


Abstract
Bromodomain-containing protein 9 (BRD9) is a specific subunit of the non-canonical SWI/SNF (ncBAF) chromatin-remodeling complex, whose function in human embryonic stem cells (hESCs) remains unclear. Here, we demonstrate that impaired BRD9 function reduces the self-renewal capacity of hESCs and alters their differentiation potential. Specifically, BRD9 depletion inhibits meso-endoderm differentiation while promoting neural ectoderm differentiation. Notably, supplementation of NODAL, TGF-β, Activin A or WNT3A rescues the differentiation defects caused by BRD9 loss. Mechanistically, BRD9 forms a complex with BRD4, SMAD2/3, β-CATENIN and P300, which regulates the expression of pluripotency genes and the activity of TGF-β/Nodal/Activin and Wnt signaling pathways. This is achieved by regulating the deposition of H3K27ac on associated genes, thus maintaining and directing hESC differentiation. BRD9-mediated regulation of the TGF-β/Activin/Nodal pathway is also demonstrated in the development of pancreatic and breast cancer cells. In summary, our study highlights the crucial role of BRD9 in the regulation of hESC self-renewal and differentiation, as well as its participation in the progression of pancreatic and breast cancers.
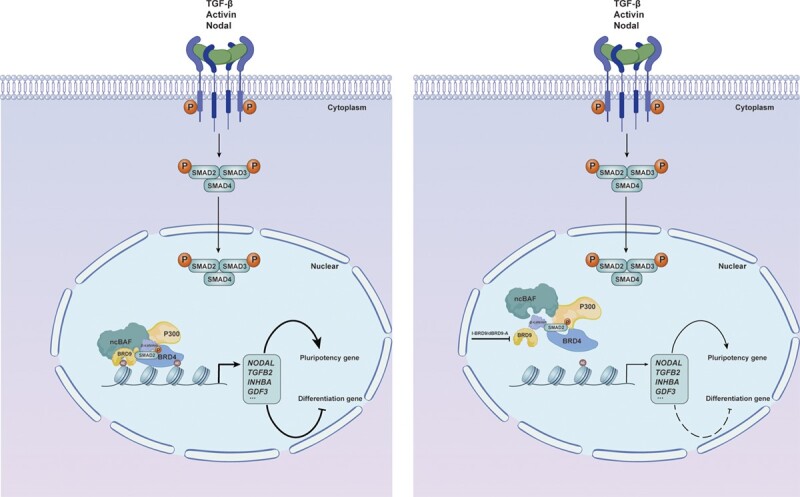
Graphical Abstract
Graphical Abstract.
Introduction
Embryonic stem cells (ESCs) possess unique characteristics, including indefinite self-renewal and pluripotency, enabling them to differentiate into all cell types under appropriate conditions (1). The maintenance of ESCs is dependent on a cooperative interplay of external signals, transcription factors and epigenetic regulators. One such epigenetic regulator is the BAF (BRG1/BRM-associated factors) complex, an ATP-dependent chromatin-remodeling complex assembled with ∼9–11 subunits that can modulate chromatin structure, thereby regulating gene expression (2). Based on distinct subunit compositions, the BAF complex is categorized into canonical BAF (cBAF), Polybromo-associated BAF (PBAF) and non-canonical BAF (ncBAF) subtypes (2,3). Multiple studies have shown that the inactivation of specific subunits of the BAF complex results in diverse aberrant phenotypes in ESCs (2). For instance, the deletion of BRG1, the catalytic subunit, leads to embryo lethality and the failure of ESC derivation from the inner cell mass (4,5). Loss of Dpf2 results in the dysregulation of Tbx3, leading to the impairment of mesoendoderm differentiation (6).
Bromodomain-containing protein 9 (BRD9) is a subunit specific to the ncBAF complex (3,7). It has a bromodomain (BD) and a DUF3512 domain, where the former serves as an epigenetic ‘reader’ that recognizes acetylated lysine residues on histones and non-histone proteins, and the latter facilitates the assembly of the ncBAF complex (8,9). Studies have highlighted the oncogenic role of BRD9 in various cancer types, influencing tumor cell growth regulation (10). In mouse ESCs, the inhibition of BRD9 has been shown to impair the maintenance of naive pluripotency (7). Recently, Sevinç et al. (11) reported that BRD9 serves as a barrier to human reprogramming, suggesting its potential role in differentiation. However, the functional and mechanistic roles of ncBAF and its specific subunits in human ESCs (hESCs) still remain poorly understood.
In this study, we combined various genomic and cell biology methods to demonstrate that inhibition of BRD9 lowered expression of pluripotency genes, and the activity of the transforming growth factor (TGF)-β/Nodal/Activin and Wnt pathways, resulting in impaired self-renewal of hESCs. Upon induction of differentiation, deficiency of BRD9 leads to defects in the differentiation of hESCs into the three germ layers, which can be rescued by supplementing NODAL, Activin A, TGF-β and WNT3A. Transcriptome analysis revealed reduced TGF-β/Nodal/Activin and Wnt signaling pathways upon suppression of BRD9. Consistently, the level of H3K27ac modification on genes associated with these pathways declined upon loss of BRD9, highlighting that BRD9 regulates these key pathways in hESCs by maintaining the level of H3K27ac. Additionally, inhibition of BRD9 significantly enhances the efficiency of cardiomyocyte differentiation in hESCs, which can be further improved in combination with a WNT inhibitor. Finally, we have shown that the BRD9-mediated regulation of the TGF-β/Activin/Nodal pathway is involved in the development of pancreatic and breast cancer cells. Therefore, our study provides novel functional and mechanistic insights into the role of BRD9 in regulating the pluripotency and differentiation of hESCs, as well as the pathogenesis of cancer.
Materials and methods
Cell lines
hESC lines H1 and H9, human induced pluripotent cell (iPSC) line NL4, pancreatic cancer cell line BxPC3 and breast cancer cell line MCF7 were used in this study.
Cell culture
Cells were maintained in home-made E8 medium [Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Thermo Fisher), recombinant human TGF-β1, recombinant human fibroblast growth factor 2 (FGF2), insulin, sodium selenite, ascorbic acid trisodium salt, transferrin and sodium bicarbonate] following the published protocol (12). Briefly, cells were cultured in E8 medium on Matrigel-coated plates (Corning) with daily medium change. When cell confluency reached 80–90%, cells were dissociated using DPBS-EDTA, and then passaged with 5 μM ROCK inhibitor Y-27632 (DC chemical).
Pancreatic and breast cancer cell lines BxPC3 and MCF7 were cultured in RPMI 1640 or DMEM (Thermo Fisher) with 10% fetal bovine serum (FBS; Gibco), respectively.
hESC differentiation
Spontaneous differentiation/neural ectoderm differentiation
hESCs were cultured on a monolayer platform using E6 medium (E8 medium without FGF2 and TGF-β) supplemented with 1× penicillin/streptomycin for 2, 4 or 6 days, starting from 30% confluence.
Mesoderm differentiation
hESCs were treated with either 20 ng/ml BMP4 (R&D Systems, 314-BP) in E8 medium for 2 or 6 days, or with 5 μM CHIR99021 (Selleck) in E6 medium for 1 day (13,14).
Endoderm differentiation
SOX17-GFP (green fluorescent protein) H9 cells at 30% confluency were treated with 5 μM CHIR99021 for 1 day, followed by the addition of 100 ng/ml activin A (R&D Systems, 338-AC) for 2 days in E5 medium (E8 medium without FGF2, TGF-β and insulin) supplemented with 1% CD lipid (15).
Cardiac differentiation
Cardiac differentiation was determined according to the methods described in (16).
Colony formation assay
To perform the colony formation assays, 500 single hESCs were seeded per well in a 12-well plate. The cells were cultured for 8 days and then stained with the alkaline phosphatase (AP) staining kit (VECTOR BCIP/NBT alkaline phosphatase SK-5300). Colonies that exhibited ≥90% AP-positive cells were categorized as undifferentiated (undiff.), colonies with ≤5% AP-positive staining cells were considered differentiated (diff.) and colonies with an intermediate number of AP-positive cells were classified as partially differentiated (mixed).
Plasmid construction
To generate short hairpin RNA (shRNA) plasmids, oligos were annealed and ligated with linearized pLKO.1 vector. Correct plasmids were confirmed by Sanger sequencing. Oligo sequences are listed in Supplementary Table S1.
The cDNA of BRD9 was cloned by polymerase chain reaction (PCR) and ligated to the LentiV_Puro vector (Addgene #111886). Correct plasmids were confirmed by DNA sequencing.
Lentivirus package
To package lentivirus, 293T cells were cultured until they reached 80% confluence, and a plasmid ratio of 4:3:1 was used for shBRD9 (or BRD9 overexpression plasmid), psPAX2 and Pmd2-vsvg. The plasmids were combined with 1 μg/ml polyethyleneimine in Opti-MEM medium at room temperature for 30 min, and then added to the cells gently. After 48 h of transfection, the supernatant containing the lentivirus was collected, and debris was removed through filtration.
Transfection and screening
To generate BRD9 knockdown hESCs or BRD9 overexpression BxPC3 cells, pre-packaged lentivirus was transduced into H1 or BxPC3 cells at ∼30% confluency. A concentration of 5 μg/ml polybrene was added to enhance the transfection efficiency. After 48 h of incubation, a concentration of 0.8 μg/ml puromycin was applied to select for BRD9 knockdown hESCs or BRD9 overexpression BxPC3 cell lines.
Western blotting
To extract proteins, cells were lysed in RIPA buffer containing a cocktail of proteinase inhibitors (Roche). The protein concentrations were determined using the BCA Protein Assay Kit (Pierce). To denature the proteins, a 1× sodium dodecylsulfate (SDS) loading buffer (Bio-Rad) was added to the protein lysate, which was then heated to 95°C for 5 min. The denatured proteins were separated by electrophoresis using a 4–12% Bis-Tris gel and transferred to a polyvinylidene fluoride (PVDF) membrane via electroblotting. The membranes were probed with primary antibodies, followed by appropriate secondary antibodies, and visualized using ECL Plus.
Apoptosis assay
Cell apoptosis was quantified using the CellEvent™ Caspase-3/7 Green Detection Reagent (C10423). Specifically, cells were initially cultured in E8 medium and treated with I-BRD9 or dBRD9-A for 48 h. Subsequently, the cells were incubated with the aforementioned reagent at a concentration of 500 nM for 30 min, followed by analysis using flow cytometry.
Cell cycle assay
The Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific, C10419H1) was utilized to perform a cell cycle analysis. hESCs were cultured in E8 medium and treated with 10 μM 5-ethynyl-2-deoxyuridine (EdU) for 30 min at 37°C. The cells were subsequently harvested using TrypLE select enzyme, fixed with 4% paraformaldehye (PFA) in phosphate-buffered saline (PBS), and permeabilized with 1× Click-iT saponin-based permeabilization and wash reagent. Thereafter, the cells were incubated with Alexa Fluor™ 647 azide, followed by staining with propidium iodide (20 mg/ml in PBS with RNase A) at 37°C for 30 min in the dark. Prior to analysis using flow cytometry, the cells were passed through a cell strainer to remove any clumps.
Fluorescence-activated cell sorting (FACS) analysis
hESCs were dissociated into single cells utilizing TrypLE select enzyme (Thermo Fisher, 12563–029), and neutralized using 10% FBS. The resulting cells were centrifuged at 300 g for 5 min and washed three times with 1× PBS. The cells were then fixed with 1% PFA (Sigma, P6148) for 10 min, followed by permeabilization with 0.5% Triton X-100 (in 1× PBS) for an additional 10 min. The primary antibodies were incubated with the cells overnight at 4°C or for 2 h at room temperature, followed by incubation with secondary antibodies at room temperature for 30 min. Prior to FACS analysis, the cells were filtered through a cell strainer to remove any clumps.
Immunofluorescence staining
The cells were fixed with 4% PFA for 10 min at room temperature, followed by blocking and permeabilization with 3% serum in PBS containing 0.3% Triton X-100. The cells were then incubated with the specified antibodies at 4°C overnight. After washing, the cells were incubated with Alex594-conjugated goat anti-mouse IgG (ThermoFisher, A-11032) and Alex488-conjugated donkey anti-goat IgG (ThermoFisher, A-11055) or Alex594-conjugated donkey anti-rat IgG (ThermoFisher, A-21209). Nuclei were visualized using 4′,6-diamidino-2-phenylindole (DAPI) staining.
Co-immunoprecipitation (Co-IP)
Cells were lysed using a lysis buffer comprising 1% IGEPAL-CA-630, 50 mM Tris–HCl pH 8.0 and 150 mM NaCl. One-tenth of the lysate was taken as input, and the remaining supernatant was pre-cleared with Protein A/G beads at 4°C for 30 min. The resulting supernatant was incubated at 4°C for 2 h with the target-specific antibody and an isotype control, followed by the addition of Protein A/G beads and overnight incubation at 4°C. The Co-IP complex was then purified, and subsequently boiled with 2× protein loading buffer for 10 min in preparation for western blot analysis.
MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay
The MTT assay was utilized to determine cell viability and the IC50. To conduct the assay, cells were seeded in a 96-well plate at a concentration of 5 × 10³ cells/well in 100 μl of culture medium. The cells were then treated with the relevant inhibitors for 24, 48 or 72 h. Next, 10 μl of MTT labeling reagent was added to each well, resulting in a final concentration of 0.5 mg/ml, and incubated for 4 h. The purple formazan crystals were then solubilized using dimethylsulfoxide (DMSO), and the spectrophotometric absorbance of the samples was measured at 570 nm.
Proliferation assay
For cell proliferation, 10 000 individual cells were seeded in each well of 24-well plates. The cells were then treated with the designated inhibitors for 1, 2 or 3 days, and the cell numbers were recorded to generate a proliferation curve.
Wound healing
The cell migration ability was assessed through a wound healing assay. Briefly, cells were seeded in a 6-well tissue culture plate at a density that allowed them to reach ∼70–80% confluence as a monolayer after 24 h of growth. A new 0.2 ml pipette tip was then used to gently and slowly scratch a straight line across the center of the well. The well was washed twice with medium to remove any detached cells and replenished with fresh medium containing or lacking I-BRD9. After ∼20 h, the cells were observed and photographed.
Quantitative real-time (qRT-PCR)
Cells were lysed using RNAios Plus reagent (Takara) for RNA exaction. Subsequently, cDNA was synthesized using Superscript III reverse transcriptase (Invitrogen), and real-time PCR was conducted using the TaqMan Gene Expression Assay (Applied Biosystems). Gene expression was measured relative to TATA-box binding protein (TBP), which is commonly used as a reference gene to normalize gene expression data, and standard deviation was calculated based on PCR triplicates. The error bars represent the standard deviation of three technical qPCR replicates from a representative experiment. The primer sequences used in the assay are provided in Supplementary Table S1.
Chromatin immunoprecipitation sequencing (ChIP-seq)
ChIP-seq was performed as previously described (17). Specifically, ∼10 × 107 cells were fixed with 1% formaldehyde at room temperature for 10 min and quenched with 0.125 M glycine. The cross-linked cells were re-suspended in sonication buffer, and subjected to sonication using a Bioruptor. Three rounds of sonication were performed with low mode pulsing settings (30 s ON; 30 s OFF), followed by four rounds of sonication with high mode pulsing settings (30 s ON; 30 s OFF). One-thirtieth of the sonicated chromatin was kept as input. The remaining chromatin was incubated with 1 μg of antibody conjugated to magnetic beads overnight at 4°C. After the immunoprecipitation, the beads were washed successively with RIPA buffer, low salt buffer, high salt buffer, LiCl buffer and 1× TE buffer. The DNA library was generated using Tn5. Finally, the DNA was extracted by reverse cross-linking at 65°C overnight with proteinase K (20 μg/ml) and sequenced using the Illumina HiSeq 2500 platform. Raw reads (FastQ files) were trimmed using Trim-Galore to remove adapter sequences at the 3′ end of the reads. Low quality reads were filtered using Cutadapt. Then the trimmed and filtered reads were aligned to the reference genome (version hg38) using bowtie2. Only mapped reads with a mapping quality >30 and properly mapped pairs (using the -q 30 and -f 2 flags in samtools) were retained. Then duplicates were removed using sambamba. The de-duplicated reads were used to call peaks using MACS2 with ‘-f BAMPE –keep-dup all -q 0.05″ options. Only peaks that were present in both replicates were used for downstream analysis. The peak annotation and Gene Ontology (GO) analysis were performed using the annotatePeaks.pl from the HOMER software suite (18) and clusterProfiler, respectively. To identify differential peaks upon BRD9 inhibition, MAnorm (19) was used with default settings, and an adjusted P-value of 0.05 and M value of 2 or –2 were used as the threshold to identify differential peaks. The full list of differential peaks can be found in Supplementary Table S2.
ChIP-qPCR
ChIP-qPCR was carried out as previously described (6). In brief, ChIP assays were performed as described above, except for the DNA library preparation step. After purification of the immunoprecipitated DNA, 1 μl of the sample was used for each qPCR . qPCR was conducted using the Applied Biosystems SYBR Green Mix (2×). qPCR was performed at least in duplicate, from at least two independent experiments, and the obtained data were normalized to input values and calculated as fold change of input. ChIP-qPCR primers were identified based on ChIP-seq analysis and are listed in Supplementary Table S1.
ChromHMM analysis
Histone modification and CTCF ChIP-seq datasets for the H1 ESCs were obtained from (20). The data were binarized using the ‘BinarizeBam’ function using the default parameter. Then 15 chromatin states were learnt using the ‘LearnModel’ function with ‘-s 42 -p 4″ settings. To calculate the enrichment of BRD9 binding events in distinct chromatin states, the BRD9 peaks coordinates were converted to hg18 using liftOver, and the ‘OverlapEnrichment’ function from ChromHMM was used as previously described (6) to compare with the 15 pre-generated chromatin states in a previous study (20).
RNA-seq
Total RNA was extracted using RNAiso Plus reagent (Takara), and sequencing was performed on Illumina HiSeq 2500 machines. Raw reads were aligned to the reference genome (version hg38) using hisat2. The count table of genes were generated using HT-Seq with default parameters. Differential gene expression was performed using DESeq2 with default parameters.
Principal component analysis (PCA)
PCA was performed using the R package FactoMineR v2.8.0 and finally the PCA results were visualized using the R package ggplot22 v3.4.2 (21,22)
GO analysis
GO analysis for enriched biological processes was performed using Metascape (http://metascape.org) to find significantly enriched terms (P-value 0.01%).
Quantitative and statistical analysis
For statistical information for each experiment, including the total number of samples analyzed and the specific statistical tests are presented as the mean ± standard error of the mean (SEM) of three independent experiments unless otherwise stated. P-values were calculated using a two-way analysis of variance (ANOVA) Dunnett's test, and statistical significance was considered when P < 0.05 (*), P < 0.01 (**), P < 0.001 (***) or P < 0.0001 (****).
Key resources
Key resources are listed in Supplementary Table S3.
Results
BRD9 deficiency impaired the maintenance of hESCs
To investigate the function of BRD9 in hESC maintenance, we examined the expression of key pluripotency factors in response to BRD9 inhibition using the BRD9-BD inhibitor, I-BRD9 (7). After 24 h of I-BRD9 exposure, both NANOG and SOX2 mRNA and protein levels were reduced in H1 hESCs, while the expression of OCT4 was only marginally affected (Figure 1A, B). Furthermore, continued I-BRD9 exposure during cell passages resulted in significant reductions in the expression of OCT4, NANOG and SOX2 (Figure 1C–E). Interestingly, the decreased expression of SOX2 upon BRD9 inhibition in hESCs returned to normal after five passages of culture (Figure 1E).
Figure 1.

BRD9 deficiency results in the instability of pluripotency in hESCs. (A) Transcript levels of pluripotency-associated genes in H1 hESCs treated with 10 μM I-BRD9 for 24 h in E8 medium, as determined by qPCR. (B) Western blot analysis of OCT4, NANOG and SOX2 protein levels in H1 hESCs treated with 10 μM I-BRD9 for 24 h in E8 medium, with glyceraldehyde phosphate dehydrogenase (GAPDH) as a loading control. (C–E) Transcript levels of OCT4 (C), NANOG (D) and SOX2 (E) in hESCs at the indicated passages treated or not with 10 μM I-BRD9 in E8 medium, as determined by qPCR. (F–H) Quantification of colony number (F) and morphology (G) of hESCs treated or not with I-BRD9 in E8 medium. Colonies were scored as undifferentiated (undiff.), mixed or differentiated (diff.) (H). (I) Transcript levels of pluripotency-associated genes in hESCs treated with 100 nM dBRD9-A for 48 h in E8 medium, as determined by qPCR. (J) Western blot analysis of BRD9, OCT4, NANOG and SOX2 protein levels in hESCs treated with 100 nM dBRD9-A for 48 h in E8 medium, with GAPDH as a loading control. (K) Transcript levels of pluripotency-associated genes in BRD9-depleted hESCs with BRD9 shRNA, as determined by qPCR.
To investigate the effects of BRD9 inhibition on the colony formation ability of hESCs, we conducted AP staining experiments using 500 single cells. The results showed that BRD9 inhibition reduced the total number of colonies formed (Figure 1F), suggesting impaired colony formation ability. Moreover, AP staining revealed a decrease in homogeneously stained, undifferentiated colonies in I-BRD9-treated hESCs, and an increase in the percentages of mixed and differentiated colonies (Figure 1G, H). Based on these findings, we conclude that BRD9 inhibition impairs the self-renewal of hESCs. To mitigate the potential non-specific inhibitory effects of I-BRD9 on other bromodomain-containing proteins, we employed a BRD9-specific degrader, namely dBRD9-A (23), to specifically degrade BRD9 protein and investigate its role in regulating hESC self-renewal (Supplementary Figure S1A). The deletion of BRD9 protein using dBRD9-A resulted in a significant down-regulation of NANOG and SOX2 at both the mRNA and the protein levels, whereas the expression of OCT4 remained unaffected (Figure 1I, J). Additionally, the knockdown of BRD9 using shRNA also led to a decrease in the expression of OCT4, SOX2 and NANOG (Figure 1K). Furthermore, the use of E6 medium, which lacks the pluripotency-promoting factors FGF2 and TGFβ-1, and inhibiting BRD9 in this medium for 24 h, resulted in a significant reduction in the mRNA and protein levels of OCT4, SOX2 and NANOG (Supplementary Figure S1B, C). These findings further support the essential role of BRD9 in maintaining hESC pluripotency.
Subsequently, we investigated the impact of BRD9 inhibition on other hESC and induced pluripotent cell lines. Consistently, exposure to either I-BRD9 or dBRD9-A for 24 h resulted in the down-regulation of pluripotency genes OCT4, SOX2 and NANOG (Supplementary Figure S1D–F). Further, expression of OCT4, NANOG and SOX2 in both NL4 iPSCs and H9 hESCs declined with increasing cell passages under I-BRD9 exposure (Supplementary Figure S1G–L). Interestingly, the decreased expression of SOX2 upon BRD9 inhibition in hESCs returned to normal after five passages of culture (Supplementary Figure S1I–L). Notably, apoptosis assays showed that BRD9 inhibition by I-BRD9 or dBRD9-A did not induce the apoptosis of hESCs (Supplementary Figure S1M). Furthermore, I-BRD9 exposure resulted in a 4.93% increase in the proportion of hESCs in the G2–M cell cycle phases, and a 12.61% reduction in the proportion of cells in the S phase (Supplementary Figure S1N). However, inhibition of BRD9 with dBRD9-A did not impact the cell cycle (Supplementary Figure S1N), indicating that BRD9 may not play a role in cell cycle regulation. Overall, our findings suggest that BRD9 inhibition impairs hESC maintenance, possibly due to the down-regulation of pluripotency-associated genes OCT4, SOX2 and NANOG in BRD9-deficient hESCs.
BRD9 deficiency suppresses meso-endoderm differentiation while it prompts neural ectoderm differentiation in hESCs
Given that the inhibition of BRD9 affects the pluripotency of hESCs (Figure 1; Supplementary Figure S1), we speculated that BRD9 may play a role in regulating hESC differentiation. To assess the influence of BRD9 on hESC differentiation, we subjected hESCs to a 4 and 6 day culture in E6 spontaneous differentiation medium with and without I-BRD9, followed by subsequent RNA-seq analysis (Supplementary Table S3). PCA revealed that I-BRD9 has a large impact on the transcriptome of hESCs, with the first principal component separating the control samples and I-BRD9-treated samples (Supplementary Figure S2A) Inhibition of BRD9 led to a significant decrease in the expression of mesoderm-associated marker genes, including TBXT, MIXL1, TBX6, HAND1 and GATA2, as well as endoderm marker genes such as EOMES and SOX7 (Figure 2A). Conversely, we observed an increase in the expression of neural ectoderm genes, notably PAX6, SOX1 and ZBTB16, upon BRD9 inhibition (Figure 2A). Intriguingly, a subset of neural crest genes, namely PAX3, TFAP2A and TFAP2B, exhibited an up-regulation trend, while FOXD3, another crest gene, showed down-regulation (Figure 2A). Subsequent validation of these findings was carried out through qPCR experiments. Consistently, the expression of typical mesoderm marker genes MIXL1, TBX6, GATA2, MESP1, HAND1 and TBXT (Figure 2B), as well as the endoderm marker genes SOX17, EOMES, Gata4, GATA6 and SOX7 (Figure 2C), decreased with the addition of dBRD9-A, whereas the expression of the neural ectoderm marker genes PAX6, SOX1, HES5 and ZBTB16 increased in hESCs (Supplementary Figure S2B). The expression of neural crest marker genes P75, SOX10 and FOXD3 decreased, while other neural crest genes PAX3, SOX9, TFAP2A, TFAP2B and AP2 increased upon the inhibition of BRD9 (Supplementary Figure S2C). Therefore, the inhibition of BRD9 may impair meso-endoderm differentiation while promoting neuronal ectoderm differentiation in hESCs. Moreover, the differentiation of hESCs into neural crest cells was also affected upon BRD9 inhibition.
Figure 2.

Deficiency of BRD9 suppresses the differentiation of hESCs into meso-endoderm, while promoting their differentiation into neural ectoderm. (A) Heatmap displaying the expression of genes associated with three germ layers during spontaneous differentiation of H1 cells in E6 medium with I-BRD9 (10 μM) treatment on days 4 and 6. (B) qPCR analysis of transcript levels of mesoderm marker genes MIXL1, TBXT6, GATA2, MESP1, HAND1 and TBXT in H1 cells in E6 medium with 100 nM dBRD9-A treatment on days 2, 4 and 6. (C) qPCR analysis of transcript levels of endoderm marker genes SOX17, EOMES, GATA4, GATA6 and SOX7 in H1 cells in E6 medium with dBRD9-A treatment on days 2, 4 and 6. (D) qPCR analysis of transcript levels of mesoderm marker genes TBXT, MIXL1, TBXT6, MESP1, HAND1 and GATA2 in BMP4-induced early mesoderm differentiation of hESCs with and without 10 μM I-BRD9 treatment for 2 days. (E) Immunostaining of TBXT in BMP4-induced early mesoderm differentiated from hESCs treated or not with 10 μM I-BRD9 for 2 days. (F) qPCR analysis of MESP1 and TBX6 transcript levels in the BMP4-induced late mesoderm differentiation stage of hESCs treated or not with 10 μM I-BRD9 for 6 days. (G) Schematic of the endoderm differentiation of hESCs. qPCR analysis of transcript levels of endoderm markers SOX17, EOMES, SOX7, GATA4 and GATA6 in hESCs differentiated into endoderm with and without I-BRD9 treatment. (H) Flow cytometry analysis of SOX17 protein expression in control and 10 μM I-BRD9-treated hESCs under endoderm differentiation conditions. (I) qPCR analysis of SOX17, EOMES, SOX7, GATA4 and GATA6 transcript levels in endoderm differentiated from hESCs with BRD9 knocked down. (J) Transcript levels of neuroectoderm markers PAX6, SOX1, HES5 and NR2E1 and neural crest marker P75, SOX10, FOXD3, PAX3 and SOX9 in H1 cells cultured in E6 medium upon BRD9 inhibition with 10 μM I-BRD9 for 4 days.
To further confirm the effect of BRD9 on mesoderm differentiation, we induced mesoderm differentiation with the addition of BMP4 (24). The expression of early mesoderm marker genes TBXT, MIXL1, TBX6, MESP1, HAND1 and GATA2 decreased in H1 and H9 hESCs, and NL4 iPSCs cultured in the presence of I-BRD9 (Figure 2D; Supplementary Figure S2D, E). Immunostaining showed a significantly reduced level of TBXT (Figure 2E), supporting the inhibition of mesoderm differentiation upon treatment with I-BRD9. Consistently, down-regulation of BRD9 with shRNA plasmids also led to reduced expression of TBXT, MIXL1, TBX6, MESP1, HAND1 and GATA2 during mesoderm differentiation (Supplementary Figure S2F). Similarly, the inhibition of BRD9 also reduced the expression of TBXT and MIXL1 in mesoderm differentiation induced by CHIR00921 treatment for 1 day (Supplementary Figure S2G). Furthermore, I-BRD9 repressed the expression of MESP1 and TBX6, the marker genes of mesoderm at a later differentiation stage (Figure 2F).
To eliminate non-specific inhibition of I-BRD9 on other BRD proteins, we employed dBRD9-A to deplete BRD9 during differentiation. The expression of TBXT, MIXL1, TBX6, MESP1, HAND1 and GATA2 decreased during BMP4- or CHIR00921-induced mesoderm differentiation upon BRD9 degradation (Supplementary Figure S2H, I). Immunostaining showed a significant decrease in TBXT expression upon treatment with dBRD9-A (Supplementary Figure S2J), supporting the inhibition of mesoderm differentiation. These results together reveal that impairing BRD9 function results in the suppression of mesoderm differentiation.
Next, we studied the role of BRD9 in endoderm differentiation of hESCs. Endoderm differentiation of hESCs was induced. The addition of I-BRD9 reduced the expression of endoderm marker genes SOX17, EOMES, SOX7, GATA4 and GATA6 in the endoderm differentiation of hESCs (Figure 2G). Decreased SOX17–GFP-positive cell number measured by FACS after treatment with I-BRD9 further confirmed that the loss of BRD9 suppressed endoderm differentiation (Figure 2H). Consistently, down-regulation of BRD9 with the shRNA plasmids reduced the expression of SOX17, EOMES, SOX7, GATA4 and GATA6 during endoderm differentiation (Figure 2I). Similarly, the inhibition of BRD9 with dBRD9-A reduced the expression of SOX17, EOMES, SOX7, GATA4 and GATA6 in the endoderm differentiation of hESCs and the percentage of SOX17–GFP-positive cells, supporting the repressive role of BRD9 on endoderm differentiation (Supplementary Figure S2K, L).
Next, we proceeded to investigate the impact of I-BRD9 on the differentiation of hESCs toward neural ectoderm. The introduction of I-BRD9 or dBRD9-A led to heightened expression levels of PAX6, SOX1, HES5 and NR2E1 (Figure 2J; Supplementary Figure S2M), indicating a promotion of neuroectoderm differentiation. However, there was a decrease in the expression of neural crest markers, namely P75, SOX10 and FOXD3, alongside an increase in other neural crest genes PAX3 and SOX9 upon BRD9 inhibition (Figure 2J; Supplementary Figure S2M). These observations collectively point towards an aberrant neural crest differentiation in hESCs under BRD9 inhibition.
Similarly, treatment with I-BRD9 induced heightened expression of neural ectoderm markers PAX6, SOX1, HES5 and NR2E1, and a disrupted expression profile of neural crest genes P75, SOX10, FOXD3, PAX3 and SOX9 in both H9 hESCs and NL4 iPSCs (Supplementary Figure S2N, O). In conclusion, our findings indicate that the inhibition of BRD9 fosters neural progenitor differentiation, while concurrently impacting the typical process of neural crest differentiation.
BRD9 regulates hESC differentiation via controlling the TGF-β signaling pathway
To investigate the mechanism by which BRD9 impacts on hESC maintenance and cell fate decisions, RNA-seq was used to assess the gene expression profile of hESCs after treatment with I-BRD9 for 12, 24 and 48 h. A significant change in gene expression was observed as early as after 12 h of treatment with I-BRD9, and prolonged treatment resulted in more pronounced changes (Figure 3A; Supplementary Figure S3A; Supplementary Table S4). We further analyzed the gene expression changes after 12 h of treatment and found that 239 genes were down-regulated and 303 genes were up-regulated (Supplementary Table S4). GO analysis showed that the down-regulated genes were functionally related to endoderm, mesoderm and neural differentiation (Figure 3B), confirming the role of BRD9 in hESC differentiation. The down-regulated genes were associated with TGF-β/Nodal and BMP4 signaling pathways (Figure 3B), suggesting that BRD9 regulates hESC maintenance by controlling these signaling pathways. The up-regulated genes were related to DNA methylation, chromatin remodeling and the Wnt signaling pathway (Supplementary Figure S3B).
Figure 3.

BRD9 regulates hESC maintenance and differentiation via controlling TGF-β and Wnt signaling pathway activity. (A) Global gene expression profiles of H1 cells upon I-BRD9 treatment for 12, 24 and 48 h. (B) GO analysis of down-regulated genes in hESCs treated with I-BRD9 for 12 h. (C) Transcript levels of TGF-β pathway-associated genes in hESCs treated with I-BRD9 for 24 h. (D) Western blot analysis of total SMAD2 and p-SMAD2 protein levels in hESCs cultured in E8 and E6 medium treated with I-BRD9 for 24 h. (E and F) Transcript levels of TGFB2, NODAL, LEFTY1, LEFTY2 and NANOG genes in hESCs cultured in E8 medium treated with I-BRD9 with and without addition of TN [5.2 ng/ml of TGF-β (T) and 100 ng/ml of NODAL (N)] proteins for 24 h. (G) Western blot analysis of SMAD2 and p-SMAD2 protein levels in hESCs cultured in E8 medium treated with 10 μM I-BRD9 with and without NODAL for 24 h; GAPDH served as a loading control. (H and I) Transcript levels of TBXT, MIXL1, TBX6, MESP1, HAND1 and GATA2 in early mesoderm differentiated from hESCs cultured in E8 medium with 20 ng/ml BMP4 upon treatment with 10 μM I-BRD9 (H) or 100 nM dBRD9-A (I) with and without TNA [5.2 ng/ml TGF-β (T), 100 ng/ml NODAL (N) and 100 ng/ml ACTIVIN-A (A)] for 2 days. (J) Transcript levels of endoderm markers SOX17, EOMES, SOX7, GATA4 and GATA6 in endoderm differentiated from hESCs upon BRD9 inhibition with and without TNA for 3 days. (K) Transcript levels of marker genes in neuroectoderm and neural crest differentiated from hESCs cultured in E6 medium upon BRD9 inhibition with and without TGF-β for 4 days. (L) Transcript levels of WNT3 and WNT3A in hESCs treated with 100 nM dBRD9-A for 2 days. (M) Western blot analysis of total β-CATENIN and active β-CATENIN protein levels in hESCs cultured in E8 medium treated with I-BRD9 for 24 h. (N) qPCR analysis of transcript levels of TBXT and MIXL1 in early mesoderm differentiated from hESCs upon BRD9 inhibition with and without 2 ng/ml WNT3A for 2 days. (O) Transcript levels of SOX17 and EOMES in endoderm differentiated from hESCs upon BRD9 inhibition with and without 2 ng/ml WNT3A. (P) qPCR analysis of transcript levels of WNT3 in I-BRD9-treated hESCs with and without TGF-β (5.2 ng/ml), NODAL (100 ng/ml) and ACTIVIN-A (100 ng/ml) for 24 h. (Q) qPCR analysis of transcript levels of TGF-β-associated genes in hESCs cultured in E8 medium upon BRD9 inhibition with and without 2 ng/ml WNT3A for 2 days.
The TGF-β/Activin/Nodal signaling pathway is essential for the maintenance of pluripotency in hESCs (25,26). Inhibition of BRD9 hinders the maintenance of hESCs, resulting in the down-regulation of genes associated with the TGF-β pathway (Figure 3B), suggesting that BRD9 regulates hESC maintenance by controlling the expression of the pluripotency factor NANOG via regulating the TGF-β pathway. Indeed, the inhibition of BRD9 by I-BRD9, dBRD9-A or shRNA down-regulated the expression of GDF3, TGFB2, NODAL, LEFTY1, LEFTY2 and INHBA in hESCs (Figure 3C; Supplementary Figure S3C, D). Recent investigations have highlighted the utilization of E8 medium to characterize hPSCs in an intermediate state between naive and primed (27). In order to evaluate the impact of BRD9 inhibition on gene expression related to the TGF-β pathway across various primed media such as TeSR, E8 + 1 μM LPA medium (28), E8 + 1.6% Alubmax medium (27), as well as the naive medium 5iLAF medium (29), we embarked on further examinations. Intriguingly, the application of I-BRD9 to inhibit BRD9 led to reduced expression of GDF3, TGFB2, NODAL, LEFTY1, LEFTY2 and INHBA in hESCs cultured in all three primed media and the 5iLAF naive medium (Supplementary Figure S3E–H). This observation underscores the diminished activity of the TGF-β pathway due to BRD9 inhibition.
Previous studies showed that activated TGF-β/Activin/Nodal signaling leads to SMAD2/3 phosphorylation, which regulates the expression of pluripotency marker genes, such as NANOG and OCT4, to maintain the pluripotency of hESCs (30). Inhibiting or depleting BRD9 with I-BRD9 or dBAD9-A did not alter the transcript and protein levels of SMAD2 but decreased p-SMAD2 levels in both E8 medium and differentiation medium E6 (Figure 3D; Supplementary Figure S3I, J). Rescue experiments using TGF-β, NODAL and/or ACTIVIN A proteins restored the impaired expression of NODAL, TGFB2, LEFTY1, LEFTY2 and NANOG upon BRD9 inhibition by both I-BRD9 (Figure 3E, F) and dBRD9-A (Supplementary Figure S3K, L). Furthermore, the addition of NODAL rescued the decreased p-SMAD2 level upon the inhibition of BRD9 (Figure 3G). Thus, it can be concluded that BRD9 regulates the activity of the TGF-β pathway and controls the expression of NANOG.
As the loss of BRD9 led to decreased activity of the TGF-β/Activin/Nodal signaling pathway (Figure 3C, D; Supplementary Figure S3C, D, J), we speculated that adding TGF-β/NODAL/ACTIVIN A could rescue the differentiation defect resulting from BRD9 inhibition. Indeed, supplementation with NODAL, ACTIVIN A or TGF-β partially restored the repressed expression of mesodermal marker genes TBXT and MIXL1 caused by BRD9 inhibition via both I-BRD9 and dBRD9-A (Supplementary Figure S3M, N). Consistently, the simultaneous addition of NODAL, ACTIVIN A and TGF-β (TNA) rescued the repressed expression of mesodermal marker genes TBXT, MIXL1, TBX6, MESP1, HAND1 and GATA2 resulting from the inhibition of BRD9 with both I-BRD9 and dBRD9-A (Figure 3H, I). Additionally, supplementation with TNA restored the repression of endodermal marker genes SOX17, EOMES, SOX7, GATA4 and GATA6 caused by BRD9 inhibition via both I-BRD9 (Figure 3J) and dBRD9-A (Supplementary Figure S3O).
Moreover, the addition of TNA restored the expression of the neuroectodermal and neural crest marker genes back to normal levels (Figure 3K; Supplementary Figure S3P). We thus conclude that BRD9 regulates the differentiation of hESCs by modulating the activity of the TGF-β/Activin/Nodal pathway.
BRD9 regulates hESC differentiation via controlling the Wnt pathway
GO analysis indicates that inhibiting BRD9 results in the deregulation of genes associated with the Wnt pathway (Supplementary Figure S3B). The expression of WNT3 and WNT3A decreased in H1 hESCs and NL4 iPSCs when BRD9 was inhibited using I-BRD9 or dBRD9-A treatment in E8 medium (Figure 3L; Supplementary Figure S3Q). Similarly, the expression of WNT3 and WNT3A decreased in H1 hESCs cultured in primed media, such as TeSR and E8 + 1 μM LPA medium, as well as in the naive 5iLAF medium (Supplementary Figure S3R–T). Notably, the protein level of active β-CATENIN decreased in E8 medium treated with I-BRD9 (Figure 3M). This observation points to impact of the inhibition of BRD9 on reducing Wnt pathway activity. The introduction of WNT3A partially reinstated the decreased expression of mesodermal marker genes TBXT and MIXL1, as well as endodermal marker genes SOX17 and EOMES caused by BRD9 inhibition (Figure 3N, O; Supplementary Figure S3U), signifying BRD9’s role in regulating hESC differentiation towards mesoderm and endoderm by modulating WNT3 and WNT3A expression. Furthermore, the decreased expression of WNT3 and WNT3A was rescued by the addition of NODAL, TGF-β or ACTIVIN A (Figure 3P; Supplementary Figure S3V). Similarly, the introduction of WNT3A also restored the reduced expression of NODAL, TGF-β, INHBA, GDF3, LEFTY1 and LEFTY2 resulting from BRD9 inhibition (Figure 3Q). To conclude, BRD9 plays a regulatory role in hESC differentiation through the modulation of the Wnt signaling pathway.
The inhibition of BRD4 hindered the differentiation of hESCs into meso-endoderm by modulating the activities of the TGF-β and Wnt pathways
Gatchalian et al. showed that BRD4 interacts with BRD9, indicating overlapping roles in regulating the naïve pluripotency transcriptional network (7). Consistent with the previous report (31), inhibition of BRD4 with JQ1 or ARV-825 decreased the expression of pluripotency genes OCT4, SOX2 and NANOG in hESCs (Supplementary Figure S4A, B). The neural ectoderm gene PAX6 increased upon the inhibition of BRD4 (Supplementary Figure S4C), consistent with the repressive role of BRD4 in neuroectodermal differentiation (31). However, the neural crest marker gene P75 was down-regulated upon BRD4 inhibition (Supplementary Figure S4C). The expression of early mesoderm marker genes TBXT and MIXL1, as well as later mesoderm marker genes MESP1 and TBX6, was observed to be down-regulated upon inhibition of BRD4 using either JQ1 or ARV-825 (Figure 4A, B; Supplementary Figure S4D). During endoderm differentiation, both qPCR and immunostaining experiments revealed that the expression of the endoderm marker gene SOX17 decreased in hESCs upon BRD4 inhibition with JQ1 or ARV-825 (Figure 4C; Supplementary Figure S4E). Our findings suggest that, similar to BRD9, BRD4 plays a critical role in mesoderm and endoderm differentiation of hESCs.
Figure 4.

Inhibition of BRD4 suppressed meso-endodermal differentiation, while promoting the differentiation of cardiomyocytes in hESCs. (A and B) qPCR analysis of transcript levels of TBXT and MIXL1 in early mesoderm (A), and MESP1 and TBX6 in late mesoderm (B) differentiated from hESCs following inhibition of BRD4 with 50 nM JQ1. (C) Transcript levels of SOX17 in endoderm differentiated from hESCs following inhibition of BRD4 with 10 or 50 nM JQ1, or 1 nM ARV-825. (D) Western blot analysis of SMAD2 and p-SMAD2 protein levels in hESCs cultured in E8 medium following inhibition of BRD4 with 50 nM JQ1 or 1 nM ARV-825 for 2 days. (Eand F) qPCR analysis of transcript levels of TGF-β pathway-associated genes (E), and WNT3 and WNT3A (F) in hESCs cultured in E8 medium following inhibition of BRD4 with 50 nM JQ1 for 2 days. (G) qPCR analysis of the transcript levels of TNNT2 and NKX2.5 in cardiomyocytes derived from hESCs following treatment with 10 μM I-BRD9, 2.5 μM IWP2 or a combination of both. (H) FACS analysis of TNNT2-positive cells in cardiomyocyte differentiation of hESCs following treatment with 10 μM I-BRD9, 2.5 μM IWP2 or a combination of both. (I and J) qPCR analysis of the transcript levels of TNNT2 (I) and NKX2.5 (J) in cardiomyocytes derived from hESCs following treatment with 2.5 μM IWP2, 1 nM ARV-825 or a combination of both.
Inhibition of BRD4 using JQ1 or ARV-825 did not alter the protein level of SMAD2 but reduced p-SMAD2 levels in hESCs (Figure 4D). qPCR analysis revealed that BRD4 inhibition with JQ1 or ARV-825 decreased the expression of GDF3, TGFB2, NODAL, LEFTY1, LEFTY2 and INHBA (Figure 4E; Supplementary Figure S4F). In addition, the expression of WNT3 and WNT3A was found to be significantly reduced in hESCs upon treatment with JQ1 (Figure 4F; Supplementary Figure S4G). Thus, similar to BRD9, inhibition of BRD4 reduced the activities of TGF-β and Wnt pathways in hESCs.
The inhibition of BRD9 and BRD4 increases the cardiomyocyte differentiation of hESCs
The Wnt pathway plays multiple roles in cardiomyocyte differentiation and development (32). Activation of the Wnt pathway with CHIR99021 induces mesoderm formation in early differentiation (33). Inhibiting the Wnt pathway with IWP2 promotes the generation of cardiomyocyte progenitor cells in the middle period of differentiation (34). Insulin supplementation enriches cardiomyocytes (33). Inhibition of the Wnt pathway with IWP2 from Day 2 to Day 5 increases the expression of TNNT2 and NXK2.5, typical markers of cardiomyocytes (Figure 4G; Supplementary Figure S4H, I), consistent with previous reports (35). Based on the observed repression of WNT3 and WNT3A upon inhibition of BRD9 (Figure 3L; Supplementary Figure S3O), we applied BRD9 inhibition to regulate cardiomyocyte differentiation. The addition of I-BRD9 led to a significant increase in the expression of TNNT2 and NXK2.5 (Figure 4G; Supplementary Figure S4I), thereby demonstrating that BRD9 inhibition promotes cardiomyocyte differentiation. Furthermore, treatment with I-BRD9 and IWP2 resulted in a synergistic effect, leading to a further increase in the expression of TNNT2 and NXK2.5 (Figure 4G; Supplementary Figure S4I). These findings were further corroborated by FACS analysis, which showed a marked increase in the percentage of TNNT2-positive cells following treatment with IWP2 (∼77.55%), I-BRD9 (∼44.72%) and I-BRD9/IWP2 (∼88.57%) (Figure 4H).
In agreement with findings for BRD9, inhibiting BRD4 suppresses the expression of WNT3 and WNT3A (Figure 4F; Supplementary Figure S4G). Treatment with JQ1, a selective BRD4 inhibitor, resulted in a dose-dependent increase in the expression of the cardiomyocyte-specific genes TNNT2 and NKX2.5 (Supplementary Figure S4J, K). Furthermore, co-treatment with JQ1 and IWP2 resulted in an even greater up-regulation of TNNT2 and NKX2.5, indicating a synergistic effect between BRD4 inhibition and Wnt pathway blockade. Similar results were obtained with the BRD4-selective degrader, ARV-825 (Figure 4I, J). Taken together, our data suggest that targeting BRD9 or BRD4 is a promising strategy for enhancing the cardiomyocyte differentiation potential of hESCs.
BRD9 collaborates with BRD4, SMAD2/3 and β-CATENIN to regulates gene expression in hESCs
To explore the mechanism by which BRD9 regulates the signaling pathways and pluripotency genes in hESCs, ChIP-seq experiments using a BRD9 antibody were performed. Two biological replicates were performed, and only peaks that are present in both replicates were considered as the high confidence binding sites. By this standard, a total of 7494 peaks were found using model-based analysis of ChIP-seq (MACS) (36). About 45% of the binding events occurred at genomic locations within ± 2 kb of transcriptional start sites (TSSs), indicating that BRD9 is mainly located around promoters (Figure 5A). To further characterize the genomic regions occupied by BRD9, we compared the BRD9 peaks with the 15 chromatin states in the H1 cell line defined by eight different histone marks and CTCF in a previous study using a hidden Markov model (ChromHMM) (37,20). Interestingly, in contrast to the classical BAF complex component DPF2 (6), BRD9 tends to appear near active promoters and strong enhancers (Figure 5B, states 1 and 4), which is consistent with the analysis of the genomic distribution of BRD9-binding sites (Figure 5A). In addition, BRD9 also appeared around weak and inactive/poised promoters, though to a lesser extent (Figure 5B, states 2 and 3). Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that BRD9-occupied sites are located close to genes associated with functions in pluripotency regulation, TGF-β, Wnt, Hippo signaling pathways and various cancers (Figure 5C), which is consistent with the function of BRD9 on ESC self-renewal maintenance and differentiation (Figures 1–4; Supplementary Figures S1–S4).
Figure 5.

BRD9 collaborates with BRD4, SMAD2/3 and β-CATENIN to regulate gene expression in hESCs. (A) Distribution of BRD9 target sites relative to TSSs in hESCs, determined by ChIP-seq. (B) Chromatin state enrichment of BRD9 target sites in hESCs. ESC chromatin states were defined in Ernst et al. (20) using ChromHMM. Rows represent chromatin states and their mnemonics. Columns give the frequency of the indicated histone marks for each chromatin state (ChromHMM emission probabilities), color-coded from blue (highest) to white (lowest). Enrichment of BRD9 in each chromatin state is shown. (C) KEGG pathway enrichment analysis with BRD9 target sites within 10 kb of their TSS. Significantly enriched KEGG pathways (P < 0.05) are presented. (Dand E) Genome browser view of ChIP-seq tracks for the histone mark H3K27ac at the NODAL (D) and NANOG (E) loci in hESCs cultured with and without 10 μM I-BRD9 for 2 days. Regions highlighted in brown signify ESC promoter and enhancer regions. The values on the y-axis represent fold enrichment over control. (F) H3K27ac levels at TGF-β-related genes in hESCs, treated or not with 100 nM dBRD9A for 2 days, determined by ChIP-qPCR. (G) Co-IP experiment with α-BRD4 antibody showing the interaction of BRD4 with P300, BRD9, RNAP II, SMAD2 and β-CATENIN. (H) Venn diagram depicting the number of H3K27ac peaks that are bound by BRD4, BRD9 and SMAD2 from H3K27ac, BRD4, BRD9 and SMAD2 ChIP-seq analyses. (Iand J) Genome browser view of BRD4, BRD9, SMAD2/3, β-CATENIN binding and H3K27ac modification at the NODAL (I) and NANOG (J) loci in hESCs. (K) BRD4 binding at the indicated genes in hESCs, treated or not with 100 nM dBRD9A for 48 h, determined by ChIP-qPCR, (L and M) SMAD2 binding at TGF-β-related genes (L) and NANOG, OCT4, WNT3 and WNT3A genes (M) in hESCs cultured in E8 medium, treated or not with 10 μM I-BRD9 for 2 days, determined by ChIP-qPCR, (N–P) BRD9 binding at TGF-β-related genes (N), and pluripotency genes NANOG, OCT4 and SOX2 (O), WNT3 and WNT3A genes (P) in hESCs cultured in E8 medium, treated or not with 50 nM JQ1 for 2 days, determined by ChIP-qPCR. (Q–S) β-CATENIN binding at TGF-β-related genes (Q), and pluripotency genes NANOG, OCT4 and SOX2 (R), WNT3 and WNT3A genes (S) in hESCs cultured in E8 medium, treated or not with 50 nM JQ1 for 2 days, determined by ChIP-qPCR.
BRD9 possesses a BD domain that can recognize acetylation of histone and non-histone proteins, especially for histone H3K27ac, a well-known active enhancer mark (9,38). Considering that the activities of TGF-β/Nodal/Activin and Wnt pathways were suppressed upon BRD9 loss, we suspected that the level of H3K27ac changed with I-BRD9 treatment, especially in the genomic regions of TGF-β/Nodal/Activin pathway- and Wnt pathway-related genes. Indeed, ChIP-seq experiments with H3K27ac antibody showed that the level of H3K27ac on regions of TGF-β-associated genes, such as NODAL, GDF3, LEFTY1 and LEFTY2, the pluripotency gene NANOG and Wnt pathway genes WNT3 and WNT3A dramatically declined in hESCs treated with I-BRD9 (Figure 5D, E; Supplementary Figure S5A–D), which was validated by ChIP-qPCR in hESCs after treatment with dBRD9-A (Figure 5F; Supplementary Figure S5E). While I-BRD9 down-regulates the expression of NANOG (Figure 1A, B), the reduction in H3K27ac levels around the NANOG locus detected through ChIP-seq is not as pronounced as that observed in the ChIP-qPCR experiment (Figure 5E; Supplementary Figure S5E). This discrepancy might suggest a moderate role for BRD9 in regulating NANOG by influencing H3K27ac deposition. P300 is the predominant H3K27 acetyltransferase in ESCs (39). We speculated that the inhibition of BRD9 may destabilize the binding of P300, and thereby reduce the levels of H3K27ac at those genes. Indeed, ChIP-qPCR experiments with P300 antibody revealed the decreased binding of P300 on regions of TGF-β-associated genes, such as TGFB2, NODAL, INHBA, LEFTY1 and LEFTY2, the pluripotency gene NANOG and Wnt pathway genes WNT3 and WNT3A in hESCs treated with dBRD9-A (Supplementary Figure S5F, G). In line with the minor decreased H3K27ac modification at the NANOG gene (Figure 5E), the binding of P300 showed a minor decrease upon BRD9 inhibition (Supplementary Figure S5G). Therefore, BRD9 may regulate the expression of pluripotency genes, the activity of TGF-β and Wnt pathways, and thereby participate in cell fate decisions and pluripotency maintenance of hESCs.
Both BRD9 and BRD4 play pivotal roles in regulating the expression of pluripotency genes and influencing the activities of the TGF-β and Wnt pathways (Figures 3D, E, L and 4D–F; Supplementary Figure S4F, G). Similarly, the introduction of WNT3A successfully reinstated the decreased expression of genes related to the TGF-β pathway upon BRD9 inhibition (Figure 3Q). These findings strongly suggest a reciprocal interplay between the TGF-β and Wnt pathways when subjected to BRD9 inhibition. Consequently, we postulate that a complex comprising BRD4, BRD9, SMAD2, β-CATENIN and P300 might collaboratively oversee the expression of pluripotency genes, along with the regulation of TGF-β and Wnt pathway activities. Encouragingly, this hypothesis gains support from Co-IP experiments, which substantiate the interaction of BRD4 with BRD9, β-CATENIN, P300, p-SMAD2, SMAD2 and RNAP-II (Figure 5G). Consistently, ChIP-seq analysis for BRD9, BRD4, SMAD2 and H3K27ac reveals 1930 co-bound sites located close to 1138 genes associated with functions in stem cell differentiation, heart development, cell population proliferation, TGF-β signaling pathway and various cancers such as breast cancer and pancreatic adenocarcinoma (Figure 5H; Supplementary Figure S5H). Enrichment analysis revealed that human BRD9-binding sites were enriched with the CTCF motif, consistent with mouse BRD9. However, they also had the OCT4/SOX2/TCF/NANOG motif, unlike mouse BRD9 (Supplementary Figure S5I) (7). This observation is in line with the role of human BRD9 as a regulator of the master pluripotency regulators and its involvement in the maintenance and differentiation of hESCs. ChIP-seq analysis shown the co-localization of BRD4, BRD9, β-CATENIN at H3K27ac deposited sites of NODAL, GDF3 and NANOG genes (Figure 5I, J; Supplementary Figure S5J). ChIP-qPCR analysis demonstrated the decreased binding of BRD4 at TGFB2, NODAL, GDF3, LEFTY1, LEFTY2, NANOG, SOX2, KLF4 and WNT3A genes upon the inhibition of BRD9 with dBRD9-A or I-BRD9 (Figure 5K; Supplementary Figure S5K). Similarly, inhibition of BRD9 reduced the binding of SMAD2 at NODAL, TGFB2, INHBA, GDF3, LEFTY1, LEFTY2, NANOG, OCT4, WNT3 and WNT3A genes (Figure 5L, M). The binding of β-CATENIN decreased at NODAL, TGFB2, GDF3, LEFTY1, LEFTY2, NANOG, OCT4, WNT3 and WNT3A genes upon BRD9 inhibition (Supplementary Figure S5L, M). Furthermore, we demonstrated that the inhibition of BRD4 with JQ1 decreased the binding of BRD9 at NODAL, TGFB2, INHBA, GDF3, LEFTY1, LEFTY2, NANOG, OCT4, WNT3 and WNT3A genes (Figure 5N–P). Similarly, the binding of both β-CATENIN and SMAD2 at NODAL, TGFB2, INHBA, GDF3, LEFTY1, LEFTY2, NANOG, OCT4, WNT3 and WNT3A genes decreased upon the inhibition of BRD4 with JQ1 or ARV825 (Figure 5Q–S; Supplementary Figure S5N, O). Therefore, we conclude that the inhibition of either BRD9 or BRD4 destabilizes the binding of a complex consisting of BRD9, BRD4, SMAD2, P300 and β-CATENIN on its target genes, thereby regulating their expression.
Inhibition of BRD9 reduces the development of cancer cells via regulating the activity of the TGF-β pathway
The deletion of BRD9 impairs the self-renewal and differentiation of hESCs by regulating the activity of the TGF-β pathway (Figure 5; Supplementary Figure S5). KEGG analysis showed the association of BRD9-occupied genes with functions in various cancers (Figure 5C). Previous studies have reported that BRD9 contributes to the development of cancer cells (40–42). Therefore, we investigated whether BRD9 regulates cancer development by controlling the TGF-β pathway. As there is a lack of functional studies on BRD9 in pancreatic carcinogenesis, we focused on understanding the role of BRD9 in pancreatic cancer. We analyzed 45 clinical samples of pancreatic cancer and their adjacent non-tumor tissues from the GSE28735 database and found that BRD9 was highly expressed in tumor samples but not in adjacent non-tumor tissues (Figure 6A). Additionally, BRD9 protein was enriched in tumor tissue compared with normal tissue (Supplementary Figure S6A). Moreover, the degree of BRD9 enrichment increased with increasing malignant grades (Figure 6B). Analysis of the association between BRD9 expression and clinical parameters in pancreatic cancer showed a positive correlation between BRD9 expression and malignant grade of pancreatic cancer (Supplementary Figure S6B, left), particularly in the elderly population (Supplementary Figure S6B, right). Patients with higher expression of BRD9 had a significantly shorter overall survival time compared with those with lower BRD9 expression in the GSE28735 datasets (Supplementary Figure S6C). We expanded the clinical analysis to include as many as 1319 cases, and the results indicated that high expression of BRD9 is associated with poor overall survival (Supplementary Figure S6D). These analyses suggest that BRD9 plays a promotive role in the progression of pancreatic cancer.
Figure 6.

BRD9 contributes to the development of cancer cells via regulating the TGF-β pathway. (A) Transcript levels of BRD9 in non-tumor tissue and pancreatic adenocarcinoma (PAAD) samples (GSE28735). (B) BRD9 protein levels in normal tissue and stage 1, 2, 3 and 4 PAAD samples in the Clinical Proteomic Tumor Analysis Consortium (CPTAC) dataset. (C) Inhibition of BRD9 with I-BRD9 reduces BxPC3 cell proliferation. The cell number was measured from day 0 to day 3 after 10 μM I-BRD9 treatment. (D) The effect of BRD9 inhibition on BxPC3 cell vitality. MTT assays were performed to measure cell vitality (OD 570 nm, n = 5). (E) I-BRD9 impairs colony formation ability of BxPC3 cells. The colony formation assay of BxPC3 with or without 10 days of I-BRD9 treatment (10 μM) was performed. (F) I-BRD9 reduces the migration ability of pancreatic cancer cells. Wound healing experiments were performed with I-BRD9 (10 μM) or DMSO treatment in the BxPC3 cell line. (G) KEGG analysis of differentially expressed genes (DEGs) in PAAD from the GSE28735 dataset. (H) Inhibition of TGF-β signaling reduces proliferation of pancreatic cancer cells. Cell numbers were measured after I-BRD9 (10 μM), SB431542 (10 μM) or SIS3 (10 μM) treatment in the BxPC3 cell line. (I) Inhibition of BRD9 reduces the expression of TGF-β signaling-associated genes in BxPC3 cells. The expression of TGF-β signaling-associated genes was measured after treatment with 10 μM I-BRD9 for 48 h. (J) Overexpression of BRD9 prompts the expression of TGF-β signaling-associated genes in pancreatic cancer cells. The expression of TGF-β signaling-associated genes was measured in control and BRD9-overexpressing BxPC3 cells. (K) Western blot for SMAD2 and p-SMAD2 protein levels in BxPC3 cells upon BRD9 inhibition with 10 μM I-BRD9 for 48 h. (L) qPCR analysis shows reduced expression of WNT3 and WNT3A genes in BxPC3 cells upon treatment with 10 μM I-BRD9 for 48 h. (M) Viability of BxPC3 cells after treatment with IWR-1, IWP-2 or DKK1. Viability was measured using the MTT assay and no significant changes were observed with 1 μM IWR-1, 5 μM IWP-2 or 10 ng/ml DKK1 treatment. (N) Inhibition of BRD9 reduces expression of TGF-β signaling-associated genes in MCF7 cells. The expression of TGF-β signaling-associated genes was measured after treatment with 10 μM I-BRD9 for 2 days. (O) Treatment with the BRD9 inhibitor I-BRD9 or the TGF-β signaling pathway inhibitor SB431542 reduces MCF7 cell proliferation. Cell numbers were measured from day 0 to day 3 after treatment with indicated inhibitors. (P) Treatment of BxPC3 cells with gemcitabine (GEM) increases expression of TGF-β signaling-associated genes. The expression of TGF-β signaling-associated genes was measured after treatment with 20 μM GEM for 2 days. (Q) Treatment of BxPC3 cells with GEM, I-BRD9 or both reduces cell proliferation. Cell numbers were measured after 48 h of treatment with 20 μM GEM, 10 or 20 μM I-BRD9 or a combination of both.
To confirm the function of BRD9 in pancreatic cancer cells, we used I-BRD9 to inhibit BRD9 in BxPC3 cells, a pancreatic cancer cell line. Firstly, we determined the IC50 of BRD9 in BxPC3 cells, which was found to be 103.8 and 17.84 μM at 24 and 48 h, respectively (Supplementary Figure S6E). For subsequent experiments, we chose 10 μM I-BRD9 unless otherwise specified. Inhibition of BRD9 significantly reduced cell number (Figure 6C) and impaired cell vitality (Figure 6D) compared with the control. The colony formation assay showed a significant decrease in the colony number after BRD9 inhibition (Figure 6E), while the wound healing assay demonstrated that the inhibition of BRD9 decreased the ability of BxPC3 cells to migrate (Figure 6F).
To investigate the function of BRD9 overexpression on BxPC3 cell proliferation and migration, we generated an overexpression BRD9 BxPC3 cell line, which was confirmed by qPCR analysis and western blot assay (Supplementary Figure S6F, G). The cell proliferation assay indicated that overexpression of BRD9 increased the cell number (Supplementary Figure S6H), while the colony formation assay showed that BRD9 overexpression increased the colony number (Supplementary Figure S6I). Moreover, the wound healing assay revealed that BRD9 overexpression increased the ability of BxPC3 cells to migrate (Supplementary Figure S6J). In summary, our results suggest that inhibition of BRD9 negatively regulates pancreatic cancer cell survival.
KEGG analysis of the GSE28735 database showed the association of DEGs with MAPK, RAS, TGF-β and Wnt signaling pathways (Figure 6G), suggesting that BRD9 inhibition may repress the proliferation of BxPC3 cells by regulating the TGF-β and Wnt pathways. To explore this, we conducted a cell proliferation assay to examine the effect of TGF-β pathway inhibition on BxPC3 growth. The number of BxPC3 cells was significantly decreased upon the addition of TGF-β inhibitors, SB431542 and SIS3 (Figure 6H). The expression of NODAL, TGFB2, TGFB3 and INHBA decreased in BxPC3 cells treated with I-BRD9 (Figure 6I), while BRD9 overexpression increased the expression of NODAL, TGFB1, TGFB2 and TGFB3 (Figure 6J). BRD9 inhibition reduced the protein levels of p-SMAD2 in BxPC3 cells (Figure 6K). Similar to the down-regulation of WNT3 and WNT3A in hESCs upon BRD9 inhibition (Figure 3L), inhibition of BRD9 also reduced the expression of WNT3 and WNT3A in BxPC3 cells (Figure 6L). However, the inhibition of the Wnt pathway with IWR-1, IWP-2 or DKK1 did not show an obvious effect on the viability of BxPC3 cells (Figure 6M). Therefore, our results suggest that BRD9 plays a significant role in regulating the proliferation and migration of BxPC3 cells through its control of the TGF-β pathway, while the effect of BRD9 on the Wnt pathway in these cells appears to be minor.
Furthermore, we investigated whether BRD9 regulates other types of cancer, such as breast cancer, by a similar mechanism. Inhibition of BRD9 with I-BRD-9 in MCF7, a breast cancer cell line, impaired proliferation (Supplementary Figure S6K), cell viability (Supplementary Figure S6L), colony formation ability (Supplementary Figure S6M) and migration (Supplementary Figure S6N). The expression of NODAL, INHBA, TGFB2 and TGFB3 in MCF7 cells decreased upon treatment with I-BRD9 (Figure 6N). The number of MCF7 cells decreased significantly upon the addition of the TGF-β inhibitor SB431542 (Figure 6O). KEGG analysis shown that the DEGs in the TCGA dataset were associated with TGF-β, RAS, MAPK, Hippo and p53 pathways (Supplementary Figure S6O). However, the inhibition of the Wnt pathway with IWR-1, IWP2 or DKK1 did not show an obvious significant effect on the viability of MCF7 cells (Supplementary Figure S6P). Our results indicate that BRD9 regulates breast cancer proliferation by controlling the activity of the TGF-β pathway.
Overall, our data and analysis suggest that BRD9 plays a pro-oncogenic role in pancreatic and breast cancers, at least in part through the regulation of the TGF-β signaling pathway.
Discussion
The ability of hESCs to self-renew indefinitely and differentiate into any cell type in the human body offers the possibility of clinical therapy (43,44). To fully harness their therapeutic potential, a comprehensive understanding of the molecular mechanisms underlying the maintenance and differentiation of hESCs is imperative. Previous studies have shown that the convergence of pluripotent stem cells to lineage specificity relies on the cooperation of transcription factors, signaling pathways and chromatin-remodeling complexes (2,45,46). This study shows that the cross-talk between the ncBAF complex and key signaling pathways contributes to pluripotency maintenance and lineage differentiation of hESCs. Specifically, our research has revealed a link between the epigenetic modification reader BRD9 and the TGF-β/Activin/Nodal and Wnt signaling pathways in hESCs, which expands our understanding of the regulation network of stem cell biology.
The TGF-β/Activin/Nodal signaling pathway is essential for pluripotency maintenance in hESCs and embryo development (47–49). This signaling pathway begins with the binding of ligands, such as TGF-β, NODAL and ACTIVIN, to type I and II receptor serine/threonine kinases, which then activate SMAD2 and SMAD3. The phosphorylated SMAD2 and SMAD3 form a complex with SMAD4, which enters the nucleus to bind to specific genome regions and regulate gene expression, including OCT4 and NANOG (50,51). Imbalances in this pathway can result in the loss of pluripotency in hESCs and the development of various cancers (52–54). The proper activation of this pathway in different developmental stages is regulated by various mechanisms, including positive feedback through auto- and cross-induced TGF-β isoforms and target transcription factors (55), negative feedback through inhibitory SMADs, SMAD6 and SMAD7 (56), recycling of activated SMADs by dephosphorylation and poly(ADP)-ribosylation (57), or removal by polyubiquitylation and proteasome-dependent degradation (58).
In this study, we revealed that the TGF-β/Activin/Nodal signaling pathway in hESCs is regulated by ncBAF, a recently defined chromatin-remodeling complex (3,7). A complex including BRD9, BRD4, β-CATENIN, SMAD2/3 and P300 regulates the enrichment of H3K27ac in TGF-β/Activin/Nodal signaling pathway-related genes and their transcription expression levels, which results in proper self-renewal and differentiation of hESCs. However, we did not detect the interaction between BRD9 and SMAD2 based on the Co-IP experiment by using BRD9 antibody (data not shown), but found that both SMAD2 and BRD9 were precipitated when the Co-IP experiment was carried out with BRD4 antibody (Figure 5G). Consistently, at mouse naive pluripotency gene targets, the BRD9 BD recognizes an acetylated form of BRD4, recruiting GBAF complexes to chromatin (7). In this study, compared with inhibition of BRD9, BRD4 inhibition led to more decreased expression of TGF-β pathway-related genes (Figures 3C and 4E; Supplementary Figures S3C and S4F). Compared with the 492 sites being significantly decreased by BRD4 binding by 1.5 fold change upon BRD9 inhibition, the inhibition of BRD4 with JQ1 led to 12 849 sites significantly losing BRD9 (7). Therefore, both our study and that of Gatchalian demonstrate that BRD4 mediated the recruitment of BRD9 to its target genes. Meanwhile, our study showed that the loss of BRD9 reduced the enrichment of BRD4 on TGF-β pathway-related genes (Figure 5K; Supplementary Figure S5K), supporting that BRD9 and BRD4 collaboratively regulate the TGF-β/Activin/Nodal signaling pathway.
Both BRD9 and TGF-β/Activin/Nodal signaling are required to maintain pluripotency of human naïve pluripotent stem cells (7,11,49). When grown in conditions that inhibit Wnt/β-catenin signaling, naïve hESCs remain undifferentiated but have a more primed-like protein expression profile, suggesting a critical role for Wnt/β-catenin signaling in regulating human naïve pluripotency (59). In this study, deletion of BRD9 reduced the activity of both TGF-β/Activin/Nodal and Wnt signaling pathways (Figure 3; Supplementary Figure S3). The addition of TGF-β/Activin/Nodal or WNT3A restored the down-regulation of the other resulting from the inhibition of BRD9 (Figure 3P, Q; Supplementary Figure S3V), indicating that BRD9 regulated the collaborative function of TGF-β/Activin/Nodal and Wnt signaling pathways on the self-renewal and differentiation of hESCs. Recently, Sevinç et al. reported the successful generation of BRD9 knockout (KO) iPSC clones through the reprogramming of fibroblast cells (11). However, the consequences of BRD9 gene KO on the maintenance of iPSCs remained unclear. Our investigation unveiled an elevated proportion of differentiated clones following BRD9 inhibition in hESCs (Figures 1G, H), thereby demonstrating impaired self-renewal of hESCs when BRD9 is inhibited. Furthermore, we endeavored to generate BRD9 KO hESCs utilizing CRISPR/Cas9 [clustered regularly interspaced palindromic repeats (CRISPR)/CRISPR-associatedprotein 9] technology, but encountered challenges in obtaining viable BRD9 KO hESC clones (data not shown). This suggests that BRD9 deletion may disrupt the maintenance of hESCs, rendering it arduous to establish stable BRD9 KO cell lines. It is noteworthy that BRD9 KO iPSCs were capable of differentiating into all three germ layers (11). Nevertheless, the impact of BRD9 KO on hESC differentiation into specific lineages necessitates further exploration, particularly considering the potential off-target effects associated with CRISPR/Cas9 technology. Hence, an investigation into the influence of BRD9 on both the self-renewal and differentiation of hESCs, utilizing BRD9 KO iPSCs or hESCs, holds particular interest.
Sevinç et al. revealed that BRD9 serves as a barrier to human reprogramming by upholding the expression of target genes MN1 and ZBTB38 in fibroblasts, thereby preserving somatic cell identity (11). Our findings highlight the critical role of BRD9 in sustaining hESC self-renewal through its collaborative regulation of essential signaling pathways and pluripotency factors. Given the congruence between our study and the work of Sevinç et al. in demonstrating BRD9’s role in maintaining cell identity, it appears that BRD9 preserves the initial cell identity by cooperating with key factors pivotal to cellular identity. An intriguing avenue for further investigation lies in elucidating the universality of BRD9’s function and mechanism in preserving the starting cell identity.
Recent studies have revealed distinct mechanisms by which BRD9 regulates the proliferation, viability and migration of cancer cells. These mechanisms include the regulation of the transcription factor MYC oncogene (40,60), glycolysis (42), androgen receptor signaling (41) and apoptosis (61,62). In this study, we demonstrated that BRD9 regulates the development of pancreatic and breast cancers by modulating the activity of the TGF-β pathway, which is aberrantly activated in various cancers. Inhibition of BRD9 decreased the expression of genes related to the TGF-β/Activin/Nodal signaling pathway and significantly suppressed the proliferation, viability, clone formation and migration of cancer cells (Figure 6; Supplementary Figure S6). Moreover, inhibition of the TGF-β/Activin/Nodal pathway with the inhibitor SB431542 impaired the progression of pancreatic and breast cancer cells (Figures 6H, K). Cancer stem cells (CSCs) have been proposed as a source of cancer cells and are primarily responsible for resistance to various treatments, such as chemotherapy and radiotherapy (63). Both CSCs and pluripotent stem cells exhibit stemness and phenotypic plasticity properties, which makes pluripotent stem cells a useful model for studying cancer development. This study uncovers a mechanism involving the BRD9-mediated TGF-β/Activin/Nodal signaling pathway that regulates human pluripotent stem cell properties and affects the self-renewal of pancreatic and breast cancer cells. It is worth noting the need to investigate the in vivo effects of the BRD9-mediated TGF-β/Activin/Nodal signaling pathway on cancer development and its potential therapeutic value.
In hESCs, the BRD9-mediated Wnt pathway has been demonstrated to be involved in regulating self-renewal and differentiation (Figure 3; Supplementary Figure S3). However, unlike in hESCs, the inhibition of the Wnt pathway did not show obvious effects on the viability of BxPC3 and MCF7 cells (Figure 6M; Supplementary Figure S6P), suggesting a distinct role for the Wnt pathway in hESCs and cancer cells. Further investigation is needed to elucidate the distinct roles and underlying mechanisms of the Wnt pathway in hESCs and cancers.
Epigenetic drugs targeting epigenetic proteins, such as readers, writers and erasers, have exhibited significant therapeutic potential in various diseases (64,65). The US Food and Drug Administration (FDA) has approved several epigenetic drugs, including DNA methyltransferase inhibitors (DNMTis) and histone deacetylase inhibitors (HDACis), for cancer treatment (66–68). Gemcitabine (GEM) is a primary chemotherapy drug used to treat pancreatic cancer by suppressing DNA synthesis and promoting apoptosis in pancreatic cancer cells (69). However, the efficacy of GEM is limited as resistance to the drug commonly arises within a few weeks of treatment initiation (70). In the current study, we observed that treatment of BxPC3 and MCF7 cells with GEM resulted in up-regulation of genes associated with the TGF-β pathway including NODAL, INHBA, GDF3, LEFTY2 and TGFB2 (Figure 6P; Supplementary Figure S6Q). Notably, co-treatment of BxPC3 and MCF7 cells with GEM and I-BRD9 led to a significant reduction in cell proliferation as compared with GEM treatment alone, suggesting a distinct mechanism of action by which BRD9 regulates the TGF-β pathway to control the growth of both cell lines (Figure 6Q; Supplementary Figure S6R). Our investigation has unveiled a promising therapeutic approach for cancers characterized by abnormal TGF-β/Activin/Nodal signal pathway activation, involving the targeting of BRD9—an epigenetic protein—through inhibitors and degraders. Notably, while preparing this manuscript, a concurrent study by Feng et al. (71) presented compelling evidence of BRD9’s collaboration with TGF-β/Activin/SMAD2/3 in regulating self-renewal, chemoresistance and invasiveness of pancreatic cancer stem cells. This finding underscores the potential significance of BRD9 inhibition or degradation as epigenetic drugs in treating cancers induced by dysregulated TGF-β/Activin/Nodal signaling. Consequently, further exploration and validation of BRD9 inhibitors or degraders as viable therapeutic options are warranted.
In brief, our study revealed that BRD9 plays a critical role in regulating the maintenance and differentiation of hESCs by modulating the activity of the TGF-β/Activin/Nodal and Wnt signaling pathways through epigenetic mechanisms. Additionally, we demonstrated that BRD9-mediated regulation of the TGF-β/Activin/Nodal pathway is also involved in the development of pancreatic and breast cancer cells. These findings provide valuable insights into the mechanisms underlying the regulation of pluripotency and differentiation in hESCs, and suggest that targeting BRD9 or the TGF-β/Activin/Nodal signaling pathway may represent a promising therapeutic approach for cancers with dysregulated TGF-β/Activin/Nodal signaling.
Supplementary Material
Acknowledgements
We would like to express our gratitude to Weimin Zuo from the University of Macau for the support in conducting cancer-related experiments, and to Qian Zhang from the Southern University of Science and Technology for the support in ChIP-seq experiments.
Contributor Information
Xuepeng Wang, The State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Taipa, Macao SAR 999078, China.
Chengcheng Song, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Ying Ye, Medical College of Soochow University, Suzhou 215123, China.
Yashi Gu, Zhejiang University–University of Edinburgh Institute (ZJE), Zhejiang University School of Medicine, Zhejiang University, Haining 314400, China.
Xuemei Li, Peninsula Cancer Research Center, School of Basic Medical Sciences, Binzhou Medical University, Yantai 264003, China.
Peixin Chen, Medical College of Soochow University, Suzhou 215123, China.
Dongliang Leng, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Jing Xiao, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Hao Wu, Medical College of Soochow University, Suzhou 215123, China.
Sisi Xie, Zhejiang University–University of Edinburgh Institute (ZJE), Zhejiang University School of Medicine, Zhejiang University, Haining 314400, China.
Weiwei Liu, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Qi Zhao, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Di Chen, Zhejiang University–University of Edinburgh Institute (ZJE), Zhejiang University School of Medicine, Zhejiang University, Haining 314400, China.
Xi Chen, Department of Biology, Southern University of Science and Technology, Shenzhen 518000, China.
Qiang Wu, The State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Taipa, Macao SAR 999078, China; The Precision Regenerative Medicine Centre, Macau University of Science and Technology, Taipa, Macao SAR 999078, China.
Guokai Chen, Centre of Reproduction, Development and Aging, Faculty of Health Sciences, University of Macau, Taipa, Macao SAR 999078, China.
Wensheng Zhang, Medical College of Soochow University, Suzhou 215123, China; Peninsula Cancer Research Center, School of Basic Medical Sciences, Binzhou Medical University, Yantai 264003, China; School of Life Sciences and Medicine, Shandong University of Technology, Zibo, 255049, China.
Data availability
The data underlying this article are available in the Sequence Read Archive at https://www.ncbi.nlm.nih.gov/sra and can be accessed under PRJNA956705; PRJNA1020906; PRJNA957053; PRJNA958615; and PRJNA1004895.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
Ministry of Science and Technology [2022YFA1104300 to W. Z.]; The National Natural Science Foundation of China [3217060054 and 31970812 to W.Z.]; Macau Science and Technology Development Fund [0072/2019/A2, SKL-QRCM (MUST)-2020-2022 and SKL-QRCM(MUST)-2023-2025 to Q.W.]; University of Macau [MYRG2015-00228-FHS, MYRG2018-00135-FHS and MYRG2019-00147-FHS to G.C]; Science and Technology Development Fund, Macau SAR [0059/2019/A1 0123/2019/A3, FDCT/0011/2019/AKP, and FDCT/0002/2021/AKP to G.C]; and Ministry of Science and Technology National Key R&D grant [2022YFA1105000 to G.C].
Conflict of interest statement. None declared.
References
- 1. Martello G., Smith A.. The nature of embryonic stem cells. Annu. Rev. Cell Dev. Biol. 2014; 30:647–675. [DOI] [PubMed] [Google Scholar]
- 2. Ye Y., Chen X., Zhang W.. Mammalian SWI/SNF chromatin remodeling complexes in embryonic stem cells: regulating the balance between pluripotency and differentiation. Front. Cell Dev. Biol. 2021; 8:626383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alpsoy A., Dykhuizen E.C.. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018; 293:3892–3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ho L., Ronan J.L., Wu J., Staahl B.T., Chen L., Kuo A., Lessard J., Nesvizhskii A.I., Ranish J., Crabtree G.R.. An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl Acad. Sci. USA. 2009; 106:5181–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kidder B.L., Palmer S., Knott J.G.. SWI/SNF–Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells. 2009; 27:317–328. [DOI] [PubMed] [Google Scholar]
- 6. Zhang W., Chronis C., Chen X., Zhang H., Spalinskas R., Pardo M., Chen L., Wu G., Zhu Z., Yu Y.et al.. The BAF and PRC2 complex subunits Dpf2 and Eed antagonistically converge on Tbx3 to control ESC differentiation. Cell Stem Cell. 2019; 24:138–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gatchalian J., Malik S., Ho J., Lee D.S., Kelso T.W.R., Shokhirev M.N., Dixon J.R., Hargreaves D.C.. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 2018; 9:5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Müller S., Pawson T.et al.. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujisawa T., Filippakopoulos P.. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017; 18:246–262. [DOI] [PubMed] [Google Scholar]
- 10. Zhu X., Liao Y., Tang L.. Targeting BRD9 for cancer treatment: a new strategy. Onco Targets Ther. 2020; 13:13191–13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sevinç K., Sevinç G.G., Cavga A.D., Philpott M., Kelekçi S., Can H., Cribbs A.P., Yıldız A.B., Yılmaz A., Ayar E.S.et al.. BRD9-containing non-canonical BAF complex maintains somatic cell transcriptome and acts as a barrier to human reprogramming. Stem Cell Rep. 2022; 17:2629–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen G., Gulbranson D.R., Hou Z., Bolin J.M., Ruotti V., Probasco M.D., Smuga-Otto K., Howden S.E., Diol N.R., Propson N.E.et al.. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods. 2011; 8:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu P., Pan G., Yu J., Thomson J.A.. FGF2 sustains NANOG and switches the outcome of BMP4-induced human embryonic stem cell differentiation. Cell Stem Cell. 2011; 8:326–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patsch C., Challet-Meylan L., Thoma E.C., Urich E., Heckel T., O'Sullivan J.F., Grainger S.J., Kapp F.G., Sun L., Christensen K.et al.. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015; 17:994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diekmann U., Lenzen S., Naujok O.. A reliable and efficient protocol for human pluripotent stem cell differentiation into the definitive endoderm based on dispersed single cells. Stem Cells Dev. 2015; 24:190–204. [DOI] [PubMed] [Google Scholar]
- 16. Lin Y., Linask K.L., Mallon B., Johnson K., Klein M., Beers J., Xie W., Du Y., Liu C., Lai Y.et al.. Heparin promotes cardiac differentiation of Human pluripotent stem cells in chemically defined albumin-free medium, enabling consistent manufacture of cardiomyocytes. Stem Cells Transl. Med. 2017; 6:527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu W., Ye Y., Sharrocks A.D., Zhang W., Chen X. Genome-wide interrogation of protein–DNA interactions in mammalian cells using ChIPmentation. STAR Protoc. 2020; 1:100187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K.. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010; 38:576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shao Z., Zhang Y., Yuan G.C., Orkin S.H., Waxman D.J.. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012; 13:R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M.et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011; 473:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lê S., Josse J., Husson F.. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 2008; 25:1–18. [Google Scholar]
- 22. Wickham H. ggplot2: elegant graphics for data analysis. 2016; NY: Springer. [Google Scholar]
- 23. Brien G.L., Remillard D., Shi J., Hemming M.L., Chabon J., Wynne K., Dillon E.T., Cagney G., Van Mierlo G., Baltissen M.P.et al.. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife. 2018; 7:e41305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bernardo A.S., Faial T., Gardner L., Niakan K.K., Ortmann D., Senner C.E., Callery E.M., Trotter M.W., Hemberger M., Smith J.C.et al.. BRACHYURY and CDX2 mediate BMP-induced differentiation of human and mouse pluripotent stem cells into embryonic and extraembryonic lineages. Cell Stem Cell. 2011; 9:144–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. James D., Levine A.J., Besser D., Hemmati-Brivanlou A.. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development. 2005; 132:1273–1282. [DOI] [PubMed] [Google Scholar]
- 26. Vallier L., Alexander M., Pedersen R.A.. Activin/nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J. Cell Sci. 2005; 118:4495–4509. [DOI] [PubMed] [Google Scholar]
- 27. Cornacchia D., Zhang C., Zimmer B., Chung S.Y., Fan Y., Soliman M.A., Tchieu J., Chambers S.M., Shah H., Paull D.et al.. Lipid deprivation induces a stable, naive-to-primed intermediate state of pluripotency in human PSCs. Cell Stem Cell. 2019; 25:120–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu F., Deng C., Ren Z., Sun L., Meng Y., Liu W., Wan J., Chen G. Lysophosphatidic acid shifts metabolic and transcriptional landscapes to induce a distinct cellular state in human pluripotent stem cells. Cell Rep. 2021; 37:110063. [DOI] [PubMed] [Google Scholar]
- 29. Bayerl J., Ayyash M., Shani T., Manor Y.S., Gafni O., Massarwa R., Kalma Y., Aguilera-Castrejon A., Zerbib M., Amir H.et al.. Principles of signaling pathway modulation for enhancing human naive pluripotency induction. Cell Stem Cell. 2021; 28:1549–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang J., Jiang W.. The role of SMAD2/3 in human embryonic stem cells. Front. Cell Dev. Biol. 2020; 8:653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Di Micco R., Fontanals-Cirera B., Low V., Ntziachristos P., Yuen S.K., Lovell C.D., Dolgalev I., Yonekubo Y., Zhang G., Rusinova E.et al.. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014; 9:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gessert S., Kühl M.. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 2010; 107:186–199. [DOI] [PubMed] [Google Scholar]
- 33. Lian X., Hsiao C., Wilson G., Zhu K., Hazeltine L.B., Azarin S.M., Raval K.K., Zhang J., Kamp T.J., Palecek S.P.. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical wnt signaling. Proc. Natl Acad. Sci. USA. 2012; 109:E1848–E1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang Y., Ren Z., Xu F., Meng Y., Zhang Y., Ai N., Long Y., Fok H.I., Deng C., Zhao X.et al.. Endogenous IGF signaling directs heterogeneous mesoderm differentiation in human embryonic stem cells. Cell Rep. 2019; 29:3374–3384. [DOI] [PubMed] [Google Scholar]
- 35. Balafkan N., Mostafavi S., Schubert M., Siller R., Liang K.X., Sullivan G., Bindoff L.A.. A method for differentiating human induced pluripotent stem cells toward functional cardiomyocytes in 96-well microplates. Sci Rep. 2020; 10:18498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ernst J., Kellis M.. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol. 2010; 28:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Martire S., Nguyen J., Sundaresan A., Banaszynski L.A.. Differential contribution of p300 and CBP to regulatory element acetylation in mESCs. BMC Mol. Cell Biol. 2020; 21:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang H., Wang Y., Li Q., Fei X., Ma H., Hu R.. miR-140-3p functions as a tumor suppressor in squamous cell lung cancer by regulating BRD9. Cancer Lett. 2019; 446:81–89. [DOI] [PubMed] [Google Scholar]
- 41. Alpsoy A., Utturkar S.M., Carter B.C., Dhiman A., Torregrosa-Allen S.E., Currie M.P., Elzey B.D., Dykhuizen E.C.. BRD9 is a critical regulator of androgen receptor signaling and prostate cancer progression. Cancer Res. 2021; 81:820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhu Q., Gu X., Wei W., Wu Z., Gong F., Dong X. BRD9 is an essential regulator of glycolysis that creates an epigenetic vulnerability in colon adenocarcinoma. Cancer Med. 2023; 12:1572–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Clevers H. Modeling development and disease with organoids. Cell. 2016; 165:1586–1597. [DOI] [PubMed] [Google Scholar]
- 44. Yamanaka S. Pluripotent stem cell-based cell therapy—promise and challenges. Cell Stem Cell. 2020; 27:523–531. [DOI] [PubMed] [Google Scholar]
- 45. Young R.A. Control of the embryonic stem cell state. Cell. 2011; 144:940–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yeo J.C., Ng H.H.. The transcriptional regulation of pluripotency. Cell Res. 2013; 23:20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu M.Y., Hill C.S.. TGF-beta superfamily signaling in embryonic development and homeostasis. Dev. Cell. 2009; 16:329–343. [DOI] [PubMed] [Google Scholar]
- 48. Jia S., Meng A.. TGFβ family signaling and development. Development. 2021; 148:dev188490. [DOI] [PubMed] [Google Scholar]
- 49. Osnato A., Brown S., Krueger C., Andrews S., Collier A.J., Nakanoh S., Quiroga Londoño M., Wesley B.T., Muraro D., Brumm A.S.et al.. TGFβ signalling is required to maintain pluripotency of human naïve pluripotent stem cells. eLife. 2021; 10:e67259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu R.H., Sampsell-Barron T.L., Gu F., Root S., Peck R.M., Pan G., Yu J., Antosiewicz-Bourget J., Tian S., Stewart R.et al.. NANOG is a direct target of TGFbeta/activin-mediated SMAD signaling in human ESCs. Cell Stem Cell. 2008; 3:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hata A., Chen Y.G.. TGF-β signaling from receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016; 8:a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Singh A.M., Reynolds D., Cliff T., Ohtsuka S., Mattheyses A.L., Sun Y., Menendez L., Kulik M., Dalton S. Signaling network crosstalk in human pluripotent cells: a Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell. 2012; 10:312–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Batlle E., Massagué J.. Transforming growth factor-β signaling in immunity and cancer. Immunity. 2019; 50:924–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Derynck R., Turley S.J., Akhurst R.J.. TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 2021; 18:9–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Derynck R., Budi E.H.. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019; 12:eaav5183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miyazawa K., Miyazono K.. Regulation of TGF-β family signaling by inhibitory smads. Cold Spring Harb. Perspect. Biol. 2017; 9:a022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lönn P., van der Heide L.P., Dahl M., Hellman U., Heldin C.H., Moustakas A.. PARP-1 attenuates Smad-mediated transcription. Mol. Cell. 2010; 40:521–532. [DOI] [PubMed] [Google Scholar]
- 58. Budi E.H., Duan D., Derynck R.. Transforming growth factor-β receptors and Smads: regulatory complexity and functional versatility. Trends Cell Biol. 2017; 27:658–672. [DOI] [PubMed] [Google Scholar]
- 59. Xu Z., Robitaille A.M., Berndt J.D., Davidson K.C., Fischer K.A., Mathieu J., Potter J.C., Ruohola-Baker H., Moon R.T.. Wnt/β-catenin signaling promotes self-renewal and inhibits the primed state transition in naïve human embryonic stem cells. Proc. Natl Acad. Sci. USA. 2016; 113:E6382–E6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hohmann A.F., Martin L.J., Minder J.L., Roe J.S., Shi J., Steurer S., Bader G., McConnell D., Pearson M., Gerstberger T.et al.. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nat. Chem. Biol. 2016; 12:672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bevill S.M., Olivares-Quintero J.F., Sciaky N., Golitz B.T., Singh D., Beltran A.S., Rashid N.U., Stuhlmiller T.J., Hale A., Moorman N.J.et al.. GSK2801, a BAZ2/BRD9 bromodomain inhibitor, synergizes with BET inhibitors to induce apoptosis in triple-negative breast cancer. Mol. Cancer Res. 2019; 17:1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mu J., Sun X., Zhao Z., Sun H., Sun P.. BRD9 inhibition promotes PUMA-dependent apoptosis and augments the effect of imatinib in gastrointestinal stromal tumors. Cell Death Dis. 2021; 12:962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Barati M., Akhondi M., Mousavi N.S., Haghparast N., Ghodsi A., Baharvand H., Ebrahimi M., Hassani S.N.. Pluripotent stem cells: cancer study, therapy, and vaccination. Stem Cell Rev. Rep. 2021; 17:1975–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dawson M.A., Kouzarides T.. Cancer epigenetics: from mechanism to therapy. Cell. 2012; 150:12–27. [DOI] [PubMed] [Google Scholar]
- 65. Ilango S., Paital B., Jayachandran P., Padma P.R., Nirmaladevi R.. Epigenetic alterations in cancer. Front. Biosci. 2020; 25:1058–1109. [DOI] [PubMed] [Google Scholar]
- 66. Raynal N.J., Da Costa E.M., Lee J.T., Gharibyan V., Ahmed S., Zhang H., Sato T., Malouf G.G., Issa J.J.. Repositioning FDA-approved drugs in combination with epigenetic drugs to reprogram colon cancer epigenome. Mol. Cancer Ther. 2017; 16:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dzobo K. Epigenomics-guided drug development: recent advances in solving the cancer treatment ‘jigsaw puzzle’. OMICS. 2019; 23:70–85. [DOI] [PubMed] [Google Scholar]
- 68. Ramaiah M.J., Tangutur A.D., Manyam R.R.. Epigenetic modulation and understanding of HDAC inhibitors in cancer therapy. Life Sci. 2021; 277:119504. [DOI] [PubMed] [Google Scholar]
- 69. Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N.et al.. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013; 369:1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Binenbaum Y., Na’ara S., Gil Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015; 23:55–68. [DOI] [PubMed] [Google Scholar]
- 71. Feng Y., Cai L., Pook M., Liu F., Chang C.H., Mouti M.A., Nibhani R., Militi S., Dunford J., Philpott M.et al.. BRD9-SMAD2/3 orchestrates stemness and tumorigenesis in pancreatic ductal adenocarcinoma. Gastroenterology. 2023; S0016-5085(23)05012-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the Sequence Read Archive at https://www.ncbi.nlm.nih.gov/sra and can be accessed under PRJNA956705; PRJNA1020906; PRJNA957053; PRJNA958615; and PRJNA1004895.