

Abstract
In the absence of cell-surface cancer-specific antigens, immunotherapies such as chimeric antigen receptor (CAR) T cells, monoclonal antibodies, or bispecific engagers must target lineage antigens. Currently, such immunotherapies are individually designed and tested for each disease. This approach is inefficient and limited to a few lineage antigens for which the on-target/off-tumor toxicities are clinically tolerated. Here, we sought to develop a universal CAR-T cell therapy directed against the pan-leukocyte marker CD45. To protect healthy hematopoietic cells, including CAR-T cells, from CD45-directed on-target/off-tumor toxicity while preserving the essential functions of CD45, we mapped the epitope on CD45 that is targeted by the CAR and used CRISPR base-editing to install a function-preserving mutation sufficient to evade CAR-T cell recognition. Epitope editing in HSCs enables the safe and effective use of CD45-directed CAR-T cells for the universal treatment of hematologic malignancies and can be exploited for other diseases requiring intensive hematopoietic ablation.
One-Sentence Summary:
Epitope editing is a novel strategy that enables the safe and effective targeting of shared antigens with immune therapies.
The era of precision and personalized medicine has ushered in major advances in cancer care, where novel therapies are selected based on the individual genomic or immunophenotypic characteristics of the cancer. Targeted immunotherapies such as antibody-drug conjugates (ADCs), bispecific T cell engagers (BTEs) or chimeric antigen receptor (CAR) T cells have been successful in the treatment of hematologic malignancies(1–8). However, they have to be individually developed for different hematologic malignancies based on the relevant lineage antigens (e.g. CD19 for B cell leukemia/lymphoma, BCMA for myeloma, CD7 for T cell leukemia/lymphoma, CD33 for acute myeloid leukemia). While this one-by-one approach has led to the effective treatment of thousands of patients, from a drug development standpoint, this fragmented approach is inefficient and limited to a few antigens for which the on-target/off-tumor toxicities are clinically tolerated. By targeting a pan-hematologic antigen, a single drug could be used for almost all indications, thereby accelerating clinical development.
CD45 is the prototypical hematopoietic lineage antigen, a receptor tyrosine phosphatase that is expressed on the surface of most hematopoietic cells, including cancer cells(9). While this makes CD45 an attractive target for a universal blood cancer immunotherapy, targeting CD45 with CAR-T cells is limited by expression of CD45 on hematopoietic stem cells (HSC) and their progeny, as well as T cells. Therefore, manufacturing of CD45 CAR-T cells would be limited by fratricide, and even if feasible would result in severe pancytopenia.
To unlock CD45-specific immunotherapy as a potentially viable pan-hematologic CAR-T option, we built upon prior work in which we and others deleted the target antigen CD33 from allogeneic donor HSCs and combined them with anti-CD33 CAR-T cells as a tandem therapy to create a cancer-specific antigen in all residual host hematopoietic cells(10–12). However, this approach requires the deleted antigen to be dispensable for hematopoietic function. Since CD45 deficiency leads to impaired T cell development, diminished antigen-induced T and B cell function, and severe combined immunodeficiency (SCID)(9, 13), we hypothesized that CD45 deletion in HSCs would not be a feasible strategy to overcome on-target/off-tumor toxicities and fratricide of anti-CD45 CAR-T cells. Therefore, instead of genetically deleting CD45 from HSCs and T cells, we sought to use CRISPR base-editing to install a non-synonymous mutation at the relevant CD45 epitope in such a way that it is not recognized by the anti-CD45 CAR-T cells while retaining CD45 expression and intracellular phosphatase function. We term this approach “epitope editing” and hypothesize that it would enable the safe and effective targeting of CD45 with fratricide-resistant CD45 CAR-T cells.
CD45 is a universal blood cancer antigen that can be targeted with CAR-T cells
CD45 is a receptor tyrosine phosphatase that is expressed on virtually all leukocytes and leukemias and lymphomas at high levels(14–16). To target CD45, we created CAR45 constructs by cloning anti-CD45 single chain variable fragments (scFvs) from five different antibody clones that recognize CD45 into a 4–1BB containing second generation CAR lentiviral vector (Fig. 1A). To evaluate the effectiveness of these novel CAR45 constructs, we used a human T cell activation reporter line in which CD45 was deleted to prevent unintended activation and “fratricide” (Fig. 1B, S1A). The CAR45 constructs that used the NOV45 and 4B2 scFvs showed the lowest levels of target binding (Fig. 1B, right panel), which corresponded with a lack of activation when co-cultured with target tumor cells (Fig. 1C, D). Based on these results, we selected the 9.4-, BC8- and Gap8.3-derived CAR constructs for further experiments using primary human T cells.
Fig. 1. CD45 is a universal blood cancer antigen that can be targeted with CD45 knockout CAR-T cells.
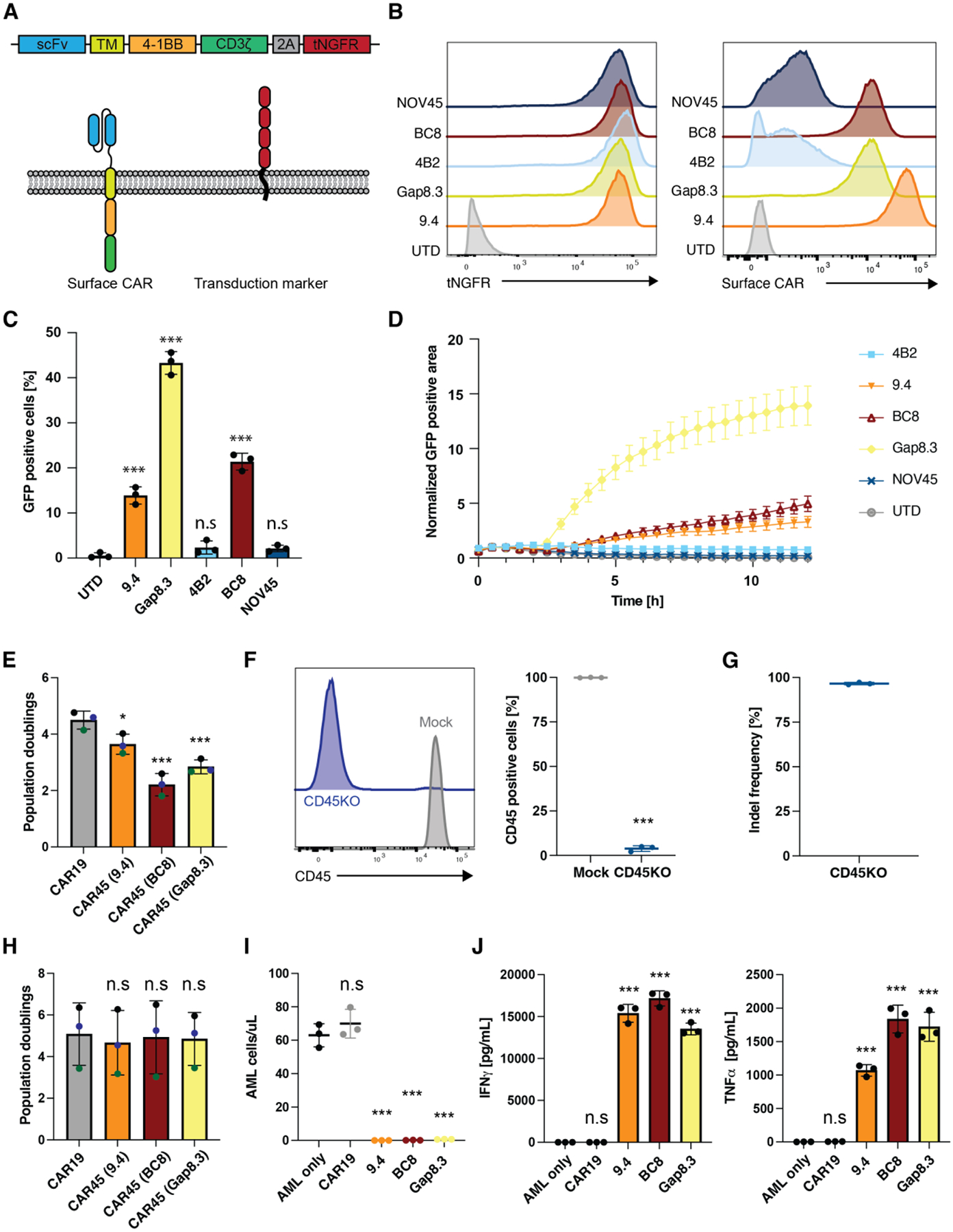
(A) Schematic overview of the tested CAR45 constructs. The heavy and light chains of different anti-CD45 antibody clones were cloned into a second-generation CAR containing a 4–1BB costimulatory domain and truncated NGFR (tNGFR) as a transduction marker. (B) CD45KO Jurkat reporter cells were transduced with CAR45 constructs and sorted for purity based on expression of tNGFR. Sorted cells were incubated with His-tagged recombinant CD45 followed by secondary staining with an anti-His antibody showing that CARs derived from clones 9.4, Gap8.3, and BC8 show highest expression of surface CAR. (C) CD45KO CAR45 Jurkat reporter cells were incubated with wild type Jurkat cells (CD45+) for 4hrs and NFAT-mediated GFP expression was measured by flow cytometry. CARs derived from antibody clones Gap8.3, BC8, and 9.4 show significant upregulation of GFP compared to untransduced reporter cells (n=3, One-way ANOVA compared to UTD, ***p<0.001). Data are represented as the mean ± SD. (D) CD45KO CAR45 Jurkat reporter cells were incubated with wild type Jurkat cells (CD45+) for 12hrs and NFAT-mediated GFP expression was measured by time lapse microscopy and the GFP positive area (normalized to t=0) was quantified. Data are represented as the mean ± SEM. (E) Population doublings of CD3/CD28 activated T cells transduced with either CAR19 or CAR45 after 13 days of ex vivo expansion. CAR45 transduced cells have significantly lower T population doublings due to fratricide (n=3 independent donors, one-ANOVA compared to CAR19, ***p<0.001; *p<0.05). Data are represented as the mean ± SD. (F) Human T cells were electroporated with SpCas9 protein and CD45 targeting gRNA (screening of CD45-specific gRNA not shown). Surface CD45 protein expression was assessed by flow cytometry (n=3 independent donors, unpaired t-test, ***p<0.001. Data are represented as the mean ± SD. (G) INDEL frequency in CD45KO T cells were quantified by TIDE following 9 days of in vitro culture. (H) T cells were electroporated with SpCas9 protein pre-complexed with CD45 gRNA followed by CD3/CD28 activation and transduction with either CAR19 or CAR45 and expansion for 13 days. CD45 deletion enables the expansion of CAR45 transduced cells similarly to CART19 cells (n=3 independent donors, one-ANOVA compared to CAR19, n.s=p>0.05). Data are represented as the mean ± SD. (I) CD45KO CART45 or CART19 cells were incubated with patient AML cells for 24hrs at a 1:4 E:T ratio and AML cells were quantified by flow cytometry. CART45 efficiently eliminates AML cells compared to CART19 (n=3 technical replicates, one way ANOVA compared to AML only, ***p<0.001; n.s=p>0.05). Data are represented as the mean ± SD. (J) CD45KO CART45 or CART19 control cells were incubated with patient AML cells for 24hrs at a 1:1 E:T ratio and cytokines in the supernatant were quantified by cytometric bead array (n=3 technical replicates, one way ANOVA compared to AML only, ***p<0.001; n.s=p>0.05). Data are represented as the mean ± SD.
Genetic disruption of PTPRC (CD45) enables expansion of CAR45 T cells but impairs CAR-T cell efficacy in vivo
Given that all T cells express CD45, anti-CD45 CAR-T cells are expected to be subject to fratricide. Expression of CAR45 on T cells indeed led to a significant reduction in T cell numbers compared to CART19 control cells (Fig. 1E, Fig. S1B). To prevent fratricide, we used CRISPR/Cas9 to delete PTPRC, the gene encoding CD45 in primary human T cells (Fig. 1F, G). As expected, CD45 disruption enabled the expansion of CART45 comparable to CART19, consistent with a lack of fratricide (Fig. 1H, Fig. S1C). This allowed us to test the activity of the three lead CAR constructs in vitro against patient-derived primary acute myeloid leukemia (AML) cells. Incubation of CART45 cells with primary AML cells resulted in potent cytotoxicity (Fig. 1I) and robust cytokine production (Fig. 1J) compared to CART19 control cells.
While these data demonstrate that CD45 disruption represents a potential strategy to prevent fratricide of CART45, given the critical role of CD45 in the T cell immune synapse(9), we hypothesized that loss of CD45 in human T cells will result in dysfunctional CAR-T cells. To investigate the functional consequences of CD45 disruption on CAR-T cells, we deleted CD45 from CART-19 and tested the function of these cells in an established model of B-cell leukemia. In short-term assays, efficient CD45 phosphatase disruption (Fig. S2A) had no impact on CART19 cytotoxicity, degranulation, or cytokine production (Fig. S2B and C), likely owing to compensation by the functionally redundant phosphatase CD148, which is expressed on T cells after CD3/CD28 activation (Fig. S2D)(17). In contrast, long-term in vivo xenograft models demonstrated that CD45KO CART19 were unable to control tumor growth (Fig. 2E). We observed significantly fewer CD4+ and CD8+ CAR-T cells in the peripheral blood of mice treated with CD45KO CAR-T cells (Fig. S2F), suggesting that CD45 deficiency impairs the in vivo expansion of CAR-T cells and likely contributes to inadequate tumor control in vivo. Therefore, even though effective at preventing fratricide, CD45 deletion is not a viable strategy to generate effective CART45 cells.
Fig. 2. Sequential epitope mapping identifies CAR45 epitopes on human CD45.
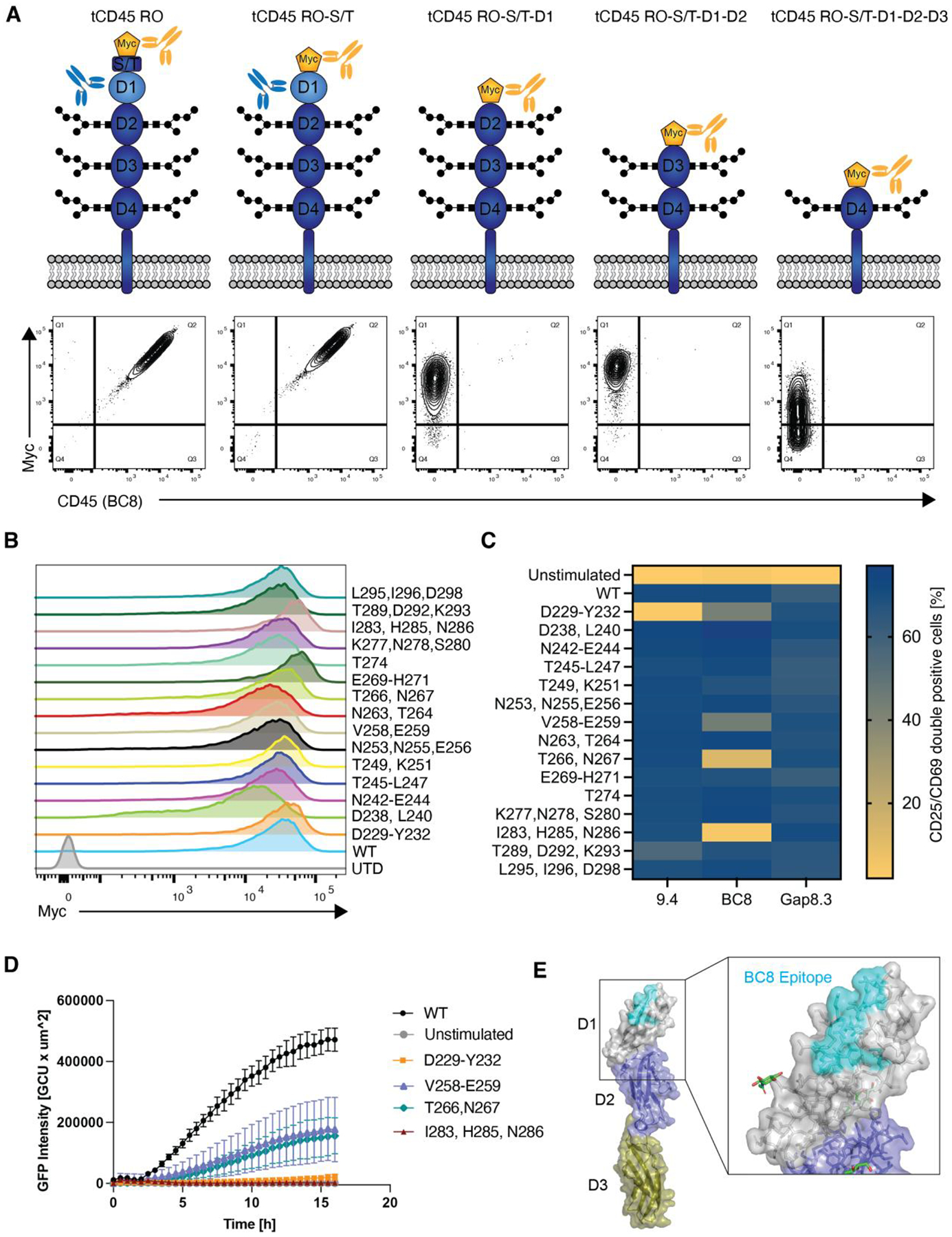
(A) Truncated human CD45 constructs that sequentially lack CD45’s extracellular subdomains were expressed in CD45 negative NALM6 cells. WT CD45RO was expressed as a positive control. Cells were stained with anti-myc to verify surface expression and with clone BC8 to determine CD45 binding. BC8 was mapped to the D1 domain of the CD45 ECD as the BC8 clone no longer bound to the CD45 constructs that lacked this domain. (B) Myc-tagged alanine mutants of the D1 domain were expressed in NALM6 cells. Alanine mutagenesis did not affect protein expression or transport to the cell surface. (C) NALM6 cells expressing either WT or alanine-mutated CD45 were co-cultured with CD45KO CART45 for 24h. Activation of CART45 cells was measured by surface expression of CD25 and CD69. Data are represented as the mean of n=2 technical replicates. (D) Jurkat NFAT-GFP reporter cells expressing the BC8-based CAR were co-cultured with NALM6 cells expressing CD45 alanine mutants and activation was measured by time lapse fluorescence microscopy (n=4 technical replicates). Data are represented as the mean ± SD. (E) Amino acid mutations that decreased BC8 CAR activation compared to CD45WT recognition were superimposed onto CD45 protein structure (PDB ID: 5FMV). BC8 recognizes a conformational epitope on the N-terminal portion of the CD45 D1 domain.
Epitope mapping identifies CAR45 target epitope on CD45
To produce fratricide-resistant, anti-CD45 CAR-T cells without CD45 deletion, we next sought to install a nonsynonymous mutation in CD45 that would abrogate recognition by the anti-CD45 clone while retaining CD45 function.
To identify the epitope in the extracellular domain (ECD) of CD45 that is targeted by CAR45, we expressed myc-tagged CD45 constructs that have been sequentially truncated from the N-terminus in CD45 negative NALM6 cells (Fig. 2A, Fig S3A). We verified surface expression of the truncated CD45 (tCD45) constructs and then used the relevant anti-CD45 antibody clone to determine binding. The BC8 clone bound to cells expressing CD45RO and CD45 in which the N-terminal serine/threonine-rich domain had been truncated, suggesting that this domain is not relevant for BC8 antibody binding (Fig. 2A). However, binding of clone BC8 was completely abolished when the D1 domain was truncated. Accordingly, CD45 in which the D1/D2 or D1/D2/D3 domains have been truncated were also not bound by clone BC8. These data suggest that clone BC8 recognizes the D1 domain. The domains for clones 9.4 and Gap8.3 have previously been mapped to the D1 domain as well, consistent with the observation that IgG-accessible CD45 epitopes are located toward the N-terminal portion of CD45’s ECD(18, 19).
To map the precise amino acids that are required for binding of the CAR45 constructs, we next performed alanine mutagenesis of the CD45 D1 domain using myc-tagged wild-type CD45RO as a template. Given the low amino acid sequence conservation between the human and murine CD45 D1 domain(20, 21) (~40%; Fig. S3B) and the lack of cross-reactivity of the CARs with murine CD45, we only mutated human-specific (non-conserved) amino acids to reduce the likelihood of causing loss-of-function mutations. We expressed alanine mutated CD45 in NALM6 cells and verified their surface expression. Importantly, none of the mutations impaired CD45 expression or trafficking to the cell surface (Fig. 2B). Next, we co-cultured NALM6 cells expressing CD45 alanine mutants with CD45KO CAR45 primary human T cells for 24hrs and measured CAR-T cell activation (Fig. 2C and Fig. S3C). Co-incubation of CART45 cells with CD45 wild-type expressing cells led to T cell activation, as expected. For the 9.4-derived CAR, mutating amino acids D229, E230, K231, and Y232 to alanine resulted in abrogation of CAR-T cell activation whereas T289A, D292A, and K293A resulted in a modest decrease in activation, suggesting that these amino acids constitute the target epitope of the 9.4-derived CAR. For the BC8 derived CAR, four different alanine mutant constructs (D229A/E230A/K231A/Y232A, V258A/E259A, T266A/N267A, and I283A/H285A/N286A) either reduced or abrogated BC8-derived CAR-T cell activation compared to that of CD45WT (Fig. 2C and Fig. S3D). Importantly, all other CD45 mutants included in the alanine scanning library activated the respective 9.4- and BC8-derived CAR-T cells comparably to CD45WT, suggesting that the above residues identify the specific epitope for these CARs. None of the tested alanine mutants affected Gap8.3-derived CAR activation. This suggests that the decrease in activation of the 9.4 and BC8 CARs after co-culture with CD45 mutant expressing cells is not due to large-scale misfolding of the mutant protein but rather that the native amino acids are critical for binding and activation of the CAR-T cells. Further validation of the hits for the BC8-derived CAR in an NFAT-GFP reporter assay confirmed that co-culture with the alanine-mutant expressing cells leads to decreased or complete abrogation of BC8 CAR activation (Fig. 2D). We superimposed the amino acids relevant for binding to the BC8 CAR onto the previously solved crystal structure of the CD45 ECD(22). Despite the amino acid hits from the alanine scan spanning almost 70 amino acids in the primary peptide sequence, these amino acids form a conformational epitope at the membrane-distal (N-terminal) portion of the D1 domain (Fig. 2E). Based on this data, all subsequent experiments were performed with the BC8-derived CAR (henceforth denoted as CAR45).
Epitope base editing CD45 enables CAR45 T cell expansion while preserving CD45 expression and CAR-T cell function
To introduce a non-synonymous mutation at the epitope on CD45 targeted by CAR45 (BC8), we used CRISPR base-editing, a technique that efficiently installs A-T to G-C or C-G to T-A base pair changes at the targeted loci without the need for DNA double-strand breaks, homology-directed repair, or donor DNA templates(23, 24).
Two promising gRNAs (BE8 and BE17) were identified that, when electroporated with mRNA encoding an adenine base editor (ABE8e,(25)) into human T cells, resulted in single amino acid substitutions at the targeted CD45 epitope (Y232C and C288R for BE8 and BE17, respectively, Fig. S4A and B). Installing these mutations in primary human T cells led to complete abrogation of CD45 binding by the BC8 antibody (Fig. 3A). Because base editing also occurs in a small window around the intended site, we quantified bystander mutations. The rate of bystander mutations for BE8 were 11.3% and 7.3% at A4 and A10 respectively. For BE17, the frequency of bystander editing at A4 was 39% (Fig. 3B). For both BE8 and BE17, detectable bystander mutations were silent mutations and therefore did not change the amino acid sequence (Fig. S4A and B).
Fig. 3. Epitope base editing CD45 enables CAR45 T cells expansion while preserving CD45 expression and CAR-T cell function.
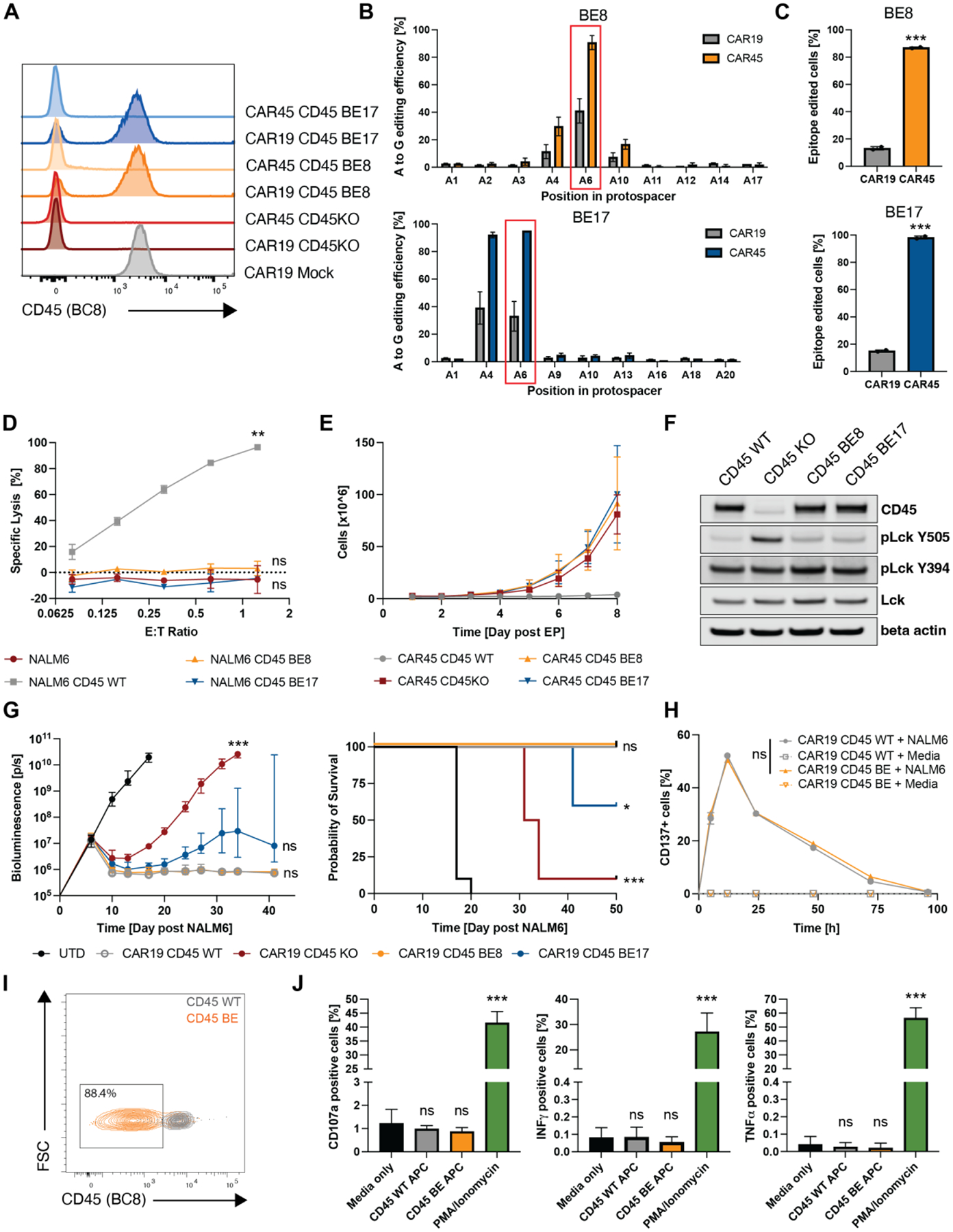
(A) Edited human T cells were transduced with either CAR19 or CAR45 and stained for CD45 expression using the BC8 antibody clone 7 days post transduction. (B) Sequencing the targeted PTPRC loci shows that the edited cells are positively selected when transduced with CAR45 compared to CAR19 at the end of CAR-T cell expansion. (C) Cells with the intended edit are positively selected over time (7 days) when transduced with CAR45 compared to CAR19 (***p<0.001, unpaired t-test, n=2 independent donors). (D) Luciferase+ NALM6 expressing WT or base edited CD45 mutants were co-cultured with CART45 for 24hrs. CART45 was able to lyse cells expressing CD45WT but not CD45BE, suggesting that cells expressing base-edited CD45 are protected from CART45. (n=3 technical replicates, two-way ANOVA, **p<0.002; ns=p>0.05). Data are represented as mean ± SD. (E) Ex vivo expansion of unedited, CD45KO, and CD45BE CART45 cells demonstrates that epitope editing and CD45 knockout prevents CART45 fratricide (n=3 independent donors). Data are represented as mean ± SD. (F) Western blots of cell lysates from CD45WT, CD45KO and CD45BE T cells show that epitope-edited cells maintain CD45 expression and a higher frequency of dephosphorylated Lck Y505 (the substrate of CD45) compared to CD45KO cells. (G) NSG mice were injected with 0.5 × 106 Luc+ NALM6 cells followed by injection of 3 × 106 CAR19+ T cells. Tumor burden was measured by bioluminescent imaging and survival was monitored. Mice that were treated with CD45KO CART19 cells rapidly relapsed and succumbed to their tumor whereas mice that received CD45BE CART19 cells cleared their tumors comparable to unedited CART19 cells (n=10 per group BLI=ANOVA, survival = log-rank test, ***p<0.001; **p<0.0021; *p<0.033; n.s=p>0.12). (H) Unedited and base edited CART19 cells were briefly stimulated with NALM6 tumor cells at a 1:1 E:T ratio and expression of the activation marker CD137 was measured over 96hrs. (n=3 technical replicates, two-way ANOVA, ns=p>0.05). Data are represented as mean ± SD. (I) M-CSF stimulated monocytes were electroporated with ABE8e mRNA + BE8 sgRNA and cultured for 5–7 days prior to staining with anti-CD45 (clone BC8). Efficient epitope base editing (>85%) was observed by flow cytometry. (J) T cells were co-cultured with unedited, or epitope edited autologous monocytes for 5hrs to assess T cell reactivity against the mutant peptide arising from base editing. PMA/Ionomycin and media only conditions served as controls. T cell reactivity was measured by degranulation (CD107a+) and intracellular cytokine production. (n=2 independent donors of different HLA type, one way ANOVA compared to media only, ***p<0.001; ns=p>0.05). Data are represented as the mean ± SD.
When a mixed population of cells containing both epitope-edited and unedited cells was transduced with CART45, the non-edited cells (CD45 clone BC8 positive) were eliminated by CART45 cells (Fig. 3A), resulting in an enrichment of edited cells (CD45 clone BC8 negative). In contrast, cells transduced with CAR19, which does not exert a selection pressure against CD45WT-expressing cells, did not show a similar enrichment of edited cells (Fig. 3A–C). To formally confirm that cells expressing the amino acid variants installed by epitope base editing are indeed protected from CAR45-mediated killing, we expressed the relevant CD45 variants and CD45WT in NALM6 cells and co-cultured them with CART45 (Fig. S4C). CART45 cells were able to lyse CD45WT-expressing cells in a dose-dependent manner, whereas untransduced cells and cells expressing the CD45 base-edited variants (CD45BE) were not killed, suggesting that a single amino acid substitution in CD45 is sufficient to prevent CAR recognition and killing (Fig. 3D).
Using ABE8e, we achieved on-target editing efficiencies above 90% in primary human T cells (Fig. S4A and B). As a result, epitope edited CART45 cells were able to expand similarly to cells in which the CD45 antigen has been deleted, whereas unedited T cells did not expand due to fratricide (Fig. 3E). Furthermore, expanded CART45 cells in which CD45 had been deleted or epitope-edited had comparable viability to CART19 control cells, whereas unedited CART45 cells had lower viability, consistent with persistent fratricide due to the presence of wild-type CD45 on the T cell surface (Fig. S4D). Thus, base editing can efficiently modulate the targeted epitope on CD45 sufficient to prevent CART45 recognition and fratricide.
Importantly, unlike CD45KO cells, epitope edited cells retain expression of the CD45 phosphatase (Fig. 3F). The expression of CD45 in T cells is essential for activating Lck by dephosphorylating an inhibitory tyrosine (Lck Y505)(26). The dephosphorylation of Y505 by CD45 relieves autoinhibition and maintains a pool of primed Lck that is necessary to initiate T cell signaling. Consistent with previous research, in the absence of CD45, phosphorylated Lck Y505 (closed-inactive) accumulates due to the lack of dephosphorylation by CD45 (Fig. 3F). In contrast, CD45WT and epitope-edited cells did not accumulate pLck Y505, indicating that CD45’s phosphatase activity remains intact after epitope editing. We hypothesize that maintaining CD45 expression and catalytic activity is crucial for proper CAR-T cell function in vivo.
As previously shown, CD45 knockout cells were not able to maintain long-term anti-tumor efficacy and the mice quickly succumbed to their tumor (Fig. 3G). In contrast, BE8 epitope-edited cells retained their anti-tumor efficacy, comparable to that of unedited cells, suggesting that epitope editing does not impair CAR-T cell function. Based on this data, we conducted subsequent experiments using the BE8 gRNA (henceforth denoted as CD45 BE). Since CD45 is involved in both activation and inhibition of T cell signaling, we wanted to assess activation and resting kinetics of base edited CAR-T cells by tracking cell number, cell size, and expression of activation markers. BE8 epitope editing did not impact CART19 activation or resting kinetics as measured by cell numbers and cell size following 48hrs of CD3/CD28 stimulation activation (Fig. S4E). Furthermore, brief restimulation of CART19 cells with CD19+ tumor cells led to a comparable activation and resting kinetics between CD45 WT and CD45 BE CAR-T cells (Fig. 3H). Additionally, base editing CD45 did not impact CD4 to CD8 ratios or T cell memory subset differentiation following expansion (Fig. S4F and S4G).
To evaluate whether the amino acid change (Y232C) caused by the BE8 gRNA could be immunogenic, we utilized in silico prediction to assess the relative ability of a peptide/MHC class I complex to elicit an immune response as well as in vitro functional using donor-matched, epitope edited antigen-presenting cells (APCs) and primary human T cells. Based on in silico prediction that uses amino acid properties as well as their position within the peptide to predict the immunogenicity of a class I peptide MHC (pMHC) complex, the mutant peptide is not predicted to be more immunogenic than the wild-type peptide sequence (Fig S4H). To experimentally validate the in silico immunogenicity prediction, we efficiently base edited antigen presenting cells (Fig. 3I) from two different donors with distinct HLA types and cultured them with autologous T cells to evaluate if T cell reactivity against the edited peptide occurs. Mutant peptides generated by epitope editing did not trigger T cell degranulation or cytokine production compared to CD45 WT peptides (Fig. 3J). This suggests that the mutant peptide is either not immunogenic or not presented by the MHC alleles tested here.
CART45 shows potent activity against multiple hematologic cancer cell lines and primary AML xenografts
To test the anti-tumor efficacy of BE CART45, we selected AML as our model, because leukemic cells universally express CD45 and remains poorly served by other immune effector cell modalities. We showed efficient killing and T cell proliferation using an AML cell line model (Fig. 4A, B) and proceeded to test primary AML samples. Immunodeficient NSG-SGM3 mice (27) were injected with primary AML samples (Fig. 4C, Table S1). Engraftment was defined as >0.5–1% circulating huCD45+ cells of live cells. Both patient AML samples were phenotypically identified as CD33+/CD45 dim (compared to lymphocytes) at baseline engraftment (Fig S5A). These mice were then treated with a single injection of BE CART45 or UTD cells (5 × 105 via tail vein injection). Disease burden and BE CART45 expansion were quantified in the peripheral blood (Fig. S5B). Leukemia was eradicated within three weeks of BE CART45 injection and long-term survival benefit was demonstrated compared to UTD cells (Fig. 4D, E, and Fig. S5C). CART45 cells proliferated extensively with peak expansion at 3 weeks post-injection, followed by contraction after the tumor had cleared (Fig. 4F). Three months after tumor clearance, mice were rechallenged with 3×106 cells of primary AML cells from the same patient samples. The AML primary samples engrafted and rapidly proliferated in naïve control mice whereas mice that previously cleared their tumors remained tumor free (Fig. 4G). This suggests that epitope base edited CART45 cells can persist and maintain anti-leukemia activity with long-term immunosurveillance.
Fig. 4. CART45 shows potent activity against multiple hematologic cancer cell lines and primary AML xenografts.
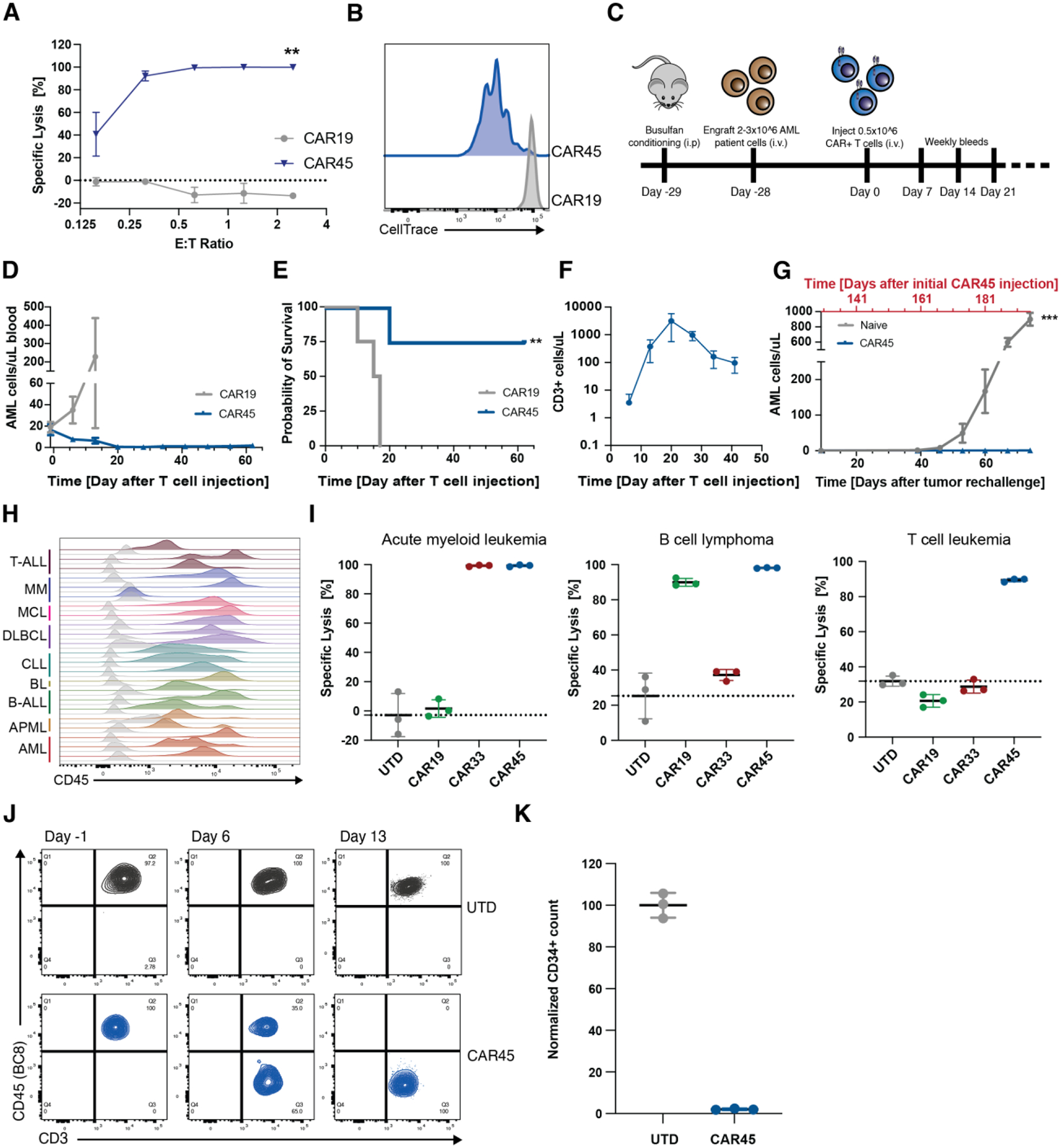
(A) Luc+ MOLM14 AML cells were incubated with CART45 cells at multiple effector:target ratios for 24hrs. CART45 efficiently lysed MOLM14 cells whereas CART19 control cells did not (n=2 independent donors, unpaired t-test, **p<0.002). Data are represented as mean ± SD. (B) MOLM14 AML cells were incubated with CellTrace labeled CART45 cells at a 1:1 E:T ratio for 5 days. CART45 cells proliferated extensively compared to CART19 control cells as measured by dye dilution. (C) Schematic overview of experimental timeline for AML patient-derived xenograft model. (D) Number of AML cells/uL peripheral blood after CART45 treatment. CART45 cells rapidly clear the primary patient tumor cells in NSG mice compared to UTD control cells. Data are represented as mean ± SEM. (E) NSGS mice that were treated with CART45 cells show improved survival compared to UTD T cells (**p<0.002, log-rank test, n=4 per group). (F) Number of CD3+ T cells in the peripheral blood. CART45 cells undergo initial expansion followed by contraction after the tumor cleared. (G) Number of AML cells/uL peripheral blood after tumor rechallenge and initial CAR45 treatment (***p<0.001, unpaired t-test, n=7 from 2 independent donors). Data are represented as mean ± SEM. Days since initial CAR45 treatment adjusted to one donor with the second donor cohort offset by 22 days. (H) Blood samples from 22 patients with nine different hematologic malignancies were stained for CD45. CD45 is expressed on all patient cells except one multiple myeloma case. Grey histograms represent fluorescence intensity of unstained controls. (I) In vitro cytotoxicity assay demonstrates that CART45 can target multiple tumor cell lineages simultaneously, whereas CAR19 and CAR33 can only target B cell or myeloid cell leukemia/lymphoma cells respectively (n=3 technical replicates). Data are represented as mean ± SD. (J) T cells (CD3+/CD45WT+) engrafted from AML patient apheresis get eliminated and replaced by CD45BE CART45 cells. (K) 48hr co-culture of unedited CD34+ HSCs with autologous CART45 cells demonstrates susceptibility of normal HSCs to CART45 on-target/off-tumor toxicity.
While targeting AML with BE CART45 cells has substantial clinical significance, CD45 is expressed on most hematologic malignancies making BE CART45 a potentially universal blood cancer immunotherapy option. Analysis of CD45 expression levels across 22 patient samples of leukemia, lymphoma, and myeloma shows that CD45 is expressed on all primary patient samples except for one multiple myeloma sample (Fig. 4H, Table S3). To demonstrate that BE CART45 is effective against a variety of hematologic malignancies, we co-cultured three hematologic cancer cell lines (Raji – B cell lymphoma, MOLM14 – AML, and Jurkat – T-ALL) with CD45 expression levels comparable or lower than that of primary tumor samples (Fig S5D) with either CART19, CART33, or BE CART45 for 24hrs. BE CART45 was able to eliminate all three hematologic cancer cell lines compared to UTD T cells, whereas the other CARTs only targeted their lineage specific target (Fig. 4I). This data also indicates that BE CART45 has comparable in vitro efficacy to the extensively tested anti-CD19 CAR.
Although malignant cells universally express CD45, they can downregulate its expression, leading to the emergence of CD45 “dim” tumor cells that may evade CART45-mediated cytotoxicity. To determine the CD45 expression level required for effective CART45 killing, we sorted NALM6 cells transduced with CD45 into three populations based on CD45 expression: dim, medium, and high (Fig. S5E). Upon co-culture with CART45, dose-dependent cytotoxicity against target tumor cells was observed in all three populations. As expected, there was small reduction in cytotoxicity against CD45 dim cells (Fig. S5F). We determined that this was due to the outgrowth of a small population of untransduced, CD45 negative NALM6 cells in the CD45 dim sorted target cells (Fig. S5G). Collectively, these data demonstrate that CD45 “dim” blood cancers likely meet the activation threshold required for effective CART45 cytotoxicity.
Lineage switching in acute leukemia from lymphoid to myeloid lineage (or vice versa) and mixed-lineage leukemias are rare instances that would benefit from a pan-hematopoietic therapy due to their resistance to single-lineage antigen targeted immunotherapies (28, 29). We hypothesized that broad targeting of both myeloid and lymphoid lineage with BE CART45 could mitigate the risk of single lineage antigen CAR-T therapy resistance. To test this, B-cell, T-cell and myeloid cell lines were co-cultured together followed by addition of either CART19, CART33, or BE CART45. Again, only BE CART45 was able to target all three cell lines simultaneously, whereas CART19 and CART33 were only able to target B cells or myeloid cells respectively (Fig. S5H and S5I).
While BE CART45 shows potent activity against various blood cancer cell lines and patient tumors, we anticipated that this activity will also lead to on-target/off-tumor toxicities against healthy hematopoietic cells. Injection of AML patient samples into immunodeficient mice is sometimes accompanied by engraftment of patient CD3+ T cells. When these mice were treated with BE CART45, the epitope edited cells quickly eliminated patient CD3+ T cells, as they too express CD45, resulting in a reduction of CD45WT T cells and an increase in epitope edited T cells (Fig. 4J). As expected, CD34+ HSCs also express CD45, making them susceptible to CART45 cytotoxicity (Fig 4K).
CD45 epitope editing does not impair hematopoietic function
To protect healthy hematopoietic cells from BE CART45 cells, we hypothesized that we could apply the same epitope base editing strategy to human CD34+ hematopoietic stem and precursor cells and expected that these shielded HSCs would differentiate and reconstitute a BE CART45 resistant hematopoietic system. The strong selective pressure towards edited cells following BE CART45 treatment requires that epitope base edited (CD45BE) HSCs maintain normal hematopoiesis and that the function of their progeny cells is retained. To test this, the function of edited human CD34+ HSCs and immune effector cells was interrogated in vitro and in vivo (Fig 5A). First, efficient base editing was confirmed in CD34+ human HSCs (Fig. 5B). Next, the ability of edited HSCs to proliferate and differentiate in vitro was assessed by measuring colony formation. After 14 days of culture, colony numbers were quantified and sequenced to determine whether edited cells are positively or negatively selected. While the number and types of colonies were comparable between CD45WT, CD45KO, and CD45BE groups (Fig. S6A, B), the indel frequency in the CD45KO HSC group declined during culture, whereas the frequency of cells harboring the epitope edit remained stable (Fig. 5C). Next, edited or control CD34+ HSCs were engrafted into NSG mice and human cell engraftment, and differentiation was measured over time by longitudinal bleeding and terminal bone marrow harvest. In the edited groups, we gated on CD45 (clone BC8) positive and negative cells separately, using the CD45WT cells in the edited groups as an internal control, while also comparing to the unedited control group. While engraftment and immune reconstitution in the peripheral blood was similar across unedited, CD45KO, and CD45BE groups (Fig. 5D, E), we observed a decline in the frequency of CD45KO cells, but not CD45BE cells over time (Fig. 5F). Similarly, the level of bone marrow chimerism was not significantly different across groups (Fig. 5G). However, the frequency of CD45KO cells was lower than the injection product, whereas the frequency of epitope edited cells remained unchanged (Fig. 5H). The percentages of the more primitive stem cells defined as CD34+ CD38- or CD34+ CD90+ were low across all groups with no statistical difference between edited or unedited cells (Fig. S6C). Next, we harvested bone marrow from the engrafted mice, sorted the CD34+ fraction, and cultured equal numbers of CD34+ cells in methocult for 14 days. After 14 days, the CD45KO cell frequency further declined compared to the injection product and at the time of bone marrow harvest (Fig. 5H). This was accompanied by a decrease in the number of colonies (Fig. S6D). On the contrary, epitope edited cells were able to maintain colony forming potential without negative selection in the frequency of edited cells (Fig. 5H and Fig. S6D). Epitope edited HSCs were able to differentiate into the major immune cell subtypes including myeloid cells (CD33), B cells (CD19), and T cells (CD3) at comparable frequencies to unedited HSCs, suggesting that epitope editing does not impair HSC differentiation (Fig. 5I). On the contrary CD45KO HSCs were not able to differentiate into human T cells and had a lower frequency of myeloid cells compared to unedited HSCs. This result is consistent with human genetics data and mouse models of CD45 deficiency(13, 30).
Fig. 5. Epitope edited hematopoietic system remains functional.
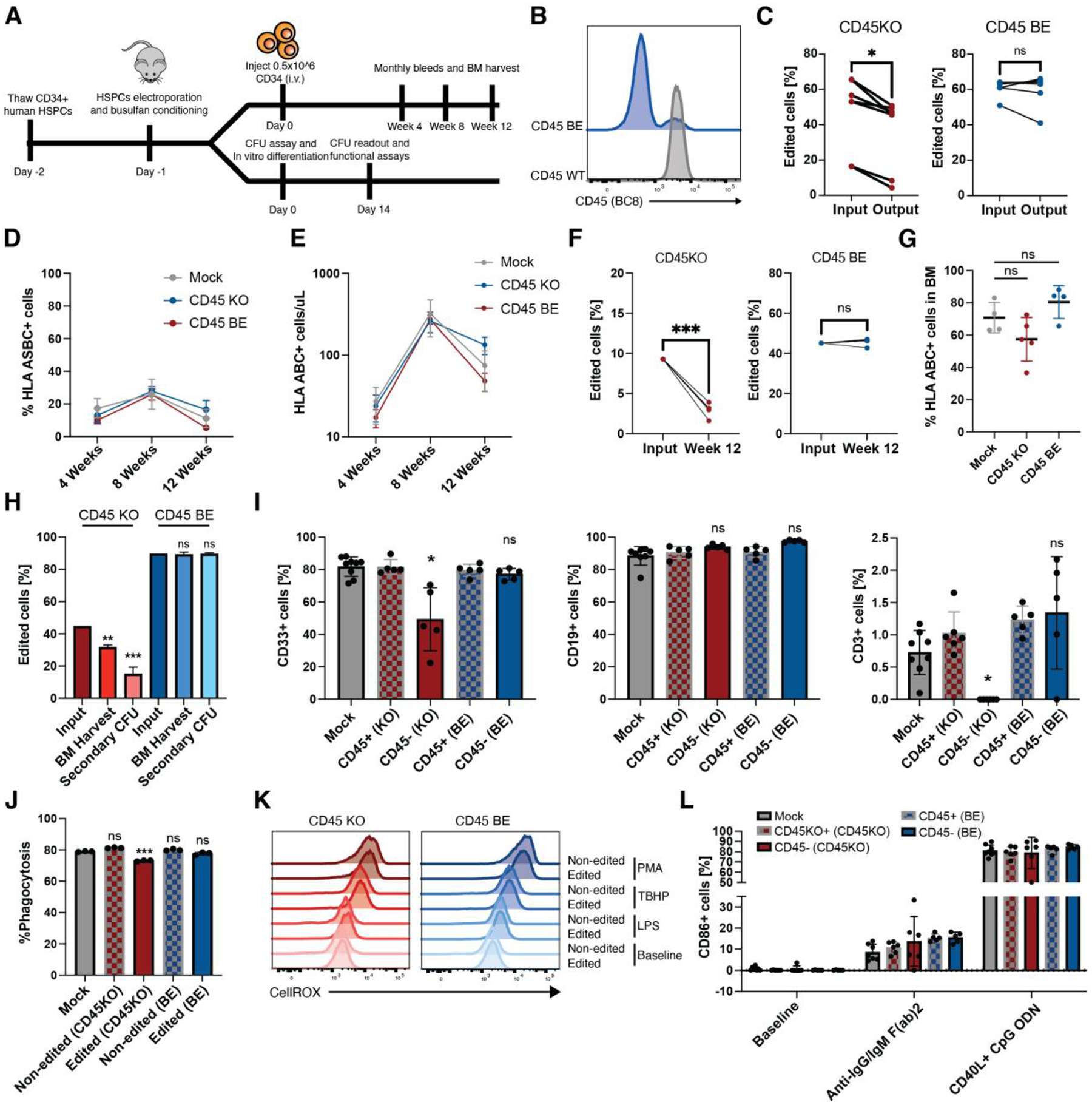
(A) Schematic overview. Human CD34+ HSCs were edited with gRNA and either ABE8e mRNA or Cas9 RNP’s. After recovery, CD34+ cells were injected into NSG mice to measure engraftment and differentiation in peripheral blood. A subset of edited CD34+ cells were plated in semi-solid methocult media for colony formation or differentiated into myeloid cells for functional assays. (B) Edited and control CD34+ HSCs were stained with BC8 antibody clone. (C) INDEL frequency in CD45KO colonies as quantified by TIDE(43) decreased after 14 days in methocult media whereas the frequency of base-edited alleles as measured by EditR remained stable (*p<0.05, paired t-test, n=6 or 7 from 4 independent donors). (D) Mice engrafted with CD45BE HSCs show similar frequency of human engrafted cells at early, mid, and late timepoints (ns=p>0.05; *p<0.05, two-way ANOVA, n=5–8). Data are represented as mean ± SD. (E) Mice engrafted with CD45BE HSCs show similar numbers of human engrafted cells at early, mid, and late timepoints (ns=p>0.05*p<0.05, two-way ANOVA, n=5–8). Data are represented as mean ± SD. (F) Longitudinal analysis of human engrafted cells shows a decrease in the frequency of CD45KO cells, while epitope edited cells show stable engraftment over 12 weeks (***p<0.001, paired t-test, n=5). (G) Mice engrafted with CD45BE HSCs show similar frequency of human engrafted in the bone marrow (ns=p>0.05, one way ANOVA, n=4–5). Data are represented as mean ± SD. (H) Mice engrafted with CD45KO HSCs show a decline in the frequency of edited cells in the BM and after secondary in vitro colony formation whereas the frequency of CD45BE edited cells remains unchanged compared to the injection input. (ns=p>0.05, **=p<0.01, ***=p<0.001, one way ANOVA compared to input, n=4–5). (I) Epitope edited HSCs show comparable myeloid, B cell, and T cell differentiation compared to unedited HSCs whereas CD45KO HSCs have a decreased frequency of myeloid cells and undetectable levels of T cells in the peripheral blood. Myeloid, B-cell, and T-cell differentiation was assessed at 4 weeks, 8 weeks, and 12 weeks respectively when peak differentiation into the corresponding lineage occurs in the NSG xenograft model. (ns=p>0.05; *=p<0.05, one way ANOVA, n=5–8). Data are represented as mean ± SD. (J) In vitro-differentiated CD45KO and CD45BE myeloid cells retain phagocytosis ability as measured by internalization of pHrodo deep red E. coli bioparticles. (ns=p>0.05; ***p<0.01, one way ANOVA, n=3 technical replicates). Data are represented as mean ± SD. (K) Levels of reactive oxygen species (ROS) production after lipopolysaccharide (LPS), tert-butyl hydroperoxide (TBHP), or phorbol myristate acetate (PMA) stimulation are similar among unedited and CD45 edited cells. ROS production was measured by fluorescence of CellROX deep red reagent. (L) In vivo differentiated B cells were harvested from peripheral blood of mice 10 weeks post-transplant and activated by either CD40L+CpG ODN or anti-IgG/IgM (Fab)2 for 48hrs. Activation as measured by surface level expression of CD86 did not differ between edited and unedited B cells and was significantly above baseline (n=5–8 mice per group). Data are represented as mean ± SD.
Next, we interrogated effector functions of in vitro differentiated myeloid cells and found that CD45BE had no effect on in vitro proliferation (Fig. S6E), phagocytosis (Fig. 5J, S6F, G), generation of reactive oxygen species (Fig. 5K), or production of inflammatory cytokines/chemokines (Fig. S6H). We then tested whether human B cells differentiated from the engrafted HSCBE can be activated through the B cell receptor, CD40 and TLR9. PBMCs from mice 10 weeks after HSC engraftment were harvested and stimulated with anti-IgG/IgM or CD40L+CpG ODN for 48hrs and B cell activation by surface expression of CD86 was quantified. B cells derived from CD45BE HSCs have similar baseline expression of CD86 and respond to both BCR and CD40/TLR9 stimulation with no significant differences compared to unedited B cells (Fig 5L). These data suggest that, unlike CD45KO cells, CD45 epitope edited hematopoietic stem cell and immune cell functions are maintained.
An epitope engineered hematopoietic system is shielded from CD45 targeted CAR-T cell therapy and bispecific T cell engagers
To demonstrate resistance of epitope edited HSCs and progeny cells to BE CART45, NSG mice were engrafted with either unedited or epitope edited CD34+ HSCs and treated the mice with BE CART45 cells (Fig. 6A). As expected, unedited HSCs and progeny cells were eliminated following BE CART45 treatment, leading to disappearance of engrafted cells in the blood and bone marrow of the unedited control mice as well as enrichment of epitope edited cells in the mice that received base edited HSCs (Fig. 6B, C). Crucially, mice that received epitope edited HSCs retained human hematopoietic cells in the peripheral blood and bone marrow (Fig. 6B, C). This data indicates that epitope editing can protect healthy human hematopoietic cells from BE CART45 mediated cytotoxicity.
Fig. 6. Epitope engineered hematopoietic system is shielded from CD45 targeted CAR-T cells and BTEs.
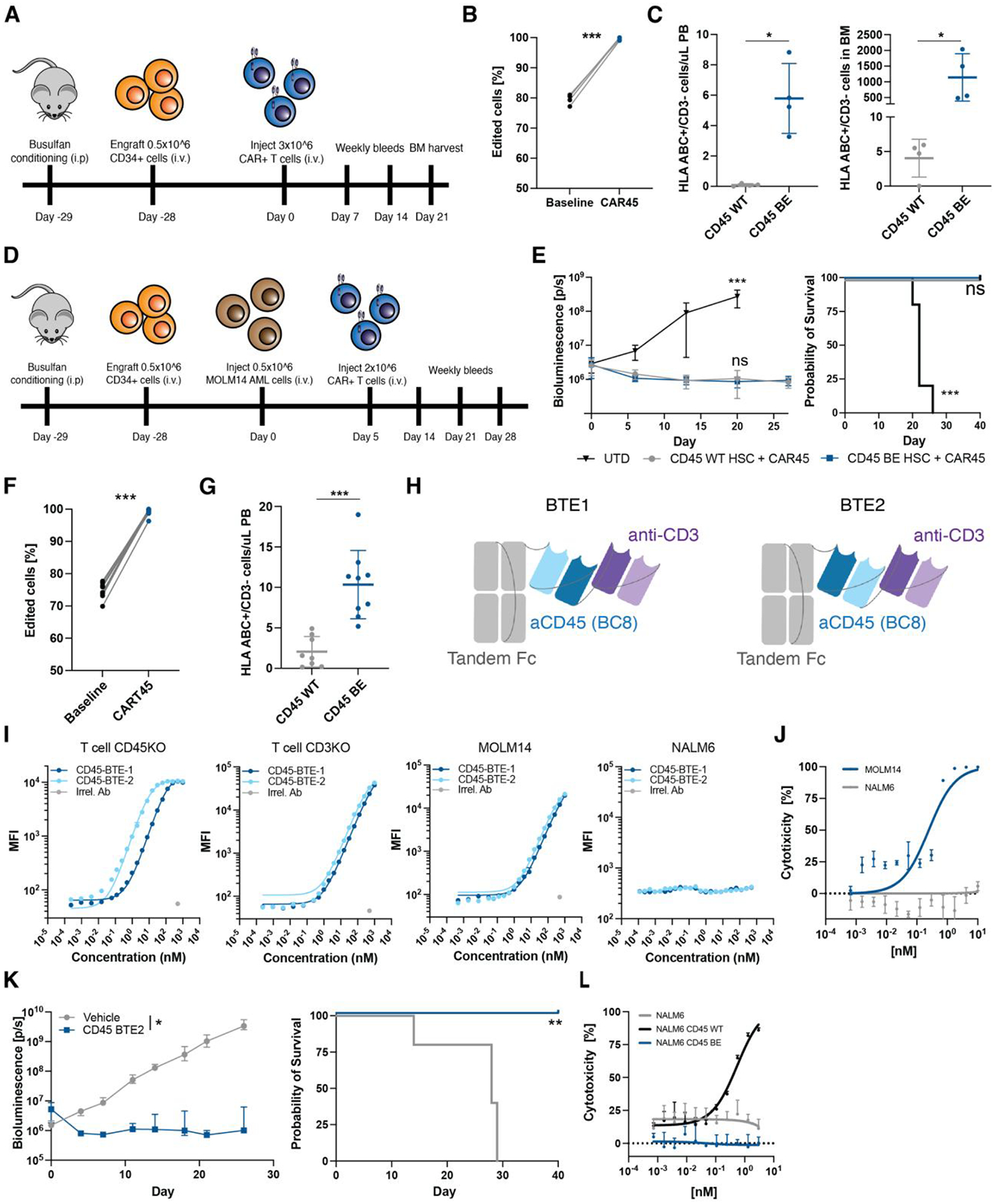
(A) Schematic overview of experimental timeline for in vivo protection of epitope edited HSC and progeny cells from CART45. (B) Frequency of edited cells in peripheral blood of NSG mice engrafted with CD45BE HSCs before and after treatment with CART45 cells (***p<0.001, paired t-test, n=4). (C) Number of human cells in peripheral blood and bone marrow of mice engrafted with CD45WT or CD45BE HSCs after CART45 injection (gated on mouse CD45-/human CD3-). n=4 mice per group, ***p<0.001, Mann-Whitney test. Data are represented as mean ± SD (D) Schematic overview of experimental timeline for in vivo protection of epitope edited HSC and progeny cells from CART45 in the presence of AML tumor cells. (E) Mice treated with CART45 clear AML cells resulting in extended survival. (n=9 for CART45 and n=4 for UTD, one-way ANOVA compared to CD45WT+CART45, ***p<0.001). Data are represented as mean ± SD. (F) Frequency of edited cells in peripheral blood of NSG mice engrafted with CD45BE HSCs before and after treatment with CART45 cells (***p<0.001, paired t-test, n=9). (G) Number of human cells in peripheral blood engrafted with CD45WT or CD45BE HSCs 3 weeks after CART45 injection (gated on mouse CD45-/human CD3-). n=9 mice per group, ***p<0.001, Mann-Whitney test. Data are represented as mean ± SD. (H) Schematic overview of BTE design. Anti-CD45 (clone BC8) and anti-CD3 (clone UCHT1) were fused to a silenced tandem Fc domain in LHHL (BTE1) and HLHL (BTE2) configurations. (I) Flow cytometry binding assay shows specific binding of BTE1 and BTE2 to CD3 and CD45 respectively. NALM6 cells (CD3 and CD45 negative) were used as a negative control. Irrelevant antibody (IgG1) was used as a staining control. (J) Dose-dependent killing of MOLM14 AML cells by T cells in the presence of BTEs. (K) Mice treated with anti-CD45 BTE clear MOLM14 AML cells resulting in extended survival. (left panel: *=p<0.05, two-way ANOVA, right panel: **=p<0.01, log-rank test, n=5). Data are represented as mean ± SD. (L) Dose-dependent killing of CD45WT expressing NALM6 cells but not CD45BE expressing NALM6 cells by T cells in the presence of BTEs suggests that CD45BE expressing cells are protected from BTE mediated cytotoxicity.
Next, we interrogated whether BE CART45 can eliminate leukemia while simultaneously sparing healthy hematopoietic cells. To test this, we engrafted unedited, or epitope edited human HSCs into NSG mice followed by injection with luciferase expressing MOLM14 AML cells (Fig. 6D). We confirmed engraftment of MOLM14 cells by bioluminescent imaging and treated the mice with BE CART45. Leukemia cells responded to BE CART45 treatment resulting in decreased tumor burden and extended survival (Fig 6E). Again, we saw rapid elimination of unedited cells in the blood resulting in leukopenia in mice that received unedited HSCs and positive selection of edited cells in mice that received epitope engineered HSCs (Fig. 6F, G). These studies show that co-infusion of BE CART45 cells with epitope edited HSCs can essentially, by a method of subtraction, create a synthetic CD45 neoantigen since wild type CD45 is exclusively expressed on cancer cells while the progeny of healthy HSCs express epitope engineered CD45.
Since epitope editing protects healthy cells by evading antibody-antigen interaction, we reasoned that epitope editing can protect cells not only from CAR-T cells but also other antibody-based therapeutics such as bispecific T cell engagers (BTEs). To test this, we generated half-life extended CD3xCD45 BTEs derived from anti-CD45 (BC8) and anti-CD3 (UCHT1) (Fig. 6H, S7A, B) and demonstrate specific binding to CD3 (kD=~11nM) and CD45 (~600nM) (Fig. 6I, S7C). To test the function of the CD45 BTEs, we co-cultured epitope edited T cells with MOLM14 tumor cells at a 5:1 E:T ratio and show that the presence of the BTEs induced killing of MOLM14 cells in a dose-dependent manner (EC50 = 0.25nM) (Fig. 6J). Next, we engrafted mice with MOLM14 tumor cells and epitope edited T cells followed by biweekly injected of 0.3mg/kg BTE or vehicle control. BTE injection led to tumor clearance in all mice, extending their survival compared to vehicle treated animals. These data indicate that CD45 BTEs in combination with epitope edited T cells have potent anti-leukemia activity (Fig. 6K). To demonstrate that epitope-edited hematopoietic cells are protected from CD45 BTEs, we cultured epitope edited T cells with either CD45WT or CD45BE expressing NALM6 cells in the presence of CD45 BTEs and show that CD45 negative or CD45BE transduced NALM6 cells are not being lysed while CD45WT expressing cells are readily killed (Fig. 6L). Our results show that the epitope editing strategy can also be combined with BTEs and therefore represents a versatile strategy to protect cells of interest from antibody-based targeted therapies.
Discussion
Here we developed a platform that combines pan-hematologic anti-CD45 CAR-T cells with an engineered hematopoietic stem cell transplant endowed with selective resistance to CD45-specific immunotherapy. Specifically, we show that editing a single nucleotide at the locus encoding the targeted epitope on CD45 is sufficient to prevent CART45 fratricide and to protect healthy hematopoietic cells from anti-CD45 immunotherapy without compromising protein expression or function, which is critical for normal hematopoietic cell function.
The absence of truly cancer specific antigens that can be targeted by monoclonal antibodies or CAR-T cells limits their use outside of B cell and plasma cell malignancies(31). We and others proposed that a leukemia specific antigen can be created de novo by combining CAR-T cells with a hematopoietic stem cell transplant that lacks the targeted antigen(10–12, 32). One limitation of this approach is that the targeted lineage antigen that will be removed must be dispensable for normal function of the respective cell lineage. We reasoned that installing a function-preserving mutation at the targeted epitope to prevent recognition, rather than completely removing the target antigen, would allow for targeting genes that are essential antigens, therefore mitigating the risk of antigen negative relapse.
In this proof-of-concept study, we selected CD45 as the target antigen for multiple reasons. Firstly, it is amongst the most abundant and stably expressed cell surface proteins on human hematopoietic cells, which positively correlates with CAR-T cell efficacy(33, 34). Secondly, CD45 is a critical regulator of signaling thresholds in immune cells, including T cells(9), making it an indispensable antigen that requires epitope editing rather than genetic deletion to shield hematopoietic cells without causing disease. Thirdly, the CD45 ECD (which is the target of antibodies and CAR-T cells) is poorly conserved across species, suggesting that editing a small portion is unlikely to cause loss of function (20). This is supported by the kinetic segregation model of T cell signaling, which postulates that size-based exclusion of large receptor tyrosine phosphatases such as CD45 are critical to initiate TCR signaling(35). Accordingly, editing CD45’s ECD is unlikely to disrupt protein function if size and expression are maintained. Lastly, it has been postulated that CD45 is essential for leukemia tumorigenesis and maintenance by regulating response to growth-promoting cytokines, suggesting that CD45 might be an “Achilles heel” molecule on leukemic cells that cannot be readily lost(36). Lastly, targeting CD45 is currently being tested in a phase III clinical trial using the same anti-CD45 monoclonal antibody used in our studies (clone BC8)(37).
Various gene-editing technologies can be used to edit the targeted epitope in such a way that it prevents CAR45 recognition. We chose CRISPR base-editing over nuclease based HDR knock-in because base-editing is largely cell-cycle independent(38), it results in lower indel formation, it has fewer off-target editing events, and yields higher purity (i.e. a high percentage of all edited alleles that contain the requisite base conversion without indels) without causing double strand breaks (DSB)(39). This mitigates the risk of chromosomal translocations, potentially toxic DNA damage responses, and activation of innate immune receptors sensing the DNA donor template(40, 41). However, base-editing is restricted to A to G and C to T transition mutations and requires a PAM that places the target nucleotide within the editing window. Therefore, not every amino acid substitution that would be suitable to abrogate CART45 recognition is achievable with base editing and would require the use of HDR knock in or prime editing. However, both HDR and prime editing are less efficient in human HSCs compared to base editing, likely restricting HSC clonal diversity and reducing the feasibility of this approach as only edited cells will survive CART45 administration(42).
The data presented in this study suggests that hematopoietic cell function is not impaired by epitope editing. However, further comprehensive studies are needed to evaluate the impact of CD45 editing. These studies should include limiting dilutions, secondary transplantation, and experiments using syngeneic animal models. Unfortunately, due to a lack of amino acid sequence conservation at the edited epitope between mice and humans, it is not feasible to develop a murine model to assess the impact of CD45 epitope editing. Instead, a non-human primate (NHP) model can be established to better understand how the epitope edit affects hematopoietic cell development and function, as the targeted epitope is conserved between humans and NHPs. Additionally, conducting these studies in an immunocompetent NHP model, along with a comprehensive analysis of human specimens with different HLA types, will be necessary to further evaluate the potential for immunogenicity after epitope editing.
Epitope editing represents a novel gene-engineering strategy to protect cellular therapies from targeted immunotherapies. The CD45 platform developed in this study could potentially be used for the treatment of all hematopoietic malignancies by virtue of completely replacing a patients hematopoietic system. We have used allogeneic HSCs in the current study. However, using autologous HSCs, this platform could be exploited for other applications such as in vivo selection of genome edited HSCs without genotoxic conditioning, autoimmune diseases and eradication of the latent HIV reservoir. While targeting CD45 with CAR-T cells or BTEs has significant therapeutic potential, the downstream applications of epitope editing can be extended to other targets such as CD117 or CD123 or other antibody based therapeutic modalities such antibody-drug-conjugates or immunotoxins.
Supplementary Material
Acknowledgments:
We would like to acknowledge the human immunology core, the cell center service core, and the stem cell and xenograft core at the University of Pennsylvania.
Funding:
CHJ: P01CA214278-05, NCI U54-CA-244711
SIG: P01CA214278-05,
KCG: 2 R01 CA177684 06A1, PICI, and U54 CA 244711
Competing interests:
Authors (NW, KCG, CHJ, SIG) have filed an invention disclosure with the University of Pennsylvania and Stanford University based on this work.
C.H.J. has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. C.H.J. is a scientific co-founder and holds equity in Capstan Therapeutics and Tmunity Therapeutics. C.H.J. serves on the board of AC Immune and is a scientific advisor to Alaunos, BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Kite, Poseida, Verismo, Viracta, and WIRB-Copernicus group.
S.G. has patents related to CAR therapy with royalties paid from Novartis to the University of Pennsylvania. S.G. is a scientific co-founder and holds equity in Interius Biotherapeutics and Carisma Therapeutics. S.G. is a scientific advisor to Carisma, Cartography, Currus, Interius, Kite, NKILT, Mission Bio, and Vor Bio.
DW has received grant funding from industry for sponsored research collaborations, he has received speaking honoraria, and received fees for consulting or serving on scientific review committees. Remunerations received by DW include direct payments and equity/options. DW also discloses the following associations with commercial partners: Geneos consultant/advisory board, AstraZeneca advisory board, speaker, Inovio board of directors, consultant, Sanofi advisory board, BBI advisory board, Pfizer advisory Board, Flagship consultant, and Advaccine consultant.
R.M.Y. is an inventor on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and Tmunity Therapeutics and receives license revenue from such licenses.
Footnotes
Data and materials availability:
All data are available in the main text or the supplementary materials.
References:
- 1.Zammarchi F et al. , ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood, The Journal of the American Society of Hematology 131, 1094–1105 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H et al. , Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. New England Journal of Medicine 376, 836–847 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL et al. , Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine 371, 1507–1517 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kantarjian HM et al. , Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. New England Journal of Medicine 375, 740–753 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Falchi L, Vardhana SA, Salles GA, Bispecific antibodies for the treatment of B-cell lymphoma: promises, unknowns, and opportunities. Blood, The Journal of the American Society of Hematology 141, 467–480 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS, Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. The Lancet Haematology 7, e816–e826 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Bachy E et al. , A real-world comparison of tisagenlecleucel and axicabtagene ciloleucel CAR T cells in relapsed or refractory diffuse large B cell lymphoma. Nature Medicine 28, 2145–2154 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen AD et al. , Efficacy and safety of cilta-cel in patients with progressive multiple myeloma after exposure to other BCMA-targeting agents. Blood, The Journal of the American Society of Hematology 141, 219–230 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hermiston ML, Xu Z, Weiss A, CD45: a critical regulator of signaling thresholds in immune cells. Annual review of immunology 21, 107–137 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kim MY et al. , Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell 173, 1439–1453. e1419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borot F et al. , Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proceedings of the National Academy of Sciences 116, 11978–11987 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Humbert O et al. , Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia 33, 762–808 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Kung C et al. , Mutations in the tyrosine phosphatase CD45 gene in a child with severe combined immunodeficiency disease. Nature medicine 6, 343–345 (2000). [DOI] [PubMed] [Google Scholar]
- 14.van der Jagt RH et al. , Localization of radiolabeled antimyeloid antibodies in a human acute leukemia xenograft tumor model. Cancer research 52, 89–94 (1992). [PubMed] [Google Scholar]
- 15.Andres TL, Kadin ME, Immunologic markers in the differential diagnosis of small round cell tumors from lymphocytic lymphoma and leukemia. American Journal of Clinical Pathology 79, 546–552 (1983). [DOI] [PubMed] [Google Scholar]
- 16.Caldwell CW, Patterson WP, Hakami N, Alterations of HLe-1 (T200) fluorescence intensity on acute lymphoblastic leukemia cells may relate to therapeutic outcome. Leukemia research 11, 103–106 (1987). [DOI] [PubMed] [Google Scholar]
- 17.Stepanek O et al. , Regulation of Src family kinases involved in T cell receptor signaling by protein-tyrosine phosphatase CD148. Journal of Biological Chemistry 286, 22101–22112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Symons A, Willis AC, Barclay AN, Domain organization of the extracellular region of CD45. Protein engineering 12, 885–892 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Rokkam D, Lupardus PJ, Discovery and characterization of llama VHH targeting the RO form of human CD45. bioRxiv, 2020.2009. 2001.278853 (2020). [Google Scholar]
- 20.Okumura M et al. , Comparison of CD45 extracellular domain sequences from divergent vertebrate species suggests the conservation of three fibronectin type III domains. Journal of immunology (Baltimore, Md.: 1950) 157, 1569–1575 (1996). [PubMed] [Google Scholar]
- 21.Holmes N, CD45: all is not yet crystal clear. Immunology 117, 145–155 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang VT et al. , Initiation of T cell signaling by CD45 segregation at’close contacts’. Nature immunology 17, 574–582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaudelli NM et al. , Programmable base editing of A• T to G• C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richter MF et al. , Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nature biotechnology 38, 883–891 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cahir McFarland ED et al. , Correlation between Src family member regulation by the protein-tyrosine-phosphatase CD45 and transmembrane signaling through the T-cell receptor. Proceedings of the National Academy of Sciences 90, 1402–1406 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wunderlich M et al. , AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 24, 1785–1788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rayes A, McMasters RL, O’Brien MM, Lineage switch in MLL‐rearranged infant leukemia following CD19‐directed therapy. Pediatric blood & cancer 63, 1113–1115 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Jacoby E et al. , CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nature communications 7, 12320 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kishihara K et al. , Normal B lymphocyte development but impaired T cell maturation in CD45-exon6 protein tyrosine phosphatase-deficient mice. Cell 74, 143–156 (1993). [DOI] [PubMed] [Google Scholar]
- 31.Mardiana S, Gill S, CAR T cells for acute myeloid leukemia: state of the art and future directions. Frontiers in oncology 10, 697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MY et al. , CD7-deleted hematopoietic stem cells can restore immunity after CAR T cell therapy. JCI insight 6, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker AJ et al. , Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Molecular Therapy 25, 2189–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Majzner RG et al. , Tuning the Antigen Density Requirement for CAR T-cell ActivityTuning the Antigen Density Requirement for CAR T Cells. Cancer discovery 10, 702–723 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cordoba S-P et al. , The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood, The Journal of the American Society of Hematology 121, 4295–4302 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saint-Paul L et al. , CD45 phosphatase is crucial for human and murine acute myeloid leukemia maintenance through its localization in lipid rafts. Oncotarget 7, 64785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gyurkocza B et al. , Clinical experience in the randomized phase 3 Sierra trial: anti-CD45 iodine (131I) apamistamab [Iomab-B] conditioning enables hematopoietic cell transplantation with successful engraftment and acceptable safety in patients with active, relapsed/refractory AML not responding to targeted therapies. Blood 138, 1791 (2021). [Google Scholar]
- 38.Burnett CA et al. , Examination of the Cell Cycle Dependence of Cytosine and Adenine Base Editors. Frontiers in Genome Editing, 39 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anzalone AV, Koblan LW, Liu DR, Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature biotechnology 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Ihry RJ et al. , p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nature medicine 24, 939–946 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Liu M et al. , Global detection of DNA repair outcomes induced by CRISPR–Cas9. Nucleic acids research 49, 8732–8742 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee B-C et al. , Clonal Dynamics of HDR-Edited HSPCs Targeting the CD33 Locus in Rhesus Macaques. Blood 140, 309–310 (2022).35737920 [Google Scholar]
- 43.Brinkman EK, Chen T, Amendola M, Van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic acids research 42, e168–e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.