

Abstract
The DELLA genes, also known as ‘Green Revolution’ genes, encode conserved master growth regulators in controlling plant development in response to internal and environmental cues. Functioning as nuclear-localized transcription regulators, DELLAs modulate expression of target genes via direct protein-protein interaction of their C-terminal GRAS domain with hundreds of transcription factors (TFs) and epigenetic regulators. However, the molecular mechanism of DELLA-mediated transcription reprogramming remains unclear. Here, by characterizing new missense alleles of an Arabidopsis DELLA, REPRESSOR OF ga1-3 (RGA) and co-IP assays, we unveil that RGA binds histone H2A via the PFYRE subdomain within its GRAS domain to form TF-RGA-H2A complex at the target chromatin. ChIP-seq analysis further shows that this activity is essential for RGA association with its target chromatin globally. Our results indicate that although DELLAs are recruited to target promoters by binding to TFs via its LHR1 subdomain, DELLA-H2A interaction via its PFYRE subdomain is necessary to stabilize the TF-DELLA-H2A complex at the target chromatin. This study provides novel insights into the two distinct key modular functions in DELLA for its genome-wide transcription regulation in plants.
INTRODUCTION
The DELLA genes are also known as ‘Green Revolution’ genes because of their pivotal role in modulating stature of the high-yielding wheat varieties, which were crucial for the success of ‘Green Revolution’ in the 1960s1–3. The DELLA genes were originally identified by genetic analyses as repressors of the phytohormone gibberellin (GA) signaling in Arabidopsis thaliana4,5. Further studies showed that DELLAs are conserved in all land plants6, and they function as pivotal integrators of multiple signaling pathways to modulate plant growth and development in response to biotic and abiotic cues3,7,8. DELLAs belong to the DELLA subfamily of the plant-specific GRAS family proteins with a conserved C-terminal GRAS domain that confers transcriptional regulator function (Fig. 1a)4,5,9,10. The unique DELLA domain in its N-terminus is required for its response to GA-induced degradation11,12, and this domain is absent in other GRAS family members. Biochemical and structural studies showed that GA triggers a conformational switch in its receptor GID1 to promote GA-GID1-DELLA domain complex formation, which in turn enhances binding of the SCFSLY1/GID2 E3 ubiquitin ligase to the GRAS domain for polyubiquitination and subsequent degradation by the 26S proteasome13–18. ChIP-qPCR and subsequent ChIP-Seq analyses showed that RGA (an AtDELLA) is associated with its target promoters, although it does not contain a canonical DNA binding motif19–21. Extensive studies indicate that DELLA proteins regulate expression of target genes by direct interaction of the GRAS domain with transcription factors/regulators and epigenetic regulators3,7,8. However, the molecular mechanism of DELLA-mediated transcription reprogramming remains unclear.
Figure 1. All missense rga hypomorphs are clustered within the GRAS domain.
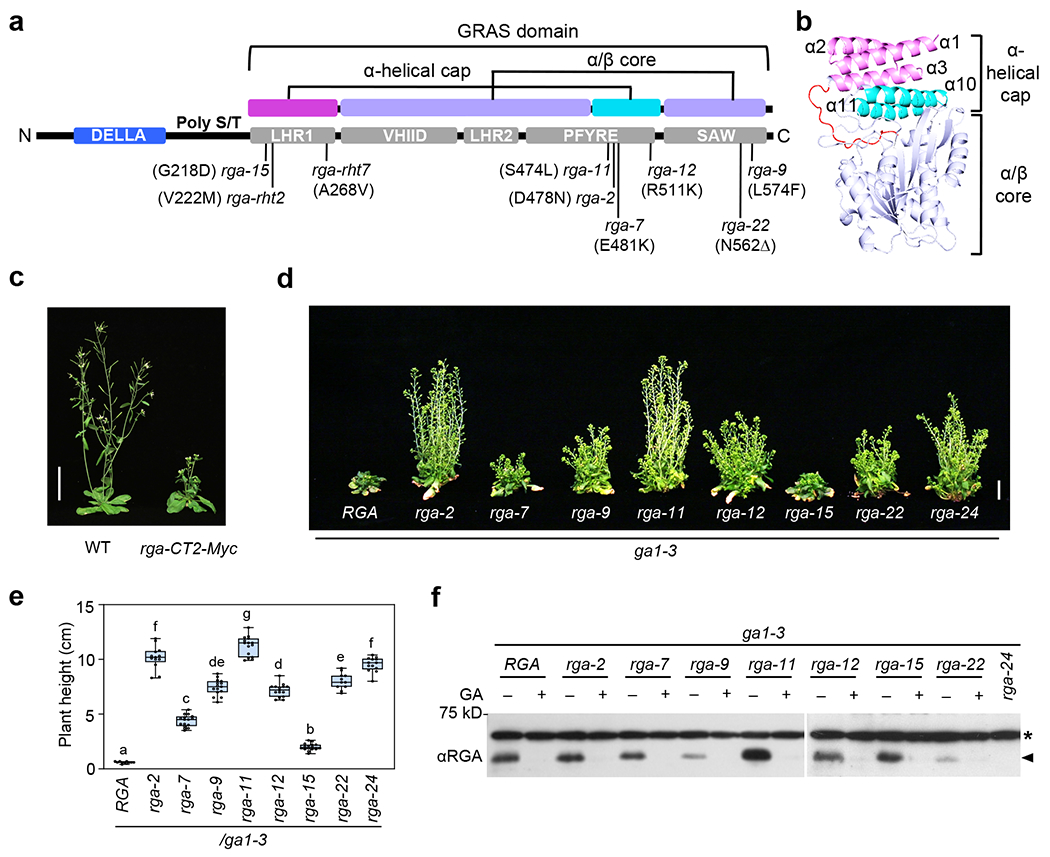
a, Domain map of the RGA protein. The missense rga alleles and corresponding amino acid substitutions or deletion are labeled. The 2 subdomains (α-helical cap and α/β core) of the GRAS domain are color-coded as in b. b, Predicted 3D structure of RGA GRAS domain using SWISS MODEL50,71 with the SCARECROW protein (PDB ID: 5B3G)49 as scaffold. The GRAS domain contains an α-helical cap (α1-α3 in magenta and α10-α11 in cyan), and an α/β core (in purple). c, Expression of PRGA:rga-CT2-Myc (rga-CT2 containing residues 207-58745) in WT Arabidopsis caused a semi-dwarf phenotype. Photo of representative 43-d-old plants in LD. Bar = 5 cm. d-e, Missense rga alleles displayed varying effects on rescuing ga1 dwarf phenotype. In d, representative 70-d-old ga1-3 (with RGA) and ga1 rga mutants as labeled. Bar = 2 cm. In e, Boxplot showing plant heights of different lines as labeled. n⩾11. Center lines and box edges are medians and the lower/upper quartiles, respectively. Whiskers extend to the lowest and highest data points within 1.5× interquartile range (IQR) below and above the lower and upper quartiles, respectively. Different letters above the bars represent significant differences (p < 0.01) as determined by two-tailed Student’s t tests. Exact n and p values are listed in Source Data Fig. 1. f, Missense rga proteins were responsive to GA-induced degradation. Immunoblot contained protein extracted from seedlings that were treated with 1 μM GA4 (+) or mock treated (−) for 1 h. The blot was probed with an anti-RGA antiserum. * Non-specific background band. Representative images of two biological repeats are shown.
Remarkably, a total of 370 potential DELLA-interacting proteins have been identified by yeast two-hybrid (Y2H) screens, and more than 40 of them have been verified with co-IP and/or genetic analyses8,22,23. Most of the DELLA-interacting proteins are transcription factors/regulators. Examples of DELLA-inhibited transcription factors/regulators include bHLH transcription factors, PIFs, in light signaling24,25; auxin signaling activators, AUXIN RESPONSE FACTORs (ARFs)26,27; BRASSINAZOLE-RESISTANT1 (BZR1), a brassinosteriod (BR) signaling activator that contains a noncanonical bHLH domain28; jasmonic acid signaling repressors, JAZs29,30; and Type I TCP (TEOSINTE BRANCHED 1 [TB1], CYCLOIDEA [CYC], and PROLIFERATING CELL FACTOR [PCF]) transcription factors31. DELLA-activated transcription factors/regulators include type-B ARABIDOPSIS RESPONSE REGULATORs in cytokinin signaling21, ABSCISIC ACID INSENSITIVE 3 (ABI3) and ABI5 (a bZIP transcription factor) in ABA signaling32, and INDETERMINATE DOMAIN (IDD) subfamily of C2H2 zinc finger transcription factors33,34. Other DELLA interactors include chromatin-remodeling complexes (SWI/SNF, and a CHD protein PICKLE (PKL))35–37, and subunits of the prefoldin complex for tubulin folding38. These findings indicate that protein-protein interaction with transcription factors/regulators is a major regulatory mechanism in DELLA-modulated plant development. DELLAs function as co-activators or co-repressors, depending on their interacting transcription factors. Current model proposes two distinct modes of DELLA action: (1) DELLA-mediated transcription activation of target genes depends on its recruiting transcription factors (e.g., IDDs), which bind to both DELLA and the target promoter sequences; (2) DELLA alters transcription by blocking DNA binding and hence sequestration of transcription activators (e.g., BZR1, PIFs and TCPs) or repressors (e.g., JAZs) from target promoters7,8.
Previous mutant and transgenic studies indicate that the GRAS domain is required for the growth suppression activity of DELLA proteins. Loss-of-function della missense mutations in several plant species are all located within the GRAS domain4,39–42. The GRAS domain contains five conserved subdomains: Leu Heptad Repeat 1 (LHR1), VHIID, LHR2, PFYRE and SAW (Fig. 1a). Evidence from previous serial deletion studies by Y2H and in vitro pulldown assays suggested that LHR1 is required for protein-protein interactions with many DELLA interactors, although C-terminal truncations of other GRAS subdomains often also abolish these interactions8. These results cannot distinguish between specific defects in the protein interaction motif vs general defects in protein conformation and structural stability. Genetic analysis and alanine scanning mutagenesis further revealed that VHIID and LHR2 are involved in the F-box protein binding43,44. However, the roles of the C-terminal PFYRE and SAW regions are unclear.
To elucidate the molecular mechanism of DELLA function in transcription regulation, it is crucial to decipher the specific roles of its GRAS subdomains. In this report, we characterized the effects of a number of missense rga alleles on plant growth and interaction with transcription factors by Y2H, pulldown and co-IP assays. Surprisingly, missense mutations in the PFYRE subdomain did not significantly affect interaction with transcription factors (BZR1, PIF, TCP14 and IDD3). We further identified a novel function of the PFYRE subdomain for binding to histone H2A. ChIP-seq analysis demonstrated that this activity is crucial for genome-wide RGA association with its target chromatin. Our results indicate that although transcription factors are required to recruit the DELLA protein via its LHR1 subdomain to target gene promoters, DELLA-H2A interaction via its PFYRE subdomain is necessary to stabilize H2A-DELLA-TF complex at the target chromatin site.
RESULTS
RGA’s PFYRE subdomain plays a key role in growth suppression
We directly examined the GRAS function of an AtDELLA RGA by expression of the RGA GRAS domain under the control of RGA promoter (PRGA:rga-CT2-Myc) in transgenic Arabidopsis45. PRGA:rga-CT2-Myc led to a semi-dwarf phenotype (Fig. 1c) that does not respond to GA treatment (Supplementary Fig. 1), supporting that the GRAS domain of DELLA alone is sufficient to confer growth suppression. Among the five conserved subdomains in the GRAS domain, LHR1 interacts with TFs8, and VHIID and LHR2 are required for F-box protein binding43,44. To elucidate the specific functions of the PFYRE and SAW subdomains at the C-terminus of the GRAS domain in RGA, we sequenced a large collection of rga mutants, which we have isolated in previous ga1-3 suppressor mutant screens (Supplementary Table 1)4,46. Among the 27 rga mutants, we found eight nonsense rga mutations, which are distributed throughout the RGA coding sequence. In contrast, all single-amino-acid mutations (seven total) are located within the GRAS domain: one missense mutation (rga-15) is located in LHR1; four missense mutations are clustered within PFYRE; and two mutations (a deletion and a missense mutation) are in SAW (Fig. 1a and Supplementary Table 1). These mutations conferred varying degrees of suppression of the GA-deficient dwarf phenotype of ga1-3 (Fig. 1d,e). Among them, rga-2 and rga-11 (PFYRE mutations) displayed the strongest phenotypes that are similar to the null allele rga-24, suggesting that the PFYRE subdomain plays a key role in RGA function. All of these rga proteins remained responsive to GA-induced degradation (Fig. 1f), indicating that they are able to interact with GID1 and SLY1.
To examine the activity of these rga alleles on target gene expression, a dual luciferase (LUC) assay47 was carried out using the transient expression system in Nicotiana benthamiana. The PSCL3:firefly LUC (fLUC) was used as the reporter for this assay because SCL3 is a direct target gene of DELLA, and its transcription is induced by DELLA19,48. 35S:Renilla LUC (rLUC) was the internal control to normalize variations in transformation efficiency. The effectors included 35S:RGA and 35S:rga constructs. As expected, when co-expressed with 35S:RGA, PSCL3:fLUC expression was induced about eight-fold in comparison to the negative control (with the empty effector construct) (Fig. 2a). The rga mutants showed reduced transactivation activity (Fig. 2a and Supplementary Fig. 2a), which correlated with the corresponding mutant phenotypes (Fig. 1d,e). RT-qPCR analysis further indicated that mRNA levels of RGA-induced genes (SCL3 and GID1B) were reduced in rga-2 and rga-11, similar to rga-24, in comparison to WT (Fig. 2b).
Figure 2. Missense rga mutant proteins showed varying degrees of reduced activities.
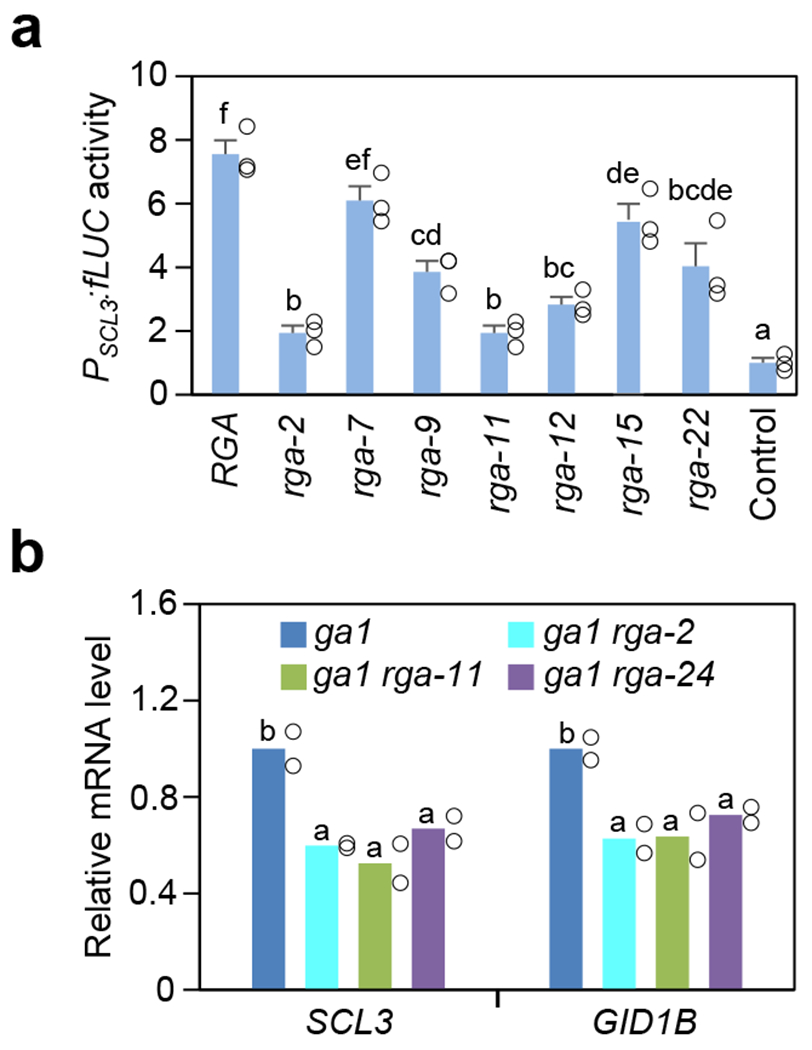
a, Dual luciferase assay in the N. benthamiana transient expression system showing rga mutant proteins were impaired in activating PSCL3:fLUC in comparison to RGA. Means ± SE of three biological replicates are shown. The reporter construct contained PSCL3:fLUC48. Effector constructs contained 35S:RGA or rga as labeled, and the empty vector was included as a negative control. RGA and rga proteins were expressed at similar levels in these assays (Supplementary Fig. 2a). b, RT-qPCR analysis showing rga-2 and rga-11 caused reduced expression of RGA-induced genes (SCL3 and GID1B) in planta, similar to the null rga-24 allele. The housekeeping gene, PP2A, was used to normalize different samples. Means of two biological replicates are shown. The level in ga1-3 was set to 1. In a and b, statistical analyses were performed with two-tailed (a) or one-tailed (b) Student’s t tests. Different letters above the bars represent significant differences, p < 0.05. Exact n and p values are listed in Source Data Fig. 2.
To understand the structure-function relationship of the DELLA GRAS domain, we generated the 3D-structure model of the RGA GRAS domain based on the crystal structure of another Arabidopsis GRAS protein, SCARECROW (SCR)49 using the online SWISS-MODEL Workspace (http://swissmodel.expasy.org/)50 (Fig. 1b). The predicted RGA GRAS domain contains one α/β core subdomain with an α-helical cap. The α-helical cap consists of five α-helixes: α1 – α3 (corresponding to LHR1) and α10 – α11 (a part of PFYRE subdomain). The remaining GRAS sequence forms the α/β core. Three missense mutations (rga-2, rga-11 and rga-7) within the PFYRE subdomain of RGA are located in the predicted α10 in the α-helical cap, suggesting that this region plays an important role in the growth suppression activity of RGA.
LHR1 subdomain, but not PFYRE, is required for TF binding
To investigate the molecular function of the PFYRE subdomain, we first tested whether rga-2 and rga-11 (containing PFYRE mutations) are impaired in binding to four DELLA-interacting transcription factors (TFs), BZR1, PIF3, TCP14 and IDD3. Surprisingly, these mutations only reduced IDD3 interaction, but did not affect binding with BZR1, PIF3 or TCP14 by Y2H assay (Fig. 3a and Supplementary Fig. 2b). To decipher the functional defect of rga-2 and rga-11 further, we generated transgenic Arabidopsis carrying PRGA:FLAG-RGA, FLAG-(rga-2) or FLAG-(rga-11) fusion genes, separately, in the ga1-3 della pentuple (ga1 dP) background. In ga1 dP, all five Arabidopsis DELLA genes (RGA, GA-INSENSITIVE (GAI), RGA-LIKE1 (RGL1), RGL2 and RGL3) were knocked out37. As expected, PRGA:FLAG-RGA restored the dwarf phenotype in the ga1 dP background (Fig. 3b,c), because RGA plays a major role in repressing vegetative growth51,52. FLAG-(rga-2) and FLAG-(rga-11) were inactive as they did not suppress growth of ga1 dP (Fig. 3b,c and Supplementary Fig. 3a,b), although the FLAG-(rga-2) and FLAG-(rga-11) protein levels in these lines were similar to the FLAG-RGA in the FLAG-RGA line (Fig. 3d). In vitro pulldown assays were performed with recombinant GST-tagged BZR1 and PIF3 expressed in E. coli, and protein extracts from transgenic Arabidopsis expressing FLAG-RGA or FLAG-rga (Fig. 4a, Supplementary Fig. 4a). Consistent with the Y2H results, FLAG-(rga-2) and FLAG-(rga-11) showed similar binding affinity to GST-BZR1/PIF3 as that of FLAG-RGA. GST-IDD3 was insoluble in E. coli. To examine the effect of rga mutations on IDD3 binding, in vitro pulldown assays were performed using recombinant Maltose-binding protein (MBP)-RGA/rga-2/rga-11 and protein extracts from N. benthamiana expressing FLAG-IDD3. We also included FLAG-PIF4 in this assay. MBP-(rga-2) and MBP-(rga-11) showed similar binding affinity to FLAG-IDD3 and FLAG-PIF4 as that of FLAG-RGA (Supplementary Fig. 4b). These results strongly support that these mutations in the PFYRE subdomain do not significantly affect RGA binding to BZR1, PIFs, IDD3 or TCP14, and indicate that PFYRE may have an unidentified novel role for RGA activity.
Figure 3. Y2H assay showed rga mutations in the LHR1 subdomain reduced interaction with BZR1, PIF3 and TCP14, whereas rga-2 and rga-11 in PFYRE did not.
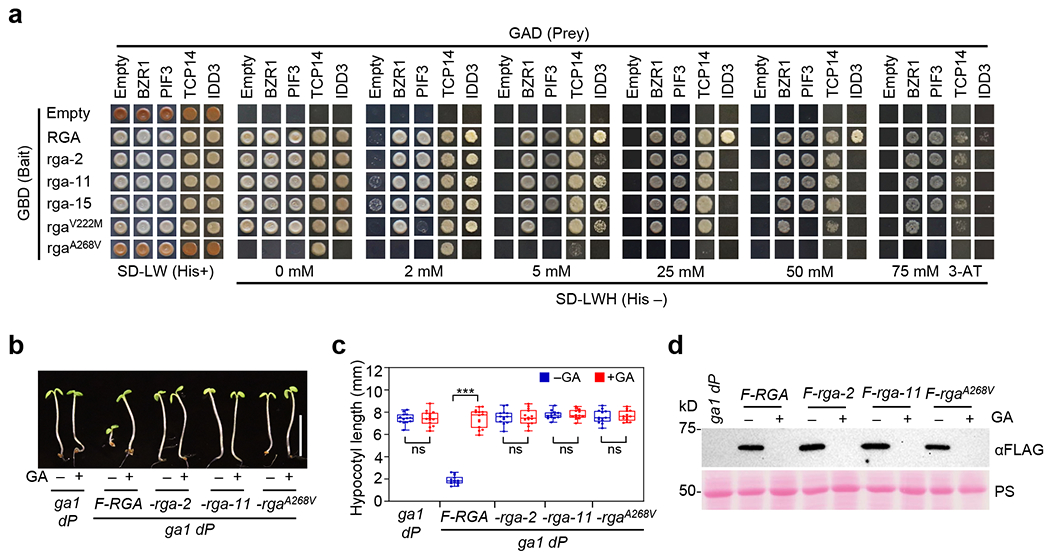
a, Y2H assay showing that rga-2 and rga-11 interacted with BZR1, PIF3 and TCP14 similarly to WT RGA, but rgaV222M reduced binding to PIF3 and rgaA268V abolished binding to BZR1, PIF3, TCP14, and IDD3. The bait constructs expressed truncated RGA/rga (amino acid residues 107-587). The strength of interaction was indicated by the ability of cells to grow on His– plates with 0 –75 mM 3-AT. The lower amounts of rga-2 protein expressed in yeast cells may contribute to the reduced growth of rga-2 + IDD3 in comparison to other rga proteins (Supplementary Fig. 2b). Representative images of three biological repeats are shown. b-c, FLAG-RGA suppressed hypocotyl growth of ga1 dP, whereas FLAG-(rga-2) and FLAG-(rga-11) did not. Transgenic seedlings containing PRGA:FLAG-RGA, FLAG-(rga-2) or FLAG-(rga-11) in the ga1 dP background were grown on media without (−) or with 25 μM GA3 (+) as labeled. In b, representative 5d-old seedlings in short-day conditions. Bar = 5 mm. In c, Boxplot showing hypocotyl lengths of different lines as labeled. n ≥ 13. *** p < 0.001. ns, no significant difference. Statistical analyses were performed with two-tailed Student’s t tests. Center lines and box edges are medians and the lower/upper quartiles, respectively. Whiskers extend to the lowest and highest data points within 1.5× interquartile range (IQR) below and above the lower and upper quartiles, respectively. Exact n and p values are listed in Source Data Fig. 3.
d, the amounts of FLAG-rga proteins were similar to FLAG-RGA in these transgenic lines. Immunoblot contained protein extracts from seedlings grown as in b, and the blot was probed with an anti-FLAG antibody. PS, Ponceau S stained gel blot. Representative image of two biological repeats are shown.
Figure 4. Pulldown and co-IP assays showed rga mutations in the LHR1 subdomain reduced interaction with BZR1, PIF3 and IDD3, whereas rga-2 and rga-11 in PFYRE did not.
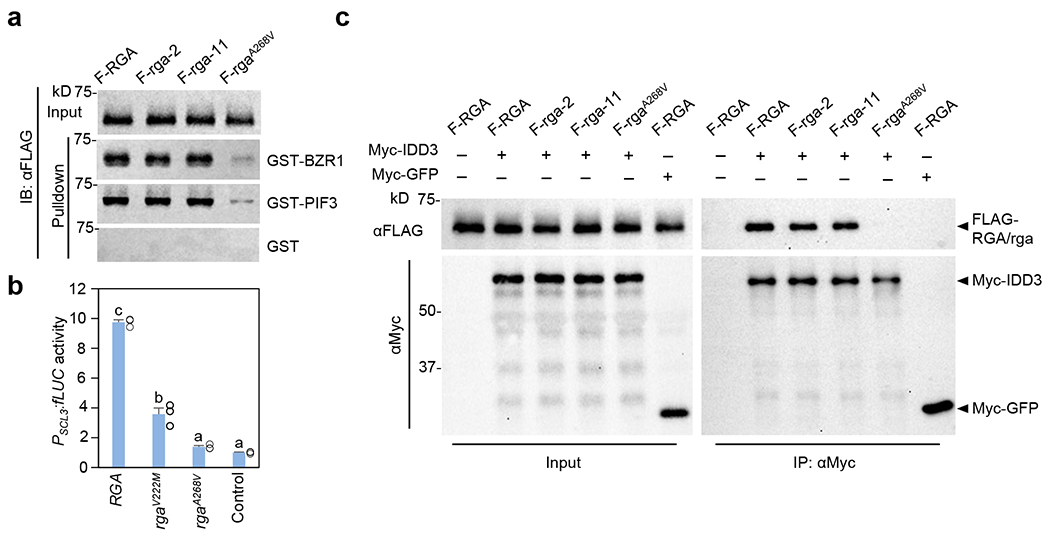
a, In vitro pulldown assay showing much reduced interaction of rgaA268V with BZR1 and PIF3. Recombinant GST, GST-BZR1 and GST-PIF3 bound to glutathione–Sepharose beads were used separately to pull down FLAG-RGA/rga from protein extracts from transgenic Arabidopsis in the ga1 dP background. Immunoblots containing input Arabidopsis extracts and pulldown samples were detected with an anti-FLAG antibody. Ponceau S-stained blots indicated that similar amounts of the GST/GST-fusion proteins were used in each set of the pulldown assays (Supplementary Fig. 4a). Representative images of three biological repeats are shown. b, rgaV222M and rgaA268V showed impaired activation of PSCL3:fLUC. The dual luciferase assay in the N. benthamiana transient expression system was performed as described in Fig. 2a. RGA and rga proteins were expressed at similar levels in these assays (Supplementary Fig. 2c). Means ± SE of three biological replicates are shown. Statistical analyses were performed with two-tailed Student’s t tests. Different letters above the bars represent significant differences, p < 0.01. Exact p values are listed in Source Data Fig. 4. c, Co-IP assay showing rgaA268V was not IP’ed by Myc-IDD3. FLAG-RGA/rga were expressed alone or co-expressed with Myc-IDD3 or Myc-GFP-NLS in N. benthamiana as indicated. An anti-Myc agarose was used for IP, and protein blots were probed with anti-Myc and anti-FLAG antibodies, separately. Representative images of two biological repeats are shown.
Previous studies suggest that the LHR1 subdomain of the DELLA proteins is required for binding many DELLA-interacting proteins (e.g., BZR1, PIFs, TCPs and IDDs). However, this conclusion was based on Y2H and in vitro pulldown assays using serial truncations of DELLA proteins, which complicated the interpretation of results because deletions of the C-terminal GRAS subdomains often also abolish protein-protein interactions8. Thus, we analyzed the role of LHR1 further using missense mutants. Among the rga mutants that we generated, the only rga mutation in the LHR1 subdomain, rga-15 displayed a relatively weak phenotype (Fig. 1d–e and Supplementary Fig. 3c). However, several missense della mutations in conserved residues within LHR1 (α1 and α3) that conferred strong phenotypes were identified in DELLA orthologs in barley (SLENDER1, SLN1) and wheat (Reduced Height, RHT)40. We created 2 new rga alleles within LHR1, rgaV222M and rgaA268V corresponding to rht-2 and rht-7 in wheat, respectively, by site-directed mutagenesis to study the role of LHR1 further. We first tested the activities of these new rga mutant proteins by assaying PSCL3:fLUC reporter expression in the transient expression system in N. benthamiana. The rgaA268V mutation (rht-7) completely abolished RGA-induced SCL3 promoter expression, whereas the rgaV222M mutation (rht-2) showed an intermediate defect in this assay (Fig. 4b and Supplementary Fig. 2c). The rgaA268V mutant protein also failed to interact with BZR1, PIF3 and IDD3 by Y2H assays, showed much weaker binding to TCP14, and abolished the self-activation activity of RGA, possibly by altering RGA interaction with unidentified yeast protein(s) (Fig. 3a). These results support that rgaA268V is a strong allele. To verify the effect of rgaA268V in planta, we generated transgenic Arabidopsis lines that carry PRGA:FLAG-rgaA268V in the ga1 dP background. Indeed, PRGA:FLAG-rgaA268V did not show any growth suppression activity, whereas PRGA:FLAG-RGA restored the dwarf phenotype in the ga1 dP background (Fig. 3b,c and Supplementary Fig. 3a–b). Pulldown assay further showed that FLAG-rgaA268V in the Arabidopsis extracts interacted very weakly with BZR1 and PIF3 in comparison to FLAG-RGA, rga-2, or rga-11 (Fig. 4a and Supplementary Fig. 4a), which is consistent with the Y2H results in Fig. 3a. Similar pulldown assay was not applicable using recombinant IDD3 fusion protein. Therefore, we performed co-IP assay to examine the effect of rgaA268V on IDD3 binding using N. benthamiana that co-expressed Myc-IDD3 and FLAG-RGA/rga (Fig. 4c). Myc-GFP was included as a negative control. FLAG-(rga-2) and FLAG-(rga-11) were co-immunoprecipitated with Myc-IDD3 similarly in comparison to FLAG-RGA, whereas FLAG-rgaA268V was not detectable in the IP’ed sample.
Taken together, the above results provided strong evidence that while both LHR1 and PFYRE subdomains are central for DELLA function, only LHR1 is required for binding to BZR1, PIF3, IDD3 and TCP14.
PFYRE subdomain is required for RGA binding to target chromatin
Although DELLA proteins do not contain canonical DNA-binding motifs, RGA was shown to associate with promoters of its target genes by ChIP-qPCR19. More recently, ChIP-seq analyses identified global RGA binding loci (2327 genes) in the inflorescence meristem using a gain-of-function GFP-rgaΔ17 transgenic line20 (rgaΔ17 contains in-frame deletion of the DELLA motif for GA/GID1-induced degradation12), and in seedlings (~400 genes) using a GFP-RGA transgenic line21. Many of the RGA binding peaks are enriched near cis-elements for TFs. These previous findings support the current model that RGA and other DELLAs are recruited to target promoters via interacting TFs that bind specific cis-elements. As described above, the rga-2 and rga-11 mutations within the PFYRE subdomain abolished the growth suppression activity of RGA in planta, although they did not affect binding of transcription factors BZR1, PIF3, IDD3 or TCP14. To decipher the molecular function of the PFYRE subdomain, we examined whether rga-2 and rga-11 affect RGA association with target chromatin in planta. ChIP-qPCR analysis was performed using transgenic Arabidopsis lines carrying PRGA:FLAG-RGA, PRGA:FLAG-(rga-2) or PRGA:FLAG-(rga-11) in the ga1 dP background. Importantly, both FLAG-(rga-2) and FLAG-(rga-11) displayed significantly reduced association with promoters of two known RGA-activated direct target genes, SCL3 and GID1B (Fig. 5a). To determine whether the PFYRE subdomain is essential for RGA association with target chromatin globally, ChIP-seq was performed using transgenic Arabidopsis lines (in the sly1 dP background) carrying PRGA:FLAG-RGA vs. PRGA:FLAG-(rga-11), as well as two negative controls sly1 dQ (quadruple della with RGA) and sly1 dP. Candidate genes were defined as those containing at least one FLAG-RGA binding peak between −3 kb 5’-upstream to 1.5 kb 3’-downstream of the coding sequences, but not in the non-transgenic sly1 dQ controls. High confidence peaks were selected by using the enrichment fold of a known RGA direct target gene GID1B as the cutoff (Supplementary Table 2), and a total of 2,228 genes near 1,558 FLAG-RGA binding peaks were identified (q < 10−3) (Supplementary Table 3). We performed ChIP-qPCR assays and the results further confirmed FLAG-RGA binding to four selected candidate genes, SAUR16, IAA16, GH3.3, and EXP8, which are RGA-repressed genes (Fig. 5a). 631 genes near FLAG-RGA binding peaks (28.3%) were also associated with GFP-rgaΔ17 binding peaks reported previously (2,327 genes near 1,677 peaks; with WT as their control; q < 10−3)20 (Fig. 5b and Supplementary Table 3). The 28.3% overlap is likely due to the differences in tissue types and RGA fusion proteins used for the two studies: young seedlings for FLAG-RGA ChIP-seq, while inflorescence meristems for GFP-rgaΔ17 ChIP-seq. Nevertheless, among the 631 overlapping genes, the two ChIP-seq datasets showed remarkable consistency in RGA vs GFP-rgaΔ17 binding peak positions with 86.1% of the relative peak summit positions within ± 200 bp (Supplementary Table 3). Genome browser images around six RGA target genes showed overlapping FLAG-RGA and GFP-rgaΔ17 binding peaks near these genes (Supplementary Fig. 5a). By binding profile analysis, we found that the majority of FLAG-RGA binding peaks were in promoter regions (Supplementary Fig. 5b), which is consistent with the function of RGA. ChIP-seq using FLAG-(rga-11) identified 196 binding peaks (associated with 313 genes) with only 79 genes that are near RGA binding sites. Strikingly, most of the RGA binding peaks (96.5%) were not detected by the mutant protein FLAG-(rga-11) (Supplementary Table 3, Supplementary Fig. 6a). Compared to the binding of RGA, the genome-wide enrichment over binding peak regions was substantially reduced by rga-11 mutation (Fig. 5c). Genome browser images around six selected RGA target genes showed that rga-11 abolished its binding to these loci (Supplementary Fig. 6b). ChIP-qPCR analysis further showed that like FLAG-(rga-11), FLAG-(rga-2) also abolished binding to all 6 selected target genes (Fig. 5a).
Figure 5. rga mutations in the PFYRE subdomain impaired association with target chromatin globally by ChIP-Seq analysis.
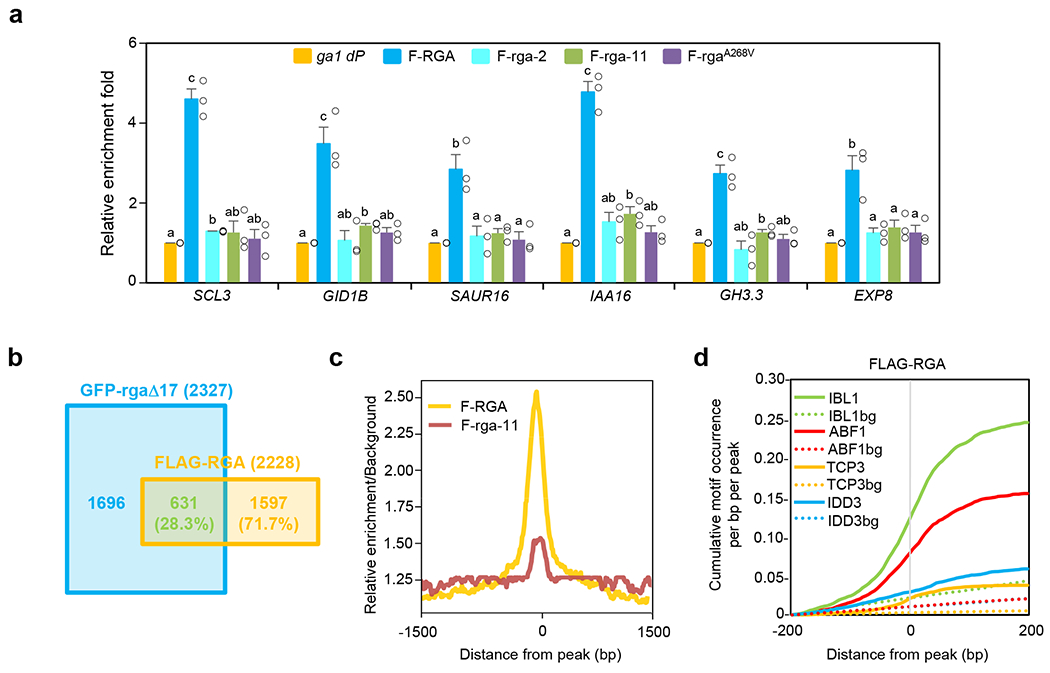
a, ChIP-qPCR analysis of six selected RGA-direct target genes showing rga-2, rga-11 and rgaA268V abolished binding to target chromatin. ChIP was performed using transgenic lines containing PRGA:FLAG-RGA/rga in the ga1 dP background as labeled. The parental line ga1 dP was included as a control. Two RGA-activated genes (SCL3 and GID1B) and four RGA-repressed genes (SAUR16, IAA16, GH3.3 and EXP8) were tested by ChIP-qPCR using primers near the RGA binding peaks. The relative enrichment fold was calculated by normalizing against ChIP-qPCR of non-transgenic ga1 dP control using PP2A. Means ± SE of three biological replicates are shown. Different letters above the bars represent significant differences (p < 0.05) by two-tailed Student’s t-test. Exact p values are listed in Source Data Fig. 5. b, A Venn diagram showing the overlap between genes adjacent to the binding sites of GFP-rgaΔ1720 and FLAG-RGA (current study). ChIP-seq was performed using transgenic lines containing PRGA:FLAG-RGA or PRGA:FLAG-(rga-11) in the sly1 dP background. The sly1 dQ (RGA) and sly1 dP lines were included as controls, respectively. All genes adjacent to a binding peak are listed in Supplementary Table 3. c, Genome-wide relative enrichment over background. The ratios of ChIP-Seq read counts compared to the background were calculated for every 5-nt bins within 1.5Kb of the high-confidence FLAG-RGA peak positions using DeepTools. We plotted the median values of the ratios of FLAG-RGA and FLAG-(rga11), compared to their respective background, for all peak positions. d, Cumulative motif occurrence in the genomic regions from −200bp to +200bp of the FLAG-RGA peak locations. All motifs significantly enriched within 200bp of the peaks (p < 0.01, hypergeometric enrichment test, one-tailed; q < 0.027 after Benjamini-Hochberg correction) are listed in Supplementary Table 4. Among bHLH, bZIP, TCP, and IDD transcription factors, IBL1, ABF1, TCP3, and IDD3, whose binding cis-elements were the most enriched, are shown as representative. Dotted lines indicate background (bg) level of motif occurrence at random genomic locations. Source Data for d are provided in Source Data Fig. 5.
cis-Elements for many TFs were enriched near RGA binding peaks
All cis-elements that were significantly enriched near binding peaks of FLAG-RGA and GFP-rgaΔ17 are listed in Supplementary Table 4. Among them, we found most significant enrichments for cis-elements of members from four TF families (bHLH, bZIP, TCP, and IDD) near both FLAG-RGA binding peaks (Fig. 5d, Supplementary Fig. 5c, and Supplementary Table 4), and GFP-rgaΔ17 binding peaks (Supplementary Table 4). Cis-elements of additional TF families were identified, including C2C2-DOFs, Homeobox, CAMTAs, AP2-EREBPs, WRKYs, MYBs, NACs, SPLs, MADS, ARF, and GRF. All of these TF families, except CAMTAs, were reported to be potential DELLA interactors, although some were previously identified only through Y2H assays23.
Because RGA is rapidly degraded upon GA treatment11, we considered genes showing both differential expression by GA treatment and FLAG-RGA binding as direct targets of RGA. Therefore, we looked for overlapping genes between the genes near the FLAG-RGA binding peaks in this study with GA-responsive genes in a previously published RNA-seq dataset37. We identified 177 and 154 genes where RGA may act as a direct activator (down-regulated by GA, i.e., higher expression with the presence of DELLA) or a repressor (up-regulated by GA), respectively (Fig. 6a, Supplementary Table 3). Almost all of these 331 RGA direct target genes, except 13, were undetectable by ChIP-seq using FLAG-(rga-11) (Supplementary Fig. 6c, Supplementary Table 3). Gene Ontology (GO) terms enriched in direct RGA targets are shown in Supplementary Table 5. RGA-activated target genes include positive regulators of GA signaling (e.g., GA receptors GID1A and GID1B), GA-repressed genes (SCL3, GASA1, GA20OX2, BOI, IDD22), ABA signaling components (e.g., ABI5, PP2C, RHA2B, AtHB6), genes that are responsive to biotic or abiotic stresses (cold, water stresses), and regulation of transcription and RNA metabolism. RGA-repressed target genes are in general involved in growth processes, including auxin metabolism and signaling (e.g., SAURs, IAAs, GH3.6), cell wall organization/biogenesis and cell growth (e.g., EXPs, PMEs, FLAs), cell division, and cell differentiation.
Figure 6. cis-Elements for bHLH, bZIP, TCP and IDD transcription factors were most enriched near RGA binding peaks associated with RGA-direct target genes.
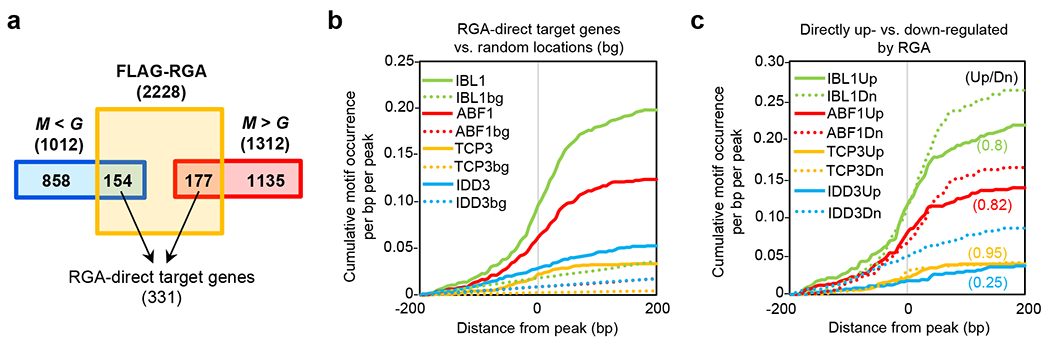
a, Identification of RGA-direct target genes. Genes located near a FLAG-RGA peak that are GA-responsive (based on an RNA-Seq dataset37) were considered as RGA-direct targets (see Methods for detail). M, mock treatment. G, GA treatment. M < G (GA-upregulated genes) and M > G (GA-downregulated genes). RGA acts as ‘direct repressor’ and ‘direct activator’ on 154 and 177 genes, respectively. The list of all RGA-direct target genes is in Supplementary Table 3. b, Cumulative occurrence of representative motifs enriched near FLAG-RGA peaks close to RGA-direct target genes. Binding motifs for representative transcription factors, IBL1 (bHLH), ABF1 (bZIP), TCP3 (TCP), and IDD3 (IDD), are shown as in Fig. 5d. c, Comparison of motif occurrence between FLAG-RGA peaks close to 177 RGA-direct activated genes (Up) and 154 RGA-direct repressed genes (Dn) identified in a. Ratios in parentheses indicate the fold difference between peaks near RGA-direct activated genes (Up) and repressed genes (Dn), after subtracting background (i.e. random locations) cumulative occurrences. Source Data for b and c are provided in Source Data Fig. 6.
FLAG-RGA binding peaks located near the 331 direct target genes of RGA showed enrichment of cis-elements for multiple transcription factors in bHLH, bZIP, TCP, and IDD families (Supplementary Table 4), represented in Fig. 6b, by IBL1, ABF1, TCP3, and IDD3, respectively. Interestingly, while IBL1, ABF1 and TCP3 showed similar motif occurrence patterns between RGA activated and repressed genes, binding motifs for IDD3 were enriched in the FLAG-RGA peaks adjacent to RGA activated genes (Fig. 6c), which is consistent with the previous report showing DELLA acting as a coactivator of IDD333. Several members of these four TF families have been shown to interact with DELLAs22–25,31–34, supporting the idea that RGA is recruited to target chromatin by binding to these TFs.
RGA binding to target genes also requires its LHR1 subdomain
Previous studies reported two distinct modes of DELLA action: (1) DELLA-mediated transcription activation of target genes (e.g., SCL3) requires its recruiting transcription factors (e.g., IDDs), which bind to both DELLA and the target promoter sequences; (2) DELLA alters transcription by sequestration of transcription activators (e.g., BZR1, PIFs and TCPs) or repressors (e.g., JAZs) from target promoters7,8. Based on this model, DELLA proteins should only be associated with chromatin of DELLA-activated genes (first mode of action), but not with DELLA-repressed/or activated genes via the second mode of action. However, our ChIP-seq and ChIP-qPCR results showed that RGA binding peaks are near both RGA-activated and -repressed genes (Fig. 5a, 6a). Similarly, GFP-rgaΔ17 binding peaks reported previously20 are also near RGA-activated and -repressed genes (Supplementary Fig. 5d). Based on these observations, we proposed an alternative model in which RGA is recruited to all its target chromatin by binding to specific TFs, regardless its additive or antagonistic role in transcriptional activation or repression. According to our model, rgaA268V (LHR1 subdomain mutation) that is impaired in binding TFs should also exhibit defect in chromatin binding globally. Indeed, ChIP-qPCR analysis showed that FLAG-rgaA268V failed to bind six selected target promoters, including two RGA-activated and four RGA-repressed genes (Fig. 5a). Our model also predicts RGA and its antagonizing TFs co-localize to target chromatin. We searched for PIF4-induced genes that are associated with both RGA and PIF4 binding peaks using published PIF4 ChIP-seq and RNA-seq datasets53, and found a total of 150 overlapping genes (Supplementary Table 6 and Supplementary Fig. 7a). Among them, we observed a high degree of co-occurrence of RGA and PIF4 binding peaks with 70.7% (106 genes) of the peak summit positions being within ± 100 bp (Supplementary Table 6). Genome browser images around six selected PIF4-induced target genes showed overlapping RGA and PIF4 binding peaks (Supplementary Fig. 7b).
Taken together, mutations in either LHR1 (rgaA268V) or PFYRE subdomain (rga-2 and rga-11) of RGA led to much reduced association with target chromatin. However, only rgaA268V was dramatically impaired in binding of transcription factors BZR1, PIF3, IDD3 or TCP14, whereas the PFYRE subdomain mutations did not significantly affect binding to these TFs. These findings suggest that additional factor(s) are required to stabilize RGA association with target chromatin, presumably after RGA binding to TFs, and α10 within the PFYRE subdomain plays a key role in this interaction.
RGA binding to histone H2A via its PFYRE subdomain
Our study on rga-2 and rga-11 (mutations in α10 within PFYRE subdomain) suggested that besides interacting TFs, RGA association with target chromatin requires additional factors. To test whether RGA directly interacts with histones, in vitro pulldown assays were performed. We found that recombinant GST-RGA was able to pulldown calf thymus histones (histone complexes containing H1, and H2A/H2B/H3/H4 core) (Fig. 7a). We then compared the binding affinity between RGA and individual histone protein by a pulldown assay using FLAG-RGA that was transiently expressed in N. benthamiana, and individual histone proteins expressed in E. coli as MBP-fusion proteins. We found that FLAG-RGA was pulled down strongly by MBP-H2A and weakly by MBP-H4, but not by MBP or other histone fusions (Fig. 7b). In addition to RGA, H2A also pulled down another Arabidopsis DELLA protein, GAI (Fig. 7b). To test whether mutations in the LHR1 (rgaA268V) and PFYRE (rga-2, rga-11) subdomains affect H2A binding, in vitro pulldown assay was performed using GST and GST-H2A, and protein extracts from Arabidopsis expressing FLAG-RGA, or FLAG-rga proteins. Importantly, FLAG-(rga-2) and FLAG-(rga-11), but not FLAG-rgaA268V, showed reduced binding to H2A in comparison to FLAG-RGA (Fig. 7c, Supplementary Fig. 8a). To confirm RGA-H2A interaction in Arabidopsis, co-IP assays were performed. FLAG-RGA in protein extracts from the PRGA:FLAG-RGA/rga transgenic plants were immunoprecipitated using anti-FLAG antibody, and then analyzed by immunoblotting using anti-H2A and anti-FLAG antibodies, separately (Fig. 7d). H2A was co-immunoprecipitated with FLAG-RGA, supporting that H2A and RGA interact in planta. But, H2A was not detected in FLAG-(rga-2) or FLAG-(rga-11) co-IP products, which is consistent with the in vitro pulldown results. In contrast, the interaction between FLAG-rgaA268V and H2A was similar to that of FLAG-RGA (Fig. 7d) These results indicate rga-2 and rga-11 (PFYRE mutations) abolish the interaction between RGA and H2A, which is distinct from the defect of rgaA268V (LHR1 mutation) in binding TFs. Our findings suggest that once TFs recruit RGA via its LHR1 subdomain to target gene promoters, RGA-H2A interaction via its PFYRE subdomain is required to stabilize H2A-RGA-TF complex at the target chromatin. To verify whether RGA binds to both H2A and its interacting TF in a complex, we performed co-IP assays by expressing epitope-tagged RGA, PIF4 and/or H2A in N. benthamiana. Myc-PIF4 was able to pulldown both HA-RGA and FLAG-H2A, whereas Myc-PIF4 did not pulldown FLAG-H2A in the absence of RGA (Fig. 7e). These results support the formation of the PIF4-RGA-H2A complex in planta. Co-IP assays were also performed using epitope-tagged RGA, IDD3 and/or H2A, and similar results were observed that support IDD3-RGA-H2A complex formation in planta (Fig. 7f).
Figure 7. rga-2 and rga-11 reduced RGA binding to histone H2A.
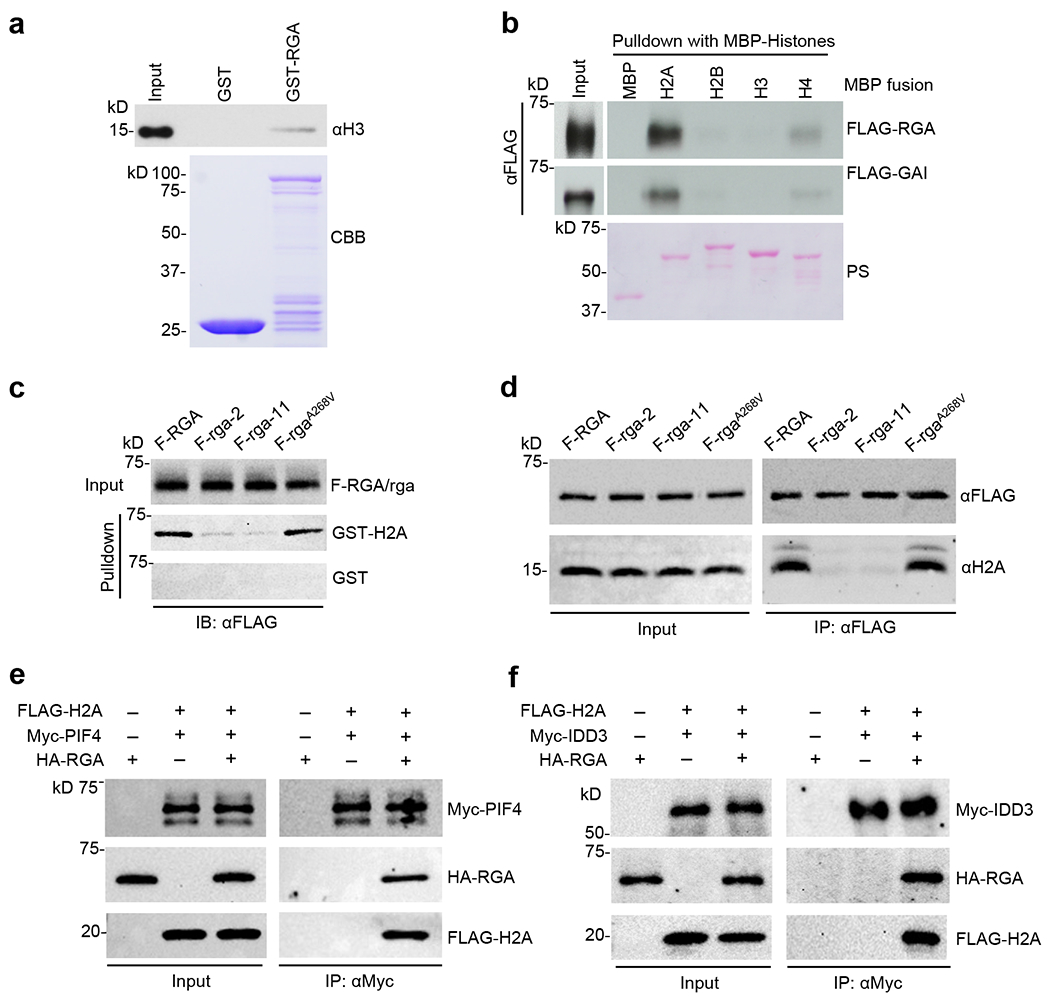
a, GST-RGA pulled down calf thymus histones. GST and GST-RGA bound to glutathione beads were mixed separately with calf thymus nucleosomes. Immunoblot containing the input (0.01 μg calf thymus histones) and pulldown samples was probed with an anti-H3 antibody. Coomassie Brilliant Blue (CBB)-stained gel shows the amounts of GST and GST-RGA used in the pulldown assay. b, MBP-H2A pulled down FLAG-RGA and FLAG-GAI from extracts of N. benthamiana expressing FLAG-RGA and FLAG-GAI. Recombinant MBP and MBP-H2A, -H2B, -H3 and -H4 bound to amylose resin were used separately in the pulldown assay. Ponceau staining showed MBP and MBP fusion proteins. c, GST-H2A pulled down FLAG-RGA more efficiently than FLAG-(rga-2) and FLAG-(rga-11) proteins from Arabidopsis extracts, but rgaA268V mutation did not affect H2A binding. GST and GST-H2A bound to glutathione beads were used separately to pull down FLAG-RGA/rga from protein extracts of transgenic Arabidopsis (in ga1 dP background) carrying PRGA:FLAG-RGA/rga as labeled. Immunoblots containing input Arabidopsis extracts and pulldown samples were detected with an anti-FLAG antibody. Ponceau S-stained blots indicated that similar amounts of the GST/GST-H2A proteins were used in each set of the pulldown assays (Supplementary Fig. 4a). d, Co-IP assay showing the endogenous H2A was co-IP’ed by FLAG-RGA and FLAG- rgaA268V, but not by FLAG-(rga-2) or FLAG-(rga-11). FLAG-RGA/rga from protein extracts of transgenic Arabidopsis (in ga1 dP background) carrying PRGA:FLAG-RGA/rga were IP’ed using an anti-FLAG antibody. Immunoblots containing input Arabidopsis extracts and IP’ed samples were detected with anti-FLAG and anti-H2A antibodies, separately. e, Detection of PIF4-RGA-H2A complex by co-IP. Myc-PIF4, HA-RGA and FLAG-H2A were transiently expressed alone or co-expressed in N. benthamiana as indicated. f, Detection of IDD3-RGA-H2A complex by co-IP. Myc-IDD3, HA-RGA and FLAG-H2A were transiently expressed alone or co-expressed in N. benthamiana as indicated. In e and f, Myc-PIF4 and Myc-IDD3 were IP’ed from protein extracts using anti-Myc agarose, respectively. Protein blots were probed with anti-Myc, anti-HA and anti-FLAG antibodies separately. Representative images of two (a, b, f) or three (c-e) biological repeats are shown, and source data are provided in Source Data Fig. 7.
H2A monoubiquitination (H2Aub1), and replacement of H2A with the H2A variant H2A.Z have been reported to regulate gene transcription in eukaryotes, including plants54–56. In Arabidopsis, H2Aub1 is enriched in many transcriptionally repressed genes. However, it has also been found recently to be located in transcriptional regulation hotspots, which have less accessibility, but are still permissive chromatin57,58. H2A.Z is enriched at +1 nucleosome [the first nucleosome downstream of the transcription start site (TSS)] in actively transcribed genes, but it can play a repressive role when located in gene bodies59,60. Monoubiquitination of H2A.Z appears to correlate with its repressive role in transcription61. Co-IP assay using transient expression in N. benthamilana showed that RGA binds to H2A and H2A.Z similarly (Supplementary Fig. 8b). To investigate the relationship between RGA and H2Aub1 or H2A.Z further, we compared genome-wide RGA binding peak positions to H2Aub1 and H2A.Z distribution using published ChIP-seq datesets58,62. (Supplementary Fig. 9a–d, Supplementary Table 7). Neither H2Aub1 nor H2A.Z peak locations co-localized with the RGA binding peak (Supplementary Fig. 9b, 9d). The average RGA binding peak is located at approximately 200 nt 5’-upstream of TSS, and then sharply decreased around TSS (Supplementary Fig. 9e), indicating that RGA does not interact with +1 nucleosome whose positioning is based on nucleosome profiling in WT seedlings detected by micrococcal nuclease sequencing (MNase-seq)63.
To investigate whether RGA binding affects target chromatin accessibility, we performed Assay for Transposase-Accessible Chromatin sequencing (ATAC-seq) using PRGA:FLAG-RGA and PRGA:FLAG-rga-11 transgenic lines in the sly1 dP background. Principle Component Analysis (PCA) showed that two biological replicates of each genotype clustered together (Supplementary Fig. 10a). Analysis of our ATAC-seq dataset identified 379 differentially accessible regions (DARs) near 689 genes (FDR ≤ 0.05) between FLAG-RGA and FLAG-rga-11 lines (Supplementary Table 8). Among them, 82 genes contain FLAG-RGA binding peaks, 108 genes are GA-responsive, and only 20 are RGA direct target genes (i.e., genes that are associated with RGA binding peaks and are GA-responsive) (Supplementary Fig. 10b). If RGA binding caused altered target chromatin accessibility, GA-repressed (RGA-induced) genes should display reduced accessibility in FLAG-rga-11 than in the FLAG-RGA line, whereas GA-induced (RGA-repressed) genes should show increased accessibility. However, scatter plot analysis of the 108 GA-responsive genes with DARs (FLAG-rga-11 vs FLAG-RGA line) did not show any correlation between chromatin accessibility and GA responsiveness (Supplementary Fig. 10c). In addition, most of RGA direct target genes (n=311, 94%) did not display differential accessibility between RGA and rga-11 mutant. The overall RGA binding peak regions for these 311 target genes co-localized with accessible peak (ATAC-seq) near TSS, although rga-11 did not alter chromatin accessibility (Supplementary Fig. 10d). Genome browser images around six selected RGA target genes further showed that chromatin accessible peak(s) near individual gene in both RGA and rga-11 backgrounds co-localized with RGA binding peak(s) (Supplementary Fig. 10e). These results indicate that RGA appears to bind to accessible chromatin, whereas RGA binding does not significantly alter target chromatin structure as detected by ATAC-seq.
DISCUSSION
Our study demonstrates that DELLA-H2A interaction is essential for DELLA-mediated global transcription reprogramming, and that DELLA proteins require at least two functionally distinct subdomains (LHR1 and PFYRE) within their C-terminal GRAS domain for transcription regulation (Fig. 8). These two subdomains form the α-helical cap of the GRAS domain (Fig. 1b), and they appear to play distinct roles in interacting with different groups of regulatory proteins. The LHR1 subdomain (α1-α3) is necessary for interactions with TFs, whereas the PFYRE subdomain (α10-α11) is essential for H2A binding. Mutations in either subdomain abolished RGA association with target chromatin globally (Fig. 8). We also confirmed the formation of PIF4-RGA-H2A, and IDD3-RGA-H2A protein complexes in planta by co-IP assays. Based on these results, we propose that RGA (and other DELLAs) are recruited to target promoters via TFs that recognize specific cis-elements, and the transient TF-RGA interaction (via the LHR1 subdomain) is stabilized by RGA-H2A binding (via its PFYRE subdomain) to form TF-RGA-H2A complexes at the target chromatin. Our meta-analysis indicated that genome-wide RGA binding peak position does not co-localize with H2Aub1, H2A.Z or +1 nucleosome. Considering that the average RGA binding peak is located at approximately 200 nt 5’-upstream of TSS, it is possible that RGA (and other DELLAs) interact with H2A of nearby nucleosomes in the promoter region after being recruited to target chromatin by specific TF.
Figure 8. Working model of DELLA-mediated transcriptional regulation.
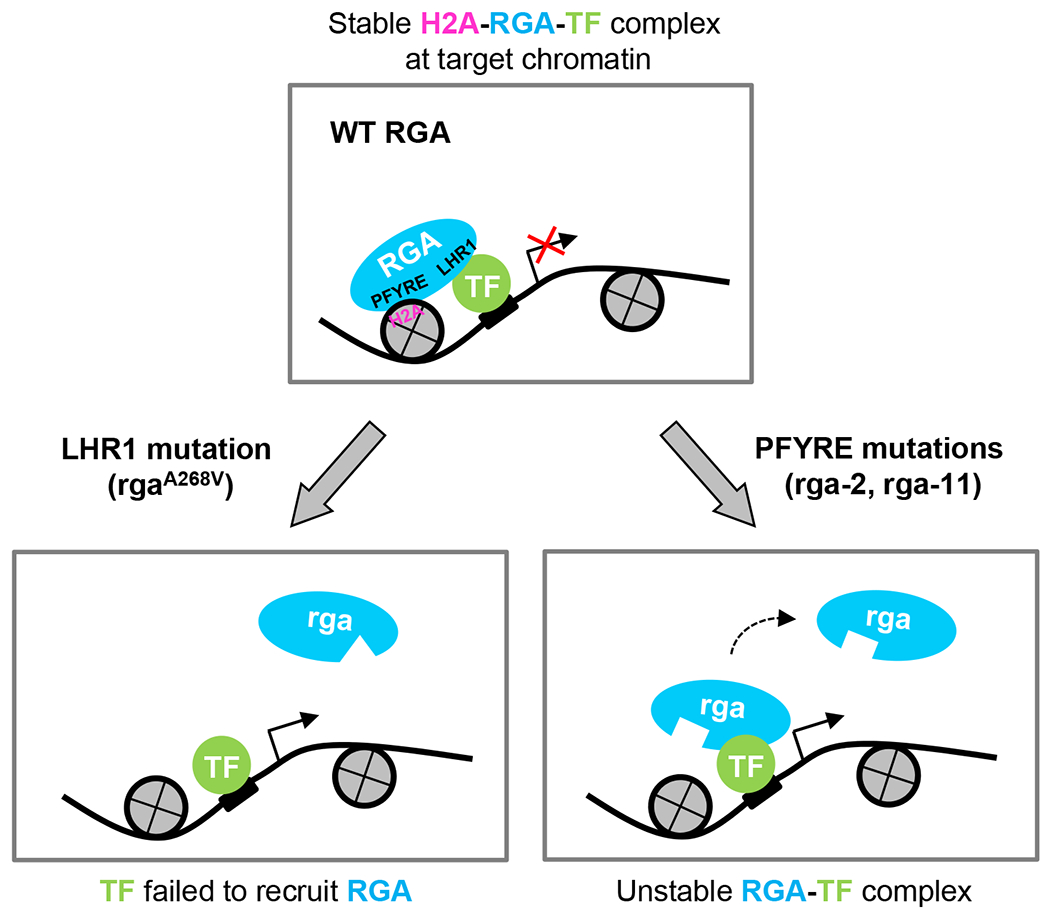
DELLA proteins (e.g., RGA) are recruited to target chromatin by interaction with TFs via the LHR1 subdomain. The transient TF-RGA interaction is stabilized by RGA-H2A binding (via its PFYRE subdomain) to form TF-RGA-H2A complexes at the target chromatin. Mutations in the LHR1 subdomain (e.g., rgaA268V) prevent recruitment to target chromatin by the TFs. In contrast, mutations in the PFYRE subdomain (e.g., rga-2 and rga-11) abolish H2A binding. Both subdomains are essential for DELLA-mediated transcription repression and activation. The diagram only depicts RGA-mediated transcription repression. A similar diagram can depict RGA-mediated transcription activation, except that the TF-RGA-H2A complex will promote transcription of target genes and that either LHR1 or PFYRE mutations will reduce transcription.
We also found that RGA binding peaks are located near both RGA-activated and RGA-repressed genes by combining the RGA ChIP-seq results (this study) and an RNA-Seq dataset (for GA-responsive genes)37. The association of RGA at RGA-repressed promoters was unexpected based on the current model for direct sequestration of transcription activators (e.g., BZR1, PIFs and TCPs) by RGA. Considering new findings in this study, it is likely that RGA (and other DELLA proteins) are recruited initially to all its target chromatin by binding to specific TFs and forming TF-DELLA-H2A complexes to repress or activate transcription of target genes. For DELLA-mediated transcription repression, DELLA either directly interferes with TF transactivation or recruits co-repressor(s) once the TF-RGA-H2A complex is formed. It remains possible that DELLA binding may result in subsequent reduction in TF binding to target DNA. For DELLA-mediated transcription activation, DELLA may function as a co-activator as it displays transactivation activity in yeast and in plant cells41,48. DELLA may also recruit other co-activator(s) after forming the TF-RGA-H2A complex at the target chromatin. Thus, DELLA-mediated transcription activation vs repression at an individual promoter is likely dependent on interacting transcription factor/co-activators or co-repressors. In addition, DELLA interacts with chromatin remodelers (SWI/SNF, and PKL)35–37. Genetic analysis indicates that most of GA-mediated developmental processes require the CHD3 chromatin remodeler PKL37, which antagonistically interacts with DELLA to promote GA responses36. Intriguingly, RNA-seq data revealed that PKL function is required for GA induction of 310 vegetative growth-related genes (in the C2 cluster, including many DELLA-repressed genes, such as EXP8, IAA19, SAURs, GH3.3)37. Conversely, 468 GA-repressed genes (in the C5 cluster, including many DELLA-induced genes, such as SCL3, GID1A, GID1B, GA20ox2) are PKL-independent. However, GA repression of another 356 genes (in C6 and C8 gene clusters) depends on PKL function37. Therefore, the precise role of PKL in regulating GA- and DELLA-responsive genes requires further investigation.
Our ATAC-seq analysis using FLAG-RGA and FLAG-rga-11 lines (in sly1 dP background) showed that most of RGA binding sites correspond to accessible regions of target chromatin whose accessibility did not change in the presence or absence of functional RGA, suggesting that RGA is not responsible for the initial opening of target chromatin. This finding is consistent with recent studies showing that recruitment of DELLA and SPL9 to the AP1 promoter for promoting flower initiation requires LEAFY64, which functions as a pioneer transcription factor for binding to target DNA in nucleosome occupied region and recruiting SWI/SNF chromatin remodelers to open chromatin65.
In summary, our study provides new insights into the complex mechanism of DELLA-mediated transcription reprogramming. We have identified a novel role of the PFYRE subdomain for binding H2A, which together with the LHR1 subdomain play two distinct modular functions in DELLA-mediated genome-wide transcription regulation in plants.
Methods
Plant materials, growth conditions, plant transformation, gibberellin (GA) treatment, and statistical analyses
Plants were grown in the growth room or on plates as described previously37. For the dim light treatment (Fig. 3b,c), seedlings were grown in 16 μmol m-2 s-1 light intensity under short-day (8 h light) conditions. The ga1-3 rga double mutants were screened and backcrossed to ga1-3 once before characterization as described previously4,46. The T-DNA rga mutants, rga28 and rga29 were reported in previous studies66,67. sly1-10 (in the Ler background)68 was introgressed into the Col-0 background by backcrossing with Col-0 6 times, and sly1-10 dellaP (sly1 dP) and sly1-10 dellaQ (with WT RGA, sly1 dQ) were generated by crossing sly1-10 (backcrossed to Col-0 6 times) with dellaP (rga-29: SALK_089146; gai-t6 (bc to Col-0 6x), rgl1: SALK_136162; rgl2: SALK_027654; rgl3-3: CS16355)67 and screening in F2 and F3 through phenotyping growth suppression and genotyping the T-DNA insertion of respective mutations. pRGA-His-3xFLAG-RGA, pRGA-His-3xFLAG-rga-2, pRGA-His-3xFLAG-rga-11 and pRGA-His-3xFLAG-rga-rht7 constructs (pCB-His-3xFLAG-RGA/rga, rht7 = rgaA268V) were transformed into ga1-3 dellaP (ga1 dP)37 by floral dipping. FLAG-RGA/rga sly1 dP were obtained by crossing sly1-10 dP with FLAG-RGA/rga ga1 dP and screening the progenies in F2 and F3. The rga-CT2 transgenic line (L78-6923) in the Ler background was described previously45. For early GA-response tests, plates having 8-day grown plants were drenched with 10 μM GA3 solution for 3 seconds and incubated in normal growth condition for indicated time. Statistical package JMP Pro 10.0.2 (SAS Institute Inc.) was used for Student’s t-tests.
Plasmid construction
The following plasmids were described previously: PSCL3:fLUC48, pEG100-3F-GAI (35S:His-3xFLAG-GAI)69, pCB-3F-RGA (pCB-His-3xFLAG-RGA), pEG100-3F-RGA (35S:His-3xFLAG-RGA), PCR8-GFPNLS and pEG3F-GW destination vector (containing His-3xFLAG-tag)70 and pDONR207-3FR45. Primers and plasmid constructs are listed in Supplementary Tables 9 and 10, respectively. All DNA constructs generated from PCR amplification were sequenced to ensure that no mutations were introduced.
In silico prediction of 3D protein structure of the RGA GRAS domain
The 3D structure model of the RGA GRAS domain was built based on the structure of SCARECROW protein (5B3G)49 as a template using the online SWISS-MODEL Workspace (http://swissmodel.expasy.org/)50,71. Pymol package v2.2.2 (https://pymol.org/) was run on Python platform to visualize and locate the mutant alleles in the RGA GRAS model.
Reverse transcription (RT)-quantitative PCR (qPCR) analyses, immunoblot analyses, and Y2H
Total RNA was isolated using the Quick-RNA MiniPrep kit (Zymo Research). Briefly, Arabidopsis seedlings (~60 mg) were ground in extraction buffer and processed following the manufacturer’s protocol. Reverse transcription was performed using M-MLV RTase (Promega) using anchored oligo dT. For qPCR, the FastStart Essential DNA Green Master mix was used with a LightCycler 96 (Roche Applied Science). Relative transcript levels were determined by normalizing with PP2A (At1g13320). Immunoblot analyses were performed using rabbit anti-RGA antiserum (DU176, 1:10,000 dilution)11, horseradish peroxidase (HRP)-conjugated anti-FLAG M2 mouse monoclonal (Sigma Aldrich A8592, 1:10,000 dilution) and mouse HRP-anti-MYC monoclonal antibodies (BioLegend #626803, 1:1,000 dilution), mouse anti-HA monoclonal antibody (BioLegend 901503, 1:1,000 dilution), rabbit anti-H2A monoclonal antibody (Abcam #ab177308, 1:1,000 dilution) and rabbit anti-Histone 3 polyclonal antibody (Abcam #ab1791). HRP-conjugated donkey anti-mouse IgG (Jackson ImmunoResearch #715-035-150) was used for anti-HA at 1:10,000 dilution. HRP-conjugated goat anti-rabbit IgG (Thermo-Fisher #31462) was used to detect anti-RGA and anti-HA at 1:10,000 dilution. Y2H assays were performed as described previously45.
Transient expression and dual luciferase assay in Nicotiana benthamiana
For dual luciferase assays and pulldown assays, transient expression of FLAG-RGA, FLAG-GAI, and FLAG-rga in N. benthamiana was performed as described with slight modifications48. The N. benthamiana leaves were harvest after 48 hr of Agro-infiltration72. Three biological repeats were conducted for each effector combination.
In vitro pulldown assay
In vitro pulldown assays using recombinant GST protein fusions expressed in E. coli BL21-CodonPlus (DE3)-RIL (Agilent Technologies) were performed following the procedures published previously with minor modifications45. To obtain the N. benthamiana lysate used for in vitro pulldown assay, leaves were infiltrated with Agrobacterium harboring the corresponding binary vectors, harvested after 2 days, ground in liquid nitrogen, and stored at −80 °C. 100 mg of the ground powder was resuspended in the N. benthamiana lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 % Triton X-100, 2.5 mM 2-mercaptoethanol, and 1x Protease inhibitor cocktail [Sigma-Aldrich]) and the slurry was passed through 70 μm strainer by quick spinning. Flow-through was collected and centrifuged at 4 °C, 15,000 rpm for 5 min. Lysate obtained after spinning the supernatant again was used for the protein binding assays. The pulldown assays between FLAG-RGA/-GAI (from N. benthamiana protein extracts) and MBP-H2A, MBP-H2B, MBP-H3 and MBP-H4 (from E. coli) were performed using the same procedures, except that amylose resin (New England BioLabs, E8021S, lot #0131305) was used to purify the MBP and MBP protein fusions and plant lysis buffer of 200 mM NaCl was used for incubation and washing. The same procedure was used for pulldown assays between FLAG-PIF4/-IDD3 (from N. benthamiana protein extracts) and MBP-RGA/-rga-2/-rga-11 (from E. coli).
The pulldown assays using protein extracts from transgenic Arabidopsis lines [FLAG-RGA ga1 dP, FLAG-(rga-2) ga1 dP, FLAG-(rga-11) ga1 dP and FLAG-(rga-rht7) ga1 dP] and recombinant proteins (GST, GST-BZR1, GST-PIF3 and GST-H2A) were performed in the same way as in FLAG-DELLA and MBP-histones binding assay, except that the ground Arabidopsis tissue powder was resuspended in the lysis buffer (50 mM Tris-HCl pH8.0, 150 mM NaCl, 1 % Triton X-100, 5 mM EDTA, 1x Protease inhibitor cocktail [Sigma-Aldrich] and 1 mM PMSF).
For the calf thymus histone binding assay, glutathione-bead charged with the GST-RGA was mixed with calf thymus histones (Sigma, H9250) in TBS buffer of 250 mM NaCl. After incubation at 4 °C with rotation for 2 h, bead was washed 5 times with the same buffer. Interaction of RGA with histones was confirmed by immunoblot detection using anti-Histone 3 antibody.
Co-IP
Total Arabidopsis protein was extracted from 0.5 g of ground powder in 2 mL of extraction buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 % Triton X-100, 5 mM EDTA, 1x Protease inhibitor cocktail [Sigma-Aldrich] and 1 mM PMSF) and centrifuged at 4 °C at maximum speed for 10 min. 50 μL input was taken from the supernatant and 1 mL protein extract were incubated with 20 μL of anti-FLAG-M2-Agarose beads (Sigma-Aldrich A2220) for 1.5 h at 4 °C. And then washed three times with wash buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween-20). Samples were analyzed by SDS-PAGE and immunoblotting using HRP-conjugated anti-FLAG antibody (Sigma-Aldrich) and anti-H2A antibody (Abcam) as described above.
To detect the PIF4-RGA-H2A or IDD3-RGA-H2A complex, MYC-PIF4 or MYC-IDD3, HA-RGA and FLAG-H2A were transiently expressed in N. benthamiana leaves, and subsequent co-IP assays using rabbit anti-Myc polyclonal antibody conjugated agarose beads (Sigma-Aldrich A7470) were performed as described previously45.
Chromatin immunoprecipitation (ChIP), ChIP-qPCR and construction of ChIP library
For ChIP-qPCR analysis, transgenic Arabidopsis seedlings carrying PRGA:FLAG-RGA/rga (in the ga1 dP background or the sly1 dP background) and the respective parental lines (ga1 dP or sly1 dP) were grown in the continuous light for 10 days, harvested, and cross-linked in 1% formaldehyde solution for 20 min. Seedlings were washed with water 3 times, snap-frozen, and ground in liquid nitrogen. Around 100-200 mg of ground tissue powder was dissolved in 2 mL of nuclear isolation buffer (NIB) (0.25 M Sucrose, 12 mM Tris-HCl pH8.0, 5 mM MgCl2, 60 mM KCl, 15 mM NaCl, 1 mM CaCl2, 0.9 % Triton X-100, and 1x Protease inhibitor cocktail) and passed through the 70 μm strainer by quick spinning. Flow-through was collected and spun at 3,000 × g for 5 min. After removing the supernatant, pellet was resuspended in 1 mL of NIB by pipetting and spun at 3,000 × g for 5 min. Rinsed pellet was resuspended in 500 μL of nuclear lysis buffer (NLB) (50 mM Tris-HCl pH8.0, 150 mM NaCl, 1 mM EDTA, 0.1 % SDS, 0.1 % Na-deoxycholate, 1 % Triton X-100, and 1x Protease inhibitor cocktail) and sonicated for 5 cycles using Bioruptor (High power, 1 cycle = on-30 sec / off-30 sec). Sonicated sample was centrifuged at 15,000 rpm for 5 min, and supernatant transferred to new tube was centrifuged at 15,000 rpm for 5 min. Sonicated chromatin was recovered and processed for immunoprecipitation of the chromatin. After saving 40 μL of chromatin as input, 10 μL of anti-FLAG-M2-Agarose beads (Sigma-Aldrich A2220) was added to 400 μL of chromatin and incubated at 4 °C with rotation for 2 hr. Beads were washed as described in the co-IP assays. Input chromatin and IP’ed chromatin bound on bead were processed as described previously48. qPCR was performed as described above, and the relative enrichment was calculated by normalizing against ChIP-qPCR of nontransgenic control samples using PP2A53. The normalized values of fold enrichment are the average ± SE of three biological replicates (two technical repeats each) from independent pools of tissues. Fold enrichment was calculated from each sample relative to the nontransgenic control (set as 1.0).
For the construction of ChIP-seq library, transgenic Arabidopsis seedlings carrying PRGA:FLAG-RGA/rga-11 (in the sly1 dP background) and their respective control constructs without FLAG tags (sly1 dQ containing WT RGA and sly1 dP) were processed for ChIP as described above, except that chromatin was obtained after 25 cycles of sonication and 200 mg of starting ground powder and 20 μL of anti-FLAG-M2-Agarose beads (Sigma-Aldrich A2220) were used instead. Two biological repeats were prepared for each genotype.
Sequencing of ChIP-seq library and data analyses
To prepare the ChIP-seq DNA library from purified DNA from ChIP, we followed the protocol published previously with minor modifications53,73. Two biological replicates of ChIP samples were pooled together for sequencing. We used the NEB’s (http://www.neb.com) Y-adapter sequences and amplification primers sequences with barcodes instead (Supplementary Table 9). DNA sequencing was performed using the Illumina platform.
After trimming adaptor sequences using Trimmomatic (v.0.39), paired-ends ChIP-seq reads were mapped to the Arabidopsis reference genome (TAIR10) with Bowtie2 (v.2.4.5). Binding peaks were identified using MACS (v.2.2.7.1)74 with default parameters except for ‘-f BAMPE’ to model peaks from paired-ends ChIP-seq data, comparing FLAG-RGA and FLAG-(rga-11) to their respective controls without FLAG tags (sdQ) for FLAG-RGA, and sdP for FLAG-(rga-11). After removing peaks from a small number of genomic regions where the mapped ChIP-seq read depth were consistently high across all samples including controls, we selected peaks with q-values < 10−3. Among them, we considered peaks with the fold enrichment values higher than GID1B as high-confidence because this gene had the lowest fold of enrichment in our dataset among all previously identified RGA target genes by ChIP-qPCR (Supplementary Table 2)19–21,28,34,37,48,64,67. The cumulative ratio of FLAG-RGA and FLAG-rga-11 read depth within 1.5Kb of all high-confidence peaks were visualized using deepTools (v.3.1.3). Following a previous study20, we identified the set of all genes with a high-confidence FLAG-RGA peak within 5’ 3Kb and 3’ 1.5Kb using BEDtools (v.2.30.0) and compared with the gene set with a GFP-rgaΔ17 peak20. To calculate the median ratio of ChIP/background read counts, DeepTools was used to plot the ratio of FLAG-RGA/sdQ, and FLAG-(rga-11)/sdP in order to remove the background.
For the detection of RGA-direct targets, we considered genes showing significant different (p<0.001) expression between mock- (M) and GA-treated samples in the ga1 mutant plants, obtained from previous RNA-seq results37, as putative DELLA-regulated genes, because DELLA is known to be degraded by GA treatment11. Among putative DELLA-regulated genes, those with a high-confidence FLAG-RGA peak within 5’ 3Kb and 3’ 1.5Kb were designated as RGA-direct target genes. GO enrichment analysis was done using GeneOntology (http://geneontology.org/).
Transcription factor-binding motifs enriched near peaks (within the region +/− 200bp) of the peak center were detected by the HOMER package v4.11 (http://homer.ucsd.edu/homer/), using motif libraries derived from published DAP-seq and ChIP-seq data75, and only displayed the motifs with p ≤0.01. To calculate the motifs enrichment from the overlap peaks between FLAG-RGA ChIP-seq and RNA-seq, the summit peak file was used to normalize the peak to be same +/−200bp size centered on the summit peak using HOMER package.
To calculate the genomic distribution of RGA binding peaks, two R package ChIPseeker (v1.36.0) and GenomicFeatures (v1.52.1) were used. The narrow peak files of FLAG-RGA or GFP-rgaΔ17 as the input file, and then input the annotation file “TAIR10_GFF3_genes_transposons.gff” (modified the file from TAIR database), set promoter region from −3kb to 1kb based on the TSS. The genome browser images were created using the Integrative Genomics Viewer (IGV) 2.12.2.
Nuclei purification and ATAC-seq libraries preparation
Nuclei were purified using sucrose sedimentation as previously reported76 with slight modifications as noted below. 10-day-old seedlings grown in liquid culture (0.5x MS with 1% sucrose) were ground to fine powder in liquid nitrogen using a mortar and pestle. For each sample, 0.2 grams of the frozen tissue powder were homogenized in prechilled 10 mL NPB buffer, and the nuclear fraction was purified as described76. The nuclei pellet was resuspended in 1 mL of cold NPB buffer. For ATAC-seq, a 25 μL nuclei aliquot was stained with DAPI (0.2 μL, 1 μg/μL) and counted using a haemocytometer.
The ATAC assay was performed as previously described77,78 with slight modifications. Approximately 50,000 nuclei were used for each ATAC-seq reaction. The purified nuclei were pelleted by swing bucket centrifugation at 1,500 × g for 7 min at 4 °C, and the supernatant was removed leaving ~10 μL at bottom of tube. The Tn5 reaction was performed using a Tagment DNA Enzyme and Buffer kit (Illumina, 20034210) as follows: 50 μL reaction mix containing 10 μL nuclei sample, 25 μL 2× TD buffer, 2 μL TDE1 was placed in an Applied Biosystems ProFlex thermocycler at 37 °C for 30 min. The tagmented DNA was purified using a MinElute PCR Purification kit (QIAGEN, 28004) and eluted in 24 μL elution buffer. The purified tagmented DNA was first amplified using Next High-Fidelity 2X PCR Master Mix (NEB, M0541S) in 50 μL reactions, and a distinct barcoded Primer 2 was used for each library (Supplementary Table 9). A 5 μL aliquot was removed from each reaction to be used for qPCR using LightCycler 96 instrument (Roche) to determine the number of additional N cycles needed to amplify the library. The remaining 45 μL of each PCR sample was then continued for additional N cycles, and resulting libraries were purified using AMPure beads (Beckman Coulter, A63881) eluting in 20 μL of elution buffer. Two biological repeats were performed for each sample. Each library was quantified using Agilent 2100 Bioanalyzer before pooling for DNA sequencing (~11 ng per library) by Illumina HiSeq 4000 (2x 150 bp paired end, ~40 million reads for each library).
Analysis of ATAC-seq data
We obtained around 20 M pair-end reads for each independent biological replicates of the different genotypes. Quality trimming and adapters removal were performed using Trim Galore v0.6.4. The reads were mapped to TAIR10 Arabidopsis reference genome using Bowtie2 v2.4.579. Subsequently, the organelle genomes were removed using samtools v1.1280. PCR duplicates were discarded from the mapped reads using samblaster v0.1.2681. The regions with an artifactual massive amount of unique mapped reads were identified as a list of blacklisted genomic regions57 and were removed from the mapped BAM files using samtools. The independent biological replicates were merged using samtools. Then, Tn5 hypersensitive sites (THS) were identified using MACS2 with the parameters: --nomodel –shift −100 –extsize 200 -q 0.05. The accessibility signals were normalized using the bamCoverage from deepTools v3.5.182. The normalized accessibility signals across whole Arabidopsis genome regions were generated using computeMatrix and plotProfile from deepTools. The differential accessibility regions (DARs) were identified using the R package DiffBind v3.8.483. The bw files were generated using the function BamCoverage from deepTools with CPM as normalization. The gene annotations were performed using BEDtools v2.30.0 with parameters: closest -D a -k 4. The metaplots were generated using the function plotProfile from deepTools. The genome browser images were created using the Integrative Genomics Viewer (IGV) 2.12.2.
Comparison between FLAG-RGA binding peaks and published ChIP-seq datasets for PIF4, H2Aub1, H2A.Z/H3
The ChIP-seq datasets were downloaded from NCBI (PIF4, accession GSM86571053; H2Aub1, accession GSE15537857; H2A.Z/H3, accession GSE9687362). Data analysis was the same as for ATAC-seq until the bam files with reads mapped to Arabidopsis genome were generated. The bw files were generated using the command BamCompare from deepTools with “ratio” as accounting for scaling. For H2A.Z/H3, the bam file with H3 coverage across genome was used as control. The positional files, which contain the coordinates of overlapped genes between the peaks from RGA and that from the corresponding dataset, were generated using narrow peak files with both TSS and AGInames. The metaplots were generated using the commands computeMatrix and plotProfile from deepTools with the bw files provided signal intensity while the positional files provided positional information.
Comparison between FLAG-RGA binding peaks and nucleosome profile
The nucleosome positioning in WT Col-0 seedlings was analyzed using micrococcal nuclease sequencing (MNase-seq) dataset (PRJNA780072)63,84 by DANPOS v2.2.285. The dpos.py command of DANPOS was used to obtain the wigs file with normalized coverage of nucleosomes. Then, the wigs file was transformed into the bw file by the command wigToBigWig v4. The metaplots of nucleosomes positioning were generated by deepTools based on 2228 genes that are associated with RGA binding peaks in our study.
Supplementary Material
Acknowledgements
We thank Giltsu Choi for helpful discussions, and for sharing Arabidopsis lines and constructs. This work was supported by the National Institutes of Health (R01 GM100051 to TPS), the National Science Foundation (MCB-1818161 to TPS, and NSF-EDGE-1923589 to DHO and MD). We also acknowledge LSU High Performance Computing services for providing computational resources.
Footnotes
Competing Interests Statements
The authors declare no competing interests.
Data Availability
Raw and processed ChIP-Seq data and ATAC-seq data have been deposited at NCBI’s Gene Expression Omnibus (accession numbers GSE220898 and GSE233124, respectively). Source Data files are provided with this paper.
REFERENCES
- 1.Peng J et al. ‘Green Revolution’ genes encode mutant gibberellin response modulators. Nature 400, 256–261 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Eshed Y & Lippman ZB Revolutions in agriculture chart a course for targeted breeding of old and new crops. Science 366 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Sun TP The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr Biol 21, R338–345 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Silverstone AL, Ciampaglio CN & Sun T.-p. The Arabidopsis RGA gene encodes a transcriptional regulator repressing the gibberellin signal transduction pathway. Plant Cell 10, 155–169 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peng J et al. The Arabidopsis GAI gene defines a signalling pathway that negatively regulates gibberellin responses. Genes Dev 11, 3194–3205 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hernandez-Garcia J, Briones-Moreno A & Blazquez MA Origin and evolution of gibberellin signaling and metabolism in plants. Semin Cell Dev Biol 109, 46–54 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Daviere JM & Achard P A pivotal role of DELLAs in regulating multiple hormone signals. Mol Plant 9, 10–20 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Van De Velde K, Ruelens P, Geuten K, Rohde A & Van Der Straeten D Exploiting DELLA Signaling in Cereals. Trends Plant Sci 22, 880–893 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Pysh LD, Wysocka-Diller JW, Camilleri C, Bouchez D & Benfey PN The GRAS gene family in Arabidopsis: sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J 18, 111–119 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Tian C, Wan P, Sun S, Li J & Chen M Genome-wide analysis of the GRAS gene family in rice and Arabidopsis. Plant Mol Biol 54, 519–532 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Silverstone AL et al. Repressing a repressor: gibberellin-induced rapid reduction of the RGA protein in Arabidopsis. Plant Cell 13, 1555–1566 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dill A, Jung H-S & Sun T.-p. The DELLA motif is essential for gibberellin-induced degradation of RGA. Proc Natl Acad Sci U S A 98, 14162–14167 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ueguchi-Tanaka M et al. GIBBERELLIN INSENSITIVE DWARF1 encodes a soluble receptor for gibberellin. Nature 437, 693–698 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Griffiths J et al. Genetic characterization and functional analysis of the GID1 gibberellin receptors in Arabidopsis. Plant Cell 18, 3399–3414 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sasaki A et al. Accumulation of phosphorylated repressor for gibberellin signaling in an F-box mutant. Science 299, 1896–1898 (2003). [DOI] [PubMed] [Google Scholar]
- 16.McGinnis KM et al. The Arabidopsis SLEEPY1 gene encodes a putative F-box subunit of an SCF E3 ubiquitin ligase. Plant Cell 15, 1120–1130 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murase K, Hirano Y, Sun T.-p. & Hakoshima T Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 456, 459–463 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Shimada A et al. Structural basis for gibberellin recognition by its receptor GID1. Nature 456, 520–523 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Zentella R et al. Global analysis of DELLA direct targets in early gibberellin signaling in Arabidopsis. Plant Cell 19, 3037–3057 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serrano-Mislata A et al. DELLA genes restrict inflorescence meristem function independently of plant height. Nat Plants 3, 749–754 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marin-de la Rosa N et al. Genome Wide Binding Site Analysis Reveals Transcriptional Coactivation of Cytokinin-Responsive Genes by DELLA Proteins. PLoS Genet 11, e1005337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marin-de la Rosa N et al. Large-scale identification of gibberellin-related transcription factors defines group VII ETHYLENE RESPONSE FACTORS as functional DELLA partners. Plant Physiol 166, 1022–1032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lantzouni O, Alkofer A, Falter-Braun P & Schwechheimer C GROWTH-REGULATING FACTORS Interact with DELLAs and Regulate Growth in Cold Stress. Plant Cell 32, 1018–1034 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng S et al. Coordinated regulation of Arabidopsis thaliana development by light and gibberellins. Nature 451, 475–479 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Lucas M et al. A molecular framework for light and gibberellin control of cell elongation. Nature 451, 480–484 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Oh E et al. Cell elongation is regulated through a central circuit of interacting transcription factors in the Arabidopsis hypocotyl. eLife 3, e03031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu J, Israeli A, Ori N & Sun TP The Interaction between DELLA and ARF/IAA mediates crosstalk between gibberellin and auxin signaling to control fruit initiation in tomato. Plant Cell 30, 1710–1728 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai MY et al. Brassinosteroid, gibberellin, and phytochrome signalling pathways impinge on a common transcription module in Arabidopsis. Nat Cell Biol 14, 810–817 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou X, Lee LY, Xia K, Yan Y & Yu H DELLAs modulate jasmonate signaling via competitive binding to JAZs. Dev Cell 19, 884–894 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Yang DL et al. Plant hormone jasmonate prioritizes defense over growth by interfering with gibberellin signaling cascade. Proc Natl Acad Sci U S A 109, E1192–1200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daviere JM et al. Class I TCP-DELLA Interactions in Inflorescence Shoot Apex Determine Plant Height. Curr Biol 24, 1923–1928 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Lim S et al. ABA-insensitive3, ABA-insensitive5, and DELLAs Interact to activate the expression of SOMNUS and other high-temperature-inducible genes in imbibed seeds in Arabidopsis. Plant Cell 25, 4863–4878 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida H et al. DELLA protein functions as a transcriptional activator through the DNA binding of the INDETERMINATE DOMAIN family proteins. Proc Natl Acad Sci USA 111, 7861–7866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fukazawa J et al. DELLAs function as coactivators of GAI-ASSOCIATED FACTOR1 in regulation of gibberellin homeostasis and signaling in Arabidopsis. Plant Cell 26, 2920–2938 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarnowska EA et al. DELLA-interacting SWI3C core subunit of switch/sucrose nonfermenting chromatin remodeling complex modulates gibberellin responses and hormonal cross talk in Arabidopsis. Plant Physiol 163, 305–317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang D, Jing Y, Jiang Z & Lin R The Chromatin-Remodeling Factor PICKLE Integrates Brassinosteroid and Gibberellin Signaling during Skotomorphogenic Growth in Arabidopsis. Plant Cell 26, 2472–2485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park J et al. GA signaling requires chromatin remodeler PICKLE to promote vegetative growth and phase transitions. Plant Physiol 173, 1463–1474 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Locascio A, Blazquez MA & Alabadi D Dynamic regulation of cortical microtubule organization through prefoldin-DELLA interaction. Curr Biol 23, 804–809 (2013). [DOI] [PubMed] [Google Scholar]
- 39.Dill A, Thomas SG, Hu J, Steber CM & Sun T.-p. The Arabidopsis F-box protein SLEEPY1 targets GA signaling repressors for GA-induced degradation. Plant Cell 16, 1392–1405 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandler PM & Harding CA ‘Overgrowth’ mutants in barley and wheat: new alleles and phenotypes of the ‘Green Revolution’ DELLA gene. J Exp Bot 64, 1603–1613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirano K et al. The suppressive function of the rice DELLA protein SLR1 is dependent on its transcriptional activation activity. Plant J 71, 443–453 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Ikeda A et al. slender rice, a constitutive gibberellin response mutant is caused by a null mutation of the SLR1 gene, an ortholog of the height-regulating gene GAI/RGA/RHT/D8. Plant Cell 13, 999–1010 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muangprom A, Thomas SG, Sun TP & Osborn TC A novel dwarfing mutation in a green revolution gene from Brassica rapa. Plant Physiol 137, 931–938 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirano K et al. Characterization of the molecular mechanism underlying gibberellin perception complex formation in rice. Plant Cell 22, 2680–2696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zentella R et al. O-GlcNAcylation of master growth repressor DELLA by SECRET AGENT modulates multiple signaling pathways in Arabidopsis. Genes Dev 30, 164–176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silverstone AL, Mak PYA, Casamitjana Martínez E & Sun T.-p. The new RGA locus encodes a negative regulator of gibberellin response in Arabidopsis thaliana. Genetics 146, 1087–1099 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuo N, Minami M, Maeda T & Hiratsuka K Dual luciferase assay for monitoring transient gene expression in higher plants. Plant Biotechnol 18, 71–75 (2001). [Google Scholar]
- 48.Zhang ZL et al. SCARECROW-LIKE 3 promotes gibberellin signaling by antagonizing DELLA in Arabidopsis. Proc Natl Acad Sci USA 108, 2160–2165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirano Y et al. Structure of the SHR-SCR heterodimer bound to the BIRD/IDD transcriptional factor JKD. Nat Plants 3, 17010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnold K, Bordoli L, Kopp J & Schwede T The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Dill A & Sun T.-p. Synergistic de-repression of gibberellin signaling by removing RGA and GAI function in Arabidopsis thaliana. Genetics 159, 777–785 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King K, Moritz T & Harberd N Gibberellins are not required for normal stem growth in Arabidopsis thaliana in the absence of GAI and RGA. Genetics 159, 767–776 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oh E, Zhu JY & Wang ZY Interaction between BZR1 and PIF4 integrates brassinosteroid and environmental responses. Nat Cell Biol 14, 802–809 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbour H, Daou S, Hendzel M & Affar EB Polycomb group-mediated histone H2A monoubiquitination in epigenome regulation and nuclear processes. Nat Commun 11, 5947 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Colino-Sanguino Y, Clark SJ & Valdes-Mora F The H2A.Z-nuclesome code in mammals: emerging functions. Trends Genet 38, 273–289 (2022). [DOI] [PubMed] [Google Scholar]
- 56.Long J, Carter B, Johnson ET & Ogas J Contribution of the histone variant H2A.Z to expression of responsive genes in plants. Semin Cell Dev Biol 135, 85–92 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin X et al. H2AK121ub in Arabidopsis associates with a less accessible chromatin state at transcriptional regulation hotspots. Nat Commun 12, 315 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y, Romero-Campero FJ, Gomez-Zambrano A, Turck F & Calonje M H2A monoubiquitination in Arabidopsis thaliana is generally independent of LHP1 and PRC2 activity. Genome Biol 18, 69 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lei B & Berger F H2A Variants in Arabidopsis: Versatile Regulators of Genome Activity. Plant Commun 1, 100015 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sura W et al. Dual Role of the Histone Variant H2A.Z in Transcriptional Regulation of Stress-Response Genes. Plant Cell 29, 791–807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gomez-Zambrano A, Merini W & Calonje M The repressive role of Arabidopsis H2A.Z in transcriptional regulation depends on AtBMI1 activity. Nat Commun 10, 2828 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wollmann H et al. The histone H3 variant H3.3 regulates gene body DNA methylation in Arabidopsis thaliana. Genome Biol 18, 94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang T et al. Chromatin remodeling complexes regulate genome architecture in Arabidopsis. Plant Cell 34, 2638–2651 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamaguchi N et al. Gibberellin acts positively then negatively to control onset of flower formation in Arabidopsis. Science 344, 638–641 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Jin R et al. LEAFY is a pioneer transcription factor and licenses cell reprogramming to floral fate. Nat Commun 12, 626 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tyler L et al. DELLA proteins and gibberellin-regulated seed germination and floral development in Arabidopsis. Plant Physiol 135, 1008–1019 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park J, Nguyen KT, Park E, Jeon JS & Choi G DELLA proteins and their interacting RING Finger proteins repress gibberellin responses by binding to the promoters of a subset of gibberellin-responsive genes in Arabidopsis. Plant Cell 25, 927–943 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steber CM & McCourt P A role for brassinosteroids in germination in Arabidopsis thaliana. Plant Physiol 125, 763–769 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zentella R et al. The Arabidopsis O-fucosyltransferase SPINDLY activates nuclear growth repressor DELLA. Nat Chem Biol 13, 479–485 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zentella R et al. SPINDLY O-fucosylates nuclear and cytoplasmic proteins involved in diverse cellular processes in plants. Plant Physiol 191, 1546–1560 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biasini M et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42, W252–258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou X et al. The ERF11 Transcription Factor Promotes Internode Elongation by Activating Gibberellin Biosynthesis and Signaling. Plant Physiol 171, 2760–2770 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong KH, Jin Y & Moqtaderi Z Multiplex Illumina sequencing using DNA barcoding. Curr Protoc Mol Biol 7, 7.11.11–17.11.11 (2013). [DOI] [PubMed] [Google Scholar]
- 74.Feng J, Liu T, Qin B, Zhang Y & Liu XS Identifying ChIP-seq enrichment using MACS. Nat Protocols 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.O’Malley RC et al. Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell 165, 1280–1292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bajic M, Maher KA & Deal RB Identification of Open Chromatin Regions in Plant Genomes Using ATAC-Seq. Methods Mol Biol 1675, 183–201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Buenrostro JD, Wu B, Chang HY & Greenleaf WJ ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol 109, 21 29 21–21 29 29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Potter KC, Wang J, Schaller GE & Kieber JJ Cytokinin modulates context-dependent chromatin accessibility through the type-B response regulators. Nat Plants 4, 1102–1111 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Danecek P et al. Twelve years of SAMtools and BCFtools. Gigascience 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Faust GG & Hall IM SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ross-Innes CS et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diego-Martin B et al. The TRIPLE PHD FINGERS proteins are required for SWI/SNF complex-mediated +1 nucleosome positioning and transcription start site determination in Arabidopsis. Nucleic Acids Res 50, 10399–10417 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen K et al. DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res 23, 341–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed ChIP-Seq data and ATAC-seq data have been deposited at NCBI’s Gene Expression Omnibus (accession numbers GSE220898 and GSE233124, respectively). Source Data files are provided with this paper.