

Abstract
This paper introduces simple analytical methods and bioassays to promptly assess the identity and function of in vitro cultured human Schwann cells (hSCs). A systematic approach is proposed to unequivocally discriminate hSCs from other glial cells, non-glial cells, and non-human SCs (authentication), identify hSCs at different stages of differentiation, and determine whether individual hSCs are proliferative or senescent. Examples of how to use distinct cell-based approaches for quality control and routine troubleshooting are provided to confirm the constitution (identity, purity, and heterogeneity) and potency (bioactivity) of hSC cultures from multiple sources. The bioassays are valuable for rapidly gauging the responses of hSCs to mitogenic and differentiating factors and ascertaining the cells’ basic properties before performing co-culture or cell grafting studies. The assays are image based and use adherent hSCs established in monoculture to simplify the experimental setup and interpretation of results. Finally, all sections contain thorough background information, notes, and references to facilitate decision making, data interpretation, and ad hoc method development for diverse applications.
Keywords: Bioassays, Proliferation, Differentiation, Senescence, Identification, Potency, Authentication, Immunodetection, Markers, Myelin, Fibroblasts
Background
Human Schwann cell (hSC) cultures are valuable in vitro models for interrogating the biology of SCs during nerve development, maturation, and regeneration in normal and disease states. These cultures are also valuable as transplantable biologics to treat the injured or dysmyelinated central (CNS) and peripheral (PNS) nervous systems [reviewed in Guest et al. (2013); Monje et al. (2021c); Vallejo et al. (2022)]. Quality control testing of the cultured hSCs is needed for numerous reasons. First, hSC cultures are seldom pure and contain other proliferative cells from the endoneurium and the connective tissue layers. Second, the cultured hSCs differ from those in the original tissue as mature hSCs rapidly dedifferentiate after isolation and adapt to the in vitro environment. Third, non-cellular impurities and factors introduced during culture, such as animal serum and coating reagents, may alter hSC function. Altogether, these issues emphasize the importance of implementing robust analytical methods to promptly (1) confirm the hSC phenotype and the level of purity of the cell cultures; (2) evaluate the state of differentiation and biological activity of the hSCs; and (3) recognize and quantify myelin debris and other visible and subvisible impurities. Selecting suitable methodologies to address these elements will depend on the intended application. Cultured cells are manipulated biologics regardless of the manufacturing method used and need to be scrutinized thoroughly. Contrary to research-grade hSC cultures, those used clinically must meet the highest possible quality standards for identity and function (Bunge et al., 2017; Khan et al., 2021).
The goal of this paper is to describe easy-to-run analytical methods to reveal the purity, bioactivity, and often-changing constitution of donor-relevant hSC cultures by proposing the following: (1) to effectively identity hSCs using live and fixed cultures and discriminate them from cellular and non-cellular contaminants such as myelin, fibroblasts, non-human cells, and glial cells of CNS origin; and (2) to determine hSC function by means of proliferation, differentiation, and senescence assays (Figure 1). As explained in Protocol 1, immunological methods are recommended to reveal the phenotype and level of purity of the hSC cultures. Once the composition of the cultures is confirmed and the cells are authenticated for the species and tissue of origin (Protocol 2), investigators are encouraged to further interrogate their products using specific bioassays designed to evaluate the cellular responses to agonists known to exert an action on SC proliferation and differentiation (Protocol 3). Each protocol contains background information and notes to guide technical or logistical decisions regarding the analysis and interpretation of results. The troubleshooting section defines issues that researchers may encounter, along with appropriate approaches for problem resolution. Assessing cell viability, metabolic activity, microbial contamination, and cell transformation is important, but assays are not described here because they can be found elsewhere. Regarding contamination with cancerous cells, it should be noted that transformation of the hSCs in culture, or amplification of tumor cells imported from the tissue of origin, are unlikely events in hSC cultures from normal tissues (Bastidas et al., 2017).
Figure 1. Quality control assessments for cultured human Schwann cells (hSCs).

Measures of identity and bioactivity described in this study were selected based on their cell type specificity and reliability for scrutinizing hSC cultures in basic and translational research applications. Image: mitogen- and serum-starved hSCs stained with CellTrackerTM Green, a vital fluorescent dye.
The experimental conditions described in the following sections were optimized for adult nerve-derived hSC monocultures (adherent cells) obtained by traditional methods, irrespective of the source, passage, and stage of differentiation of the cells. However, our approach may be equally suitable for the analysis of other types of hSC cultures if enough cells (1 million or more) are available for the initial seeding. The use of multi-well plates is recommended for direct image analysis by fluorescence or light microscopy to reduce time, labor, and cost during experimentation and facilitate data interpretation. However, it is advisable to implement a systematic investigative approach (explained above) including more than one measure (or assay) to confirm the results. The suggested strategy is not intended to replace longer term in vitro (e.g., traditional co-culture systems) or in vivo studies (e.g., cell grafting). Some complex cellular responses requiring the interactions of hSCs with other cell types and the physical environment cannot be recapitulated under the simplified conditions of these experiments. Notwithstanding, the assays described here can be used as screening or discovery platforms to tackle various questions, since no consensus has been reached on how to approach quality control measures for SCs in culture. Future directions include scaling up the assays for a larger number of samples and developing quantitative approaches for specific readouts (e.g., myelin gene expression) relevant to nerve regeneration, therapy, or disease.
Materials and reagents
Our cell culture protocols use research-grade materials, reagents, and solutions that are endotoxin-free and suitable for cell culture. Still, researchers are encouraged to assess the quality of relevant materials by appropriate methods to identify potentially toxic or incompatible elements in the culture medium, coating reagents, and plasticware that can inadvertently impair the health of the hSCs.
All assays can be implemented using basic cell culture equipment and labware typically available in research labs. The description of products provided below is for reference only. Researchers may find comparable items from other providers. An exception is the primary antibodies (Table 1) because they have been validated carefully in our lab using cultured hSCs and nerve tissues. If other antibodies are selected, take into consideration that the ones used for rodent SC research may fail to recognize the respective antigens from humans. For this reason, it is important to confirm the specificity and reactivity of all antibodies in-house using proper positive and negative controls. Some of the suggested monoclonal antibodies were produced in our laboratory from hybridoma cell cultures but alternative ones are available from commercial sources (see Table 1). Lastly, the list below is not fully comprehensive. Additional technical details can be found in the accompanying papers (Aparicio and Monje, 2023; Monje, 2023).
Table 1. Useful antibodies to characterize human Schwann cells (hSCs) and non-glial cells established in cell culture.
These antibodies were validated using traditional adult nerve-derived hSC monocultures. Staining with NGFR, Sox10, and S100B antibodies, alone or together with fibronectin, SMA (α-smooth muscle actin), and FAP (fibroblast activation protein/seprase) antibodies, is recommended for the initial characterization of hSCs and non-glial cells. We have not found a ubiquitous non-glial cell marker. Protocol 1C is suitable for staining with all antibodies except for anti-O1 and anti-O4, which can be accomplished only in live cells (Protocol 1B). A 1:200–1:500 starting dilution is suitable for most antibodies; the optimal concentration should be determined by the end user in the target cells. (*) Indicates that the antibodies are produced from hybridoma cell lines. MBP: myelin basic protein; MAG: myelin-associated glycoprotein; MPZ: myelin protein zero; GFAP: glial fibrillary acidic protein.
| Name of marker & subcellular localization | Product information | Expected results and notes |
|
NGFR Cell membrane |
Mouse monoclonal 8737-IgG. ATCC # HB8737* Rabbit monoclonal, EP1039Y. Abcam, catalog number: ab52987. |
Highly specific hSC marker. Expressed at high, homogeneous levels in all cultured hSCs. |
|
Sox10 Nuclear |
Rabbit monoclonal. Abcam catalog number: ab155279. | Highly specific hSC marker. Expressed at high, homogeneous levels in all hSCs. |
|
S100β Cytoplasmic (typical) and nuclear (rare) |
Rabbit polyclonal. DAKO, catalog number: Z0311. Mouse monoclonal. Sigma, catalog number: S2657 | Highly specific hSC marker. High, homogeneous levels in all hSCs with occasional nuclear localization. |
|
Sox2 Nuclear |
Rabbit polyclonal. Santa Cruz, catalog number: sc-20088. | Highly specific hSC marker, heterogenously expressed in individual hSCs and reduced by CPT-cAMP stimulation. |
|
Nestin Cytoplasmic |
Mouse monoclonal. EMD Millipore, catalog number: MAB353. | Highly specific hSC marker. Heterogeneously expressed (donor- or batch-dependent) and reduced by CPT-cAMP stimulation. |
|
GFAP Cytoplasmic |
Rabbit polyclonal. DAKO, catalog number: Z0334. | Specific hSC marker expressed at low levels in expanded hSCs. |
|
MAG Cell membrane and myelin |
Mouse monoclonal. Chemicon, catalog number: MAB1567. | Mostly in myelin debris. Expressed at low or undetectable levels in expanded hSCs. Induced with CPT-cAMP. |
|
MBP Myelin |
Rat monoclonal. MAB386 (Millipore, former Chemicon) | Mostly in myelin debris. Non-myelin-associated MBP is rarely detectable in expanded hSC cultures. |
|
MPZ/P0 Cell membrane and myelin |
Chicken polyclonal. AB9352 (Millipore, former Chemicon) | Mostly in myelin debris. Non-myelin-associated MPZ is rarely detectable in expanded hSC cultures. |
|
Erg2/Krox20 Nuclear |
Rabbit polyclonal (non-commercial) |
Expressed at low levels in hSCs. Enhanced with CPT-cAMP. |
|
O4 Cell membrane and myelin |
Mouse monoclonal. O4-IgM * | Expressed in all hSCs right after isolation but only in a proportion of the expanded hSCs. Enhanced with CPT-cAMP. |
|
O1 Cell membrane and myelin |
Mouse monoclonal. O1-IgM * | Expressed in hSCs right after isolation. Expanded hSCs are O1- regardless of cAMP levels. |
|
Vimentin Cytoplasmic |
Rabbit monoclonal. Cell Signaling, catalog number: D21H3, 5741. | Equally expressed in hSCs and fibroblasts at high, homogeneous levels. |
|
CD44 Cell membrane |
Mouse monoclonal. Cell Signaling, catalog number: 156-3C11. | Equally expressed in hSCs and fibroblasts at high, homogeneous levels. |
|
Fibronectin Extracellular (typical) and cytoplasmic |
Mouse monoclonal. Santa Cruz, catalog number: sc-8422. Mouse monoclonal. Sigma, catalog number: HFN 36.3 (89062006) * |
Filamentous or punctuated staining with significantly higher levels in fibroblasts as compared to hSCs. |
|
SMA Cytoplasmic |
Mouse monoclonal. Thermo Fisher: catalog number: MS113-PO. Rabbit monoclonal. Cell Signaling, catalog number: D4K9N 19245. |
Expressed at high levels in a proportion of non-glial cells, possibly pericytes. hSCs display low levels of SMA. |
|
FAP Cell membrane |
Rabbit Monoclonal. Cell Signaling, catalog number: 66562. | Expressed at high levels in a proportion of non-glial cells. hSCs do not typically express FAP. |
|
Thy1/CD90 Cell membrane |
Rabbit monoclonal. Abcam. Catalog number: 92574 Abcam | Expressed at variable levels in a proportion of non-glial cells. Hard to detect by simple immunostaining. hSCs do not typically express Thy1. |
Supplies and consumables
Polypropylene conical-bottom centrifuge tubes, 15 and 50 mL (Corning, catalog numbers: 430791 and 430290)
Serological pipettes, 5, 10, and 25 mL, polystyrene, sterile (VWR)
Pasteur pipettes, polystyrene, individually wrapped for liquid disposal (VWR, Argos Technology, catalog number: 10122-560)
Laminin-coated cell culture dishes for cell expansion. 100 mm × 20 mm plates, polystyrene (Corning, catalog number: 353003) coated with a laminin substrate, as described in Andersen and Monje (2018)
Laminin-coated multi-well plates for analytical assays. Cell culture–treated 24-well plates, flat bottom, polystyrene (Corning, catalog number: 3524) coated sequentially with PLL and laminin, as described in (Andersen and Monje, 2018). (Optional) Use commercially available 24-well plates coated with poly-L-ornitine (PO) and laminin (BD Biosciences, catalog number 354659). Do not plate hSCs on uncoated surfaces. Polystyrene plates or chamber slides are preferred. Coverslips are not suitable since the hSCs are unstable and display an abnormal morphology on any glass surface
Paraformaldehyde (PFA) 20% stock solution (Electron Microscopy Sciences, catalog number: 15713)
Methanol (Sigma, catalog number: 154903) maintained at -20 °C for cell permeabilization. (Optional) 0.1% (v/v) Triton X-100 (Sigma, catalog number: 11332481001) prepared in D-PBS and stored at 4 °C
Laboratory wrapping film (Parafilm, catalog number: PM-996) and aluminum foil
Media, supplements, and other cell culture products
Distilled water, cell culture grade (Fisher Scientific, Gibco, catalog number: 15-230-147)
Dulbecco’s phosphate-buffered saline (DPBS) with calcium and magnesium, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14190)
Hank’s balanced salt solution (HBSS) formulated without calcium or magnesium and containing phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 14170-112)
Dulbecco’s modified Eagle’s medium (DMEM) with high glucose and phenol red, pH 7.2 (Thermo Fisher Scientific, Gibco, catalog number: 11965092)
DMEM, Nutrient Mixture F-12 (DMEM/F-12), no glutamine, with phenol red (Thermo Fisher Scientific, Gibco, catalog number: 21331020)
De-complemented fetal bovine serum (FBS) (HyClone, catalog number: SV 30014.03), stored in aliquots at -80 °C
100× GlutaMAX supplement (Thermo Fisher Scientific, Gibco, catalog number: 35050061)
Gentamycin 50 mg/mL, 1,000× stock solution (Thermo Fisher Scientific, Gibco, catalog number: 15750-060)
HEPES buffer solution 1 M (Thermo Fisher Scientific, Gibco, catalog number: 15630-080)
Normal goat serum (GeneTex, catalog number: GTX73206), stored in aliquots at -80 °C
Forskolin (Sigma-Aldrich, catalog number: F68861); for a detailed protocol on preparation, storage, and use of forskolin stock solution, see Andersen and Monje (2018)
Heregulin-β1 (referred to as heregulin), HRG1-B1177-244 recombinant peptide (Preprotech, catalog number: G-100-03); for a detailed protocol on preparation, storage, and use of heregulin stock solution, see Andersen and Monje (2018)
CPT-cAMP stock solution (5 mM in DMEM), prepared from adenosine 3′,5′-cyclic monophosphate, 8-(4-chlorophenylthio), sodium salt (Calbiochem, catalog number: 116812); for a detailed protocol on preparation, storage, and use of CPT-cAMP stock solution, see Monje (2018)
Low proliferation medium (LP) (see Recipes)
High proliferation medium (HP) (see Recipes)
Starvation or D1 medium (DMEM/F12 - 1% FBS) (see Recipes)
PFA-based fixation solution (see Recipes)
Blocking solution (see Recipes)
Antibodies, dyes, and commercially available detection kits
Mouse monoclonal antibodies from hybridoma cell lines. Anti-nerve growth factor receptor (NGFR), anti-O4, and anti-O1 (Sommer and Schachner, 1981) in the form of conditioned medium produced from HB-8737 cells (also known as 200-3-G6-4, obtained from the American Type Culture Collection, ATCC), O4 cells, and O1 cells (kindly provided by Dr. Melitta Schachner), respectively. Researchers can refer to our publication (Ravelo et al., 2018) for technical details on our hybridoma culture protocols. Briefly, transfer the cell content of a hybridoma stock (1 × 106–2 × 106 cells/cryovial) directly into a T-75 flask containing Iscove’s modified Dulbecco’s medium (with phenol red) supplemented with 10% FBS and antibiotics. Culture the cells in suspension inside a CO2 incubator until the cultures are sufficiently dense and the medium becomes slightly acidic. Next, separate the culture supernatant from the cellular content by centrifugation to obtain conditioned medium enriched in monoclonal antibodies. The conditioned medium is often used without dilution, but the specificity and reactivity of each batch should be tested using appropriate positive control cells or tissues
Primary antibodies. pERK1/2/MAPK mouse monoclonal antibody (Santa Cruz, catalog number: sc7383); pAkt-Ser-473 rabbit polyclonal antibody (Santa Cruz, catalog number: sc7985); Akt rabbit polyclonal (Cell Signaling, catalog number: 9272); ERK2/MAPK rabbit polyclonal (Santa Cruz, catalog number: sc154); human nuclei (HNA), mouse monoclonal (Sigma, MAB1281, clone 235-1) or HNA mouse monoclonal (Abcam, ab191181). Other antibodies are listed in Table 1
Fluorescent secondary antibodies of the appropriate species and class, Alexa FluorTM-conjugated (Molecular Probes). Fluorochromes should be chosen and combined as optimal for visualization
FM4-64FX, fixable membrane stain (Invitrogen, catalog number: F34653). (Optional) FluoroMyelinTM (red or green) myelin stain (Invitrogen, catalog number: F34652)
Hoeschst-34580 (Molecular probes, catalog number: H21486). (Optional) 4′,6-Diamidino-2-Phenylindole, Dilactate, (DAPI, Invitrogen, catalog number: D3571)
Click-iT EdU (5-ethynyl-2′-deoxyuridine) Alexa-Fluor-594 Imaging Kit (Life Technologies, catalog number: C10339)
Senescence-associated (SA) β-Galactosidase (SA-β-Gal) staining kit (Cell Signaling Tech, catalog number: 9860)
VectaShieldTM antifade liquid mounting reagent for fluorescence (Vector Laboratories, catalog number: H-1000). (Optional) Prepare a homemade mounting reagent, as suggested in Ravelo et al. (2018), to control photobleaching and preserve the stained cultures. Add a sufficient volume of mounting reagent to fully cover the fixed cells (300 μL/well in a 24-well plate) or a couple of drops if a glass coverslip is mounted on top
Equipment
Biological safety cabinet, BL2 level (Thermo Scientific 1300 series class II, 1300 series, type A2)
Cell incubator set at 37 °C and 8%–9% CO2 (Thermo Scientific Forma, series II, water-jacketed)
Inverted phase contrast microscope (VWR) equipped with 10× to 40× phase contrast objectives and attached digital camera (VWR, V5MP)
Benchtop refrigerated centrifuge (Beckman CoulterTM, Allegra X-12R) equipped with swing bucket rotor (SX4750) and adapters for 50 and 15 mL tubes
Automated cell counter for the image-based counting of cells in suspension (Bio-Rad, TC20) or a hemocytometer for manual cell counting
Inverted fluorescence microscope (Olympus, IX71) for brightfield, phase contrast, and fluorescence microscopy; equipped with standard UV, FITC, and TRITC filter sets and attached digital camera
Procedure
Protocol 1: Analysis of identity and purity
The characterization of cultured SCs has changed over time. Early investigations relied on histochemical and ultrastructural visualization of cells (Askanas et al., 1980) and biochemical measurements for the enzymatic activity of 2′,3′-cyclic nucleotide 3′-phosphohydrolase (Reddy et al., 1982). Currently, investigators can implement various methods to unequivocally understand the constitution of cell cultures. High-throughput sequencing methodologies, such as RNA-seq performed on whole populations or single cells, are thus far the most sensitive and comprehensive approaches. However, simple image-based immunological tests using cell type–specific antibodies are fast, informative, and cost effective to analyze cultured cells. These tests can aid in making crucial decisions, such as changing the culture conditions, expanding the cells further, or subjecting them to purification.
The hSC phenotype from normal mature peripheral nerves can be identified based on the expression of markers such as NGFR (also known as the Low-Affinity Neurotrophin Receptor p75), the S100 Calcium Binding Protein, Beta (S100B), and the SRY-Box Transcription Factor 10 (Sox10) (Scarpini et al., 1986; Assouline and Pantazis, 1989a and 1989b). It is important to estimate the type and proportion of non-glial cells, because endoneurial fibroblasts, endothelial cells, and pericytes can contaminate the hSC cultures in various proportions (Hoyng et al., 2015; Weiss et al., 2016; Peng et al., 2020; Khan et al., 2021). hSCs in vitro derived from non-pathological human nerves or the skin encompass defined phenotypic characteristics (Stratton et al., 2017; Chu et al., 2022); however, the cultured hSCs can change if passaged extensively or subjected to certain experimental treatments. A rigorous phenotyping analysis cannot be overlooked when the source of hSCs is unknown, the tissue material used to derive the cells is abnormal, or the hSCs are obtained artificially by in vitro differentiation or other methods [discussed in Monje (2020)].
The sections below include basic protocols for the routine analysis of hSC cultures. Procedure A describes a generic protocol to plate hSCs in multi-well dishes for direct image analysis. Procedure B encompasses a quick protocol (i.e., < 1 h long) to identify hSCs based on live-cell immunostaining. Procedure C uses fixed cells to detect hSC markers alone or together with markers of non-glial cells (see Table 1). Procedure D enables the detection of extracellular and intracellular myelin debris, which may be undesirable in certain applications. Representative results from these procedures are shown in Figures 2–3 and 5–7.
Figure 2. Identification of human Schwann cell (hSC) cultures using distinct antibody combinations.

Low (A–B) and high (C–G) magnification images of typical hSC cultures are shown to reveal the reactivity and specificity of some recommended antibody combinations. Cultures containing a proportion of non-glial cells (white arrowheads) were selected for display. The morphology of hSCs is varied according to the plating density, the formulation of the culture medium, and the level of fibroblast contamination, but the antibody reactivity and specificity are unchanged. All cultures were grown in HP medium except for those in panel G, which were grown in differentiation medium for seven days (Protocol 3C). Refer to Table 1 for technical details and interpretation of results. Nuclei were stained with DAPI (blue) in all images.
Finally, it should be mentioned that the methods for quantification of fluorescent cells and the parameters for assay design, such as the selection of positive and negative controls, are not described here. Please refer to publications from our group for additional experimental data and information (Monje et al., 2018; Monje, 2020; Peng et al., 2020).
-
Cell plating (common to all protocols)
Prepare a single-cell suspension of hSCs, count the cells, and estimate their viability by the method of choice. For a reference to detailed protocols, see Ravelo et al. (2018).
Plate the cell suspensions at mid-density in HP medium (see Recipe 2) inside a 24-well plate using 50,000 cells/well or an equivalent density (25,000 cells/cm2) as determined by the available surface. Use plastic dishes coated with PLL (or PO) and laminin to ensure prompt adhesion and survival of the cells (see Materials and Reagents). Consider that enough replicates are needed to achieve statistical significance in all measurements that compare control vs. treatment conditions. Triplicate samples are sufficient for most assays.
Culture the cells in the CO2 incubator for 24–72 h to obtain a culture at 40%–60% confluence, as determined empirically by periodic image analysis by phase contrast microscopy.
When the cells are ready to be stained (or fixed), remove the culture medium by gentle aspiration inside a biosafety cabinet, working quickly to avoid unnecessary exposure of cells to the air flow.
(Optional) Wash the cells with D-PBS or LP medium (see Recipe 1) if floating myelin or cellular debris is evident by phase contrast microscopy.
Proceed with the description provided in Protocol 1B–1D.
-
Live cell immunolabeling
Add a sufficient volume of conditioned medium containing NGFR or O4 antibodies (e.g., 500 μL for each well of a 24-well plate). Alternatively, use an appropriate dilution of a commercially available antibody (see Table 1). Properly scale up or down the suggested measures if other plate formats are used in this and all subsequent steps.
Incubate the cells at room temperature (RT) for 20 min and leave them undisturbed inside the biosafety cabinet (see Note a).
Remove the conditioned medium and quickly wash the cells 2× with an excess of D-PBS to eliminate unbound antibodies while ensuring that cells do not detach.
Fix the cells for 20 min with fixation solution (see Recipe 4) and wash them 3× with D-PBS to remove traces of PFA.
Prepare a 1:300–1:1,000 dilution of Alexa-conjugated secondary antibody (as determined by the primary antibody) in blocking solution (see Recipe 5) and add it to the cells in an adequate volume (see Protocol 1B, step 1). For instance, use Alexa Fluor 488-conjugated anti-mouse IgG to detect NGFR (from HB-8737 conditioned medium) or anti-mouse IgM to detect O4 (see Note b).
Add a general nuclear stain, such as DAPI or Hoechst-33342 (1:1,000) to the secondary antibody solution. Alternatively, the nuclear staining can be done independently before mounting after a brief 10 min incubation at RT using dyes (1:1,000) diluted in DPBS.
Cover the plates with aluminum foil and place them on an orbital shaker for 30–60 min at slow motion (20–30 rpm), as the cells can peel off from the dishes even after fixation.
Wash the cells 3× with D-PBS to eliminate unbound secondary antibodies.
Add mounting reagent in a sufficient volume to cover the cells (see Materials and Reagents). Mount the cells with a glass coverslip if visualization is performed at 40× or higher power objectives.
Image the cultures using an inverted fluorescence microscope and a suitable filter set (see Equipment) starting at 10× to obtain a panoramic view of the cultures (Figure 2A–2B) before visualization at higher magnification (Figure 2C–2G).
Estimate the proportion of NGFR+ and O4+ cells in relationship to total cells (DAPI+ or Hoechst+) to inform on the percentage of glia/Schwann (NGFR+) and non-glial cells (NGFR-), and of mature (O4+) and immature (O4-) hSCs. This can be achieved by manual or automated counting methods.
-
Immunolabeling of fixed cells
Gently aspirate the culture medium, fix the cells for 20 min with fixation solution (see Recipe 4), and wash them 3× with D-PBS.
Permeabilize the cells by incubation with cold methanol (preferred) or 0.1% Triton X-100 in D-PBS for 10 min. Permeabilization of the cell membranes is required only for the staining of intracellular antigens exhibiting cytoplasmic or nuclear localization. Methanol is preferred because detergents can damage the cellular structure if timing is not carefully controlled.
Aspirate the methanol (or the Triton X-100 solution) quickly and wash the cells 3× with D-PBS.
Add blocking solution (see Recipe 5) for at least 30 min at RT.
Remove the blocking solution and replace it with either of the following: (1) conditioned medium from NGFR hybridoma cells (see above); (2) anti-S100B (1:300 in blocking solution), or (3) anti-Sox10 (1:300 in blocking solution) to identify hSC-specific markers with localization to the plasma membrane, the cytoplasm, and the nucleus, respectively. (Optional) Refer to Table 1 for an alternative selection of primary antibodies.
Incubate the cells overnight at 4 °C with gentle agitation (20–30 rpm in an orbital shaker) to maximize immunolabeling detection.
Proceed as described in Protocol 1B, steps 5–11, using a corresponding selection of secondary antibodies and a nuclear stain to estimate the proportion of hSCs (NGFR+, S100B+, Sox10+) and non-glial cells (NGFR-, S100B-, Sox10-) by imaging analysis.
-
Labeling of myelin debris
Perform a live staining of the cell cultures using anti-O1 antibodies (see Materials and Reagents) following the protocol described for anti-O4 (Protocol 1B, steps 1–3) (see Note c).
Fix the cells with fixation solution (see Recipe 4), as described in Protocol 1B, step 4.
Prepare a labeling solution containing AlexaFluor488-conjugated anti-mouse IgM antibodies (1:500) in combination with FM4-64FX (1:1,000) or FluoroMyelinTM-red (1:500) in an adequate volume of D-PBS (see Notes c and d). (Optional) Incorporate a nuclear stain such as DAPI or Hoechst-33342 to this mixture.
Add the abovementioned labeling solution directly to the cells, cover the plates with aluminum foil, and incubate them for 30–60 min with gentle agitation (20–30 rpm in an orbital shaker). Monitor the cells under the fluorescence microscope to confirm the levels of staining, as larger diameter myelin granules require a longer labeling time with FM4-64FX or FluoroMyelinTM.
Remove the solution and wash the cells 3× with D-PBS to remove traces of the fluorophores.
-
Perform image analysis to discriminate and quantify extracellular (O1+) and total (FM4-64FX+/FluoroMyelinTM+) myelin granules. The observation of granular spots heavily stained with O1 antibodies may be interpreted as extracellular myelin. In such cases, confirm that O1+ granules are non-cellular by showing they are not associated with intact nuclei. Total myelin can also be revealed by immunostaining with antibodies against myelin proteins (see Table 1) in fixed cell cultures (see Protocol 1C).
Notes:
Prolonged incubation with primary antibodies (> 30 min) leads to internalization or capping of the antibodies, which is seen as patchy, granular (instead of smooth) staining. Placing the cells on ice reduces the capping effect; however, lowering the temperature should be done with caution because the temperature shock can impair hSC adhesion. More information on troubleshooting and staining protocols can be found in Ravelo et al. (2018).
NGFR immunostaining can be performed using live or fixed cells (Protocol 1C) with similar results. However, a live-cell labeling protocol is required for lipid antigens, as PFA fixation leads to non-specific membrane binding of O4 and O1 (galactocerebroside/GALC) antibodies. Whereas NGFR expression is homogeneous and constant in all hSCs regardless of the culture conditions, O4 expression is heterogeneous and depends on cAMP stimulation (Peng et al., 2020). For best results, culture hSCs in CPT-cAMP-containing medium for at least three days (Protocol 3) before performing O4 immunostaining. The expression of myelin lipids declines rapidly after hSC isolation from the nerves. It has been reported that hSCs from explant cultures can maintain GALC expression in 30% of the cells two weeks after isolation (Turnbull et al., 2001). Yet, GALC+ and O1+ cells are rarely found in established hSC cultures even under optimal conditions for differentiation (Monje et al., 2018).
hSC cultures from mature nerves contain a variable proportion of myelin debris in the extracellular environment and within the cells themselves, because hSCs effectively engulf myelin fragments before and after isolation from the nerve tissue. Extracellular myelin can be washed out easily; however, intracellular myelin is retained inside the hSC’s cytoplasm. Whereas extracellular myelin stains heavily with O1 and O4 antibodies, intracellular myelin requires staining with myelin fluorophores. An important point to take into consideration is that cultured, isolated hSCs do not contain intact or newly formed myelin sheaths. If myelin-like structures are observed in primary or established hSC cultures, they are likely derived from the original tissue. Myelin debris do not interfere with hSC function in vitro but can inhibit axon growth and trigger an immune response if myelin-containing cultures are used for cell grafting in vivo.
FM4-64FX and FluoroMyelinTM enable quick and selective myelin labeling in live and fixed hSC cultures and can be used in combination with antibody staining. FM4-64FX is preferred because it emits strong fluorescence and can be fixed permanently with aldehyde-based buffers. These dyes are believed to work via lipophilic interactions. However, membranes other than myelin and intracellular lipid droplets can be stained as well.
Protocol 2: Authentication of hSC cultures
Authentication is performed to correctly identify the cell material by a series of established methods. An authentication guide has been established for most common mammalian cell lines. However, a similar guide for cultured hSCs has not yet been developed. Traditionally, SC cultures were derived directly from a nerve fascicle (or ganglia), which simplified the identification of the neural (Schwann) phenotypes by virtue of knowing the tissue of origin. At present, however, culturing technologies have expanded, and hSC-like cells can be created by various in vitro techniques (Huang et al., 2020). This shift in culturing technologies represents a major challenge in identifying the hSCs and discriminating them from other glial and non-glial cell types.
State-of-the-art technologies, such as RNAseq, can readily determine the constitution of cell cultures. However, the time and cost associated with these high-resolution approaches can be limiting. Immunodetection methods are highly reliable for quickly confirming (or disproving) the species and tissue of origin of individual cells within mixed populations. For instance, antibodies with selective reactivity to human NGFR and MPZ proteins have been used to differentiate human from non-human SCs in cell culture and within grafted tissues (Levi and Bunge, 1994). Alternatively, this discernment has been achieved by concomitantly detecting a SC-specific marker (e.g., with MPZ or NGFR antibodies) and a ubiquitous human marker with nuclear localization (Bastidas et al., 2017). Researchers have discriminated hSCs from central glial cells (namely, astrocytes) based on NGFR immunodetection, as this membrane receptor is expressed only in PNS glia (Assouline and Pantazis, 1989a). Individual myelin-associated hSCs, i.e., either mature (myelin-forming) or repair (myelin-engulfing), can be discriminated from CNS glia (oligodendrocytes) by immunodetection of the major peripheral myelin glycoprotein MPZ (Levi and Bunge, 1994).
Here, we suggest a simple approach to validate hSC cultures by incorporating at least two elements of support, (1) for the human origin and (2) the PNS origin of the hSCs, using combined antibody- and ligand-based fluorescent labeling methods (Table 2). This approach may be useful in the following scenarios: (1) when the cells deemed to be SCs are not obtained from human peripheral nerves or ganglia; (2) when the origin of the tissue or the cell cultures is dubious or unknown; (3) when contamination with spinal cord or CNS tissue cannot be ruled out; and (4) when there is a known or suspected pathology affecting the donor tissue used to derive the hSCs. Authentication is also recommended when cells initially confirmed to be normal hSCs depart from the expected phenotype while in culture. This observation may signal an underlying issue, such as fibroblast overgrowth, transformation of the hSCs, or cross-contamination with cancerous cell lines. Importantly, results from our suggested bioassays (Protocol 3) should be interpreted under the assumption that the cells under investigation faithfully represent the PNS-derived hSC phenotype. This author recommends a stepwise approach to cell identification (Protocol 1) and authentication (Protocol 2) when researchers believe that this general assumption should be challenged.
Table 2. Quick staining protocols for human Schwann cell (hSC) validation in culture.
Table 2 presents simple strategies for verifying both the human and the SC background of cultured cells. Different antibody combinations using human-selective and multispecies-selective NGFR antibodies in combination with other stains are proposed. NGFR-8737 (mouse monoclonal), S100B (rabbit polyclonal), and GFAP (rabbit polyclonal) antibodies are described in Table 1. HNA monoclonal antibodies are described in Materials and Reagents.
| Antibody or fluorophore combination | Purpose of the staining | Interpretation of results |
| NGFR-8737 (human-specific) + S100B (multispecies) | Discriminate SCs that are human (NGFR+) from total SCs (S100B+) regardless of species |
Cells that are S100B+/NGFR+ can be considered hSCs if derived from peripheral nerve NGFR-/S100B+ cells should not be regarded hSCs. Suspect of contamination with CNS glia |
| NGFR (multispecies) + HNA (human nuclei only) | Discriminate hSCs (NGFR+/HNA+) from other human cells (NGFR-/HNA+) |
Cells that exhibit an NGFR-/HNA+ phenotype cannot be considered hSCs NGFR+/HNA- cells may consist of nonhuman SCs |
| NGFR-8737 + GFAP (multispecies) | Discriminate hSCs (NGFR+) from astrocytes (GFAP+) | NGFR-/GFAP+ cells may consist of CNS glia (astrocytic) |
| NGFR-8737 + FM4-64X (or FluoroMyelinTM) | Confirm the hSC phenotype among NGFR+ cells | FM4-64X+ granules inside NGFR+ cells are strong evidence of the adult, repair-like hSC phenotype |
Plate cells from a mixed culture or unknown source as described in Protocol 1A. Limit the numbers to 20,000 cells/well (approx. 10,000 cells/cm2) to achieve a low-density culture. Ideally, the cells should be separated from each other to identify them individually.
Gently aspirate the culture medium, fix the cells for 20 min with fixation solution (see Recipe 4), and wash them 3× with D-PBS.
Permeabilize the cells by incubation with cold (-20 °C) methanol.
Aspirate the methanol quickly and wash the cells 3× with D-PBS.
Add blocking solution (see Recipe 5) for at least 30 min at RT.
Replace the blocking solution with the selected antibody combinations presented in Table 2.
-
Proceed as described in Protocol 1C, steps 6–7, using the appropriate selection of secondary antibodies and fluorescent dyes (Table 2), followed by image analysis (see Notes a and b).
Notes:
NGFR-8737 reacts strongly and specifically with human/primate NGFR without cross-reacting with NGFR from other species. Cultured NGFR-8737+ cells can be considered human Schwann-like glia if they originate from PNS tissues. Nevertheless, refer to Table 1 to develop alternative antibody combinations to confirm the hSC phenotype. As shown in Figure 3C, S100B+ cells may not be considered Schwann-like without confirming double-labeling with NGFR antibodies. We have not identified the presence of NGFR+, S100B- in established hSC cultures (Peng et al., 2020).
The hSC phenotype can be confirmed by co-immunostaining with HNA and SC-specific antibodies (Tables 1 and 2, Figure 3B). An alternative human-specific nuclear protein is NuMA, which has proven useful for localizing hSCs in the environment of a xenografted host (Bastidas et al., 2017).
Figure 3. Quality control testing of known and unknown cell cultures.

A–B. Discrimination of human and rat Schwann cells (SCs) using human-specific NGFR-8737 (A) and HNA (B) antibodies. The proportion of hSCs and rat SCs has differed in A (1 human: 9 rat) and B (9 rat: 1 human). C–I. Immunological assessment of an unidentified cell culture using multiple antibody combinations against glia (NGFR, S100B, and GFAP) and non-glial cell markers (SMA, FN). Cells were treated in the absence (control, C–G) and presence of CPT-cAMP (H–I), an agent that drives SC differentiation (see Protocol 3C). The unidentified human-derived (HNA+) cell culture contained only a few hSC-like cells, as evidenced by co-expression of S100B and NGFR (arrowheads in C2). Contamination with CNS glia was suspected considering the presence of S100B+ (C1–C2) and GFAP+ (G–I) cells that did not co-express NGFR. Notice the strong staining for FN (E) and the induction of SMA in the CPT-cAMP condition, which is a response not expected of hSCs.
Protocol 3: Analysis of bioactivity
For over five decades, researchers have relied on in vitro systems to address the neuron-supportive function and other typical SCs responses during nerve development, maturation, and repair. Traditional bioassays have involved placing hSCs in co-culture with dorsal root ganglia (DRG) neurons to evaluate SC–neuron interactions, such as SC-elicited axon growth (Stratton et al., 2017), axon contact-driven SC proliferation (Morrissey et al., 1995c), and myelin sheath differentiation (Morrissey et al., 1995b). However, researchers noticed that cultured hSCs reduced the viability of neuronal cells and did not differentiate effectively under conditions that supported myelination by non-human (rat) SCs (Morrissey et al., 1995c), and that expanded hSCs failed to align along axons, proliferate, and differentiate when placed in co-culture with DRG neurons despite their strong pro-regenerative phenotype (Monje et al., 2018). Other studies indicated that only a subset of nerve-derived hSCs activated the promyelinating factor POU3F1 and myelinated PNS axons after transplantation (Stratton et al., 2017), and that myelination of axons in the spinal cord was achieved when grafting cultured SCs from rats rather than humans (Bastidas et al., 2017).
The determination of potency for cultured hSCs is complex because there is no common analytical method or bioassay able to address the multiple functions of SCs in vivo. FDA regulations state that surrogate markers that correlate with bioactivity may be used alone or together with cell-based assays for the functional characterization of cellular products (FDA, 2008). In this scenario, an argument is made that the levels of expression and activation of key membrane receptors, intracellular molecules (e.g., kinases), and transcription factors deemed relevant to SC lineage specification and differentiation may be considered surrogate bioactivity indicators for hSCs in vitro. One example from the literature is the consistent use of the transcriptional regulators Egr2/Krox20 and cJun, which drive and inhibit myelination, respectively, to highlight the properties of SCs from intact and injured nerves (Arthur-Farraj et al., 2012).
Therefore, this author recommends a stepwise characterization of hSC identity and function by implementing simple, neuron-free assays prior to launching more sophisticated studies involving co-culture systems or xenotransplantation. We have found that kinase activation (phosphorylation), proliferation, differentiation, and senescence assays (Monje et al., 2018) provide useful information on the characteristics of donor-relevant hSC populations. As explained in the following sections, kinase (Protocol A) and proliferation (Protocol B) assays are designed to evaluate the magnitude of cellular responses to stimulation with heregulin alone and in combination with forskolin, a reversible adenylyl cyclase activator. Differentiation assays (Protocol C) evaluate the expression of myelination-associated markers in response to high doses of cAMP analogs, a potent pharmacological trigger for myelin-related SC differentiation in vitro (Monje, 2018). Senescence assays (Protocol D) evaluate senescence-associated growth arrest, which can be linked to extended passaging or stress-inducing treatments in cultured hSCs.
The proposed cell-based assays share the following features: (1) they are SC-specific, i.e., they inform on the properties expected of SCs rather than other cell types; (2) they are quantitative or semi-quantitative, i.e., the output from the assays is (or can be converted into) a measurable readout; and (3) they assess the magnitude of a cellular or molecular response as a function of a stimulatory signal or experimental condition (i.e., heregulin in kinase activation assays, cAMP in differentiation assays, and rounds of subculture in senescence assays) in reference to a negative control condition (i.e., vehicle-stimulated cells or cells not subjected to passaging). Bioactivity assays should be performed only with purified hSC cultures or cultures whose identity is thoroughly understood (Protocols 1 and 2). Deviations from the normal hSC phenotype, or the presence of contaminating cells, may alter or misrepresent the results. Please, refer to our publications (Monje et al., 2018; Peng et al., 2020) for additional data, technical details, and alternative approaches regarding the optimization and use of the bioassays. Whilst image analysis using fluorescence or light microscopy (this protocol) is useful as the first approach, other methods (western blot, ELISA, and q-RT-PCR) may be advantageous to confirm the results or increase the detection sensitivity.
-
Determination of β1-heregulin/ErbB-elicited ERK and Akt activation
This procedure describes how to evaluate ERK and Akt phosphorylation using pathway-selective, phospho-specific antibodies in β1-heregulin-stimulated hSC cultures, as reported previously (Monje et al., 2006; Monje et al., 2021b). The responsiveness to β1-heregulin, and the consequent activation of ErbB/HER2 and ErbB/HER3 receptors, are key bioactivity measures for cultured SCs in general. β1-heregulin (a member of the NRG family) is the most potent mitogenic factor described for hSCs (Levi et al., 1995) and induces proliferation by promptly activating ErbB-mediated signaling via Ras-ERK and PI3K-Akt pathways (Monje et al., 2006 and 2008). ERK and Akt phosphorylation are equally reliable readouts for (and correlate with) β1-heregulin-induced cell cycle progression (Monje et al., 2006). This response is expected to be SC-specific because other nerve-resident cell types, such as endoneurial fibroblasts, lack the expression of the ligand binding partner ErbB3 and are unresponsive to the β1-heregulin stimuli. In these assays, a recombinant β1-heregulin peptide is used as a mimetic of the natural axon-bound heregulin/NRG family members known to mediate axon-contact driven SC mitogenesis (Morrissey et al., 1995c). Detection of activated (phosphorylated) ERK and Akt can be completed within 3 h by immunofluorescence microscopy imaging, as per the protocol described below.
Plate the cells as explained in Protocol 1A.
Starve the cells by progressively removing mitogenic factors and serum from the culture medium, as follows: the day after plating, remove the HP medium and replace it with an equal volume of LP medium; 24 or 48 h later, replace the LP medium with starvation medium (D1, see Recipe 3), and incubate the cells overnight before stimulation (see Note a).
Induce ERK and Akt activation by stimulating the cells with recombinant β1-heregulin. To do so, replace the culture medium with DMEM/F12 containing vehicle (control) or β1-heregulin at 10 nM (treatment, single dose). (Optional) Provide β1-heregulin in a range of concentrations from 0.1 to 10 nM to ascertain dose-dependent changes (see Note b).
Incubate the cells for 10 min (fixed time point) in a CO2 incubator. The activation of ERK and Akt by β1-heregulin is maximal at 5–10 min and declines thereafter. However, P-ERK is maintained for 24 h after β1-heregulin addition in hSCs (Monje et al., 2006), so the time course of these experiments can be extended for several hours or even days (Monje et al., 2008).
Rapidly remove the medium by gentle aspiration and fix the cells with methanol (-20 °C) for 15 min.
Proceed as described in Protocol 2C, steps 3–7, except use anti-Phospho(P)-ERK/Total ERK and/or anti-P-Akt/Total Akt antibodies, 1:200–1:300 each in blocking solution. (Optional) If an expedited reading is needed, incubation with primary antibodies can be reduced to 1–2 h at RT.
Analyze the cells by fluorescence microscopy to identify ERK and Akt phosphorylation levels in reference to total kinase levels in β1-heregulin-treated and control cells. Refer to Table 3 for data interpretation and Figure 4 for representative images. See Monje et al. (2006) for additional results.
-
Determination of heregulin-dependent proliferation with and without forskolin
As mentioned above, cultured hSCs are responsive to heregulin/NRG because they constitutively express ErbB2-ErbB3 receptors in the absence of other ErbB/EGFR family members (Morrissey et al., 1995c; Peng et al., 2020). SCs are among the few known cellular types whose proliferation is enhanced, rather than reduced, by pharmacological agonists of intracellular cAMP at low doses. In addition, the effect of cAMP-elevating agents on heregulin-induced mitogenesis is synergistic in SCs. This synergism was discovered during early investigations using in vitro cultured SCs from rats (Stewart et al., 1991) and humans (Levi et al., 1995) and can be regarded as a distinctive SC response. Nerve-derived fibroblasts are not heregulin-sensitive and respond to cAMP agonists by decreasing their rate of cell division (Levi et al., 1995).
The assays described below are designed to determine cell cycle progression (DNA synthesis) in response to β1-heregulin and forskolin, alone and in combination, as reported in Monje et al. (2006, 2008 and 2018), and Peng et al. (2020). These assays are well-suited to determine a hSC-specific outcome in populations confirmed to be pure and non-senescent. The requirement of β1-heregulin as a mitogenic factor is a defining feature of the cultured hSC phenotype. Cells that do not require exogenous β1-heregulin for propagation in vitro should be further scrutinized for their identity or transformed phenotype.
Plate the cells as described in Protocol 1A and starve them of mitogenic factors (Video 1) as described in Protocol 3A.
Stimulate the cells with β1-heregulin and forskolin. Replace the culture medium with D1 medium containing vehicle (control), 10 nM β1-heregulin (main mitogen), 2 μM forskolin (adjuvant), and 10 nM β1-heregulin plus 2 μM forskolin (growth factor combination to observe a synergistic effect in the number of proliferating cells). (Optional) These assays can be performed in the presence of 10% FBS for maximal hSC viability without hindering the synergistic effect of forskolin (Monje et al., 2018) (see Note c).
Label the newly synthesized DNA with the thymidine analog EdU. Add EdU (1 μM) to all wells to label cells undergoing S-phase entry 4–18 h post stimulation with β1-heregulin and forskolin. (Optional) Add EdU concurrently with the stimulating factors in Protocol 3B step 2 (see Note d).
Incubate the cells in a CO2 incubator for 48–72 h.
Fix the cells and detect DNA-bound EdU by following the manufacturer’s instructions for fluorescent detection of EdU+ nuclei. Add a nuclear dye such as Hoechst-33342 or DAPI to label all nuclei.
Image the cultures by fluorescence microscopy and count the number of EdU+ cells in reference to the total number of cells (DAPI+ or Hoechst+) to estimate the percentage of cell division. Refer to Table 3 for data interpretation, and Videos 1 and 2 for representative results.
-
Determination cAMP-dependent differentiation
hSCs growing in the absence of neurons maintain a highly immature, proliferative phenotype, unless induced to differentiate with a potent stimulus, such as prolonged treatment with high doses of cAMP analogs. This unique response of hSCs allows the measurement of myelin-associated gene expression in response to a single stimulatory signal without the need to introduce neurons. Researchers should consider that SCs established in culture express negligible or undetectable levels of myelin proteins and lipids. Thus, a strong rationale exists for challenging the hSCs with differentiating factors such as cAMP inducers before measuring the expression of myelin markers.
Our hSC differentiation protocol is based on prolonged incubation with high doses of the cell-permeable, phosphodiesterase-resistant analog of cAMP, CPT-cAMP (Monje et al., 2018). These simple bioassays can substitute for the laborious assessment of myelin sheath formation in neuron-SC culture systems. Use highly viable, non-senescent populations and fresh reagents (e.g., CPT-cAMP stocks) for optimal results. Additional technical details on the preparation of cells, substrates, and media, and the optimization of experimental variables can be found in Monje (2018).
Plate the cells in HP medium as explained in Protocol 1A.
The day after plating, replace the HP medium with an equal volume of D1 or LP medium containing vehicle (DMEM) or CPT-cAMP (250 μM in DMEM), to maintain the cells exhibiting an immature phenotype (control) or to promote differentiation, respectively.
Incubate the cells in a CO2 incubator for 3–5 days with daily observations to identify morphological changes associated with CPT-cAMP stimulation (see Note e). (Optional) Add EdU labeling reagent by the second- or third-day post-CPT-cAMP addition, to concurrently determine myelin gene elevation and cell cycle exit.
Proceed to stain the cells with antibodies that recognize the state of differentiation of the hSCs, e.g., by O4 labeling (Protocol 1B) or immunostaining for myelin-related markers (Protocol 1C). See Table 1 and Figure 5 for antibody options.
Add a nuclear dye such as Hoechst-33342 or DAPI to label all nuclei.
Image the cultures by fluorescence microscopy and count the number of O4+ cells (or the marker of choice) in reference to the total cells to estimate the differentiation efficiency.
Refer to Table 3 for data interpretation. See Figure 5 and Videos 1 (control) and 3 (CPT-cAMP) for representative results.
-
Determination of passage-dependent senescence
SCs of human origin undergo senescence as they are passaged in vitro or exposed to various stressors, including environmental changes. The causes of senescence are ill-defined. Yet, senescence can be considered an hSC-specific attribute, as senescence does not affect human fibroblasts from nerve tissues (Peng et al., 2020). Intriguingly, cultured SCs established from rat nerves can expand almost indefinitely without getting transformed or acquiring senescence (Mathon et al., 2001). A hypothetical hSC culture that fails to senesce under standard in vitro conditions is likely not human or not SC-related. By determining the proportion of senescent hSCs, one can predict the progression (expandability) of donor-relevant cell cultures.
We recommend a generic enzymatic SA-β-Gal test for senescence detection because markers indicative of hSC senescence are elusive (Monje et al., 2021a). Senescent and non-senescent cultures are hard to discriminate (Figure 6 and Video 4). Perform senescence assays on populations that have been expanded for several passages, become unresponsive to proliferate with SC-specific mitogens, or manifest morphological or functional changes, such as excessive cell clumping, detachment, or floating debris.
Plate the cells in HP medium as explained in Protocol 1A. Include a known population of senescent cells in all SA-β-Gal experiments to serve as positive control. hSCs from passage-4 or higher and/or hSCs treated with H2O2 for >3 days are suitable, as environmental stressors readily drive hSC senescence.
Incubate the cells in a CO2 incubator for at least 24 h to ensure all cells are well-adhered and extend processes on the substrate (see Video 4).
Fix the cells with fixation solution (see Recipe 4) for 20 min as explained in Protocol 1B.
Reveal the enzymatic activity of SA-β-Gal by following the manufacturer's recommendations.
Add a nuclear dye such as Hoechst-33342 or DAPI to label all nuclei.
Image the cultures by light microscopy (SA-β-Gal) and fluorescence microscopy (nuclei) and count the number of SA-β-Gal+ cells in reference to the total number of cells. Refer to Table 3 for data interpretation. Figures 6 and 7, and Video 4 show representative results.
Notes:
Progressive removal of mitogenic factors and serum is needed to lower the endogenous levels of phosphorylated kinases and induce hSC quiescence without massive apoptotic cell death triggered by loss of trophic support. Starved hSCs maintain their alignment and general morphology, even though their processes become thinner (Figure 1 and Video 2).
This protocol is equally suitable to determine the activation status of other β1-heregulin-responsive kinases such as ErbB-2, ErbB-3, Raf, MEK, and RSK (Monje, Bartlett Bunge et al. 2006). Always include a positive control condition to activate ERK and Akt. The phorbol ester PMA, an activator of PKC, can be used to elicit maximal ERK activation in all cells. Purified PIP2,3 can be used to activate Akt in a membrane receptor-independent manner (Monje, Athauda, et al.2008).
Alternative ErbB- and cAMP-inducers may be considered. ErbB-dependent hSC proliferation can be elicited with axolemmal preparations enriched in membrane-bound NRG, a mitogen for cultured hSCs (Sobue et al., 1984). Cholera toxin, pertussis toxin, and phosphodiesterase-resistant analogs of cAMP (e.g., db-cAMP) are effective in promoting a synergistic effect on heregulin-induced hSC proliferation (Levi et al., 1995; Monje et al., 2006)
EdU incorporation assays are a suitable alternative to radioactive (e.g., tritiated thymidine) and antibody-based (e.g., BrDU) methods. EdU detection uses mild fixation and can be multiplexed with antibody labeling. Results from EdU incorporation assays are best interpreted when done in conjunction with immunostaining (Protocols 1B–C) and senescence assays (Protocol 3D). We routinely stain cells with antibodies (e.g., anti-NGFR or anti-O4) before developing the EdU reaction to readily discriminate proliferative hSCs from other cell types (Monje t et al, 2018).
Typically, hSCs lose their spindle-shaped morphology and acquire a large, reticulated shape coincidently with cell cycle exit within 2–3 days after CPT-cAMP treatment (Video 3). If morphological differentiation is confirmed by phase contrast microscopy, the cells can be considered ready for analysis. If morphological differentiation is not appreciated by the third or fourth day of treatment, new medium containing CPT-cAMP should be added, and additional time should be allowed for differentiation to occur.
Table 3. Schwann cells (SC)-specific bioassays.
Table 3 summarizes our suggested assays to reveal the unique responses of cultured hSCs. All assays rely on the detection of one or more inducible molecular readouts while exploiting the experimental fine-tuning of an activating signal (input) driving a measurable change (output) through a SC-specific mechanism of action.
| Bioassay | Expected results | Interpretation |
|
Kinase activation (heregulin- dependent) |
Cultured hSCs respond to β1-heregulin by rapidly activating ERK and Akt. The differential immunolabeling between control (without heregulin) and treated cells (heregulin-stimulated) should be obvious in all hSCs. |
Inter-experimental variability is expected due to the rapid kinetics of ERK and Akt phosphorylation, but the overall responses should be consistent across hSC populations. For a reference, include positive controls to reveal maximal ERK and Akt activation. |
| Proliferation (heregulin and forskolin-dependent) |
β1 heregulin is sufficient to increase the percentage of dividing hSCs. This response is synergistically enhanced by forskolin but forskolin alone does not increase hSC proliferation. hSCs incorporate EdU in a nonsynchronous manner starting roughly at 18–20 h post stimulation under these conditions. |
The proportion of proliferating (EdU+) cells is donor- and passage-dependent, but it is usually < 40% in early-passage cultures under the suggested conditions. High basal proliferation (in the control condition) may indicate an excess of fibroblasts or, in rare cases, cancerous (heregulin-independent) hSC proliferation. |
|
Differentiation (CPT-cAMP dependent) |
CPT-cAMP induces upregulation of certain myelin-associated markers (e.g., O4, Krox20, PRX) and downregulation of immature hSC markers (e.g., cJun) within 3–5 days. Corresponding changes in mRNA expression occur at earlier time points. CPT-cAMP treated hSCs become post-mitotic and morphologically dissimilar to untreated cells. |
Select the markers that provide the highest resolution for detection. Assessment of myelin gene expression is only relevant when comparing control (no CPT-cAMP) and CPT-cAMP-treated conditions. The magnitude and kinetics of the expression of different gene products are variable. Substantial batch variability is also expected. Molecular changes may be evident even when morphological differentiation is not apparent. |
| Senescence (passage-dependent) |
hSC populations contain a proportion of senescent cells even at passage-zero. Late-passage cultures may consist only of senescent hSCs. Fibroblasts do not get senescent under these conditions and can be discriminated from hSCs by their negative SA-β-Gal staining. |
Normal hSC cultures (nerve-derived) become senescent, usually after four rounds of passaging. The percentage of senescent cells varies from culture to culture or donor to donor, even when same-passage cultures are compared. |
Figure 4. Heregulin-elicited ERK activation in human Schwann cells (hSCs).

hSCs were plated, deprived of mitogenic factors and serum, and stimulated as per Protocol 3A. The cells were stained with antibodies against the phosphorylated (P-ERK) and total forms of ERK, respectively. hSCs rapidly respond to β1-heregulin by inducing P-ERK and its shuttling to the nucleus. ERK staining is strong and homogeneous in the hSC’s cytoplasm (right panels, control).
Video 1. Human Schwann cells (hSCs) deprived of mitogenic factors.
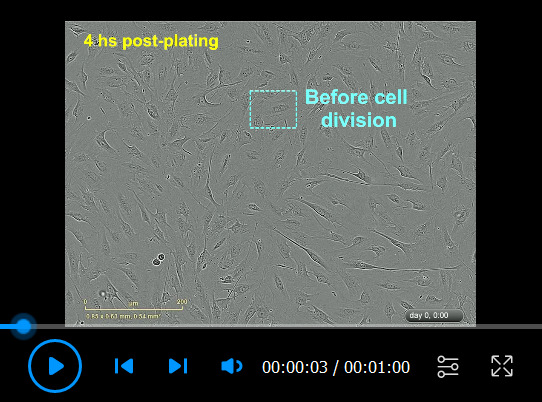
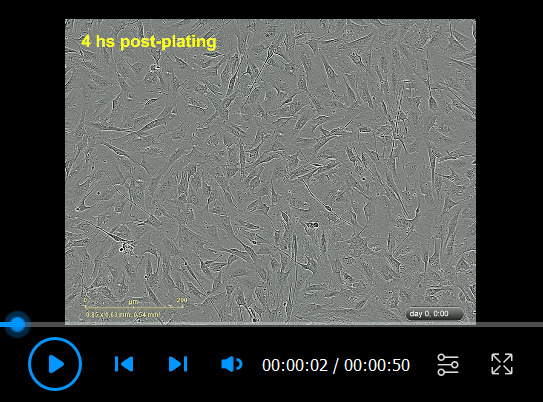
hSCs were plated on a PLL-laminin-coated dish in LP medium and imaged using IncuCyte ZOOMTM using 20× objective lenses. This condition serves as a common control for the proliferation (Video 2) and differentiation (Video 3) assays. Notice the fast adhesion of the cells, the changes in cell morphology, and the progressive cell-cell alignment. hSCs proliferated moderately (see mitotic figures) due to serum factors present in the LP medium. Individual phase contrast images were taken every 30 min starting 4 h post-plating. Some relevant elements were highlighted in the video.
Figure 5. Differentiation assays.

Human Schwann cells (hSCs) were stimulated and analyzed per Protocol 3C and stained with antibodies per Table 1. Low (A–B) and high magnification (C–J) images of control (non-stimulated cells in D1) and cAMP-differentiated hSCs (cells treated with 250 μM CPT-cAMP in D1 for 7 days) are shown. Notice the phenotypic conversion of the hSCs from an elongated, roughly bipolar shape to an enlarged, flattened morphology in the CPT-cAMP condition. CPT-cAMP treatment does not change S100B (A–D) and NGFR (C–D, I–J) expression. However, a proportion of the CPT-cAMP-treated hSCs express higher levels of Krox20 (G–H), periaxin (PRX, E–F), and O4 (E–H) along with lower levels of nuclear cJun (I–J), as denoted by the white arrowheads. Heterogeneity in the expression levels of myelin markers is expected (H–F). The S100B negative cells highlighted by the black arrowheads in panels A–B are fibroblasts. Nuclei were stained with DAPI (blue, C–F). The PRX antibody was courtesy of Peter Brophy. The Krox20 antibody was courtesy of Dies Meijer.
Figure 6. Discrimination of expandable and non-expandable (senescent) human Schwann cell (hSC) cultures.
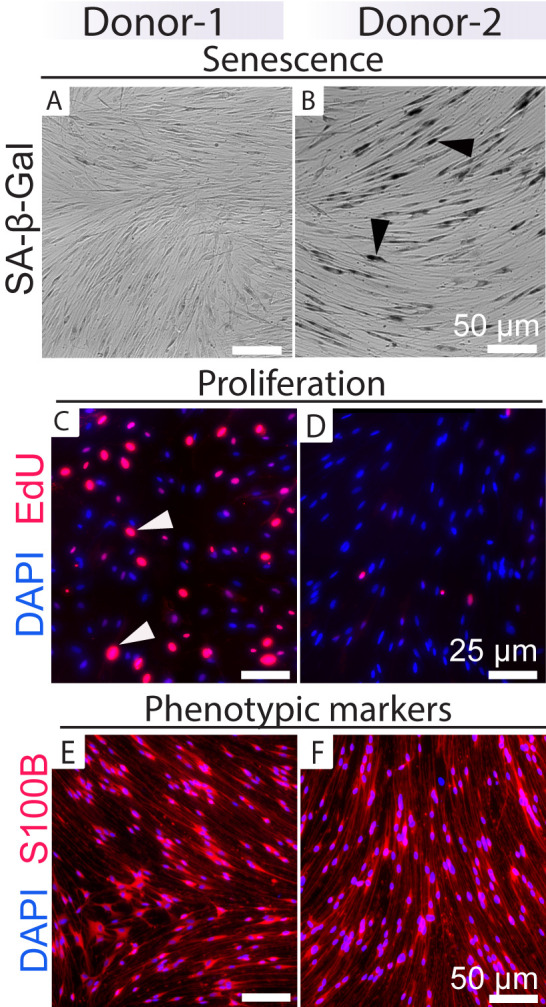
This figure shows an example of how to functionally discriminate non-senescent (Donor-1) vs. senescent (Donor-2) cultures based on results from SA-β-Gal (A–B) and EdU incorporation (C–D) assays in cultures growing in HP medium. hSCs from these two donors are visually undistinguishable based on their morphology, pattern of alignment, and levels of SC-specific markers such as S100B (E–F). Most cells from Donor-2 are non-proliferative and stain positive for SA-β-Gal (black arrowheads in B). Estimating the percentage of senescent cells, alone or together with EdU staining, can help to predict the characteristics of cells from subsequent rounds (Monje et al., 2018). Notice the dissimilar levels of EdU labeling in individual cells, indicating the asynchronous incorporation of DNA. Some individual cells (white arrowheads) have undergone more than one cycle of cell division.
Video 4. Senescent human Schwann cell (hSC) cultures.
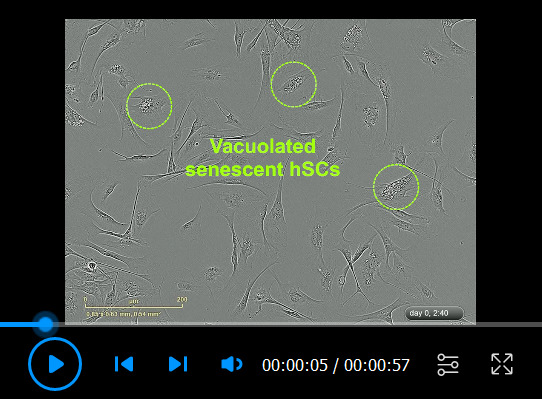
The cells were plated in HP medium and recorded via IncuCyte ZOOMTM every 20 min. Notice that senescent hSCs are highly viable and migratory but fail to efficiently expand in number despite the presence of heregulin and forskolin. The appearance of vacuoles (seen clearly at the onset), the expanded cytoplasm, and the lack of cell-cell alignment are morphological evidence of senescence. However, senescence is defined by the lack of proliferation and should be confirmed with appropriate assays. The senescent phenotype is irreversible. Some relevant elements were highlighted in the video. For a reference, compare this video with Video 2, featuring a highly proliferative hSC culture.
Figure 7. Identification of fibroblast contamination and senescent cells.

Examples are shown of how to recognize cultures enriched in fibroblasts (left panels) and senescent cells (right panels) by immunostaining with cell type–specific markers (FN, green; S100B, red) and enzymatic SA-β-Gal staining (blue precipitate inside the hSCs), respectively. The cultures were grown in LP medium in the absence (control, A–B) or presence of 250 μM CPT-cAMP (Protocol 3C, C–D) for seven days before fixation and immunostaining (Protocol 1C). Even though hSCs differentiated effectively as denoted by S100B staining, the fibroblasts proliferated extensively (C–D). In E–H, hSCs (passage-5) were plated in HP medium, fixed, and stained with SA-β-Gal after three days (Protocol 3D). Observe the differential behavior of the cells in areas of low (E–F) and high (G–H) density. Senescent hSCs are migratory and have the capacity to form aggregates that are prone to detachment. Recommendations: (1) purify the cells from A–D; (2) use or cryopreserve the cells from E–H but do not continue the culturing, as nearly all hSCs are senescent.
Video 2. Human Schwann cell (hSC) proliferation in response to the mitogenic factors heregulin and forskolin.
Cells were plated in LP medium for 4 h before being stimulated with heregulin and forskolin (Protocol 3B) and recorded via IncuCyte ZOOMTM, as described in Video 1. Notice the high levels of proliferation (mitotic figures), cell-cell alignment, and migration of hSCs under the influence of mitogenic factors. This culture can be considered confluent and ready for analysis by the end of the recording period.
Video 3. Human Schwann cell (hSC) differentiation in response to CPT-cAMP.
Cells were plated in LP medium for 4 h before being stimulated with CPT-cAMP (250 µM) per (Protocol 3C). The plates were transferred to IncuCyte ZOOMTM for live cell imaging immediately after CPT-cAMP stimulation. Notice that CPT-cAMP inhibits hSC proliferation, alignment, and migration while inducing a flat, expanded cytoplasm that features transient vacuoles of different sizes. This culture is ready to be analyzed for the presence of myelin-related markers by the end of the incubation period. Some relevant elements were highlighted in the video. For a reference, compare this video with Video 1 (control condition).
Recommendations and troubleshooting
Clinical investigations have revealed that with optimized standard operating procedures, clinical-grade reagents, and systematic approaches, it is possible to consistently achieve hSC cultures at high yields (e.g., 108 cells at passage-2) and purity (> 90% hSCs) without introducing purification steps (Khan et al., 2021). In non-clinical settings, however, the level of control is much lower, starting with the conditions for tissue procurement, which are usually incomplete or unknown to the investigators. The importance of applying good cell culture practices cannot be more emphasized. Routine monitoring and periodic testing of the cultures using benchmark verification tests and assays can allow researchers to understand and correct problematic issues as soon as they arise. Unlike rat SCs, hSC cultures are sensitive to various experimental variables, such as changes in the media or substrate. Considerable donor- or batch-related variability is expected. Additionally, the cells within single cultures are heterogenous, thus emphasizing the need to use various analytical tools to evaluate the quality and stability of donor-relevant hSC cultures.
This section was elaborated to assist in the detection and resolution of technical problems that are commonly seen in hSC cultures (Table 4). Running appropriate tests (Protocols 1–2) and bioassays (Protocol 3) is most important in the following scenarios: (1) when the cultures acquire an unusual appearance or show a distorted alignment pattern, (2) when there is excessive floating debris, and (3) when the rate of proliferation changes or cell growth becomes density independent. For a reference, Figures 6–7 and Videos 4–5 present examples of cultures that merit further scrutiny due to their atypical features attributable to senescence and fibroblast contamination.
Table 4. Troubleshooting guide for hSC cultures.
| Potential problem | Possible Cause | Corrective measure |
| Cultures do not expand at the expected rate | Ineffective mitogenic cocktail, or arrival to senescence (Figures 6-7). | Test the activity of heregulin and forskolin using specific bioassays (Monje et al., 2021b) or simply replace the stock solutions and perform a senescence test. |
| Cultures do not acquire the typical pattern of growth or bipolar morphology | This is normal if cultures are not yet confluent but may reflect problems such as over-proliferation of fibroblasts (Figure 7), cytotoxicity, or senescence. | Continue the culturing and provide new HP media to expedite hSC division. If this problem persists, perform a purity check along with viability assays. |
| Cells do not adhere or extend membrane processes after plating | This is likely the result of poor adhesion and may indicate a problem with the substrate. Short or retracted processes may indicate stress and poor viability (e.g., due to over-trypsinization). | Perform a quick viability test. If cells are alive, transfer the cell suspensions to a fresh laminin-coated dish within 2–4 h post-plating. Otherwise, hSCs would undergo apoptosis triggered by lack of adhesion (anoikis). |
| There is excessive floating debris, and the medium contains particulate material | This is a usual phenomenon in some hSC cultures but may also indicate cytotoxicity or microbial contamination. | Wash the cells with DPBS to remove the debris, add new HP medium, and perform a sterility test to discriminate this condition from microbial contamination. |
| Signs of microbial contamination become clear | This can be manifested at any time due to unknown contamination of the nerve explant or an improper aseptic technique during the culturing steps. | Discard the contaminated hSC cultures. Perform proper tests to identify the type and source of contamination. Decontamination attempts are futile. Discard the stocks if contamination is traced to the stocks. |
| The cells migrate to certain areas, form clumps, and detach from the substrate | This is usual in hSC cultures and may indicate stress due to factors in the medium or substrate. Clumping may result from over confluence or senescence (Figure 7). | This may be corrected by replating the cells into a new dish. The culture may be terminated if the cells are confirmed to be senescent. |
| Cultures become enriched in senescent cells | Cells have reached their expansion limit, or environmental factors have triggered premature senescence. | Discard the cultures or use them as such. Senescent cells are highly viable and bioactive, but proliferation is irreversibly halted (Figures 6 and 7). |
| Cells do not adhere after being seeded from a cryogenic stock, or there is an excess of apoptotic cells | This can indicate an adhesion problem (substrate), cytotoxicity (to the DMSO) during and after plating, or poor cryopreservation (i.e., suboptimal storage conditions). | Confirm the viability of the stocks. Take precautions to work fast and eliminate traces of DMSO after plating. Recover the culture after removing the apoptotic cells (e.g., via MACS). |
| Cultures do not contain O4+ hSCs | This is usual. O4+ cells decline with expansion. O4 expression is not constitutive but induced in response to cAMP agonists and axon contact. | Confirm the identity of the cultures by immunostaining with NFGR, Sox10, or S100B. Add forskolin or other cAMP-stimulating agents for at least 72 h to restitute O4 expression (Figure 5). |
| Fibroblasts proliferate fast and overtake the culture | This issue occurs when the selective pressure of cAMP-stimulating agents is removed (Figure 7 and Video 5). Fibroblasts proliferate fast and are resistant to undergo senescence. | Restitute the mitogenic combination to maintain high levels of cAMP and reduce fibroblast growth. Purify the cells if fibroblast contamination becomes problematic. |
| Cells align well and cover the available surface, but cell counts remain low | This common observation indicates poor proliferation or enrichment in senescent cells. | Test the bioactivity of the mitogenic factors and perform a senescence test. |
| Cells proliferate in the absence of heregulin and lose density-dependent control of proliferation | This is unusual in normal hSC cultures. It can indicate transformation or cross-contamination with a highly proliferative, cancerous cell line. | Perform identity and authentication tests to verify the source of the proliferative cells. If the cells are confirmed to be hSCs, evaluate the possibility of transformation via appropriate tests. |
Video 5. Fibroblast contamination.
Human Schwann cells (hSCs) were plated in LP medium and imaged via IncuCyte ZOOMTM every 30 min. This culture contained a high proportion of fibroblasts, which can be recognized by their expanded (low contrast) cytoplasm and conspicuous nuclei. The hSCs extended processes but failed to align with one another, possibly due to the presence of fibroblasts concentrating in the central area. This culture has abundant cellular debris. The over-proliferation of fibroblasts and the debris can signal an underlying issue with the hSC culture (see Table 4). Some relevant elements were highlighted in the video. For a reference, compare this video with Video 1, which displays a pure hSC culture under the same culture conditions.
Recipes
-
Low proliferation medium (LP)
Reagent [stock concentration] Final concentration Volume DMEM (or DMEM/F12) n/a 445 mL FBS (100%) 10% 50 mL GlutaMax 200 mM (100×) 1× 5 mL Gentamycin (1,000×) 1× 0.5 mL Total n/a 500 mL Note: Balance the pH to make it slightly acidic (pH = 6), sterilize it using a 0.22 μm filter, and store it at 4 °C. Follow the guidelines provided in the accompanying paper (Monje, 2023).
-
High proliferation medium (HP)
Reagent [stock concentration] Final concentration Volume Low proliferation medium n/a 500 mL Heregulin-β1177-244 (25 μM) 10 nM 200 μL Forskolin (15 mM) 2 μM 69.25 μL Total 500 mL Note: Balance the pH to 7, filter, and store the HP medium as indicated for the LP medium. More information can be found in Monje (2023).
-
Starvation or D1 medium (DMEM/F12 – 1% FBS)
Reagent [stock concentration] Final concentration Volume DMEM/F12 n/a 45 mL FBS (100%)GlutaMax 200 mM (100×)HEPES 1 M (100×)Gentamycin 50 mg/mL (1,000×)Total1%1×1× (10 mM)1×n/a0.5 mL0.5 mL0.5 mL0.05 mL50 mLNote: Balance the pH to 7, filter, and store as indicated for the LP medium. This medium is used only for controlled starvation, as hSCs are affected by the absence of serum. Do not freeze.
-
PFA-based fixation solution
Reagent [stock concentration] Final concentration Volume PFA (20%) 4% 4 mL DPBS 1× 16 mL Total 20 mL Note: The stocks are maintained frozen at -20 °C until use. Avoid repeated freeze-thaw cycles. Working solutions can be stored at 4 °C.
-
Blocking solution
Reagent [stock concentration] Final concentration Volume Normal goat serum 5% (v/v) 5 mL DPBS 1× 1× 45 mL Total 50 mL Note: The stocks are maintained frozen at -20 °C until use. Avoid repeated freeze–thaw cycles.
Acknowledgments
Natalia Andersen, Ketty Bacallao and Kaiwen Peng contributed to the experimental work. Patrick M. Wood, Mary Bunge, and Linda White provided expert guidance, troubleshooting tips, and banked cells during the initial investigations. The development of these protocols was supported by grants (to P.V.M.) from the National Institutes of Health (NS084326), the Craig H Neilsen Foundation (339576), The Miami Project to Cure Paralysis and the Buoniconti Foundation from the University of Miami, the Charcot Marie Tooth Association, and the Indiana State Department of Health (33997, 43547). Grant NIH/NS009923 (to M.B., P.M.W and P.V.M) provided seminal funding for developing proliferation and differentiation assays and the creation of cell stocks. The contents of this article are the responsibility of the author and do not necessarily represent the official views of the funding agencies. Valeria Nogueira assisted with illustrations, Gabriela Aparicio with manuscript and figure formatting, Thomas Dolan with video editing, and Louise Pay with English editing. Lingxiao Deng contributed fruitful scientific discussions and laboratory support. We are greatly indebted to the generosity of the anonymous individuals and their families for providing tissues for research.
Competing interests
P.V.M. is the founder of GliaBio LLC., a consulting company that focuses on glial cell research. The author declares that the research described here was conducted in the absence of commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical considerations
The experiments were performed using anonymized, banked hSC cultures from non-pathological tissues without regard to the gender or age of the donor. This research was deemed to constitute non-human-subject research by the Institutional Human Subjects Research Offices of the University of Miami (2003–2018) and Indiana University (2019–2022). The human cells used in these studies were derived from postmortem tissues made available by the Life Alliance Organ Recovery Agency (LAORA) of the University of Miami to the laboratory of P.M.W. Requests for information and materials (cells, antibodies, other) should be directed to P.V.M. Investigators are encouraged to contact P.V.M. for feedback on the reported protocols.
Citation
Readers should cite both the Bio-protocol article and the original research article where this protocol was used.
Q&A
Post your question about this protocol in Q&A and get help from the authors of the protocol and some of its users.
References
- 1.Andersen N. D. and Monje P. V.(2018). Isolation, Culture, and Cryopreservation of Adult Rodent Schwann Cells Derived from Immediately Dissociated Teased Fibers. In: Monje, P. and Kim, H.(Eds.). Schwann Cells(pp. 49–66). Methods in Molecular Biology. Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- 2.Aparicio G.I. and Monje P. V.(2023). Human Schwann Cells in vitro I. Nerve Tissue Processing, Pre-degeneration, Isolation, and Culturing of Primary Cells. Bio-protocol 13: e4748. DOI: 10.21769/BioProtoc.4748.
- 3.Arthur-Farraj P. J., Latouche M., Wilton D. K., Quintes S., Chabrol E., Banerjee A., Woodhoo A., Jenkins B., Rahman M., Turmaine M., et al.(2012). c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75(4): 633-647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Askanas V., Engel W. K., Dalakas M. C., Lawrence J. V. and Carter L. S.(1980). Human schwann cells in tissue culture: histochemical and ultrastructural studies. Arch. Neurol. 37(6): 329-337. [DOI] [PubMed] [Google Scholar]
- 5.Assouline J. G. and Pantazis N. J.(1989). Detection of a nerve growth factor receptor on fetal human Schwann cells in culture: absence of the receptor on fetal human astrocytes. Brain Res. Dev. Brain Res. 45(1): 1-14. [DOI] [PubMed] [Google Scholar]
- 6.Assouline J. G. and Pantazis N. J.(1989). Localization of the nerve growth factor receptor on fetal human Schwann cells in culture. Exp. Cell Res. 182(2): 499-512. [DOI] [PubMed] [Google Scholar]
- 7.Bastidas J., Athauda G., G. De La Cruz, Chan W. M., Golshani R., Berrocal Y., Henao M., Lalwani A., Mannoji C., Assi M., et al.(2017). Human Schwann cells exhibit long-term cell survival, are not tumorigenic and promote repair when transplanted into the contused spinal cord. Glia 65(8): 1278-1301. [DOI] [PubMed] [Google Scholar]
- 8.Bunge M. B., Monje P. V., Khan A. and Wood P. M.(2017). From transplanting Schwann cells in experimental rat spinal cord injury to their transplantation into human injured spinal cord in clinical trials. Prog. Brain Res. 231: 107-133. [DOI] [PubMed] [Google Scholar]
- 9.Chu T. H., Baral K., Labit E., Rosin N., Sinha S., Umansky D., Alzahrani S., Arora R., Cao L., Rancourt D., et al.(2022). Comparison of human skin- and nerve-derived Schwann cells reveals many similarities and subtle genomic and functional differences. Glia 70(11): 2131-2156. [DOI] [PubMed] [Google Scholar]
- 10.Food and Drug Administration(FDA)(2008). Guidance for Industry: Potency tests for cellular and gene therapy products. U.S. Department of Health and Human Services. Center for Biologics Evaluation and Research: 1–15.
- 11.Guest J., Santamaria A. J. and Benavides F. D.(2013). Clinical translation of autologous Schwann cell transplantation for the treatment of spinal cord injury. Curr. Opin. Organ Transplant. 18(6): 682-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoyng S. A., De Winter F., Gnavi S., van Egmond L., Attwell C. L., Tannemaat M. R., Verhaagen J. and Malessy M. J.(2015). Gene delivery to rat and human Schwann cells and nerve segments: a comparison of AAV 1-9 and lentiviral vectors. Gene Ther. 22(10): 767-780. [DOI] [PubMed] [Google Scholar]
- 13.Huang Z., Powell R., Phillips J. B. and Haastert-Talini K.(2020). Perspective on Schwann Cells Derived from Induced Pluripotent Stem Cells in Peripheral Nerve Tissue Engineering. Cells 9(11): 2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan A., Diaz A., Brooks A. E., Burks S. S., Athauda G., Wood P., Lee Y. S., Silvera R., Donaldson M., Pressman Y., et al.(2021). Scalable culture techniques to generate large numbers of purified human Schwann cells for clinical trials in human spinal cord and peripheral nerve injuries. J. Neurosurg. Spine 36(1): 135-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levi A. D. and Bunge R. P.(1994). Studies of myelin formation after transplantation of human Schwann cells into the severe combined immunodeficient mouse. Exp. Neurol. 130(1): 41-52. [DOI] [PubMed] [Google Scholar]
- 16.Levi A. D., Bunge R. P., Lofgren J. A., Meima L., Hefti F., Nikolics K. and Sliwkowski M. X.(1995). The influence of heregulins on human Schwann cell proliferation. J. Neurosci. 15(2): 1329-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathon N. F., Malcolm D. S., Harrisingh M. C., Cheng L. and Lloyd A. C.(2001). Lack of replicative senescence in normal rodent glia. Science 291(5505): 872-875. [DOI] [PubMed] [Google Scholar]
- 18.Monje P. V.(2018). Scalable Differentiation and Dedifferentiation Assays Using Neuron-Free Schwann Cell Cultures. In: Monje, P. and Kim, H.(Eds.). Schwann Cells(pp. 213–232). Methods in Molecular Biology. Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- 19.Monje P. V.(2020). The properties of human Schwann cells: Lessons from in vitro culture and transplantation studies. Glia 68(4): 797-810. [DOI] [PubMed] [Google Scholar]
- 20.Monje P. V.(2023). Human Schwann Cells in vitro II. Passaging, Purification, Banking and Labeling of Established Cultures.. Bio-protocol 13(22)13(22): e4882. DOI: 10.21769/BioProtoc.4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monje P. V., Andersen N., Sant D., Peng K., Wang G. and Xu X-M.(2021). Stability of lineage-specific attributes in senescent human peripheral glia. XV European Meeting on Glial Cells in Health and Disease(Poster presentation).
- 22.Monje P. V., Athauda G. and Wood P. M.(2008). Protein kinase A-mediated gating of neuregulin-dependent ErbB2-ErbB3 activation underlies the synergistic action of cAMP on Schwann cell proliferation. J. Biol. Chem. 283(49): 34087-34100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monje P. V., Bacallao K., Aparicio G. I. and Lalwani A.(2021). Heregulin Activity Assays for Residual Testing of Cell Therapy Products. Biol. Proced. Online 23(1): 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monje P. V., Bartlett Bunge M. and Wood P. M.(2006). Cyclic AMP synergistically enhances neuregulin-dependent ERK and Akt activation and cell cycle progression in Schwann cells. Glia 53(6): 649-659. [DOI] [PubMed] [Google Scholar]
- 25.Monje P. V., Deng L. and Xu X. M.(2021). Human Schwann Cell Transplantation for Spinal Cord Injury: Prospects and Challenges in Translational Medicine. Front. Cell. Neurosci. 15: 690894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monje P. V., Sant D. and Wang G.(2018). Phenotypic and Functional Characteristics of Human Schwann Cells as Revealed by Cell-Based Assays and RNA-SEQ. Mol. Neurobiol. 55(8): 6637-6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morrissey T. K., Bunge R. P. and Kleitman N.(1995). Human Schwann cells in vitro. I. Failure to differentiate and support neuronal health under co-culture conditions that promote full function of rodent cells. J. Neurobiol., 28(2): 171–189.
- 28.Morrissey T. K., Kleitman N. and Bunge R. P.(1995). Human Schwann cells in vitro. II. Myelination of sensory axons following extensive purification and heregulin-induced expansion. J. Neurobiol. 28(2): 190-201. [DOI] [PubMed] [Google Scholar]
- 29.Morrissey T. K., Levi A. D., Nuijens A., Sliwkowski M. X. and Bunge R. P.(1995). Axon-induced mitogenesis of human Schwann cells involves heregulin and p185erbB2. Proc. Natl. Acad. Sci. U. S. A. 92(5): 1431-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng K., Sant D., Andersen N., Silvera R., Camarena V., Piñero G., Graham R., Khan A., Xu X. M., Wang G., et al.(2020). Magnetic separation of peripheral nerve-resident cells underscores key molecular features of human Schwann cells and fibroblasts: an immunochemical and transcriptomics approach. Sci. Rep. 10(1): 18433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravelo K. M., Andersen N. D. and Monje P. V.(2018). Magnetic-Activated Cell Sorting for the Fast and Efficient Separation of Human and Rodent Schwann Cells from Mixed Cell Populations. In: Monje, P. and Kim, H.(Eds.). Schwann Cells(pp. 87–109). Methods in Molecular Biology. Humana Press, New York. [DOI] [PubMed] [Google Scholar]
- 32.Reddy N. B., Askanas V. and Engel W. K.(1982). Demonstration of 2’,3’-Cyclic Nucleotide 3’-Phosphohydrolase in Cultured Human Schwann Cells. J. Neurochem. 39(3): 887-889. [DOI] [PubMed] [Google Scholar]
- 33.Scarpini E., Meola G., Baron P., Beretta S., Velicogna M. and Scarlato G.(1986). S-100 protein and laminin: Immunocytochemical markers for human Schwann cells in vitro. Exp. Neurol. 93(1): 77-83. [DOI] [PubMed] [Google Scholar]
- 34.Sobue G., Brown M. J., Kim S. U. and Pleasure D.(1984). Axolemma is a mitogen for human Schwann cells. Ann. Neurol. 15(5): 449-452. [DOI] [PubMed] [Google Scholar]
- 35.Sommer I. and Schachner M.(1981). Monoclonal antibodies(O1 to O4) to oligodendrocyte cell surfaces: An immunocytological study in the central nervous system. Dev. Biol. 83(2): 311-327. [DOI] [PubMed] [Google Scholar]
- 36.Stewart H. J., Eccleston P. A., Jessen K. R. and Mirsky R.(1991). Interaction between cAMP elevation, identified growth factors, and serum components in regulating Schwann cell growth. J Neurosci Res 30: 346-352. [DOI] [PubMed] [Google Scholar]
- 37.Stratton J. A., Kumar R., Sinha S., Shah P., Stykel M., Shapira Y., Midha R. and Biernaskie J.(2017). Purification and Characterization of Schwann Cells from Adult Human Skin and Nerve. eneuro 4(3): ENEURO.0307–16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turnbull V. J., Petratos S., Papadopoulos R., Gonzales M. F. and Ayers M.(2001). Variable galactocerebroside expression by human Schwann cells in dissociated and peripheral nerve explant cultures. J. Neurosci. Res. 65(4): 318-321. [DOI] [PubMed] [Google Scholar]
- 39.Vallejo F. A., Diaz A., Errante E. L., Smartz T., Khan A., Silvera R., Brooks A. E., Lee Y. S., Burks S. S., Levi A. D., et al.(2022). Systematic review of the therapeutic use of Schwann cells in the repair of peripheral nerve injuries: Advancements from animal studies to clinical trials. Front. Cell. Neurosci. 16: e929593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiss T., Taschner-Mandl S., Bileck A., Slany A., Kromp F., Rifatbegovic F., Frech C., Windhager R., Kitzinger H., Tzou C. H., et al.(2016). Proteomics and transcriptomics of peripheral nerve tissue and cells unravel new aspects of the human Schwann cell repair phenotype. Glia 64(12): 2133-2153. [DOI] [PubMed] [Google Scholar]