

Abstract

Metastases to the brain remain a significant problem in lung cancer, as treatment by most small-molecule targeted therapies is severely limited by efflux transporters at the blood–brain barrier (BBB). Here, we report the discovery of a selective, orally bioavailable, epidermal growth factor receptor (EGFR) inhibitor, 9, that exhibits high brain penetration and potent activity in osimertinib-resistant cell lines bearing L858R/C797S and exon19del/C797S EGFR resistance mutations. In vivo, 9 induced tumor regression in an intracranial patient-derived xenograft (PDX) murine model suggesting it as a potential lead for the treatment of localized and metastatic non-small-cell lung cancer (NSCLC) driven by activating mutant bearing EGFR. Overall, we demonstrate that an underrepresented functional group in medicinal chemistry, the trisubstituted hydroxylamine moiety, can be incorporated into a drug scaffold without the toxicity commonly surmised to accompany these units, all while maintaining potent biological activity and without the molecular weight creep common to drug optimization campaigns.
Introduction
Non-small-cell lung cancer (NSCLC) is a disease that comprises approximately 85% of all newly diagnosed lung cancers (Figure 1a).1−3 Activating mutations in epidermal growth factor receptor (EGFR, ERBB1, HER1), which are detected in 10 to 30% of patients with NSCLC, such as in-frame exon 19 deletions (del E746_A750) and a point mutation on exon 21 (L858R), confer sensitivity to reversible first-generation EGFR-targeted tyrosine kinase inhibitors (TKIs) such as gefitinib (1).2−5 Resistance to first-generation TKIs is mainly due to the acquisition of a secondary T790M “gatekeeper” mutation on exon 20, detected in 60% of drug-resistant NSCLC, which enhances the binding of adenosine triphosphate (ATP) to the EGFR kinase.6−8 Subsequent second- and third-generation irreversible covalent inhibitors overcome T790M resistance by targeting a conserved cysteine (C797).9−11 They are inactivated, however, through a C797S point mutation that converts the nucleophilic cysteine to a serine and blocks covalent binding.12,13 Additionally, patients with activating EGFR mutations who are treated with frontline osimertinib can develop unique secondary mutations such as L858R/C797S or del E746_A750/C797S, which confer resistance.14,15 Unfortunately in NSCLC, up to 40% of patients will also develop brain metastases (BM), and this number is likely to increase as treatment options continue to improve life expectancy for patients with advanced disease.16 As such, BM are a significant risk and result in a poor prognosis for NSCLC patients being treated with poorly central nervous system (CNS) penetrant TKIs.16−20 Multiple studies have revealed the low CNS penetrability of a majority of marketed EGFR TKIs due to active efflux by transporters such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), which are highly enriched at the blood–brain barrier (BBB), and in combination with tight junctions, ultimately exclude up to 98% of all drugs from the CNS.17−20 Thus, future treatment options for NSCLC would benefit from improved CNS disposition, thereby enabling treatment of the local disease and of ensuing BMs. Numerous strategies and guidelines have been proposed to reduce drug efflux and increase CNS permeability, such as the cLogBB optimization approach, that incorporates lipophilicity and total polar surface area (TPSA), or the multiparameter optimization (MPO) method, but frequently a resource-intensive iterative optimization strategy is employed.20−23 Instead, we were drawn to the differences in pKa values between tertiary amines and even weakly basic ones such as N-methylmorpholine (pKa = 7.4)24 and hydroxylamine itself (pKa = 5.9).25 We hypothesized that the bioisosteric replacement of the N-alkylmorpholine unit in 1 by an N-noralkoxy morpholine moiety, effectively converting the morpholine group into a hydroxylamine, would attenuate the basicity and modify the lipophilicity of the compound leading in turn to a reduction in efflux propensity and increased CNS disposition (Figure 1b).
Figure 1.

Overview of selected EGFR inhibitors and bioisosteric strategy used in this study. (A) Chemical structures of select approved EGFR inhibitors and their efflux status against common efflux transporters. (B) Proposed bioisosteric replacement.
The use of nonclassical bioisosteres in modern-day drug design to modify potency and key pharmacokinetic parameters has had a profound impact on how small-molecule lead optimization campaigns are approached.26−28 Underrepresented functional groups are a rich reservoir of nonclassical bioisosteres that enhance small-molecule compound collections by providing access to novel chemical space and conferring unique biological properties.29−31 Despite this, the uptake of an underrepresented functional group in medicinal chemistry is often thwarted by synthetic tractability or by its designation as a “structural alert”.32 Such alerts are a feature of all high-throughput screening campaigns and refer to functional groups that are excluded in the earliest stages of the development process to minimize false positives and inherent toxicities.32,33 While it is true that exclusion of promiscuous functional groups and adherence to established drug-like properties in drug discovery programs has reduced clinical attrition rates, uncritical application of structural alert “rules” across broad functional group classes can preclude the development of structurally innovative lead candidates residing in novel chemical space.34
The presence of a heteroatom-heteroatom bond is one such structural alert, of which the hydroxylamine N–O bond, with its bond dissociation energy (BDE) of 55–65 kcal·mol–1 and reputation for inherent mutagenicity and genotoxicity, is a pertinent example.35−37 This broad moratorium typically excludes trisubstituted hydroxylamines from small-molecule optimization schemes even though they should not harbor the same mutagenic potential as lesser substituted hydroxylamines.38,39 This is because the mutagenicity of less substituted hydroxylamines arises from sulfation or acetylation by cytosolic acetyltransferases and sulfotransferases and subsequent elimination to reactive nitroso compounds.38,39 The related hydroxamates undergo Lossen rearrangement to form reactive isocyanates that can subsequently act as electrophiles for DNA.40,41 In contrast, trisubstituted hydroxylamines are not susceptible to these modes of activation and their consequent toxicity; nevertheless, they are routinely excluded from compound optimization schemes. To our knowledge, there is only one actual small-molecule pharmaceutical containing the trisubstituted hydroxylamine moiety: the tetracycline, Sarecycline.42 To access novel chemical space accessible to trisubstituted hydroxylamines, we have developed synthetic strategies for their assembly by direct N–O bond formation.43−47 Using this method, we conducted a matched molecular pair (MMP) analysis48 to assess the early absorption, distribution, metabolism, and excretion (ADME) parameters of trisubstituted hydroxylamines and found them to exhibit unique properties that include similarities in lipophilicity to morpholine units.47 This suggested that trisubstituted hydroxylamines could serve as a novel bioisosteric modification of nitrogen heterocycles, which have received little attention in bioisosteric design compared to carbocycles,49−52 but which are present in roughly 60% of unique small-molecule drugs.53 Herein, we report on the success of this novel approach to bioisosterism and describe our efforts that culminated in the discovery of compound 9: a promising lead for the treatment of CNS metastases in EGFR+ NSCLC that bears an atypical amine bioisostere, the trisubstituted hydroxylamine, and that lacks the mutagenicity and genotoxicity commonly surmised to accompany these units.
Results and Discussion
We began by evaluating inhibitor-binding constants and biochemical inhibition of the relevant EGFR kinase forms of 1 and a direct hydroxylamine analogue 6; both 1 and 6 exhibited single-digit nanomolar activity against activating mutant bearing EGFR, indicating that an N-(noralkoxy)morpholine unit is indeed an effective bioisostere of the N-alkylmorpholine unit (Tables S1 and S3). We then assessed in vitro ADME properties, beginning with a colon carcinoma (Caco-2) cell permeability assay, which expresses both P-gp and BCRP; 6 displayed a 14-fold enhancement in permeability and greatly decreased efflux compared with 1 (Figure 2b). Substantiating this effect, 6 also showed a roughly 4-fold increase in permeability and decreased efflux compared to 1 in a Madin-Darby canine kidney (MDCK) MDCKII-MDR1 cell permeability assay that is commonly used to mimic the BBB through overexpression of the active efflux transporter P-gp, or MDR1. These results indicated that replacement of the N-alkylmorpholine unit by an N-(noralkoxy)morpholine unit might overcome the known54 poor BBB permeability and active efflux observed with 1. Of note, currently osimertinib (5), a third-generation EGFR TKI, is the only approved EGFR TKI that shows promise in treating BM in EGFR+ NSCLC despite it being a substrate for both P-gp and BCRP.18 We then performed an AMES fluctuation assay both with and without metabolic activation by rat liver S9 (± S9) across 4 Salmonella strains (TA98, TA100, TA1537, and TA1535) and found that neither 1 or 6 were mutagenic.55 Taken in combination with a similar stability profile across multiple species in liver microsomes and plasma as compared to 1, these findings dispelled any early concerns regarding perceived mutagenicity and instability toward oxidative metabolism of the trisubstituted hydroxylamine unit in 6 (see the Supporting Information for details).
Figure 2.

In vitro antiproliferative activity and early absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile of the inhibitors. (A) Chemical structures of the hydroxylamine-bearing EGFR inhibitors used in this study. (B) In vitro ADMET properties of gefitinib and the hydroxylamine-based EGFR inhibitors. Values represent the mean of n = 2 independent replicates unless otherwise stated. Negative AMES results on 9 valid up to 50 μM, after which bacterial cytotoxicity was observed. Aq. Sol., aqueous solubility; LMClint, intrinsic clearance in liver microsomes; HEPClint, intrinsic clearance in hepatocytes; t1/2, half-life; Papp, apparent permeability; MDCK, Madine-Darby-canine kidney; MDR1, multidrug resistance 1 (or P-glycoprotein). Abbreviations: H, human; R, rat; and nd, not determined. (C) 9 displays potent antiproliferative activity in patient-derived non-small-cell lung cancer cell lines HCC827 (EGFR mutation = exon19del), NCI-H3255 (EGFR mutation = L858R), and osimertinib-resistant engineered cell lines (Ba/F3-L858R/C797S; Ba/F3 del E746_A750/C797S) while displaying no activity in NCI-H1975 (EGFR mutation = L858R/T790M) and minimal activity in A431 cell line (overexpressed EGFRwt) (72 h dosing period). For all antiproliferative assays, points indicate mean and error bars indicate standard deviation (SD); n = 3 independent replicates; IC50 values (nM) are reported beside the dose–response curves and represent mean ± standard error of the mean (SEM). IC50 values (nM) are unadjusted for fetal bovine serum (FBS).
We sought to further minimize efflux and improve drug-like properties, particularly the aqueous solubility of 6, by synthesizing additional trisubstituted hydroxylamine-bearing inhibitors (7–9) (vide infra). In view of the importance of hydrogen bond donors (HBD) on efflux transporter substrate recognition, and noting the improved CNS penetrability of EGFR inhibitors AZD3759 and JCN037, 7 was designed with a para- to ortho-fluorine switch to minimize the HBD capability of the adjacent aniline (Figures 2a and 3).56−59 Additionally, 8 and 9 were prepared by exchange of the morpholine unit with an N-methylpiperazine group, all while maintaining the key trisubstituted hydroxylamine moiety, with the intent of improving aqueous solubility and potential in vivo exposure as borne out by subsequent pharmacokinetic (PK) measurements (vide infra) (Figure 2a). All newly prepared analogues maintained single-digit to subnanomolar binding and biochemical inhibition of activating mutant bearing EGFR (Tables S1 and S3). In the Caco-2 cellular assay, 7 and 9, which bear the ortho-fluorine, exhibited remarkably enhanced permeability and reduced efflux ratios compared to 1 and even improved to that seen with 6 (Figure 2b). Notably, 8, which contains a more basic nitrogen heterocycle than 1, 6, and 7 and a para-fluoro substitution pattern on the aniline ring, showed high efflux and low permeability, highlighting the synergistic improvement of modified HBD and reduced pKa on efflux transporter substrate recognition. In the MDCKII-MDR1 cellular assays, 7 and 9 showed excellent permeability and low efflux as compared to 1, 8, and even 6. Importantly, 9 also displayed good stability in human and rat microsomes and hepatocytes while exhibiting markedly improved aqueous solubility compared to both 1 and 6 (Figure 4b).
Figure 3.

Chemical structures of investigational EGFR inhibitors AZD3759 and JCN037. Kp,uu brain (AUC0–7h) values refer to the unbound brain-to-unbound plasma partitioning coefficient.
Figure 4.

Pharmacokinetic profile of 9 and additional in vitro ADMET parameters. (A) Total plasma vs time profile (0–24 h) of 9 after administration into Sprague-Dawley rats at a single dose of 2 mg/kg IV and 20 mg/kg PO and BALB/c nude mice at 3 mg/kg IV and 30 mg/kg PO. AUC0-inf (nM·h), area under concentration–time curve from 0 to ∞; t1/2 (h), mean elimination half-life obtained from either intravenous infusion (IV) or oral gavage (PO); F(%), bioavailability (%); Tmax (h), time to reach peak plasma concentration; Cmax (nM), peak plasma concentration; CL (mL·min–1·kg), clearance obtained from intravenous infusion. For pharmacokinetic profiles, points indicate mean and error bars indicate SD; n = 3 animals per route (n = 6 total). Values represent mean ± SD. (B) Additional in vitro ADMET profile of 9. Values represent the mean of n = ≥2 independent replicates. In vitro micronucleus negative test results valid to 31 μM + rat liver S9 and 8 μM − rat liver S9, after which cytotoxicity was observed. Aq. Sol., aqueous solubility; fu,plasma%, percent fraction unbound in plasma; fu,brain%, percent fraction unbound in brain; HEPClint, intrinsic clearance in hepatocytes. Abbreviations: H, human; R, rat; C, cynomolgus monkey; and D, dog. (C) 9 has low hERG inhibitory potential; points indicate mean, and error bars indicate the SD; n = 3 independent replicates; IC50 values (μM) are reported beside the dose–response curves and represent mean ± SEM. Low DDI is predicted with only minimal inhibition of CYP2D6 observed. Points indicate mean; n = 2 independent replicates. IC50 values (μM) are reported beside the dose–response curves and represent the mean.
We profiled the anticancer activity of the hydroxylamine-bearing inhibitors against four patient-derived cell lines harboring differing EGFR status with cisplatin as the positive control throughout (Figure 2c). Notably, 9 displayed excellent activity against the NCI-H3255 NSCLC cell line bearing the common EGFRL858R mutation, with an IC50 of 7.2 nM representing a roughly 12-fold improvement over 6. Compound 9 also had potent activity against HCC827 NSCLC cells harboring EGFRdel E746_A750 with an IC50 of 3.1 nM. In osimertinib-resistant engineered Ba/F3 cell lines bearing EGFRL858R/C797S and EGFRdel E746_A750/C797S, which block covalent inhibitor binding by a C797S point mutation, like 1,609 exhibited strong activity, with IC50 values of 4.6 and 2.5 nM, respectively.12,13 In the skin-derived A431 cell line bearing overexpressed EGFRwt9 exhibited a comparatively high IC50 of 83 nM, affording 12- to 33-fold selectivity for activating mutant EGFR over wild-type EGFR. Overall, 9 is a potent inhibitor in osimertinib-resistant engineered cell lines and patient-derived NSCLC cell lines NCI-H3255 and HCC827, which harbor mutations in EGFR that encompass approximately 85% of all newly diagnosed mutant EGFR+ NSCLC cases.
Moving forward with 9, we determined the unbound fractions in plasma and brain tissue (Figure 4b).20 With regard to plasma protein binding, 9 exhibited good unbound fractions in plasma and excellent unbound rodent brain tissue fractions (fu,brain% = 4.5 and 4.1) (Table S8). With respect to potential toxicity, only moderate human Ether-à-go-go-Related Gene (hERG) potassium ion channel inhibition by 9 was observed with an IC50 of 6.48 μM and a maximal inhibition of approximately 60% at 10 μM (Figure 4c). We also assayed for CYP inhibition across all major isoforms (3A4, 1A2, 2C9, 2D6, 2C19) and saw none (IC50 = >30 μM) except for moderate inhibition of CYP2D6 (IC50 = 1.1 μM) (Figure 4c). In a follow-up CYP2D6 time-dependent inhibition (TDI) IC50-shift experiment in human liver microsomes and primary human hepatocytes, this latter activity was shown not to be time-dependent, thus dispelling concerns of potential drug–drug interactions (DDI) (Figure 4c).61 In the AMES fluctuation assay across 4 Salmonella strains (TA98, TA100, TA1537, and TA1535), and an in vitro micronucleus test in Chinese hamster ovary (CHO-K1) cells both with and without metabolic activation by rat liver S9 (± S9), 9 exhibited neither mutagenic nor genotoxic potential, contrary to the popular belief that hydroxylamines are inherently mutagenic and genotoxic (Tables S33 and S35).34,35,55,62 Finally, to dispel concerns regarding the perceived metabolic instability of the N–O bond, we conducted an in vitro metabolite identification (MetID) study in both human and rat hepatocytes after 4 h incubation. Gratifyingly, the metabolism of 9 closely followed that of 1,63 with N–O bond cleavage only contributing to minor metabolites identified (<5%) (Figures S1–S2). Overall, these experiments indicate that 9 is a potent inhibitor of activating mutant bearing EGFR but exhibits much reduced efflux compared to those of most currently approved targeted EGFR therapies.
Using KINOMEScan technology, we determined the selectivity of 9 against a panel of >400 human kinases at a concentration of 1 μM; exquisite kinase selectivity was found with an S(10) score of 0.015 (6/403 nonmutant kinases showing ≤10% activity at 1 μM) (Figure 5a; Table S38).64 Apart from EGFRwt (0.30%), 9 only showed high affinity for ERBB2 (HER2) (0.45%). Moderate affinity for ABL1 (7.7%), DRAK1 (8.3%), LYN (7.4%), and PIKFYVE (1.6%) was also observed but confirmed in a follow-up binding experiment to be of minor significance considering the potent binding to EGFR (Kd = <0.2 nM) (Tables S1 and S3). As a result of the moderate affinity for HER2 that was confirmed in a follow-up binding experiment (Kd = 21 nM) and considering the lack of efficient treatments for metastatic HER2+ breast cancer,65 we further profiled 9 against HER2 using additional biochemical and cellular assays (Tables S2 and S4; Figure 5b). We found that 9 exhibited low antiproliferative activity in HER2+ breast cancer cell lines in comparison to the activating mutant bearing EGFR+ cells, with IC50’s ranging from 0.99 to 1.6 μM.
Figure 5.

Kinase selectivity profile for 9 and extended in vitro assays. (A) KINOMEScan nonmutant or lipid kinase screening results of 9 at a screening concentration of 1 μM. The size of circles mapped onto the kinase phylogenetic tree using DiscoverX TREEspot corresponds to strength of binding affinity. Full KINOMEScan results are listed in the Supporting Information. (B) 9 displays moderate antiproliferative activity in patient-derived HER2-positive breast cancer cell lines: AU565, SK-BR-3, and ZR-75–30. For all antiproliferative assays, points indicate mean and error bars indicate SD; n = 3 independent replicates; IC50 values (nM) are reported beside the dose–response curve and represent mean ± SEM. IC50 values (nM) are unadjusted for FBS.
To qualify 9 as a candidate for further studies, we performed a head-to-head pharmacokinetic study with 7 by administration into Sprague–Dawley rats (Figures S3–S4). No adverse events or signs of toxicity were observed for the compound at the tested doses, contrary to the common belief that hydroxylamines are inherently toxic.34,35 As we had hypothesized (vide supra), 9 exhibited excellent exposure (AUCinf = 279 h·ng/mL) and half-life (t1/2 = 2.29 h) compared to 7 (AUCinf = 89 h·ng/mL) (t1/2 = 0.56 h) at 1 mg/kg intravenous (IV) and 5 mg/kg oral (PO) dosing, that we attribute in part to the increased solubility wrought by the piperazine for morpholine exchange (Figure 2a; Figures S3–S4). To further demonstrate the excellent pharmacokinetic properties of 9 we increased the dose to 2 mg/kg IV and 20 mg/kg PO dosing in Sprague-Dawley rats and saw excellent oral bioavailability (F = 64%), exposure (AUCinf = 3907 h·ng/mL) and an acceptable half-life (t1/2 = 2.37 h) with a high volume of distribution (Vss = 6.67 L/kg) (Figure 4a). Furthermore, after administration of a single oral dose of 20 mg/kg in Sprague-Dawley rats, 9 exhibited a Kp,uu (unbound brain-to-unbound plasma partitioning coefficient) (AUCinf) of 0.33 and a Kp (brain-to-plasma partitioning coefficient) (AUCinf) of 0.95, indicating excellent brain penetration (Figure 6a).20,66 Substantiating this effect, after administration of a single oral dose of 40 mg/kg in CD1 mice, a Kp,uu (AUCinf) of 0.45 and a Kp (AUCinf) of 0.77 was obtained (Figure 6a). This stands in contrast, to 1 which is reported18 to have a Kp,uu (AUC0−16h) of 0.0092 after administration of a single oral dose of 10 mg/kg in Han Wistar rats, and underlines the potential of 9 in the treatment of CNS metastatic NSCLC.
Figure 6.

Central nervous system pharmacokinetic profile and intracranial efficacy assessment of 9. (A) Unbound plasma and brain concentration vs time profile (0–24 h) of 9 after administration into Sprague-Dawley rats at a single dose of 20 mg/kg and CD1 mice at 40 mg/kg by oral gavage (PO) shows excellent unbound brain penetration. Unbound concentrations were calculated by multiplying total concentrations at a given time point by the unbound fraction values in plasma or brain. Kp brain (AUCinf) refers to the brain-to-plasma partitioning coefficient. Kp,uu brain (AUCinf) refers to the unbound brain-to-unbound plasma partitioning coefficient. For pharmacokinetic profiles, points indicate mean and error bars indicate SD; n = 3 animals at each time point (n = 21 total). (B) Bioluminescence images of vehicle control and 9 (10 mg/kg PO b.i.d.) dosing groups indicating a change in tumor volume over 21 days of treatment. (C) Intracranial bioluminescence over time. Points indicate mean, and error bars indicate the SEM; P = 0.0059; n = 10 mice per group. PDX, patient-derived xenograft. p value was obtained from an unpaired two-tailed t-test comparing the means of vehicle control and 9 (10 mg/kg PO b.i.d.) study arms after 21 days of treatment. *P < 0.05; **P < 0.01. (D) Mean body weight of mice during the efficacy study. Points indicate mean, and error bars indicate the SEM; P = 0.2275; n = 10 mice per group. P value was obtained from an unpaired two-tailed t-test comparing the means of vehicle control and 9 (10 mg/kg PO b.i.d.) study arms after 21 days of treatment. ns, not significant.
Finally, to assess the viability of 9 as a potential treatment for BM in EGFR+ NSCLC, we performed an intracranial patient-derived xenograft (PDX) model with luciferase-tagged HCC827 cells implanted into the brains of BALB/c nude mice (Figure 6b). At 10 mg/kg PO b.i.d. (twice daily)67 dosing, profound tumor regression (P = 0.0059) relative to vehicle control (1% methylcellulose) was observed, confirming that 9 has intracranial antitumor activity (Figure 6c). Additionally, over the 21-day treatment window, mean body weight loss never exceeded 10% and no adverse clinical events were observed at the studied dose, thereby demonstrating the potential of 9 as a candidate for CNS metastatic EGFR+ NSCLC (Figure 6d).
Chemistry
The synthesis of all inhibitors commenced with a modified version of our N–O bond-forming reaction as the key step (Figure 7a).43,68 To this end, the 2-methyltetrahydropyranyl (MTHP) monoperoxyacetal69−71 (11) derived from commercially available 2-((tert-butyldimethylsilyl)oxy)ethanol (10) was exposed to a morpholine (12) or N-methylpiperazine (13) derived magnesium amide affording the hydroxylamines (14–15) in 65–67% yields, respectively, on multigram scales. Notably, we discovered that the use of Knochel’s68 turbo-Grignard (iPrMgCl·LiCl) as base proved optimal for N–O bond formation on a larger scale (Figure S6; see the Supporting Information for details). Deprotection of the tert-butyldimethylsilyl groups in 14 and 15 afforded alcohols (16 and 17) in 71–79% yields, that on conversion to the chloride by reaction with thionyl chloride and subsequent displacement with commercially available phenols (18–19) under basic conditions, afforded inhibitors 6–9 in 20–35% yields over 2 steps. A crystal structure was also obtained to support the structure of the direct hydroxylamine analogue derivative (6) (see the Supporting Information for details).
Figure 7.
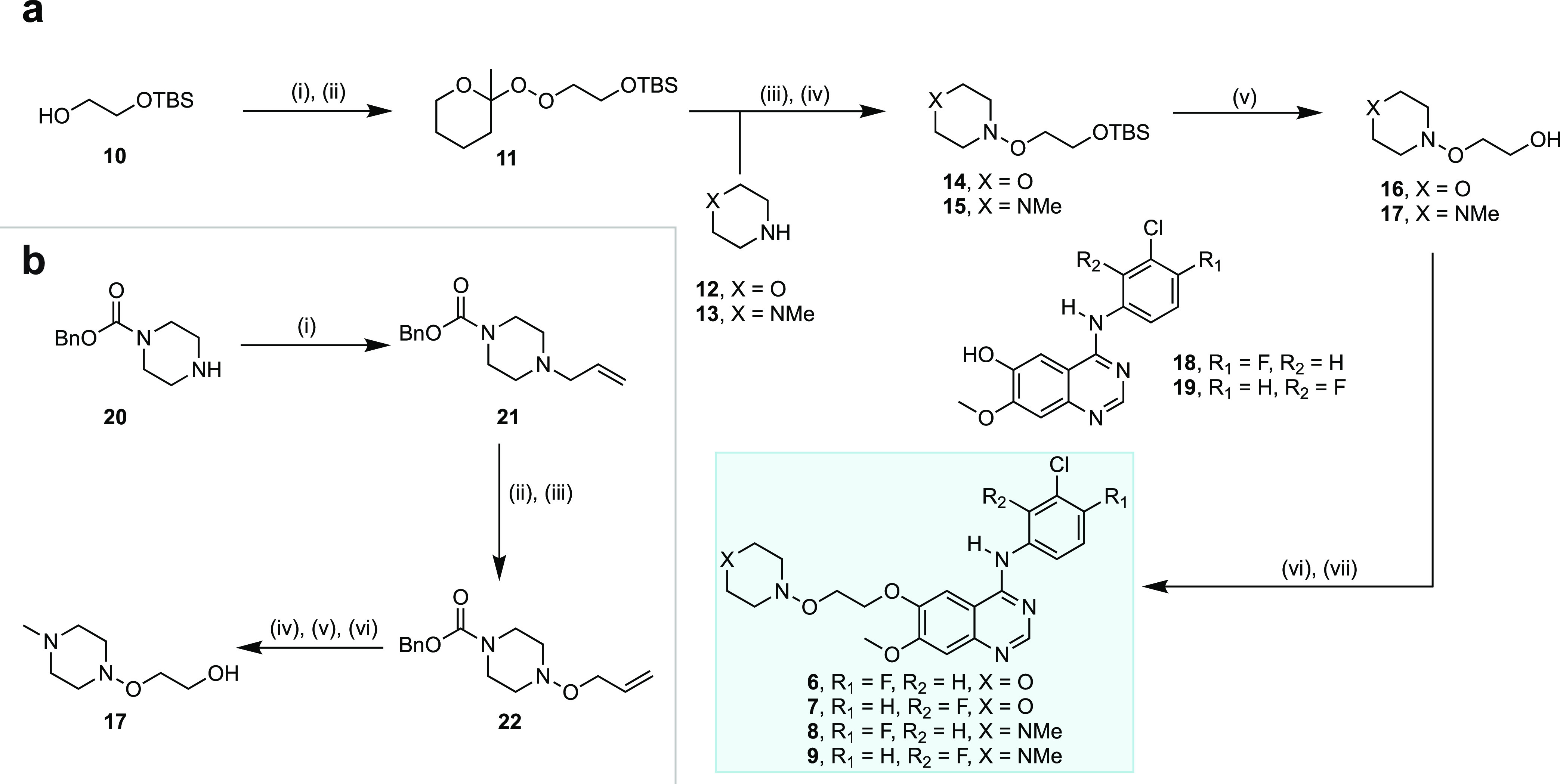
Chemical synthesis of hydroxylamine-bearing EGFR inhibitors and precursors. (A) Chemical synthesis of inhibitors 6–9. Reagents and conditions: (i) Triflic anhydride (Tf2O), pyridine, dichloromethane (DCM), 0 °C, 45 min; (ii) 2-methyltetrahydropyranyl hydroperoxide (MTHPOOH), potassium tert-butoxide (KOtBu), tetrahydrofuran (THF), 0 °C to r.t., 90 min; (iii) 12 (morpholine) or 13 (N-methylpiperazine), iPrMgCl·LiCl (isopropylmagnesium chloride lithium chloride), THF, 0 °C to r.t., 45 min; (iv) 11, THF, r.t., 3 h; (v) tetra-N-butylammonium fluoride (TBAF), THF, 0 °C to r.t., 1−1.5 h; (vi) thionyl chloride (SOCl2), toluene, r.t. to 60 °C, 3 h; (vii) 18 or 19, sodium hydride (NaH, 60% dispersion in mineral oil), THF, 0–80 °C, 16 h. (B) Alternative scaled synthesis of the key hydroxylamine precursor (17). Reagents and conditions: (i) Allyl bromide, potassium carbonate (K2CO3), THF, r.t. to 65 °C, 16 h; (ii) meta-chloroperoxybenzoic acid (MCPBA), DCM, −30 °C, 1 h; (iii) toluene, 80 °C, 14 h; (iv) O3, DCM/MeOH, −78 °C, 30 min; (v) sodium borohyride (NaBH4), DCM/MeOH, −78 °C to r.t., 2 h; (vi) lithium aluminum hydride (LiAlH4), THF, 0 °C to r.t., 2 h.
While we have encountered no issues with our direct N–O bond-forming reaction43 up to the decagram scale, an alternative scalable approach to the key N-methylpiperazine-derived hydroxylamine precursor (17) was also developed (Figure 7b). Thus, the allylation of commercially available 20 under basic conditions gave the N-allyl derivative (21) in 72% yield. Multigram scale quantities of 22 were then obtained in 47% yield by N-oxidation of 21 with meta-chloroperoxybenzoic acid (MCPBA) followed by [2,3]-Meisenheimer rearrangement.72,73 Compound 22 was then subjected to ozonolysis followed by reductive workup with sodium borohydride (NaBH4), after which, the carboxybenzyl group was reduced with lithium aluminum hydride74 (LiAlH4) to give the intended hydroxylamine precursor 17 in 50% yield over 3 steps. This alternative route to 17 proceeds in 17% overall yield and the longest linear sequence of only 3 steps from commercially available benzyl 1-piperazinecarboxylate (20).
Conclusions
A highly selective, orally bioavailable, brain-penetrant EGFR inhibitor, 9, bearing a novel amine bioisostere, a trisubstituted hydroxylamine, has been reported. In contrast to the common expectation for hydroxylamines in medicinal chemistry, 9 lacks mutagenic or genotoxic potential and exhibits good stability in vitro and in vivo. An intracranial PDX murine model revealed profound tumor regression on oral dosing with 9 suggesting this novel compound as a potential lead in the treatment of localized and CNS metastatic NSCLC driven by activating mutant bearing EGFR and for osimertinib-resistant EGFR+ NSCLC bearing the C797S resistance mutation profile (L858R/C797S and exon19del/C797S). All told, our results show that the trisubstituted hydroxylamine moiety, a perceived “structural alert” in medicinal chemistry, can be incorporated into drug scaffolds to improve drug properties while maintaining potent biological activity and avoiding molecular weight creep. These findings support the broader application of trisubstituted hydroxylamines as bioisosteres in drug discovery programs for lead optimization and in patent life-cycle management.
Experimental Section
Chemistry
General Experiment and Information
All reactions were conducted in single-neck oven-dried glassware fitted with a rubber septum under an argon atmosphere, unless otherwise stated. All organic solutions were concentrated under reduced pressure on a rotary evaporator and a water bath. Flash column chromatography was performed using silica gel (Fischer Silica Gel Sorbent (230–400 Mesh, grade 60)).75 Thin-layer chromatography (TLC) was carried out with 250 μM glass back silica (XHL) plates with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet light (UV) and/or submersion in ceric ammonium molybdate (CAM) in ethanol, followed by heating on a hot plate (120 °C, 10–15 s). Solvents were purchased from Sigma-Aldrich and used without further purification. Starting material 2-hydroperoxy-2-methyltetrahydro-2H-pyran (MTHPOOH) was prepared according to literature procedures43,69,70 and spectral data were in accord with that previously reported. 2-(tert-Butyldimethylsilyloxy)-ethan-1-ol was purchased from AK Scientific. Morpholine was purchased from Sigma-Aldrich. N-Methylpiperazine was purchased from Sigma-Aldrich. Isopropylmagnesium chloride lithium chloride complex solution (1.3 M in THF) was purchased from Sigma-Aldrich. Ethylmagnesium bromide (3 M in THF) was purchased from Sigma-Aldrich. Tetrabutylammonium fluoride (1 M in THF) was purchased from TCI. Thionyl chloride was purchased from Alfa Aesar. Sodium Hydride (60% dispersion in mineral oil) was purchased from Sigma-Aldrich. Pyridine was purchased from OakWood Chemicals. 4-(3-Chloro-4-fluorophenylamino)-7-methoxyquinazolin-6-ol was purchased from AK Scientific. 4-(3-Chloro-2-fluorophenylamino)-7-methoxyquinazolin-6-ol was purchased from AmBeed. Gefitinib (1) (≥98% (HPLC), SML1657–50MG) was purchased from Sigma-Aldrich for in vitro assays. Nuclear magnetic resonance (NMR) spectra of all compounds were obtained in either CDCl3 (δH 7.26 and δC 77.16 ppm, respectively) or C6D6 (δH 7.16 and δC 128.06 ppm, respectively), Toluene-D8 (δH 7.09, 6.98, 7.00, 2.09 and δC 137.86, 129.24, 128.33, 125.49, 20.40 ppm, respectively), or DMSO–D6 (δH 2.50 and δC 39.52 ppm, respectively) using a 500 MHz, EZC500 JEOL instrument at 298 K unless otherwise specified. The chemical shifts (δ) are calculated with respect to residual solvent peak and are given in ppm. Multiplicities are abbreviated as follows: s (singlet), m (multiplet), b (broad), d (doublet), t (triplet), q (quartet), and hept (heptet). High-resolution mass spectra were obtained on a ThermoFisher Orbitrap Q-Exactive instrument using electrospray ionization (ESI). Melting point of solids 6, 7, 8, and 9 were determined on a Barstead Electrothermal 9100. Ultra high-performance liquid chromatography (UHPLC) traces of compounds 6, 7, 8, and 9 were obtained using a ThermoFisher Vanguish UHPLC with PDA detector and an Acclaim 120 18C 4.6 mm × 50 mm column, and the %purity was determined using the Avalon peak area algorithm. Purities of all final compounds were confirmed to be >95% by UHPLC. UHPLC conditions for 6, 7, 8, and 9 are given in the catalogue of spectra section on pages S109–S113 in the Supporting Information. The purity of commercial gefitinib (1) was confirmed by NMR and high-resolution mass spectrometry (HRMS) prior to in vitro study initiation.
tert-Butyldimethyl(2-((2-methyltetrahydro-2H-pyran-2-yl)peroxy)ethoxy)silane (11)
To a stirred solution of 2-(tert-butyldimethylsilyloxy)-ethan-1-ol (10) (13.33 g, 75.71 mmol, 1.0 equiv) in anhydrous DCM (250 mL) at 0 °C was added pyridine (9.66 mL, 113.57 mmol, 1.5 equiv) followed by dropwise addition of Tf2O (15.26 mL, 90.85 mmol, 1.2 equiv), and the solution was stirred for 45 min at 0 °C. After such time, the mixture was diluted with DCM (100 mL) and washed successively with 1 N HCl (1×, 150 mL), aq. NaHCO3 (1×, 150 mL), and brine (1×, 150 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The crude triflate was used directly in the next step without further purification. To a stirred solution of MTHPOOH43,69,70 (12 g, 90.85 mmol, 1.2 equiv) in anhydrous THF (260 mL) at 0 °C was added KOtBu (10.19 g, 90.85 mmol, 1.2 equiv), and the solution was stirred for 30 min, after which the crude triflate obtained in the previous step was added dropwise in anhydrous THF (40 mL). The solution was warmed to rt and stirred for 90 min. After such time, the mixture was diluted with EtOAc (100 mL) and quenched via the addition of aq. NaHCO3 (1×, 200 mL). The layers were separated, and the aqueous layer was extracted with EtOAc (2×, 100 mL). The combined organic layers were washed with brine (1×, 100 mL), dried over Na2SO4, filtered and concentrated in vacuo. The residue obtained was purified by flash column chromatography on silica (eluent: 10:90 EtOAc/Hexanes) to afford the title compound 11 (11.19 g, 38.56 mmol, 51%) as an off-yellow oil. Spectral data are in accord with that previously reported in the literature.47 TLC Rf = 0.50 (20:80 EtOAc/Hexanes; CAM). 1H NMR (500 MHz, CDCl3) δ 4.13–4.07 (m, 2H), 3.92 (td, J = 11.4, 2.8 Hz, 1H), 3.85 (t, J = 5.4 Hz, 2H), 3.71–3.68 (m, 1H), 1.79–1.50 (m, 6H), 1.43 (s, 3H), 0.90 (s, 9H), 0.08 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 102.5, 76.7, 61.8, 60.8, 33.3, 26.0, 24.9, 24.6, 19.2, 18.5, −5.2. CAUTION: While we have not encountered any decomposition, standard caution should be exercised when working with peroxides (i.e., avoid exposure to light, reducing agents, excessive heat, and work behind a protective shield).
4-(2-((tert-Butyldimethylsilyl)oxy)ethoxy)morpholine (14)
To a stirred solution of morpholine (12) (7.64 mL, 87.33 mmol, 3.0 equiv) in anhydrous THF (73 mL) at 0 °C was added iPrMgCl·LiCl (1.3 M in THF) (56 mL, 72.78 mmol, 2.5 equiv) and the solution was brought to r.t. and stirred for 45 min. After such time, 11 (8.45 g, 29.11 mmol, 1.0 equiv) was added dropwise in anhydrous THF (73 mL) and the solution was stirred for 3 h. After this time, the solution was quenched with aq. NaHCO3 (100 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3×, 75 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was purified by flash column chromatography on silica (eluent: 15:85 EtOAc/Hexanes) to afford the title compound 14 (4.95 g, 18.95 mmol, 65%) as a light yellow oil. TLC Rf = 0.20 (10:90 EtOAc/Hexanes; CAM). 1H NMR (500 MHz, CDCl3) δ 3.88 (d, J = 11.8 Hz, 2H), 3.79–3.72 (m, 4H), 3.62–3.54 (m, 2H), 3.15 (d, J = 10.6 Hz, 2H), 2.66 (td, J = 10.9, 3.3 Hz, 2H), 0.89 (s, 9H), 0.06 (s, 6H). 13C NMR (126 MHz, CDCl3) δ: 73.1, 66.4, 61.6, 56.4, 26.0, 18.5, −5.1. HRMS-ESI (m/z): [M + H]+ calculated for [C12H28O3NSi]+: 262.1833, found: 262.1835. See Figure S6 for the reaction setup.
1-(2-((tert-Butyldimethylsilyl)oxy)ethoxy)-4-methylpiperazine (15)
To a stirred solution of N-methylpiperazine (13) (12.62 mL, 113.73 mmol, 3.0 equiv) in anhydrous THF (95 mL) at 0 °C was added iPrMgCl·LiCl (1.3 M in THF) (72.9 mL, 94.78 mmol, 2.5 equiv) and the solution was brought to r.t. and stirred for 45 min. After such time, 11 (11 g, 37.91 mmol, 1.0 equiv) was added dropwise in anhydrous THF (95 mL) and the solution was stirred for 3 h. After this time, the solution was quenched with aq. NaHCO3 (75 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3×, 50 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was purified by flash column chromatography on silica (eluent: 40:55:5 Hexanes/EtOAc/Et3N) to afford the title compound 15 (6.99 g, 25.5 mmol, 67%) as a yellow oil. TLC Rf = 0.30 (40:55:5 Hexanes/EtOAc/Et3N; CAM). 1H NMR (500 MHz, C6D6) δ 3.83 (t, J = 5.2 Hz, 2H), 3.75 (t, J = 5.5 Hz, 2H), 3.15–3.13 (m, 2H), 2.84 (brs, 2H), 2.49–2.46 (m, 2H), 2.10–2.06 (m, 2H), 2.02 (s, 3H), 0.99 (s, 9H), 0.09 (s, 6H). 13C NMR (126 MHz, C6D6) δ: 73.2, 62.0, 55.9, 54.4, 45.6, 26.2, 18.6, −5.1. HRMS-ESI (m/z): [M + H]+ calculated for [C13H31O2N2Si]+: 275.2149, found: 275.2146. See Figure S8 for the reaction setup.
2-(Mopholinooxy)ethan-1-ol (16)
To a stirred solution of 14 (4.90 g, 18.80 mmol, 1.0 equiv) in anhydrous THF (150 mL) at 0 °C was added TBAF (1 M in THF) (37.60 mL, 37.60 mmol, 2.0 equiv) dropwise, and the solution was brought to r.t. and stirred for 1 h. After such a time, the mixture was concentrated in vacuo and the residue obtained was purified by flash column chromatography (eluent: 95:5 EtOAc/Et3N) to afford the title compound 16 (2.17 g, 14.75 mmol, 79%) as a yellow oil. TLC Rf = 0.50 (95:5 EtOAc/Et3N; CAM). 1H NMR (500 MHz, CDCl3) δ 3.91 (d, J = 12.3 Hz, 2H), 3.86–3.79 (m, 4H), 3.56 (t, J = 12.6 Hz, 2H), 3.34 (br s, 1H), 3.21 (d, J = 11.9 Hz, 2H), 2.67 (td, J = 10.9, 3.3 Hz, 2H). 13 C NMR (126 MHz, CDCl3) δ: 71.6, 66.3, 63.6, 56.2. HRMS-ESI (m/z): [M + H]+ calculated for [C6H14O3N]+: 148.0968, found: 148.0964.
2-((4-Methylpiperazin-1-yl)oxy)ethan-1-ol (17)
To a stirred solution of 15 (6.35 g, 23.16 mmol, 1.0 equiv) in anhydrous THF (300 mL) at 0 °C was added TBAF (1 M in THF) (46.32 mL, 46.32 mmol, 2.0 equiv) dropwise, and the solution was brought to rt and stirred for 90 min. After such time, the mixture was concentrated in vacuo and the residue obtained was purified by flash column chromatography (eluent: 90:10 EtOAc/Et3N) to afford the title compound 17 (2.18 g, 13.61 mmol, 71%) as a yellow oil. TLC Rf = 0.10 (90:10 EtOAc/Et3N; CAM).1H NMR (500 MHz, Toluene-D8) δ 3.69–3.66 (m, 2H), 3.65–3.60 (m, 2H), 3.01 (br d, J = 10.3 Hz, 2H), 2.73–2.59 (m, 2H), 2.41 (br d, J = 11.3 Hz, 2H), 1.96 (s, 3H), 1.94–1.90 (m, 2H).13 C NMR (126 MHz, Toluene-D8) δ 72.2, 63.2, 55.4, 54.2, 45.3. HRMS-ESI (m/z): [M + H]+ calculated for [C7H17O2N2]+: 161.1285, found: 161.1282.
N-(3-Chloro-4-fluorophenyl)-7-methoxy-6-(2-(morpholinooxy)ethoxy)quinazolin-4-amine (6)
To a stirred solution of 16 (1 g, 6.80 mmol, 1.0 equiv) in anhydrous toluene (20 mL) at 0 °C was added SOCl2 (1.23 mL, 17.00 mmol, 2.5 equiv) dropwise. The reaction mixture was stirred until no longer exothermic and then brought to 60 °C and stirred for 3 h. After such time, the mixture was concentrated in vacuo and the resulting residue was used directly in the next step without further purification. To a stirred solution of 18 (3.26 g, 10.20 mmol, 1.5 equiv) in anhydrous N,N-dimethylformamide (DMF, 35 mL) at 0 °C was added NaH (60% dispersion in mineral oil) (406 mg, 10.20 mmol, 1.5 equiv) slowly, and the solution was warmed to r.t. and stirred for 45 min. Following this, the crude chloride obtained in the previous step was added dropwise in DMF (5 mL) and the solution was brought to 80 °C and stirred for 16 h. After such time the mixture was concentrated in vacuo and coconcentrated with toluene (3×, 50 mL). The resulting residue was then redissolved in EtOAc (200 mL) and washed successively with 1 M NaOH (2×, 100 mL), brine (2×, 100 mL), then the combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The residue obtained was purified by flash column chromatography (eluent: 10:90 Et3N/EtOAc) to afford the title compound 6 (1.08 g, 2.41 mmol, 35% over 2 steps) as a light yellow powder. TLC Rf = 0.50 (10:90 Et3N/EtOAc; UV, CAM). 1H NMR (500 MHz, DMSO–D6) δ 9.51 (s, 1H), 8.50 (s, 1H), 8.12 (dd, J = 6.8, 2.7 Hz, 1H), 7.83–7.78 (m, 2H), 7.44 (t, J = 9.1 Hz, 1H), 7.20 (s, 1H), 4.29 (t, J = 4.5 Hz, 2H), 4.08 (t, J = 4.5 Hz, 2H), 3.94 (s, 3H), 3.81 (d, J = 11.6 Hz, 2H), 3.44 (t, J = 11.4 Hz, 2H) 3.18 (d, J = 10.4 Hz, 2H), 2.54 (d, J = 11.8 Hz, 2H). 13C NMR (126 MHz, DMSO–D6) δ 156.0, 154.5, 153.1 (d, 1JC–F = 243.2 Hz), 152.7, 148.2, 147.0, 136.8 (d, 3JC–F = 3.8 Hz), 123.4, 122.2 (d, 3JC–F = 6.3 Hz), 118.8 (d, 2JC–F = 18.9 Hz), 116.5 (d, 2JC–F = 21.4 Hz), 108.7, 107.4, 102.7, 69.0, 67.4, 65.5, 56.2, 55.8. 13C NMR {19F} (126 MHz, DMSO–D6) δ 156.0, 154.5, 153.1, 152.7, 148.2, 147.0, 136.8, 123.4, 122.2, 118.8, 116.5, 108.7, 107.4, 102.7, 69.0, 67.4, 65.5, 56.2, 55.8.19F {1H} (470 MHz, DMSO–D6) δ −123.2. HRMS-ESI (m/z) [M + H]+ calculated for [C21H23O4N4ClF]+: 449.1386, found: 449.1378. Mp 186.4–187.5 °C (mean of n = 3 determinations).
N-(3-Chloro-2-fluorophenyl)-7-methoxy-6-(2-(morpholinooxy)ethoxy)quinazolin-4-amine (7)
To a stirred solution of 16 (1.90 g, 12.9 mmol, 1.0 equiv) in anhydrous toluene (40 mL) at 0 °C was added SOCl2 (3.84 mL, 32.25 mmol, 2.5 equiv) dropwise. The reaction mixture was stirred until no longer exothermic and then brought to 60 °C and stirred for 3 h. After such time, the mixture was concentrated in vacuo and the resulting residue was used directly in the next step without further purification.
To a stirred solution of 19 (6.19 g, 19.35 mmol, 1.5 equiv) in anhydrous DMF (70 mL) at 0 °C was added NaH (60% dispersion in mineral oil) (770 mg, 19.35 mmol, 1.5 equiv) slowly, and the solution was warmed to rt and stirred for 45 min. Following this, the crude chloride obtained in the previous step was added dropwise in DMF (10 mL) and the solution was brought to 80 °C and stirred for 16 h. After such time the mixture was concentrated in vacuo and coconcentrated with toluene (3×, 75 mL). The resulting residue was then redissolved in EtOAc (150 mL) and washed successively with 1 M NaOH (2×, 75 mL), brine (2×, 75 mL), then the combined organic layers were dried over Na2SO4, filtered and concentrated in vacuo. The residue obtained was purified by flash column chromatography (eluent: 10:90 Et3N/EtOAc) to afford the title compound 7 (1.67 g, 3.73 mmol, 29% over 2 steps) as an off-white powder. TLC Rf = 0.50 (10:90 Et3N/EtOAc; UV, CAM). 1H NMR (500 MHz, DMSO–D6) δ 9.61 (s, 1H), 8.38 (s, 1H), 7.82 (s, 1H), 7.54–7.52 (m, 1H), 7.49–7.46 (m, 1H), 7.28 (t, J = 8.3 Hz, 1H), 7.21 (s, 1H), 4.28 (t, J = 4.6 Hz, 2H), 4.07 (t, J = 4.6 Hz, 2H), 3.94 (s, 3H), 3.81 (d, J = 12.0 Hz, 2H), 3.44 (t, J = 11.3 Hz, 2H), 3.17 (d, J = 10.4 Hz, 2H), 2.55–2.52 (m, 2H). 13C NMR (126 MHz, DMSO–D6) δ 156.9, 154.6, 153.0, 152.4 (d, 1JC–F = 249.5 Hz), 148.2, 147.0, 128.4 (d, 2JC–F = 12.6 Hz), 127.1, 126.9, 124.9 (d, 3JC–F = 5.0 Hz), 120.2 (d, 2JC–F = 16.4 Hz), 108.6, 107.2, 102.8, 69.0, 67.3, 65.5, 56.2, 55.9. 13C NMR {19F} (126 MHz, DMSO–D6) δ: 156.9, 154.6, 153.0, 152.4, 148.2, 147.0, 128.4, 127.1, 126.9, 124.9, 120.2, 108.6, 107.2, 102.8, 69.0, 67.3, 65.5, 56.2, 55.9. 19F {1H} (470 MHz, DMSO–D6) δ −120.4. HRMS-ESI (m/z): [M + H]+ calculated for [C21H23O4N4ClF]+: 449.1386, found:449.1376. Mp 141.3–142.9 °C (mean of n = 3 determinations).
N-(3-Chloro-4-fluorophenyl)-7-methoxy-6-(2-((4-methylpiperazin-1-yl)oxy)ethoxy)quinazolin-4-amine (8)
To a stirred solution of 17 (300 mg, 1.87 mmol, 1.0 equiv) in anhydrous toluene (7 mL) at 0 °C was added SOCl2 (340 μL, 4.68 mmol, 2.5 equiv) dropwise. The reaction mixture was stirred until no longer exothermic and then brought to 60 °C and stirred for 3 h. After such time, the mixture was concentrated in vacuo, and the resulting residue was dissolved in EtOAc (20 mL) and washed with an aq. K2CO3 solution (2×, 15 mL). The combined organic layers were then dried over Na2SO4, filtered, and concentrated. The residue obtained was used without further purification.
To a stirred solution of 18 (898 mg, 2.81 mmol, 1.5 equiv) in anhydrous DMF (10 mL) at 0 °C was added NaH (60% dispersion in mineral oil) (112 mg, 2.81 mmol, 1.5 equiv) slowly, and the solution was warmed to r.t. and stirred for 45 min. Following this, the crude chloride obtained in the previous step was added dropwise in DMF (5 mL) and the solution was brought to 80 °C and stirred for 16 h. After such time, the mixture was concentrated in vacuo and coconcentrated with toluene (3×, 20 mL). The resulting residue was then redissolved in EtOAc (30 mL) and washed successively with 1 M NaOH (2×, 20 mL), brine (2×, 20 mL), then the combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was purified by flash column chromatography (eluent: 85:10:5 EtOAc/MeOH/Et3N) to afford the title compound 8 (286 mg, 0.687 mmol, 33% over 2 steps) as a light orange powder. TLC Rf = 0.40 (85:10:5 EtOAc/MeOH/Et3N; UV, CAM). 1H NMR (500 MHz, DMSO–D6) δ 9.53 (s, 1H), 8.50 (s, 1H), 8.12 (dd, J = 6.8, 2.6 Hz, 1H), 7.84–7.78 (m, 2H), 7.44 (t, J = 9.1 Hz, 1H), 7.21 (s, 1H), 4.28 (t, J = 5.0 Hz, 2H), 4.04 (t, J = 5.0 Hz, 2H), 3.94 (s, 3H), 3.15–3.13 (m, 2H), 2.70–2.67 (m, 2H), 2.57 (br s, 2H), 2.13–2.09 (m, 5H). 13C NMR (126 MHz, DMSO–D6) δ 156.0, 154.5, 153.1 (d, 1JC–F = 243.2 Hz), 152.7, 148.2, 147.0, 136.8, 126, 123.4, 122.2 (d, 3JC–F = 6.3 Hz), 118.8 (d, 2JC–F = 18.9 Hz), 116.5 (d, 2JC–F = 21.4 Hz), 108.7, 107.4, 102.7, 69.1, 67.4, 55.9, 55.0, 53.5, 45.0. 13C NMR {19F} (126 MHz, DMSO–D6) δ: 156.0, 154.5, 153.1, 152.7, 148.2, 147.0, 136.8, 123.4, 122.2, 118.8, 116.5, 108.7, 107.4, 102.7, 69.0, 67.4, 55.9, 55.0, 53.5, 45.0. 19F {1H} (470 MHz, DMSO–D6): δ −123.2. HRMS-ESI (m/z): [M + H]+ calculated for [C22H26O3N5ClF]+: 462.1703, found: 462.1691. Mp 64.0–66.5 °C (mean of n = 3 determinations).
N-(3-Chloro-2-fluorophenyl)-7-methoxy-6-(2-((4-methylpiperazin-1-yl)oxy)ethoxy)quinazolin-4-amine (9)
To a stirred solution of 17 (2.10 g, 13.12 mmol, 1.0 equiv) in anhydrous toluene (50 mL) at 0 °C was added SOCl2 (2.38, mL, 32.80 mmol, 2.5 equiv) dropwise. The reaction mixture was stirred until no longer exothermic, and then brought to 60 °C and stirred for 3 h. After such time, the mixture was concentrated in vacuo, and the resulting residue was dissolved in EtOAc (100 mL) and washed with an aq. K2CO3 solution (2×, 75 mL). The combined organic layers were then dried over Na2SO4, filtered, and concentrated. The residue obtained was used without further purification.
To a stirred solution of 19 (6.29 g, 19.68 mmol, 1.5 equiv) in anhydrous DMF (90 mL) at 0 °C was added NaH (60% dispersion in mineral oil) (783 mg, 19.68 mmol, 1.5 equiv) slowly, and the solution was warmed to rt and stirred for 45 min. Following this, the crude chloride obtained in the previous step was added dropwise in DMF (10 mL) and the solution was brought to 80 °C and stirred for 16 h. After such time the mixture was concentrated in vacuo and coconcentrated with toluene (3×, 25 mL). The resulting residue was then redissolved in EtOAc (150 mL) and washed successively with 1 M NaOH (2×, 100 mL), brine (2×, 100 mL), then the combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was purified by flash column chromatography (eluent: 85:10:5 EtOAc/Et3N/MeOH) to afford the title compound 9 (1.19 g, 2.58 mmol, 20% over 2 steps) as a light yellow powder. TLC Rf = 0.30 (85:10:5 EtOAc/MeOH/Et3N; UV, CAM). 1H NMR (500 MHz, DMSO–D6) δ 9.61 (s, 1H), 8.39 (s, 1H), 7.83 (s, 1H), 7.57–7.51 (m, 1H), 7.48–7.46 (m, 1H), 7.29–7.26 (m, 1H), 7.21 (s, 1H), 4.27 (t, J = 4.6 Hz, 2H), 4.03 (t, J = 4.6 Hz, 2H), 3.94 (s, 3H), 3.15–3.12 (m, 2H), 2.69–2.67 (m, 2H), 2.57 (br s, 2H), 2.13–2.06 (m, 5H). 13C NMR (126 MHz, DMSO–D6) δ 156.9, 154.6, 153.0, 152.4 (d, 1JC–F = 249.5 Hz), 148.2, 147.0, 128.4 (d, 2JC–F = 11.3 Hz), 127.1, 126.8, 124.9 (d, 3JC–F = 5.0 Hz), 120.2 (d, 2JC–F = 16.4 Hz), 108.6, 107.2, 102.8, 69.0, 67.2, 55.9, 55.0, 53.6, 45.0. 13C NMR {19F} (126 MHz, DMSO–D6) δ 156.9, 154.6, 153.0, 152.4, 148.2, 147.0, 128.4, 127.1, 126.8, 124.9, 120.1, 108.6, 107.2, 102.8, 69.0, 67.2, 55.9, 55.0, 53.6, 45.0.19F {1H} (470 MHz, DMSO–D6) δ −120.4. HRMS-ESI (m/z): [M + H]+ calculated for [C22H26O3N5ClF]+: 462.1703, found: 462.1695. Mp 143.5–144.9 °C (mean of n = 3 determinations).
Benzyl 4-Allylpiperazine-1-carboxylate (21)
To a stirred solution of benzyl piperazine-1-carboxylate (20) (15 g, 68.14 mmol, 1.0 equiv) in anhydrous THF (250 mL) at r.t. was added K2CO3 (18.8 g, 136.28 mmol, 2.0 equiv) followed by slow addition of allyl bromide (11.8 mL, 136.28 mmol, 2.0 equiv). The solution was brought to 65 °C and stirred for 16 h, after which time the mixture was cooled to rt and quenched via the addition of 1 M NaOH (200 mL). The layers were separated and the aqueous layer was extracted with EtOAc (3×, 100 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was purified by flash column chromatography on silica (eluent: 50:45:5 Hexanes/EtOAc/Et3N) to afford the title compound 21 (12.81 g, 49.2 mmol, 72%) as a yellow oil. TLC Rf = 0.60 (50:45:5 Hexanes/EtOAc/Et3N; CAM, UV). 1H NMR (500 MHz, CDCl3) δ 7.37–7.29 (m, 5H), 5.89–5.78 (m, 1H), 5.23–5.14 (m, 2H), 5.13 (br s, 2H), 3.56 (m, 4H), 3.00 (d, J = 6.6 Hz, 2H), 2.40 (br s, 4H). 13C NMR (126 MHz, CDCl3) δ: 155.4, 136.9, 134.7, 128.6, 128.1, 128.0, 118.5, 67.2, 61.8, 52.8, 43.9. HRMS-ESI (m/z) [M + H]+ calculated for [C15H21N2O2]+: 261.1598, found: 261.1594.
Benzyl 4-(Allyoxy)piperazine-1-carboxylate (22)
To a stirred solution of 21 (12.2 g, 46.9 mmol, 1.0 equiv) in anhydrous DCM at −30 °C was added MCPBA (70% mixture with 3-chlorobenzoic acid and water) (11.56 g, 46.9 mmol, 1.0 equiv) portion-wise for 5 min. After full addition, the reaction mixture was stirred at −30 °C for 1 h, then was quenched with aq. NaHCO3 (150 mL), and the layers were separated. The organic layer was washed with NaHCO3 (3×, 150 mL) after which the organic layer was dried over Na2SO4, filtered, and concentrated in vacuo. The residue obtained was dissolved in toluene and heated to 80 °C with stirring for 14 h. After such time, the mixture was cooled to r.t. and concentrated in vacuo. The residue obtained was purified by flash column chromatography on silica (eluent: 80:15:5 Hexanes/EtOAc/Et3N) to afford the title compound 22 (6.13 g, 22.2 mmol, 47%) as a clear oil. TLC Rf = 0.50 (30:70 EtOAc/Hexanes; CAM, UV). 1H NMR (500 MHz, CDCl3) δ 7.39–7.30 (m, 5H) 5.99–5.88 (m, 1H), 5.27 (d, J = 17.3 Hz, 1H), 5.18 (d, J = 10.3 Hz, 1H), 5.13 (s, 2H), 4.21 (d, J = 6.1 Hz, 2H), 4.03 (s, 2H), 3.19–3.11 (m, 4H), 2.57 (s, 2H). 13C NMR (126 MHz, CDCl3): δ 155.3, 136.8, 134.6, 128.7, 128.2, 128.1, 118.0, 73.2, 67.4, 55.3, 42.7. HRMS-ESI (m/z): [M + H]+ calculated for [C15H21N2O3]+: 277.1547, found: 277.1540.
2-((4-Methylpiperazin-1-yl)oxy)ethan-1-ol (17)
Compound 22 (5.6 g, 20.27 mmol, 1.0 equiv) was dissolved in a mixture of DCM (200 mL) and methanol (50 mL) and cooled to −78 °C. Ozone was bubbled through the reaction mixture for 30 min until full consumption of the starting material. On completion, as indicated by ESIMS, argon was bubbled through the reaction mixture to disperse residual ozone. NaBH4 (1.53 g, 40.54 mmol, 2.0 equiv) was then added portion-wise at −78 °C and the solution was gradually warmed to rt and stirred for 1 h. After such time, the mixture was quenched with aq. NaHCO3 (200 mL) and the layers separated. The aqueous layer was extracted with DCM (2×, 150 mL), and the combined organic layers were washed with brine (2×, 150 mL), dried over Na2SO4, filtered, and concentrated in vacuo to afford the alcohol as a clear oil (5.26 g, 18.8 mmol, 93% crude yield). TLC Rf = 0.10 (70:30 EtOAc/Hexanes; CAM, UV). 1H NMR (500 MHz, CDCl3) δ: 7.38–7.30 (m, 5H), 5.13 (s, 2H), 4.07 (s, 2H), 3.84–3.81 (m, 4H), 3.24–3.10 (m, 5H), 2.60–2.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ: 155.2, 136.6, 128.7, 128.3, 128.1, 71.9, 67.5, 65.3, 55.0, 42.7. HRMS-ESI (m/z) [M + H]+ calculated for [C14H21O4N2]+: 281.1496, found: 281.1489.
The so-obtained crude alcohol (5.26 g, 18.8 mmol, 1.0 equiv) obtained in the previous step was dissolved in anhydrous THF (68 mL) and cooled to 0 °C. LiAlH4 (2.31 g, 60.81 mmol, 3.2 equiv) was then added portion-wise for 5 min. After full addition, the solution was warmed to r.t. and stirred for 2 h. After such time, the reaction mixture was cooled back to 0 °C and quenched by slow addition of H2O. After effervescence ceased, the solution was acidified with concentrated HCl until pH ≈ 2. The aqueous layer was then extracted with EtOAc (4×, 150 mL), after which, the aqueous layer was cooled to 0 °C and basified to pH ≈ 10 by portion-wise addition of solid NaOH. The basified aqueous layer was extracted with EtOAc (4×, 100 mL) and the resulting organic layer was dried over Na2SO4, filtered, and concentrated in vacuo to afford the title compound 17 (1.63 g, 10.2 mmol, 50% yield over 3 steps from 22) as a yellow oil with spectral data identical to that of the above sample.
Kinase Activity
In vitro kinase activity (inhibitor binding constants (Kd) and biochemical inhibition (IC50’s)) were assessed by Eurofins DiscoverX (Kd determinations) and Eurofins Cerep (biochemical IC50 determinations). For Kd determinations, compounds were run in duplicate (n = 2) and assayed using an 11-point, 3-fold dilution series at a top compound testing concentration of 10 μM with Eurofin’s KINOMEScan KdELECT assay. For biochemical IC50 determinations, compounds were run in duplicate (n = 2) and tested in an enzymatic radiometric assay using a 9-point, half-log dilution series at a top compound testing concentration of 10 μM and an ATP concentration of 10 μM with Eurofin’s KinaseProfiler technology.
Cell Lines
The A431, HCC827, SK-BR-3, ZR-75–30, AU565, and Caco-2 cell lines were obtained from ATCC. The NCI-H1975 cell line was obtained from SIBS. The NCI-H3255 cell line was obtained from CoBioer. Engineered Ba/F3 cell lines were obtained from Crown Bioscience. MCKII-MDR1 cells were obtained from The Netherlands Cancer Institute. HEK293 cells were obtained from Invitrogen. A431 cells were cultured in DMEM (Life Technologies) with 10% FBS. HCC827, NCI-H1975, and AU565 cells were cultured in RPMI1640 (Invitrogen) with 10% FBS. NCI-H3255 cells were cultured in BEGM (Lonza) with 10% FBS. Ba/F3 EGFR-del E746_A750/C797S and Ba/F3 EGFR-L858R/C797S cells were cultured in RPMI (Invitrogen) with 10% FBS. SK-BR-3 cells were cultured in McCoy’s 5a (Invitrogen) with 10% FBS. ZR-75-30 cells were cultured in RPMI1640 (Invitrogen) with 20% FBS. Caco-2 and MCKII-MDR1 culture information is given in the Permeability Studies section. HEK293 cells were cultured in DMEM (Gibco) with 10% FBS, 0.1 mM NEAA, 25 mM HEPES, 100 U/mL penicillin-streptomycin, μg/mL blasticidin, and 400 μg/mL Geneticin. All cells were cultured in a humidified incubator with 5% CO2 at 37 °C.
Cell Viability Assay
Viability assays using A431, HCC827, NCI-H1975, NCI-H3255, Ba/F3 EGFR-del E746_A750/C797S, Ba/F3 EGFR-L858R/C797S, AU565, SK-BR-3, and ZR-75-30 cells were performed at Crown BioScience. Cells were plated into 96-well plates at 1500–7000 cells per well and dosed in triplicate (n = 3) in a nine-point, 4-fold dilution series with compounds (0.15 nM to 10 μM) in DMSO and incubated for 72 h. After 72 h, cell viability was assayed by CellTiter-Glo Luminescent Viability Assay (Promega). Dose–response curves were generated and used to calculate the IC50 values which were calculated on GraphPad Prism from the nonlinear regression equation fitted with a sigmoidal dose response and are presented as mean ± SEM.
Lipophilicity Assay
Compound lipophilicity was determined by Pharmaron, Inc. using the shake-flask method with 1-octanol and PBS (pH 7.4). Each compound was assessed in duplicate (n = 2) at 1 μM in a 96-well plate shaken at 25 °C and 2000 rpm for 2 h. Samples were analyzed by liquid chromatography and tandem mass spectrometry (LC-MS/MS).
Solubility Assay
Solubility was determined by Pharmaron, Inc. and each compound was assessed in duplicate (n = 2) by adding 15 μL of compound stock solution (10 mM in DMSO) into a 96-well plate along with 485 μL of buffer followed by shaking at 25 °C, 1100 rpm for 2 h. Wells were filtered, and samples (5 μL) were taken followed by dilution with an equal volume of DMSO (5 μL) and 490 μL of aqueous solution. Samples were analyzed by LC-MS/MS.
Plasma Protein Binding and Brain Tissue Binding Assays
Plasma protein binding and brain tissue binding were determined by Pharmaron, Inc. using the equilibrium dialysis method. Each compound was assessed in duplicate (n = 2) at 5 μM with the final percent volume of the organic solvent at 0.5%. Compounds were incubated at 37 °C, 5% CO2 for 6 h at 100 rpm, and samples (50 μL) were taken at the beginning and end of the incubation. Following incubation, the samples were analyzed by LC-MS/MS and the concentrations of the compounds were determined in the buffer and plasma solution chambers.
Metabolic Stability Assay
Assessment of the compound’s metabolic stability in incubations containing liver microsomes and hepatocytes of human and rat using the compound depletion approach was performed by Pharmaron, Inc. Compound 9 underwent additional stability evaluation in monkey and dog hepatocytes. Microsome stability was assessed in duplicate (n = 2) in the presence and absence of nicotinamide adenine dinucleotide phosphate (NADPH) with liver microsomes (0.5 mg/mL) from human (BD Gentest) and Sprague-Dawley rats (BD Gentest). Compounds were incubated in liver microsomes at 1 μM and samples (30 μL) were taken at 0.5, 15, 30, 45, and 60 min time points for analysis by LC-MS/MS. Hepatocyte stability was assessed in duplicate (n = 2) at a working cell density of 0.5 × 106 cells/mL with hepatocytes from human (BiolVT), Sprague-Dawley rat (BiolVT), cynomolgus monkey (RILD) or beagle dog (BiolVT). Compounds were incubated in hepatocytes at 1 μM and samples (25 μL) were taken at 0.5, 15, 30, 60, 90, and 120 min time points for analysis by LC-MS/MS. The in vitro half-life and intrinsic clearances were determined as previously described.76
In vitro half-life (in vitro t1/2) was determined from the slope value in
where k was determined by linear regression of the natural logarithm of the remaining percentage of the parent drug vs the incubation time curve.
Plasma Stability Assay
Assessment of compounds to determine stability in incubations containing plasma of human (Pharmaron) and Sprague-Dawley rat (IPHASE) was performed by Pharmaron, Inc. using the compound deletion approach. Compounds were assessed in duplicate (n = 2) and incubated in plasma at 5 μM and samples (50 μL) were taken at 0, 15, 30, 60, and 120 min time points for analysis by LC-MS/MS. Peak area ratios were determined from the extracted ion chromatogram and the percent compound remaining at each time point was calculated by the following equation:
where peak area ratiot min is the peak area ratio of control and test compound at t min and peak area ratio0 min is the peak area ratio of control and test compound at zero time point.
Permeability Studies
Compounds were evaluated at Pharmaron, Inc. for their ability to permeate Caco-2 cells and MDCKII-MDR1 cells. A 96-well HTS Transwell plate (Corning) was used for Caco-2 and MDCKII-MDR1 cell seeding. Caco-2 cells were seeded at a density of 6.86 × 105 cells/mL and cultivated for 14–18 days prior to assays. MDCKII-MDR1 cells were seeded at a density of 1.56 × 106 cells/mL and cultivated for 4–8 days prior to assays. To determine the rate of drug transport in both the absorptive (apical to basolateral (A–B)) and secretory (basolateral to apical (B–A)) directions, compounds (5 μM in DMSO for Caco-2 cells, and 1 μM in DMSO for MDCKII-MDR1 cells) were added to the donor wells. Plates were incubated at 37 °C for 2 h, and samples (50 μL) were taken at the beginning and end of the incubation in both the donor and acceptor wells with the assay run in duplicate (n = 2). Samples were analyzed by LC-MS/MS. The apparent permeability coefficient (Papp) in units (cm/s × 10–6) was calculated using the following equation:
where VA represents the volume (mL) in the acceptor well, Area is the surface of the membrane (0.143 cm2 for Transwell 96-well plate), and Time is the total transport time in seconds.
The efflux ratio was determined by using the following equation:
where Papp (B–A) indicates the apparent permeability in the basolateral to apical direction and Papp (A–B) indicates the apparent permeability in the apical to basolateral direction.
hERG Channel Inhibition Assay
The potential inhibitory effect of compounds on the hERG channel was assessed by Pharmaron, Inc. using the manual patch clamp system as previously described.77 The HEK293 cell line (Invitrogen) stably transfected with the hERG gene was employed. Compounds were tested at 5 concentrations (0.37, 1.11, 3.33, 10, and 30 μM) and run in triplicate (n = 3). IC50 values were determined by plotting the % inhibition against the concentration of compounds using GraphPad Prism from the nonlinear regression equation fitted with a sigmoidal dose–response curve, and the IC50 values are presented as mean ± SEM.
Determination of Human CYP450 Inhibition by 9
Assessment of compound 9 was carried out at Pharmaron, Inc. for its potential to inhibit cytochrome P450 (CYP) isoforms using human liver microsomes. The activities tested were CYP1A2-mediated phenacetin O-demethylation, CYP2C19-mediated (S)-mephenyotin 4′-hydroxylation, CYP2C9-mediated diclofenac 4′-hydroxylation, CYP2D6-mediated dextromethorphan O-demethylation, and CYP3A4-mediated midazolam 1′-hydroxylation. Concentrations of substrates were phenacetin (40 μM), mephenytoin (50 μM), diclofenac (6 μM), dextromethorphan (2 μM), and midazolam (1 μM). Probe substrates phenacetin, mephenytoin, and dextromethorphan were incubated at 37 °C for 20 min. Probe substrates diclofenac and midazolam were incubated at 37 °C for 5 min. Compound (9) was tested in an eight-point, 3-fold dilution series (0.01–30 μM) in DMSO and incubated with pooled human liver microsomes (0.5 mg/mL) (BD Gentest) and a cocktail of the probe substrates for selective CYP isoform. The reactions were initiated by adding NADPH (1 mM final concentration) after a 5 min preincubation, and the assay was run in duplicate (n = 2). Samples were analyzed by ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS). Inhibition of each P450 enzyme was measured as the percentage decrease in the activity of marker metabolite formation compared to noninhibited controls. IC50 values were determined on GraphPad Prism with remaining activity (%) and logarithm of inhibitor concentrations fitted with a nonlinear fit [inhibitor] vs normalized response with variable slope.
Determination of CYP2D6 Time-Dependent Inhibition by 9
Assessment of compound 9 was carried out at Pharmaron, Inc. for the potential time-dependent inhibition of CYP2D6 using human liver microsomes (BD Gentest) and primary human hepatocytes (BiolVT). Bufuralol (2 μM) was used as a substrate for liver microsomes, and dextromethorphan (40 μM) was used for hepatocytes. The activities tested were CYP2D6-mediated bufuralol 1′-hydroxylation and CYP2D6-mediated dextromethorphan O-demethylation. Compound 9 was assayed in duplicate (n = 2) and tested in a six-point, 3-fold dilution series (0.03–10 μM) in DMSO and incubated with either pooled human liver microsomes (0.5 mg/mL) or human hepatocytes (0.3 × 106 cells/mL). Incubations in pooled human liver microsomes were carried out with NADPH (1 mM final concentration) with or without 30 min preincubation at 37 °C, 5% CO2. Incubations in human hepatocytes were carried out without preincubation or with 30 min preincubation at 37 °C, 5% CO2. The experiment was carried out at 37 °C in 5% CO2 for 5 min. Inhibition of CYP2D6 was measured as the percentage decrease in the activity of marker metabolite formation compared to noninhibited controls. IC50 values were determined on GraphPad Prism with remaining activity (%) and logarithm of inhibitor concentrations fitted with a nonlinear fit [inhibitor] vs normalized response with variable slope.
AMES Fluctuation Assay
Compounds were assessed for mutagenicity at the Eurofins Panlabs in 384-well plates using four Salmonella strains: TA98, to probe for frameshift mutation (with quercetin as control), TA100 and TA1535, to probe for base pair insertions/deletions (with streptozotocin as control), and TA1537, to probe for frameshift mutations (with aminacridine as control) as previously described.78 Each compound was incubated at 37 °C for 96 h at four different concentrations (5, 10, 50, and 100 μM), with each concentration tested using 48 replicates, both with and without rat liver S9 metabolic activation. A concurrent bacterial cytotoxicity assay was run at 0.6, 1.2, 2.5, 5, 10, 25, 50, and 100 μM to rule out false negatives. Bacterial cytotoxicity was expressed as percent of control growth (OD650). Compounds with growth of less than 60% control were flagged and considered bacterial cytotoxic. Wells that displayed bacterial growth due to the reversion of the histidine mutation (as judged by the ratio of OD430/OD570 being greater than 1.0) were counted and recorded as positive counts. Significances of the positive counts between the treatment and control were determined using a one-tailed Fisher’s exact test.
In Vitro Micronucleus Test
Compound 9 was assessed for genotoxicity by Eurofins Panlabs using the in vitro micronucleus test in Chinese hamster ovary (CHO-K1) cells as previously described.79 Compound 9 was incubated in a 96-well plate format at varying concentrations (8, 16, 31, 62, 125, 250, 500, and 1000 μM) for 4 h at 37 °C with metabolic activation by rat liver S9 and 24 h at 37 °C without metabolic activation by rat liver S9. High content analysis and fluorescence imaging were used to detect micronuclei and compared against positive controls (cyclophosphamide (ran at 7.2 μM) and mitomycin C (ran at 0.3 μM)). Significances of the positive counts between the treatment and control were determined by one-tailed t test with two-sample equal variance.
In Vitro Metabolite Identification of 9
Compound 9 underwent in vitro metabolite identification by Pharmaron, Inc. A 200 μL sample of compound 9 was tested at 10 μM in human and rat hepatocytes (1.0 × 106 cells/mL) and incubated for 0, 120, and 240 min at 37 °C. After incubation, reactions were quenched with 2 volumes of acetonitrile followed by centrifugation for 15 min at 16,000g. Supernatants were then analyzed by UHPLC-MS/MS. A figure of identified metabolites is provided in the Supporting Information (Figures S1–S2).
Kinase Selectivity Determination of 9
Profiling of a 468-member human kinase panel was performed with Eurofins DiscoverX using the KINOMEScan platform. A panel of 468 kinases was assayed at a single concentration of 1 μM for 9. Percent control was mapped onto the kinome tree using TREEspot. The S scores were calculated as previously described64 and are reflective of the number of kinases bound by 9 over the total number of wild-type kinases.
Animal Studies
Animal experiments were performed at Pharmaron, Inc. and animal use was approved by Pharmaron’s Institutional Animal Care and Use Committee (IACUC) in Pharmaron (Pharmaron IACUC, Protocol #PK-M-07182022, #PK-R-06012022, and #ON-CELL-XEN-06012022) following the guidance of AAALAC. Six- to eight-week-old male Sprague-Dawley rats (approximately 200–300 g) obtained from Si Bei Fu Laboratory Animal Technology Co., six- to eight-week-old male CD1 mice (approximately 20–30 g) obtained from Si Bei Fu Laboratory Animal Technology Co., and six- to eight-week-old female BALB/c nude mice (approximately 20–30 g) obtained from Si Bei Fu Laboratory Animal Technology Co., were used in the pharmacokinetic studies. Six- to eight-week-old female BALB/c nude mice (approximately 18–22 g) obtained from Beijing Anikeeper Biotech Co., Ltd. were used in the intracranial PDX study. Animals were housed at 20–25 °C with humidity ranging from 40 to 70% relative humidity, were exposed to 12 h light and dark cycles, and were supplied with food and water ab libitum.
Pharmacokinetic Studies of 9
Standard pharmacokinetic assessment of 9 was done by IV (intravenous) tail vein injection (formulation: DMSO: 10% captisol in saline = 1:99) and PO (oral) oral gavage (formulation: 1% methylcellulose) followed by blood sampling at 8 time points for IV (0.0833, 0.25, 0.5, 1,2, 4, 7, 24 h post dose) and 7 time points for PO (0.25, 0.5, 1, 2, 4, 8, 24 h post dose) with n = 3 animals per dosing route (n = 6 total). Approximately 0.03 mL of blood was collected from the dorsal metatarsal vein (in the case of CD1 mice), 0.03 mL of blood from the orbit vein (in the case of BALB/c nude mice), or 0.2 mL of blood from the jugular vein (in the case of Sprague-Dawley rats) at each time point. Blood at each sampling point was transferred into a plastic micro centrifuge tube containing K2-EDTA, and collection tubes with blood samples and anticoagulant were inverted several times for proper mixing of the tube contents and placed on ice prior to centrifugation for plasma. Blood samples were centrifuged at 4 °C, 4000g for 5 min to obtain plasma. Samples were stored in a freezer at −75 °C prior to analysis. Concentrations of test articles in the plasma samples were determined using LC-MS/MS and WinNonlin 8.3 (Phoenix) was used for pharmacokinetic calculations. The values obtained were plotted on GraphPad Prism software and are presented as mean ± SD.
Oral CNS pharmacokinetic assessment of 9 was done by oral gavage (formulation: 1% methylcellulose) followed by blood and brain sampling at 7 time points (0.25, 0.5, 1, 2, 4, 8, 24 h post dose) with n = 3 animals per time point (n = 21 total). For plasma collection, approximately 0.03 mL of blood was collected from the dorsal metatarsal vein (in the case of CD1 mice) or 0.2 mL from the jugular vein (in the case of Sprague-Dawley rats) at each time point. Blood at each sampling point was transferred into a plastic micro centrifuge tube containing K2-EDTA, and collection tubes with blood samples and anticoagulant were inverted several times for proper mixing of the tube contents and placed on ice prior to centrifugation for plasma. Blood samples were then centrifuged at 4 °C, 4000g for 5 min to obtain plasma, and the samples were stored in a freezer at −75 °C prior to analysis. For brain sample collection, rodents were fully exsanguinated prior to collection, and the thoracic cavity was opened exposing the heart, which was catheterized from the left ventricular. A small incision was then made at the right atrial appendage and a gentle administration of saline via syringe was performed. Brain samples were collected at each time point and quickly frozen in an ice box. Samples were stored at −75 °C prior to analysis. All brain samples were prepared with water to achieve a brain weight (g)/water volume (mL) ratio of 1:4 prior to analysis. Concentrations of 9 in the plasma and brain samples were determined using LC-MS/MS and WinNonlin 8.3 (Phoenix) was used for pharmacokinetic calculations. The values obtained were plotted on GraphPad Prism software and are presented as mean ± SD.
Intracranial PDX Model of 9
HCC827 cells stably expressing luciferase (HCC827-luc) were injected intracranially. Briefly, 3 × 105 HCC827-luc tumor cells suspended in 2 μL of RPMI1640 medium were injected into the right forebrain of anesthetized mice (anesthetic: intramuscular injection of ZoletlTM 50 (Virbac S.A)). Mice were imaged biweekly using IVIS Lumina III (PerkinElmer). Images were acquired 10 min post intraperitoneal (IP) injection with 15 mg/mL (at 5 μL/g body weight) of D-luciferin in anesthetized mice (anesthetic: 1–2% isoflurane inhalation). On day 20 postcellular inoculation, mice were randomized into 2 treatment groups (n = 10 mice per group): (1) 1% methylcellulose as vehicle control and (2) 9 (10 mg/kg) (formulation: 1% methylcellulose), PO, b.i.d. (twice daily) dosing (12 h intervals) × 7 days for 21 days. Body weights of all mice were measured biweekly throughout the study and BW change, expressed in % was calculated using the following formula:
where BWDay X is the BW on a given day and BWDay 0 is BW on day 0 (initiation of treatment).
Statistical Analyses
Statistical analysis was performed by using GraphPad Prism 9.0. Data are presented as mean ± SD or SEM as indicated when n = ≥3, or as mean when n = 2. For in vitro ADME and kinase activity studies, data is presented as a mean of n = 2 independent replicates. For in vivo PK experiments, data is presented as mean ± SD, n ≥ 3 animals per study arm. For in vitro short-term growth delay experiments, IC50 values were determined from the nonlinear regression equation fitted with a sigmoidal dose response curve and are presented as mean ± SEM, n = 3 independent replicates. Intracranial PDX study data (bioluminescence and body weight means on day-21 post-treatment initiation, n = 10 animals per study arm) were compared using a two-tailed unpaired t-test. A P value of <0.05 was considered statistically significant.
Acknowledgments
The authors thank Dr. Pingrong Wei (UGA) for X-ray crystallography of 6. They thank the scientists at Pharmaron, Eurofins, and Crown Biosciences for their expert help in conducting the in vitro and in vivo assays. D.C. gratefully acknowledges financial support from the National Institutes of Health (GM144753), the University of Georgia, and the Georgia Research Alliance (GRA). J.H. was supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) PGS-D scholarship.
Glossary
Abbreviations Used
- ABL1
Abelson murine leukemia viral oncogene homologue 1
- Aq. Sol.
aqueous solubility at pH 7.4
- BCRP
breast cancer resistance protein
- BDE
bond dissociation energy
- BM
brain metastases
- C
cynomolgus monkey
- Cmax
maximal concentration
- Caco-2
colon carcinoma cell line
- DRAK1
death receptor-associated kinase 1
- FBS
fetal bovine serum
- fu
fraction unbound
- H
human
- HEPClint
intrinsic clearance in hepatocytes
- HER2
human epidermal growth factor receptor 2
- iPrMgCl·LiCl
isopropylmagnesium chloride lithium chloride
- Kd
dissociation constant
- Kp
brain-to-plasma partitioning coefficient
- Kp,uu
unbound brain-to-unbound plasma partitioning
- LiAlH4
lithium aluminum hydride
- LMClint
intrinsic clearance in liver microsomes
- logD7.4
logarithm of distribution at pH 7.4
- LYN
Lck/Yes novel kinase
- M
mouse
- MDCK
madine-darby canine kidney cell line
- MetID
metabolite identification
- MMP
matched molecular pair
- MPO
multiparameter optimization
- MTHP
2-methyltetrahydropyranyl
- nd
not determined
- NaBH4
sodium borohydride
- Papp
apparent permeability
- PDA
photo diode array
- PDX
patient-derived xenograft
- PIKFYVE
phosphoinositide kinase, FYVE-type zinc finger containing
- R
rat
- SD
standard deviation
- SD Rat
Sprague–Dawley Rat
- SEM
standard error of the mean
- TDI
time-dependent inhibition
- TKI
tyrosine kinase inhibitor
- Tmax
time to reach maximal concentration
- Vss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01669.
The authors declare the following competing financial interest(s): J.H., R.M.J., and D.C. are cofounders of and hold equity in Quintet Pharmaceuticals, a startup exploring the use of hydroxylamines in medicinal chemistry. J.H. and D.C. are inventors on US Provisional Applications 63/375,622 and 63/507,876 submitted by the University of Georgia Research Foundation (UGARF), that cover the compounds described in this report for use in the treatment of EGFR mutation positive local and metastatic NSCLC.
Supplementary Material
References
- Thai A. A.; Solomon B. J.; Sequist L. V.; Gainor J. F.; Heist R. S. Lung cancer. Lancet 2021, 398 (10299), 535–554. 10.1016/S0140-6736(21)00312-3. [DOI] [PubMed] [Google Scholar]
- Paez J. G.; Jänne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304 (5676), 1497–1500. 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Lynch T. J.; Bell D. W.; Sordella R.; Burubhagavatula S.; Okimoto R. A.; Brannigan B. W.; Harris P. L.; Haserlat S. M.; Supko J. G.; Haluska F. G.; Louis D. N.; Christiani D. C.; Settleman J.; Haber D. A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350 (21), 2129–2139. 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Pao W.; Miller V.; Zakowski M.; Doherty J.; Politi K.; Sarkaria I.; Singh B.; Heelan R.; Rusch V.; Fulton L.; Mardis E.; Kupfer D.; Wilson R.; Kris M.; Varmus H. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (36), 13306–13311. 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosell R.; Moran T.; Queralt C.; Porta R.; Cardenal F.; Camps C.; Majem M.; Lopez-Vivanco G.; Isla D.; Provencio M.; Insa A.; Massuti B.; Gonzalez-Larriba J. L.; Paz-Ares L.; Bover I.; Garcia-Campelo R.; Moreno M. A.; Catot S.; Rolfo C.; Reguart N.; Palmero R.; Sánchez J. M.; Bastus R.; Mayo C.; Bertran-Alamillo J.; Molina M. A.; Sanchez J. J.; Taron M. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361 (10), 958–967. 10.1056/NEJMoa0904554. [DOI] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19 (8), 2240–2247. 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun C. H.; Mengwasser L. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K. K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (6), 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S.; Boggon T. J.; Dayaram T.; Jänne P. A.; Kocher O.; Meyerson M.; Johnson B. E.; Eck M. J.; Tenen D. G.; Halmos B. EGFR Mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352 (8), 786–792. 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Miller V. A.; Hirsh V.; Cadranel J.; Chen Y.-M.; Park J.; Kin S.-W.; Zhou C.; Su W.-C.; Wang M.; Sun Y.; Heo D. S.; Crino L.; Tan E.-H.; Chao T.-Y.; Shahidi M.; Cong X. J.; Lorence R. M.; Yang J. C.-H. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol. 2012, 13 (5), 528–538. 10.1016/S1470-2045(12)70087-6. [DOI] [PubMed] [Google Scholar]
- Engelman J. A.; Zejnullahu K.; Gale C.-M.; Lifshits E.; Gonzales A. J.; Shimamura T.; Zhao F.; Cinvent P. W.; Naumov G. N.; Bradner J. E.; Althaus I. W.; Gandhi L.; Shapiro G. I.; Nelson J. M.; Heymach J. V.; Meyerson M.; Wong K.-K.; Jänne P. A. PF00299804, An irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007, 67, 11924–11932. 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- Finlay M. R. V.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D. A. E.; Currie G. S.; Grist M.; Hassall L.; Hill G. B.; James D.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamot S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. L. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57 (20), 8249–8267. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Chul Cho B.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Jänne P. A.; Oxnard G. R. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21 (6), 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti A.; Sharma S.; Minari R.; Perego P.; Giovannetti E.; Tiseo M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121 (9), 725–737. 10.1038/s41416-019-0573-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalingam S. S.; Yang J. C.-H.; Lee C. K.; Kurata T.; Kim D.-W.; John T.; Nogami N.; Ohe Y.; Mann H.; Rukazenkov Y.; Ghiorghiu S.; Stetson D.; Markovets A.; Barrett J. C.; Thress K. S.; Jänne P. A. Osimertinib as first-line treatment of EGFR mutation-positive advanced non-small-cell lung cancer. J. Clin. Oncol. 2018, 36 (9), 841–849. 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- Chmielecki J.; Gray J. E.; Cheng Y.; Ohe Y.; Imamura F.; Cho B. C.; Lin M.-C.; Majem M.; Shah R.; Rukazenkov Y.; Todd A.; Markovets A.; Barrett J. C.; Harmaier R. J.; Ramalingam S. S. Candidate mechanisms of acquired resistance to first-line osimertinib in EGFR-mutated advanced non-small cell lung cancer. Nat. Commun. 2023, 14, 1070 10.1038/s41467-023-35961-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak L.; Lee E. Q.; Wen P. Y. Epidemiology of brain metastases. Curr. Oncol. Rep. 2012, 14 (1), 48–54. 10.1007/s11912-011-0203-y. [DOI] [PubMed] [Google Scholar]
- Rangachari D.; Yamaguchi N.; VanderLaan P. A.; Folch E.; Mahadevan A.; Floyd S. R.; Uhlmann E. J.; Wong E. T.; Dahlberg S. E.; Huberman M. S.; Costa D. B. Brain metastases in patients with EGFR-mutated or ALK-rearranged non-small-cell lung cancers. Lung Cancer 2015, 88 (1), 108–111. 10.1016/j.lungcan.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colclough N.; Chen K.; Johnström P.; Strittmatter N.; Yan Y.; Wrigley G. L.; Schou M.; Goodwin R.; Varnäs K.; Adua S. J.; Zhao M.; Nguyen D. X.; Maglennon G.; Barton P.; Atkinson J.; Zhang L.; Jandefeldt A.; Wilson J.; Smith A.; Takano A.; Arakawa R.; Kondrashov M.; Malmquist J.; Revunov E.; Vazquez-Romero A.; Moein M. M.; Windhorst A. D.; Karp N. A.; Finlay M. R. V.; Ward R. A.; Yates J. W. T.; Smith P. D.; Farde L.; Cheng Z.; Cross D. A. E. Preclinical comparison of the blood-brain barrier permeability of osimertinib with other EGFR TKIs. Clin. Cancer Res. 2021, 27 (1), 189–201. 10.1158/1078-0432.CCR-19-1871. [DOI] [PubMed] [Google Scholar]
- Heon S.; Yeap B. Y.; Britt G. J.; Costa D. B.; Rabin M. S.; Jackman D. M.; Johnson B. E. Development of central nervous system metastases in patients with advanced non-small cell lung cancer and somatic EGFR mutations treated with gefitinib or erlotinib. Clin. Cancer Res. 2010, 16 (23), 5873–5882. 10.1158/1078-0432.CCR-10-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffron T. P. Small molecule kinase inhibitors for the treatment of brain cancer. J. Med. Chem. 2016, 59 (22), 10030–10066. 10.1021/acs.jmedchem.6b00618. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Huo X.; Verhoest P. R.; Villalobos A. Moving beyond rules: the development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci. 2010, 1 (6), 435–449. 10.1021/cn100008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong B.; Wang Y.; Chen Y.; Xing S.; Liao Q.; Chen Y.; Li Q.; Li W.; Sun H. Strategies for structural modification of small molecules to improve blood-brain barrier penetration: a recent perspective. J. Med. Chem. 2021, 64 (18), 13152–13173. 10.1021/acs.jmedchem.1c00910. [DOI] [PubMed] [Google Scholar]
- Young R. C.; Mitchell R. C.; Brown T. H.; Ganellin C. R.; Griffiths R.; Jones M.; Rana K. K.; Saunders D.; Smith I. R. Development of a new physicochemical model for brain penetration and its application to the design of centrally acting H2 receptor histamine antagonists. J. Med. Chem. 1988, 31 (3), 656–671. 10.1021/jm00398a028. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Röder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. Predicting and tuning physicochemical properties in lead optimization: Amine basicities. ChemMedChem 2007, 2 (8), 1100–1115. 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- Mollin J.; Kaspárek F.; Lasovský J. On the basicity of hydroxylamine and its derivatives. Chem. Zvesti 1975, 29 (1), 39–43. [Google Scholar]
- Meanwell N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54 (8), 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- Subbaiah M. A. M.; Meanwell N. A. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J. Med. Chem. 2021, 64 (19), 14046–14128. 10.1021/acs.jmedchem.1c01215. [DOI] [PubMed] [Google Scholar]
- Levterov V. V.; Panasyuk Y.; Pivnytska V. O.; Mykhailiuk P. K. Water-soluble non-classical benzene mimetics. Angew. Chem., Int. Ed. 2020, 59 (18), 7161–7167. 10.1002/anie.202000548. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 2016, 29 (4), 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Mäder P.; Kattner L. Sulfoximines as rising stars in modern drug discovery? Current status and perspective on an emerging functional group in medicinal chemistry. J. Med. Chem. 2020, 63 (23), 14243–14275. 10.1021/acs.jmedchem.0c00960. [DOI] [PubMed] [Google Scholar]
- Wuitschik G.; Rogers-Evans M.; Müller K.; Fischer H.; Wagner B.; Schuler F.; Polonchuk L.; Carreira E. M. Oxetanes as promising modules in drug discovery. Angew. Chem., Int. Ed. 2006, 45 (46), 7736–7739. 10.1002/anie.200602343. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Walker D. P.; Bauman J.; Price D. A.; Baillie T. A.; Kalgutkar A. S.; Aleo M. D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24 (9), 1345–1410. 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S. Designing around structural alerts in drug discovery. J. Med. Chem. 2020, 63 (12), 6276–6302. 10.1021/acs.jmedchem.9b00917. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L. The chemical space project. Acc. Chem. Res. 2015, 48 (3), 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Yu B.; Reynisson J. Bond stability of the “undesirable” heteroatom-heteroatom molecular moieties for high-throughput screening libraries. Eur. J. Med. Chem. 2011, 46 (11), 5833–5837. 10.1016/j.ejmech.2011.09.044. [DOI] [PubMed] [Google Scholar]
- Bruns R. F.; Watson I. A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 2012, 55 (22), 9763–9772. 10.1021/jm301008n. [DOI] [PubMed] [Google Scholar]
- Bach R. D.; Schlegel H. B. The bond dissociation energy of the N-O bond. J. Phys. Chem. A 2021, 125 (23), 5014–5021. 10.1021/acs.jpca.1c02741. [DOI] [PubMed] [Google Scholar]
- Davis C. D.; Schut H. A. J.; Snyderwine E. G. Enzymatic phase II activation of the N-hydroxylamines of IQ and MeIQx and PhIP by various organs of monkeys and rats. Carcinogenesis 1993, 14 (10), 2091–2096. 10.1093/carcin/14.10.2091. [DOI] [PubMed] [Google Scholar]
- Davis C. D.; Ghoshal A.; Schut H. A. J.; Snyderwine E. G. Metabolic activation of heterocycle amine food mutagens in the mammary gland of lactating fischer 344 rats. Cancer Lett. 1994, 84 (1), 67–73. 10.1016/0304-3835(94)90359-X. [DOI] [PubMed] [Google Scholar]
- Shen S.; Kozikowski A. P. Why hydroxamates may not be the best histone deacetylase inhibitors – what some may have forgotten or would rather forget?. ChemMedChem 2016, 11 (1), 15–21. 10.1002/cmdc.201500486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citarella A.; Moi D.; Pinzi L.; Bonanni D.; Rastelli G. Hydroxamic acid derivatives: From synthetic strategies to medicinal chemistry applications. ACS Omega 2021, 6 (34), 21843–21849. 10.1021/acsomega.1c03628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batool Z.; Lomakin I. B.; Polikanov Y. S.; Bunik C. G. Sarecycline interferes with tRNA accommodation and tethers mRNA to the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (34), 20530–20537. 10.1073/pnas.2008671117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.; Hettikankanamalage A. A.; Crich D. Diversity-oriented synthesis of N,N,O-trisubstituted hydroxylamines from alcohols and amines by direct N-O bond formation. J. Am. Chem. Soc. 2020, 142 (35), 14820–14825. 10.1021/jacs.0c05991. [DOI] [PubMed] [Google Scholar]
- Hill J.; Crich D. Synthesis of O-tert-butyl-N,N-disubstituted hydroxylamines by direct N-O bond formation. Org. Lett. 2021, 23 (16), 6396–6400. 10.1021/acs.orglett.1c02215. [DOI] [PubMed] [Google Scholar]
- Hill J.; Beckler T. D.; Crich D. Recent advances in the synthesis of di- and trisubstituted hydroxylamines. Molecules 2023, 28 (6), 2816. 10.3390/molecules28062816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhyaya K.; Crich D. Synthesis of 10-aza-9-oxakalkitoxin by N-O bond formation. Org. Lett. 2022, 24 (9), 1833–1836. 10.1021/acs.orglett.2c00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J.; Crich D. The N,N,O-trisubstituted hydroxylamine isostere and its influence on lipophilicity and related parameters. ACS Med. Chem. Lett. 2022, 13 (5), 799–806. 10.1021/acsmedchemlett.1c00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen E.; Leach A. G.; Robb G. R.; Warner D. J. Matched molecular pairs as a medicinal chemistry tool. J. Med. Chem. 2011, 54 (22), 7739–7750. 10.1021/jm200452d. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Subramanyam C.; Efremov I. V.; Dutra J. K.; O’Sullivan T. J.; DiRico K. J.; McDonald S. W.; Won A.; Dorff P. H.; Nolan C. E.; Becker S. L.; Pustilnik L. R.; Riddell D. R.; Kauffman G. W.; Kormos B. L.; Zhang L.; Lu Y.; Capetta S. H.; Green M. E.; Karki K.; Sibley E.; Atchison K. P.; Hallgren A. J.; Oborski C. E.; Robshaw A. E.; Sneed B.; O’Donnell C. J. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 2012, 55 (7), 3414–3424. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Smith R. T.; Le C.; McCarver S. J.; Shireman B. T.; Carruthers N. I.; MacMillan D. W. C. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 2020, 580 (7802), 220–226. 10.1038/s41586-020-2060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank N.; Nugent J.; Shire B. R.; Pickford H. D.; Rabe P.; Sterling A. J.; Zarganes-Tzitzikas T.; Grimes T.; Thompson A. L.; Smith R. C.; Schofield C. J.; Brennan P. E.; Duarte F.; Anderson E. A. Synthesis of meta-substituted arene bioisosteres from [3.1.1]propellane. Nature 2022, 611 (7937), 721–726. 10.1038/s41586-022-05290-z. [DOI] [PubMed] [Google Scholar]
- Wiesenfeldt M. P.; Rossi-Ashton J. A.; Perry I. B.; Diesel J.; Garry O. L.; Bartels F.; Coote S. C.; Ma X.; Yeung C. S.; Bennett D. J.; MacMillan D. W. C. General access to cubanes as benzene bioisosteres. Nature 2023, 618 (7965), 513–518. 10.1038/s41586-023-06021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Agarwal S.; Sane R.; Gallardo J. L.; Ohlfest J. R.; Elmquist W. F. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J. Pharmacol. Exp. Ther. 2010, 334 (1), 147–155. 10.1124/jpet.110.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames B. N.; McCann J.; Yamasaki E. Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome 40 utagenicity test. Mutat. Res. 1975, 31 (6), 347–364. 10.1016/0165-1161(75)90046-1. [DOI] [PubMed] [Google Scholar]
- Zeng Q.; Wang J.; Cheng Z.; Chen K.; Johnström P.; Varnäs K.; Li D. Y.; Yang Z. F.; Zhang X. Discovery and evaluation of clinical candidate AZD3759, a potent, oral active, central nervous system-penetrant, epidermal growth factor receptor tyrosine kinase inhibitor. J. Med. Chem. 2015, 58 (20), 8200–8215. 10.1021/acs.jmedchem.5b01073. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Guo Q.; Wang Y.; Chen K.; Zhang L.; Cheng Q.; Xu Y.; Yin X.; Bai Y.; Rabbie S.; Kim D.-W.; Ahn M.-J.; Yang J. C.-H.; Zhang X. AZD3759, a BBB-penetrating EGFR inhibitor for the treatment of EGFR mutant NSCLC with CNS metastases. Sci. Transl. Med. 2016, 8 (368), 368ra172 10.1126/scitranslmed.aag0976. [DOI] [PubMed] [Google Scholar]
- Tsang J. E.; Urner L. M.; Kim G.; Chow K.; Baufeld L.; Faull K.; Cloughesy T. F.; Clark P. M.; Jung M. E.; Nathanson D. A. Development of a potent brain-penetrant EGFR tyrosine kinase inhibitor against malignant brain tumors. ACS Med. Chem. Lett. 2020, 11 (10), 1799–1809. 10.1021/acsmedchemlett.9b00599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss M. M.; Williamson T.; Babu-Khan S.; Bartberger M. D.; Brown J.; Chen K.; Cheng Y.; Citron M.; Croghan M. D.; Dineen T. A.; Esmay J.; Graceffa R. F.; Harried S. S.; Hickman D.; Hitchcock S. A.; Horne D. B.; Huang H.; Imbeah-Ampiah R.; Judd T.; Kaller M. R.; Kreiman C. R.; La D. S.; Li V.; Lopez P.; Louie S.; Monenschein H.; Nguyen T. T.; Pennington L. D.; Rattan C.; Miguel T. S.; Sickmier E. A.; Wahl R. C.; Wen P. H.; Wood S.; Xue Q.; Yang B. H.; Patel V. F.; Zhong W. Design and preparation of a potent series of hydroxyethylamine containing β-secretase inhibitors that demonstrate robust reduction of central β-amyloid. J. Med. Chem. 2012, 55 (21), 9009–9024. 10.1021/jm300119p. [DOI] [PubMed] [Google Scholar]
- Ercan D.; Choi H. G.; Yun C.-H.; Capelletti M.; Xie T.; Eck M. J.; Gray N. S.; Jänne P. A. EGFR Mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 2015, 21 (17), 3913–3923. 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm S. W.; Einolf H. J.; Hall S. D.; He K.; Lim H.-L.; Ling K.-H. J.; Lu C.; Nomeir A. A.; Seibert E.; Skordos K. W.; Tonn G. R.; Horn R. V.; Wang R. W.; Wong Y. N.; Yang T. J.; Obach R. S. The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America. Drug Metab. Dispos. 2009, 37 (7), 1355–1370. 10.1124/dmd.109.026716. [DOI] [PubMed] [Google Scholar]
- Corvi R.; Albertini S.; Hartung T.; Hoffmann S.; Maurici D.; Pfuhler S.; Benthem J. V.; Vanparys P. ECVAM retrospective validation of in vitro micronucleus test (MNT). Mutagenesis 2008, 23 (4), 271–283. 10.1093/mutage/gen010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Zhang J.; Zhou S.; Yu L.; Han F.; Ling R.; Ling J. Tentative identification of gefitinib metabolites in non-small-cell lung cancer patient plasma using ultra-performance liquid chromatography coupled with triple quadrupole time-of-flight mass spectrometry. PLoS One 2020, 15 (7), e0236523 10.1371/journal.pone.0236523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. I.; Hunt J. P.; Herrgard S.; Ciceri P.; Wodicka L. M.; Pallares G.; Hocker M.; Treiber D. K.; Zarrinkar P. P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29 (11), 1046–1051. 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Winer E. P.; Lin N. U. Brain metastases: the HER2 paradigm. Clin. Cancer Res. 2007, 13 (6), 1648–1655. 10.1158/1078-0432.CCR-06-2478. [DOI] [PubMed] [Google Scholar]
- Di L.; Rong H.; Feng B. Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem. 2013, 56 (1), 2–12. 10.1021/jm301297f. [DOI] [PubMed] [Google Scholar]
- A b.i.d. dosing schedule was chosen to ensure 24 h target coverage as after administration of 30 mg/kg PO of compound 9 to BALC/c nude mice no compound was detected at the 24 h time point (Figure 4a)
- Krasovskiy A.; Knochel P. A LiCl-mediated Br/Mg exchange reaction for the preparation of functionalized aryl- and heteroarylmagnesium compounds from organic bromides. Angew. Chem., Int. Ed. 2004, 43 (25), 3333–3336. 10.1002/anie.200454084. [DOI] [PubMed] [Google Scholar]
- Kyasa S.; Meier R. N.; Pardini R. A.; Truttmann T. K.; Kuwata K. T.; Dussault P. H. Synthesis of ethers via reaction of carbanions and monoperoxyacetals. J. Org. Chem. 2015, 80 (24), 12100–12114. 10.1021/acs.joc.5b02043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang K. M.; Lees N. R.; Herzon S. B. Programmable synthesis of 2-deoxyglycosides. J. Am. Chem. Soc. 2019, 141 (20), 8098–8103. 10.1021/jacs.9b03982. [DOI] [PubMed] [Google Scholar]
- Hoang K. M.; Lees N. R.; Herzon S. B. General method for the synthesis of α- or β-deoxyaminoglycosides bearing basic nitrogen. J. Am. Chem. Soc. 2021, 143 (7), 2777–2783. 10.1021/jacs.0c11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisenheimer J. Über eine eigenartige Umlagerung des Methyl-allyl-anilin-N-oxyds. Ber. Dtsch. Chem. Ges. 1919, 52, 1667–1677. 10.1002/cber.19190520830. [DOI] [Google Scholar]
- Albini A. Synthetic utility of amine N-oxides. Synthesis 1993, 1993, 263–277. 10.1055/s-1993-25843. [DOI] [Google Scholar]
- Vallakati R.; May J. A. Biomimetic synthesis of the antimalarial flindersial alkaloids. J. Am. Chem. Soc. 2012, 134 (16), 6936–6939. 10.1021/ja301387k. [DOI] [PubMed] [Google Scholar]
- Still W. C.; Kahn M.; Mitra A. Rapid chromatographic technique for preparative separations with moderate resolution. J. Org. Chem. 1978, 43 (14), 2923–2925. 10.1021/jo00408a041. [DOI] [Google Scholar]
- Kalvass J. C.; Tess D. A.; Giragossian C.; Linhares M. C.; Maurer T. S. Influence of microsomal concentration on apparent intrinsic clearance: implications for scaling in vitro data. Drug Metab. Dispos. 2001, 29 (10), 1332–1336. [PubMed] [Google Scholar]
- Danker T.; Möller C. Early identification of hERG liability in drug discovery programs by automated patch clamp. Front. Pharmacol. 2014, 5, 203. 10.3389/fphar.2014.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maron D. M.; Ames B. N. Revised methods for the Salmonella mutagenicity test. Mutat. Res., Environ. Mutagen. Relat. Subj. 1983, 113, 173–215. 10.1016/0165-1161(83)90010-9. [DOI] [PubMed] [Google Scholar]
- Diaz D.; Scott A.; Carmichael P.; Shi W.; Costales C. Evaluation of an automated in vitro micronucleus assay in CHO-K1 cells. Mutat. Res., Genet. Toxicol. Environ. Mutagen. 2007, 630, 1–13. 10.1016/j.mrgentox.2007.02.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.