

Abstract
Bispecific antibodies exhibit proven clinical benefit, and many bispecifics are currently in clinical development for oncology. Cytokine release syndrome (CRS) is a common clinical adverse effect observed following CD3‐based bispecific dosing. However, the pathophysiology of CRS is not fully understood, and no computational model mechanistically describing clinical cytokine dynamics following bispecific dosing in solid tumors exists. Here, a quantitative systems pharmacology (QSP) model describing peripheral clinical cytokine dynamics following bispecific dosing in solid tumors is presented. Using tebentafusp as a case study, a CD3‐bispecific approved for uveal melanoma, the model successfully captures the dynamics of five cytokines. The QSP model was shown to predict observed phenomena, such as cytokine maximum concentration suppression using step‐up dosing regimens and the importance of on‐target off‐tumor binding toward CRS and toxicity. Furthermore, the QSP model provides rationale for these biological phenomena based on dynamics of immune cell activation and desensitization in tumors and healthy tissues. Overall, the QSP model structure presented here serves as a basis to infer cytokine dynamics for other CD3‐based bispecifics or tumor types by altering model parameters to capture the scenario of interest, supporting applications including dose selection, candidate nomination, and disease area selection.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Cytokine release syndrome (CRS) is a common clinical adverse effect following CD3‐bispecific dosing. However, CRS pathophysiology is not fully understood, and no computational model mechanistically describing clinical cytokine dynamics following bispecific dosing in solid tumors exists.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study addresses how peripheral cytokine dynamics, a clinical biomarker, can be mechanistically modeled for CD3‐bispecific treated solid tumors. In addition, the model provides insight into biological mechanisms driving cytokine biomarker dynamics.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This study provides biological rationale for clinical observations in bispecific treated tumors. Specifically, clinical observations of cytokine attenuation with repeated bispecific dosing and the importance of on‐target off‐tumor effects toward CRS are linked, through model application, to immune cell activation and desensitization in tumors and healthy tissues.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The model presented serves as a basis to predict safety outcomes via cytokine biomarker dynamics for CD3‐based bispecifics, supporting development decisions including dose and regimen selections, candidate nomination, and disease area selection.
INTRODUCTION
Bispecific antibodies are a promising class of therapeutics that are currently being explored for the treatment of many different malignancies, 1 and many bispecifics are currently in clinical development for oncology. 2 T cell–engager bispecifics for oncology typically work by binding a target antigen on the tumor cells and a T cell–specific antigen, such as CD3. These bispecifics create a cytolytic immunologic synapse between T cells and tumor cells, resulting in T cell activation and expansion and tumor cell death. Bispecifics are thus attractive therapies in oncology as they can provide superior cytotoxic effects and lower resistance rates, due to antigen recognition by T cell receptors not being necessary, 3 compared with other therapy types. 4 , 5 Cytokine release syndrome (CRS) is one of the most common side effects observed during CD3 bispecific clinical trials. 6 Although the pathophysiology of CRS is not fully understood, current understanding indicates on‐target bispecific binding activates bystander immune and nonimmune cells causing systemic proinflammatory cytokine release. 6 Approaches to better understand CRS risk and reduce CRS likelihood for CD3‐targeting bispecifics are currently being explored. 7
Currently, four CD3‐targeting bispecifics have been approved: blinatumomab is US Food and Drug Administration (FDA) approved for relapsed or refractory (R/R) B cell precursor acute lymphoblastic leukemia, mosunetuzumab is European Medicines Agency approved for R/R follicular lymphoma, tebentafusp‐tebn is FDA approved for metastatic uveal melanoma, and teclistamab‐cqyv is FDA approved for R/R multiple myeloma. CRS was a common adverse event for these approved CD3 bispecifics, with overall incidence rates in registrational trials of 14%, 8 44%, 9 89%, 10 and 72%, 11 respectively. CRS severity in these trials were graded using a grading system 12 based on therapy interruption and medical interventions required to manage patient response, with higher grade severity (Grades 3–4) becoming life threating and requiring hospitalization. Although severe CRS (≥ grade 3) occurred at low incidence rates for the approved CD3 bispecifics (4.9%, 1%, 1%, and 0.6%, respectively, in the trials cited previously), CRS is still an important dose‐limiting toxicity and should be reduced to provide the best possible patient experience.
Tebentafusp‐tebn, the only approved CD3 bispecific targeting solid tumors, exhibits the greatest overall CRS incidence of all four approved CD3 bispecifics. Preliminary studies from nonapproved therapies indicate that higher CRS rates may occur in solid tumor–targeting bi/trispecifics compared with hematological‐targeting therapies. 13 Therefore, better understanding mechanisms driving CRS in solid tumor–treated therapies is desired. Clinical studies with tebentafusp‐tebn have shown that peripheral cytokines transiently increase shortly after dosing and that patients manifesting CRS have greater cytokine increases than patients without CRS. 14 With this observation in mind, this article aims to provide a mathematical model to capture peripheral cytokine increases following bispecific dosing as a first step toward understanding CRS incidence. Such a model would allow predictive insight into how cytokine dynamics are affected by bispecific dosing, supporting dose optimization to reduce CRS risk. Specifically, this article focuses on capturing clinical cytokine dynamics following tebentafusp‐tebn (henceforth termed tebentafusp) dosing. Tebentafusp is chosen for this article as it is currently the only CD3 bispecific approved for solid tumors.
Quantitative systems pharmacology (QSP) models seek to mathematically represent disease biology and pharmacology to provide holistic quantitative predictions of treatment effects. 15 Betts et al. 16 and Ma et al. 17 applied QSP modeling to show how processes such as biodistribution, target binding, and solid tumor growth dynamics can be modeled to capture treatment efficacy in tumor growth inhibition. Hosseini et al. 18 developed a QSP model to understand how cytokine release from hematological‐targeting bispecifics can be mitigated. Such previous studies show that QSP is well poised to model cytokine dynamics from bispecific‐treated solid tumors and provide model structure and parameter precedence (Table S1) employed in model development here.
Here, a QSP model to describe peripheral cytokine dynamics following tebentafusp dosing is developed (Figure 1). Five cytokines are included in the model (interferon γ [IFNγ], interleukin [IL]‐6, IL‐10, C‐X‐C motif chemokine ligand [CXCL] 10 [CXCL10], and CXCL11) based on the availability of published tebentafusp clinical data. 19 Because gp100 is expressed both in uveal melanoma cells and melanocytes, 20 trimer formation and subsequent cytokine release both in tumor and skin tissue are modeled. Following successful model calibration, simulations and sensitivity analysis are performed to glean insight into how bispecific and target parameters direct cytokine release, contributions of tumor and healthy tissue to cytokine release, and dosing considerations such as run‐in dosing.
FIGURE 1.
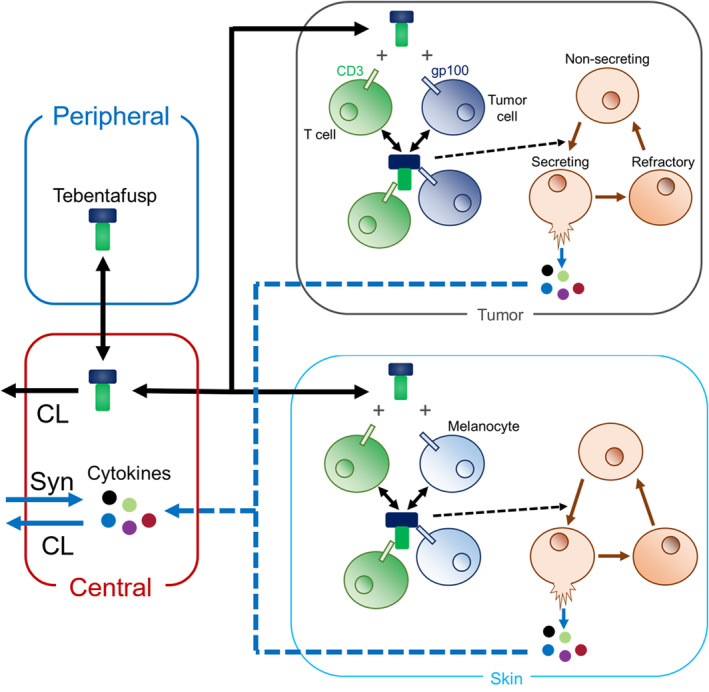
Quantitative systems pharmacology model schematic to describe changes in cytokine dynamics following bispecific dosing in a solid tumor. The model is built to capture clinical cytokine data from tebentafusp‐treated uveal melanoma patients. Tebentafusp binds CD3 on T cells and gp100 on tumor cells and melanocytes present in skin. Immune cells in the tumor and skin reside in a nonsecreting, secreting, or refractory state. All immune cells reside in the nonsecreting state before bispecific dosing. Once trimer (CD3‐tebentafusp‐gp100 complex) forms, immune cells in the nonsecreting state transition through a secreting state to a refractory state. While in the secreting state, immune cells release cytokines into the tissue that will undergo distribution to the central compartment, whereas nonsecreting and refractory immune cells do not release cytokines. Refractory immune cells transition slowly back to the nonsecreting state. Basal cytokine production (Syn), cytokine clearance (CL), and tebentafusp clearance occur in the central compartment, and thus any changes in central cytokine dynamics observed from model simulations are solely an effect of bispecific dosing. Five cytokines (interferon γ [IFNγ], interleukin [IL]‐6, IL‐10, C‐X‐C motif chemokine ligand [CXCL] 10 [CXCL10], and CXCL11) are modeled based on public tebentafusp clinical data. 19
METHODS
Model structure
The model was developed in MATLAB SimBiology R2021b. The QSP model contains the following four compartments representing physiological spaces: (1) a central compartment, (2) a peripheral compartment, (3) a tumor compartment, and (4) a skin compartment (Figure 1). The central compartment represents systemic circulation where clinical biomarker collection occurs (i.e., tebentafusp pharmacokinetics [PK], peripheral biomarkers). The peripheral compartment included as a two‐compartment model best captures tebentafusp clinical PK based on population PK analysis. 21 Tumor and skin compartments represent tissues relevant to gp100 expression, 20 where trimer formation and subsequent cytokine release occurs. Model equations and parameters are given in the supplementary material (Table S1).
In the tumor and skin compartments, tebentafusp binds CD3 expressed on T cells and gp100 expressed on tumor cells or melanocytes, respectively. Once tebentafusp forms a dimer complex with CD3 or gp100, it can bind the other antigen to form a trimer complex composed of tebentafusp‐CD3‐gp100. It is assumed that this trimer complex is the only species that triggers cytokine secretion in both tumor and skin tissue.
In the tumor and skin compartments, immune cells are modeled as being present in the following three states: nonsecreting, secreting, and refractory. Nonsecreting cells represent baseline immune cells prior to tebentafusp dosing that are assumed to be trigger sensitive for cytokine secretion. Secreting cells represent immune cells that have been triggered and are actively secreting cytokines. Refractory cells represent immune cells that have stopped secreting cytokines and are no longer trigger sensitive. Once trimer complex forms, nonsecreting cells are triggered and become secreting cells based on trimer concentration. Secreting cells transition to refractory cells rapidly. Refractory cells transition back to nonsecreting cells slowly. It is important to note that immune cells are classified only with respect to cytokine secretion and do not represent potential to perform other functions such as antitumor activities. In addition, the model assumes generalized immune cells that can represent any immune cell (i.e., monocytes, natural killer cells, the T cells themselves, etc.).
Once cytokines are secreted in the tumor or skin, they are assumed only to move to systemic circulation (central compartment) and do not undergo any reactions in tissue. In the central compartment, cytokines undergo basal production and degradation. Cytokine distribution from the central compartment to tissue compartments was assumed to not occur as these parameters were not identifiable and including these processes did not improve model optimization to available data. Thus, any changes in cytokine dynamics in the central compartment are strictly the result of trimer formation in tumor or skin tissue. The following five cytokines are included in the model: IFNγ, IL‐6, IL‐10, CXCL10, and CXCL11. These cytokines were chosen based on publicly available clinical tebentafusp data. 19
Model assumptions and parameterization
Tebentafusp dosing
Tebentafusp is dosed at 50 μg intravenously for 20 min once weekly (q.w.) to match the clinical dose regimen. 19 All simulated scenarios use this dosing regimen unless otherwise stated. After intravenous administration in the central compartment, tebentafusp undergoes biodistribution to the peripheral, tumor, and skin compartments. Parameters describing tebentafusp disposition between central and peripheral compartments were taken from the published population PK model. 21 Although high antidrug antibody titers significantly decrease tebentafusp exposure, safety and efficacy were not impacted by antidrug antibody status, 21 and thus the model here does not incorporate immunogenicity. Tebentafusp biodistributions to tumor and skin compartments were assumed to follow typical antibody kinetics such that the total tebentafusp concentration in tumor and skin is 5% the central compartment concentration. 22
Target binding
Tebentafusp binds CD3 with an affinity (KD) of 38 nM and binds gp100 with an affinity of 24 pM. 21 The on and off rates of tebentafusp binding to CD3 and gp100 are not published, so it is assumed that the off rate of tebentafusp binding to CD3 and gp100 is 1 h−1 and the on rate is set to provide the respective KD values for each target. All T cells present in the model are assumed to express 100,000 CD3/cell, 23 , 24 and all tumor cells are assumed to express 100,000 gp100/cell. 25 Expression of gp100 on melanocytes is assumed to be 100‐fold lower than on tumor cells based on RNA levels in human normal skin versus melanoma measured by Northern blot analysis. 26
Tissue parameters
Skin parameters were derived from average adult values. Skin compartment volume was assumed to be 10% of total skin volume, 27 as melanocytes are contained within the cellular epidermis, which comprises 10% of total skin volume. 28 Likewise, the baseline number of CD3+ T cells in the skin compartment was taken as 10% of the total in skin. 29 The baseline number of melanocytes was calculated assuming the skin surface area is 1.7 m2 30 and melanocyte skin density is 1000 melanocytes/mm2. 31
Tumor parameters were derived to represent uveal melanoma. Tumor volume was calculated by assuming the tumor is spherical with an 11‐mm diameter (r tumor = 5.5 mm). 32 The baseline number of tumor cells was calculated assuming the tumor cells have a volume of 6 × 10−12 L/cell. 33 The baseline number of T cells in the tumor was calculated from the CD3+ cell density in the tumor being ~1000 cells/mm2. 19 CD3+ cells/mm2 is converted to cells/liter in the tumor by assuming the tumor is spherical and there are 1000 cells/mm2 at the tumor cross‐section. Thus, the volume of the tumor is ‐fold greater than the cross‐sectional surface area, or 7.3‐fold greater using an 11‐mm diameter.
Immune cell and cytokine parameters
At baseline, all immune cells are assumed to be in the nonsecreting state. The baseline number of nonsecreting immune cells is assumed to be twofold greater than the number of T cells within each tissue. This is derived from CD3+ T cells comprising approximately half of peripheral blood mononuclear cells (PBMCs) 34 ; here, it is assumed that PBMCs represent all immune cells in a tissue and CD3+ T cells also comprise half the PBMCs in that tissue.
Each cytokine was modeled undergoing basal production and degradation in the central compartment. Baseline concentrations for each cytokine were assumed to be equal to plasma concentrations in healthy adults, and the basal clearance rate for each cytokine was set to published plasma half‐lives (Table S1). Cytokines were assumed to be at steady state prior to tebentafusp dosing, and thus the basal synthesis rate within the central compartment is calculated as
where ksyn is the cytokine synthesis rate, BL is the baseline cytokine concentration, and kdeg is the cytokine degradation rate.
Parameters describing immune cell transition rates (Table S1) were optimized to the published clinical tebentafusp data. 19 The rate at which refractory immune cells transition back to nonsecreting immune cells (parameter k_refract) could not be parameterized with available data; available data are taken across 8 weeks during which no dose interruptions occur and cytokine maximum concentration (Cmax) values continually decrease. These data imply the rate of refractory to nonsecreting immune cell transition is so slow that k_refract approaches zero or that this transition requires drug washout. Either way, this transition rate cannot be parameterized from available data, and setting k_refract = 0 provides the best model fit. The release rates of the five cytokines from secreting immune cells was also optimized to the published data. Once cytokines are released in tumor or skin tissue, they are assumed to undergo no biological reactions other than efflux from the tissue to the central compartment. The efflux rates from tumor and skin were fixed to a high value (10 h−1) to assume cytokine efflux is not a rate‐limiting step in peripheral cytokine increases following tebentafusp dosing.
Virtual populations
A virtual patient population was established by placing variability on 15 model parameters (Table S2). Variability for four of the model parameters were taken from the tebentafusp population PK model. 35 Two model parameters represent tumor size and skin surface area, and variability was incorporated to capture the range of adult values. 30 , 32 Note that tumor size is used to calculate tumor volume, number of tumor cells, and number of T cells in the tumor. Likewise, skin surface area is used to calculate skin volume, number of melanocytes, and number of T cells in the skin. Two model parameters represent variability in CD3 and gp100 expression. 23 , 32 The other seven model parameters are related to immune cell dynamics and cytokine secretion; these parameters were assumed lognormally distributed with means given by parameter optimization and percent coefficients of variation chosen to reasonably capture the clinically observed variability. 19
Model acceptance
To determine whether the model was sufficiently calibrated to the observed data, the percent prediction errors (%PEs) between mean observed and simulated peak cytokine levels (Cmax) and area under the cytokine curve (AUC) after the first, fourth, and eighth tebentafusp doses were calculated. Based on model acceptance criteria defined for predicting accuracy of PK parameters, 36 a 50% target %PE was preselected for goodness‐of‐fit criteria.
RESULTS
The bispecific cytokine QSP model was able to be successfully calibrated to tebentafusp clinical cytokine data (Figure 2, Figure S1). Based on goodness‐of‐fit criteria, the model successfully captured 13/15 cytokine Cmax and 12/15 cytokine AUC values (Table S3). The Cmax and AUC values that were not successfully captured with the prespecified goodness‐of‐fit criteria are still considered sufficiently represented by the model, discussed in the “Model Calibration” section of the supplemental information (Appendix S1). Thus, the model is deemed appropriate at representing clinical cytokine dynamics following tebentafusp dosing.
FIGURE 2.
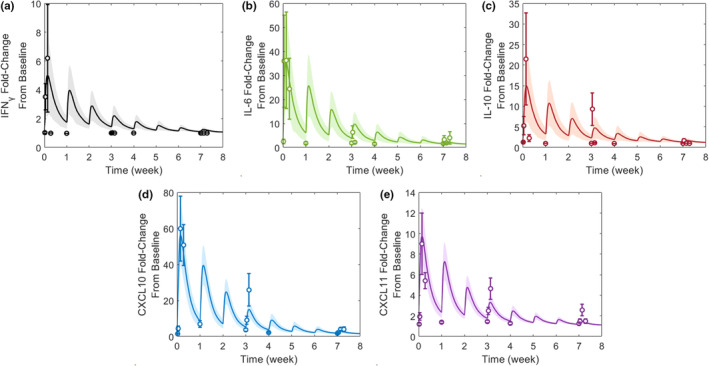
Observed versus model‐calibrated peripheral cytokine dynamics given 50‐μg tebentafusp once weekly for eight doses. The observed data are represented as mean ± standard error of the mean from 15 patients. 19 The simulated data are represented as mean (solid line) ± standard error of the mean (shaded region) across 100 cohorts of 15 virtual patients. The cytokines included in the model are (a) IFNγ, (b) IL‐6, (c) IL‐10, (d) CXCL10, and (e) CXCL11 based on published data. 19 CXCL, chemokine (C‐X‐C motif) ligand; IFNγ, interferon γ; IL, interleukin.
Contribution of tumor and healthy tissues to cytokine release
QSP model simulations predict that increased peripheral cytokines following tebentafusp dosing is primarily driven by cytokine production in skin rather than tumor tissue (Figure 3). Model simulations found that trimer complexes (CD3‐tebentafusp‐gp100) form in both tumor and skin tissue (Figure 3a), causing immune cell transitions to secret cytokines (Figure 3b,c). Although there are approximately threefold as many trimer complexes forming in tumor than skin (Figure 3a), a larger quantity of immune cells transition into the secretory state in skin compared with tumor (Figure 3b) due to the larger immune cell pool present in skin (~3000‐fold greater). This small and rapidly refracted immune cell pool in tumor results in peripheral cytokine dynamics being driven almost entirely by cytokine formation in skin. Indeed, the maximal mass of CXCL10, used as representative of all cytokines, produced in the tumor is ~1000‐fold lower than the mass produced in the skin.
FIGURE 3.
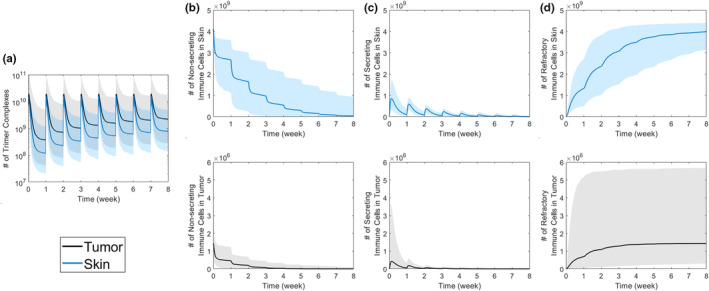
Impact of bispecific dosing on trimer and immune cell dynamics. In response to a flat, 50‐μg, once‐weekly tebentafusp dose, the dynamics of (a) trimer complex, (b) nonsecreting immune cells, (c) secreting immune cells, and (d) refractory immune cells in tumors and skin are given. Simulated outputs are shown as median (solid line) and 5th to 95th coefficients of variation (shaded region) across 1000 virtual patients.
Immune cell dynamics in the tissues reveal that cytokine Cmax attenuation over time is a consequence of immune cell desensitization by transitioning to the refractory state (Figure 3c,d). With each tebentafusp dose, the number of immune cells sensitive to triggering for cytokine release (nonsecreting immune cells) decreases via transitioning from secretory to refractory states (Figure 3). The number of immune cells that transition to the secretory state is based on (1) the number of nonsecreting immune cells available for transitioning and (2) the number of trimer complexes formed. In this scenario, tebentafusp is administered at a flat 50‐μg dose q.w., and the number of trimers does not rapidly accumulate (Figure 3a), whereas the nonsecreting immune cell pool rapidly decreases (Figure 3b). This indicates that the cytokine Cmax attenuation over time is driven by a depleting pool of nonsecreting immune cells, indicating immune cell desensitization to tebentafusp over time.
Dosing considerations: step‐up dosing
Step‐up dosing is becoming a common dosing regimen for bispecific therapies in order to alleviate CRS incidence. 37 , 38 , 39 To better understand how to select bispecific step‐up dosing regimens, and the biological mechanisms driving reduced CRS incidence with step‐up dosing, the QSP model was used to simulate cytokine dynamics across zero to five administered step‐up doses (Figure 4a). Here, CXCL10 dynamics are shown as representative for all five cytokines (the other four cytokines are shown in Figure S2). Model simulations found that CXCL10 Cmax decreased as the number of step‐up doses increased (Figure 4b). CXCL10 Cmax is considered to plateau at three step‐up doses, as Cmax decreases less than 10% with more than three step‐up doses. To explore whether step‐up dosing may impact treatment efficacy, tumor trimer formation across these step‐up dose regimens was examined (Figure S3). Simulations show that although the AUC of tumor trimer formation after the first tebentafusp dose is reduced 10‐fold using five step‐ups compared with zero step‐ups, AUC after the eighth dose is only reduced ~20% (Figure S3c).
FIGURE 4.
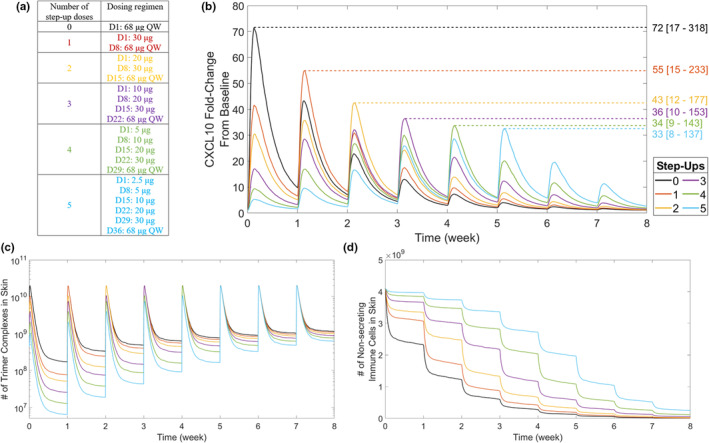
Effect of bispecific step‐up dosing on CXCL10 production. (a) Step‐up dosing was applied using 0 to 5 step‐up dose regimens. The step‐up dosing was based on the approved recommended tebentafusp dose regimen, which has two step‐up doses. 21 For regimens with greater than two step‐up doses, each step‐up dose was set as half the proceeding dose. The effect of step‐up dosing is shown for (b) simulated peripheral CXCL10 dynamics, (c) trimer complex formation in skin, and (d) number of immune cells in the nonsecreting state. Colored numbers on the right‐hand side of the graph in b indicate CXCL10 maximum concentration for each step‐up regimen represented as median [5%–95% coefficients of variation]. Simulated outputs are shown as median across 1000 virtual patients for each dose regimen. Variability in simulated outputs is not plotted for visualization ease. CXCL, C‐X‐C motif chemokine ligand; D, Day; QW, once weekly.
Interestingly, time to reach maximum concentration (Tmax) increased as the number of step‐up doses increased and always corresponded with the first 68‐μg dose administered (Figure 4b). For example, zero step‐ups gave Tmax within Day 1, three step‐ups gave Tmax within Day 22, and five step‐ups gave Tmax within Day 36. This Tmax elongation is attributed to the number of trimer complexes substantially increasing with each step‐up dose until the top 68‐μg dose is reached (Figure 4c). However, cytokine Cmax is decreased with increased step‐up dosing because fewer nonsecreting immune cells are available by the time the top 68‐μg dose is reached (Figure 4d). Cytokine Tmax can be varied by altering step‐up dose amounts; using a three step‐up dose regimen with a 68‐μg q.w. top dose while increasing the first dose amount shifts Tmax lower (Figure 5). A three step‐up dose regimen starting at 10 μg gives Tmax = 22 days (after the fourth dose); however, increasing the first dose to 20 μg or 32 μg gives Tmax = 8 days (after the second dose), and further increasing the first dose to 44 μg or 56 μg gives Tmax = 1 day (after the first dose; Figure 5). Importantly, a lower shift in Tmax is associated with an increase in CXCL10 Cmax, although every simulated three step‐up dose regimen provided CXCL10 Cmax values lower than when no step‐up dosing was applied (Figures 4b and 5). Furthermore, Tmax is dependent on model parameterization; increasing the nonsecreting to secreting immune cell transition rate gives Tmax = 1 day for zero, one, and two step‐up dose regimens (Figure S4). This finding indicates that cytokine release and CRS are highly variable based on immune cell dynamics.
FIGURE 5.
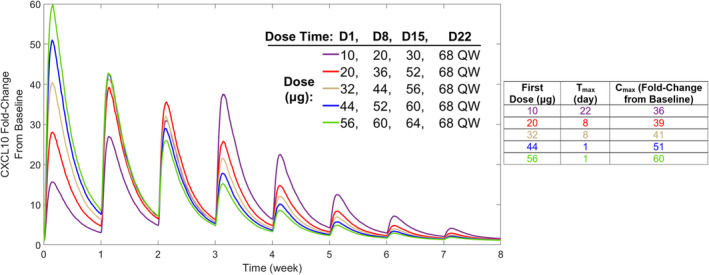
Effect of dose amount on CXCL10 production using a set step‐up dose regimen. Five dose scenarios, all applying three step‐up doses with a 68‐μg, once‐weekly top dose but varying first dose and step‐up dose amounts, were simulated. In each scenario, tebentafusp is administered on Days 1, 8, and 15 at different dose amounts, followed by 68 μg once weekly starting on Day 22. CXCL10 Tmax and Cmax across the five dose scenarios are given in the table. Cmax, maximum concentration; CXCL, chemokine (C‐X‐C motif) ligand; D, Day; QW, once weekly; Tmax, time to reach maximum concentration.
Based on findings that cytokine release is based on immune cell parameters, local sensitivity analysis was performed to better understand how disease and bispecific characteristics direct cytokine release. Peripheral CXCL10 Cmax values in response to changing these parameters across four orders of magnitude (100‐fold lower to 100‐fold higher than baseline values) were calculated from model simulations (Figure 6). Sensitivity analysis results for the other four cytokines are provided in the supplemental information (Figure S5). Sensitivity analysis found that although bispecific PK dictates CXCL10 dynamics, PK is less critical than CD3 and tumor antigen parameters. Changing bispecific clearance across four orders of magnitude provided an ~15‐fold range of CXCL10 Cmax values (4.73–72.7 fold change from baseline) compared with >90‐fold ranges provided from changing CD3 and gp100 parameters (affinity and expression per cell). With respect to immune cell dynamics, the transition rate from nonsecreting immune cells dominates over the transition rate from secreting immune cells in determining peripheral cytokine dynamics.
FIGURE 6.
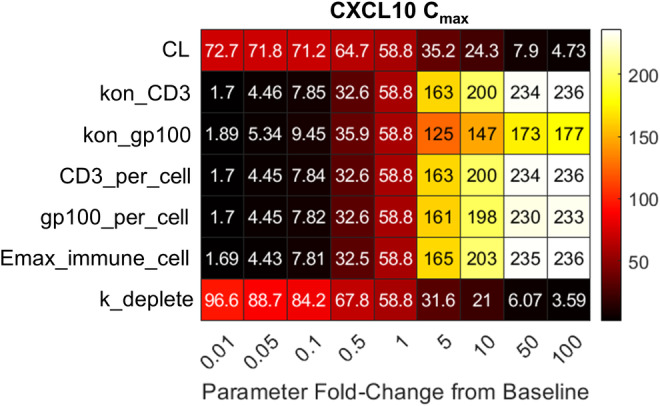
CXCL10 Cmax values in response to local sensitivity analysis across seven parameters related to bispecific or tumor characteristics. Seven parameters were changed across four orders of magnitude (100‐fold lower to 100‐fold higher than the baseline parameter value), and the peripheral CXCL10 Cmax was determined from model simulations. Colored squares and associated numbers in the table provide the CXCL10 Cmax given changing a parameter (rows) by a fold amount relative to baseline (columns). CXCL10 Cmax values are the median value across 1000 virtual patients administered a single 50‐μg tebentafusp dose. Cmax, maximum concentration; CL, clearance; CXCL, C‐X‐C motif chemokine ligand; Emax_immune_cell, transition rate from nonsecreting immune cells; k_deplete, transition rate from secreting immune cells; kon, tebentafusp binding on‐rate.
DISCUSSION
The QSP bispecific model presented here was shown to successfully represent tebentafusp clinical cytokine biomarker data. The model structure showcased here can be translated to explore cytokine dynamics from other CD3‐based bispecifics via altering tebentafusp‐specific parameters to represent the other bispecific of interest. In addition, the skin and uveal melanoma tumor compartments can be reparameterized to represent other tissues/tumors of interest, and additional cytokines can be incorporated using the same equation structure as the five cytokines modeled here. One key application of this QSP model is dose and dose‐regimen selection. Model simulations and sensitivity analyses show that cytokine response to bispecific therapy in solid tumors is dependent on bispecific parameters (PK, antigen affinity), target parameters (expression in tumor and healthy tissue), and immune cell parameters (activation and depletion rates). All these considerations can be holistically accounted for with this QSP model to explore cytokine response to different dosing scenarios. QSP‐predicted cytokine dynamics can be compared with acceptance thresholds to select the dose amount, dosing schedule, and whether run‐in dosing should be applied as well as how many step‐up doses should be selected. Such an application would provide stronger scientific rationale for first‐in‐human study designs and provide additional confidence that administered doses will be safe for patients.
A key finding here is that peripheral cytokine dynamics following bispecific dosing is dominated by cytokine production in healthy tissue. This finding is consistent with current scientific understanding: on‐target off‐tumor effects are a large contributor to toxicity from cell therapies and T cell engagers. 40 , 41 , 42 , 43 This QSP model thus presents a modeling framework to predict on‐target off‐tumor binding and cytokine production for CD3 bispecifics. This QSP model can be a powerful tool to assist in dose selection with increased confidence in safety outcomes.
Model application found that the step‐up dose regimen is a critical consideration for bispecific cytokine release. Model simulations show that cytokine Cmax is reduced with an increasing number of step‐up doses. However, increasing the number of step‐up doses may increase cytokine Tmax, shifting CRS likelihood and hospitalization requirements to later dosing cycles, which would make clinical trials more burdensome on patients. 44 Model simulations showed that Tmax is further impacted by the dose range applied across step‐up dosing; as the range between first and top dose increases in a step‐up dose regimen, increased cytokine Tmax is likely. Conversely, model simulations imply that efficacy is not likely to be impacted by step‐up dosing; step‐up dosing reduced tumor trimer formation <20% at 8 weeks compared with flat dosing, with tumor trimer formation between the two dose regimens becoming more similar as the number of doses increases. Given the median follow‐up duration for a phase II tebentafusp study was 19.5 months, 45 it is therefore likely that step‐up dosing would not impact tebentafusp efficacy. Indeed, clinical studies with tebentafusp and other bispecifics have found that step‐up dosing improves safety without compromising efficacy. 46 , 47 Note that tumor trimer formation is used as a surrogate efficacy end point here as incorporating mechanistic tumor growth inhibition is outside the scope of this study. Future work linking tumor trimer formation to tumor growth inhibition would provide greater insight into the safety–efficacy relationship provided by step‐up dosing. Overall, the QSP model shows how both the number of step‐up doses and the dose range across step‐ups are important toxicity considerations and serves as a tool to support step‐up dose optimization.
Sensitivity analysis found that parameters related to CD3 and gp100 affinity and expression highly influenced cytokine dynamics following tebentafusp dosing. Particularly, CD3 affinity is the most sensitive parameter influencing cytokine dynamics that can also be controlled during bispecific engineering. This model result agrees with preclinical findings that tuning CD3 affinity improves bispecific efficacy and safety through reducing cytokine release. 7 , 48 , 49 , 50 The agreement between QSP and empirically observed findings indicates that the QSP model can provide biologically relevant insights into the relationship between bispecific characteristics and cytokine dynamics. This highlights the QSP model utility during the candidate selection stage, where bispecific manufacturing decisions can be made based on model projected safety.
The QSP model was shown to successfully represent clinical cytokine biomarker data following tebentafusp dosing in uveal melanoma. The model was shown to predict observed biological phenomena, such as cytokine Cmax suppression, using step‐up dosing and the importance of on‐target off‐tumor binding toward CRS and toxicity. In addition, the model provides rationale for these biological phenomena that can be explored more experimentally; cytokine Cmax suppression with step‐up dosing occurs due to immune cell desensitization, and skin is the major contributor to cytokine release following tebentafusp dosing due to the large immune cell pool present in skin. Overall, the QSP model structure presented here serves as a basis to infer cytokine dynamics for any bispecific therapy or tumor type by altering model parameters to represent the scenario of interest.
AUTHOR CONTRIBUTIONS
J.W. wrote the manuscript, designed the research, performed the research, and analyzed the data.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST
The author declared no competing interests for this work.
Supporting information
Appendix S1
Weddell J. Mechanistically modeling peripheral cytokine dynamics following bispecific dosing in solid tumors. CPT Pharmacometrics Syst Pharmacol. 2023;12:1726‐1737. doi: 10.1002/psp4.12928
REFERENCES
- 1. Wang S, Chen K, Lei Q, et al. The state of the art of bispecific antibodies for treating human malignancies. EMBO Mol Med. 2021;13(9):e14291. doi: 10.15252/emmm.202114291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou S, Liu M, Ren F, Meng X, Yu J. The landscape of bispecific T cell engager in cancer treatment. Biomark Res. 2021;9(1):38. doi: 10.1186/s40364-021-00294-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zugmaier G, Klinger M, Schmidt M, Subklewe M. Clinical overview of anti‐CD19 BiTE(®) and ex vivo data from anti‐CD33 BiTE(®) as examples for retargeting T cells in hematologic malignancies. Mol Immunol. 2015;67(2 Pt A):58‐66. doi: 10.1016/j.molimm.2015.02.033 [DOI] [PubMed] [Google Scholar]
- 4. Duell J, Lammers PE, Djuretic I, et al. Bispecific antibodies in the treatment of hematologic malignancies. Clin Pharmacol Ther. 2019;106(4):781‐791. doi: 10.1002/cpt.1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ma J, Mo Y, Tang M, et al. Bispecific antibodies: from research to clinical application. Front Immunol. 2021;12:626616. doi: 10.3389/fimmu.2021.626616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shimabukuro‐Vornhagen A, Gödel P, Subklewe M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. doi: 10.1186/s40425-018-0343-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haber L, Olson K, Kelly MP, et al. Generation of T‐cell‐redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci Rep. 2021;11(1):14397. doi: 10.1038/s41598-021-93842-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kantarjian H, Stein A, Gökbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836‐847. doi: 10.1056/NEJMoa1609783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Budde LE, Sehn LH, Matasar M, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single‐arm, multicentre, phase 2 study. Lancet Oncol. 2022;23(8):1055‐1065. doi: 10.1016/S1470-2045(22)00335-7 [DOI] [PubMed] [Google Scholar]
- 10. Nathan P, Hassel JC, Rutkowski P, et al. Overall survival benefit with Tebentafusp in metastatic uveal melanoma. N Engl J Med. 2021;385(13):1196‐1206. doi: 10.1056/NEJMoa2103485 [DOI] [PubMed] [Google Scholar]
- 11. Moreau P, Garfall AL, van de Donk NWCJ, et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med. 2022;387(6):495‐505. doi: 10.1056/NEJMoa2203478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625‐638. doi: 10.1016/j.bbmt.2018.12.758 [DOI] [PubMed] [Google Scholar]
- 13. Arvedson T, Bailis JM, Britten CD, et al. Targeting solid tumors with bispecific T cell engager immune therapy. Annu Rev Cancer Biol. 2022;6(1):17‐34. doi: 10.1146/annurev-cancerbio-070620-104325 [DOI] [Google Scholar]
- 14. Carvajal RD, Sato T, Butler MO, et al. Characterization of cytokine release syndrome (CRS) following treatment with tebentafusp in patients (pts) with previously treated (2L+) metastatic uveal melanoma (mUM). J Clin Oncol. 2021;39(15_suppl):9531. doi: 10.1200/JCO.2021.39.15_suppl.9531 [DOI] [Google Scholar]
- 15. Zineh I. Quantitative systems pharmacology: a regulatory perspective on translation. CPT Pharmacometrics Syst Pharmacol. 2019;8(6):336‐339. doi: 10.1002/psp4.12403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Betts A, Haddish‐Berhane N, Shah DK, et al. A translational quantitative systems pharmacology model for CD3 bispecific molecules: application to quantify T cell‐mediated tumor cell killing by P‐cadherin LP DART(®). AAPS J. 2019;21(4):66. doi: 10.1208/s12248-019-0332-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ma H, Wang H, Sove RJ, et al. A quantitative systems pharmacology model of T cell engager applied to solid tumor. AAPS J. 2020;22(4):85. doi: 10.1208/s12248-020-00450-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hosseini I, Gadkar K, Stefanich E, et al. Mitigating the risk of cytokine release syndrome in a phase I trial of CD20/CD3 bispecific antibody mosunetuzumab in NHL: impact of translational system modeling. Npj Syst. Biol Appl. 2020;6(1):28. doi: 10.1038/s41540-020-00145-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Middleton MR, McAlpine C, Woodcock VK, et al. Tebentafusp, a TCR/anti‐CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res. 2020;26(22):5869‐5878. doi: 10.1158/1078-0432.CCR-20-1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bakker AB, Schreurs MW, de Boer AJ, et al. Melanocyte lineage‐specific antigen gp100 is recognized by melanoma‐derived tumor‐infiltrating lymphocytes. J Exp Med. 1994;179(3):1005‐1009. doi: 10.1084/jem.179.3.1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.“Kimmtrack Assessment Report (EMA/206916/2022).” https://www.ema.europa.eu/en/documents/assessment‐report/kimmtrak‐epar‐public‐assessment‐report_en.pdf.
- 22. Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67‐86. doi: 10.1007/s10928-011-9232-2 [DOI] [PubMed] [Google Scholar]
- 23. Carpentier B, Pierobon P, Hivroz C, Henry N. T‐cell artificial focal triggering tools: linking surface interactions with cell response. PLoS One. 2009;4(3):e4784. doi: 10.1371/journal.pone.0004784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicolas L, Monneret G, Debard AL, et al. Human gammadelta T cells express a higher TCR/CD3 complex density than alphabeta T cells. Clin Immunol. 2001;98(3):358‐363. doi: 10.1006/clim.2000.4978 [DOI] [PubMed] [Google Scholar]
- 25. Lepage S, Lapointe R. Melanosomal targeting sequences from gp100 are essential for MHC class II‐restricted endogenous epitope presentation and mobilization to endosomal compartments. Cancer Res. 2006;66(4):2423‐2432. doi: 10.1158/0008-5472.CAN-05-2516 [DOI] [PubMed] [Google Scholar]
- 26. Wagner SN, Wagner C, Schultewolter T, Goos M. Analysis of Pmel17/gp100 expression in primary human tissue specimens: implications for melanoma immuno‐ and gene‐therapy. Cancer Immunol Immunother. 1997;44(4):239‐247. doi: 10.1007/s002620050379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leider M. On the weight of the skin**from the New York skin and cancer unit, Department of Dermatology and Syphilology (Marion B. Sulzberger, M.D. director) and the post graduate medical School of the new York University‐Bellevue Medical Center. J Invest Dermatol. 1949;12(3):187‐191. doi: 10.1038/jid.1949.28 18117764 [DOI] [Google Scholar]
- 28. Samant PP, Prausnitz MR. Mechanisms of sampling interstitial fluid from skin using a microneedle patch. Proc Natl Acad Sci. 2018;115(18):4583‐4588. doi: 10.1073/pnas.1716772115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Clark RA, Chong B, Mirchandani N, et al. The vast majority of CLA+ T cells are resident in Normal skin. J Immunol. 2006;176(7):4431‐4439. doi: 10.4049/jimmunol.176.7.4431 [DOI] [PubMed] [Google Scholar]
- 30. Bender DA. A Dictionary of Food and Nutrition. Oxford University Press; 1995. [Google Scholar]
- 31. Jorizzo JL, Bolognia JL, Rapini RP. Dermatology: 2‐Volume Set: MOSBY. Elsevier; 2008. [Google Scholar]
- 32. Shields CL et al. Metastasis of uveal melanoma millimeter‐by‐millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009;127(8):989‐998. doi: 10.1001/archophthalmol.2009.208 [DOI] [PubMed] [Google Scholar]
- 33. Szalai E, Jiang Y, van Poppelen NM, et al. Association of uveal melanoma metastatic rate with stochastic mutation rate and type of mutation. JAMA Ophthalmol. 2018;136(10):1115‐1120. doi: 10.1001/jamaophthalmol.2018.2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kleiveland CR. In: Verhoeckx K, Cotter P, López‐Expósito I, et al., eds. Peripheral Blood Mononuclear Cells BT ‐ the Impact of Food Bioactives on Health: in Vitro and Ex Vivo Models. Springer International Publishing; 2015:161‐167. [PubMed] [Google Scholar]
- 35. U. F. and D. A. C. for D. E. and Research , “Kimmtrak Drug Approval Package”.
- 36. Abduljalil K, Cain T, Humphries H, Rostami‐Hodjegan A. Deciding on success criteria for predictability of pharmacokinetic parameters from in vitro studies: an analysis based on in vivo observations. Drug Metab Dispos. 2014;42(9):1478‐1484. doi: 10.1124/dmd.114.058099 [DOI] [PubMed] [Google Scholar]
- 37. Bartlett NL, Sehn LH, Assouline SE, et al. Managing cytokine release syndrome (CRS) and neurotoxicity with step‐fractionated dosing of mosunetuzumab in relapsed/refractory (R/R) B‐cell non‐Hodgkin lymphoma (NHL). J Clin Oncol. 2019;37(15_suppl):7518. doi: 10.1200/JCO.2019.37.15_suppl.7518 [DOI] [Google Scholar]
- 38. Leclercq G, Servera LA, Danilin S, et al. Dissecting the mechanism of cytokine release induced by T‐cell engagers highlights the contribution of neutrophils. Onco Targets Ther. 2022;11(1):2039432. doi: 10.1080/2162402X.2022.2039432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bannerji R et al. Clinical activity of REGN1979, a bispecific human, anti‐CD20 x anti‐CD3 antibody, in patients with relapsed/refractory (R/R) B‐cell non‐Hodgkin lymphoma (B‐NHL). Blood. 2019;134(Supplement_1):762. doi: 10.1182/blood-2019-122451 [DOI] [Google Scholar]
- 40. Edeline J, Houot R, Marabelle A, Alcantara M. CAR‐T cells and BiTEs in solid tumors: challenges and perspectives. J Hematol Oncol. 2021;14(1):65. doi: 10.1186/s13045-021-01067-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khadka RH, Sakemura R, Kenderian SS, Johnson AJ. Management of cytokine release syndrome: an update on emerging antigen‐specific T cell engaging immunotherapies. Immunotherapy. 2019;11(10):851‐857. doi: 10.2217/imt-2019-0074 [DOI] [PubMed] [Google Scholar]
- 42. Siegler EL, Kenderian SS. Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies. Front Immunol. 2020;11:1973. doi: 10.3389/fimmu.2020.01973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leclercq G, Steinhoff N, Haegel H, De Marco D, Bacac M, Klein C. Novel strategies for the mitigation of cytokine release syndrome induced by T cell engaging therapies with a focus on the use of kinase inhibitors. Onco Targets Ther. 2022;11(1):2083479. doi: 10.1080/2162402X.2022.2083479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Salvaris R, Ong J, Gregory GP. Bispecific antibodies: a review of development, clinical efficacy and toxicity in B‐cell lymphomas. J Pers Med. 2021;11(5):355. doi: 10.3390/jpm11050355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Carvajal RD, Butler MO, Shoushtari AN, et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: a phase 2 trial. Nat Med. 2022;28(11):2364‐2373. doi: 10.1038/s41591-022-02015-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carvajal RD, Nathan P, Sacco JJ, et al. Phase I study of safety, tolerability, and efficacy of Tebentafusp using a step‐up dosing regimen and expansion in patients with metastatic uveal melanoma. J Clin Oncol. 2022;40(17):1939‐1948. doi: 10.1200/JCO.21.01805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trudel S, Cohen AD, Krishnan AY, et al. Cevostamab monotherapy continues to show clinically meaningful activity and manageable safety in patients with heavily pre‐treated relapsed/refractory multiple myeloma (RRMM): updated results from an ongoing phase I study. Blood. 2021;138(Supplement 1):157. doi: 10.1182/blood-2021-147983 [DOI] [Google Scholar]
- 48. Dang K, Castello G, Clarke SC, et al. Attenuating CD3 affinity in a PSMAxCD3 bispecific antibody enables killing of prostate tumor cells with reduced cytokine release. J Immunother Cancer. 2021;9(6):e002488. doi: 10.1136/jitc-2021-002488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Staflin K et al. Target arm affinities determine preclinical efficacy and safety of anti‐HER2/CD3 bispecific antibody. JCI Insight. 2020;5(7):e133757. doi: 10.1172/jci.insight.133757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moore GL, Lee SH, Schubbert S, et al. Tuning T cell affinity improves efficacy and safety of anti‐CD38 × anti‐CD3 bispecific antibodies in monkeys—a potential therapy for multiple myeloma. Blood. 2015;126(23):1798. doi: 10.1182/blood.V126.23.1798.1798 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1