

Abstract
Lafora disease (LD; OMIM#254780) is a rare form of progressive myoclonus epilepsy (prevalence less than 1<1,000,000) characterized by the accumulation of insoluble deposits of aberrant glycogen (polyglucosans), named Lafora bodies, in the brain but also in peripheral tissues. LD is the most severe form of the group of progressive myoclonus epilepsies, since patients present a rapid deterioration and dementia with amplification of seizures, leading to death after a decade from the onset of the first symptoms. We have recently described that reactive glia-derived neuroinflammation should be considered a novel hallmark of LD since we observed a florid upregulation of differentially expressed genes in both LD mouse lines, which were mainly related to mediators of inflammatory response. In this work, we define an upregulation of the expression of mediators of the TNF and IL6/JAK2 signaling pathways in LD. In addition, we describe the activation of the non-canonical form of the inflammasome. Furthermore, we describe the infiltration of peripheral immune cells in the brain parenchyma, which could aggravate glia-derived neuroinflammation. Finally, we describe CXCL10 and S100b as blood biomarkers of the disease, which will allow the study of the progression of the disease using serum blood samples. We consider that the identification of these initial inflammatory changes in LD will be very important to implement possible anti-inflammatory therapeutic strategies to prevent the development of the disease.
Keywords: Lafora disease, astrocytes, microglia, neuroinflammation, TNF, IL6, lymphocyte infiltration
INTRODUCTION
Lafora disease (LD; OMIM#254780) is a rare form of progressive myoclonus epilepsy (EPM2; prevalence less than 1<1,000,000) characterized by the accumulation of insoluble deposits of aberrant glycogen (polyglucosans), named Lafora bodies (LBs), in the brain but also in peripheral tissues (Turnbull et al., 2016), (Garcia-Gimeno et al., 2018). LD occurs during childhood or early adolescence. Clinically, LD shares some symptoms with other progressive myoclonus epilepsies (EPMs), such as generalized tonic-clonic seizures, myoclonus, absences, and visual hallucinations. However, LD is the most severe form of EPM since patients present a rapid deterioration and dementia with amplification of seizures, leading to death after around a decade from the onset of the first symptoms (Turnbull et al., 2016), (Garcia-Gimeno et al., 2018), (Pondrelli et al., 2021). Unfortunately, no treatment is available yet for this devastating disease. Patients are treated initially with regular anti-epileptic drugs but soon they become resistant to the beneficial effects of these drugs. LD is caused by mutations in two genes, namely EPM2A, encoding the glucan phosphatase laforin, and EPM2B, encoding the E3-ubiquitin ligase malin (Minassian et al., 1998), (Chan et al., 2003). Both proteins form a functional complex where laforin recognizes possible substrates that are then ubiquitinated by malin (Gentry et al., 2005), (Solaz-Fuster et al., 2008). Perhaps, this is the reason why patients carrying mutations in either the EPM2A or the EPM2B have a similar clinical presentation (Gómez-Abad et al., 2005). Furthermore, the laforin/malin complex regulates glycogen synthesis by modulating the activity of glycogen synthase either directly or through the negative modulation of activators of the enzyme, such as the protein phosphatase 1 targeting subunit PPP1R3C. This could explain why glycogen synthesis is enhanced in LD patients, leading to the accumulation of LBs. In addition, the laforin/malin complex is also involved in the regulation of glucose transporters, the maintenance of proteostasis, and the response to oxidative stress (Roma-Mateo et al., 2015), (Garcia-Gimeno et al., 2018)
To gain knowledge on the pathophysiological determinants of LD, several animal models of the disease are currently used, being the mouse models the ones that have been utilized more extensively: Epm2a−/− mice, which lacks exon 4 from the Epm2a gene (Ganesh et al., 2002); and Epm2b−/− mice, which lacks the single exon present in the Epm2b gene (Criado et al., 2012). Both mouse models present similar pathophysiological phenotypes, which resemble the ones present in LD patients: they show an accumulation of LBs in the brain and peripheral tissues (Criado et al., 2012); they are more sensitive to the effects of pro-epileptic drugs such as kainate of pentylenetetrazole (PTZ) (Garcia-Cabrero et al., 2014); they show altered proteostasis with decreased autophagy (Aguado et al., 2010), (Puri and Ganesh, 2010), (Criado et al., 2012), and decreased mitophagy (Lahuerta et al., 2018); they manifest behavioral impairments (Garcia-Cabrero et al., 2012), and they exhibit decreased ability to glutamate uptake, which could lead to excitotoxicity (Perez-Jimenez et al., 2021).
In addition, the brain of LD mice presents reactive astrocytes and microglia, which are responsible for the pro-inflammatory phenotype present in LD (Lopez-Gonzalez et al., 2017). In this sense, we recently described that reactive glia-derived neuroinflammation should be considered a novel hallmark of LD. By performing RNAseq analyses of the brain of both Epm2a−/− and Epm2b−/− mice at 16 months of age and comparing them with samples from control mice of the same age, our results indicated a florid upregulation of differentially expressed genes in both LD mice, which were related to mediators of inflammatory response. This upregulated expression progressed with age, starting at 3 months. Finally, by performing immunofluorescence co-localization experiments, we were able to identify astrocytes and microglia as the main neuroglial cells involved in the expression of these inflammatory mediators (Lahuerta et al., 2020).
Taking into account these previous results (Lahuerta et al., 2020), we decided to analyze which were the putative upstream regulators responsible for the enhanced expression of the corresponding pro-inflammatory mediators. This identification would allow us to define the main inflammatory signaling pathways that operate in LD, and therefore would allow the identification of putative therapeutic targets. Thus, we decided to perform an in silico analysis of the upstream regulators and validate them by using antibodies against key intermediate mediators of the selected inflammatory signaling pathways. In this work, we present evidence indicating that the tumor necrosis factor alpha (TNF) and the interleukin6 (IL6) signaling pathways play a key role in triggering the inflammatory reaction present in LD, and progress in the understanding of additional causes of the neuroinflammatory phenotype present in LD.
METHODS
Ethics statement, animal care, mice and husbandry
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Consejo Superior de Investigaciones Científicas (CSIC, Spain) and approved by the Consellería de Agricultura, Medio Ambiente, Cambio Climatico y Desarrollo Rural from The Generalitat Valenciana. All procedures were approved by the animal committee of the Instituto de Biomedicina de Valencia, CSIC (authorization number IBV-51). All efforts were made to minimize animal suffering. Male and female homozygous Epm2b−/− mice in a pure C57BL/6JRccHsd background and the corresponding wild type (wt) of 7, 12, and 16 months of age were used in this study. Mice were maintained in the IBV-CSIC facility on a 12/12 light/dark cycle under constant temperature (23°C) with food and water provided ad libitum. The presence of polyglucosan inclusions (the hallmark of LD) in the brain of the Epm2b−/− and wt mouse lines were analyzed by Periodic acid–Schiff (PAS) staining (Supplementary Figure S1A). Mice used for immunofluorescence were perfused as described below. Mice used for western blot analyses were sacrificed by cervical dislocation and brains were rinsed twice with cold phosphate buffer saline (PBS) (0.1 M, pH 7.4) and frozen immediately in liquid nitrogen. When planning the experiments, the principles outlined in the ARRIVE guidelines and the Basel declaration including the 3R concept have been considered.
Preparing brain tissue by conventional cardiac perfusion.
The animals were deeply anesthetized with sodium pentobarbital 80 mg/Kg by intraperitoneal injection and perfused via the heart, using a perfusion pump, at a speed of 7 ml/min with 4% paraformaldehyde (PFA), in PBS (40–48 ml per animal). In the beginning, blood removal from the circulatory system was performed with 0.9% saline solution (24 ml per animal) before the fixative entered. After perfusion, brains were removed and incubated in 15 ml of 4% PFA at 4°C with shaking up for 2h. Then, they were washed three times with PBS at room temperature for 10 min. Next, brains were incubated for 30 min with 50% ethanol, and then they were incubated overnight in 70% ethanol. The next day, the two hemispheres were separated, dehydrated, cleared, and embedded in paraffin.
Immunofluorescence analyses
Five individuals (including two males and three females) from each genotype of 16 months-old mice were perfused as described above. The samples embedded in paraffin were sagittally sectioned at 4 μm using a microtome HistoCore Biocut (Leica, Madrid, Spain). Sections were deparaffined, rehydrated, and microwave antigen retrieval was performed for 10 min in 10 mM citrate buffer pH 6.0, or 10 mM Tris 1 mM EDTA pH 9.0, following the antibody manufacturer’s instructions in each case. Sections were immersed in blocking buffer (1% BSA, 10% FBS, 0.2% Triton X-100, in PBS) and incubated overnight at 4°C with the corresponding primary antibodies listed in Supplementary Table S1 diluted in blocking buffer. Some primary antibodies required a pre-incubation with a 400 mM glucose solution at 4°C overnight to avoid cross-reaction with polyglucosan bodies (Riba et al., 2021). After three washes of 10 min in PBS, sections were incubated for one hour at room temperature with the appropriate secondary antibody diluted at 1/500 (Supplementary Table S1) in blocking buffer without Triton X-100, washed once with PBS, incubated with DAPI (Sigma, Madrid, Spain), washed twice with PBS and mounted in AquaPolymount (Polysciences Inc., USA). Images were acquired in a Confocal Spectral Leica TCS SP8 microscope (Leica, Wetzlar, Germany). Pictures of each area of the whole hippocampus were taken with a 40X objective. Image stitching of all the images was performed using the LasX (Leica) software. Five independent samples (one slice per mouse) were analyzed for each staining and condition (wt and Epm2b−/− mice). For each staining, the percentage of the corresponding lasers and gain settings were maintained between samples. All images were acquired at one unit Airy pinhole, with a resolution of 512 × 512 pixels, zoom 0.75, and z stacks of 0.8 μm. For image analysis, the background was subtracted at 50 pixels using the image-processing package Fiji-ImageJ. The selected area was the same for all channels in the same image and the intensity signal was normalized by the size of the studied area. Particular areas within the hippocampus were: A, containing the radiatum, the lacunosum-moleculare, and the molecular dentate gyrus layers (RLM+MoDG area); B, the granular layer of the dentate gyrus (GrDG area); C, Pyramidal layer of the Cornus ammonis 1–3 and the polymorph layer of the dentate gyrus (Py+PoDG area); (Supplementary Figure S1B). Antibodies that gave no differences between Epm2b−/− and wild type samples are listed in the right column of Supplementary Table S1.
Western blot
Mouse brain hippocampi were lysed in RIPA buffer [50 mM Tris-HCl, pH 8; 150 mM NaCl; 0.5% sodium deoxycholate; 0.1% SDS; 1% Nonidet P40; 1mM PMSF; and complete protease inhibitor cocktail (Roche, Barcelona, Spain)] for 30 min at 4°C with occasional vortexing. The lysates were passed ten times through a 25 gauge needle in a 1 ml syringe and centrifuged at 13,000 × g for 15 min at 4°C. Supernatants were collected and a total of 35 μg protein was subjected to SDS-PAGE and transferred onto a PVDF membrane. Membranes were blocked in 5% (w/v) nonfat milk in Tris-buffered saline (TBS-T: 50 mM Tris-HCl, 150 mM NaCl, pH 7.4; with 0.1% Tween-20) for 1 h at room temperature and incubated overnight at 4°C with the corresponding primary antibodies listed in Supplementary Table S1. Mouse anti-GAPDH (Santa Cruz Biotechnologies, sc-32233) was used as a housekeeping antibody. Then, membranes were probed with suitable secondary antibodies for 1 h at room temperature. Signals were obtained by chemiluminescence using ECL Prime Western Blotting Detection Reagents (Cytiva-Amersham, RPN2232), and the image reader Fuji-LAS-4000 (GE Healthcare, Barcelona, Spain). The results were analyzed using the software Image Studio Lite version 5.2 (LI-COR Biosciences, Germany). Experiments were performed on at least three individuals from each genotype (males and females). Results are shown as median with range.
Analysis of the presence of inflammatory biomarkers in mice blood serum
Male and female mice at 7, 12, and 16 months of age were anesthetized with isoflurane gas 4%, and blood was collected from the retro-orbital sinus. Blood samples were incubated for 2h at room temperature for clotting and then centrifuged at 1,800 × g for 5 min twice. The serum samples were aliquoted and stored at −80°C until analysis. The detection of inflammatory mediators in serum was performed by using commercial enzyme-linked immunosorbent assay (ELISA) kits: LCN2 (Abcam, #ab199083), CXCL10 (Abcam, #ab260067), CCL5 (Abcam, #ab100739), C3 (Abcam, #ab157711), S100b (Abcam, #ab234573), CCL20 (Abcam, #ab100728) and HMGB1 (Cloud-Clone Corporation #SEA399). Samples were diluted 1/2 – 1/3 and analyzed by replicates following the manufacturer’s instructions. Mouse cytokine arrays were performed using the Proteome Profiler Mouse XL cytokine array kit (R&D systems, #ARY028), which contains 111 captured antibodies against inflammatory mediators, in two samples from 16 month-old mice of each genotype. Cytokine profiles were performed as instructed in the manufacturer’s directions. Membrane quantifications were performed with HLImage software (Western Vision Software).
Statistical analyses
Experiments were performed in at least three mice from each genotype for the western blot analyses and at least five mice for immunofluorescence analyses. The data was previously analyzed for normal (Gaussian) distribution by D’Agostino-Pearson Omnibus normality as well as Shapiro-Wilk normality tests, using GraphPad Prims version 6.0 statistical software (La Jolla, CA, USA). The alpha value was less than 0.05 for both tests, meaning that the data did not follow a normal Gaussian distribution. Consequently, an unpaired and non-parametric Mann-Whitney test was performed, according to Gradhpath software. P-values have been considered as *p<0.05, **p<0.01, and ****p<0.0001; ns, corresponds to no significant differences.
RESULTS
Complementary in silico analysis of RNAseq data from the brain of Epm2a−/− and Epm2b−/− mice of 16 months of age.
We have recently described the identification of differentially expressed genes in the brain of 16-month-old Epm2a−/− and Epm2b−/− mice compared to wild type animals. We reported that the most upregulated genes were related to inflammatory processes (Lahuerta et al., 2020). To understand if there was any common upstream regulator that could drive the expression of the upregulated genes, we performed a new in silico analysis of the data using the Qiagen Ingenuity Pathway Analysis (IPA) (QIAGEN Inc., https://digitalinsights.qiagen.com/IPA, (Kramer et al., 2014)). This analysis indicated that the top upstream regulators of the upregulated genes were TNF, IFNgamma, IL1beta, CSF2, NFkB, IL33, IL1alpha, and IL6 (Table I). In addition, this new analysis indicated that the top biological functions of the dataset were related to immune and inflammatory responses, confirming previously reported results (Lahuerta et al., 2020), but also cell movement of granulocytes, homeostasis of leukocytes, and cell infiltration (Table II). It is worth pointing out that the expression of some genes was observed in all the identified biological functions (e.g., C3, CCL2, CCL5, CXCL10, IL1RL1, and LCN2), highlighting the contribution of these genes in these pathways.
Table I:
Top upstream regulators of RNAseq dataset from the brain of LD mice in comparison to wild type, ordered by z-score (z-score >2 was defined as the threshold of significant activation). The p-value and the regulated genes are indicated.
| Upstream mediator | z-score | p-value | Regulated genes |
|---|---|---|---|
| TNF | 5.07 | 2.11 E-17 | ARG1, C3, CASP4, CCL2, CCL5, CCN5, CST7, CXCL6, CXCL10, IL1RL1, ITGAX, LCN2, LGALS3, MMP12, MMP3, STEAP4, TIMP1 |
| IFNgamma | 4.61 | 1.52 E-31 | ARG1, C3, CCL2, CCL5, CCL8, CD74, CHIL4, CXCL6, CXCL10, FCGR3A, HCAR2, IL1RL1, ITGAX, LCN2, MMP3, MMP12, RENTLA, TGM1, TIMP1 |
| IL1beta | 4.51 | 1.84 E-17 | ARG1, C3, CASP4, CCL2, CCL5, CCL17, CD74, CXCL6, CXCL10, FCGR2B, IL1RL1, LCN2, MMP12, MMP3, TGM1, TIMP1, TNFRSF9 |
| CSF2 | 4.40 | 4.34 E-19 | ACP5, ARG1, C3, C4A, CLEC7A, CCL2, CCL5, CCL17, CD74, CHIL4, CXCL6, CXCL10, FCGR3A, GPNMB, IL1RL1, ITGAX, MMP12, RENTLA, TNFSF8 |
| NFkB | 4.23 | 6.23 E-15 | BCL2A1, C3, CCL2, CASP4, CCL5, CD74, CXCL6, CXCL10, IL1B, LCN2, RENTLA, TNFRS9 |
| IL33 | 4.11 | 4.32 E-25 | CCL2, CCL5, CCL11, CCL17, CHIL4, CXCL10, IL1RL1, MMP12, TIMP1, TNFRSF9 |
| IL1alpha | 3.76 | 1.04 E-14 | ACP5, C3, CCL2, CCL5, CXCL6, CXCL10, LCN2, MMP3, MMP12, TIMP1 |
| IL6 | 3.74 | 2.01 E-16 | ACP5, ARG1, C3, CCL2, CCL5, CD74, CHIL4, CXCL6, CXCL10, HLA-DQA1, HLA-DRB5, IL1RL1, LCN2, LYZ, MMP3, MMP12, RENTLA, STEAP4, TIMP1 |
Table II:
Top biological functions of RNAseq dataset from the brain of LD mice in comparison to wild type ordered by z-score (z-score >2 was defined as the threshold of significant activation). The p-value and the genes involved in the corresponding biological functions are indicated.
| Diseases and Biological functions | z-score | p-value | Genes from the data set |
|---|---|---|---|
| Cell movement of granulocytes | 3.51 | 1.33 E-16 | C3, CCL2, CCL5, CD74, CHIL4, CXCL6, CXCL10, IL1RL1, LCN2, MMP12, TIMP1 |
| Homeostasis of leukocytes | 3.42 | 4.42 E-16 | ARG1, C3, CCL2, CCL5, CD74, CXCL6, CXCL10, IL1RL1, LCN2 |
| Cellular infiltration | 3.27 | 1.24 E-16 | ARG1, C3, CCL2, CCL5, CHIL4, CXCL10, IL1RL1, LCN2, MMP12, TIMP1 |
| Inflammatory response | 2.89 | 1.25 E-14 | C3, CCL2, CCL5, CD74, CHIL4, CLEC7A, CXCL6, CXCL10, FCGR3A, HCAR2, IL1RL1, LCN2, LYZ, RENTLA |
| Phagocytosis | 2.81 | 5.37 E-11 | C3, CCL2, CCL5, CLEC7A, CXCL10, FCGR3A, IL1RL1, LCN2 |
| Immune response of cells | 2.78 | 1.05 E-19 | ARG1, C3, CCL2, CCL5, CD74, CLEC7A, CXCL10, FCGR3A, IL1RL1, ITGAX, LCN2 |
TNF is a master upstream regulator of the inflammatory pathways present in 16 months-old Epm2b−/− mice
To validate the in silico results, we performed immunofluorescence analyses of selected inflammatory mediators to define the most compromised inflammatory signaling pathways in LD. Since the inflammatory response is similar in Epm2a−/− and Epm2b−/− mice (Lahuerta et al., 2020), we performed these analyses only in 16 months-old Epm2b−/− animals. We focused the study on the hippocampus since this is the area of the brain more related to LD pathophysiology (Rubio-Villena et al., 2018), (Sun et al., 2021). In fact, it is in this area where we observed higher intensity signals for all the inflammatory markers analyzed in this study (see below).
Since the highest z-score in the in silico analysis (Table I) pointed out TNF as a putative upstream regulator that could be responsible for the inflammatory response present in Epm2b−/− mice, we first analyzed the expression of TNF and its downstream-related mediators (Figure 1A). The protein levels for TNF were analyzed by immunofluorescence using brain slices from five mice (including males and females) from Epm2b−/− or wt animals of 16 months of age. Full hippocampus confocal images were obtained by image stitching, and immunofluorescence signal for each region [A, containing the radiatum, the lacunosum-moleculare and the molecular dentate gyrus layers (RLM+MoDG area); B, the granular layer of the dentate gyrus (GrDG area); C, Pyramidal layer of the Cornus ammonis 1–3 and the polymorph layer of the dentate gyrus (Py+PoDG area); (Supplementary Figure S1B)] was analyzed as described in Methods. Since no differences were observed between genders, we quantified and analyzed the images corresponding to males and females together. Interestingly, the intensity of the signal for TNF was higher (p<0.05) in the RLM+MoDG area of Epm2b−/− compared to wt mice (Figure 1B and Supplementary Figure S2A), but not in the other studied areas (Supplementary Fig. S3, panels A and B).
Figure 1: TNF is a master upstream regulator of the p65-NFkB and MAPKs inflammatory pathways in 16 months-old Epm2b−/− mice.
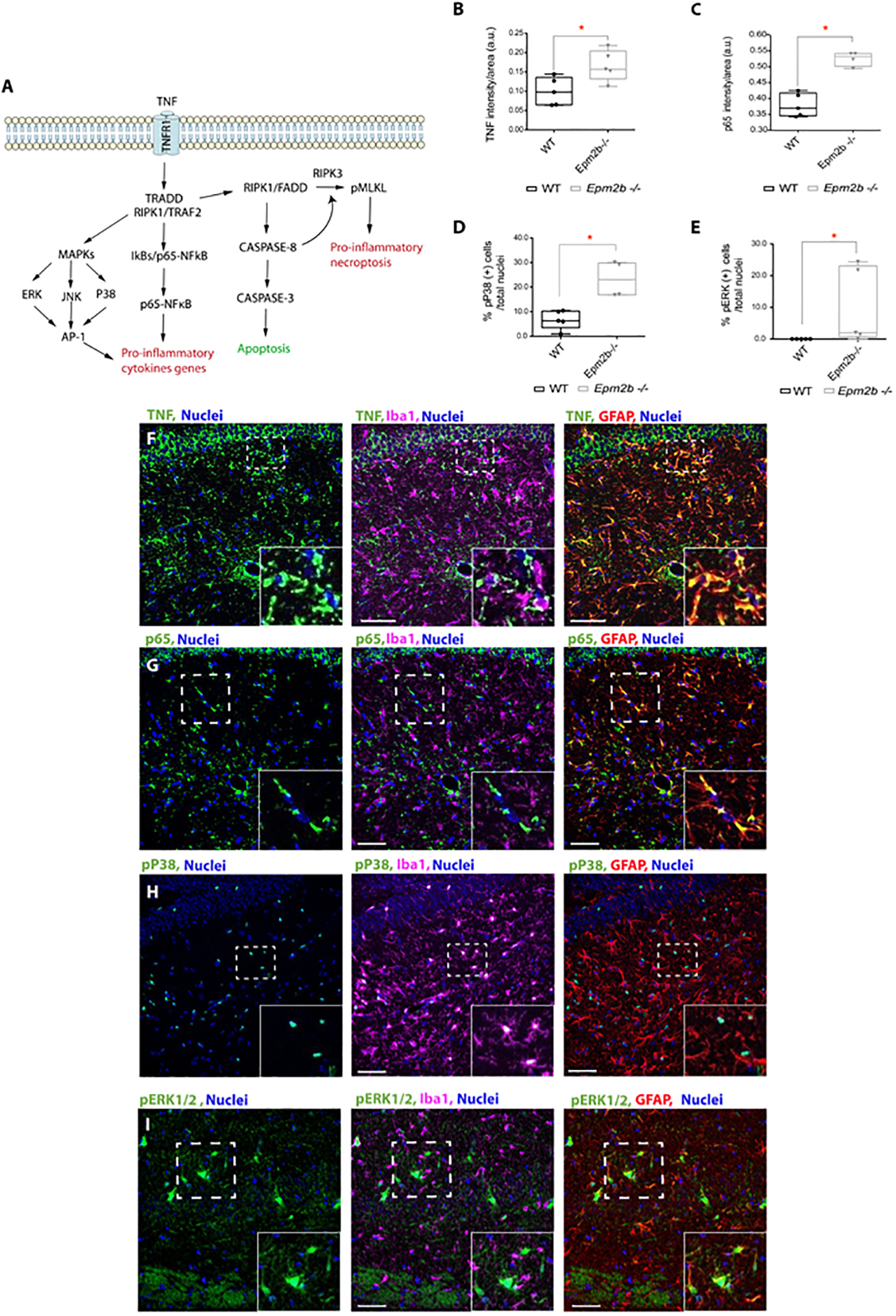
(A) Schematic representation of TNF-dependent inflammatory pathways. (B-E) The levels of TNF, p65-NFkB (p65), phospho (Thr180/Tyr182)-P38 (pP38) and phospho (Thr202/Tyr204)-ERK1/2 (pER1/2) proteins were analyzed by immunofluorescence in hippocampus from 16-month-old Epm2b−/− and wt mice. Quantification of the intensity signal for TNF (B) and p65 (C), or the percentage of positive cells for pP38 (D) and pERK1/2 (E), referred to the total nuclei in the RLM+MoDG area from each genotype is indicated and represented as arbitrary units (a.u.). Results are expressed as median with a range of five independent samples including male and female mice from both genotypes. The differences between the two groups (wt and Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as *p<0.05. (F-I) Representative confocal images of the immunofluorescence analyses of the RLM+MoDG area from 16-month-old Epm2b−/− mice using anti-GFAP (red), anti-Iba1 (magenta), and (F) anti-TNF (green), (G) anti-p65 (green), (H) anti-phospho-P38 (green) or (I) anti-phospho-ERK1/2 (green) (See Supplementary Table S1). Nuclei (blue) were stained with DAPI. The scale bar, shown in the merged images (central and right panels), corresponds to 25μm. A squared dashed line indicates the area that is magnified.
It is known that TNF triggers a signaling pathway by interacting with the TNFR1 and TNFR2 receptors (Amin et al., 2022), (Lousa et al., 2022) (Figure 1A). In the case of TNFR1, TNF induces the trimerization of the receptor, which is then able to recruit several adaptors such as TNF receptor type1-associated death domain protein (TRADD), TNF receptor-associated factor 2 (TRAF2), and Receptor-interacting Ser/Thr protein kinase 1 (RIPK1), resulting in the activation of RIPK1 kinase, which activates both, the p65-NFkB (p65) and the MAPKs signaling pathways (Figure 1A). This leads, on the one hand, to the nuclear import of p65, and on the other hand, to the phosphorylation and activation of the major MAPKs, such as P38, ERK1/2, and JNK (Courtois and Fauvarque, 2018). The combined action of p65 and phospho-MAPKs results in the upregulation of the expression of pro-inflammatory cytokines (e.g., IL1beta, IL6, etc.) (Figure 1A). To test if these downstream inflammatory pathways were playing a role in LD, the levels of p65 were analyzed by immunofluorescence as described above for TNF. As expected, the protein levels for p65 were higher (p<0.05) in the RLM+MoDG area of the Epm2b−/− compared to wt mice (Figure 1C and Supplementary Figure S2B). No statistically significant differences were observed in the other studied areas (Supplementary Fig. S3, panels C and D).
We explored next the activation status for members of the MAPKs pathways in the same way. To perform this analysis, the hippocampus from 16 months-old Epm2b−/− and wt mice were stained with antibodies against phospho (Thr180/Tyr182)-P38, phospho (Thr183/Tyr185)-JNK, and phospho (Thr202/Tyr204)-ERK1/2 MAPK kinases. Interestingly, the signal of the phosphorylated (activated forms) of P38 and ERK1/2 were higher (p<0.05) in the RLM+MoDG area of the Epm2b−/− compared to wt mice (Figure 1D and 1E, respectively, and Supplementary Figure S2C and S2D). However, no differences were detected for pJNK (Supplementary Table S1). As for the other markers included in this study, no statistically significant differences were observed in the other studied areas (Supplementary Fig. S3, panels E to H).
To figure out which cells were producing these inflammatory mediators, brain sections from 16 months-old mice were assessed by immunofluorescence, combining the use of anti-TNF, anti-p65, anti-pP38, and anti-pERK1/2 antibodies, with the use of specific astrocytic (GFAP) and microglia (Iba1) markers. As shown in Figures 1F, 1G, and 1I, reactive astrocytes, which were positive for GFAP, were also positive for the expression of TNF, p65, and pERK1/2, respectively. Meanwhile, pP38 was detected in the nuclei of microglia (Figure 1H). These results pointed out again that in the Epm2b−/− mice reactive glia was involved in the inflammatory process described here.
The TNF signaling pathway has also a branch, which is involved in the regulation of either apoptosis or necroptosis (Figure 1A). In this branch, caspase-8 plays a major role since, when it is cleaved and then activated, it triggers apoptosis through the activation of caspase-3 (Shi and Sun, 2018), (Lousa et al., 2022), (Richard and Mousa, 2022). However, when caspase-8 is not cleaved, it gets engaged in the formation of a complex with RIPK1, which will recruit and activate Receptor Interacting Protein Kinase 3 (RIPK3), and this will activate by phosphorylation Mixed linage kinase domain-like pseudokinase (MLKL), leading to necroptosis (Lousa et al., 2022), (Richard and Mousa, 2022), (Fritsch et al., 2019). So, we analyzed caspase-8 levels as above. Interestingly, higher levels of caspase-8 were detected in the RLM+MoDG area of the 16 months-old Epm2b−/− mice compared to wt (P<0.05) (Figure 2A and Supplementary Figure S4, panel A). No statistically significant differences were observed in the other studied areas (Supplementary Fig. S4, panels B and C). In addition, we analyzed the levels of caspase-8 by western blot. This technique lets us distinguish between the full-length caspase-8 and cleaved-caspase-8 (active form). As shown in Figure 2B and Figure 2C–2D, the levels of full-length caspase-8 and cleaved caspase-8 were higher (p<0.05, in both cases) in samples from Epm2b−/− mice compared to wt. We also analyzed the levels of p65 in the same way and observed higher (p<0.01) levels of p65 in Epm2b−/− mice compared to wt (Figure 2B and 2E), confirming that the activation of the TNF pathway increased the levels of this pro-inflammatory mediator (see above, Figure 1).
Figure 2: Pro-inflammatory necroptosis is activated in the brain of Epm2b−/− mice.
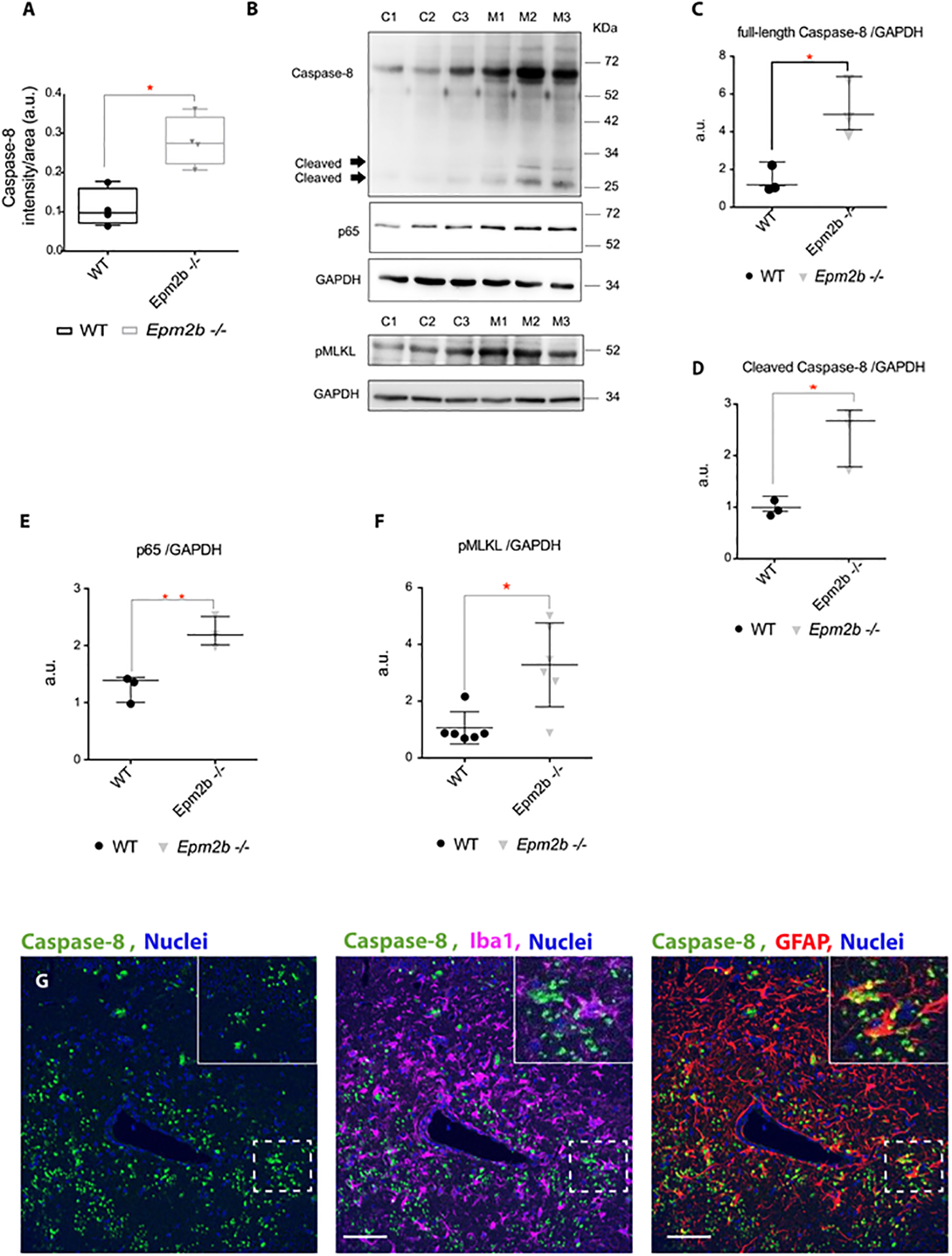
(A) Quantification of the intensity signal obtained by immunofluorescence with anti-Caspase-8 in the RLM+MoDG area of Epm2b−/− and wt 16 months old mice. (B) Protein levels of Caspase-8 (full length and cleaved), p65, and phospho (Ser345)-MLKL (pMLKL) were assessed by western blot of hippocampi extracts from 16-month-old wt (C1-C3) and Epm2b−/− (M1-M3) mice using the indicated antibodies. Molecular weight standards are on the right. Densitometric quantification of the corresponding blots was carried out as described in Methods. Protein levels of full-length Caspase-8 (C), cleaved Caspase-8 (D), p65 (E), and pMLKL (F) were related to the levels of GAPDH and represented as arbitrary units (a.u.). Results are expressed as median with a range of three independent samples from each genotype (C-E) or six independent samples for F. Differences between the two groups (wt and Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as *p<0.05 and **p<0.01. (G) Representative confocal images of the immunofluorescence analysis of the RLM+MoDG area from 16-month-old Epm2b−/− mice using anti-GFAP (red), anti-Iba1 (magenta), and anti-Caspase-8 (green) (see Supplementary Table S1). Nuclei (blue) were stained with DAPI. The scale bar, shown in the merged images (central and right panels), corresponds to 25μm. A squared dashed line indicates the area that is magnified.
As shown in Figures 2B and 2D, although cleaved caspase-8 is detectable in Epm2b−/− samples but not in the wt, the highest levels of caspase-8 were detected as full-length protein form (Figure 2B and 2C). This result suggests that full-length caspase-8 would be activating the necroptosis pathway in the hippocampus of the Epm2b−/− mice but not the apoptosis pathway. Confirming these results, we did not observe differences in the levels of the caspase-3 related to apoptosis (either in the full-length and the cleaved activated forms), between the hippocampus derived from Epm2b−/− and wt mice (Supplementary Table S1). Since our results suggested an activation of the necroptosis pathway in the Epm2b−/− mice model, we analyzed the levels of the activated form of MLKL protein (phospho Ser345), a necroptosis-related executioner, by western blot (Figure 2B and 2F), and observed higher levels of this protein (p<0.05) in the hippocampus of the 16 months-old Epm2b−/− mice compared to wt mice.
To figure out which neuroglial cells were expressing caspase-8 in Epm2b−/− mice, brain sections from 16 months-old mice were assessed by immunofluorescence combining the use of anti-caspase-8, with the use of specific astrocytic (GFAP) and microglia (Iba1) markers. As shown in Figure 2G, astrocytes were the main neuroglial cell expressing caspase-8.
In summary, all these results confirmed that the TNF signaling pathway is upregulated in the hippocampus of the 16 months-old Epm2b−/− mice, triggering inflammatory p65-NFkB- and MAPKs-dependent pathways. At the same time, we observed higher levels of necroptosis-related mediators such as caspase-8 and pMLKL. More importantly, the analysis by immunofluorescence also pointed out that these inflammatory proteins were mainly produced by reactive glia.
The non-canonical pathway of the inflammasome is upregulated in the hippocampus of the 16 months-old Epm2b−/− mice
Taking into account the increased levels of p65-NFkB observed in Epm2b−/− mice brains (see above) and since this mediator is a positive regulator of the inflammasome pathway (Chen et al., 2021), next, we investigated whether the activity of this pathway was affected in the brain of Epm2b−/− mice. The inflammasome pathway is mainly activated through NLRP3 and caspase-1 (canonical pathway), but recently a non-canonical branch has been described, which is independent of NLRP3 but dependent on caspase-11 (caspase-4 in humans) (Figure 3A). Activation of caspase-11 drives the cleavage of Gasdermin-D (GSDM-D); then, the N-terminal fragment of GSDM-D (GSDM-D NT) localizes in the plasma membrane to facilitate the exit of cytokines and other inflammatory molecules (De Nardo, 2017), (Rathinam et al., 2019), (Agnew et al., 2021). We analyzed by immunofluorescence the levels of NLRP3, caspase-1, and some other components of the canonical inflammasome pathway, such as Absent in Melanoma 2 (AIM2) or NLR Family Apoptosis Inhibitor Protein (NAIP) (Supplementary Table S1), and we found no differences between Epm2b−/− samples and wt, indicating that the canonical pathway of the inflammasome was not affected. Interestingly, analysis of the hippocampus of 16 months-old Epm2b−/− and wt mice by immunofluorescence showed an increase in GSDM-D levels (p<0.05) in the RLM+MoDG area (Figure 3C and Supplementary Figure S5B). No statistically significant differences were observed in the other studied areas (Supplementary Fig. 5, panels C and D). Surprisingly, we did not detect statistical immunofluorescence differences in the levels of caspase-11 (Figure 3B and Supplementary Figure S5A). Since caspase-11 is activated by cleavage, we explored the activation status of this caspase by western blot analysis. Figure 3D and 3E show that cleaved caspase-11 was detected in the hippocampus derived from the Epm2b−/− samples and not in the wt mice (p<0.05). Moreover, this experiment let us confirm that the levels of GSDM-D were also increased in Epm2b−/− samples (Figure 3D and 3F) (p<0.01). On the contrary, no differences were detected for NLRP3 and caspase-1 (Figures 3D, 3G, and 3H), confirming the results obtained by immunofluorescence indicated above.
Figure 3: The non-canonical pathway of the inflammasome is upregulated in the hippocampus of 16 months-old Epm2b−/− mice.
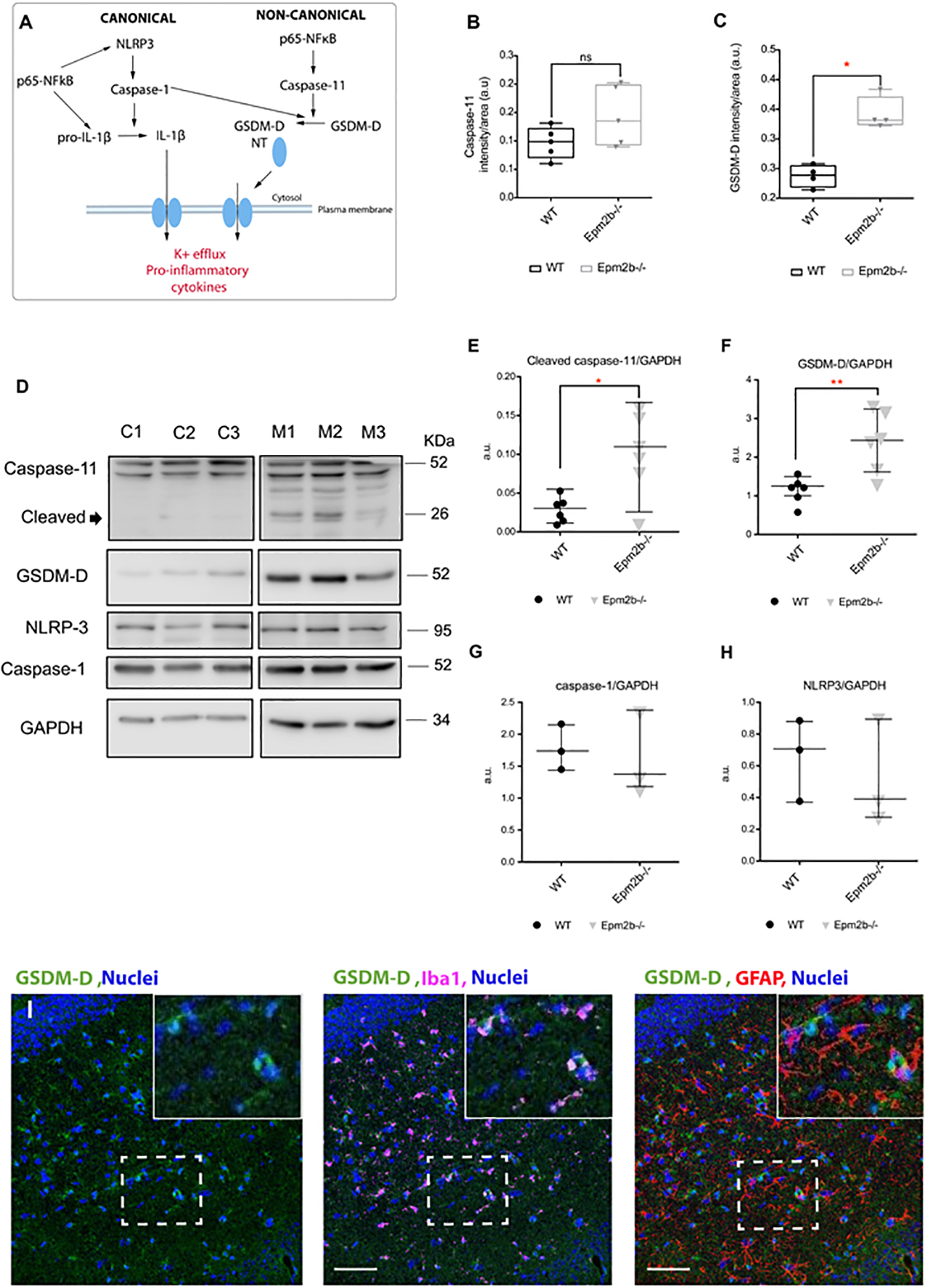
(A) Schematic representation of the canonical (left) and non-canonical (right) inflammasome pathways. (B and C) Quantification of the intensity signal detected by immunofluorescence in the RLM+MoDG area of 16 months-old Epm2b−/− and wt mice for caspase-11 (B) or GSDM-D (C). Results are expressed as median with a range of four to six different samples from each genotype, including male and female mice. (D) Protein levels of caspase-11 (cleaved and full-length protein), GSDM-D, caspase-1, and NLRP3 proteins, were analyzed in hippocampi extracts from 16 months-old wt (C1-C3) and Epm2b−/− (M1-M3) mice by western blot using the indicated antibodies. Molecular weight standards are on the right. Densitometric quantification of the corresponding blots for cleaved caspase-11 (E), GSDM-D (F), caspase-1 (G), and NLRP3 (H), were related to GAPDH levels and represented as arbitrary units (a.u.). Results are expressed as median with a range of at least three independent samples. Differences between the two groups (wt and Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as *p<0.05 and **p<0.01; ns corresponds to no significant differences. (I), Representative confocal images of the immunofluorescence analysis of the RLM+MoDG area from 16 months-old Epm2b−/− mice using anti-GFAP (red), anti-Iba1 (magenta), and anti-GSDM-D (green). Nuclei (blue) were stained with DAPI. The scale bar, shown in the merged images (central and right panels), corresponds to 25μm. A squared dashed line indicates the area that is magnified.
All these results together suggest that the non-canonical inflammasome pathway is upregulated in the brain of Epm2b−/− mice. To further investigate the role of this pathway in the pathophysiology of LD, next, we analyzed the neuroglial cells in which GSDM-D was expressed. With this aim, hippocampi from 16 months-old Epm2b−/− mice were co-stained with anti-GSDM-D together with anti-GFAP (astrocytes) and anti-Iba1 (microglia) markers. Figure 3I shows that GSDM-D was mainly expressed in microglia.
IL6/JAK2 inflammation pathway is upregulated in the hippocampus of the 16 months-old Epm2b−/− mice
We then moved to the validation of alternative upstream regulators defined in Table I. We analyzed the levels of components of the Toll-like receptor (TLR)/IL1RL1 signaling pathways (e.g., HMGB1, TLR4, MyD88, TLR7, IRF3, IRF7, IL1RL1, IL1beta), and the levels of INFgamma and INFalpha (Supplementary Table S1), but we did not detect changes in Epm2b−/− samples in comparison to wt.
Additionally, we investigate the role of IL6 in LD pathophysiology, as a putative upstream regulator (Table I). IL6 interacts with its receptor IL6R (gpr130), which has an associated Janus kinase 2 (JAK2), leading to the activation of JAK2 by phosphorylation. This triggers the phosphorylation of cytosolic proteins such as Signal transducer and activator of transcription-1 (STAT1) and STAT3, which undergo dimerization and nuclear translocation, to regulate the expression of pro-inflammatory cytokines, as well as suppressor of cytokine signaling 3 (SOCS3), an inhibitor of IL6/JAK2 signaling pathway (Figure 4A) (Dey et al., 2016). To understand if IL6 played a role in LD pathophysiology, 16 months-old samples from Epm2b−/− and wt mice were stained with antibodies against the main proteins involved in this pathway. Interestingly, the intensity signal for IL6 (p<0.05), and phospho (Tyr1007-Tyr1008)-JAK2 (p<0.05) were higher in the RLM+MoDG area of Epm2b−/− mice compared to wt mice (Figure 4B and 4C, respectively, and Supplementary Figure S6A and S6B). No statistically significant differences were observed in the other studied areas (Supplementary Fig. S7, panels A to D). As expected (Figure 4A), higher levels of phospho (Tyr701)-STAT1 (p<0.01) and phospho (Tyr705)-STAT3 (p<0.01) were found in the RLM+MoDG area of the Epm2b−/− samples (Figure 4D and 4E, respectively, and Supplementary Figure S6C and S6D). In the case of pSTAT1 and pSTAT3, we noticed that the signal was localized in the nuclei (Figure 4I and 4J, respectively). In the case of pSTAT3, significant differences were also observed in the GrDG area (p<0.01) (Supplementary Fig S7 panel G). Finally, to confirm the upregulation of this pathway, we analyzed the expression of SOCS3. As shown in Figure 4F and Supplementary Figure S6E, higher levels of SOCS3 (p<0.05) were detected by immunofluorescence in the RLM+MoDG area of Epm2b−/− samples compared to wt. No statistically significant differences were observed in the other studied areas (Supplementary Fig. S7, panels I and J).
Figure 4: IL6-JAK/STAT pathway is upregulated in the hippocampus of 16-month-old Epm2b−/− mice.
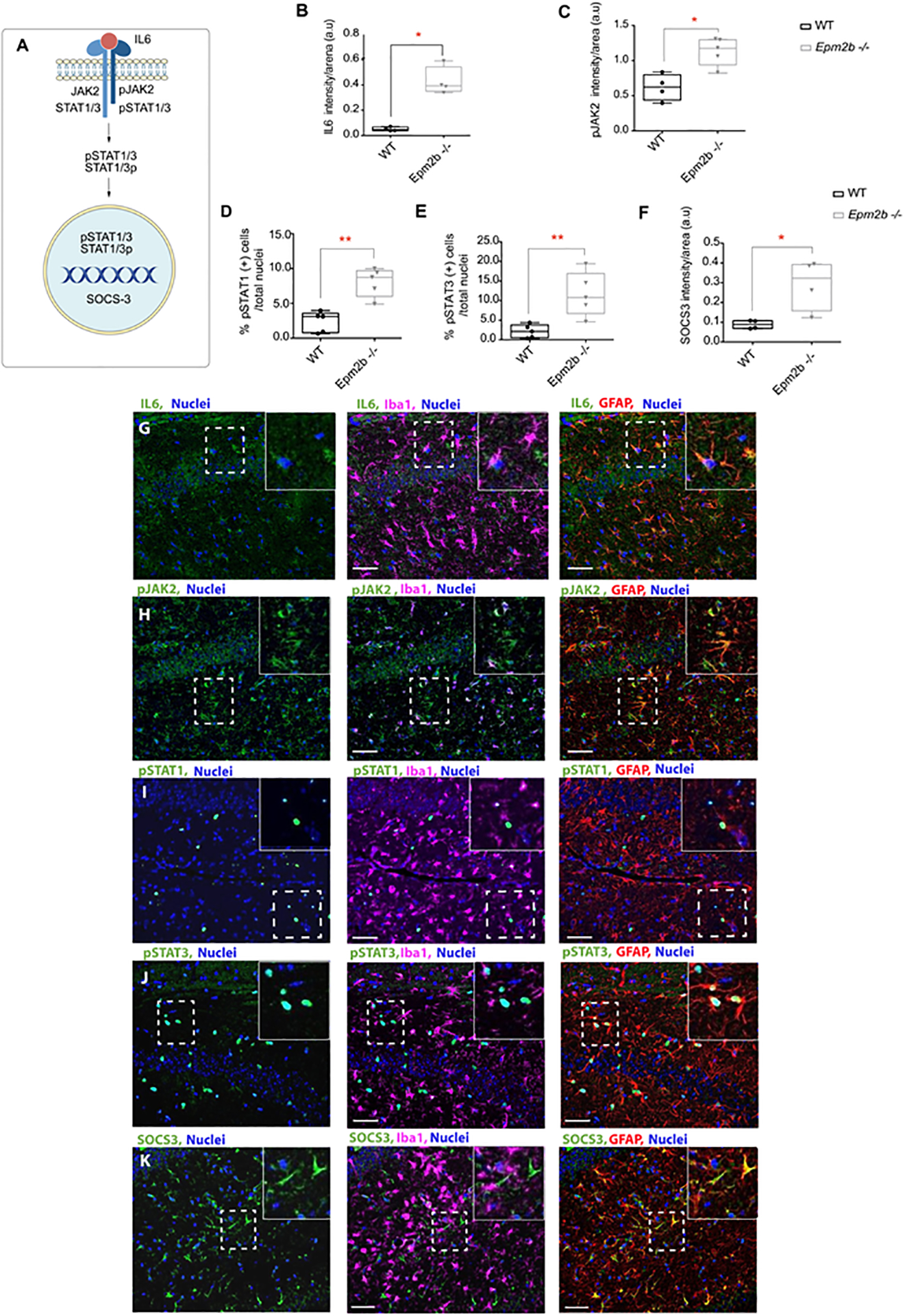
(A) Schematic representation of the IL6/JAK2-STAT signaling pathway. (B-F) Quantification of the intensity signal detected by immunofluorescence in the RLM+MoDG area of Epm2b−/− and wt of 16 months-old mice. IL6 (B), phospho (Tyr1007/Tyr1008)-JAK2 (pJAK2) (C), or SOCS3 (F) showed increased levels of these proteins in Epm2b−/− samples. In the case of proteins that were localized in the nucleus, the percentage of phospho (Tyr-701)-STAT1 (pSTAT1) (D) or phospho (Tyr-705)-STAT3 (pSTAT3) (E), referred to the total nuclei, showed also higher levels in the Epm2b−/− samples. Results are expressed as median with a range of five independent samples including male and female mice, from both genotypes, and represented as arbitrary units (a.u.). Differences between the two groups (wt and Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as *p<0.05 and **p<0.01. (G-K) Representative images of the immunofluorescence analysis of the RLM+MoDG area of Epm2b−/− 16 months old mice using anti-GFAP (red), anti-Iba1 (magenta), and anti-IL6 (G), anti-phospho-JAK2 (pJAK2) (H), anti-phospho-STAT1 (pSTAT1) (I), anti-phospho-STAT3 (pSTAT3) (J), or anti-SOCS3 (K), all in green. Nuclei (blue) were stained with DAPI. The scale bar, shown in the merged images (central and right panels), corresponds to 25μm. A squared dashed line indicates the area that is magnified.
As for the other pathways analyzed in this work, next, we investigated the neuroglial cell types where the IL6/JAK2 pathway is upregulated in Epm2b−/− brain mice. Samples were stained with antibodies against IL6, pJAK2, pSTAT1, pSTAT3, and SOCS3 in combination with astrocytes and microglia markers. This analysis pointed out again astrocytes as the main cell type involved in the inflammation landscape depicted by the Epm2b−/− LD mice model since all these proteins were mainly expressed in these cells (Figure 4 G–K).
Infiltration of peripheral lymphocytes into the brain parenchyma occurs in the 16 months-old Epm2b−/− mice without major blood barrier disruption
Since TNF and IL6 pathways are involved in the extravasation of immunocompetent cells into the central nervous system (CNS) during inflammation processes (Dey et al., 2016), (Mukhtar, 2020), (Amin et al., 2022), next, we analyzed the possible presence of infiltrating lymphocytes in the brain parenchyma of Epm2b−/− mice. Figure 5A and 5B show an increase in the levels of CD3+ cells (T-lymphocytes) (p<0.01), which correlated with an increase in CD4+ T-helper cells (p<0.01) (Figure 5C and 5D) and increased levels of the CD8+ T-cytotoxic lymphocytes (p<0.01) (Figure 5E and 5F) in the 16 months-old Epm2b−/− mice hippocampus. These results validated the in silico analysis (see above) where cell movement of granulocytes and cellular infiltration were among the top biological functions related to LD (Table II).
Figure 5: Hippocampus of 16 months-old Epm2b−/− mice present infiltration of T-lymphocytes.
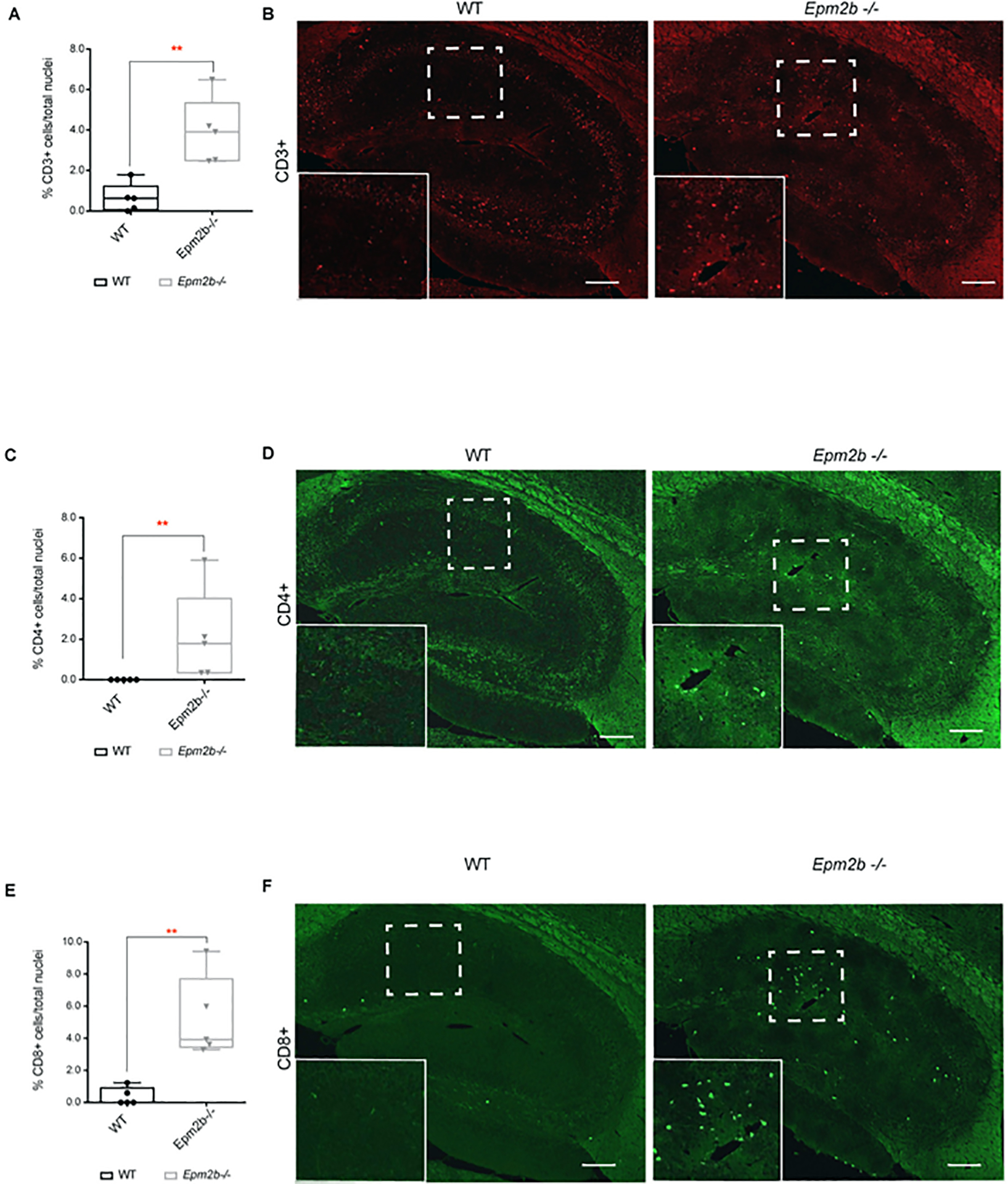
(A, C, and E) Quantification of the relative number of infiltrating cells related to the total nuclei in the corresponding area. (A) CD3+ (T-cell marker) cells, (C) CD4+ (helper T-cell marker) cells, and (E) CD8+ (cytotoxic T-cell marker) cells in the whole hippocampus area of 16 months-old Epm2b−/− and wt mice detected by immunofluorescence. The results are represented as a median with a range of five samples for each genotype including male and female mice. Differences between the two groups (wt and Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as **p<0.01. (B, D, and F) representative confocal images of the whole hippocampus from 16 months-old Epm2b−/− and wt mice stained with the following antibodies: Rat anti-CD3 (red) (B), mouse anti-CD4 (D) or rabbit anti-CD8 (green) (F). The scale bar corresponds to 100μm. A squared dashed line indicates the area that is magnified.
Taking into account these results, we analyzed whether the infiltration of peripheral lymphocytes was due to a massive disruption of the blood-brain barrier (BBB). Interestingly, no differences were observed in the levels of granulocytes (Ly-6G+), macrophages (F4/80+) and albumin in the Epm2b−/− mice brain compared to wt (Supplementary Table S1), confirming that there was not a massive leakage of the BBB, but a specific infiltration of T-lymphocytes.
Analysis of the levels of selected pro-inflammatory mediators in serum samples from mouse models of LD.
It is known that when the expression levels of some inflammatory mediators reach a certain threshold, they may diffuse to blood, serving as mechanistic disease biomarkers for the progression of the disease (Ravizza and Vezzani, 2018), (Simonato et al., 2021). Thus, we decided to explore the presence of some of the markers defined in this, and previous work (Lahuerta et al., 2020), in the serum of male and female Epm2b−/− mice at 16 months of age. We initiated our study by measuring by ELISA the levels of the proteins corresponding to four of the most expressed genes in LD models, namely lipocalin 2 (LCN2), CXCL10, CCL5, and C3 (Lahuerta et al., 2020). In each case, as no differences between males and females were found, we plotted the results as a mixture of males and females of each genotype. As it is shown in Figure 6A, we found similar levels of LCN2, CCL5, and C3 in serum samples from Epm2b−/− animals in comparison to wt mice. However, the levels of CXCL10 were significantly higher in the samples from Epm2b−/− animals (p<0.0001). As it has been described that the levels of alarmins such as S100b and HMGB1 are higher in blood samples from several models of epilepsy (Terrone et al., 2017), (Terrone et al., 2019), (Simonato et al., 2021), (Miller and Blanco, 2021), we also measured by ELISA the levels of these two proteins in the serum of the LD mouse model at 16 months of age. As it is shown in Figure 6A, we found higher levels of S100b in the Epm2b−/− mice (p<0.01) in comparison to wt animals. However, no differences in the levels of HMGB1 in comparison to wt mice were observed.
Figure 6: Levels of CXCL10 and S100b in serum could serve as a biomarker of the progression of Lafora disease in mice.
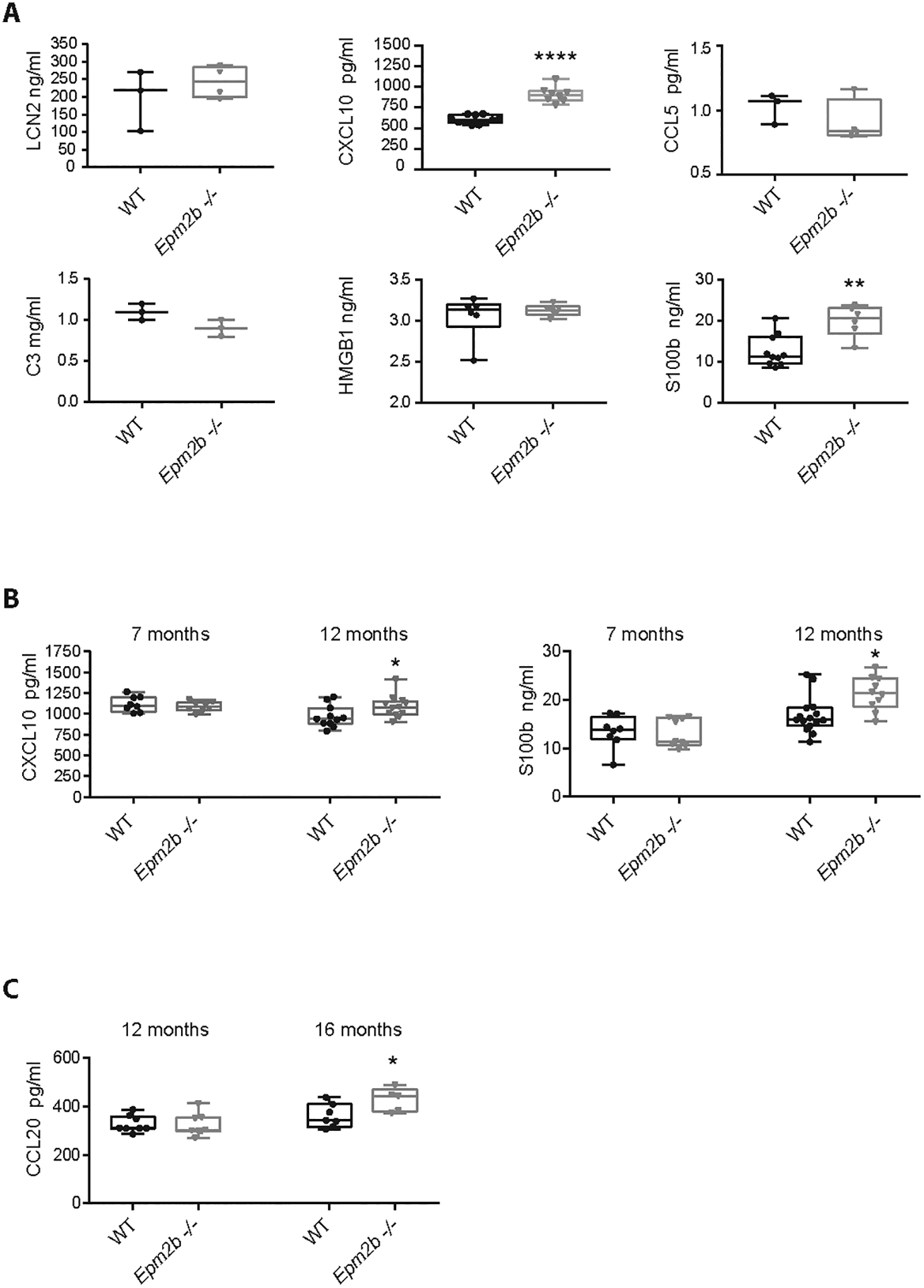
(A) The levels of LCN2, CXCL10, CCL5, C3, HMGB1, and S100b were measured in serum samples from Epm2b−/− and wt of 16 months-old mice by using specific ELISA tests (see Methods). (B) The levels of CXCL10 and S100b were measured as above in serum samples from 12 and 7 months-old mice. (C) The levels of CCL20 were measured in serum from 16 and 12 months-old mice using a specific ELISA test. Values represent the mean +/− standard error of the mean (SEM) of the following number of animals: At 16 months of age, LCN2 (wt 3 males; Epm2b−/− 4 males); CXCL10 (wt 7 males and 4 females; Epm2b−/− 5 males and 3 females); CCL5 (wt 3 males; Epm2b−/− 4 males); C3 (wt 3 males; Epm2b−/− 3 males); HMGB1 (wt 3 males and 3 females; Epm2b−/− 3 males and 3 females); S100b (wt 6 males and 4 females; Epm2b−/− 4 males and 2 females); CCL20 (wt 4 males and 3 females; Epm2b−/− 3 males and 2 females). At 12 months of age, CXCL10 (wt 5 males and 6 females; Epm2b−/− 4 males and 8 females); S100b (wt 8 males and 7 females; Epm2b−/− 4 males and 6 females); CCL20 (wt 5 males and 6 females; Epm2b−/− 4 males and 8 females). At 7 months of age, CXCL10 (wt 4 males and 4 females; Epm2b−/− 4 males and 4 females); S100b (wt 5 males and 3 females; Epm2b−/− 4 males and 5 females). Differences between the groups (wt vs Epm2b−/− mice) were analyzed by Mann-Whitney non-parametric t-test. P-values have been considered as *p<0.05, **p<0.01 and ****p<0.0001.
Next, we decided to measure the levels of CXCL10 and S100b in samples from younger animals. As it is shown in Figure 6B, higher levels of CXCL10 and S100b were found in samples from Epm2b−/− mice at 12 months of age (p<0.05 in both cases), but no differences in the levels of these mediators were found in samples from 7 months old animals. Taken together, our results suggest that the levels of CXCL10 and S100b could be used as biomarkers of Epm2b−/− mice when they age.
Since by the ELISA techniques described above we only measured the levels of six selected mediators, we decided to extend our analysis to additional candidates by using a high throughput mouse cytokine array (see Methods). Serum samples from Epm2b−/− and wt mice at 16 months of age were used in the analysis. In Supplementary Table S2, we show the crude average intensity of all the spots, which was measured by using the Quick Spot software provided by the manufacturer, and the fold change between Epm2b−/− and wt mice-derived samples. Among those, only the levels of CCL20 and IL15 reached statistical significance (p≤0.05).
Since the fold change for CCL20 (FC 2.4) was higher than for IL15 (FC 1.6), we validated by ELISA only the levels of CCL20 in serum samples from LD and wt mice at different ages. As it is shown in Figure 6C, at 16 months of age, the levels of CCL20 were higher (p<0.05) in samples from Epm2b−/− mice in comparison to wt samples of the same age, but at 12 months of age, we found no differences in LD mice in comparison to wt mice (Figure 6C).
DISCUSSION
Lafora disease (LD, OMIM#254780) is a rare form of progressive myoclonus epilepsy characterized by the accumulation of polyglucosan inclusions named Lafora bodies (LBs) in the brain and other peripheral tissues. Using animal models of LD, we have recently described that neuroinflammation is another hallmark of the disease, starting at three months of age and getting more severe as the animals get older (Lahuerta et al., 2020). By RNAseq analysis, we described a general pro-inflammatory landscape in the brain of LD animals. However, at that time we were not able to identify the putative upstream regulators responsible for the up-regulation of the expression of the corresponding pro-inflammatory mediators. In this work, we first performed an in silico analysis of the RNAseq datasets using the Qiagen Ingenuity Pathway Analysis (IPA) software (Kramer et al., 2014). This analysis identified some candidates as upstream regulators of the expression of the mediators that were upregulated in LD (Table I). Interestingly, these upstream regulators were similar to the ones found in the analysis of the epileptic EL mouse model (Lee et al., 2021), suggesting a close connection between LD and EL mouse models.
Our results indicate that in the brain of 16 months-old Epm2b−/− mice there were higher levels of TNF, which correlated with higher levels of p65-NFkB. In both cases, astrocytes were the main neuroglial cells involved in the expression of these two proteins, highlighting the key role of astrocytes in triggering LD inflammation. These results are in agreement with previous reports indicating that TNF is released by astrocytes and microglia in response to neurodegeneration (Amin et al., 2022).
The TNF signaling pathway has also a branch, which is involved in the regulation of either apoptosis or necroptosis (Figure 1A). In this branch, we observed higher levels of caspase 8 and the necroptosis executioner pMLKL, suggesting that the TNF signaling might lead to this form of programmed cell death.
Taking all these results together, we suggest that TNF is a major upstream regulator of the expression of pro-inflammatory mediators in LD. This result is in agreement with previous reports, which suggest that activation of TNF occurs in the first stages of several neurodegenerative diseases (Lalani et al., 2018), (Rodgers et al., 2020), (Amin et al., 2022). The activation of the TNF signaling pathway, mainly in astrocytes, could lead to necroptosis, a programmed form of necrosis. These results would confirm previous reports which indicated that necrosis but not apoptosis played a role in the neurodegeneration present in LD (Ganesh et al., 2002), (Delgado-Escueta, 2007), (Machado-Salas et al., 2012).
Since we detected an increase in the levels of p65-NFkB (see above), which is a positive regulator of the inflammasome pathway (Chen et al., 2021), we decided to check whether the activity of this pathway was affected in the brain of Epm2b−/− mice. We found higher levels of cleaved and activated caspase-11, and GSDM-D (Figure 3B–3F), two key components of the non-canonical inflammasome pathway (Rathinam et al., 2019), (Chen et al., 2021), (Agnew et al., 2021). Probably, the increase in the levels of p65, pP38, and pSTAT1 present in Epm2b−/− mice, which are recognized stimulators of caspase-11 expression (Agnew et al., 2021), could account for the upregulation of the non-canonical inflammasome pathway in these animals. As the activation of this non-canonical pathway may result in an alternative form of cell death named pyroptosis (Rathinam et al., 2019), (Chen et al., 2021), (Agnew et al., 2021), our results would suggest that the neurodegeneration present in the brain of Epm2b−/− mice could also be due to pyroptosis. We would like to point out that this form of cell death has also been described in another form of progressive myoclonus epilepsy (EPM1). Mouse models of this disease (Cstb−/− mice) present higher levels of caspase-11 as a consequence of elevated levels of p65 and secrete higher amounts of pro-inflammatory cytokines (Maher et al., 2014).
Our work also highlights the role of the IL6 signaling pathway in LD pathophysiology. As in the case of the TNF signaling pathway, astrocytes were again the main neuroglial cells involved in the IL6/JAK2 signaling pathway, which is in agreement with reports which indicate that this pathway is one of the first signaling pathways initiating astrocyte reactivity (Lee et al., 2022).
In addition to the TNF and IL6/JAK2 signaling pathways, we would like to indicate that our previous work highlighted the role of astrocytic-released chemokines such as CXCL10, CCL5, and LCN2 in LD pathophysiology (Lahuerta et al., 2020). Thus, we suggest that these signaling pathways play a main role in the neuroinflammation present in LD.
Finally, although in this study we have identified that astrocytes and microglia are the main cells responsible for the expression of the upregulated pro-inflammatory mediators, we have also defined for the first time the presence of infiltrating lymphocytes in the brain parenchyma of Epm2b−/− mice. Our results show an increase in the presence of CD3+ cells (related to T-lymphocytes), which were mainly CD8+ (related to cytotoxic T-lymphocytes). Interestingly, the numbers of CD3+ and CD8+ T-cells are higher in the hippocampus of epileptic patients and these numbers correlate with the degree of neuronal loss (Troscher et al., 2021). The infiltration of peripheral lymphocytes was not due to a massive disruption of the blood-brain barrier (BBB), since no changes in the presence of other alternative peripheral immune cells were detected (granulocytes Ly-6G+, macrophages F4/80+), and we did not detect the presence of albumin in the brain parenchyma (Supplementary Table S1). This is in agreement with reports which indicate that a cross-talk between the periphery and the brain occurs in the absence of a leakage of the BBB (Cervellati et al., 2020), (Passaro et al., 2021), (Yamanaka et al., 2021). In our opinion, this infiltration could be due to the combined action of TNF and IL6, which have a recognized role in the extravasation of immunocompetent cells into the CNS during systemic inflammation (Dey et al., 2016), (Mukhtar, 2020), (Amin et al., 2022). Interestingly, a similar leukocyte infiltration has been reported in the mouse model of another form of progressive myoclonus epilepsy (Cstb−/− mice; EPM1 disease). In that case, the authors reported an infiltration of CD3+ cells in the brain parenchyma not accompanied by BBB disruption, despite increased brain vascularization, and suggested that peripheral immune cells could have a role in brain inflammation present in their model (Okuneva et al., 2016).
The presence of infiltrated peripheral immune cells could indicate a possible diffusion of inflammatory mediators and cells in both directions, blood to the brain and vice versa. So, we wanted to know whether the increase in the levels of some of the pro-inflammatory mediators present in the brain of LD mice could promote their diffusion to blood, serving as mechanistic disease biomarkers for the progression of the disease (Ravizza and Vezzani, 2018), (Simonato et al., 2021). Several reports have defined the use of different parameters as possible biomarkers in different epilepsy-related pathologies. For example, higher serum levels of GFAP (Elhady et al., 2021), (Nass et al., 2021), (Heimfarth et al., 2022), (Abdelhak et al., 2022), and ubiquitin carboxyterminal hydrolase L1 (UCHL-1) (Elhady et al., 2021), (Nass et al., 2021), have been detected in different types of epileptic seizures (e.g., juvenile epilepsy), and other neurological diseases [traumatic brain injury (TBI), multiple sclerosis (MS), etc.]. Serum levels of S100b, progranulin, and pro-inflammatory mediators (TNF, IL6, IL1beta, and HMGB1) were also high in status epilepticus conditions (Janigro et al., 2021), (Hanin et al., 2022), (Terrone et al., 2017), (Terrone et al., 2019), (Simonato et al., 2021), (Miller and Blanco, 2021). In our case, after analyzing the levels of several cytokines and chemokines by ELISA in serum (LCN2, CXCL10, CCL5, C3, CCL20, S100b, and HMGB1), only the levels of CXCL10 and S100b were higher in samples from LD animals at 16 and 12 months of age, but no differences in comparison to controls at 7 months of age were found. Probably this is due to differences in the expression of these mediators in the brain. Perhaps at 7 months of age, the levels of CXCL10 and S100b were not high enough to diffuse from the brain parenchyma to the blood.
In any case, these results indicate that CXCL10 and S100b could be considered biomarkers of LD progression. Interestingly, it has been recently reported a correlation between the production of CXCL10 by astrocytes and the attraction of peripheral immune cells, favoring infiltration (Linnerbauer et al., 2020), (Lee et al., 2022). Remarkably, in another form of progressive myoclonus epilepsy (EPM1), higher levels of CXCL1, CXCL10, CXCL13, IL1alpha, and IL18 were found in serum from an EPM1 mouse model (Cstb−/− mice) (Okuneva et al., 2016), but in that case, the authors proposed the expression of CXCL13 as the hallmark of the disease.
In conclusion, in this work, we define an upregulation of the expression of mediators of the TNF and IL6/JAK2 signaling pathways in LD. In addition, we describe the activation of the non-canonical form of the inflammasome. As these signaling pathways occurred mainly in astrocytes and microglia, these results confirmed again the main role that these neuroglial cells have in LD. The activation of these signaling pathways could trigger the activation of alternative forms of programmed cell death such as necroptosis (TNF-dependent) and pyroptosis (non-canonical inflammasome-dependent), which could explain the neurodegeneration present in LD. Furthermore, we have shown the infiltration of peripheral immune cells in the brain parenchyma, which could aggravate glia-derived neuroinflammation. Finally, we describe CXCL10 and S100b as possible blood biomarkers of the disease, which will allow the study of the progression of the disease using serum blood samples.
We would like to extend our studies to human samples of LD patients to confirm our findings. In the meantime, we consider that the identification of these initial inflammatory changes in LD will be very important to implement possible anti-inflammatory therapeutic strategies to prevent the development of the disease. In this sense, we have recently reported the beneficial effect of this type of strategy in Epm2b−/− mice. We defined the beneficial effect of metformin (Berthier et al., 2016), which allowed us to obtain the orphan designation of this drug for the treatment of LD by the European Medicines Agency and the Food Drug Administration from the USA. In addition, and as a follow-up of these studies, we have recently reported the beneficial effect of propranolol (Molla et al., 2021) and memantine (Molla et al., 2022). Finally, in a pilot assay, LD patients receiving metformin had a slower progression of the disease (Burgos et al., 2022), suggesting the feasibility of using anti-inflammatory therapies for the treatment of LD.
Supplementary Material
Highlights.
TNF and IL6 pathways are the main contributors to LD neuroinflammation.
The non-canonical inflammasome pathway is activated in LD samples.
Cytotoxic T-cell infiltration is present in the brain of LD.
CXCL10 and S100b are blood biomarkers of the progression of LD.
ACKNOWLEDGMENTS
We thank Dr. Maria Adelaida Garcia-Gimeno for the critical reading of the manuscript. This work was supported by a grant from the Spanish Ministry of Science and Innovation PID2020-112972RB-I00, an intramural grant ACCI2020 from CIBERER, a grant from la Fundació La Marató TV3 (202032), and a grant from the National Institutes of Health P01NS097197, which established the Lafora Epilepsy Cure Initiative (LECI), to P.S.
Footnotes
Conflict of interest: Authors declare that none of them has any type of conflict of interest.
REFERENCES
- Abdelhak A, et al. , 2022. Blood GFAP as an emerging biomarker in brain and spinal cord disorders. Nat Rev Neurol. 18, 158–172. [DOI] [PubMed] [Google Scholar]
- Agnew A, et al. , 2021. Regulation, Activation and Function of Caspase-11 during Health and Disease. Int J Mol Sci. 22, 1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado C, et al. , 2010. Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet. 19, 2867–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin R, et al. , 2022. The role of Tumour Necrosis Factor in neuroinflammation associated with Parkinson’s disease and targeted therapies. Neurochem Int. 158, 105376. [DOI] [PubMed] [Google Scholar]
- Berthier A, et al. , 2016. Pharmacological Interventions to Ameliorate Neuropathological Symptoms in a Mouse Model of Lafora Disease. Mol Neurobiol. 53, 1296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos DF, et al. , 2022. Early Treatment with Metformin Improves Neurological Outcomes in Lafora Disease. Neurotherapeutics. 10.1007/s13311-022-01304-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervellati C, et al. , 2020. Inflammation in Neurological Disorders: The Thin Boundary Between Brain and Periphery. Antioxid Redox Signal. 33, 191–210. [DOI] [PubMed] [Google Scholar]
- Courtois G, Fauvarque MO, 2018. The Many Roles of Ubiquitin in NF-kappaB Signaling. Biomedicines. 6, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criado O, et al. , 2012. Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet. 21, 1521–33. [DOI] [PubMed] [Google Scholar]
- Chan EM, et al. , 2003. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 35, 125–7. [DOI] [PubMed] [Google Scholar]
- Chen MY, et al. , 2021. The Signaling Pathways Regulating NLRP3 Inflammasome Activation. Inflammation. 44, 1229–1245. [DOI] [PubMed] [Google Scholar]
- De Nardo D, 2017. Activation of the Innate Immune Receptors: Guardians of the Micro Galaxy : Activation and Functions of the Innate Immune Receptors. Adv Exp Med Biol. 1024, 1–35. [DOI] [PubMed] [Google Scholar]
- Delgado-Escueta AV, 2007. Advances in lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 7, 428–33. [DOI] [PubMed] [Google Scholar]
- Dey A, et al. , 2016. Anti-Inflammatory Small Molecules To Treat Seizures and Epilepsy: From Bench to Bedside. Trends Pharmacol Sci. 37, 463–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhady M, et al. , 2021. Circulating glial fibrillary acidic protein and ubiquitin carboxy-terminal hydrolase-L1 as markers of neuronal damage in children with epileptic seizures. Childs Nerv Syst. 37, 879–884. [DOI] [PubMed] [Google Scholar]
- Fritsch M, et al. , 2019. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 575, 683–687. [DOI] [PubMed] [Google Scholar]
- Ganesh S, et al. , 2002. Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet. 11, 1251–62. [DOI] [PubMed] [Google Scholar]
- Garcia-Cabrero AM, et al. , 2012. Laforin and malin deletions in mice produce similar neurologic impairments. J Neuropathol Exp Neurol. 71, 413–21. [DOI] [PubMed] [Google Scholar]
- Garcia-Cabrero AM, et al. , 2014. Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Front Neurosci. 8, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gimeno MA, et al. , 2018. Lafora Disease: A Ubiquitination-Related Pathology. Cells. 7, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, et al. , 2005. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci U S A. 102, 8501–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Abad C, et al. , 2005. Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology. 64, 982–986. [DOI] [PubMed] [Google Scholar]
- Hanin A, et al. , 2022. Neuron Specific Enolase, S100-beta protein and progranulin as diagnostic biomarkers of status epilepticus. J Neurol. 269, 3752–3760. [DOI] [PubMed] [Google Scholar]
- Heimfarth L, et al. , 2022. Serum glial fibrillary acidic protein is a body fluid biomarker: A valuable prognostic for neurological disease - A systematic review. Int Immunopharmacol. 107, 108624. [DOI] [PubMed] [Google Scholar]
- Janigro D, et al. , 2021. Peripheral Blood and Salivary Biomarkers of Blood-Brain Barrier Permeability and Neuronal Damage: Clinical and Applied Concepts. Front Neurol. 11, 577312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer A, et al. , 2014. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 30, 523–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahuerta M, et al. , 2018. Degradation of altered mitochondria by autophagy is impaired in Lafora disease. FEBS J. 285, 2071–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahuerta M, et al. , 2020. Reactive Glia-Derived Neuroinflammation: a Novel Hallmark in Lafora Progressive Myoclonus Epilepsy That Progresses with Age. Mol Neurobiol. 57, 1607–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalani AI, et al. , 2018. TRAF molecules in inflammation and inflammatory diseases. Curr Pharmacol Rep. 4, 64–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HG, et al. , 2022. Function and therapeutic value of astrocytes in neurological diseases. Nat Rev Drug Discov. 21, 339–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TS, et al. , 2021. Gene expression in the epileptic (EL) mouse hippocampus. Neurobiol Dis. 147, 105152. [DOI] [PubMed] [Google Scholar]
- Linnerbauer M, et al. , 2020. Astrocyte Crosstalk in CNS Inflammation. Neuron. 108, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez I, et al. , 2017. Inflammation in Lafora Disease: Evolution with Disease Progression in Laforin and Malin Knock-out Mouse Models. Mol Neurobiol. 54, 3119–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lousa I, et al. , 2022. The Signaling Pathway of TNF Receptors: Linking Animal Models of Renal Disease to Human CKD. Int J Mol Sci. 23, 3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Salas J, et al. , 2012. Ontogeny of Lafora bodies and neurocytoskeleton changes in Laforin-deficient mice. Exp Neurol. 236, 131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher K, et al. , 2014. A role for stefin B (cystatin B) in inflammation and endotoxemia. J Biol Chem. 289, 31736–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Blanco MJ, 2021. Small molecule therapeutics for neuroinflammation-mediated neurodegenerative disorders. RSC Med Chem. 12, 871–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian BA, et al. , 1998. Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet. 20, 171–4. [DOI] [PubMed] [Google Scholar]
- Molla B, et al. , 2022. Pharmacological Modulation of Glutamatergic and Neuroinflammatory Pathways in a Lafora Disease Mouse Model. Mol Neurobiol. 59, 6018–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molla B, et al. , 2021. Modulators of Neuroinflammation Have a Beneficial Effect in a Lafora Disease Mouse Model. Mol Neurobiol. 58, 2508–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhtar I, 2020. Inflammatory and immune mechanisms underlying epileptogenesis and epilepsy: From pathogenesis to treatment target. Seizure. 82, 65–79. [DOI] [PubMed] [Google Scholar]
- Nass RD, et al. , 2021. Serum biomarkers of cerebral cellular stress after self-limiting tonic clonic seizures: An exploratory study. Seizure. 85, 1–5. [DOI] [PubMed] [Google Scholar]
- Okuneva O, et al. , 2016. Brain inflammation is accompanied by peripheral inflammation in Cstb (−/−) mice, a model for progressive myoclonus epilepsy. J Neuroinflammation. 13, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passaro AP, et al. , 2021. Immune Response in Neurological Pathology: Emerging Role of Central and Peripheral Immune Crosstalk. Front Immunol. 12, 676621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Jimenez E, et al. , 2021. Endocytosis of the glutamate transporter 1 is regulated by laforin and malin: Implications in Lafora disease. Glia. 69, 1170–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pondrelli F, et al. , 2021. Natural history of Lafora disease: a prognostic systematic review and individual participant data meta-analysis. Orphanet J Rare Dis. 16, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri R, Ganesh S, 2010. Laforin in autophagy: a possible link between carbohydrate and protein in Lafora disease? Autophagy. 6, 1229–31. [DOI] [PubMed] [Google Scholar]
- Rathinam VAK, et al. , 2019. Innate immunity to intracellular LPS. Nat Immunol. 20, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravizza T, Vezzani A, 2018. Pharmacological targeting of brain inflammation in epilepsy: Therapeutic perspectives from experimental and clinical studies. Epilepsia Open. 3, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riba M, et al. , 2021. Corpora Amylacea in the Human Brain Exhibit Neoepitopes of a Carbohydrate Nature. Front Immunol. 12, 618193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard R, Mousa S, 2022. Necroptosis in Alzheimer’s disease: Potential therapeutic target. Biomed Pharmacother. 152, 113203. [DOI] [PubMed] [Google Scholar]
- Rodgers KR, et al. , 2020. Innate Immune Functions of Astrocytes are Dependent Upon Tumor Necrosis Factor-Alpha. Sci Rep. 10, 7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma-Mateo C, et al. , 2015. Oxidative stress, a new hallmark in the pathophysiology of Lafora progressive myoclonus epilepsy. Free Radic Biol Med. 88, 30–41. [DOI] [PubMed] [Google Scholar]
- Rubio-Villena C, et al. , 2018. Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet. 27, 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi JH, Sun SC, 2018. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor kappaB and Mitogen-Activated Protein Kinase Pathways. Front Immunol. 9, 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonato M, et al. , 2021. Identification of clinically relevant biomarkers of epileptogenesis - a strategic roadmap. Nat Rev Neurol. 17, 231–242. [DOI] [PubMed] [Google Scholar]
- Solaz-Fuster MC, et al. , 2008. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 17, 667–78. [DOI] [PubMed] [Google Scholar]
- Sun RC, et al. , 2021. Brain glycogen serves as a critical glucosamine cache required for protein glycosylation. Cell Metab. 33, 1404–1417 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrone G, et al. , 2019. Inflammation and reactive oxygen species in status epilepticus: Biomarkers and implications for therapy. Epilepsy Behav. 101, 106275. [DOI] [PubMed] [Google Scholar]
- Terrone G, et al. , 2017. Inflammation and Epilepsy: Preclinical Findings and Potential Clinical Translation. Curr Pharm Des. 23, 5569–5576. [DOI] [PubMed] [Google Scholar]
- Troscher AR, et al. , 2021. T cell numbers correlate with neuronal loss rather than with seizure activity in medial temporal lobe epilepsy. Epilepsia. 62, 1343–1353. [DOI] [PubMed] [Google Scholar]
- Turnbull J, et al. , 2016. Lafora disease. Epileptic Disord. 18, 38–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka G, et al. , 2021. Links between Immune Cells from the Periphery and the Brain in the Pathogenesis of Epilepsy: A Narrative Review. Int J Mol Sci. 22, 4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.