable seizure frequency was observed, with one patient experiencing four seizures over five years and others experiencing multiple daily seizures. Six patients (37.5%) meet criteria for drug-resistant epilepsy. Based on interviews with caregivers and previous medical reports, it is not possible to determine which antiseizure medications are more effective. P6 was given a ketogenic diet, which produced a positive but temporary response. Other non-pharmaceutical treatments, such as vagal nerve stimulation, were not utilized in any patient.
Neurodevelopmental outcome.
Neurodevelopmental delay was present in all cases. Notably, five patients had a relatively mild phenotype, manifesting normal head growth, independent ambulation, limited spoken language, and purposeful hand function. In contrast, seven patients tended to be more severely affected, with a phenotype more consistent with the classically described GNAO1-related disorder (Table 3). Neurodevelopmental assessment scores of Participants (Bayley-III, Vineland-II, and GMFM-88) can be found in Table S2.
Feeding.
Five patients (31.2%) required gastrostomies, and one patient (6.3%) required a transpyloric tube, while the majority of patients were fed by mouth with food of normal or mashed consistency (Table 3).
Brain MRI.
Normal brain MRIs were reported for 11 patients (68.6%). Unspecific white matter lesions (1 patient, 6.3%) and global atrophy or widening of the extra axial spaces and ventricular system (2 patients, 12.5%) were frequent findings. Interestingly, P5 showed cortical cytotoxic edema, probably due to status epilepticus versus hypernatremia. No obvious radiologic abnormalities of the basal ganglia were detected.
CSF analysis.
CSF neurotransmitter analysis was previously undertaken in five patients. Abnormalities, namely low homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA), were seen in one patient (P15). This patient began L-dopa and 5-OH-thryptophan supplementation with doubtful benefit. It is currently unclear as to why this finding has been observed, and it is possible that this may be a coincidental occurrence.
GNAO1-related disorders severity score.
The range of total severity scores was between 2 and 13 (Table 4). Five patients had a score of mild (from 0 to 3.9 points), four had a score of moderate (from 4 to 7.9 points), and seven had a score of severe (> 8 points). The mean score for each subcategory was as follows: 1) epilepsy 0.9 (range 0 to 3), 2) movement disorders 1.6 (range 0.3 to 3), 3) gross motor development 2.3 (range 0 to 4), 4) language development 1.8, and 5) feeding 0.3.
Table 4. Clinical Severity scores and Experimental Measurement scores for each patient in the study.
1A = frequency of seizures; 1B = intensity and duration of seizures; 1C = falls or injuries during seizures; 1D = amount of antiseizure medications (ASMs)/therapy for epilepsy; 2A = frequency of movement disorders; 2B = intensity and duration of dyskinetic crises; 2C = falls or injuries experienced during dyskinetic crises or as a result of any movement disorders; 2D = amount of medication/therapy for control of the movement disorders.
| Patient | GNAO1 Variant | Current age | 1A | 1B | 1C | 1D | Epilepsy (1A+1B+ 1C+1D) /4 | 2A | 2B | 2C | 2D | Movement disorders (2A+2B+ 2C+2D) /4 | Gross motor development | Language development | Feeding | GNAO1-related disorders severity score | mild (0–3.9), moderate (4,0–7.9), severe (>8) | Expression | Trimer formation | Loss of function | Dominant negative activity | Receptor interaction | Experimental measure Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | G40R | 8,6 | 0 | 0 | 0 | 1 | 0,3 | 1 | 1 | 1 | 0 | 0,8 | 2 | 2 | 0 | 5,0 | moderate | 3 | 2 | 3 | 1 | −2 | 7 |
| P2 | K46R | 4,7 | 0 | 0 | 0 | 0 | 0,0 | 1 | 0 | 1 | 0 | 0,5 | 0 | 2 | 0 | 2,5 | mild | 0 | 0 | 2 | 0 | 3 | 5 |
| P3 | T48I | 0,91 | 3 | 3 | 2 | 3 | 2,8 | 1 | 2 | 1 | 0 | 1,0 | 4 | 2 | 0 | 9,8 | severe | 3 | 3 | 3 | 2 | −1 | 10 |
| P4 | T182I | 12,2 | 1 | 2 | 1 | 1 | 1,3 | 3 | 3 | 2 | 3 | 2,8 | 4 | 2 | 1 | 11,0 | severe | 2 | 3 | 3 | 2 | 0 | 10 |
| P5 | T182I | Exitus | 3 | 3 | 3 | 3 | 3,0 | 3 | 3 | 3 | 3 | 3,0 | 4 | 2 | 1 | 13,0 | severe | 2 | 3 | 3 | 2 | 0 | 10 |
| P6 | L199P | Exitus | 2 | 2 | 3 | 3 | 2,5 | 3 | 3 | 3 | 3 | 3,0 | 4 | 2 | 1 | 12,5 | severe | 3 | 3 | 3 | 1 | −3 | 7 |
| P7 | R209P | 8,1 | 0 | 0 | 0 | 0 | 0,0 | 1 | 1 | 0 | 2 | 1,0 | 0 | 1 | 0 | 2,0 | mild | 0 | 0 | 3 | 3 | 1 | 7 |
| P8 | Y231C | 12,9 | 3 | 1 | 1 | 2 | 1,8 | 1 | 0 | 0 | 1 | 0,5 | 3 | 2 | 0 | 7,3 | moderate | 1 | 2 | 2 | 0 | −1 | 4 |
| P9 | L235P | 3,6 | 0 | 0 | 0 | 0 | 0,0 | 1 | 0 | 1 | 0 | 0,5 | 0 | 2 | 0 | 2,5 | mild | 0 | 0 | 2 | 0 | 0 | 2 |
| P10 | Y291R | 15,9 | 0 | 0 | 0 | 0 | 0,0 | 3 | 2 | 2 | 1 | 2,0 | 0 | 2 | 1 | 5,0 | moderate | 1 | 0 | 2 | 0 | 0 | 3 |
| P11 | G203R | 3,9 | 3 | 1 | 1 | 2 | 1,8 | 3 | 3 | 2 | 3 | 2,8 | 4 | 2 | 0 | 10,5 | severe | 0 | 1 | 2 | 1 | 3 | 7 |
| P12 | G203R | 0,91 | 1 | 2 | 1 | 3 | 1,8 | 1 | 0 | 0 | 0 | 0,3 | 4 | 2 | 0 | 8,0 | severe | 0 | 1 | 2 | 1 | 3 | 7 |
| P13 | G203R | 1,25 | 0 | 0 | 0 | 0 | 0,0 | 3 | 3 | 2 | 2 | 2,5 | 4 | 2 | 1 | 9,5 | severe | 0 | 1 | 2 | 1 | 3 | 7 |
| P14 | R209C | 11,5 | 0 | 0 | 0 | 0 | 0,0 | 1 | 0 | 2 | 3 | 1,5 | 0 | 1 | 0 | 2,5 | mild | 0 | 0 | 2 | 1 | 0 | 3 |
| P15 | R209C | 9 | 0 | 0 | 0 | 0 | 0,0 | 3 | 1 | 1 | 2 | 1,8 | 0 | 1 | 0 | 2,8 | mild | 0 | 0 | 2 | 1 | 0 | 3 |
| P16 | E237K | 8,8 | 0 | 0 | 0 | 0 | 0,0 | 3 | 2 | 1 | 2 | 2,0 | 3 | 1 | 0 | 6,0 | moderate | 0 | 0 | 1 | 0 | 2 | 3 |
Patients with the same GNAO1 variant had comparable total severity scores (P4 and P5 with p.T182I variant (11 and 13, severe), P11, P12, and P13 with p.G203R variant (10.5, 8 and 9, severe), and P14 and P15 with p.R209C variant (2.5 and 2.8, mild) and were categorized as having the same severity. Video 2 displays examples of patients with varying degrees of severity in the GNAO1-related disorders severity score.
Structure - Functional studies.
To understand the molecular mechanism behind the pathology of each uncharacterized variant, we began by mapping their locations onto the structure of Gαo (Figure 1A). We found several variants (p.G40R, p.K46R, and p.T84I) located in the P-loop region. Another variant, p.T182I, directly coordinates the Mg2+ ion. Several other variants include proline substitutions. These could be predicted to disrupt secondary structure in regions such as β3, Switch II and Switch III, in the cases of p.L199P, p.R209P, and p.L235P, respectively. The remaining two variants, p.Y231C and p.Y391N, are both substitutions of tyrosine residues, with unclear implications for Gαo organization based on the structural considerations.
Figure 1. All variants exhibit an inhibited agonist response.
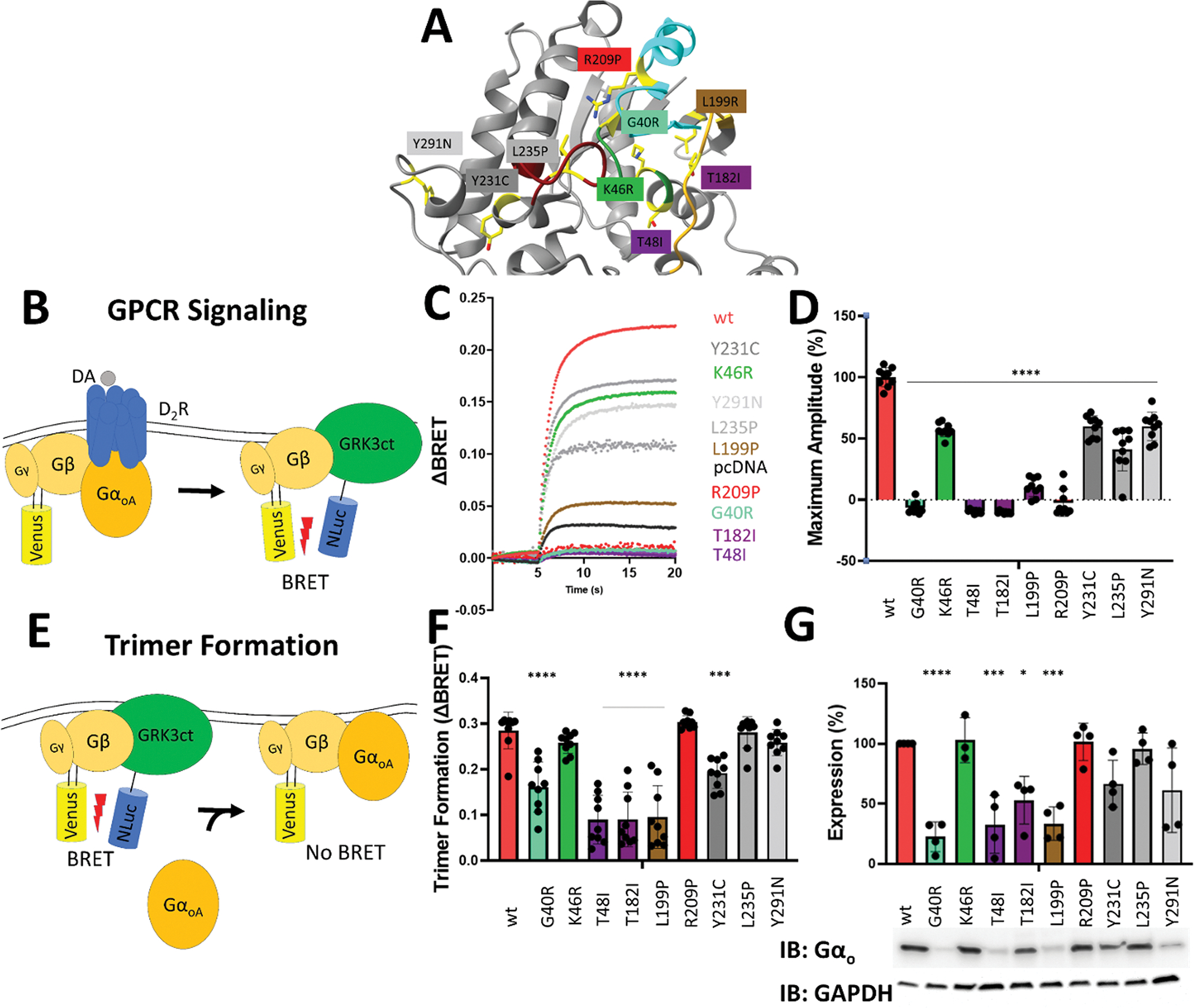
A. Location of each variant on Gαo (PDB: 3C7K). Regions are indicated as follows: Green – P-loop; Goldenrod – Switch I; Cyan – Switch II; Magenta – Switch III.
B. Schematic of GPCR Signaling Assay. Dopamine stimulation induces the release of venus-Gβγ from Gαo, allowing venus-Gβγ to associate with NLuc-GRK3 and increase the BRET signal.
C. Representative traces of variant dopamine BRET responses.
D. The effect of GNAO1 variants on dopamine induced BRET signal. 0% response was defined as the amount of BRET signal upon dopamine activation of the Gαo-free control. 100% response was defined as the amount of BRET signal upon dopamine activation of the WT Gαo control.
E. Schematic of Trimer formation assay. In the absence of Gαo, venus-Gβγ and NLuc-GRK3 have a high level of basal BRET. The binding of Gαo to Gβγ interferes with BRET between venus-Gβγ and NLuc-GRK3.
F. The effect of GNAO1 variants on basal BRET. The amount of basal BRET without Gαo expressed was defined as 0. The amount of trimer formation is determined by subtracting the basal BRET of each variant from the Gαo-free control.
G. Expression levels of each variant. Western blot analysis of each variant blotted with α-Gαo antibody.
We have previously developed a pipeline for mechanistic evaluation of disease variants in Gα using a suite of BRET assays that monitor transitions in the G protein cycle 11,23. We applied this approach to understand the molecular mechanisms underlying the novel Gαo variants uncovered in this study.
We started our examination by assessing the overall ability of the Gαo variants to transmit signals using D2R as a model GPCR that prominently couples to Gαo and is involved in striatal motor control (Figure 1B). For each variant, we observed a significantly reduced maximum response amplitude in response to dopamine stimulation as compared to WT (Figure 1C). Five mutants (p.G40R, p.T48I, p.T182I, p.L199P, p.R209P) completely failed to respond to dopamine stimulation, while others displayed varying degrees of deficiency (Figure 1D).
To probe the mechanisms underlying the signaling deficits, we first tested Gαo variants for an ability to form heterotrimers with the Gβγ subunits. This was accomplished by comparing the baseline BRET ratio between NanoLuc-tagged GRK and the Venus-tagged Gβγ (Figure 1E). As BRET between these two molecules is precluded by association of Gαo with Gβγ, we determined the amount of heterotrimer formation based on changes in baseline BRET values induced upon the introduction of Gαo. This analysis revealed that all of the mutants that were completely unable to transduce GPCR signals (p.G40R, p.T48I, p.T182I, p.L199P) also had significantly reduced ability to form G protein heterotrimers (Figure 1F).
We questioned if there were any folding and/or stability defects in the variants that could cause their reduced ability to form heterotrimers. Western blotting of transfected HEK293FT cell lysates indicated that p.G40R, p.T48I, pT182I, and p.L199P were indeed expressed at significantly lower levels than WT Gαo (Figure 1G). This decrease in expression closely correlates with the reduction in heterotrimer formation observed in these variants, suggesting that loss of protein stability likely underlies the functional deficits in trimer formation and, consequently, transduction of GPCR signals.
We next tested GNAO1 variants for their dominant negative activity given that it was shown to be present in some previously reported variants 11. To test this, we expressed the variants alongside WT Gαo and measured any decrease in dopamine-induced BRET response (Figure 2A). Three variants (p.T48I, p.T182I and p.R209P) displayed a significant decrease in dopamine response, indicating that they interfere with normal activation of Gαo by D2R (Figure 2B, 2C).
Figure 2. Some GNAO1 variants exhibit dominant negative activity.
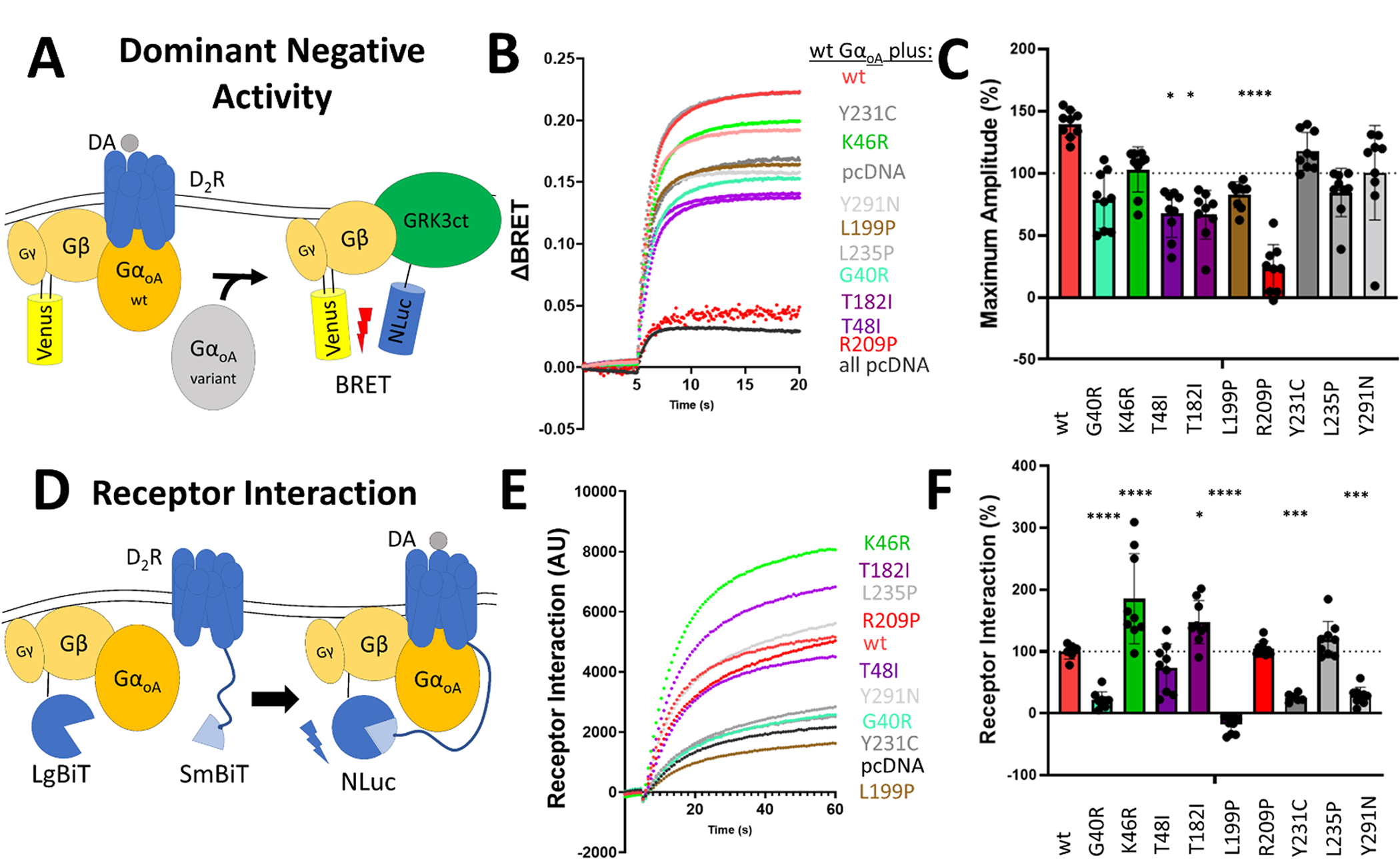
A. Schematic of the Dominant Negative Activity Assay. Variants of Gαo are expressed alongside WT Gαo. Variants with dominant negative activity are able to suppress WT Gαo dopamine induced activation.
B. Representative traces of dopamine BRET responses of GNAO1 variants expressed alongside WT Gαo.
C. The effect of GNAO1 variants on WT Gαo dopamine induced BRET signal. Any condition with a max amplitude below the condition expressing pcDNA3.1+ alongside WT Gαo indicates that the variant has dominant negative activity.
D. Schematic of the Receptor Interaction Assay. Dopamine stimulation induces the recruitment of G protein heterotrimer to the receptor where the SmBiT tag on the receptor can interact with the LgBiT tag on Gβ. This reconstitutes NLuc and gives a luminescent readout.
E. Representative luminescence traces of the interaction of G protein heterotrimer with D2R receptor.
F. The effect of GNAO1 variants on dopamine-induced receptor interaction with G protein heterotrimer. 0% interaction is defined as the amount of luminescence detected when the Gαo-free control was stimulated with dopamine. 100% interaction is defined as the amount of luminescence detected when WT Gαo was stimulated with dopamine.
We further interrogated possible mechanisms behind dominant negative effects by studying the recruitment of G protein heterotrimer to the D2R receptor upon its activation by an agonist. This was achieved using an assay that measures complementation between SmBit-tagged D2R and LgBit-tagged Gβγ which reconstitutes Nluc, producing luminescence upon Gαo- mediated interaction of Gβγ with the D2R (Figure 2D). Interestingly, with the single exception of p.L199P, we detected interaction with D2R for all mutants with a varying degree of efficiency (Figure 2E, 2F). The p.G40R, p.Y321C, and p.Y291N variants showed a significantly decreased interaction with the receptor compared to WT, likely explained by the decreased expression of the variants. On the other hand, the p.K46R and p.T182I variants displayed a significant increase in interaction with the receptor compared to WT, suggesting that these variants have more non-productive interactions with the receptor.
Correlation Between Clinical and Experimental Measurements
To assess the strength of our experimental measurements in predicting the clinical severity of the different variants, we gave each variant a score ranging from 0 to 3 in the categories of Gαo Expression, Heterotrimer Formation, Loss-of-function, and Dominant Negative phenotype, with 0 indicating no departure from WT values and 3 being the most severe departure from WT values (Table 4). We also gave a score ranging from −3 to +3 for Receptor interaction, with positive values indicating increased interaction with the receptor and negative values indicating decreased interaction with the receptor. These five categories were then combined into an overall Experimental Measurement score. The variants in Muntean et al.11 were also given an Experimental Measurement score.
Overall, there was a strong correlation between the Clinical Severity score and the Experimental Measurement score (R2 = 0.486) (Figure 3A). To further dissect what molecular mechanisms underpinned the clinical severity, we compared the overall Clinical Severity score with the scores of each individual functional experiment. We discovered a correlation between the Clinical Severity score and the variant Expression Levels (R2 = 0.301) (Figure 3B) and Trimer Formation (R2 = 0.685) (Figure 3C). The correlations for the Loss-of-function, Dominant Negative, and Receptor Interaction experiments were much weaker (R2 = 0.139, 0.079, and 0.001, respectively) (Figure 3D–F). We further analyzed relationship between the highest correlating functional metric of Trimer Formation and individual clinical traits that contribute to Clinical Severity score. This analysis revealed the greatest level of correlation with epilepsy measures (Figure S1 A–E) and weak, if any, correlation with other measures (Figure S1 F–M).
Figure 3. Correlations between Scores for GNAO1-related disorders severity score and Experimental Measurement.
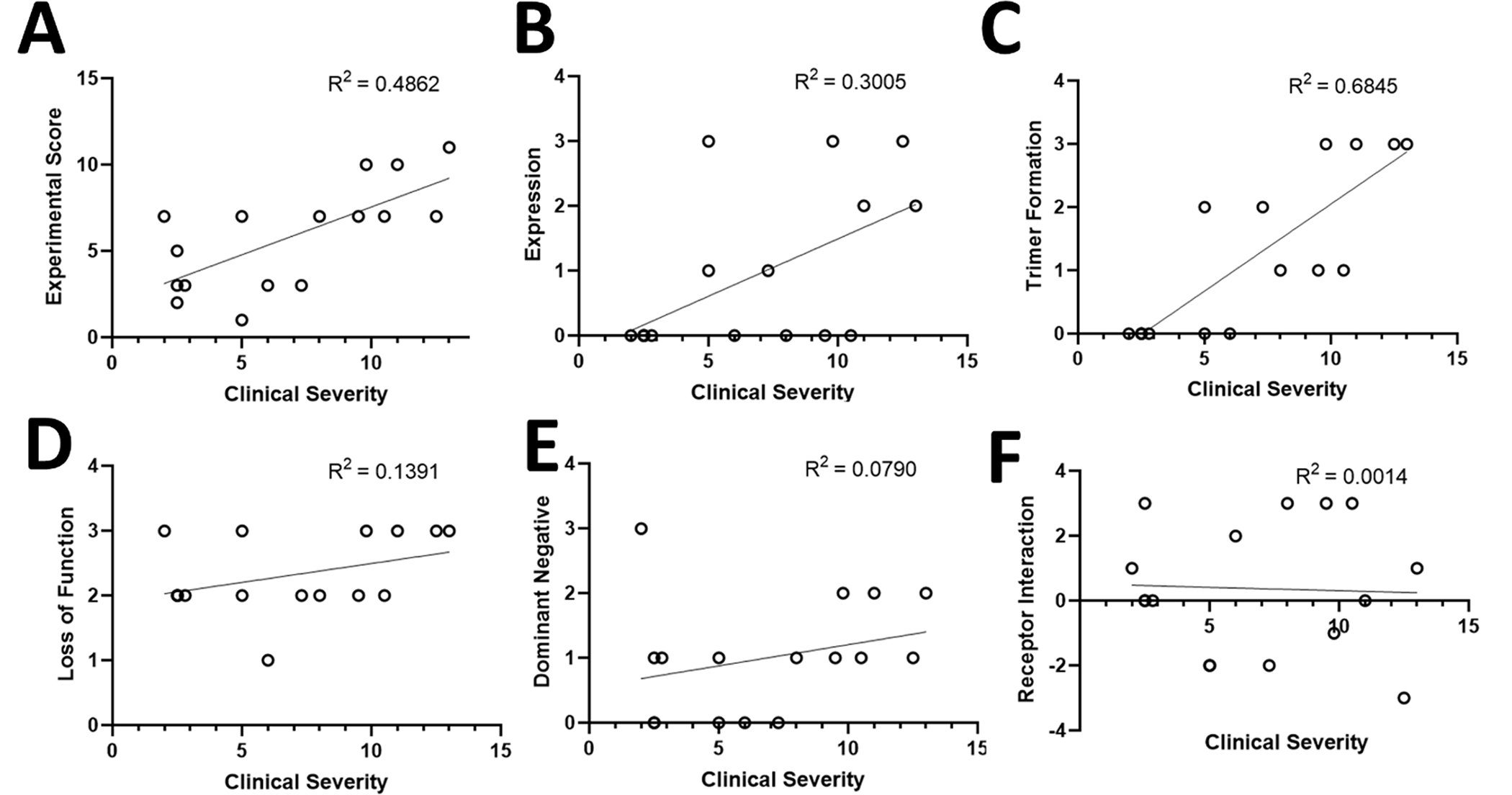
A-F. Comparisons between clinical and experimental measurements do include repeated measures for patients with the same variant.
Correlation Analysis with Other Standardized Scales.
We used Spearman’s rank correlation and found a significant negative correlation between the GNAO1-related disorders severity score and Vineland-II (r=−.553*, p=0.032), Bayley-III Cognitive (r=−.811**, p=0.008), Bayley-III Receptive Language (r=−.868**, p<0.001), Bayley-III Expressive Language (r=−.777**, p=0.002), GMFM-88 (r=−.955**, p<0.001). On the other hand, we found only a weak non-significant positive correlation between GNAO1-related disorders severity score and the Burke-Fahn-Marsden Dystonia Rating Scale (r=.191, p=0.532), and the Abnormal Involuntary Movement Scale (r=.127, p=0.680). Table S3 displays the correlations observed between the GNAO1-related disorders severity score and other standardized scales.
Discussion
We report detailed phenotypic data for 16 individuals with GNAO1-related disorders harboring 12 distinct missense variants, including four novel variants (p.K46R, p.T48I, p.R209P, and p.L235P), thus expanding the genetic variability of the disease. Furthermore, we develop a clinical severity score system in order to standardize the assessment of individuals with GNAO1-related disorders. Additionally, we delineate the molecular mechanisms (receptor-mediated activation of the G protein, trimer formation, dominant negative activity, and receptor interaction) of each variant included in this study, develop an experimental measurement score for missense GNAO1 variants, and analyze its correlation with the clinical score.
Disease-causing missense variants in GNAO1 are associated with a neurodevelopmental syndrome that ranges from mild to severe and is characterized by epilepsy, developmental delay or intellectual disability, hypotonia, and movement disorders 6–10. Using a disease-specific composite score to quantify the severity among different individuals is necessary for establishing truthful phenotype-genotype correlations. The GNAO1-related disorders severity score proposed here incorporates the most relevant aspects of this condition, including epilepsy, movement disorders, neurodevelopmental issues, and the need for a gastrostomy. Our team, comprised of pediatric neurologists specialized in both movement disorders and epilepsy, developed the GNAO1-related disorders severity score after conducting a thorough examination of the phenotype of individuals with GNAO1-related disorders. In addition, we carefully reviewed and analyzed other scales utilized in the assessment of neurodevelopmental disorders and epilepsy to inform our design process.30 In regards to items 1) epilepsy and 2) movement disorders, we have taken into account the most significant factors that influence how these conditions impact the quality of life. This encompasses the frequency, duration, and intensity of the condition, as well as the potential for disruptive events such as falls, sleep disturbances, pain, or injury. Furthermore, we have also considered the response of the condition to treatment. Despite the absence of falls related to movement disorders or seizures in our cohort, we have designed the scale to be universally applicable to all individuals affected by GNAO1-related disorders. Given the possibility that movement disorders and epilepsy may manifest or fluctuate over the clinical course of GNAO1-related disorders, it is necessary to undertake additional investigations to confirm the reliability and usefulness of the GNAO1-related disorders severity score in larger patient cohorts over an extended time frame. Nonetheless, we observed that in our sample, the severity score appropriately captured the heterogeneous clinical phenotypes and overall severity of each patient, with a wide range of scores and elevated scores consistently correlated with greater disease severity across all domains. Furthermore, we did not observe any substantial variations in severity levels during the follow-up period of our prospective study. The GNAO1-related disorders severity score obtained from our cohort did not exhibit any correlation with age, as we did not observe higher scores in older children. Additionally, in the retrospective analysis of different individuals, we did not identify any significant changes in the severity scores over time. This observation leads us to hypothesize that a significant modification in the severity score of the disorder is more likely to be associated with a therapeutic intervention rather than the natural progression of the condition. We here present an initial stage in the creation of the GNAO1-related disorders severity score. Our aim is to enhance this framework by testing it on a more diverse population, including individuals with varying GNAO1 variants, to improve the precision of scoring subscales. Additionally, we are interested in exploring the possibility of applying our severity ranking to individuals with other DEEs, which would be a fascinating avenue of investigation. Limited information on the natural history of specific DEEs 31, makes disease progression poorly traceable for the majority of DEEs. Yet, as more information becomes available, common severity scoring systems could delineate cross-disorder differences and similarities. Overall, we hope that our GNAO1-related disorders severity score will advance the development of therapies for this and related conditions.
The results of our analysis revealed significant correlations between the GNAO1-related disorders severity score and several standardized scales commonly used to assess developmental and movement disorders (Table S3). Specifically, we found a strong negative correlation between the GNAO1-related disorders severity score and the Vineland-II and Bayley-III (cognitive, receptive language, and expressive language sub scores). These findings suggest that as the GNAO1-related disorders severity score increases, individuals with GNAO1-related disorders tend to exhibit more severe cognitive and language impairments, as well as greater difficulties in adaptive behaviors. In addition, we observed a very strong negative correlation between the GNAO1-related disorders severity score and GMFM-88 scale, indicating that as the GNAO1-related disorders severity score increases, individuals with GNAO1-related disorders tend to have more severe gross motor impairments. Interestingly, we did not find a significant correlation between the GNAO1-related disorders severity score and the Burke-Fahn-Marsden Dystonia Rating Scale or the Abnormal Involuntary Movement Scale, suggesting that the severity of dystonia or involuntary movements may not be strongly related to overall disease severity in this population. In our study, we expected to find no correlation with the Burke-Fahn-Marsden Dystonia Rating Scale or the Abnormal Involuntary Movement Scale, as we have previously observed in our natural history study that dystonia does not necessarily correlate with the ability to walk (Table S1). There are “mild” phenotypes that are highly dystonic but still have preserved walking ability. Additionally, we believe that axial hypotonia is a sign that influences motor development more significantly. Another finding that requires further confirmation in a larger cohort is that children with preserved walking ability and generalized dystonia exhibit fewer or no dyskinetic crises compared to hypotonic children with more severe motor impairment.
Remarkably, in our cohort, we have indeed identified three patients with a very severe phenotype, characterized by marked hypotonia (GMFM-88, 0%), similar to that observed in patients with neuromuscular disease. These patients have very poor voluntary motor control and profound intellectual disabilities, which hinder their ability to follow commands and participate in motor testing. These severe phenotypes are so debilitating that two out of the three patients have unfortunately passed away.
Our study is the first to utilize an integrative severity score for each individual with GNAO1-related disorders based on a comprehensive clinical scoring system. Similar disease-specific scores have been developed for other rare disorders that have allowed to guide the prognosis of the disease by recognizing clinical or molecular findings as well as establish causal relationships. Brock et al. found a significant inverse correlation between cerebral visual impairment and neurodevelopmental outcome in individuals with DEE 2 due to CDKL5 variants32. Balagura et al. found that the age at the onset of seizures correlates with a poor developmental outcome, as measured by the STXBP1 (DEE 4) composite developmental score33. In our study, we observed that video-EEG in patients with the most severe GNAO1-related disorders showed a remarkable absence of physiological sleep elements and periods of diffuse low-voltage activity during sleep. These characteristics have been identified in other severe DEEs 34,35, as well as in the previous report of P6 27. Although more data are needed, this characteristic might be included in the assessment of the severity score for this disorder, or it may even be a potential neurophysiologic biomarker for the diagnosis of a GNAO1-related disorder. Along the same lines, the mother of P4 and P5, with a severe phenotype, described increased fetal movement. Although this is somewhat subjective, we believe that rather than epileptic seizures, these movements corresponded to dyskinetic episodes, as both patients with a severe phenotype (11 and 13 points in the severity score, respectively) experience daily dyskinetic episodes. Moreover, P5 died due to dyskinetic status. While this is a subjective symptom and needs to be interpreted with caution, this would be the first report of the prenatal onset of movement disorders in GNAO1-related disorders. Increased fetal movements suggestive of epileptic seizures have been reported in some neurological disorders, e.g., 3-methylglutaconic aciduria36. Given these observations, it is likely that increased fetal movement is associated with a more severe phenotype, making the application of prenatal exome sequencing 37 worth considering.
Regarding molecular findings, we have found that individuals with the same variant have comparable scores on the GNAO1-related disorder severity score (P4 and P5; P11, P12, and P13; and P14 and P15). Moreover, we found a significant correlation between the clinical severity score and the Experimental Measurement score of missense GNAO1 variants. It will be highly interesting if these findings are replicated with a larger sample size, or if genetic modifiers 38,39 or epigenetic factors 40 influence clinical expression as they do for other DEEs. Since we have not found significant changes in the severity of symptoms throughout the evolution, it does not appear that changes in the natural history of the disease can significantly modify the correlation with the pathophysiological findings. We have established three levels of severity, namely mild, moderate, and severe, in our GNAO1-related disorders severity score. This stratification takes into account the score variability and aims to group patients in the same range of severity, even if their symptoms may have fluctuated over time.
The results described here support previous findings that any variants in Gαo interfere with receptor-mediated activation of the G protein (Figure 4A). Specifically, it reinforces the assumption that the variants in the P-loop of Gαo reduce the stability of the G protein heterotrimer. Two previously uncharacterized Gαo variants reported here, p.G40R and p.T48I, are located near the P-loop and display decreased interaction with Gβγ. One exception to this observation is the novel variant p.K46R. Although this variant is located in the P-loop, it does not show any significant variation in heterotrimer formation from WT. This is likely due to the conserved nature of the lysine to arginine variant. Additionally, the variants p.T182I, which is located in the Switch I, and p.L199R, which is located on β3, also display a significant decrease in heterotrimer formation. This indicates that in addition to the P-loop, variants in these regions also interfere with heterotrimer formation. These variants are related structurally to the P-loop, as changes in β3 are passed via α1 to the nucleotide binding pocket41. Intriguingly, variants in the switch II region that directly interact with Gβ, such as p.G203R, p.R209C, and p.R209P, do not interfere with heterotrimer formation. Remarkably, among the categories that comprise the Experimental Measure score, Heterotrimer Formation showed the highest correlation with the Clinical Severity score (R2 = 0.685), thus making it really interesting for predicting the clinical severity of the different variants.
Figure 4. Each GNAO1 variant presents a unique functional profile and can be placed on a spectrum of Loss-of-function/Dominant Negative activity.
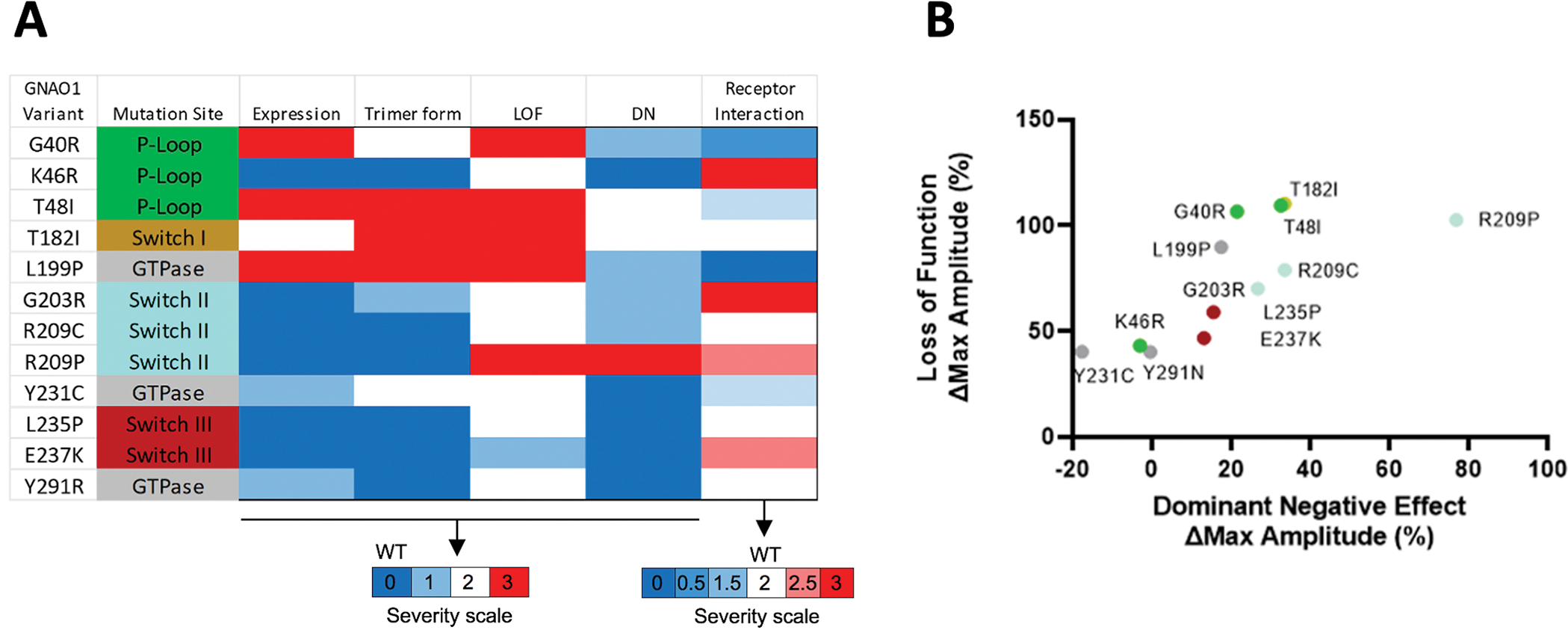
A. Meta-analysis of all GNAO1 variants, combining the results of all functional experiments.
B. Each variant can be placed on a spectrum with loss-of-function and dominant negative properties. Dots are colored according to mutation site as in panel A.
Notably, all the variants studied here interfere significantly with agonist-induced signal propagation. The amount of reduced signal propagation (loss-of-function) gave the strongest correlation with the amount of dominant negative activity presented by a variant (Figure 4B). Surprisingly, the dominant negative activity of a variant had little to do with how strongly it interacted with the receptor. This indicates that the molecular mechanisms of the dominant negative activity for many variants studied remain to be determined. Furthermore, according to our observations, the degree of dominant negative activity is a poor indicator of clinical severity. Indeed, the individual with the only variant showing the most pronounced dominant negative phenotype, p.R209P, only had a mild overall clinical severity score. It is also worth noting that in our study we used the D2 dopamine receptor to model the behavior of Gαo variants. Given the conserved mechanism of G protein activation by GPCRs, we expect similar behavior from Gαo variants with other Gi/o-coupled receptors; however, this assumption remains to be tested.
In our previous studies, we characterized the behavior of p.G203R and p.R209C variants in the endogenous neuronal setting11. We found that p.G203R lowered the efficacy of dopamine signaling in indirect-pathway medium spiny neurons (iMSNs) and increased the potency of dopamine signaling in direct-pathway medium spiny neurons (dMSNs). Contrastingly, it reduced adenosine efficacy in dMSNs and increased potency in iMSNs. On the other hand, p.R209C exclusively affected iMSNs by lowering dopamine efficacy and exclusively affected dMSNs by lowering adenosine efficacy. This difference in response is intriguing because p.R209C has a mild clinical severity outcome while p.G203R has a severe outcome. It is conceivable that reductions in efficacy in dMSNs and iMSNs are not as pathogenic as increases in agonist potency in these neuronal populations.
Major limitations of our study include selection bias toward individuals with missense variants, the restricted number of individuals recruited, the limited number of different variants in the GNAO1 gene (12 out of the 60 described to date) included, and the relatively brief period of clinical follow-up. Therefore, further studies are needed to validate the GNAO1-related disorders severity score and verify its usefulness in a generalized clinical routine. Nevertheless, the prospective standardization of data collection and the utilization of the GNAO1-related disorders severity score allow us to address the heterogeneity of these individuals, compare clinical and molecular pathology data, and identify significant correlations.
The majority of current treatments for DEEs focus on individual symptoms, such as seizures or movement disorders, rather than the underlying disease mechanisms 4. Many individuals with GNAO1-related disorders do not achieve seizure or movement disorder control, and even in patients who achieve partial control, neurodevelopmental impairments and other comorbidities frequently continue to be severe 7. The GNAO1-related disorders severity score incorporates the most relevant aspects of this condition, including epilepsy, movement disorders, neurodevelopmental issues, and the need for gastrostomy, and it evaluates the most fundamental aspects of DEEs: epilepsy and movement issues, in a manner that is sensitive to the temporal evolution of these cardinal clinical signs. Our scoring system, together with our molecular findings, can contribute to the design of future trials and studies focusing on natural history. These studies should take into account all of the major aspects of the GNAO1-related disorders at different stages among the various subgroups, identify the beneficial endpoints and windows for therapeutic interventions, and determine the optimal timing for therapeutic interventions 33.
In conclusion, we found that each GNAO1 variant has a unique profile of clinical and functional phenotypes. Although there is overlap in the clinical outcomes of each variant, each appears to have a unique pathological molecular mechanism. Moreover, patients harboring the same variants have similar clinical outcomes, indicating that the differences in disease profiles are not due to inter-patient variability but rather to unique disease mechanisms. As posited previously, each variant lies on a spectrum of loss-of-function and dominant negative activity. Finally, it is likely that the present mechanistic findings will aid in the development of pharmacological interventions for the treatment of GNAO1-related disorders. Additionally, the GNAO1-related disorders severity score will facilitate standardization of the categorization of patients according to clinical severity and assessment of response to therapies in development.
Supplementary Material
Table S1. Evaluation of Dystonia and Chorea using Standardized Rating Scales.
Figure S1. Correlations between the Trimer Formation Score and aspects of the Clinical Severity Score A-M. Comparisons between individual aspects of the Trimer Formation score and each aspect of the Clinical Severity subscore. Comparisons include repeated measures for patients with the same variant.
Table S2. Neurodevelopmental Assessment Scores of Participants: Bayley-III, Vineland-II, and GMFM-88.
Table S3. Correlations between GNAO1-related disorders severity score and other standardized scales. Asterisks indicate statistical significance (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001).
Video 1. The first part of the video showcases P4 during a dyskinetic crises. The crises had an abrupt onset and was clearly distinguishable from the patient’s baseline state. During this dyskinetic episode, ballistic and unpredictable movements of all four limbs were observed, which were non-intentional and non-predictable. The video shows approximately 30 seconds of this dyskinetic crises. There was no disconnection from the environment during the crises. In the second part of the video, P16 is shown experiencing another dyskinetic crises. In this case, the movements were less violent than in P4, but were still clearly distinguishable by the family as an episode with sudden onset and termination. The patient displayed chorea and dystonia of all four limbs and trunk, along with facial dyskinesias. These crises usually last between 1–2 minutes. Sometimes parents report a trigger such as stress, exhaustion, movement, bathing, fever, etc.
Video 2. showcases three patients with different severity levels. P7 has a mild phenotype (2 points), presenting with generalized dystonia and preserved independent gait. P16 displays an intermediate phenotype (6 points) with head control and sitting balance, but without gait. Marked axial hypotonia with action dystonia in the upper and lower extremities is observed. P4 has a severe phenotype (11 points) with marked generalized hypotonia and isolated dystonic movements. P4 carries a deep brain stimulation device (DBS). It is noteworthy that in Video 1, the dyskinetic crises of P16 and P4 can be observed, allowing for a comparison with their baseline state.
Acknowledgement
We wish to express our gratitude to the parents and patients who participated in this study, as well as Asociación GNAO1 España (https.//gnao1.es) and Aurrera Markelekin (https://aurreramarkelekin.org/wordpress/). The authors would also like to thank Esther Alvarez, MD and Javier Aparicio, MD, PhD for their assistance with study coordination. This work was funded by the NIH grants DA036596 to KAM and NS124758 to WGL.
GNAO1-Study Group:
Judith Armstrong (Department of Genetic and Molecular Medicine and Pediatric Institute of Rare Diseases. Hospital Sant Joan de Déu, Barcelona, Spain. judith.armstrong@sjd.es).
Raquel Blanco-Lago (Pediatric Neurology Department, Hospital Universitario Central de Asturias (HUCA), Oviedo, Spain. rablabul81@gmail.com).
Rosa Bou (Pediatric Rheumatology Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. rosa.bou@sjd.es).
Cristina Cáceres-Marzal (Department of Pediatric Neurology, Materno Infantil Hospital, Badajoz, Spain. cristina.caceres@salud-juntaex.es).
Ramón Cancho-Candela (Pediatric Neurology Unit of Hospital Universitario Río Hortega, Valladolid, Spain. rcanchoc@saludcastillayleon.es).
Santiago Candela (Pediatric Neurosurgery Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. santiago.candela@sjd.es).
Alejandra Darling (Movement Disorders Unit, Department of Child Neurology, Institut de Recerca Sant Joan de Déu, Barcelona, Spain. alejandra.darling@sjd.es).
Mariela Mercedes de Los Santos (Pediatric Gastroenterology Hepatology and Nutrition Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. marielamercedes.santos@sjd.es).
Angels García-Cazorla (Neurometabolic Unit, Department of Child Neurology, Institut de Recerca Sant Joan de Déu, Barcelona, Spain. angeles.garcia@sjd.es).
Itxaso Martí-Carrera. (Biodonostia Health Research institute, Pediatric Group; Donostia University Hospital, Department of pediatrics; University of the Basque Country UPV/EHU, San Sebastian, Spain. itxasomarti@gmail.com).
Loreto Martorell (Department of Genetic and Molecular Medicine and Pediatric Institute of Rare Diseases. Hospital Sant Joan de Déu, Barcelona, Spain. loreto.martorell@sjd.es).
Rosario Mateos Checa (Department of Pediatric Neurology, Hospital Juan Ramón Jimenez, Huelva, Spain. rmateoscheca@gmail.com).
Silvia Meavilla (Pediatric Gastroenterology Hepatology and Nutrition Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. silviamaria.meavilla@sjd.es).
Beatriz Mínguez Rodríguez (Pediatric Gastroenterology Hepatology and Nutrition Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. beatriz.minguez@sjd.es).
Jorge Pantoja (Department of Pediatrics, University Hospital De la Plana, Vila-Real, Castellón, Spain. pantoja_jor@gva.es).
Leticia Pías (Department of Genetic and Molecular Medicine and Pediatric Institute of Rare Diseases. Hospital Sant Joan de Déu, Barcelona, Spain. leticiadiana.pias@sjd.es).
Fátima Parra Plantagenet-Whyte (Pediatric Palliative Care Unit of Aragon. Miguel Servet University Hospital, Zaragoza, Spain. fparrap@salud.aragon.es).
Sergio Pinillos (Pediatric Gastroenterology Hepatology and Nutrition Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. sergio.pinillos@sjd.es).
Carlos José Ruiz-Hernández (Pediatric Gastroenterology Hepatology and Nutrition Unit, Hospital Sant Joan de Déu, Esplugues de Llobregat, Spain. carlosjose.ruiz@sjd.es).
Jordi Rumià (Department of Neurological Surgery, Hospital Clínic de Barcelona, Barcelona, Spain. jordi.rumia@sjd.es).
Victoria Caballero Pérez (Pediatric Palliative Care Unit of Aragón. Miguel Servet University Hospital, Zaragoza, Spain. vcaballero@salud.aragon.es).
Maria Salvador Cañibano (Unit of Child Neurology, Hospital Universitario de Canarias, San Cristóbal de La Laguna, Spain. msalcan@gobiernodecanarias.org).
Delia Yubero (Department of Genetic and Molecular Medicine and Pediatric Institute of Rare Diseases. Hospital Sant Joan de Déu, Barcelona, Spain. delia.yubero@sjd.es).
Footnotes
Potential Conflicts of interest
Nothing to report.
Data availability
The datasets that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL, Nordli DR, Perucca E, Tomson T, Wiebe S, Zhang YH ZS. ILAE Classification of the Epilepsies Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raga S, Specchio N, Rheims S, Wilmshurst JM. Developmental and epileptic encephalopathies: recognition and approaches to care. Epileptic Disord. 2021;23(1):40–52. [DOI] [PubMed] [Google Scholar]
- 3.Gorodetsky C, Fasano A. Developmental and Epileptic Encephalopathies in Adults: An Evolving Field. Neurology 2022;99(3):89–91. [DOI] [PubMed] [Google Scholar]
- 4.Guerrini R, Conti V, Mantegazza M, Balestrini S, Galanopoulou AS BF. Developmental and epileptic encephalopathies : from genetic heterogeneity to phenotypic continuum. Physiol Rev. 2022;Aug 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura K, Kodera H, Akita T, et al. De novo mutations in GNAO1, encoding a gαo subunit of heterotrimeric g proteins, cause epileptic encephalopathy. Am. J. Hum. Genet. 2013;93(3):496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schirinzi T, Garone G, Travaglini L, et al. Phenomenology and clinical course of movement disorder in GNAO1 variants: Results from an analytical review. Park. Relat. Disord. 2019;61(August 2018):19–25. [DOI] [PubMed] [Google Scholar]
- 7.Axeen E, Bell E, Robichaux Viehoever A, et al. Results of the First GNAO1-Related Neurodevelopmental Disorders Caregiver Survey. Pediatr. Neurol. 2021;121:28–32. [DOI] [PubMed] [Google Scholar]
- 8.Danti FR, Galosi S, Romani M, et al. GNAO1 encephalopathy: Broadening the phenotype and evaluating treatment and outcome. Neurol. Genet. 2017;3(2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelly M, Park M, Mihalek I, et al. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate–binding region. Epilepsia 2019;60(3):406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saitsu H, Fukai R, Ben-Zeev B, et al. Phenotypic spectrum of GNAO1 variants: Epileptic encephalopathy to involuntary movements with severe developmental delay. Eur. J. Hum. Genet. 2016;24(1):129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muntean BS, Masuho I, Dao M, Sutton LP, Zucca S, Iwamoto H, Patil DN, Wang D, Birnbaumer L, Blakely RD, Grill B MK. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep. 2021;34(5):108718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akamine S, Okuzono S, Yamamoto H, et al. GNAO1 organizes the cytoskeletal remodeling and firing of developing neurons. FASEB J. 2020;34(12):16601–16621. [DOI] [PubMed] [Google Scholar]
- 13.Cha HL, Choi JM, Oh HH, et al. Deletion of the α subunit of the heterotrimeric Go protein impairs cerebellar cortical development in mice. Mol. Brain 2019;12(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulkarni NEIL, Tang SHA, Bhardwaj RATAN, et al. Progressive Movement Disorder in Brothers Carrying a GNAO1 Mutation Responsive to Deep Brain Stimulation. J. Child Neurol. 2016;31(2):211–214. [DOI] [PubMed] [Google Scholar]
- 15.Galosi S, Pollini L, Novelli M, Bernardi K, Di Rocco M, Martinelli SLV. Motor, epileptic, and developmental phenotypes in genetic disorders affecting G protein coupled receptors-cAMP signaling. Front Neurol. 2022;13:886751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wirth T, Garone G, Kurian MA, et al. Highlighting the Dystonic Phenotype Related to GNAO1. Mov. Disord. 2022;37(7):1547–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim SY, Shim YK, Ko YJ, et al. Spectrum of movement disorders in GNAO1 encephalopathy: in-depth phenotyping and case-by-case analysis. Orphanet J. Rare Dis. 2020;15(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng H, Khalil S, Neubig RR, Sidiropoulos C. A mechanistic review on GNAO1-associated movement disorder. Neurobiol. Dis. 2018;116(January):131–141. [DOI] [PubMed] [Google Scholar]
- 19.Feng H, Yuan Y, Williams MR, et al. Mice with monoallelic GNAO1 loss exhibit reduced inhibitory synaptic input to cerebellar Purkinje cells. J. Neurophysiol. 2022;127(3):607–622. [DOI] [PubMed] [Google Scholar]
- 20.Feng H, Sjögren B, Karaj B, et al. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 2017;89(8):762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smrcka AV FI. G-protein βγ subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell Mol Life Sci. 2019;76(22):4447–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabrera-Vera TM, Vanhauwe J, Thomas TO, et al. Insights into G Protein Structure, Function, and Regulation. Endocr. Rev. 2003;24(6):765–781. [DOI] [PubMed] [Google Scholar]
- 23.Masuho I, Chavali S, Muntean BS, et al. Molecular Deconvolution Platform to Establish Disease Mechanisms by Surveying GPCR Signaling. Cell Rep. 2018;24(3):557–568.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang D, Dao M, Muntean BS, et al. Genetic modeling of GNAO1 disorder delineates mechanisms of Gαo dysfunction. Hum. Mol. Genet. 2022;31(4):510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Di Rocco M, Galosi S, Lanza E, et al. Caenorhabditis elegans provides an efficient drug screening platform for GNAO1-related disorders and highlights the potential role of caffeine in controlling dyskinesia. Hum. Mol. Genet. 2022;31(6):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Solis GP, Kozhanova TV, Koval A, et al. Pediatric Encephalopathy: Clinical, Biochemical and Cellular Insights into the Role of Gln52 of GNAO1 and GNAI1 for the Dominant Disease. Cells 2021;10(10):2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcé-Grau A, Dalton J, López-Pisón J, García-Jiménez MC, Monge-Galindo L, Cuenca-León E, Giraldo J MA. GNAO1 encephalopathy: Further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J. Rare Dis. 2016;11:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masuho I, Martemyanov KA, Lambert NA. Monitoring G protein activation in cells with BRET. Methods Mol. Biol. 2015;1335:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K MK. Distinct profiles of functional discrimination among G proteins determine the actions of G protein–coupled receptors. Sci Signal. 2015;8(405):ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thurman DJ, Beghi E, Begley CE, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia 2011;52(SUPPL. 7):2–26. [DOI] [PubMed] [Google Scholar]
- 31.Stamberger H, Crosiers D, Balagura G, et al. Natural History Study of STXBP1-Developmental and Epileptic Encephalopathy into Adulthood. Neurology 2022;99(3):E221–E233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brock D, Fidell A, Thomas J, et al. Cerebral Visual Impairment in CDKL5 Deficiency Disorder Correlates With Developmental Achievement. J. Child Neurol. 2021;36(11):974–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balagura G, Xian J, Riva A, et al. Epilepsy Course and Developmental Trajectories in STXBP1 -DEE. Neurol. Genet. 2022;8(3):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicita F, Ulgiati F, Bernardini L, et al. Early Myoclonic Encephalopathy in 9q33-q34 Deletion Encompassing STXBP1 and SPTAN1. Ann. Hum. Genet. 2015;79(3):209–217. [DOI] [PubMed] [Google Scholar]
- 35.Elsaadany L, El-Said M, Ali R, et al. W44X mutation in the WWOX gene causes intractable seizures and developmental delay: A case report. BMC Med. Genet. 2016;17(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pronicka E, Ropacka-Lesiak M, Trubicka J, et al. A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients. J. Inherit. Metab. Dis. 2017;40(6):853–860. [DOI] [PubMed] [Google Scholar]
- 37.de Koning MA, Hoffer MJV, Nibbeling EAR, et al. Prenatal exome sequencing: A useful tool for the fetal neurologist. Clin. Genet. 2022;101(1):65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malerba F, Alberini G, Balagura G, et al. Genotype-phenotype correlations in patients with de novo KCNQ2 pathogenic variants. Neurol. Genet. 2020;6(6):0–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helbig I, Abou Tayoun AN. Understanding genotypes and phenotypes in epileptic encephalopathies. Mol. Syndromol. 2016;7(4):172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Loo KMJ, Carvill GL, Becker AJ, Conboy K, Goldman AM, Kobow K, Lopes-Cendes I, Reid CA, van Vliet EA HD. Epigenetic genes and epilepsy — emerging mechanisms and clinical applications. Nat Rev Neurol. 2022;18(9):530–543. [DOI] [PubMed] [Google Scholar]
- 41.Sun D, Flock T, Deupi X, et al. Probing Gα i1 protein activation at single-amino acid resolution. Nat. Struct. Mol. Biol. 2015;22(9):686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Evaluation of Dystonia and Chorea using Standardized Rating Scales.
Figure S1. Correlations between the Trimer Formation Score and aspects of the Clinical Severity Score A-M. Comparisons between individual aspects of the Trimer Formation score and each aspect of the Clinical Severity subscore. Comparisons include repeated measures for patients with the same variant.
Table S2. Neurodevelopmental Assessment Scores of Participants: Bayley-III, Vineland-II, and GMFM-88.
Table S3. Correlations between GNAO1-related disorders severity score and other standardized scales. Asterisks indicate statistical significance (* = p < 0.05, ** = p < 0.01, *** = p < 0.001, **** = p < 0.0001).
Video 1. The first part of the video showcases P4 during a dyskinetic crises. The crises had an abrupt onset and was clearly distinguishable from the patient’s baseline state. During this dyskinetic episode, ballistic and unpredictable movements of all four limbs were observed, which were non-intentional and non-predictable. The video shows approximately 30 seconds of this dyskinetic crises. There was no disconnection from the environment during the crises. In the second part of the video, P16 is shown experiencing another dyskinetic crises. In this case, the movements were less violent than in P4, but were still clearly distinguishable by the family as an episode with sudden onset and termination. The patient displayed chorea and dystonia of all four limbs and trunk, along with facial dyskinesias. These crises usually last between 1–2 minutes. Sometimes parents report a trigger such as stress, exhaustion, movement, bathing, fever, etc.
Video 2. showcases three patients with different severity levels. P7 has a mild phenotype (2 points), presenting with generalized dystonia and preserved independent gait. P16 displays an intermediate phenotype (6 points) with head control and sitting balance, but without gait. Marked axial hypotonia with action dystonia in the upper and lower extremities is observed. P4 has a severe phenotype (11 points) with marked generalized hypotonia and isolated dystonic movements. P4 carries a deep brain stimulation device (DBS). It is noteworthy that in Video 1, the dyskinetic crises of P16 and P4 can be observed, allowing for a comparison with their baseline state.
Data Availability Statement
The datasets that support the findings of this study are available from the corresponding authors upon reasonable request.