

Abstract
Angiotensin-converting enzyme 2 (ACE2) is a key component of the renin-angiotensin-aldosterone system. Yet, little is known about the clinical and biologic correlates of circulating ACE2 levels in humans. We assessed the clinical and proteomic correlates of plasma (soluble) ACE2 protein levels in human heart failure. We measured plasma ACE2 using a modified aptamer assay among Penn Heart Failure Study (PHFS) participants (n=2,248). We performed an association study of ACE2 against ~5000 other plasma proteins measured with the SomaScan platform. Plasma ACE2 was not associated with angiotensin-converting enzyme inhibitor and/or angiotensin-receptor blocker use. Plasma ACE2 was associated with older age, male sex, diabetes mellitus, a lower estimated glomerular filtration rate, worse NYHA class, a history of coronary artery bypass surgery, and higher pro-BNP levels. Plasma ACE2 exhibited associations with 1,011 other plasma proteins. In pathway overrepresentation analyses, top canonical pathways associated with plasma ACE2 included clathrin-mediated endocytosis signaling, actin cytoskeleton signaling, mechanisms of viral exit from host cells, eukaryotic initiation factor 2 (EIF2) signaling and the protein ubiquitination pathway. In conclusion, in humans with HF, plasma ACE2 is associated with various clinical factors known to be associated with severe COVID-19, including older age, male sex, and diabetes, but is not associated with angiotensin-converting enzyme inhibitor and/or angiotensin-receptor blocker use. Plasma ACE2 protein levels are prominently associated with multiple cellular pathways involved in cellular endocytosis, exocytosis, and intracellular protein trafficking. Whether these have a causal relationship with ACE2 or are relevant to novel coronavirus-2 infection remains to be assessed in future studies.
Keywords: Angiotensin converting enzyme 2, renin-angiotensin system, ACE inhibitors, angiotensin receptor blocker, proteomics
Introduction
Angiotensin-converting enzyme 2 (ACE2) is a key counter-regulatory component of the renin-angiotensin-aldosterone system (RAAS).1 Renin metabolizes angiotensinogen to angiotensin (Ang)-I, a decapeptide which is further cleaved by angiotensin-converting enzyme (ACE) to the octapeptide Ang-II. ACE2, a membrane-bound or secreted soluble enzyme, is a mono-carboxypeptidase that functions as a central regulator of the RAAS mainly by converting angiotensin (Ang) I and Ang II into Ang 1–9 and Ang 1–7, respectively. Ang II Type 1 receptor (AT1R) activation by Ang-II promotes hypertension, left ventricular hypertrophy and fibrosis. In contrast, Ang-(1–7) mediates opposite effects (vasodilation, growth inhibition, and antifibrotic effects) via activation of Mas receptors (MasR). Therefore, ACE2-mediated degradation of Ang I and Ang II opposes RAAS activation and mitigates the deleterious actions mediated by Ang II and AT1R. Experimental models of human diseases have defined a critical role for ACE2 in heart failure (HF), hypertension and various other cardiovascular conditions.1
In addition to its key cardiovascular effects, ACE2 has recently received widespread interest due to its role in the pathogenesis of coronavirus disease 2019 (COVID-19). ACE2 is the cellular receptor for SARS-CoV-2, the virus that causes COVID-192, 3. Increased ACE2 expression in various cell lines correlates with susceptibility to infection by SARS-CoV-1, a closely related coronavirus responsible for the severe acute respiratory syndrome (SARS).1 However, the ACE2/Ang 1–7 axis also mitigates SARS-induced tissue injury. Moreover, soluble ACE2 can act as a decoy for SARS-CoV-2 4 and SARS-CoV-15 in experimental conditions, presumably by competing with membrane-bound cellular receptors. Despite the apparent importance of ACE2 in cardiovascular and infectious diseases and the large body of knowledge accumulated from pre-clinical studies since its discovery approximately 20 years ago, little is known about the biologic correlates and genetic determinants of plasma ACE2 levels in humans.
In this study we aimed to assess the clinical and proteomic correlates of plasma ACE2 levels in human heart failure (HF).
Methods
The data, analytic methods, and study materials are not publicly available for purposes of reproducing the results or replicating the procedures. Such data may be made available to other researchers for collaborative research, through the establishment of appropriate data sharing agreements and regulatory approvals.
Study population
We analyzed plasma samples and data from participants enrolled in the Penn Heart Failure Study (PHFS). The PHFS design has been previously published.6–9 Briefly, the PHFS was a prospective cohort study of ambulatory patients with chronic HF recruited between 2003–2011 at the University of Pennsylvania (Philadelphia, PA), Case Western Reserve University (Cleveland, OH), and the University of Wisconsin (Madison, WI). Patients with a clinical diagnosis of HF as determined by a HF specialist were enrolled. Each participant provided written informed consent. At the time of study entry, standardized questionnaires were administered to participants and their physicians to obtain detailed clinical data. Participants with life expectancy <6 months from a non-cardiac condition, on mechanical circulatory support, or unable to provide informed consent were excluded. Venous blood samples were obtained at enrollment and stored at −80 °C for later analysis. An institutional review board from each participating center approved the protocol.
Measurement of ACE2 protein levels and proteomic analyses
We measured plasma ACE2 protein levels with a modified aptamer assay (SomaScan® assay, version 4), as previously described.10–14 The SomaScan assay version 4 is a multiplexed, modified aptamer-based binding-assay that includes 4,979 modified aptamer reagents to 4,776 unique protein targets. The SomaScan assay utilizes Slow-Off-rate Modified Aptamer (SOMAmer) reagents, which are chemically modified nucleotides, to bind and quantify target proteins in relative fluorescent units directly proportional to the amount of target protein in the sample. The ACE2 aptamer validated as previously described by Emilsson et al 10 as further detailed in the Online Supplemental Methods Section.
Statistical analysis
Participant characteristics were summarized using mean (SD) for continuous variables with a symmetric distribution and median (interquartile range) for continuous variables with a skewed distribution. Categorical variables are expressed as counts (percentages).
We stratified the study populations according to tertiles of plasma ACE2 and compared various clinical characteristics across the strata. We used analysis of variance (ANOVA) for normally distributed variables, the Kruskal-Wallis test for skewed variables, and the chi-square or Fisher’s exact test, as appropriate, for categorical data. We assessed for independent correlation with ACE2 using linear regression adjusted for all factors associated with ACE2 levels.
We performed knowledge-based pathway analysis to assess the correlates of plasma ACE2. First, we assessed the correlation between levels of ACE2 and all proteins measured in the SomaScan assay using linear regression, adjusting for age, sex, race/ethnicity and all variables found to be associated with plasma ACE2 levels. We utilized Box-Cox transformation as needed to improve the normality of the distributions of protein levels. To perform unit-independent and readily comparable metrics of association between various proteins and plasma ACE2, we computed standardized regression coefficients (β), which represents the regression coefficient obtained after variables are scaled to Z scores. We adjusted for multiple comparisons based on the principal components underlying the variability of all measured proteins, as previously described 15–19. Associations between ACE2 and individual proteins that were significant, with a multiplicity-corrected P value <0.01 were then utilized to perform pathway analyses. In this approach, the statistical overrepresentation in multiple biologic pathways of proteins that are associated with plasma ACE2, using the entire list of measured proteins as the reference set, is analyzed. Proteins were identified according to their UniProt ID annotation. During pathway analysis, a P value is computed for each canonical pathway, followed by correction of multiple testing using the Benjamini & Hochberg method 20, to determine whether a specific canonical pathway is significantly enriched among the proteins of interest.
All probability values presented are 2-tailed. Analyses were performed using the MATLAB statistics and machine learning toolbox (Matlab 2016b, the Mathworks; Natick, MA) and R Statistical Software v3.5.2 (Foundation for Statistical Computing, Vienna, Austria). Pathway analyses were performed using Ingenuity® Pathway Analysis software (Qiagen; Hilden, Germany).
Results
The general characteristics of PHFS participants stratified by tertiles of plasma ACE2 levels are shown in Table 1. Lower levels of plasma ACE2 were significantly associated with older age, male sex, history of diabetes mellitus, lower estimated glomerular filtration rate, the presence of New York Heart Association (NYHA) class 3–4, history of coronary artery bypass surgery, and higher pro-B-type natriuretic peptide (pro-BNP) levels.
Table 1.
Comparisons of general Characteristics of PHFS trial participants stratified by tertiles of plasma ACE2
| General Characteristic | Lowest Tertile | Mid Tertile | Highest Tertile | P value |
|---|---|---|---|---|
| Age, years | 56.3 (55.1 to 57.5) | 54.5 (53.3 to 55.6) | 52.9 (51.8 to 54) | 0.0001 |
| Male Sex | 521 (69.84%) | 508 (68.46%) | 454 (60.78%) | 0.0003 |
| Race | 0.2283 | |||
| White | 546 (74.79%) | 545 (76.65%) | 550 (76.82%) | |
| African American | 174 (23.84%) | 147 (20.68%) | 150 (20.95%) | |
| Hispanic | 12 (1.94%) | 19 (3.03%) | 11 (1.77%) | |
| Other | 10 (1.37%) | 19 (2.67%) | 16 (2.23%) | |
| BMI, kg/m 2 | 29.1 (28.7 to 29.6) | 29.5 (29 to 30) | 29.5 (29 to 30) | 0.5003 |
| Systolic BP, mmHg | 112 (100,128) | 112 (100,128) | 110 (100,125) | 0.1965 |
| Diastolic BP, mmHg | 70 (60,78) | 70 (61.3,78) | 70 (60,78) | 0.5764 |
| LV Ejection Fraction, % | 28.5 (27.3 to 29.7) | 29.1 (27.8 to 30.3) | 28.5 (27.4 to 29.7) | 0.7597 |
|
| ||||
| Medical History | ||||
|
| ||||
| Atrial Fibrillation/flutter | 289 (38.58%) | 263 (35.11%) | 269 (35.87%) | 0.3405 |
| Ischemic etiology | 248 (33.42%) | 222 (30.20%) | 230 (31.08%) | 0.3875 |
| Coronary Artery Bypass Graft | 169 (22.56%) | 129 (17.22%) | 119 (15.87%) | 0.0020 |
| Percutaneous Coronary Intervention | 162 (21.63%) | 174 (23.23%) | 162 (21.60%) | 0.6849 |
| Diabetes Mellitus | 249 (33.24%) | 201 (26.84%) | 192 (25.60%) | 0.0021 |
| History of Smoking | 64 (8.54%) | 81 (10.81%) | 58 (7.73%) | 0.0976 |
| HFpEF | 91 (12.28%) | 86 (11.70%) | 76 (10.22%) | 0.4340 |
| NYHA Class | 0.0041 | |||
| 1 to 2 | 426 (57.34%) | 467 (63.36%) | 483 (65.36%) | |
| 3 to 4 | 317 (42.66%) | 270 (36.64%) | 256 (34.64%) | |
|
| ||||
| Laboratory Parameters | ||||
|
| ||||
| eGFR | 50.8 (37,67) | 58.4 (43.6,71.1) | 60.5 (45.9,74) | <0.0001 |
| ProBNP | 232 (75,704) | 136 (41,448) | 140 (41,523) | <0.0001 |
|
| ||||
| Medication Uses | ||||
|
| ||||
| Beta Blockers | 646 (86.25%) | 659 (87.98%) | 668 (89.07%) | 0.2439 |
| Calcium Channel Blockers | 69 (9.21%) | 77 (10.28%) | 59 (7.87%) | 0.2663 |
| ACE Inhibitors | 482 (64.35%) | 515 (68.76%) | 494 (65.87%) | 0.1863 |
| ARBs | 173 (23.10%) | 152 (20.29%) | 158 (21.07%) | 0.3941 |
| ACE Inhibitors or ARBs | 626 (83.58%) | 646 (86.25%) | 631 (84.13%) | 0.3183 |
| Aldosterone antagonists | 239 (31.91%) | 266 (35.51%) | 268 (35.73%) | 0.2163 |
| Aspirin | 432 (57.68%) | 434 (57.94%) | 421 (56.13%) | 0.7463 |
| Hydralazine | 61 (8.14%) | 71 (9.48%) | 59 (7.87%) | 0.4885 |
| Nitrates | 129 (17.22%) | 118 (15.75%) | 110 (14.67%) | 0.3972 |
| Statins | 377 (50.33%) | 411 (54.87%) | 381 (50.80%) | 0.1539 |
| Insulin | 97 (12.95%) | 92 (12.28%) | 95 (12.67%) | 0.9267 |
LV=left ventricle; BP=blood pressure; NYHA=New York Heart Association; eGFR=estimated glomerular filtration rate; ACE=angiotensin-converting enzyme; ARBs=angiotensin receptor blockers; BMI=body mass index; HFpEF=heart failure with preserved ejection fraction.
Multivariable clinical correlates of plasma ACE2
A linear regression model including all factors associated with plasma ACE2 levels, with the latter included as the dependent variable, is shown in S1 (please see http://hyper.ahajournals.org). Given the intrinsic mathematical dependency of eGFR on age, which may confound the association between age and plasma ACE2, in this model we included serum creatinine (rather than eGFR) and adjusted for height, weight, and sex. In this model, older age, higher serum creatinine and the presence of NYHA class 3–4 were all independently and negatively associated with higher plasma ACE2 levels (S1), whereas sex, diabetes, a history of coronary artery bypass surgery, and pro-BNP levels were not. However, this model explained only a small proportion (1.9%) of the variability in plasma ACE2 levels (model R2=0.019).
Association between ACE inhibitor and Ang II receptor blocker (ARB) use and plasma ACE2
Plasma ACE2 did not significantly differ between participants who were taking (277 AU; 95%CI=274 to 281) vs. those who were not taking (274; 95%CI=265 to 283) an ACE inhibitor or ARB (P=0.51). In a multivariable model adjusted for factors associated with plasma ACE2 (age, sex, diabetes mellitus, history of CABG, NYHA class 3–4 and eGFR), there was no independent association between ACE inhibitor (β=−0.002; P=0.93) or ARB use (β=−0.016; P=0.57) and plasma ACE2. In stratified analyses, plasma ACE2 was not associated with ACE inhibitor or ARB use in either patients with reduced or preserved ejection fraction.
Association between ACE2 and the plasma proteome
The association between plasma ACE2 and other examined plasma proteins is shown in Figure 2; standardized regression coefficients and the -log (P values) are shown. After correction for multiple comparisons, 1011 proteins were found to be significantly associated with plasma ACE2. The proteins most strongly associated with ACE2 are shown in Table 2.
Figure 2. Volcano plot demonstrating associations between all plasma proteins measured and plasma ACE2.
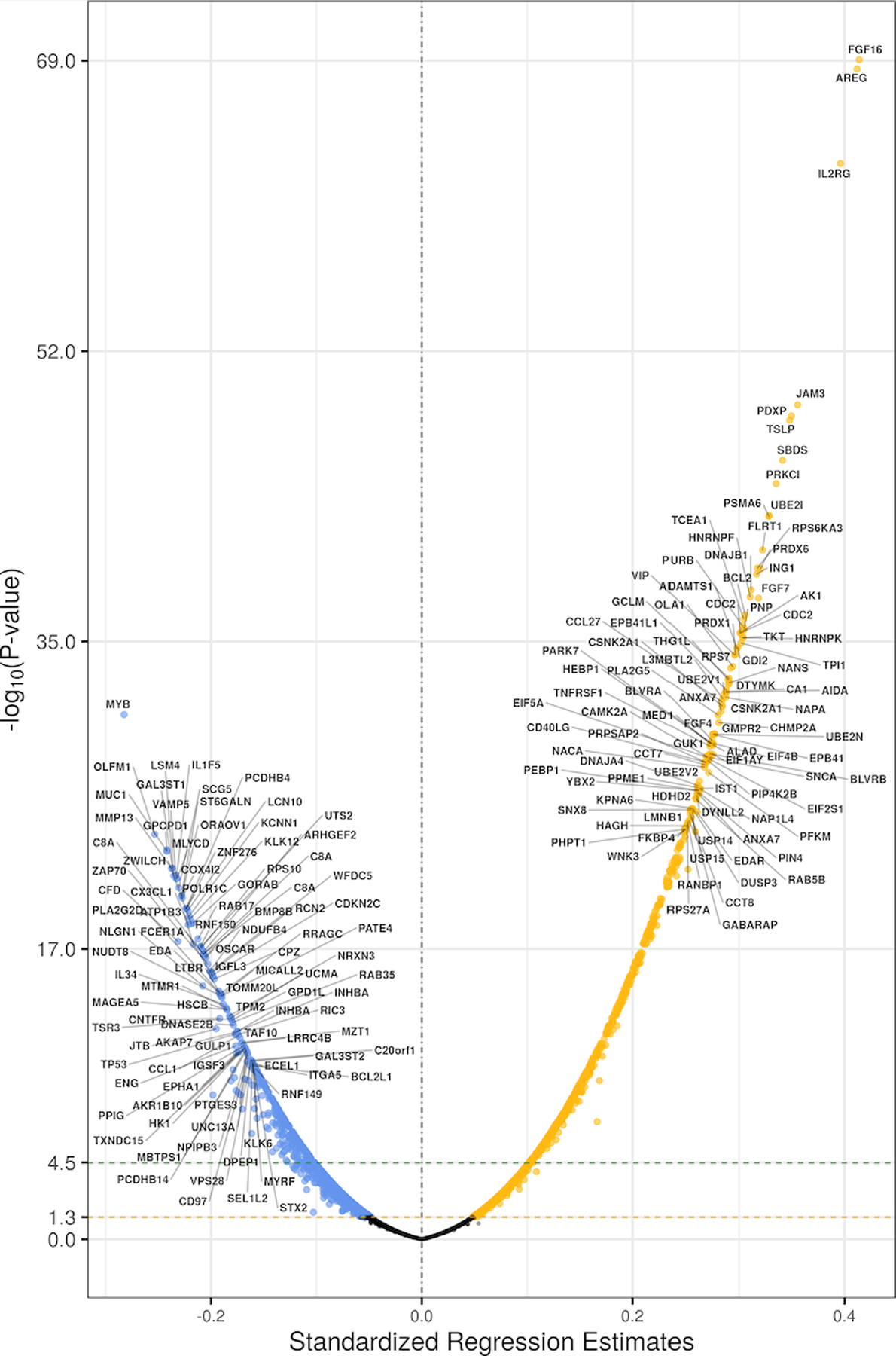
Only the top 100 proteins positively or negatively associated with plasma ACE2 are labeled.
Table 2.
List of top proteins associated with plasma ACE2 with a standardized regression coefficient >0.3 in the PHFS.
| Entrez Gene Name | Standardized Regression Coefficient | P value | UniProt ID | Symbol |
|---|---|---|---|---|
| Fibroblast growth factor 16 | 0.414 | <10−13 | O43320 | FGF16 |
| Amphiregulin | 0.412 | <10−13 | P15514 | AREG |
| Interleukin 2 receptor Subunit gamma | 0.396 | <10−13 | P31785 | IL2RG |
| junctional adhesion molecule 3 | 0.355 | <10−13 | Q9BX67 | JAM3 |
| Pyridoxal phosphatase | 0.35 | <10−13 | Q96GD0 | PDXP |
| Thymic stromal lymphopoietin | 0.348 | <10−13 | Q969D9 | TSLP |
| SBDS ribosome maturation factor | 0.341 | <10−13 | Q9Y3A5 | SBDS |
| protein kinase C iota | 0.335 | <10−13 | P41743 | PRKCI |
| proteasome subunit alpha 6 | 0.328 | <10−13 | P60900 | PSMA6 |
| ubiquitin conjugating enzyme E2 I | 0.328 | <10−13 | P63279 | UBE2I |
| fibronectin leucine rich transmembrane protein 1 | 0.322 | <10−13 | Q9NZU1 | FLRT1 |
| peroxiredoxin 6 | 0.32 | <10−13 | P30041 | PRDX6 |
| fibroblast growth factor 7 | 0.318 | <10−13 | P21781 | FGF7 |
| inhibitor of growth family member 1 | 0.317 | <10−13 | Q9UK53 | ING1 |
| ribosomal protein S6 kinase A3 | 0.317 | <10−13 | P51812 | RPS6KA3 |
| DnaJ heat shock protein family (Hsp40) member B1 | 0.311 | <10−13 | P25685 | DNAJB1 |
| heterogeneous nuclear ribonucleoprotein F | 0.31 | <10−13 | P52597 | HNRNPF |
| BCL2 apoptosis regulator | 0.306 | <10−13 | P10415 | BCL2 |
| purine nucleoside phosphorylase | 0.305 | <10−13 | P00491 | PNP |
| adenylate kinase 1 | 0.304 | <10−13 | P00568 | AK1 |
| heterogeneous nuclear ribonucleoprotein K | 0.304 | <10−13 | P61978 | HNRNPK |
| transcription elongation factor A1 | 0.304 | <10−13 | P23193 | TCEA1 |
| transketolase | 0.304 | <10−13 | P29401 | TKT |
| purine rich element binding protein B | 0.303 | <10−13 | Q96QR8 | PURB |
| triosephosphate isomerase 1 | 0.302 | <10−13 | P60174 | TPI1 |
| cyclin B1 | 0.301 | <10−13 | P14635 | CCNB1 |
| cyclin dependent kinase 1 | 0.301 | <10−13 | P06493 | CDK1 |
All proteins that mapped into pathway analysis, along with their Uniprot ID, standardized regression coefficient, P value for association with plasma ACE2, Entrez gene name and cellular location, are shown in S2 (please see http://hyper.ahajournals.org). In pathway overrepresentation analyses, we found plasma ACE2 to be significantly associated with multiple canonical pathways. A full list of canonical pathways examined, along with -log P value for association with ACE2, the Z score for directionality and the list of proteins in each particular pathway, are listed in S1.
The top canonical pathways associated with plasma ACE2 via pathway analysis are shown in Figure 3, and included clathrin-mediated endocytosis signaling, actin cytoskeleton signaling, mechanisms of viral exit from host cells, eukaryotic initiation factor 2 (EIF2) signaling and the protein ubiquitination pathway. A list of proteins in each of these pathways, along with -log P value and the standardized regression coefficient for association with ACE2, are listed in S2–7 (please see http://hyper.ahajournals.org).
Figure 3. Pathway Enrichment Analysis from plasma proteomics.
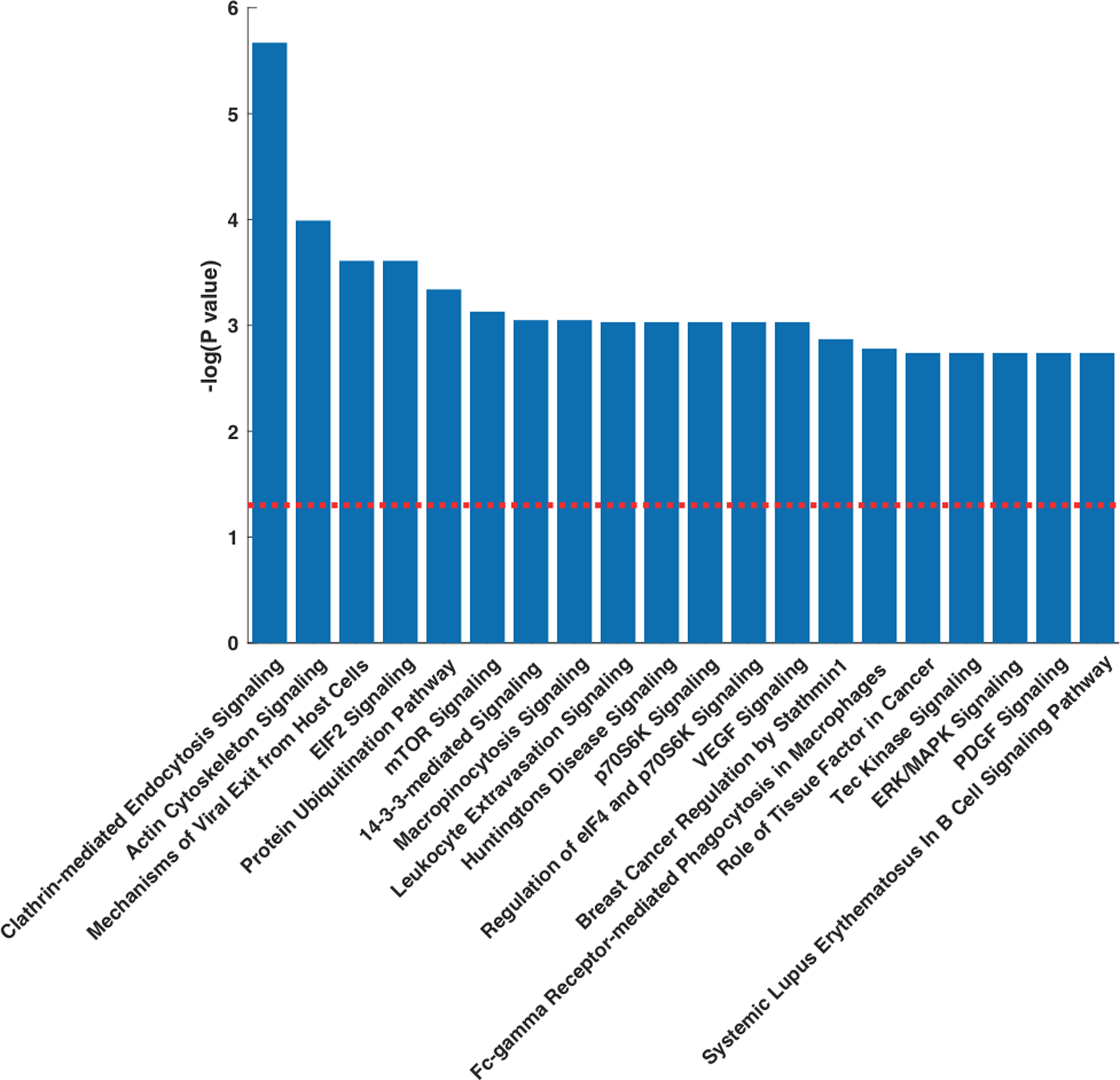
Top canonical pathways associated with plasma ACE2 in the PHFS. (A) Pathways ranked according to statistical significance. The redline represents the threshold for statistical significance (P=0.05, corrected for multiple comparisons).
Discussion
Our study assessed the clinical and proteomic correlates of plasma ACE2 protein levels in a large cohort of patients with HF. In this population, we found lower levels of plasma ACE2 to be associated with older age, male sex, diabetes mellitus, a lower estimated glomerular filtration rate, worse NYHA class, a history of coronary artery bypass surgery, and higher pro-BNP levels. In contrast, plasma ACE2 levels were not associated with angiotensin-converting enzyme inhibitor and/or angiotensin-receptor blocker use. We demonstrate that plasma ACE2 exhibited multiple proteomic correlations, including significant associations with 1,011 other plasma proteins. In pathway overrepresentation analyses, we identified canonical pathways that exhibit the most prominent associations with plasma ACE2, including clathrin-mediated endocytosis, actin cytoskeleton signaling, mechanisms of viral exit from host cells, eukaryotic initiation factor 2 (EIF2) signaling and protein ubiquitination. Our study reports for the first time, pathways related to ACE2 in HF patients, as assessed by plasma proteomics in HF patients.
The main physiologic function of ACE2 is thought to relate to its carboxypeptidase activity, which results in: (A) the degradation of Ang I to Ang 1–9, limiting ACE-mediated Ang-II formation; (B) the degradation of Ang II, thus reducing its biologic effects; and, (C) the formation of Ang 1–7, which exerts protective effects. These mechanisms tend to oppose multiple deleterious effects of Ang II, which may be of particular importance in pathological conditions where the RAAS is overstimulated. Ang 1–7, in particular, exhibits a variety of biologic effects through MasR activation, which are opposite to those of AT1R activation by Ang II. ACE2 also has RAAS-independent functions, such as regulation of intestinal amino acid homeostasis and the gut microbiome. 21 Despite the importance of ACE2 and its potentially high relevance in HF and other clinical conditions in which the RAAS is important, little is still known about the clinical and biologic correlates of plasma ACE2 levels in humans.
In this relatively large HF cohort, we found several clinical correlates of lower plasma ACE2, including older age, male sex, a history of diabetes mellitus, a lower estimated glomerular filtration rate, the presence of New York Heart Association (NYHA) class 3–4, a history of coronary artery bypass surgery, and higher pro-B-type natriuretic peptide (pro-BNP) levels. Interestingly, several of these clinical factors have been found to be associated with severe COVID-19, including older age, male sex, and diabetes.22 However, these clinical factors explained a small proportion of the variability in plasma ACE2 levels. Of note, we did not find an association between ACE inhibitor/ARB use and plasma ACE2. Although ACE2 is insensitive to direct inhibitory effects by conventional ACE inhibitors, 23, 24 ACE inhibitor and ARBs could impact ACE2 through downstream effects on the RAAS system. For instance, AT1R activation by Ang II can lead to downregulation of ACE2 expression via ERK1/2 and p38 MAPK signaling pathways, 1, 25 and can also induce internalization of membrane-bound ACE2 26. In a number of experimental models, ACEIs and ARBs increased ACE2 expression in the heart and kidneys. 27–30 For example, Ferrario et al. observed an increase in cardiac ACE2 mRNA expression in Lewis normotensive rats that received 12 days of lisinopril or losartan 27. Similarly, Ishiyama et al. administered losartan (an ARB), olmesartan (an ARB), or normal saline for 28 days after coronary artery ligation in Lewis normotensive rats 29. Those rats that received saline demonstrated no change in cardiac ACE2 expression, while those that received losartan and olmesartan experienced a 3-fold increase in ACE2 expression, accompanied by increases in Ang I, Ang II, and Ang-(1–7) concentration and AT1R expression.
The lack of association between ACE inhibitor or ARB use and circulating ACE2 in our study is in contrast to animal studies described above, but consistent with several other experimental and human studies evaluating the association of RAAS blockade and ACE2 activity and expression.31–34 In a previous study of 58 patients with paroxysmal atrial fibrillation, 20 patients with persistent atrial fibrillation, and 25 patients without atrial fibrillation, no association between ACE inhibitor or ARB use with ACE2 was found.33 Similarly, in a study of 79 subjects with obstructive coronary artery disease, Ramchand et al. found no association of ACE inhibitor or ARB use with plasma ACE2 levels 34. A recent publication by Sama, et al.35 reported no association between ACE inhibitor or ARB use and plasma ACE2 in one cohort of patients with heart failure; in a second cohort, the same publication reported an association between ACE inhibitor and ARB use with lower plasma ACE2 levels. Thus, our findings add to the body of human studies suggesting that ACE inhibitors and ARBs may not upregulate ACE2, particularly among individuals with underlying cardiovascular disease. However, the vast majority of our study population was receiving ACE inhibitor or ARBs, which may limit the interpretability of our findings in this regard. Moreover, the causality between ACE inhibitor or ARB use and plasma ACE2 levels cannot established from our cross-sectional data. Amidst the COVID-19 pandemic, there has been increasing interest in the potential relationship between ACE inhibitors, ARBs, ACE2 activity/expression, and COVID-19 severity. Specifically, there is speculation that ACE inhibitor or ARB-induced upregulation of ACE2 may increase the risk of contracting and have more severe COVID-19, 36 a controversial issue that is being addressed in several ongoing randomized trials.
Our study provides novel data regarding the top canonical pathways associated with plasma ACE2 in human HF. It is important to recognize that these pathway associations are observational findings and are limited by their derivation from measurements in human plasma alone at one point in time. That said, the specific pathways identified have plausible biologic relevance to viral infection and cardiovascular disease. As such, they may be of value in suggesting specific hypothesis for experimental studies.
The top associated pathway, clathrin-mediated endocytosis, is a key process in vesicular trafficking that transports a wide range of cargo molecules from the plasma membrane of eukaryotic cells into the cytoplasm. 37 The cargo consists mainly of transmembrane proteins and their extracellular ligands. Clathrin-mediated endocytosis is considered to be the major endocytic route for internalization of many cargoes.37 Over 50 proteins have been shown to be part of the molecular machinery that generates the clathrin-coated endocytic vesicles. We found as many as 46 plasma proteins involved this pathway to be significantly associated with plasma ACE2 (S2), consistent with a marked overrepresentation of this canonical pathway relative to the background of measured proteins. The mechanisms behind the pronounced relationship between ACE2 and clathrin-mediated endocytosis are unclear. ACE2 itself may undergo internalization in response to stimuli such as Ang-II; in addition, pathways downstream of ACE2 may be related to clathrin-mediated endocytosis. For instance, Ang (1–7) has been shown to trigger Mas receptor-mediated clathrin-dependent internalization.38
The retrieval and recycling of internalized cargo proteins are known to play critical roles in important human diseases, particularly neurodegenerative diseases and viral infections. The entry of enveloped viruses into cells is occurs via 2 main mechanisms: some viruses deliver their genomes to the cytoplasm after fusion of their envelopes with the plasma membrane at the cell surface, whereas others utilize the host cell’s endocytic machinery.39 Interestingly, ACE2 expression is reduced after SARS-CoV infection, suggesting a regulated internalization mechanism. To our knowledge, the main cellular mechanism of viral entry of SARS-CoV2 has not yet been firmly established 39, 40. The relevance of these findings, derived from a HF population, to SARS-CoV2 infection, is unknown.
We also found prominent relationships between plasma ACE2 and actin cytoskeleton signaling, mechanisms of viral exit from host cells, and the protein ubiquitination pathway. Following endocytosis, internalized cargos first enter the early endosomes where the cargos are sorted for either recycling to the plasma membrane (secretory pathway), or for lysosomal degradation through delivery to late endosomes. In a mouse model of Ang-II-mediated hypertension, Ang-II led to AT1R-dependent internalization of ACE2, followed by lysosomal degradation. 26 ACE2 ubiquitination, a post-translational modification essential for lysosomal targeting and degradation, was found to be involved in this process. However, the precise mechanisms that regulate the cellular trafficking of internalized ACE2 remain incompletely characterized and require further research. These pathways are also important in viral cellular replication. Actin signaling has been shown to participate in the cycle of a wide variety of viruses.41 Similarly, endosomal sorting complex required for transport (ESCRT)-dependent membrane dynamics are known to be highjacked by multiple viruses to support their propagation and exit from cells.42 We stress that we studied a general HF population, and therefore the role of these mechanisms in the setting populations with viral infections (including SARS-CoV and SARS-CoV-2) is unknown.
We also observed a relationship of plasma ACE2 levels with EIF2 signaling and mTOR signaling. EIF2 signaling is involved in interferon-mediated inflammation/innate immunity and endoplasmic reticulum stress response in the heart. Protein kinase R-like endoplasmic reticulum kinase (PERK), a EIF2α kinase, is upregulated in heart failure and has been demonstrated to be important in protecting against pressure overload-induced heart failure in experimental studies 43. This pathway is also important in SARS-CoV viral infection 44, 45. In an in vitro study, Krahling et al. demonstrated that ACE2-mediated SARS-CoV cellular infection results in activation of EIF2α kinase protein kinase R and PERK as well as sustained inactivation of EIF2α 44, which promotes SARS-CoV viral replication. However, there is also evidence that coronaviruses prevent activation of protein kinase activated by double-stranded RNA (PKR, a kinase that phosphorylates EF2α), which may lead to evasion of early innate and intrinsic antiviral host cell responses.46
The source of plasma ACE2 protein levels is an important issue requiring further research. ACE2 is widely expressed in the lungs, kidney, heart, gastrointestinal tract and endothelium. Its plasma concentrations are likely the result of transcriptional regulation and post-translational regulation, including shedding from the cell surface. The latter appears to be an important aspect of ACE2 biology, regulated by A disintegrin and metalloproteinase 17 (ADAM17). 26 However, the physiologic role of soluble ACE2 in healthy subjects or in patients with chronic disease is not well-defined, and the mechanisms regulating shedding of plasma-bound ACE2 are not well understood. Membrane-bound cardiac ACE2 may be released into the plasma as a counter-regulatory response to Ang II activity 47 and it has been suggested to be depleted in more advanced heart failure, 48 which may explain our finding of lower plasma ACE2 levels in patients with NYHA class 3–4. As pointed out before, another important mechanism of ACE2 regulation may be internalization and lisosomal degradation, in response to stimuli such as Ang-II.26 A better understanding of the physiologic interactions between these cellular processes and regarding the physiologic role of circulating ACE2 vs. plasma-bound ACE2 is needed, which will require further research in experimental models and human populations.
It is critical to emphasize that our findings apply only to ACE2 protein levels, which are not equivalent to soluble ACE2 activity. Previous studies have assessed soluble ACE2 activity in HF. Epelman demonstrated that among adults with HF, plasma sACE2 activity is elevated in increased relative to healthy controls (in both ischemic and non-ischemic etiology) and correlates with worsening left ventricular ejection fraction, functional class, and greater BNP levels49, right ventricular systolic dysfunction, a higher estimated pulmonary artery systolic pressure and adverse outcomes during follow-up.50 Interestingly, in patients with acute decompensated heart failure, Shao et al demonstrated that increasing serum sACE2 activity levels during intensive medical therapy predict improved outcomes independently from underlying cardiac indices.51 Soluble is also increased in the acute phase of St-elevation myocardial infarction, and correlates with the extent of jeopardized myocardium and infarct size moreover, soluble ACE2 activity measured acutely seemed to be an independent predictor of late LV systolic function and it is also associated with the occurrence of adverse remodeling.52 Clearly, further research regarding the physiologic and clinical significance of plasma ACE2 activity, plasma ACE2 protein levels and their relationship to plasma-bound ACE2 are required.
Our study should be interpreted in the context of its strengths and limitations. Strengths of our study included our large sample size, the prospective enrollment with strict case adjudication, and the use of state-of-the-art proteomics analytic and analytic approaches. Our proteome-wide association study using >5,000 proteins, rather than more targeted approaches, minimizes biases in the discovery of biologic pathways associated with ACE2. Our study also had several limitations. We did not measure ACE2 gene expression in specific tissues or membrane bound ACE2. Moreover, whereas the significant overrepresentation of proteins correlated with ACE2 in these pathways suggests a biologic relationship, the causal relationship with ACE2 activation remains to be determined in mechanistic studies. Moreover, several proteins in the analyzed pathways were intracellular. It is not uncommon to find proteins that are not predicted to be secreted in plasma samples. This may happen as a result of normal cell turnover, disease processes that result in release of cellular content, soluble alternative splice forms, and cellular lysis in a sample. Therefore, relationships with cellular pathways require validation in mechanistic studies. Although our aptamer-based measurement of ACE2 was validating with the pull-down assay (as detailed in the supplemental section), we did not compare the performance of this methods against various other available methods. Future studies should assess ACE2 protein levels measured by various methods (including ELISA), as well as ACE2 activity. Our proteomics analyses were made with aptamers, which have advantages and limitations. Reproduction of our findings in future studies using alternative techniques to characterize proteomic patterns in relation to plasma ACE2 would be beneficial. Our study did not include data from healthy controls and our findings therefore applies only to HF patients. The addition of control samples without HF was lacking and would have enhanced the interpretation of our findings; comparisons of proteomic signatures associated with ACE2 in patients with vs. without HF, which should be the focus of future studies. Our sample collection was not standardized in terms of time of the day or relationship with meals, which can alter the plasma proteome, and it is possible that variables delays in the processing sampling and/or various medications may have confounded our findings. Finally, we emphasize that our study assessed the correlation of the plasma proteome with plasma ACE2 in a HF population, rather than a COVID-19 population or the general population. Therefore, our findings should not be extrapolated to COVID-19, and the potential relevance of the observed relationships and their directionality in the setting of COVID-19 should not be overinterpreted. However, to the degree that physiologic processes related to ACE2 may also be utilized by ACE2-mediated viral entry into cells, our findings should stimulate research regarding the relevance of these cellular pathways to SARS-CoV-2 infection.
Perspectives
Our study represents the first assessment of the clinical and plasma proteomic correlates of ACE2 in a human population. We demonstrate that, in a large cohort of patients with HF, plasma ACE2 was prominently associated with multiple cellular pathways involved in cellular endocytosis, exocytosis, and intracellular protein trafficking. Although these pathways are potentially relevant for cardiovascular disease and are known to be involved in various viral infections, whether they have a causal relationship with ACE2 or are relevant to novel coronavirus-2 infection remains to be assessed.
Supplementary Material
{kind=link}
Figure 1. Key clinical and proteomic correlates of plasma ACE2.
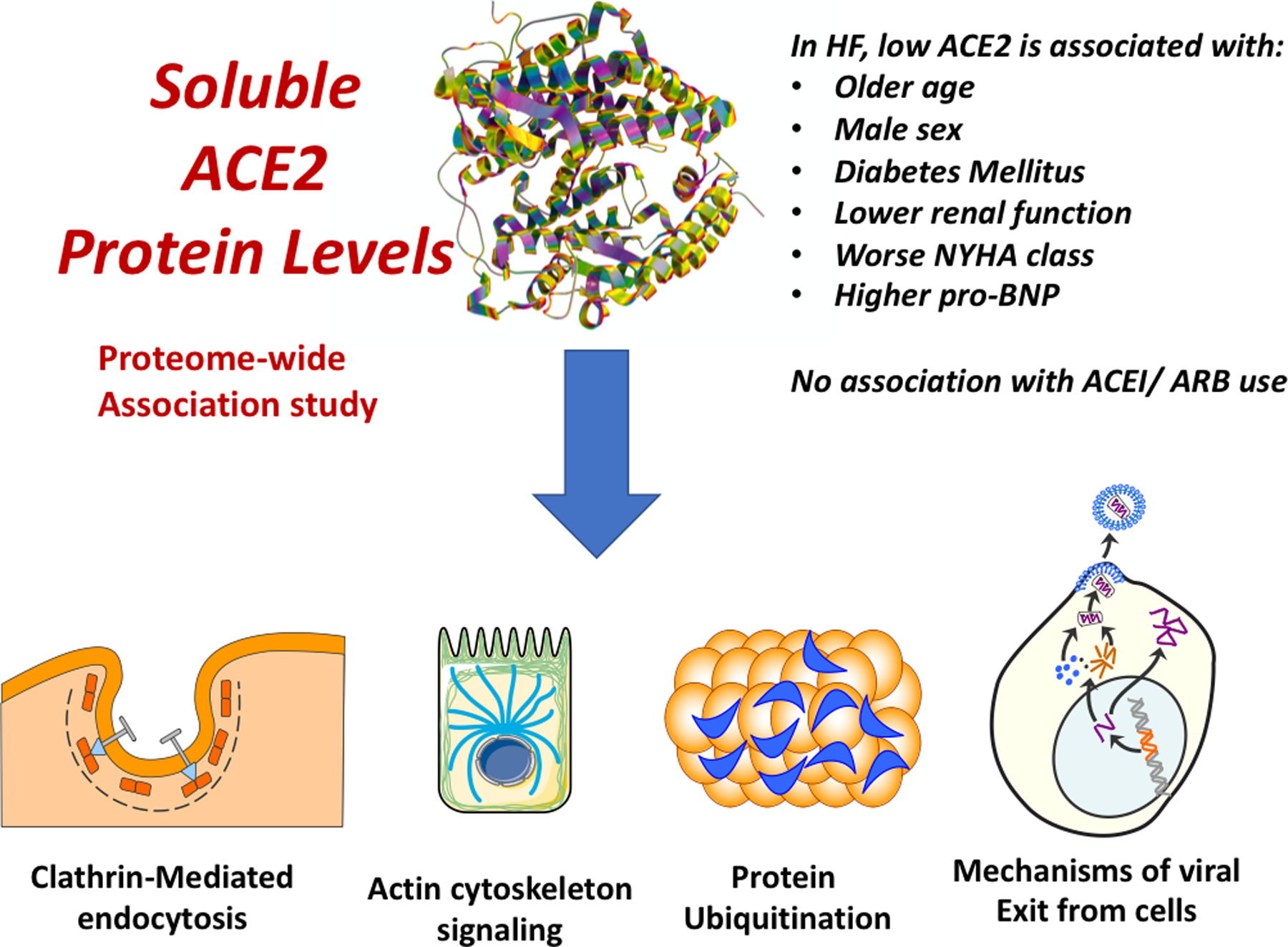
Lower plasma ACE2 was associated with various clinical factors, including older age, male sex, diabetes mellitus, a lower estimated glomerular filtration rate, worse New York Heart Association (NYHA) class and higher pro-BNP levels, but were not associated with angiotensin-converting enzyme inhibitor (ACEI) and/or angiotensin-receptor blocker (ARB) use. In a broad proteome-wide association study, plasma ACE2 was associated with various canonical pathways, including clathrin-mediated endocytosis signaling, actin cytoskeleton signaling, mechanisms of viral exit from host cells, the protein ubiquitination pathway.
Novelty and Significance.
What is new?
Despite its biologic importance, little is known about the clinical and biologic correlates of circulating soluble ACE2 levels in humans.
We assessed the clinical and proteomic correlates of plasma (soluble) ACE2 protein levels among 2,248 participants with heart failure and performed an association study of ACE2 against ~5000 other plasma proteins.
What is relevant?
Plasma ACE2 protein levels are prominently associated with multiple cellular pathways involved in cellular endocytosis, exocytosis, and intracellular protein trafficking.
Plasma ACE2 was not associated with angiotensin-converting enzyme inhibitor and/or angiotensin-receptor blocker use.
Summary
We describe novel proteomics associations of plasma soluble ACE2 in human heart failure.
Acknowledgements
Terrye Delmonte, Karl Kammerhoff, Sheri Wilcox and Erik S. Zimmerman are acknowledged for their excellent technical support.
Sources of Funding
This study was funded by a research grant from Bristol Myers Squibb (JAC).
J.A.C. is supported by NIH grants R01-HL 121510-01A1, R61-HL-146390, R01-AG058969, 1R01-HL104106, P01-HL094307, R03-HL146874-01 and R56-HL136730. He has received consulting honoraria from Sanifit, Microsoft, Fukuda-Denshi, Bristol Myers Squibb, OPKO Healthcare, Ironwood Pharmaceuticals, Pfizer, Akros Pharma, Merck, Edwards Lifesciences, Bayer and JNJ. He has received research grants from the NIH, Microsoft, Fukuda-Denshi and Bristol Myers Squibb. He is named as inventor in an UPenn patent for the use of inorganic nitrates/nitrites for the treatment of HF and Preserved Ejection Fraction. P.Z. has consulted for Vyaire. TC is supported by R61HL146390 and R01HL141232. JBC is supported by K23-HL133843 and R01HL153646. Other authors have nothing to disclose.
References
- 1.Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB and Oudit GY. Angiotensin Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ Res 2020;126:1456–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020;181:271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monteil V, Kwon H, Prado P, et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020;181:905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, Gramberg T and Pohlmann S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun 2004;319:1216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basuray A, French B, Ky B, Vorovich E, Olt C, Sweitzer NK, Cappola TP and Fang JC. Heart failure with recovered ejection fraction: clinical description, biomarkers, and outcomes. Circulation 2014;129:2380–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ky B, French B, Ruparel K, Sweitzer NK, Fang JC, Levy WC, Sawyer DB and Cappola TP. The vascular marker soluble fms-like tyrosine kinase 1 is associated with disease severity and adverse outcomes in chronic heart failure. J Am Coll Cardiol 2011;58:386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ky B, Kimmel SE, Safa RN, Putt ME, Sweitzer NK, Fang JC, Sawyer DB and Cappola TP. Neuregulin-1 beta is associated with disease severity and adverse outcomes in chronic heart failure. Circulation 2009;120:310–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lakshmi Kannan ARC. Thyroid Dysfunction in Heart Failure and Cardiovascular Outcomes. Circ Heart Fail 2018;11. [DOI] [PMC free article] [PubMed]
- 10.Emilsson V, Ilkov M, Lamb JR, et al. Co-regulatory networks of human serum proteins link genetics to disease. Science 2018;361:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brody E, Gold L, Mehan M, Ostroff R, Rohloff J, Walker J and Zichi D. Life’s simple measures: unlocking the proteome. J Mol Biol 2012;422:595–606. [DOI] [PubMed] [Google Scholar]
- 12.Gold L, Ayers D, Bertino J, et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 2010;5:e15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganz P, Heidecker B, Hveem K, Jonasson C, Kato S, Segal MR, Sterling DG and Williams SA. Development and Validation of a Protein-Based Risk Score for Cardiovascular Outcomes Among Patients With Stable Coronary Heart Disease. JAMA 2016;315:2532–41. [DOI] [PubMed] [Google Scholar]
- 14.Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature 2018;558:73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao X, Starmer J and Martin ER. A multiple testing correction method for genetic association studies using correlated single nucleotide polymorphisms. Genet Epidemiol 2008;32:361–9. [DOI] [PubMed] [Google Scholar]
- 16.Auro K, Joensuu A, Fischer K, et al. A metabolic view on menopause and ageing. Nat Commun 2014;5:4708. [DOI] [PubMed] [Google Scholar]
- 17.Tromp J, Khan MA, Klip IT, et al. Biomarker Profiles in Heart Failure Patients With Preserved and Reduced Ejection Fraction. J Am Heart Assoc 2017;6. [DOI] [PMC free article] [PubMed]
- 18.Javaheri A, Allegood JC, Cowart LA and Chirinos JA. Circulating Ceramide 16:0 in Heart Failure With Preserved Ejection Fraction. J Am Coll Cardiol 2020;75:2273–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chirinos JA, Zhao L, Jia Y, et al. Reduced Apolipoprotein M and Adverse Outcomes Across the Spectrum of Human Heart Failure. Circulation 2020;141:1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini YHY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B 1995:11.
- 21.Hashimoto T, Perlot T, Rehman A, et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012;487:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zheng Z, Peng F, Xu B, et al. Risk factors of critical & mortal COVID-19 cases: A systematic literature review and meta-analysis. J Infect 2020;81:e16–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G and Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 2000;275:33238–43. [DOI] [PubMed] [Google Scholar]
- 24.Rice GI, Thomas DA, Grant PJ, Turner AJ and Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 2004;383:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koka V, Huang XR, Chung AC, Wang W, Truong LD and Lan HY. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol 2008;172:1174–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deshotels MR, Xia H, Sriramula S, Lazartigues E and Filipeanu CM. Angiotensin II mediates angiotensin converting enzyme type 2 internalization and degradation through an angiotensin II type I receptor-dependent mechanism. Hypertension 2014;64:1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI and Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005;111:2605–10. [DOI] [PubMed] [Google Scholar]
- 28.Ocaranza MP, Godoy I, Jalil JE, et al. Enalapril attenuates downregulation of Angiotensin-converting enzyme 2 in the late phase of ventricular dysfunction in myocardial infarcted rat. Hypertension 2006;48:572–8. [DOI] [PubMed] [Google Scholar]
- 29.Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB and Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension 2004;43:970–6. [DOI] [PubMed] [Google Scholar]
- 30.Soler MJ, Ye M, Wysocki J, William J, Lloveras J and Batlle D. Localization of ACE2 in the renal vasculature: amplification by angiotensin II type 1 receptor blockade using telmisartan. Am J Physiol Renal Physiol 2009;296:F398–405. [DOI] [PubMed] [Google Scholar]
- 31.Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J 2005;26:369–75; discussion 322–4. [DOI] [PubMed] [Google Scholar]
- 32.Burchill LJ, Velkoska E, Dean RG, Griggs K, Patel SK and Burrell LM. Combination renin-angiotensin system blockade and angiotensin-converting enzyme 2 in experimental myocardial infarction: implications for future therapeutic directions. Clin Sci (Lond) 2012;123:649–58. [DOI] [PubMed] [Google Scholar]
- 33.Walters TE, Kalman JM, Patel SK, Mearns M, Velkoska E and Burrell LM. Angiotensin converting enzyme 2 activity and human atrial fibrillation: increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017;19:1280–1287. [DOI] [PubMed] [Google Scholar]
- 34.Ramchand J, Patel SK, Srivastava PM, Farouque O and Burrell LM. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS One 2018;13:e0198144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sama IE, Ravera A, Santema BT, et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin-angiotensin-aldosterone inhibitors. Eur Heart J 2020;41:1810–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sparks MA, South A, Welling P, et al. Sound Science before Quick Judgement Regarding RAS Blockade in COVID-19. Clin J Am Soc Nephrol 2020;15:714–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaksonen M and Roux A. Mechanisms of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 2018;19:313–326. [DOI] [PubMed] [Google Scholar]
- 38.Gironacci MM, Adamo HP, Corradi G, Santos RA, Ortiz P and Carretero OA. Angiotensin (1–7) induces MAS receptor internalization. Hypertension 2011;58:176–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H, Yang P, Liu K, Guo F, Zhang Y, Zhang G and Jiang C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res 2008;18:290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Inoue Y, Tanaka N, Tanaka Y, Inoue S, Morita K, Zhuang M, Hattori T and Sugamura K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J Virol 2007;81:8722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor MP, Koyuncu OO and Enquist LW. Subversion of the actin cytoskeleton during viral infection. Nat Rev Microbiol 2011;9:427–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vietri M, Radulovic M and Stenmark H. The many functions of ESCRTs. Nat Rev Mol Cell Biol 2020;21:25–42. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Kwak D, Lu Z, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension 2014;64:738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Krahling V, Stein DA, Spiegel M, Weber F and Muhlberger E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J Virol 2009;83:2298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minakshi R, Padhan K, Rani M, Khan N, Ahmad F and Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One 2009;4:e8342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kindler E, Gil-Cruz C, Spanier J, et al. Early endonuclease-mediated evasion of RNA sensing ensures efficient coronavirus replication. PLoS Pathog 2017;13:e1006195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danilczyk U and Penninger JM. Angiotensin-converting enzyme II in the heart and the kidney. Circ Res 2006;98:463–71. [DOI] [PubMed] [Google Scholar]
- 48.Kassiri Z, Zhong J, Guo D, Basu R, Wang X, Liu PP, Scholey JW, Penninger JM and Oudit GY. Loss of angiotensin-converting enzyme 2 accelerates maladaptive left ventricular remodeling in response to myocardial infarction. Circ Heart Fail 2009;2:446–55. [DOI] [PubMed] [Google Scholar]
- 49.Epelman S, Tang WH, Chen SY, Van Lente F, Francis GS and Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol 2008;52:750–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Epelman S, Shrestha K, Troughton RW, Francis GS, Sen S, Klein AL and Tang WH. Soluble angiotensin-converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail 2009;15:565–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shao Z, Shrestha K, Borowski AG, Kennedy DJ, Epelman S, Thomas JD and Tang WH. Increasing serum soluble angiotensin-converting enzyme 2 activity after intensive medical therapy is associated with better prognosis in acute decompensated heart failure. J Card Fail 2013;19:605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ortiz-Perez JT, Riera M, Bosch X, De Caralt TM, Perea RJ, Pascual J and Soler MJ. Role of circulating angiotensin converting enzyme 2 in left ventricular remodeling following myocardial infarction: a prospective controlled study. PLoS One 2013;8:e61695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.