

Abstract
Despite the notoriously poor membrane permeability of peptides in general, many cyclic peptide natural products show high passive membrane permeability and potently inhibit a variety of “undruggable” intracellular targets. A major impediment to designing cyclic peptides with good permeability is the high desolvation energy associated with the peptide backbone amide NH groups. While several strategies have been proposed to mitigate this deleterious effect, only few studies have used polar side chains to sequester backbone NH groups. We investigated the ability of N,N-pyrrolidinyl glutamine (Pye), whose side chain contains a powerful hydrogen bond accepting C=O amide group but no hydrogen bond donors, to sequester exposed backbone NH groups in a series of cyclic hexapeptide diastereomers. Analyses revealed that specific Leu-to-Pye substitutions conferred dramatic improvements in aqueous solubility and permeability in a scaffold- and position-dependent manner. Therefore, this approach offers a complementary tool for improving membrane permeability and solubility in cyclic peptides.
Graphical Abstract
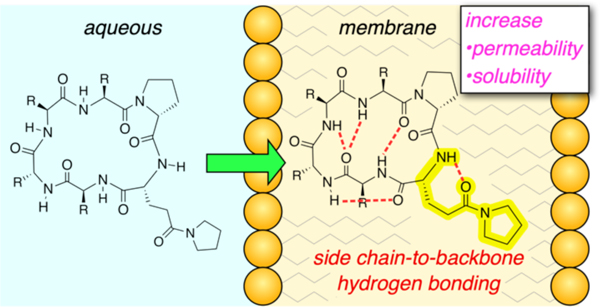
Introduction
Advances in medicine and molecular cell biology continue to generate therapeutic targets whose large, flat binding interfaces make them challenging to drug with traditional, Rule of 5 (Ro5)-compliant small molecules.1–6 Although large biomolecules can often bind with high affinity to such undruggable sites, due to their inability to cross the cell membrane these drugs are generally restricted to parenteral delivery against extracellular targets.7–9 Thus the inverse relationship between molecular weight and cell permeability has focused the search for new chemical matter against undruggable intracellular targets within the chemical space that lies between small molecules and biologics, where there are a growing number of drugs, leads, and model systems that can engage classically undruggable sites yet exhibit the favorable membrane permeability and oral absorption typical of small molecule drugs.10–13 Among the chemotypes that are in the “beyond Rule of 5 (bRo5)” chemical space, cyclic peptides are gaining attention as they can potentially interact with protein targets but still present some of the physicochemical properties of traditional small molecule drugs. The influence of emerging bRo5 scaffolds has already driven novel approved oral drugs toward larger molecular weights over the past two decades.14, 15
Cyclic peptides have several favorable pharmacological characteristics as therapeutic agents and chemical probes. 16–18 Cyclization of peptides reduces conformational flexibility, enhances proteolytic stability and increases binding affinity and selectivity.16–18 The relatively larger surface areas of cyclic peptides, compared to conventional small molecules, allow them to engage large protein surfaces, leading to a high affinity toward targets that lack well-defined binding pockets.18–20 Moreover, cyclic peptides can be diversified enormously by the addition of non-proteinogenic amino acids (e.g., D-amino acids, N-methyl amino acids, peptoids, etc.) and by varying ring size and backbone linkages (e.g., lariats, depsipeptides, inclusion of polyketide fragments).
Lessons learned from the orally bioavailable cyclic peptide cyclosporine A (CsA)9, 21–23, along with numerous other model systems17, 19, 24, have confirmed the importance of overall lipophilic character, as well as the degree to which hydrogen bond donors can be shielded from solvent, in determining membrane permeability. Several methods have been proposed to minimize exposed amide protons, such as (i) stabilization of intramolecular hydrogen bonding through stereochemistry of amino acid residues24–28, (ii) replacement of polar NH residues that cannot participate in intramolecular hydrogen bonding (IMHB) with N-methyl amino acids or peptoids9, 29–33, (iii) α-methylation of amino acid residues to reinforce the IMHB network34 and (iv) steric shielding with nearby hydrophobic side chains.1, 30, 35, 36
While these methods (which have been summarized in a recent review18) successfully enhance membrane permeability, some drawbacks impede their application. N-methylation removes amide protons that can potentially interact with protein targets, and N-methylation at some residues may disrupt IMHB networks leading to a decrease in membrane permeability.30 Furthermore, steric occlusion of HBD requires the introduction of bulky side chains, which can impede target binding and increase overall lipophilicity beyond acceptable limits. Consequently, new strategies for sequestering polar amide protons that are orthogonal to N-methylation and steric occlusion would be useful.
Our interest in the use of side chain hydrogen bond acceptors (HBAs) to sequester exposed amide hydrogen bond donors (HBDs) was motivated in part by the observation that these types of side chain-to-backbone (SC-BB) interactions are present in some crystal structures of natural product cyclic peptides that are known to have intracellular targets and whose structures suggest that they may be passively permeable. For example, argyrin B, 37 which inhibits the prokaryotic and mitochondrial elongation factor G (EF-G), contains an IMHB between the MeO-of its 4’-methoxytryptophan and its backbone amide NH (Figure 1A).38
Figure 1.
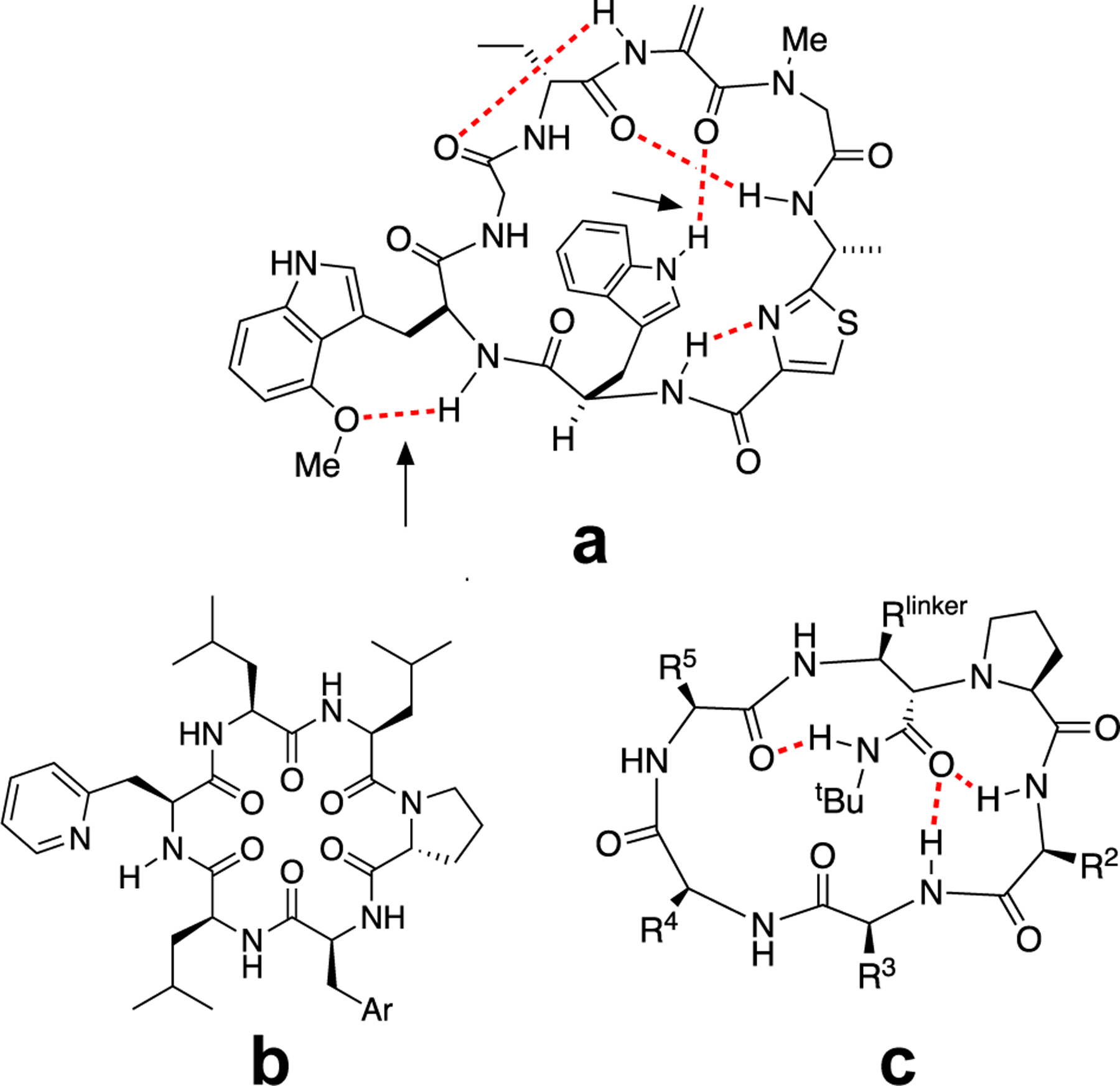
Structures of (a) argyrin B, (b) Simpson’s 2-pyridinylalanine cyclic hexapeptide and (c) Yudin’s 18-membered ring macrocycle with exocyclic amide. Hydrogen bonds are indicated with dashed red lines.
A few synthetic cyclic peptides have been reported that utilize exocyclic HBAs to sequester exposed backbone amides. Thansandote, et al.,34 incorporated a 2-pyridinylalanine side chain into a cyclic hexapeptide, which improved solubility but failed to improve permeability, possibly due to the deleterious effect of the pyridyl side chain on hydrocarbon-water partitioning. Another system reported by Yudin, et al., integrated an exocyclic amide motif into a peptide macrocycle, which formed hydrogen bonds with two backbone NH groups, resulting in an improvement of passive membrane permeability. However, the exocyclic amide introduced an additional hydrogen bond, thus potentially limiting the generality of this approach.39–42 Although other cyclic peptide systems have been described that show side chain HBAs interacting with backbone amide NH groups, the impact of these interactions on the compounds' drug-like properties were not investigated systematically.43–45
Herein, we set out to investigate the interaction between side chain HBAs and backbone amides in a series of cyclic hexapeptide model systems, and, in turn, evaluate their effect on conformation and their ability to improve membrane permeability. By substituting a side chain HBA on each position in three different congeneric series, we found that side chain HBAs can sequester exposed amide protons by forming SC-BB H-bonds, supported by X-ray crystal and NMR solution structures. As a result, we observed an increase in lipophilicity and membrane permeability compared to isomeric scaffolds in which the position and stereochemistry of the polar side chain did not favor SC-BB H-bonding. Moreover, the substitution of a polar, HBA-containing side chain dramatically increased aqueous solubility, thereby offsetting the combined deleterious effect of multiple lipophilic residues.
Results and Discussion
Side chain-to-backbone Hydrogen Bonding of Model Structures
In order to identify side chains that could sequester backbone amide NH groups through IMHB interactions, we performed computational studies on a series of model compounds bearing various side-chain HBAs. We performed conformational searches in an implicit, low-dielectric solvent continuum to mimic the membrane dielectric, and plotted the distance between the side chain HBA and backbone amide NH vs. energy, relative to the lowest-energy conformation (Figure S1). The results showed that N,N-disubstituted Gln residues had low-energy conformers with short HBA-HBD distances (< 2.0 Å; Figure S1a, d, e), while the distances from N,N-disubstituted Asn were slightly longer (Figure S1f, h). Similar HBA-HBD distances were observed when replacing an amide functional group with an ester (Figure S1c).
The encouraging in silico data prompted us to investigate the degree of SC-BB H-bonding in the set of synthetic model structures 4 shown in Table 1. For commercially available amino acids (4-Ala, 4-Val, 4-Leu, 4-Ile, 4-Gln, 4-Glu(OMe)), Boc-protected amino acids were esterified with 4-phenyl-1-butanol, deprotected, and acetylated to obtain compounds 4 (Scheme S1a). Substituted amides (4-Gln(NMe2), 4-Gln(Pyr), 4-Asn(Pyr), 4-Asn(NMe2)) were synthesized from corresponding N- and C-protected Glu or Asp amino acids (Boc-Glu-OtBu and Boc-Asp-OtBu; Scheme S1b). After amide coupling with pyrrolidine (Pyr) or N,N-dimethylamine, the intermediates were treated in TFA to deprotect all acid-labile protecting groups, the Boc-protecting group was reintroduced, and the compounds were carried forward as described above.
Table 1.
Chemical Shifts and Amide Temperature Coefficients of Model Structures in CDCl3a.
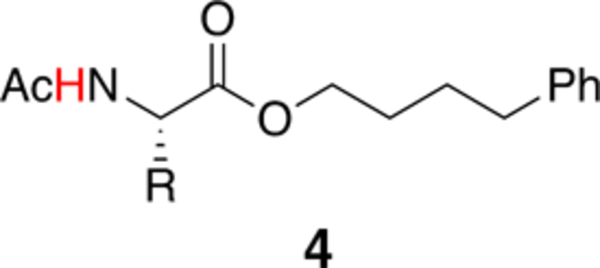
| |||
|
| |||
|---|---|---|---|
| ID | R | δNH (300 K) | ΔδNH /ΔT (ppb/K) |
|
| |||
| 4-Ala | Me | 6.08 | −3.54 |
| 4-Val |
|
5.97 | −3.31 |
| 4-Leu |
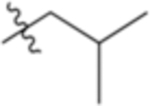
|
5.86 | −3.02 |
| 4-Ile |
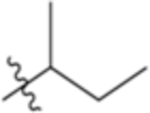
|
5.96 | −2.95 |
| 4-Gln |
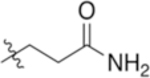
|
6.59 | −5.37 |
| 4-Glu(OMe) |
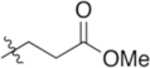
|
6.18 | −3.51 |
| 4-Gln(NMe2) |
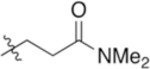
|
6.88 | −6.48 |
| 4-Gln(Pyr) (Pye) |
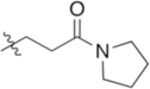
|
7.11 | −7.84 |
| 4-Asn(Pyr) |
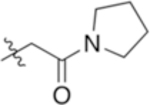
|
6.81 | −1.66 |
| 4-Asn(NMe2) |
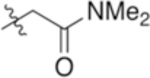
|
6.89 | −1.52 |
|
| |||
All experiments were performed at a concentration of 50 mM or below.
NMR chemical shifts (δ) and temperature coefficients (ΔδNH /ΔT) are widely used to gain structural information in peptides and proteins.26, 46, 47 Temperature coefficients of backbone amide protons have been used to report on their degree of solvent exposure; for well-structured proteins in aqueous solution, amide NH protons with temperature shifts above (more positive than) −4.0 ppb/K are considered to be involved in IMHB, whereas more negative values indicate greater solvent exposure.46 This relationship has been attributed to the strong (1/r3) dependence of NH chemical shifts on the NH–O distance, where the NH becomes more deshielded and shifts downfield as the H-bond becomes shorter.48
In proteins, solvent-exposed amides have relatively large thermal shifts due to the weaker H-bond to water compared to the NH–to–carbonyl H-bond. However, in nonpolar solvents49, 50, fully exposed NH groups can have small temperature shifts because they interact with neither solvent nor acceptor atoms within the molecule. For NH groups that are involved in IMHB in nonpolar solvents, temperature shifts can either be large, if the IMHB interaction has a strong negative entropic component, e.g., with multiple rotatable bonds between the donor and acceptor, or small, if the enthalpic gain outweighs the entropic penalty of H-bond formation and the interaction remains stable with increasing temperature. Therefore, interpreting thermal shift data is challenging, especially in small conformationally dynamic peptides, where additional information such as NH chemical shifts derived from model systems can be useful.51
To avoid artifacts due to intermolecular H-bonding, we determined the NH chemical shifts of 4-Ala and 4-Gln(Pyr) as a function of concentration (Figure S4). At concentrations below 50 mM the NH chemical shift remained nearly constant, indicating minimal intermolecular H-bonding. The model compounds with aliphatic side chains (4-Ala, 4-Val, 4-Leu, 4-Ile; Table 1 and Figure S3) showed chemical shifts of ~6.0 ppm and temperature shift coefficients between −2.95 and −3.54 ppb/K. These values are consistent with observations from Gellman, et al., who saw comparable chemical shifts and temperature coefficients in CD2Cl2 for similar model systems lacking in IMHB.50 For the substituted Gln amides (4-Gln(NMe2), 4-Gln(Pyr)), the amide proton chemical shifts appeared approximately 1 ppm downfield of the NH protons in the aliphatic controls and showed significantly larger negative ΔδNH /ΔT values (−6.48 and −7.84 ppb/K). Both the chemical shifts and temperature coefficients of ester 4-Glu(OMe) were similar to the aliphatic controls, consistent with the weaker hydrogen bond-accepting ability of esters compared to amides.50 Taken together, these results are consistent with previous studies finding comparably upfield chemical shifts in nonpolar media for weakly hydrogen-bonding NH groups, and downfield chemical shifts for NH groups that are significantly hydrogen-bonded. Interestingly, while the chemical shifts of substituted Asn amides (4-Asn(Pyr), 4-Asn(NMe2)) were similarly downfield (6.81 and 6.89 ppm) to the Gln compounds, their ΔδNH/ΔT values (−1.66 and −1.52 ppb/K) were much less negative than either the Gln derivatives or the aliphatic controls. The larger temperature coefficients of the Gln compared to Asn residues are consistent with the larger entropic penalty for H-bond formation of the more flexible Gln side chain. Combined with the computational studies, these NMR data prompted us to investigate the 4-Gln(Pyr) residue, which we abbreviate as “Pye” (pyrrolidine amide of glutamic acid ‘E’), as the most promising candidate for eliciting permeability-enhancing SC-BB interactions in cyclic peptides.
NMR Studies of Cyclic Hexapeptides
Both L and D-Pye were synthesized on a multigram scale (Scheme S2) first using EDC and HOBT to couple pyrrolidine to the side chain of Fmoc-Glu-OtBu, followed by removal of the tert-butyl ester with TFA and diethyl ether precipitation. Previously we identified a series of diastereomeric cyclic hexapeptide scaffolds whose membrane permeabilities52 varied significantly depending on their ability to sequester exposed amide NH groups in IMHB.53 In order to determine the impact of Pye on passive permeability, we selected three of the least permeable diastereomers, 1, 2 and 3, which differ in stereochemistry at positions Leu3 and Leu5 (we replaced the Tyr6 residue from the previous study with Leu to avoid the deleterious effect of the phenolic OH on permeability). Herein, compounds are named by the type of polar residue (Asn, Pye, etc) preceded by the scaffold number and followed by a superscript indicating the position of the substitution (e.g., 1-Pye2 refers to scaffold 1 in which Leu2 is replaced with Pye). Substituting each Leu with Pye in the three scaffolds yielded a total of 18 compounds, including the three parental compounds 1, 2 and 3.
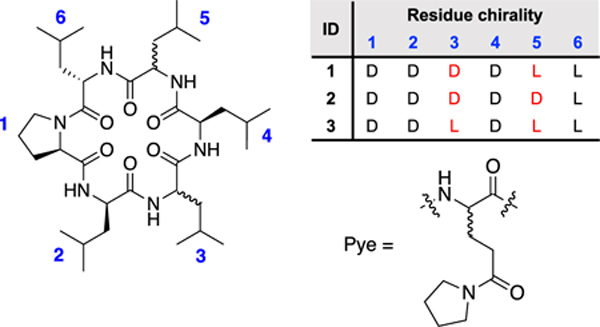
For the series based on 1 and 2, we were unable to assign all of the resonances due to line broadening and/or peak overlap. However, for the series based on diastereomer 3, the peaks were sharp and dispersed enough to allow complete assignment of the parental compound as well as all of the Pye positional variants (Figure 2). For 3-Pye2 and 3-Pye5 (Figure 2), the amide proton of the Pye residue fell significantly downfield of the other NH resonances, even as far as 10 ppm for 3-Pye2. Based on the chemical shifts and temperature coefficients of the model peptides (Table 1), we hypothesized that the Pye side chain in these compounds form IMHB to their own backbone amide NH groups. For many of the other compounds, including 3-Pye3, 3-Pye4, and 3-Pye6 (Figure 2), (as well as 1-Pye4, 1-Pye5, and 1-Pye6) (Figure S5b), the chemical shift of the Pye NH was in the same range as that of the other amide NH resonances, between 6 and 8.5 ppm.
Figure 2.
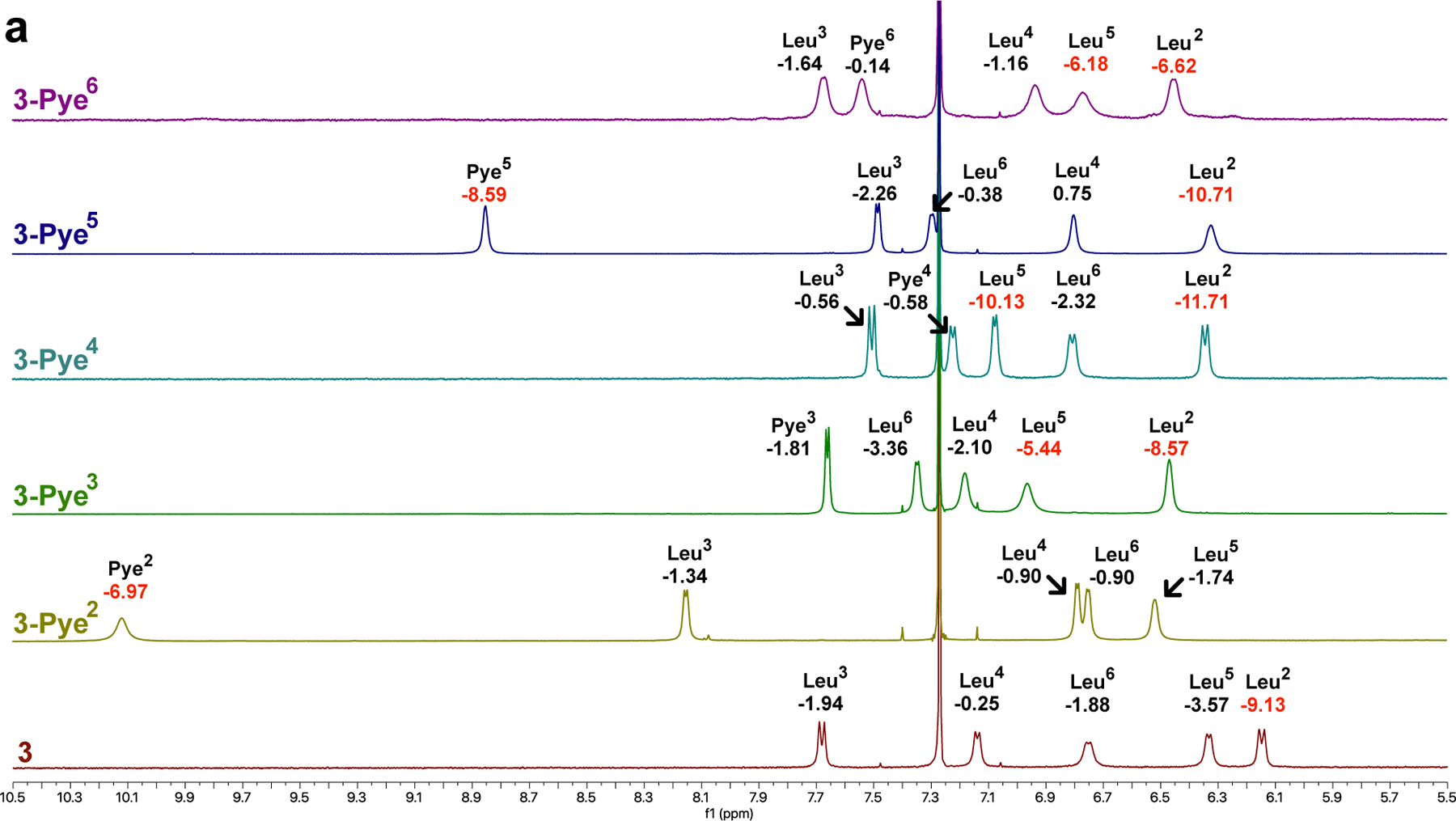
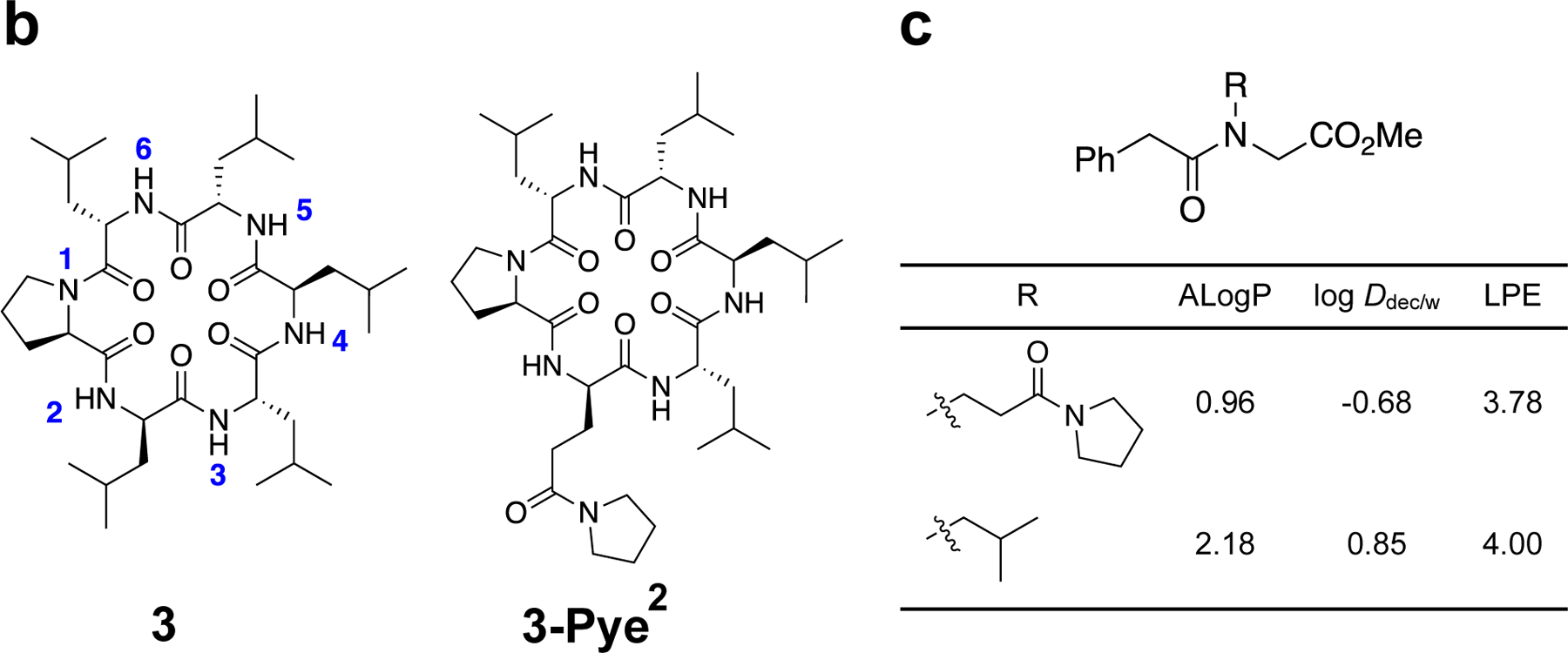
(a) 1H NMR of amide protons of 3 series at a concentration of ~10 mM. Values under labels indicate the ΔδNH /ΔT in ppb/K. Amide temperature coefficients less than −4.0 are highlighted in red. (b) Structure of 3 and 3-Pye2. (c) The intrinsic effect of Pye and isobutyl side chains on in the absence of HBD.
The temperature coefficients of 1, 2 and 3, showed at least one amide proton exposed to solvent (Figure 2 and S4b, d); for the fully assigned compound 3, the NH with the high temperature coefficient corresponded to Leu2 (-9.13 ppb/K). For all other 3-Pye positional variants (excluding 3-Pye2, in which Leu2 is replaced with Pye), the Leu2 NH was the most shielded and also had the largest temperature coefficient compared to the other NH peaks. For 3-Pye3, 3-Pye4, and 3-Pye6, Leu5 also showed a relatively high temperature coefficient. The high variability in both chemical shifts and temperature coefficients indicates that the position of the Pye residue in the scaffold has a significant effect on its ability to participate in IMHB and demonstrates that substitution of a single aliphatic residue with an HBA-containing residue can have a strong position-dependent effect on the scaffold’s low-dielectric conformation.
Lipophilicity, Passive Permeability and Solubility of Pye-scanning Cyclic Hexapeptides
Previously we introduced a metric called lipophilic permeability efficiency () for assessing the efficiency with which a compound achieves membrane permeability at a given lipophilicity.53 is defined as log - 1.06() + 5.47, where log is the experimental partition coefficient between 1,9-decadiene and PBS buffer at pH 7.4, and 54 is a calculated, atomistic 2D octanol/water partition coefficient (a slight variation on the more commonly used cLogP). Hydrocarbons provide a good model of the membrane interior, and thus partitioning between water and 1,9-decadiene reflects the energic penalty of desolvating exposed polar functionality, in particular HBDs, as they cross the barrier region of the phospholipid bilayer. Therefore, for compounds that are water-soluble, log correlates positively with passive permeability. On the other hand, since octanol is capable of forming hydrogen bonds with solute, does not penalize exposed HBD but rather reflects a compound’s minimum lipophilic character in the aqueous environment. Therefore, for highly lipophilic compounds (with above ~4), shows a good negative correlation with solubility as well as a negative correlation with permeability in the insoluble regime.55–57 By subtracting from log , thus provides a measure of a scaffold’s ability to sequester polar functionality (in particular HBD) and therefore its potential to achieve high permeability at a given -defined lipophilicity.
To determine the of the Pye residue (relative to an aliphatic control) in the absence of IMHB, we synthesized a simple model compound in which the Pye functional group was introduced as a peptoid side chain (Figure 2c). The Pye substitution decreased by 0.22 units relative to the isobutyl control.53 Next, and thermodynamic aqueous solubilities were determined for parent compounds 1-3 and their Pye substitutions (Table 2). Replacing Leu with Pye decreases by 1.28 units. The same substitution also decreased log , by a variable amount depending on both the scaffold and the position of the Pye residue within the scaffold. For most compounds, substitution of Leu for Pye caused a significant decrease in (by up to 1 unit); however, for 1-Pye2 and 3-Pye2, increased upon Pye substitution. This observation is consistent with the upfield chemical shift and large temperature coefficient of Leu2 in parent compound 3, indicating solvent exposure of the Leu2 NH. Although the NH resonances in parent compound 1 could not be assigned due to peak overlap, there is a single upfield amide peak with a large temperature coefficient that, based on the other similarities to scaffold 1, likely corresponds to Leu2. The far downfield chemical shifts of the Pye residue in 1-Pye2 and 3-Pye2 suggest that the Pye side chain’s C=O is able to sequester these exposed NH groups via an SC-BB IMHB. The temperature shift of the Pye2 amide in 1-Pye2, however, is small (-1.1 ppb/K), while that of 3-Pye2 is much larger (-7.0 ppb/K). Since the Pye2 substitution’s effect on is greater for scaffold 1 than scaffold 3 (suggesting that the corresponding SC-BB IMHB is stronger for 1-Pye2), perhaps the SC-BB interaction in 1-Pye2 is enthalpically favorable enough to offset the entropic penalty of H-bond formation, thus lowering the temperature coefficient for this amide.
Table 2.
Pye-scanning Cyclic Hexapeptides Experimental Data
| ID | log | a | P app b | solubilityc |
|---|---|---|---|---|
| 1 | 2.0 | 3.5 | 8.1 | 16 |
| 1-Pye 2 | 1.3 | 4.1 | 1.7 | 640 |
| 1-Pye 3 | −0.41 | 2.5 | 0.66 | 550 |
| 1-Pye 4 | 0.02 | 2.9 | 0.30 | 580 |
| 1-Pye 5 | −0.42 | 2.5 | 0.32 | 510 |
| 1-Pye 6 | −0.30 | 2.6 | 0.66 | 720 |
| 2 | 2.0 | 3.5 | 5.3 | 240 |
| 2-Pye 2 | −0.29 | 2.6 | 0.56 | 610 |
| 2-Pye 3 | −0.29 | 2.6 | 1.1 | 450 |
| 2-Pye 4 | 0.040 | 2.9 | 1.3 | 460 |
| 2-Pye 5 | −0.82 | 2.0 | 0.13 | 550 |
| 2-Pye 6 | −0.29 | 2.6 | 1.5 | 560 |
| 3 | 2.0 | 3.6 | 8.0 | 63 |
| 3-Pye 2 | 1.1 | 3.9 | 1.5 | 730 |
| 3-Pye 3 | −0.22 | 2.7 | 0.23 | 140 |
| 3-Pye 4 | 0.21 | 3.1 | 0.14 | 580 |
| 3-Pye 5 | 0.14 | 3.0 | 0.28 | 550 |
| 3-Pye 6 | 0.16 | 3.0 | 0.18 | 600 |
= log – 1.06 x + 5.4753; = 3.74 for 1, 2 and 3; 2.46 for Pye-containing cyclic hexapeptides.
PAMPA x 10−6 cm/s, N=4.
μM, pH 7.4, N=3.
The parallel artificial membrane permeability assay (PAMPA)58 is a cell-free permeation tool for screening passive permeabilities (Papp). PAMPA measures the rate of diffusion of test compounds from donor to acceptor compartments across a solution of 1% lecithin in n-dodecane trapped in a polyvinyldifluoroethane membrane. The positive correlation between permeation and lipophilicity generally holds for compounds in the soluble regime, generally below ~ 4. At higher lipophilicities, effective permeability decreases due to factors such as membrane sequestration and poor aqueous solubility, a trend that has been observed in both cyclic peptides and small molecule drugs.27, 59, 60 Thus, a plot between log Papp and produces a downward-facing parabola with a vertex near ~ 4.25
Compounds 1, 2 and 3 showed excellent PAMPA permeabilities but had modest solubilities, consistent with their relatively high lipophilicity ( = 3.74, Table 2). Most of the Pye-substituted compounds had Papp values below 1 x 10−6 cm/s except 1-Pye2, 2-Pye3, 2-Pye4, 2-Pye6 and 3-Pye2 (Figure 3a). Permeabilities and of 1-Pye2 and 3-Pye2 were clearly higher than the other Pye-substitutions. In addition, selected compounds in the 3 series were measured for their rate of flux across the human colon carcinoma cell (Caco-2) line, and their permeability rates were similar to those observed in PAMPA (Figure 3b).
Figure 3.
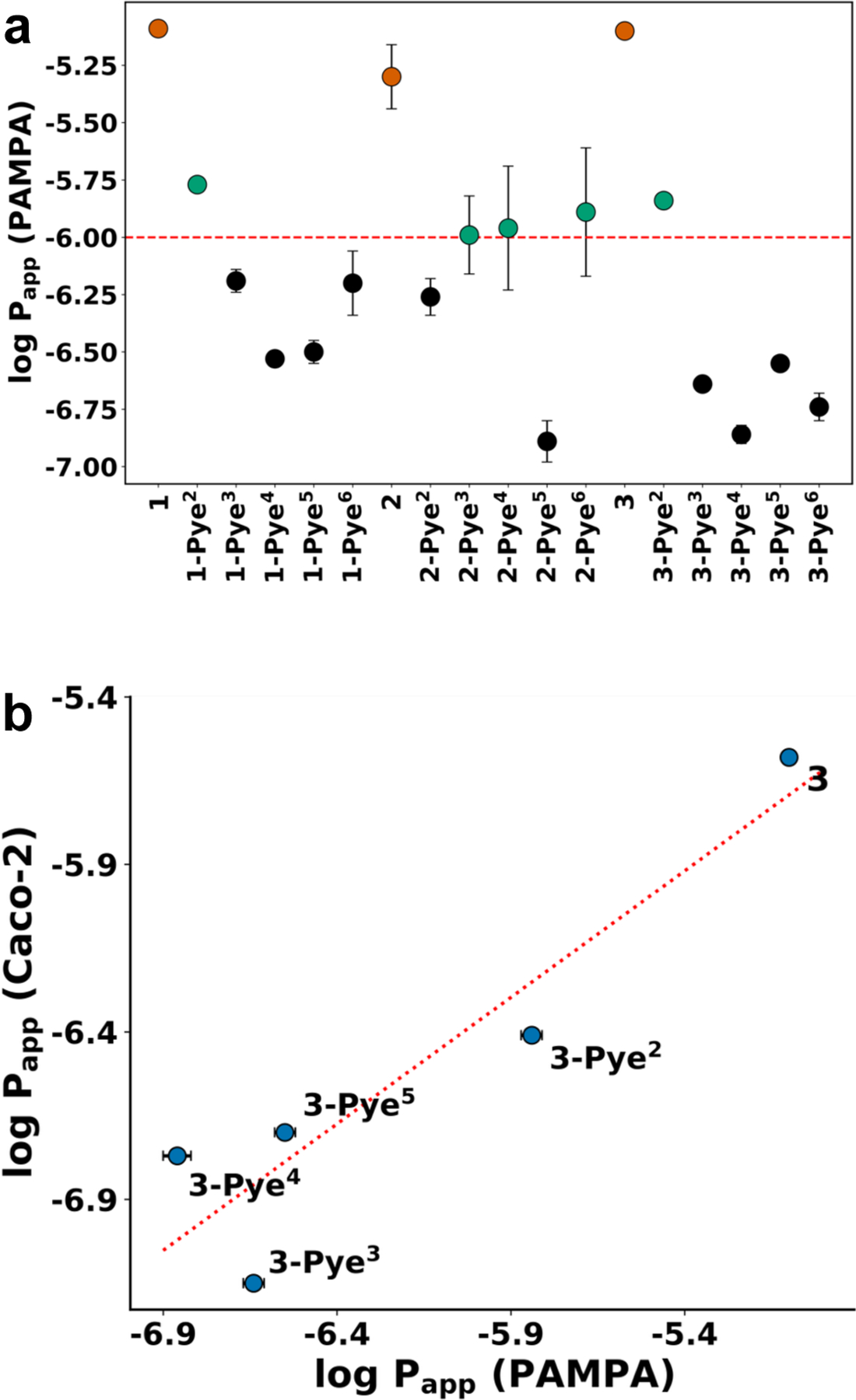
(a) PAMPA permeabilities of cyclic hexapeptides 1, 2, 3 and their derivatives. Minimum acceptable oral permeability is shown in dashed line. (b) PAMPA and Caco-2 experiments of compounds 3 and their derivatives show similar permeability rate (R2 = 0.86). High and low permeability standards are measured along with test compounds in Caco-2 experiment to ensure the accuracy; N=4 for PAMPA experiment; N=1 for Caco-2 experiment.
Besides their ability to sequester backbone NH groups, another advantage of polar side chains is their potential for increasing a compound’s aqueous solubility. Indeed, in all cases Pye substitution led to a significant improvement in solubility over the parental cyclic hexapeptides (Table 2). Interestingly, the degree to which introduction of the Pye residue improved permeability was also dependent on scaffold and position. For example, when Leu6 was replaced with Pye on scaffold 1, solubility improved 45-fold, whereas replacing Leu3 for Pye on scaffold 2 improved solubility only two-fold. Interestingly, the degree to which Pye substitution improved aqueous solubility did not correlate with the effect on . For example, the two compounds that showed an increase in upon Pye substitution (1-Pye2 and 3-Pye2) also saw substantial improvements in solubility, consistent with a degree to which structural/conformational factors that contribute to solubility and permeability can be considered independently.
Membrane Permeability of 3-Pye2 and 3-Pye3 Libraries
To investigate the effect of Pye on lipophilicity and membrane permeability in the context of various amino acid combinations, we synthesized 3-Pye2 and 3-Pye3 libraries using the split-pool approach in which two residues were mutated to different hydrophobic side chains (Figure 4a). We chose these two scaffolds as they had the largest log difference between Pye positional variants on the same scaffold, and the 1H NMR spectra in the scaffold 3 series were interpretable. We selected side chain combinations that would cover a broad range, in this case from 0.5 to 4.7. The experimental hydrocarbon/water coefficients shown in Figure 4b agreed with Pye-scanning data in Table 2, showing that the log values for the compounds based on the 3-Pye2 positional variant were greater than those based on 3-Pye3. Matched pairs bearing the same combination of side chains from the 3-Pye2 and 3-Pye3 libraries had log values that differed on average by one log unit, highlighting the importance of the Pye residue’s position in determining its effect on physico-chemical properties.
Figure 4.
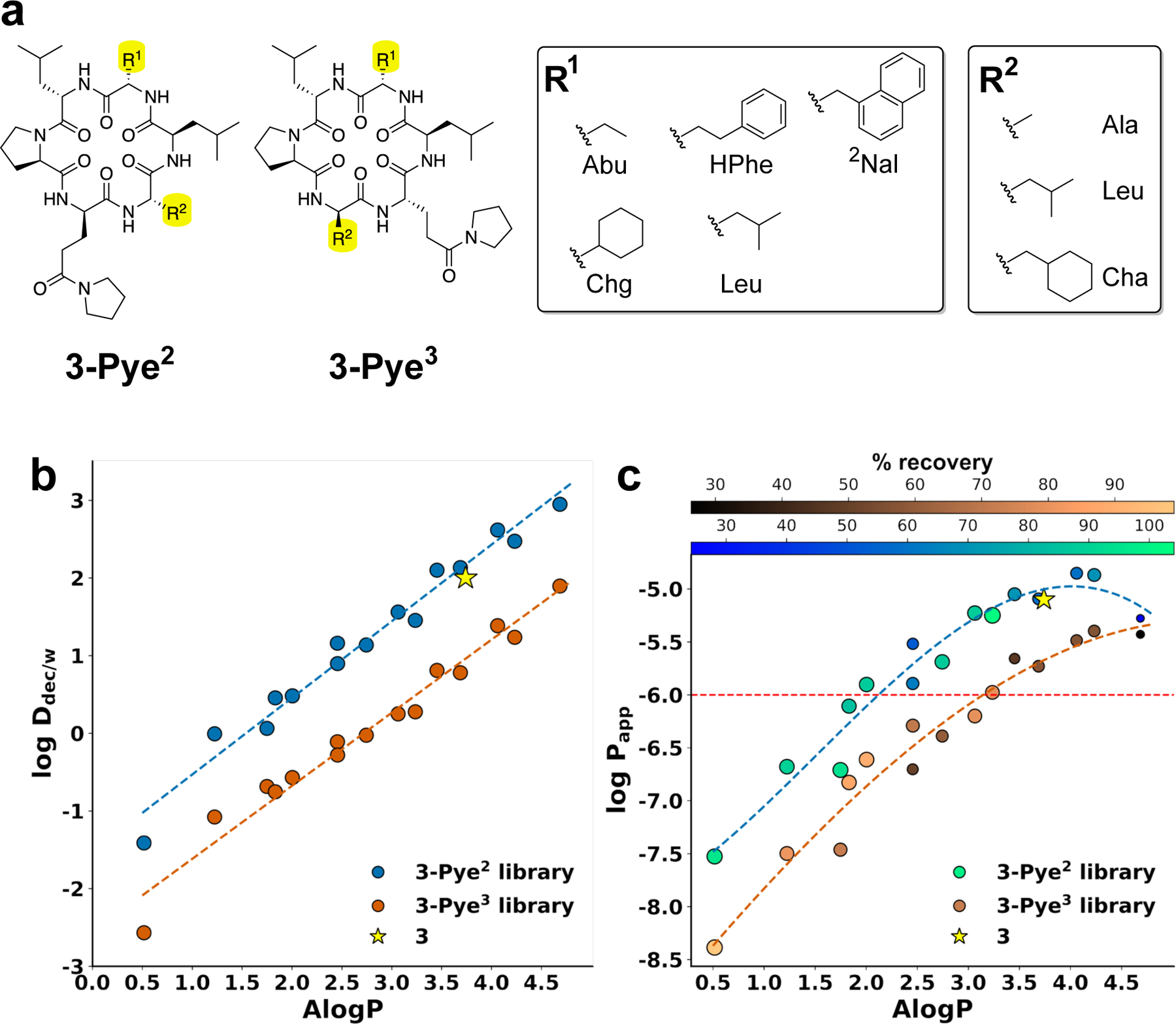
(a) Structures of 3-Pye2 and 3-Pye3 libraries and side chain composition. (b) Log vs of 3-Pye2 and 3-Pye3 libraries. (c) Log Papp vs of 3-Pye2 and 3-Pye3 libraries. Red-dashed line indicates the minimum acceptable oral absorption. Compound 3 is shown as a yellow star.
The plot of PAMPA permeability (log Papp) versus for the 3-Pye2 and 3-Pye3 libraries revealed a similar trend, in which the 3-Pye2 derivatives were generally more permeable than the 3-Pye3 derivatives of the same (i.e., comparing pairs of compounds bearing the same side chain combinations; Figure 4c). At values below 4, the 3-Pye2 library was 0.5–1.0 log unit higher in permeability than the 3-Pye3 library. Above = 4, PAMPA permeabilities for both libraries showed the characteristic downward trend as aqueous solubilities fall below the threshold required to sustain permeability (Table S2–3). The two naphthyl-Ala derivatives 3-Pye2(Nal5) and 3-Pye3(Nal5) have nearly the same as the parent compound 3, with the increased lipophilicity introduced by the Nal residue offsetting the decrease in lipophilicity upon substitution of Leu2 or Leu3 with Pye. But whereas the permeability of 3-Pye2(Nal5) was comparable to that of 3, the Papp of 3-Pye3(Nal5) was 0.6 log units lower than the Papp of 3. In addition, two of the 3-Pye2 derivatives (Cha3Chg5 and Cha3HPhe5; = 4.1 and 4.2, respectively) achieved permeabilities greater than that of the all-Leu parent scaffold, whereas none of the 3-Pye3-based derivatives matched the permeability of the parent scaffold. Because of the lower of the 3-Pye3 scaffold, its derivatives require a higher to achieve the same of permeability as scaffolds of higher (e.g.,3-Pye2); in this case, the lipophilicity required for the 3-Pye3 -based compounds to achieve the permeability of 3 puts them on the descending (low solubility) part of the Papp vs. curve (Figure 4c and Table S2 and S3).
Overall, the results from scaffold 3 suggest that substitution with Pye can enable SC-BB interactions which preserve the of the parent scaffold, thus allowing for incorporation of larger, more lipophilic side chains without compromising water solubility. For example, 3-Pye2(Cha3Chg5) ( = 4.1, Papp = 14 x 10−6 cm/s, solubility = 74 μM; Table S2) has better solubility and permeability than 3 ( = 3.7, Papp = 8.0 x 10−6 cm/s, solubility = 63 μM; Table 2).
Structural Elucidation of 3-Pye2 derivatives
The exceptional permeability of the 3-Pye2 library encouraged us to explore the structural basis of the Pye residue’s effect on permeability. We obtained structures by single crystal X-ray diffraction of two side chain variants on 3-Pye2, 3-Pye2(Ala3Nal5) and 3-Pye2(HPhe5), crystallized by vapor-diffusion in THF-pentane (Figure 5). These two crystals showed virtually identical backbone conformations (RMSD = 0.07 Å) and had nearly identical Cα-Cβ vectors (RMSD = 0.09 Å). As expected from the NMR studies, the Pye2 side chain C=O showed a strong hydrogen bond to its own backbone amide NH (Figure 5a–b, black arrow). The distance and N–H--O angle were 1.9 Å and 155° for both crystals, respectively, which is an optimal hydrogen bond geometry.50 Three other backbone amides participated in IMHB as β-turn motifs (i.e., 3rd NH to 6th CO and 6th NH to 3rd CO; Figure 5a–b). The NH groups of residues 4 and 5 formed intermolecular hydrogen bonds with other molecules in the crystal lattice (Figure S13).
Figure 5.
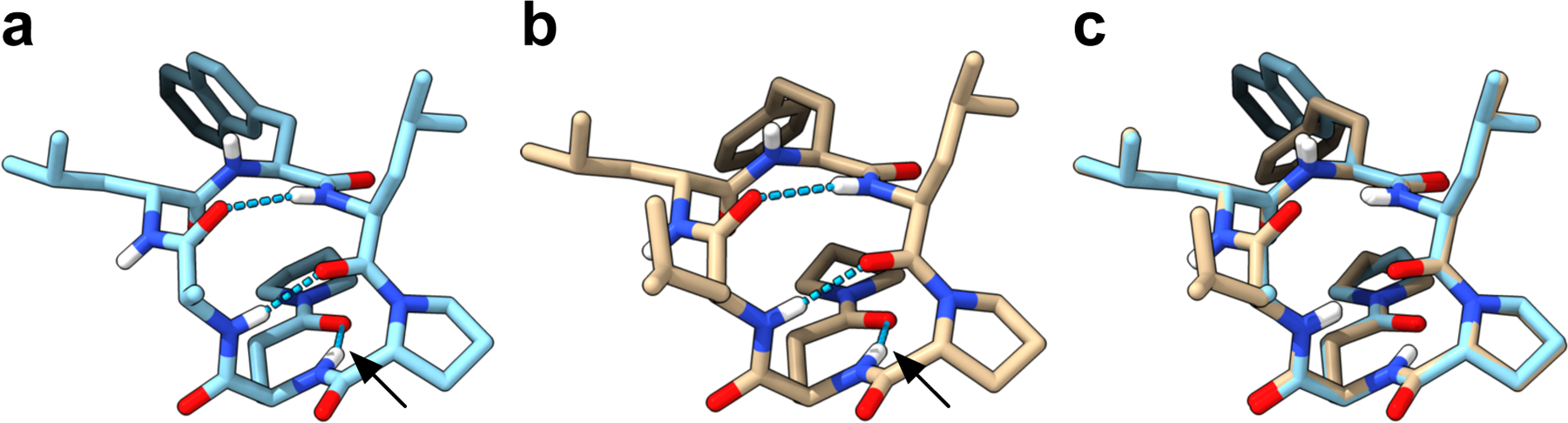
X-ray crystal structures of (a) 3-Pye2(Ala3Nal5) and (b) 3-Pye2(HPhe5). Hydrogen bond between Pye and 2nd amide is indicated with black arrow. Putative hydrogen bonds are indicated with dashed blue lines. (c) Overlay between 3-Pye2(Ala3Nal5) and 3-Pye2(HPhe5).
These crystal contacts suggested that these amide NH groups might be exposed in low-dielectric solvent; however, this hypothesis was inconsistent with the small ΔδNH /ΔT NMR temperature shifts of these NH groups (Figure S6) as well as the high and PAMPA permeabilities of these compounds (Table S2). We hypothesized that crystal packing forces may impact their conformation, prompting us to investigate the solution structure of 3-Pye2(Ala3Nal5) in CDCl3. By collecting NOESY spectra at various mixing times and performing a careful analysis of each crosspeak volume, we obtained a total of 20 crosspeaks that complied with the initial rate approximation.61–63 The distance between the δ-proline geminal protons (1.78 Å) was used as a reference to calculate other interproton distances (Table S5). After applying 10% random noise to the calculated distances, 17 out of the 19 distances matched the crystal structure, and the two remaining distances were close to the predicted distance which suggested the overall structural similarity between the crystallographic and solution structures.
We did not detect a CαH-to-CαH NOESY cross peak between Pro1 and Leu6, indicating that the Pro1 amide is most likely in the trans conformation. To obtain the solution NMR ensembles from the NOESY-derived distances, we performed an unrestrained conformational search using multicanonical molecular dynamics (McMD) simulations.64 The 20 structures from the ensemble obtained by McMD at T = 300 K with the fewest violations from the NOESY-derived distances closely resembled the crystal structure (RMSD = 0.27 – 0.64 Å; Figure 6a–c) and were in a cluster that made up 9% of the unrestrained ensemble, although NOE violations averaged over the entire unrestrained McMD ensemble were also relatively low, indicating that the McMD simulation accurately captured the solution conformation in CDCl3 (Table S5). The side chains were flexible except for the Pye2 side chain, which uniformly converged to form the expected SC-BB hydrogen bond.
Figure 6.
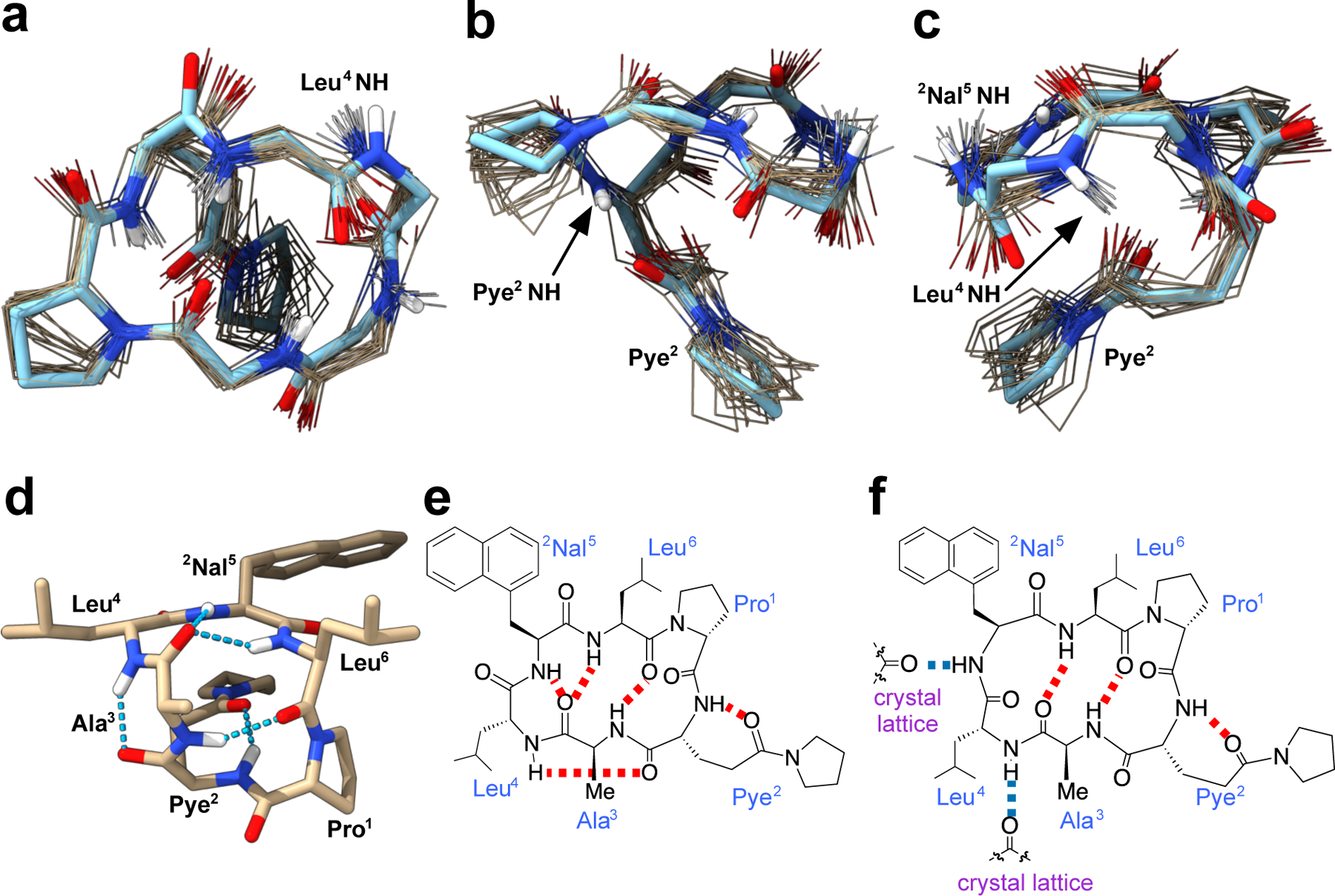
(a-c) Overlays of 3-Pye2(Ala3Nal5) crystal structure (blue) and 20 simulated McMD structures (gold) derived from NOESY information. Only the backbone and Pye side chain are shown for clarity. (d) Structure of simulated 3-Pye2(Ala3Nal5) having 5 IMHBs. (e) IMHB pattern of (d). (f) IMHB pattern of 3-Pye2(Ala3Nal5) crystallographic structure.
The backbone NH groups of Ala3 and Leu6 formed the same transannular hydrogen bonding pattern as found in the crystal structure (Figure S14). The amides of Leu4 and Nal5, however, which formed crystal contacts in the X-ray structure, rotated inward in the solution ensemble to orientate toward the center of the macrocycle (Figure 6c). Specifically, the Leu4 NH reoriented to form a hydrogen bond with the C=O of Pye2, and the Nal5 NH reoriented to form a hydrogen bond with the C=O of Ala3 (Figure 6d–e), interactions that were absent in the crystal structure (Figure 6f). These reorientations around the 4th and 5th amide protons between the solid state and solution conformations amounted to a relatively subtle shift in overall backbone geometry, which is perhaps surprising given the net change in hydrogen bonding between the two structures. In solution all of the amide NH groups of 3-Pye2(Ala3Nal5) are involved in IMHB, consistent with the high and PAMPA permeability of 3-Pye2 and its derivatives. The large discrepancy in overall 3D polar surface area between the solid state and solution conformations despite similar backbone geometries suggests that dramatic shifts in 3D polar surface area can be modulated by relatively subtle shifts in amide rotamers.
Effect of Different HBAs on Lipophilicity and Permeability
Data from various experiments above confirmed the lipophilicity and permeability improvements are due to the hydrogen bond from side chain HBA to the exposed HBD. To further expand the scope of HBAs that can be employed, we replaced Pye2 with four other HBAs, including N,N-dimethylamine substituent {3-Gln(NMe2)2}, methyl ester functional group {3-Glu(OMe)2} and two short side chain amides {3-Asn(Pyr)2 and 3-Asn(NMe2)2} (Figure 7a). Amide temperature coefficient experiments (Figure 7b) showed that changing substituent from pyrrolidine to N,N-dimethylamine {3-Pye2 to 3-Gln(NMe2)2} did not significantly change the chemical shift or the temperature coefficient of its backbone NH. In contrast, replacing the amide side chain with an ester {3-Pye2 to 3-Glu(OMe)2} caused a significant upfield shift and much more negative ΔδNH /ΔT (-12.8 ppb/K), consistent with the decreased electron density at the ester carbonyl and the enthalpically weaker nature of the NH-to-ester hydrogen bond.
Figure 7.
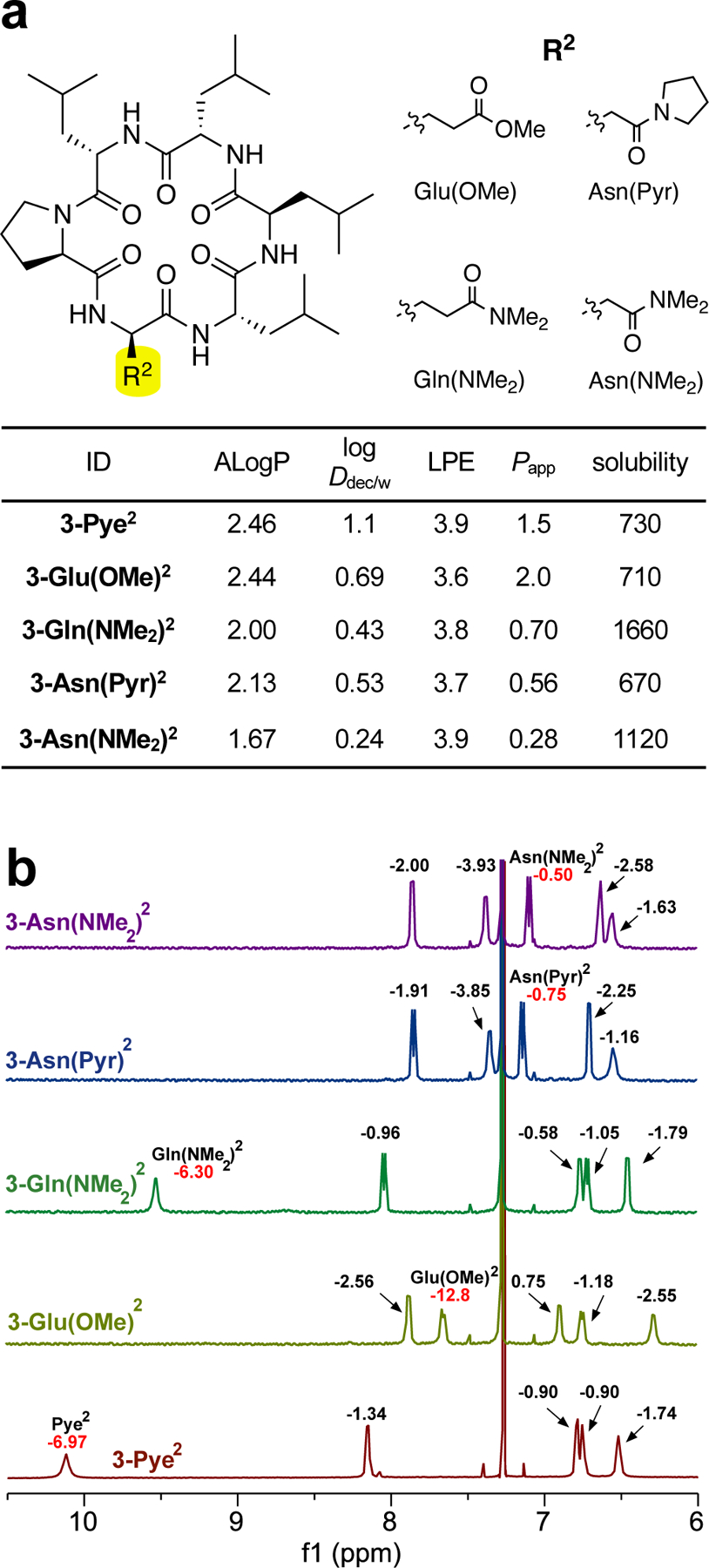
(a) Compound 3 derivatives bearing different HBAs on 2nd position and their experimental data. Papp x 10−6 cm/s, solubility in μM. (b) Amide-proton NMR spectra of compounds shown in (a). Amide NHs corresponding to the side-chain HBA residues are labeled, and the values indicate the ΔδNH /ΔT in ppb/K.
Shortening the Pye side chain by one methylene group {3-Pye2 to 3-Asn(Pyr)2 or 3-Asn(NMe2)2} shifted the backbone NH upfield and resulted in a much smaller (less negative) ΔδNH /ΔT. These shifts relative to the longer-chain Gln derivatives is consistent with the pattern observed in the simple model compounds, with the decreased temperature shift resulting from the lower entropic penalty associated with the shorter Asn chain and the upfield shift resulting from the differences in the H-bond geometry between the two sets. However, McMD simulations of 3-Asn(Pyr)2 in CHCl3 offer another explanation: The shorter Asn side chain cannot reach its own backbone amide, although its close proximity to the NH groups of both residues 4 and 5 result in a bidentate H-bond with these two HBDs. Although this results in a completely new SC-BB H-bonding pattern, the overall backbone geometry is similar to that of 3-Pye2 (Figure S12). The McMD ensemble of 3-Asn(Pyr)2 shows no hydrogen bond to the amide NH of residue 2, consistent with its upfield shift and small temperature shift compared to the Gln derivatives (Figure S11). Testing these hypotheses will require further structural analysis.
Lipophilicity and permeability of all four chemotypes were similar to the 3-Pye2 scaffold (Figure 7a). of amide HBAs were within 0.2 log unit difference, while the of the ester was slightly lower. Moreover, all compounds had PAMPA permeabilities within the same trend of 3-Pye2 library (Figure S2). Notably, N,N-dimethylamine substituent improved cyclic hexapeptide’s solubility as expected from higher intrinsic hydrophilicity than pyrrolidine {3-Pye2 vs 3-Gln(NMe2)2 and 3-Asn(Pyr)2 vs 3-Asn(NMe2)2} More surprisingly, considering amides with same substituents {3-Pye2 vs 3-Asn(Pyr)2 and 3-Gln(NMe2)2 vs 3-Asn(NMe2)2 }, cyclic hexapeptides bearing Gln side chain, despite having higher , were more soluble than Asn side chain. A short Asn side chain may form IMHB even in the polar solvent unlike a flexible Gln side chain that may have an equilibrium between IMHB and solvent-exposed formations. Alternatively, the alternate low-dielectric conformation calculated for 3-Asn(Pyr)2 may be slightly less chameleonic, i.e., less able to access more polar conformations in aqueous solution. These experimental results broaden the opportunity to fine-tune lipophilicity of cyclic hexapeptide, for instance, by introducing a small amide substituent to enhance solubility of a highly hydrophobic cyclic hexapeptide, or a large amide substituent to slightly ameliorate solubility penalty while preserving overall permeability.
Conclusion
The inability of traditional small molecules to target large protein interfaces steers medicinal chemists to search for novel scaffolds in the bRo5 chemical space. Cyclic peptides have the potential to bind previously “undruggable” protein targets, but poor membrane permeability due to their bRo5 and peptidic nature has impeded the application toward intracellular targets. In this work, we introduce a potentially general approach to mask the amide HBD using SC-BB from simple N,N-dialkylated derivatives of Gln and Asn. Our studies in cyclic hexapeptide models showed that N,N-disubstituted Gln derivatives can serve as a side chain HBA to sequester the exposed backbone NH group in scaffold 1 and 3 when these units are substituted next to the proline on the C-terminus. Moreover, the SC-BB hydrogen bonding helps promote the IMHB network within the cyclic hexapeptides by restricting backbone conformation based on the McMD calculations. As a result, we observed an improvement in permeability in both artificial and cell-based assays. This method can be applied to any side chain combinations, and the HBA can be modified extensively by varying the functional group, amide substituents, or length of side chain. While polar and/or charged residues are often used to improve solubility in peptides, every polar proteinogenic residue also contains HBD, which are intrinsically deleterious to passive permeability. As a polar, non-proteinogenic residue that contains a strong HBA moiety but that lacks HBD, the Pye residue may allow medicinal chemists to modulate the chemical properties of cyclic peptides to balance their passive permeabilities, solubilities in the context of maintaining favorable target binding.
Masking exposed HBD by SC-BB hydrogen bonding could be used in conjunction with other methods to improve physico-chemical properties cyclic peptides. HBA-containing side chains can be introduced as pro-drugs, for
example, masking both the side chain functional group as well as the backbone amide in cases where SC-BB interactions are favorable.65–67 However, since the Pye residue itself dramatically improves solubility while only slightly diminishing , it may be generally useful for improving the properties of lipophilic cyclic peptide leads even in the absence of favorable SC-BB interactions. Therefore, this approach opens an opportunity to deliver cyclic peptides containing polar amino acids and backbone amide NH groups through the passive permeability pathway. Future studies will be directed toward evaluating the scope of Pye and similar residues in the context of improving the properties of bioactive compounds without abrogating their biochemical potency.
Experimental Section
All reactions were carried out under an inert atmosphere (nitrogen, or argon where stated) with dry solvents under anhydrous conditions. Glassware for anhydrous reactions was dried in an oven at 140 °C for minimum 6 h prior to use. Dry solvents were obtained by passing the previously degassed solvents through activated alumina columns. Reagents were purchased at a high commercial quality (typically 97 % or higher) and used without further purification, unless otherwise stated. Amino acids and amines were purchased from Combi- Blocks, Oakwood, Sigma-Aldrich, or Chem-Impex. COMU and HATU were purchased from Combi-Blocks or Chem-Impex. Piperidine was purchased from Spectrum Chemical. 1,9-Decadiene was purchased from TCI Chemicals. 2-Chlorotrityl chloride polystyrene resin was purchased from Rapp-Polymere. High field NMR spectra were recorded with Bruker Avance III HD at 500 MHz for 1H and 126 MHz for 13C or Bruker Ascend™ with cryoprobe at 800 MHz for 1H and 201 MHz for 13C. NMR spectra were calibrated using residual non-deuterated solvent as an internal reference (CDCl3: 1H NMR = 7.27, 13C NMR = 77.0). The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, dd = double doublet, dt = double triplet, dq = double quartet, m = multiplet, br = broad. Purity of synthesized compounds were determined on an Advion AVANT HPLC-expression® CMS system using a C18 Kinetex® colum (30 x 2.1 mm, 2.6 μm 100 Å) at a flow rate of 0.4 mL/min. The mobile phase was composed of 0.1% (v/v) formic acid in Milli-Q water (solvent A) and 0.1% formic acid in CH3CN (solvent B). The gradient elution was ramped from 20 to 100% B over 6 minutes and held at 100% B for 1 minute. The detection was performed at 200 nm and 254 nm and the column temperature in an oven was 50 °C. All individual synthesized compounds are ≥ 95% pure by HPLC or NMR analysis. 3-Pye2 and 3-Pye3 libraries were synthesized as purified mixtures and quantified via UPLC-MS and selected-mass monitoring.
Loading Pye on 2-Chlorotrityl Resin.
2-Chlorotrityl chloride resin was swelled in dried CH2Cl2 for 45 minutes. The solution of Pye and 2 eq of DIPEA in CH2Cl2 (0.16 M) was prepared 10 minutes prior adding to the resin. The resin was shaken in the solution for 3 h and drained. The mixture of 1:2:17 iPr2NEt:MeOH:CH2Cl2 was added to cap any remaining active site. Resin was washed with CH2Cl2 (3x), DMF (3x) and CH2Cl2 (3x) and dried under vacuum overnight. The loading capacity was quantified by UV absorbance of the dibenzofulvene byproduct at 301 nm in ethanol by microcleavage of weighted resin.
Automatic Linear Peptide Synthesis.
Linear peptides were synthesized using an automated peptide synthesizer (Prelude X, Protein Technologies). Commercial pre-loaded 2-chlorotrityl resin was used in 0.05 mmol scale. Fmoc deprotection was carried out with 20% piperidine in DMF for 3 minutes at 90 °C twice. Couplings were performed using Fmoc-protected amino acids (4 eq), HATU (3.8 eq), and iPr2NEt (6 eq) in DMF (0.4 M with respect to amino acid) for 10 minutes at 90 °C. A capping step was performed after each amide coupling with a 1:1 mixture of acetic anhydride and iPr2NEt in DMF. Each coupling, deprotection, and capping step was followed by a wash with DMF (6x), CH2Cl2 (3x) and DMF (6x). Complete linear peptides were cleaved off resin with 30% HFIP in CH2Cl2 for 1 h three times with a CH2Cl2 wash equivalent to 5 resin volumes in between each step. Solvent was removed with Biotage V10 evaporator to obtain linear peptides as solid.
Peptide Cyclization.
Crude linear peptides were dissolved in dry THF with 4 eq of iPr2NEt and added dropwise to a solution of 1:1 THF/MeCN containing 2 eq of COMU for a final concentration of 2 mM under argon atmosphere. Reactions were stirred for 12–24 h until complete cyclization was achieved as monitored by LC-MS. The reaction was reduced in vacuo and purified via reverse-phased column chromatography using a Biotage Isolera Prime.
Purification of Cyclic Hexapeptides.
Purification of cyclic peptides were carried by Biotage Isolera Prime equipped with Biotage Sfär Bio C18 D 25 g column eluting with the 20–100% MeCN/H2O gradient modified with 0.1% TFA at a flow rate of 40 mL/min.
Synthesis for 3-Pye2 and 3-Pye3 Libraries.
Cyclic hexapeptide libraries were synthesize manually by split-pool approach (Scheme S3 and S4). Linear peptide sequences were synthesized from commercial pre-loaded 2-chlorotrityl-L-leucine resin starting with 200 mg for each R1 amino acids (1 g total; 1.13 mmol). Fmoc deprotections were carried out with 20% piperidine in DMF for 20 minutes twice. Couplings were performed using Fmoc-protected amino acids (4 eq), HATU (4 eq), HOAt (4 eq) and iPr2NEt (8 eq) in DMF (0.3 M with respect to amino acid) for 1 h. After each coupling and deprotection step, the resin was washed with DMF (5x), CH2Cl2 (3x) and DMF (5x), and monitored the completeness of reaction by Kaiser test with small amount of resin. After coupling the R2 amino acids, the resin was kept separately as the sub library. Linear peptides were cleaved in 30% HFIP / CH2Cl2 solution. Cyclization was performed same as mentioned above.
Synthesis of Pye peptoid monomer.
2-Chlorotrityl chloride resin was swelled in CH2Cl2 1 h before charging with 1M bromoacetic acid, 1M DIPEA in CH2Cl2. Resin was shaken for 1 h, then rinsed with DMF (3x) and CH2Cl2 (3x). 3-Amino-1-(pyrrolidin-1-yl)propan-1-one (4 eq) was first free-base by shaking in NMP in the presence of powdered KOH (12 eq). The KOH powder was separated by centrifuging at 16,000 x g for 5 minutes. Resin was shaken in this supernatant for 2 h and washed with DMF (3x) and CH2Cl2 (3x). Phenylacetic acid (3 eq), COMU (2.8 eq) and DIPEA (6 eq) were added to cap the resin. After successively wash with DMF (3x) and CH2Cl2 (3x), 3M HCl generated by mixing acetyl chloride in anhydrous MeOH.68 was added and the resin was shaken overnight to cleave the peptoid off the resin as well as undergoing methyl esterification simultaneously. The crude was dried under nitrogen stream and used for the experiment without further purification (scheme S5).
General Protocol of UPLC-MS Analysis.
UPLC-MS analyses of cyclic peptides were performed via a Thermo Scientific Ultimate 3000 UPLC system, using a Thermo Hypersil GOLD C18 30 x 2.1 (1.9u) column (#25002–032130). Flow rate was set to 1 mL/min and a gradient method was as followed: 0.0–0.5 min, 5% MeCN; 0.5–0.75 min, ramp to 95% MeCN; 0.75–3.0 min, 95% MeCN; 3.0–3.5 min, 5% MeCN. Mass identification and quantification used an inline Thermo Scientific Orbitrap VelosPro (FTMS mode), tuned for maximum ionization of cyclosporin A, background ion locking on octyl phthalate, 200–2000 AMU mass windows, using +/− 0.02 AMU windows for integration.
Shake Flask Partition Experiment.
Test compounds (400 μM, 8 μL) in DMSO were mixed in the solution of 800 μL 1,9-deacdiene and 800 μL PBS buffer in Eppendorf tubes and agitated by vortex (30 min) and sonication (30 min). Tubes were centrifuged for 10 min at 16,000 x g to separate two layers. 150 μL of each layer was transferred to the 96-well plate in quadruplicate and evaporated overnight in a Genevac centrifugal evaporator (60 °C). Test compounds were resuspended in 150 μL of 1:1 MeCN/H2O and analyzed by UPLC-MS as described above.
calculation:
was calculated from the equation published previously.53
is a 2D molecular descriptor to represent the octanol/water partition coefficient determined from atoms in a molecule, which was calculated from Discovery Studio software.
PAMPA Assays:
A 96-well donor plate with 0.45 μm hydrophobic Immobilon-P membrane supports (Millipore MAIPNTR10) was loaded with 5 μL of 1% lecithin in n-dodecane. Cyclic peptides in PBS solution containing 5% DMSO were loaded into donor plate (150 μL) and attached to the acceptor plate having 300 μL of 5% DMSO in PBS buffer. Each sample was run in quadruplicate for ~15 h at 20 °C. The concentration of each compound in the donor and acceptor wells was quantified by UPLC-MS to calculate Papp.
Thermodynamic Solubility Assay:
20 μL of 10 mM stock solutions were dispensed into a 96-well conical plate and evaporated overnight in a Genevac centrifugal evaporator (60 °C). PBS (100 μL) was reintroduced to the plate to yield a maximum 2 mM concentration, then the plate was sealed and sonicated for 1 h. The plate was then gently agitated at 37 °C for ~24 h. The mixtures were filtered through a 0.7 μm glass fiber filter plate (Agilent 200965–100) into a 96-well conical plate. The filtrate was further diluted up to 40 fold with MeCN in a new 96-well plate for quantification via UPLC-MS. Standard curves of each compound were acquired from serial dilution of stock solution with DMSO (50 μM to 0.1 μM) and used to calculate concentrations of analytes. All standards and analytes were performed in triplicate and averaged.
Amide Proton Temperature Coefficient Experiment:
Approximately 3 mg of model compounds or cyclic peptides samples were dissolved in 550 μL CDCl3 and 1H NMR spectra were recorded at temperatures 300, 305, 310, 315, 320 and 323 K. Chloroform residue was used for calibration, and temperature coefficients of amide protons were calculated from best fit of experimental data points.
Caco-2 Cell Permeability:
The Caco-2 assay was performed by Axcelead Drug Discovery Partners, Inc. (Kanagawa, Japan).
Conformational Search McMD Simulations:
The procedure of the conformational search was same as previous reports.52 Briefly, flat potential energy was obtained corresponding to the temperature between at T = 280 and 1505 K, and after production run, resampling method was used to obtain the ensemble at T = 300 K. From this ensemble, 5,000 conformers were used to further analysis.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by funding from the National Institutes of Health (GM131135). X-ray diffraction studies were performed on an instrument purchased with NSF MRI grant #2018501. We also thank Prof. Sam Gellman for helpful suggestions during the preparation of this manuscript.
ABBREVIATIONS USED
- AA
arbitrary amino acid
- ALogP
atomistic calculated octanol/water partition coefficient
- Boc2O
di-tert-butyl decarbonate
- bRo5
beyond-Rule of 5
- Caco-2
human colorectal adenocarcinoma cells
- COMU
(1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylaminomorpholino-carbenium hexafluorophosphate
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethylsulfoxide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- Fmoc
9-fluorenylmethoxycarbonyl
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- HB
hydrogen bond
- HBA
hydrogen bond acceptor
- HBD
hydrogen bond donor
- HFIP
1,1,1,3,3,3-hexafluoro-2-propanol
- HOBt
hydroxybenzotriazole
- IMHB
intramolecular hydrogen bond
- log D
shake flask distribution coefficient at pH 7.4
- LPE
Lipophilic Permeability Efficiency
- MeCN
acetonitrile
- MeOH
methanol
- MW
molecular weight
- NMM
N-methylmorpholine
- PAMPA
cell-free parallel artificial membrane permeability assay
- Papp
apparent permeation rate (A-to-B, x10−6 cm/s)
- PBS
phosphate-buffered saline
- Ro5
Rule of 5
- SPPS
solid phase peptide synthesis
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- UPLC-MS
ultra-high performance liquid chromatography mass spectrometry
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Syntheses of model compounds 4 and amino acid building blocks; Details of solid-phase synthesis, In silico SC-BB hydrogen bonding, shake flask partition experiment, PAMPA assay, thermodynamic solubility assay, amide proton temperature coefficient experiment, NOE buildups and interproton distances, IMHB patterns, crystal structure of 3-Pye2(Ala3Nal5) and 3-Pye2(HPhe5) (PDF)
The 20 structures of 3-Pye2(Ala3Nal5) from the McMD ensemble at T = 300K with the fewest violations from the NOESY-derived distances (PDB)
CCDC 2122986–2122987 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033
The authors declare no competing financial interest.
References
- (1).Hewitt WM; Leung SS; Pye CR; Ponkey AR; Bednarek M; Jacobson MP; Lokey RS Cell-Permeable Cyclic Peptides from Synthetic Libraries Inspired by Natural Products. J. Am. Chem. Soc 2015, 137, 715–721. [DOI] [PubMed] [Google Scholar]
- (2).Shiroma Y; Takahashi R -u.; Yamamoto, Y.; Tahara, H. Targeting DNA Binding Proteins for Cancer Therapy. Cancer Sci 2020, 111, 1058–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wu P Inhibition of RNA-Binding Proteins with Small Molecules. Nat. Rev. Chem 2020, 4, 441–458. [DOI] [PubMed] [Google Scholar]
- (4).Hong S RNA Binding Protein as an Emerging Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev 2017, 22, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Overington JP; Al-Lazikani B; Hopkins AL How Many Drug Targets Are There? Nat. Rev. Drug Discov 2006, 5, 993–996. [DOI] [PubMed] [Google Scholar]
- (6).Lu H; Zhou Q; He J; Jiang Z; Peng C; Tong R; Shi J Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther 2020, 5, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Naylor MR; Bockus AT; Blanco M-J; Lokey RS Cyclic Peptide Natural Products Chart the Frontier of Oral Bioavailability in the Pursuit of Undruggable Targets. Curr. Opin. Chem. Biol 2017, 38, 141–147. [DOI] [PubMed] [Google Scholar]
- (8).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- (9).Li Y; Li W; Xu Z Improvement on Permeability of Cyclic Peptide/Peptidomimetic: Backbone N-Methylation as a Useful Tool. Mar. Drugs 2021, 19, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Villar EA; Beglov D; Chennamadhavuni S; Porco JA; Kozakov D; Vajda S; Whitty A How Proteins Bind Macrocycles. Nat. Chem. Biol 2014, 10, 723–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Doak BC; Zheng J; Dobritzsch D; Kihlberg J How Beyond Rule of 5 Drugs and Clinical Candidates Bind to Their Targets. J. Med. Chem 2016, 59, 2312–2327. [DOI] [PubMed] [Google Scholar]
- (12).Doak Bradley C.; Over B; Giordanetto F; Kihlberg J Oral Druggable Space Beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol 2014, 21, 1115–1142. [DOI] [PubMed] [Google Scholar]
- (13).Caron G; Kihlberg J; Goetz G; Ratkova E; Poongavanam V; Ermondi G Steering New Drug Discovery Campaigns: Permeability, Solubility, and Physicochemical Properties in the BRo5 Chemical Space. ACS Med. Chem. Lett 2021, 12, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Shultz MD Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem 2019, 62, 1701–1714. [DOI] [PubMed] [Google Scholar]
- (15).Brown DG; Wobst HJ A Decade of FDA-Approved Drugs (2010–2019): Trends and Future Directions. J. Med. Chem 2021, 64, 2312–2338. [DOI] [PubMed] [Google Scholar]
- (16).Jing X; Jin K A Gold Mine for Drug Discovery: Strategies to Develop Cyclic Peptides into Therapies. Med. Res. Rev 2020, 40, 753–810. [DOI] [PubMed] [Google Scholar]
- (17).Dougherty PG; Sahni A; Pei D Understanding Cell Penetration of Cyclic Peptides. Chem. Rev 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Buckton LK; Rahimi MN; McAlpine SR Cyclic Peptides as Drugs for Intracellular Targets: The Next Frontier in Peptide Therapeutic Development. Chem. Eur. J 2021, 27, 1487–1513. [DOI] [PubMed] [Google Scholar]
- (19).Nielsen DS; Shepherd NE; Xu W; Lucke AJ; Stoermer MJ; Fairlie DP Orally Absorbed Cyclic Peptides. Chem. Rev 2017, 117, 8094–8128. [DOI] [PubMed] [Google Scholar]
- (20).Guillen Schlippe YV; Hartman MCT; Josephson K; Szostak JW In Vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc 2012, 134, 10469–10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Witek J; Keller BG; Blatter M; Meissner A; Wagner T; Riniker S Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model 2016, 56, 1547–1562. [DOI] [PubMed] [Google Scholar]
- (22).Wang CK; Swedberg JE; Harvey PJ; Kaas Q; Craik DJ Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J. Phys. Chem. B 2018, 122, 2261–2276. [DOI] [PubMed] [Google Scholar]
- (23).Loosli H-R; Kessler H; Oschkinat H; Weber H-P; Petcher TJ; Widmer A Peptide Conformations. Part 31. The Conformation of Cyclosporin A in the Crystal and in Solution. Helv. Chim. Acta 1985, 68, 682–704. [Google Scholar]
- (24).Caron G; Kihlberg J; Ermondi G Intramolecular Hydrogen Bonding: An Opportunity for Improved Design in Medicinal Chemistry. Med. Res. Rev 2019, 39, 1707–1729. [DOI] [PubMed] [Google Scholar]
- (25).Furukawa A; Townsend CE; Schwochert J; Pye CR; Bednarek MA; Lokey RS Passive Membrane Permeability in Cyclic Peptomer Scaffolds Is Robust to Extensive Variation in Side Chain Functionality and Backbone Geometry. J. Med. Chem 2016, 59, 9503–9512. [DOI] [PubMed] [Google Scholar]
- (26).Wang CK; Northfield SE; Colless B; Chaousis S; Hamernig I; Lohman R-J; Nielsen DS; Schroeder CI; Liras S; Price DA; Fairlie DP; Craik DJ Rational Design and Synthesis of an Orally Bioavailable Peptide Guided by NMR Amide Temperature Coefficients. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 17504–17509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang CK; Northfield SE; Swedberg JE; Colless B; Chaousis S; Price DA; Liras S; Craik DJ Exploring Experimental and Computational Markers of Cyclic Peptides: Charting Islands of Permeability. Eur. J. Med. Chem 2015, 97, 202–213. [DOI] [PubMed] [Google Scholar]
- (28).Schwochert J; Lao Y; Pye CR; Naylor MR; Desai PV; Gonzalez Valcarcel IC; Barrett JA; Sawada G; Blanco MJ; Lokey RS Stereochemistry Balances Cell Permeability and Solubility in the Naturally Derived Phepropeptin Cyclic Peptides. ACS Med. Chem. Lett 2016, 7, 757–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Biron E; Chatterjee J; Ovadia O; Langenegger D; Brueggen J; Hoyer D; Schmid HA; Jelinek R; Gilon C; Hoffman A; Kessler H Improving Oral Bioavailability of Peptides by Multiple N-Methylation: Somatostatin Analogues. Angew. Chem. Int. Ed 2008, 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- (30).Bockus AT; Schwochert JA; Pye CR; Townsend CE; Sok V; Bednarek MA; Lokey RS Going out on a Limb: Delineating the Effects of β-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide a Analogues. J. Med. Chem 2015, 58, 7409–7418. [DOI] [PubMed] [Google Scholar]
- (31).Kwon Y-U; Kodadek T Quantitative Comparison of the Relative Cell Permeability of Cyclic and Linear Peptides. Chem. Biol 2007, 14, 671–677. [DOI] [PubMed] [Google Scholar]
- (32).Tan NC; Yu P; Kwon Y-U; Kodadek T High-Throughput Evaluation of Relative Cell Permeability between Peptoids and Peptides. Bioorg. Med. Chem 2008, 16, 5853–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Räder AFB; Reichart F; Weinmüller M; Kessler H Improving Oral Bioavailability of Cyclic Peptides by N-Methylation. Bioorg. Med. Chem 2018, 26, 2766–2773. [DOI] [PubMed] [Google Scholar]
- (34).Thansandote P; Harris RM; Dexter HL; Simpson GL; Pal S; Upton RJ; Valko K Improving the Passive Permeability of Macrocyclic Peptides: Balancing Permeability with Other Physicochemical Properties. Bioorg. Med. Chem 2015, 23, 322–327. [DOI] [PubMed] [Google Scholar]
- (35).Hill TA; Lohman R-J; Hoang HN; Nielsen DS; Scully CCG; Kok WM; Liu L; Lucke AJ; Stoermer MJ; Schroeder CI; Chaousis S; Colless B; Bernhardt PV; Edmonds DJ; Griffith DA; Rotter CJ; Ruggeri RB; Price DA; Liras S; Craik DJ; Fairlie DP Cyclic Penta- and Hexaleucine Peptides without N-Methylation Are Orally Absorbed. ACS Med. Chem. Lett 2014, 5, 1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Nielsen DS; Hoang HN; Lohman R-J; Hill TA; Lucke AJ; Craik DJ; Edmonds DJ; Griffith DA; Rotter CJ; Ruggeri RB; Price DA; Liras S; Fairlie DP Improving on Nature: Making a Cyclic Heptapeptide Orally Bioavailable. Angew. Chem. Int. Ed 2014, 53, 12059–12063. [DOI] [PubMed] [Google Scholar]
- (37).Vollbrecht L; Steinmetz H; Hofle G; Oberer L; Rihs G; Bovermann G; von Matt P Argyrins, Immunosuppressive Cyclic Peptides from Myxobacteria. II. Structure Elucidation and Stereochemistry. J Antibiot (Tokyo) 2002, 55, 715–721. [DOI] [PubMed] [Google Scholar]
- (38).Nyfeler B; Hoepfner D; Palestrant D; Kirby CA; Whitehead L; Yu R; Deng G; Caughlan RE; Woods AL; Jones AK; Barnes SW; Walker JR; Gaulis S; Hauy E; Brachmann SM; Krastel P; Studer C; Riedl R; Estoppey D; Aust T; Movva NR; Wang Z; Salcius M; Michaud GA; McAllister G; Murphy LO; Tallarico JA; Wilson CJ; Dean CR Identification of Elongation Factor G as the Conserved Cellular Target of Argyrin B. PLOS ONE 2012, 7, e42657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zaretsky S; Hickey JL; Tan J; Pichugin D; Denis MA St.; Ler S; Chung BKW; Scully CCG; Yudin AK Mechanistic Investigation of Aziridine Aldehyde-Driven Peptide Macrocyclization: The Imidoanhydride Pathway. Chem. Sci 2015, 6, 5446–5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hickey JL; Zaretsky S; Denis MA St.; Kumar Chakka S; Morshed MM; Scully CCG; Roughton AL; Yudin AK Passive Membrane Permeability of Macrocycles Can Be Controlled by Exocyclic Amide Bonds. J. Med. Chem 2016, 59, 5368–5376. [DOI] [PubMed] [Google Scholar]
- (41).Zaretsky S; Scully CCG; Lough AJ; Yudin AK Exocyclic Control of Turn Induction in Macrocyclic Peptide Scaffolds. Chem. Eur. J 2013, 19, 17668–17672. [DOI] [PubMed] [Google Scholar]
- (42).Appavoo SD; Huh S; Diaz DB; Yudin AK Conformational Control of Macrocycles by Remote Structural Modification. Chem. Rev 2019, 119, 9724–9752. [DOI] [PubMed] [Google Scholar]
- (43).Driver RW; Hoang HN; Abbenante G; Fairlie DP A Cyclic β-Strand Tripeptide with an α-Helix Like CD Spectrum. Org. Lett 2009, 11, 3092–3095. [DOI] [PubMed] [Google Scholar]
- (44).Grotenbreg GM; Timmer MSM; Llamas-Saiz AL; Verdoes M; van der Marel GA; van Raaij MJ; Overkleeft HS; Overhand M An Unusual Reverse Turn Structure Adopted by a Furanoid Sugar Amino Acid Incorporated in Gramicidin S. J. Am. Chem. Soc 2004, 126, 3444–3446. [DOI] [PubMed] [Google Scholar]
- (45).Wang S; König G; Roth H-J; Fouché M; Rodde S; Riniker S Effect of Flexibility, Lipophilicity, and the Location of Polar Residues on the Passive Membrane Permeability of a Series of Cyclic Decapeptides. J. Med. Chem 2021, 64, 12761–12773. [DOI] [PubMed] [Google Scholar]
- (46).Cierpicki T; Otlewski J Amide Proton Temperature Coefficients as Hydrogen Bond Indicators in Proteins. J. Biomol. NMR 2001, 21, 249–261. [DOI] [PubMed] [Google Scholar]
- (47).Baxter NJ; Williamson MP Temperature Dependence of 1H Chemical Shifts in Proteins. J. Biomol. NMR 1997, 9, 359–369. [DOI] [PubMed] [Google Scholar]
- (48).Wagner G; Pardi A; Wuethrich K Hydrogen Bond Length and Proton NMR Chemical Shifts in Proteins. J. Am. Chem. Soc 1983, 105, 5948–5949. [Google Scholar]
- (49).Gellman SH; Adams BR Intramolecular Hydrogen Bonding in Simple Diamides. Tetrahedron Lett 1989, 30, 3381–3384. [Google Scholar]
- (50).Gellman SH; Dado GP; Liang GB; Adams BR Conformation-Directing Effects of a Single Intramolecular Amide-Amide Hydrogen Bond: Variable-Temperature NMR and IR Studies on a Homologous Diamide Series. J. Am. Chem. Soc 1991, 113, 1164–1173. [Google Scholar]
- (51).Andersen NH; Neidigh JW; Harris SM; Lee GM; Liu Z; Tong H Extracting Information from the Temperature Gradients of Polypeptide NH Chemical Shifts. 1. The Importance of Conformational Averaging. J. Am. Chem. Soc 1997, 119, 8547–8561. [Google Scholar]
- (52).Ono S; Naylor MR; Townsend CE; Okumura C; Okada O; Lokey RS Conformation and Permeability: Cyclic Hexapeptide Diastereomers. J. Chem. Inf. Model 2019, 59, 2952–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Naylor MR; Ly AM; Handford MJ; Ramos DP; Pye CR; Furukawa A; Klein VG; Noland RP; Edmondson Q; Turmon AC; Hewitt WM; Schwochert J; Townsend CE; Kelly CN; Blanco MJ; Lokey RS Lipophilic Permeability Efficiency Reconciles the Opposing Roles of Lipophilicity in Membrane Permeability and Aqueous Solubility. J. Med. Chem 2018, 61, 11169–11182. [DOI] [PubMed] [Google Scholar]
- (54).Ghose AK; Crippen GM Atomic Physicochemical Parameters for Three-Dimensional-Structure-Directed Quantitative Structure-Activity Relationships. 2. Modeling Dispersive and Hydrophobic Interactions. J. Chem. Inf. Comput. Sci 1987, 27, 21–35. [DOI] [PubMed] [Google Scholar]
- (55).Thompson SJ; Hattotuwagama CK; Holliday JD; Flower DR On the Hydrophobicity of Peptides: Comparing Empirical Predictions of Peptide Log P Values. Bioinformation 2006, 1, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duban ME; Bures MG; DeLazzer J; Martin YC Virtual Screening of Molecular Properties: A Comparison of Log P Calculators. In Pharmacokinetic Optimization in Drug Research, 2001; pp 483–497. [Google Scholar]
- (57).Ghose AK; Viswanadhan VN; Wendoloski JJ Prediction of Hydrophobic (Lipophilic) Properties of Small Organic Molecules Using Fragmental Methods: An Analysis of AlogP and ClogP Methods. J. Phys. Chem. A 1998, 102, 3762–3772. [Google Scholar]
- (58).Kansy M; Senner F; Gubernator K Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (59).Fujikawa M; Nakao K; Shimizu R; Akamatsu M QSAR Study on Permeability of Hydrophobic Compounds with Artificial Membranes. Bioorg. Med. Chem 2007, 15, 3756–3767. [DOI] [PubMed] [Google Scholar]
- (60).Sawada GA; Barsuhn CL; Lutzke BS; Houghton ME; Padbury GE; Ho NFH; Raub TJ Increased Lipophilicity and Subsequent Cell Partitioning Decrease Passive Transcellular Diffusion of Novel, Highly Lipophilic Antioxidants. J. Pharmacol. Exp. Ther 1999, 288, 1317–1326. [PubMed] [Google Scholar]
- (61).Hu H; Krishnamurthy K Revisiting the Initial Rate Approximation in Kinetic Noe Measurements. J. Magn. Reson 2006, 182, 173–177. [DOI] [PubMed] [Google Scholar]
- (62).Butts CP; Jones CR; Towers EC; Flynn JL; Appleby L; Barron NJ Interproton Distance Determinations by NOE – Surprising Accuracy and Precision in a Rigid Organic Molecule. Org. Biomol. Chem 2011, 9, 177–184. [DOI] [PubMed] [Google Scholar]
- (63).Begnini F; Poongavanam V; Atilaw Y; Erdelyi M; Schiesser S; Kihlberg J Cell Permeability of Isomeric Macrocycles: Predictions and NMR Studies. ACS Med. Chem. Lett 2021, 12, 983–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Nakajima N; Nakamura H; Kidera A Multicanonical Ensemble Generated by Molecular Dynamics Simulation for Enhanced Conformational Sampling of Peptides. J. Phys. Chem. B 1997, 101, 817–824. [Google Scholar]
- (65).Barlow N; Chalmers DK; Williams-Noonan BJ; Thompson PE; Norton RS Improving Membrane Permeation in the Beyond Rule-of-Five Space by Using Prodrugs to Mask Hydrogen Bond Donors. ACS Chem. Biol 2020, 15, 2070–2078. [DOI] [PubMed] [Google Scholar]
- (66).Simplício AL; Clancy JM; Gilmer JF Prodrugs for Amines. Molecules 2008, 13, 519–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Wang D; Zou L; Jin Q; Hou J; Ge G; Yang L Human Carboxylesterases: A Comprehensive Review. Acta Pharm. Sin. B 2018, 8, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Turner RA; Weber RJ; Lokey RS Direct Conversion of Resin-Bound Peptides to C-Terminal Esters. Org. Lett 2010, 12, 1852–1855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.