

Abstract
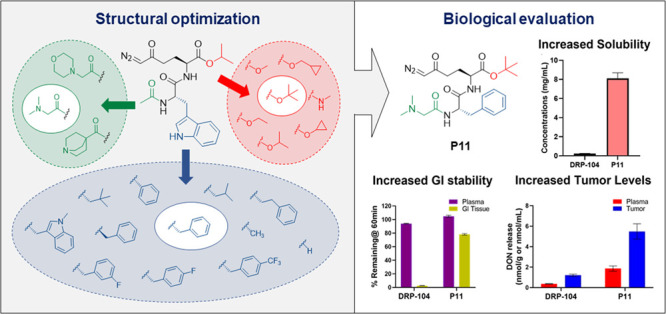
The glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON) exhibits remarkable anticancer efficacy; however, its therapeutic potential is hindered by its toxicity to gastrointestinal (GI) tissues. We recently reported the discovery of DRP-104, a tumor-targeted DON prodrug with excellent efficacy and tolerability, which is currently in clinical trials. However, DRP-104 exhibits limited aqueous solubility, and the instability of its isopropyl ester promoiety leads to the formation of an inactive M1-metabolite, reducing overall systemic prodrug exposure. Herein, we aimed to synthesize DON prodrugs with various ester and amide promoieties with improved solubility, GI stability, and DON tumor delivery. Twenty-one prodrugs were synthesized and characterized in stability and pharmacokinetics studies. Of these, P11, tert-butyl-(S)-6-diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-phenylpropanamido)-5-oxo-hexanoate, showed excellent metabolic stability in plasma and intestinal homogenate, high aqueous solubility, and high tumor DON exposures and preserved the ideal tumor-targeting profile of DRP-104. In conclusion, we report a new generation of glutamine antagonist prodrugs with improved physicochemical and pharmacokinetic attributes.
Introduction
Glutamine is the most abundant amino acid in the mammalian body. Its metabolism serves as a fundamental source of nitrogen and carbon, providing the essential building blocks for the biosynthesis of amino acids, nucleotides, fatty acids, and coenzymes.1 Glutamine uptake and utilization are greatly increased in cancer cells due to the increased energy demand required for rapid proliferation2 and can lead to an oncogene-dependent addiction to glutamine.3 Thus, blocking glutamine metabolism, particularly in cancer cells, serves as a rational therapeutic approach for cancer.
6-Diazo-5-oxo-l-norleucine (DON; Figure 1) is a glutamine antagonist with antitumor efficacy demonstrated in multiple preclinical studies4−7 as well as in several clinical trials.8−15 In one of the earliest clinical studies, 66% of patients demonstrated disease stability or regression following 2 weeks or more of DON therapy.16 Further, in children with hematologic malignancies on standard 6-mercaptopurine (6-MP) therapy, DON combination led to complete bone marrow remissions in 42% of patients, showing remarkable superiority to 6-MP monotherapy.17 However, its further clinical evaluation was aborted due to dose-limiting gastrointestinal (GI) toxicity, as GI cells are highly glutamine-utilizing. To revamp DON’s clinical translation, prodrug strategies have been employed to develop GI-stable analogues that remain intact and inactive in the gut while preferentially bioactivating to DON within the cancer cells.18,19
Figure 1.

Chemical structures of DON (6-diazo-5-oxo-l-norleucine), DRP-104 (Sirpiglenastat), and DRP-104 (M1) metabolite.
For example, the previously reported DON prodrug termed JHU-083 was shown to cause significant tumor regression in several mouse models at doses that were well-tolerated and lacked GI toxicities.20−22 In addition, JHU-083 was shown to markedly increase endogenous antitumor immunity and provide robust and durable antitumor effects when combined with anti-PD-1 therapy.21,23,24 Recently, we reported the discovery of DRP-104 (Figure 1), a dipeptide prodrug consisting of an N-acetyl tryptophan moiety on the amino group of DON isopropyl ester.25 DRP-104 was shown to be preferentially transformed to DON in tumor cells resulting in an 11-fold greater delivery of DON to tumor versus GI tissues. DRP-104 caused robust inhibition of tumor growth in mice, similar to equimolar DON, but with markedly reduced GI side effects. Additionally, DRP-104 showed added benefits when combined with PD-1 therapy.25 Given this promising profile, DRP-104 was selected for clinical development as a single agent, as well as in combination with immunotherapy (identifier NCT04471415). While DRP-104 showed promising pharmacokinetics and robust efficacy in preclinical studies, it was metabolized to a charged, inactive metabolite, M1: (S)-2-((S)-2-acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoic acid.25 In addition, DRP-104 showed poor aqueous solubility (<1 mg/mL), necessitating formulation approaches for systemic administration.
In an attempt to discover prodrugs with improved stability, solubility, and DON tumor delivery, we designed and evaluated a series of tripeptide-based prodrugs of DON. We initially optimized moieties on DON’s carboxylate employing simple alkyl esters, cyclic esters, and amides. Next, using the GI-stable tert-butyl ester, we explored various acyl moieties at the amino group of the tryptophan residue on DRP-104. Lastly, we replaced the tryptophan on DRP-104 with smaller aromatic and aliphatic amino acids. These systematic structural changes improved the prodrugs’ physicochemical and pharmacokinetic properties.
Chemistry
DRP-104 (isopropyl (S)-2-((S)-2-acetamido-3-(1H-indol-3-yl)-propanamido)-6-diazo-5-oxo-hexanoate; also known as Sirpiglenastat) was identified as a lead glutamine antagonist with efficacy in multiple murine cancer models, including enhancement of immunotherapy.25−28 Metabolite identification (MET ID) studies revealed formation of the charged M1 metabolite via metabolism of the isopropyl ester.25 This metabolite was shown to be inert and inactive presumably leading to reduced systemic intact prodrug exposure.25 In an attempt to increase the stability of the ester moiety, we systematically replaced the isopropyl ester with several simple alkyl esters, cyclic esters, and amide. We introduced methyl (P1) and ethyl (P2) esters, which are commonly found in FDA-approved prodrugs.29−31 To enhance the metabolic stability of the ester promoiety, we synthesized sterically hindered cyclic ester based prodrugs, including cyclopropyl (P3) and methyl cyclopropyl (P4), and branched tert-butyl ester (P5).32,33 Prodrugs P1–P5 were synthesized by a seven-step procedure similar to our previously reported method (Scheme 1).18 Briefly, l-pyroglutamic acid 1 was converted to the respective pyroglutamate esters 2a–2e by reaction with thionyl chloride in methanol (2a) or ethanol (2b), Steglich esterification (2c and 2d), or acid catalyzed transesterification (2e). Ester intermediates 2a–2e were protected as Fmoc carbamates (3a–3e) using Fmoc-Cl and LiHMDS. The reaction of diazo(trimethylsilyl)methyllithium salt with protected pyroglutamate esters 3a–3e afforded the corresponding diazo ketones 4a–4e. Piperidine-mediated deprotection of the Fmoc group in 4a–4e gave free amines 5a–5e, which were coupled to Fmoc-l-Trp-OH activated with HATU in the presence of DIPEA to yield the corresponding dipeptides 6a–6e. The Fmoc protecting group was then removed by piperidine, resulting in amines 7a–7e, which were subsequently acetylated with acetic anhydride to afford the final prodrugs P1–P5. Notably, as illustrated in Table 1, there was a consistent increase in lipophilicity, as measured by cLogP (calculated using ChemDraw Professional 16.0), with the extension of ester chain length. This increase is also supported by cLogD7.4 (Table S1). For the sake of simplicity, we will primarily discuss cLogP in this context. The lipophilicity values for P1–P5 ranged from −0.05 to 1.19, with the tert-butyl variant exhibiting the highest cLogP of approximately 1.2. Notably, this value exceeded that of DRP-104, which measured 0.79. Next, the simple aliphatic methylamide prodrug P6 was synthesized by modifying DON’s carboxylic acid portion to an amide, as amides are known to be typically more resistant to cleavage compared to esters.34,35 However, this modification reduced the cLogP to a negative value of −0.91, suggesting high polarity and poor penetration to cellular membranes including tumor cells. As outlined in Scheme 1, amide analogue P6 was prepared from prodrug P2 in a one-step procedure using a methanolic solution of methylamine. The aim was to maximize DON delivery to the tumor while maintaining stability at off-target sites.
Scheme 1. Synthesis of Prodrugs P1–P6.
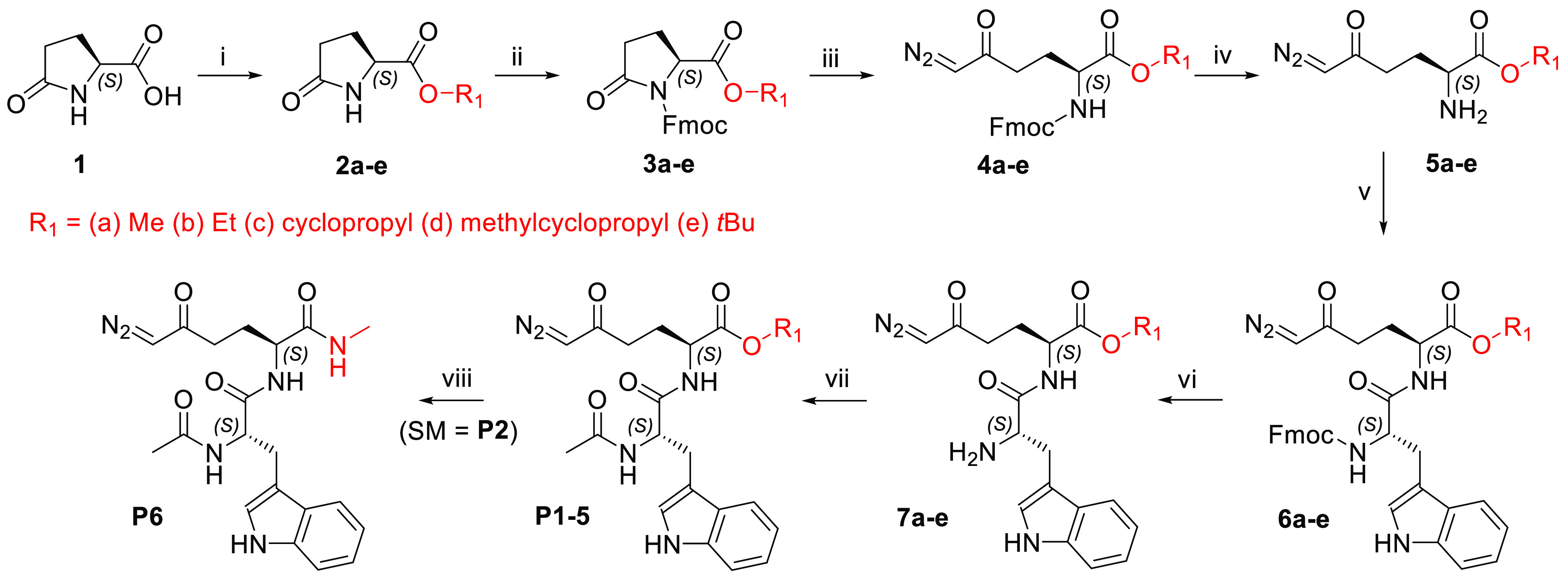
Reagents and conditions: (i) for 2a and 2b, R1–OH (MeOH or EtOH), SOCl2, 0 °C to rt, 16 h, 93–98%; for 2c and 2d, R1–OH (cyclopropanol or cyclopropylmethanol), DCC, DMAP, DCM, rt, 16 h, 96–97%; for 2e, tert-butyl acetate, perchloric acid, rt, 48 h, 90%; (ii) Fmoc-Cl, LiHMDS, THF, −78 °C to rt, 16 h, 63–95%; (iii) TMSCHN2, n-BuLi, THF, −78 °C, 3 h, 47–67%; (iv) piperidine, DCM, rt, 3 h, 49–67%; (v) Fmoc-l-Trp-OH, HATU, DIPEA, DCM, or DCM/DMF 4:1, 0 °C to rt, 1.5 h, 73–95%; (vi) diethylamine, DCM, rt, 3–6 h, 90–95%; (vii) Ac2O, py, DMF, rt, 3–15 h, 66–92%; (viii) (SM = P2), 2 M methylamine in MeOH, 60 °C, 20 h, 65%.
Table 1. cLogP and Stability of Prodrugs P1–P21 in Mouse Plasma and Intestinal Homogenate (GIh)a.

#, calculated using ChemDraw professional 16.0. ∗, CES1–\– mice intestinal homogenate (GIh) and plasma were used for stability assay.
Following optimization of the carboxylate moiety for stability, modifications were made to the acyl moiety of the tryptophan residue to enhance tumor delivery and prodrug solubility. To achieve this, the acetyl group of DRP-104 was replaced with morpholinomethyl (P7), quinuclidinyl (P8), and dimethylglycinyl (P9) (Scheme 2) to enhance cLogP to 1.61–1.70. These prodrugs were synthesized in one step from intermediate 7e using conditions for amide coupling in the presence of the appropriate carboxylic acid, i.e., morpholinoacetic acid, quinuclidine-4-carboxylic acid, or dimethylglycine, for P7, P8, and P9, respectively, in the presence of HATU and DIPEA.
Scheme 2. Synthesis of Prodrugs P7–P9.

Reagents and conditions: (i) R–COOH (for P7, morpholinoacetic acid hydrochloride; for P8, quinuclidine-4-carboxylic acid hydrochloride; for P9, dimethylglycine), HATU, DIPEA, DMF, 0 °C to rt, 2.5 h, 82–89%.
In the last part of our structure–property optimization study, we changed the structure by focusing on the amino acid of our tripeptide prodrugs. DRP-104 has low intrinsic aqueous solubility (<1 mg/mL). Thus, we aimed to identify the minimum structural requirements for tumor-targeted delivery with enhanced solubility, stability, and pharmacokinetic properties. As outlined in Scheme 3, we synthesized the prodrugs by replacing the tryptophan on DRP-104 with aromatic (P10–P17) and aliphatic amino acids (P18–P21), including standard (P11, P18–P20) and nonstandard amino acids (P10, P13–P17, and P21), fluorinated amino acids (P15–P17), and d-amino acid (P12). Prodrugs P10–P21 were prepared in a three-step synthetic procedure starting with intermediate 5e. Dipeptides 8a–8l were synthesized by a standard HATU coupling reaction between the appropriate Fmoc-protected amino acids and compound 5e. The Fmoc group was removed by diethylamine to afford intermediates 9a–9l in good to excellent yields. Final prodrugs P10–P21 were prepared by two different coupling conditions—with dimethylglycine activated with HATU in the presence of DIPEA (P10, P11, P18, P19) or with 2,5-dioxopyrrolidin-1-yl dimethylglycinate36 (P12–P17, P20, P21). Most of the synthesized prodrugs (P10–P17, P20, P21) retained a degree of lipophilicity (cLogP from 1.48 to 2.50) similar to that of DRP-104, except for prodrugs P18 (0.20) and P19 (−0.11) containing a smaller glycine or alanine moiety.
Scheme 3. Synthesis of Prodrugs P10–P21.
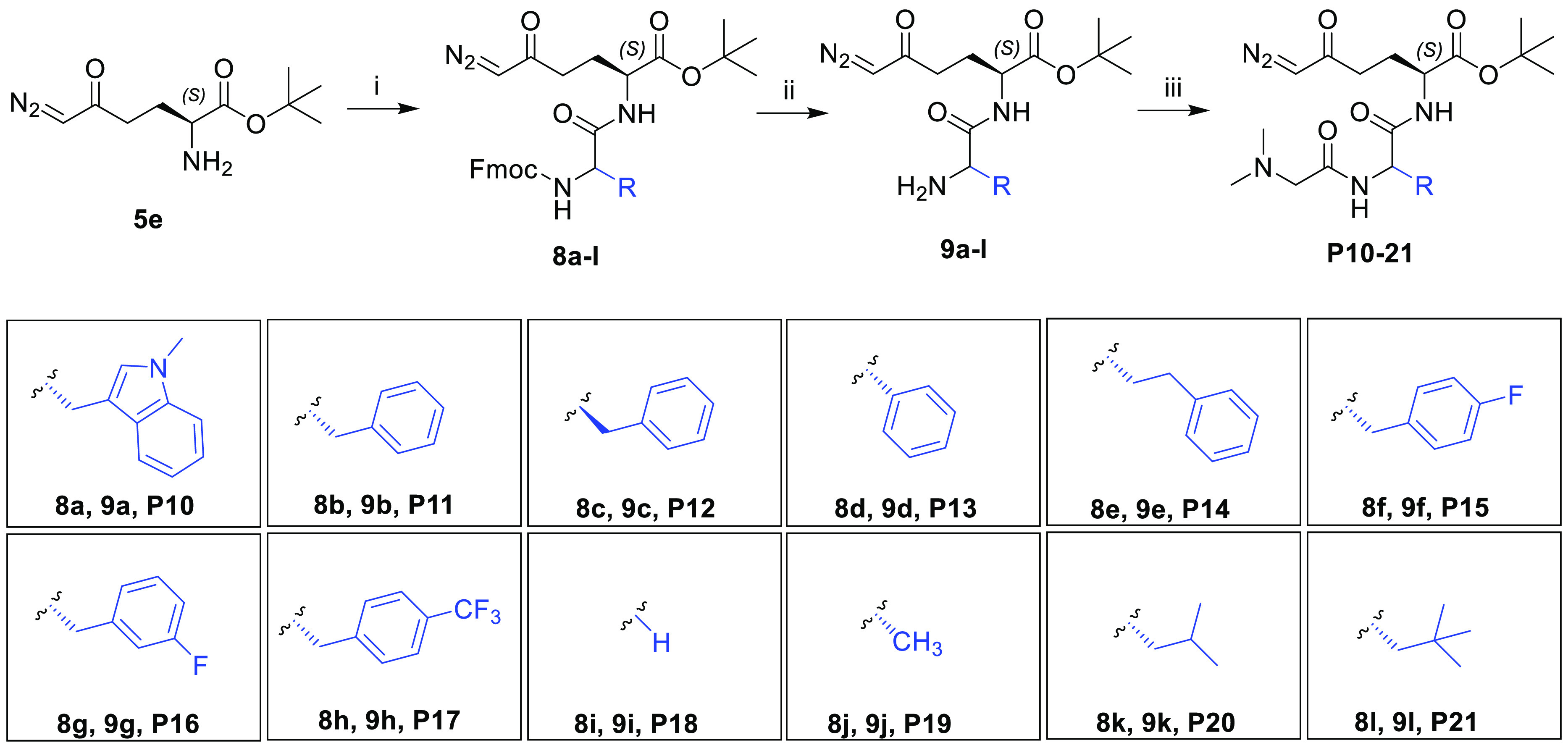
Reagents and conditions: (i) Fmoc-AA-OH (8a, Fmoc-l-Trp(N-Me)-OH; 8b, Fmoc-l-Phe-OH; 8c, Fmoc-d-Phe-OH; 8d, Fmoc-l-Phg-OH; 8e, Fmoc-l-HomoPhe-OH; 8f, Fmoc-l-Phe(4-F)-OH; 8g, Fmoc-l-Phe(3-F)-OH; 8h, Fmoc-l-Phe(4-CF3)-OH; 8i, Fmoc-Gly-OH; 8j, Fmoc-l-Ala-OH·H2O; 8k, Fmoc-l-Leu-OH; 8l, Fmoc-l-Ala(β-tBu)-OH), HATU, DIPEA, DCM, 0 °C to rt, 1.5–16 h, 68–98%; (ii) diethylamine, DCM, rt, 1.5–7 h, 76–96%; (iii) for P10, P11, P18, and P19, dimethylglycine, HATU, DIPEA, DCM, or DMF, 0 °C to rt, 1.5–2.5 h, 64–73%; for P12–P17, P20, and P21, 2,5-dioxopyrrolidin-1-yl dimethylglycinate, DCM, rt, 2–20 h, 51–92%.
Results and Discussion
Screening Strategy
The goal herein was to obtain prodrugs that could be effectively delivered to tumor cells while retaining stability in both the GI tract and plasma. To accomplish this, all prodrugs were systematically tested using a predefined screening paradigm. Drugs that were found to be stable in mice intestinal homogenate (GIh; >50% remaining at 1 h) were evaluated for stability in mice plasma (Table 1). Prodrugs showing stability in both matrices (>50% remaining at 1 h) were next evaluated in a single time point pharmacokinetic study in mice where plasma and tumor levels of DON were quantified (Figure 2). The prodrug with the best tumor DON levels and tumor/plasma ratio was then characterized in a full pharmacokinetic study in mice with functional tumor target engagement assessment (Figure 3). Selected prodrugs were also assessed for solubility, human tumor cell partitioning in a human plasma/tumor cell suspension assay, and human tumor cell viability assay (Figure 4) as detailed below.
Figure 2.

Single time point pharmacokinetic screening of selected prodrugs. Prodrugs (1 mg/kg DON equivalent) were administered subcutaneously (SC) to C57BL/6/CES1–/– mice and (A) DON levels released in plasma (red) and tumor (blue) were measured 30 min post dose and (B) the tumor to plasma ratio of released DON was calculated. Data expressed as mean ± SEM, n = 3. ∗, p < 0.05; ∗∗, p < 0.01; and ∗∗∗, p < 0.001, versus DRP-104 tumor levels (one-way ANOVA with Dunnett’s post hoc test).
Figure 3.
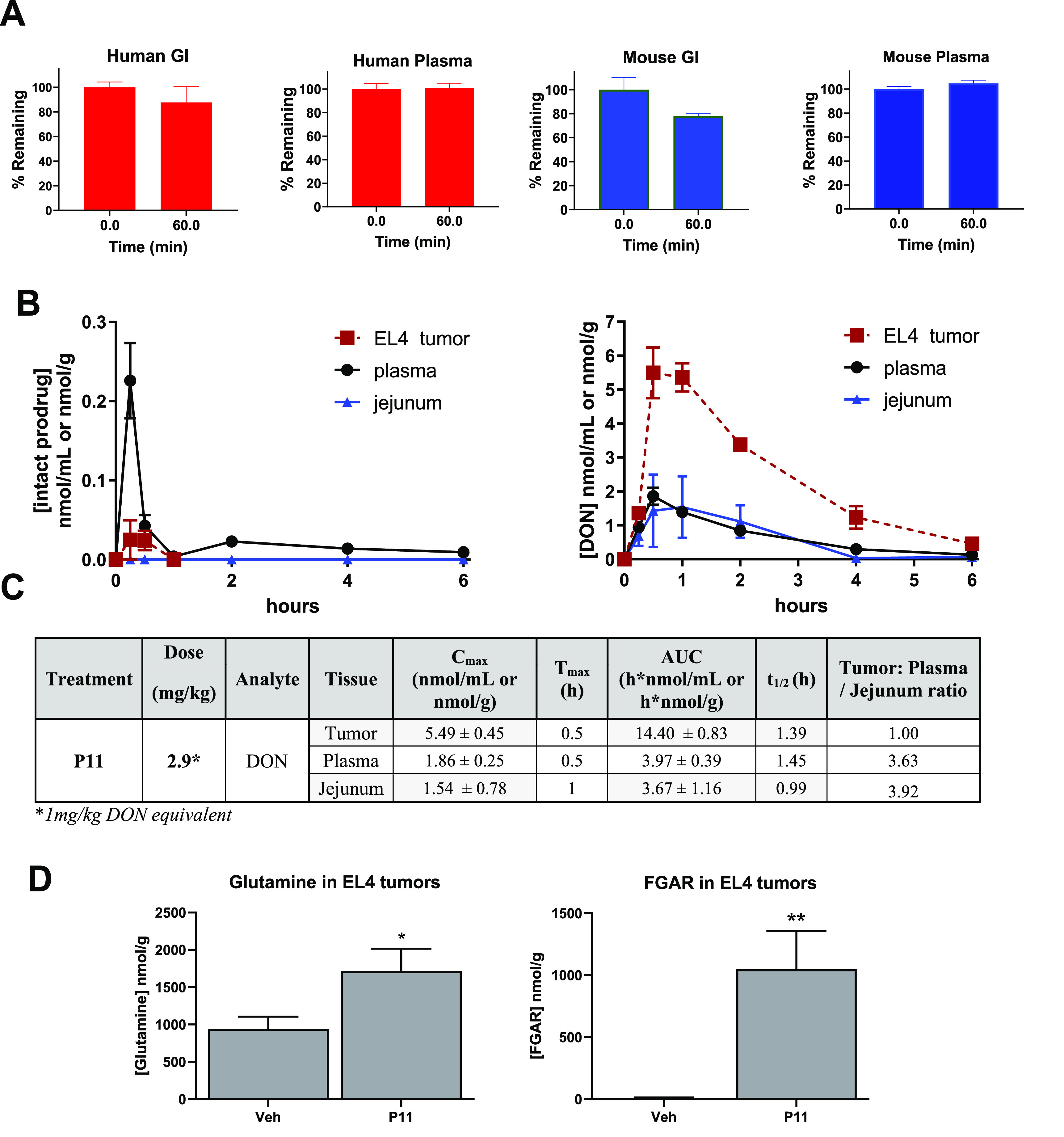
Stability, pharmacokinetic analysis, and tumor target engagement of P11. (A) Stability of P11 in human GI microsomes and plasma and mouse GI homogenate and mouse plasma. (B, C) P11 (2.9 mg/kg) was administered subcutaneously (SC) to C57BL/6/CES1–/– mice bearing EL4 tumors, and tissues were harvested and analyzed for (B) intact (P11) and released DON in tumor, plasma, and jejunum. (C) PK parameters of released DON. Data expressed as mean ± SEM, n = 3. EL4 tumors collected from these mice at Tmax were used for quantification of (D) tumor glutamine and FGAR quantification at 30 min post dose for target engagement evaluation (mean ± SD; ∗, p > 0.01; ∗∗, p > 0.001; unpaired two-tailed t test).
Figure 4.

Comparison of solubility, human tumor cell to plasma partitioning of DRP-104 and P11, and their antiproliferative effect. (A) Aqueous solubility of DRP-104 and P11 in buffer at pH 7.4. (B) Human tumor cell to plasma partitioning of DRP-104 and P11 were conducted by incubating either compound for 1 h in P493B lymphoma cells suspended in human plasma. Both intact DRP-104 and P11 levels were measured in human plasma and tumor cells, as well as prodrug-derived DON release in human plasma and tumor cells. (C) Cell viability assay was performed using P493B lymphoma cells incubated with DRP-104 and P11 for 72 h. Nonlinear regression analysis of the log-transformed data gave EC50 values.
Characterization of Metabolic Stability and Single Time Point Pharmacokinetics of Prodrugs P1–P21
Considering that the GI tract was the primary site of DON toxicity in clinical studies,8,14,16 minimizing DON release at this site was crucial. Thus, we sought to improve the GI stability of newly designed prodrugs. P1 with methyl, P2 with ethyl, P3 with cyclopropyl, and P4 with cyclopropylmethyl esters were all found to be unstable (<10% remaining at 1 h) in the GI homogenate as shown in Table 1. In contrast, P5 (tert-butyl ester) and P6 (methyl amide) were found to be stable (>50% remaining at 1 h). All subsequent prodrugs P7–P21 synthesized with the tert-butyl esters at the DON carboxylate, irrespective of the moieties at positions R2 and R3, were stable in the GI homogenate. For the stability assay, CES–/– mice37 were used, as these mice are generated by inactivating the CES1 gene such that there is undetectable CES activity in plasma but normal activity in tissues including the GI tissue. These data indicated that the primary site of metabolism for all prodrugs in the GI tract was the ester hydrolysis that occurred likely by the action of carboxylesterase enzyme CES1, as we have previously demonstrated.25 Interestingly, the tert-butyl ester was resistant to hydrolysis in GI tissue. Next, all the GI-stable prodrugs were evaluated in mouse plasma. Interestingly, all GI-stable prodrugs also exhibited stability in plasma with >50% remaining after a 1 h incubation. We next evaluated the plasma- and GI-stable prodrugs in a single time point pharmacokinetic (PK) study in CES1–/– mice bearing EL4 tumor. The C57BL/6/CES1–/– mice were generated by inactivating the CES1 gene such that there is undetectable CES activity in plasma but normal activity in tissues.37 CES1–/– mice were used as they mimic the distribution of CES1 in humans.38 These mice are often used in preclinical prodrug studies, including prior studies with DRP-104.25 The prodrugs were dosed subcutaneously (SC) at a dose of 1 mg/kg DON equivalent (n = 3 mice/group). After 30 min, the mice were sacrificed, and plasma and tumor samples were collected to measure the levels of released DON. This 30 min time point was selected as it corresponded to the time resulting in the maximal concentration of DON release following DRP-104 administration.25 Because maintaining a high tumor-to-plasma ratio was important, we quantified the release of DON in both plasma and tumor. The results from the single time point analysis of the DON release from prodrugs are shown in Figure 2. Of the 15 prodrugs evaluated, administration of P11 (5.49 ± 1.3 μM; p < 0.001), P14 (4.52 ± 0.90 μM; p < 0.01), and P17 (4.16 ± 1.13 μM; p < 0.05) led to significantly higher tumor concentrations of DON compared to administration of equimolar DRP-104 (1.21 ± 0.18 μM). Of these, P11 showed the highest tumor DON delivery with >4.5 higher DON levels compared to equimolar DRP-104. Most prodrugs, except for P5, P6, P18, and P19, also showed higher DON plasma levels compared to DRP-104. Notably, P11 and P14 maintained the preferential DON tumor versus plasma delivery as was observed for DRP-104. Given that P11 exhibited preferential tumor delivery and provided the highest DON tumor levels, it was selected to undergo a full-time-course pharmacokinetic evaluation as well as target engagement in EL4 tumor-bearing mice.
Stability, Pharmacokinetics, and Tumor Target Engagement of P11
The stability of P11 was confirmed in mouse and human plasma, as well as in GI matrices (Figure 3A). The results indicate similar stability between the two species, validating that the mouse model was suitable for PK studies. PK evaluation of P11 was performed in CES1–/– mice bearing flank murine EL4 lymphoma tumors. P11 was dosed via a subcutaneous (SC) route at 2.9 mg/kg (1 mg/kg DON equivalent dose), and plasma, tumor, and GI tissues were collected 0–6 h post dose. Tissues were analyzed for both the intact prodrug and DON release from the prodrug, using liquid chromatography with tandem mass spectrometry as we have previously described, with minor modifications.19,25Figure 3B,C illustrate the pharmacokinetic profile of P11 following subcutaneous dosing. P11 exhibited excellent pharmacokinetics, delivering DON preferentially to tumor cells with a maximum concentration (Cmax) of 5.49 ± 0.75 nmol/g compared to plasma (1.86 ± 0.25 nmol/mL) and intestinal tissue (1.54 ± 0.91 nmol/g), which were approximately 3-fold lower. In terms of overall exposure, P11 delivered approximately 3.6-fold higher tumor exposure of DON (area under curve, AUC0–t = 13.7 ± 0.90 h·nmol/g) versus that of plasma (3.8 ± 0.37 h·nmol/mL) and 4.4-fold higher tumor exposure versus that of jejunum (AUC = 3.13 ± 0.87 h·nmol/g). Intact prodrug P11 showed low levels in all matrices including plasma (AUC = 0.15 h·nmol/mL) and tumor (0.092 h·nmol/g). All intestinal tissue levels for the intact prodrug were below the limit of quantification (0.01 nmol/mL). These in vivo results confirmed preferential tumor distribution and efficient conversion of P11 to DON. We further confirmed target engagement of P11 by assessing the levels of glutamine and formylglycinamide ribonucleotide (FGAR) at the Tmax in tumor (30 min) (Figure 3D). These biomarkers were previously demonstrated to be significantly affected by DON treatment serving as efficient target engagement tools.39,40 We observed a significant, nearly 2-fold, rise in glutamine (from 941 ± 95 to 1710 ± 173 nmol/g) in tumors treated with P11 compared with tumor treated with vehicle. Similarly, there was a substantial 150-fold increase in FGAR (from 7.00 ± 3.00 to 1040 ± 179 nmol/g) in tumors treated with P11, as we have previously reported with other glutamine antagonist prodrugs.39,40 The increase in FGAR is observed due to DON’s inhibition of the enzyme FGAR amidotransferase (FGAR-AT) that catalyzes the ATP-dependent amidation of FGAR to formylglycinamidine ribonucleotide (FGAM) using glutamine as source of the amidic nitrogen.25 These data confirmed that P11 was effective at delivering DON to tumor and inhibiting the relevant mechanistic pathways.
Solubility, Human Tumor Cell Partitioning, and Antiproliferation Efficacy Assessment of P11
Next, P11 and DRP-104 were evaluated for their solubility, their ability to permeate and be cleaved to DON in human P493B lymphoma cells incubated in human plasma, and their ability to inhibit proliferation of human P493B lymphoma cells. Figure 4A illustrates the aqueous solubility of P11 (8.1 ± 1.1 mg/mL), which was 33 times greater than that of DRP-104 (0.24 ± 0.03 mg/mL). The chemical stability of prodrugs P11 and DRP-104 was evaluated in tandem using high resolution mass spectrometry (HRMS), confirming both prodrugs remained intact without any degradation during the solubility assay (Figure S1A–D). Figure 4B shows the tumor cell partitioning results where both DRP-104 and P11 were stable in human plasma with no DON release. In contrast, in the tumor cells, both DRP-104 and P11 showed both partitioning into and biotransformation to DON with tumor cell to plasma partitioning ratios of 180 and 140, respectively. Similar to the in vivo mouse studies, P11 provided a 5-fold increase in DON tumor cell levels when compared to DRP-104 (46.7 ± 1.2 μM versus 9.1 ± 0.15 μM). Moreover, consistent with their high cell partitioning, DRP-104 and P11 both exhibited excellent antiproliferative activity in a P493B lymphoma cell viability assay. A dose-dependent decrease in cell proliferation was observed following 72 h of incubation (Figure 4C). P11 caused a leftward shift in the viability curve where nonlinear regression analysis of the log-transformed data gave EC50 values for DRP-104 and P11 at 1 ± 0.2 and 0.30 ± 0.05 μM, respectively.
Conclusions
Over 20 prodrugs were systematically synthesized and characterized; among these, prodrug P11 emerged as the most promising. P11 showed metabolic stability in the GI tract and plasma and exhibited a >30-fold solubility improvement when compared to DRP-104. Additionally, in mice bearing flank EL4 lymphoma tumors, administration of P11 led to enhanced tumor DON exposure as well as significant increases in glutamine and FGAR levels, confirming target engagement. Furthermore, we evaluated the prodrug P11 in a human P493 lymphoma cell partitioning assay, where we confirmed the preferential tumor distribution and bioactivation of P11 to DON, with minimal DON release in plasma. Lastly, P11 exhibited excellent potency in a human tumor cell viability assay. In sum, we present the discovery of a new generation of DON prodrugs with improved biopharmaceutic and pharmacokinetic properties.
Importantly, it is crucial to highlight that even though we achieved significant progress in enhancing gastrointestinal (GI) stability, increasing overall DON tumor exposure, and successfully attaining a preferential tumor-targeting effect with P11, our study did not encompass in vivo assessments for dose-dependent toxicity or efficacy testing in tumor models. These investigations, which would substantiate that our findings translate into an enhanced therapeutic window, will be explored in our future research. In line with this, it should be noted that the DON AUCtumor:plasma ratio with P11 was similar to that with DRP-104. Furthermore, the AUCtumor:GI tissue ratio of ∼4 achieved by P11 is about 2–3-fold lower than those observed for other prodrugs, including DRP-104.25,41 Nonetheless, the systematic prodrug design strategies employed to identify P11 in this study can serve as a valuable blueprint for enhancing the pharmacokinetic profile and stability of other prodrugs with suboptimal properties and enable their clinical development.
Experimental Section
Commercially available reagents or HPLC grade solvents and materials were used for the synthesis of compounds described. All chemicals were reagent grade, purchased from Sigma-Aldrich, TCI, Combi-Blocks, AK Scientific, AstaTech. or Iris Biotech GmbH, and were used without further purification. TLC was performed on silica gel 60 F254 coated aluminum sheets (Merck), and spots were visualized with UV light and by the solution of Ce(SO4)2 × 4H2O (1%) and H3P(Mo3O10)4 (2%) in sulfuric acid (10%). Column chromatography was performed on silica gel 60 (0.040–0.063 mm, Fluka) or on a Biotage Isolera One Flash Chromatography System using SiliCycle SiliaSep cartridges with silica gel grade 40–63 μm. NMR spectra were measured on Bruker AVANCE 400 or Varian Oxford 500 instruments. 1H NMR were recorded at 401 or 500 MHz, and signals of TMS (δ 0.0, CDCl3), CDCl3 (δ 7.26), and d6-DMSO (δ 2.50, 3.33) were used for standardization. 13C NMR spectra were recorded at 101 or 125 MHz, and the signal of CDCl3 (δ 77.16) or d6-DMSO (δ 39.52) was used for standardization. The chemical shifts are given in δ scale; the coupling constants J are given in hertz. The low resolution ESI mass spectra were recorded using a ZQ micromass mass spectrometer (Waters) or an Agilent 1200 series HPLC system. High resolution ESI mass spectra were recorded using an LTQ Orbitrap XL spectrometer (Thermo Fisher Scientific). Preparative HPLC purification was performed on an Agilent 1200 series HPLC system with an Agilent G1315D DAD detector (methods). All compounds subjected to biological testing were >95% pure by HPLC analysis.
Methyl (S)-5-Oxopyrrolidine-2-carboxylate (2a)
Compound 2a was synthesized according to the published procedure,42 and the 1H NMR spectrum aligned with published data.
Ethyl (S)-5-Oxopyrrolidine-2-carboxylate (2b)
Compound 2b was synthesized according to the published procedure,43 and the 1H NMR spectrum aligned with published data.
General Method for Synthesis of Esters 2c and 2d
l-Pyroglutamic acid 2 (2.00 g, 15.5 mmol, 1 equiv) was dissolved in anhydrous DCM (30 mL) and the corresponding alcohol (46.5 mmol, 3 equiv) was added, followed by DMAP (94.6 mg, 0.775 mmol, 0.05 equiv) and DCC (3.52 g, 17.0 mmol, 1.1 equiv). The resulting mixture was stirred at rt under nitrogen atmosphere for 20 h. The precipitate (DCU) was filtered off and volatiles were removed under reduced pressure. The residue was redissolved in a small amount of cold EtOAc (10 mL), and the remaining precipitate was removed by a second filtration. Solvent was evaporated and the crude material was purified by LC on silica gel (DCM/MeOH, 30:1) to afford desired products 2c and 2d.
Cyclopropyl (S)-5-Oxopyrrolidine-2-carboxylate (2c)
Cyclopropanol (2.70 g, 3.03 mL). Product 2c was isolated as a colorless oil (2.40 g) in 92% yield. 1H NMR (500 MHz, CDCl3): δ 0.67–0.78 (m, 4H), 2.12–2.22 (m, 1H), 2.26–2.49 (m, 3H), 4.09–4.32 (m, 2H), 6.68 (bs, 1H). ESI MS: 170.1 ([M + H]+).
Cyclopropylmethyl (S)-5-Oxopyrrolidine-2-carboxylate (2d)
Cyclopropylmethanol (3.35 g, 3.76 mL). Product 2d was isolated as a colorless oil (2.51 g) in 88% yield. 1H NMR (500 MHz, CDCl3): δ 0.21–0.37 (m, 2H), 0.49–0.65 (m, 2H), 1.06–1.21 (m, 1H), 2.20–2.28 (m, 1H), 2.30–2.46 (m, 2H), 2.46–2.54 (m, 1H), 3.99 (d, J = 7.4, 2H), 4.26 (ddd, J = 8.9, 5.1, 0.7, 1H), 6.26 (bs, 1H). ESI MS: 184.2 ([M + H]+).
tert-Butyl (S)-5-Oxopyrrolidine-2-carboxylate (2e)
Compound 2e was synthesized according to the published procedure,44 and the 1H NMR spectrum aligned with published data.
General Method for Synthesis of Fmoc-Protected Compounds 3a–3e
A solution of esters 2a–2e (14.0 mmol, 1 equiv) in anhydrous THF (40 mL) was cooled to −78 °C under inert nitrogen atmosphere. LiHMDS (1 M in THF; 13.3 mL, 13.3 mmol, 0.95 equiv) was added dropwise during 10 min, and the mixture was stirred for an additional 15 min at the same temperature. Then it was transferred via cannula to a solution of Fmoc-Cl (4.34 g, 16.8 mmol, 1.2 equiv) in anhydrous THF (60 mL) cooled to −78 °C. The resulting mixture was stirred at −78 °C for 2 h and at rt for 16 h. The reaction was quenched with saturated NH4Cl (100 mL), and the aqueous phase was extracted with EtOAc (3 × 100 mL). Combined organic phases were washed with brine (2 × 100 mL) and dried over anhydrous MgSO4. Volatiles were evaporated in vacuo, and the residue was chromatographed on a Biotage Flash chromatography (CHCl3/0–100% EtOAc).
1-((9H-Fluoren-9-yl)methyl) 2-Methyl (S)-5-Oxopyrrolidine-1,2-dicarboxylate (3a)
Starting material 2a (2.00 g). Product 3a was isolated as a colorless solid (4.85 g) in 95% yield. 1H NMR (500 MHz, CDCl3): δ 2.07–2.24 (m, 1H), 2.39 (ddt, J = 13.4, 10.7, 9.4, 1H), 2.57 (ddd, J = 17.6, 9.3, 3.1, 1H), 2.71 (ddd, J = 17.5, 10.8, 9.4, 1H), 3.74 (s, 3H), 4.30 (t, J = 7.2, 1H), 4.46 (dd, J = 10.6, 7.3, 1H), 4.58 (dd, J = 10.6, 7.2, 1H), 4.65 (dd, J = 9.5, 2.5, 1H), 7.28–7.38 (m, 2H), 7.41 (t, J = 7.4, 2H), 7.70 (d, J = 7.5, 1H), 7.75 (d, J = 7.5, 1H), 7.77 (d, J = 7.5, 2H). ESI MS: 366.1 ([M + H]+).
1-((9H-Fluoren-9-yl)methyl) 2-Ethyl (S)-5-Oxopyrrolidine-1,2-dicarboxylate (3b)
Compound 3b was synthesized according to the published procedure,18 and the 1H NMR spectrum aligned with published data.
1-((9H-Fluoren-9-yl)methyl) 2-Cyclopropyl (S)-5-Oxopyrrolidine-1,2-dicarboxylate (3c)
Starting material 2c (2.37 g). Product 3c was isolated as a light-yellow solid (3.42 g) in 63% yield. 1H NMR (500 MHz, CDCl3): δ 0.66–0.79 (m, 4H), 2.09 (ddt, J = 13.4, 9.5, 3.0, 1H), 2.38 (ddt, J = 13.5, 10.6, 9.4, 1H), 2.57 (ddd, J = 17.6, 9.4, 3.2, 1H), 2.72 (ddd, J = 17.6, 10.6, 9.5, 1H), 4.17–4.26 (m, 1H), 4.31 (t, J = 7.4, 1H), 4.43 (dd, J = 10.6, 7.5, 1H), 4.49–4.68 (m, 2H), 7.33 (tdd, J = 7.5, 3.0, 1.2, 2H), 7.37–7.46 (m, 2H), 7.71 (dd, J = 7.5, 1.0, 1H), 7.74–7.79 (m, 3H). ESI MS: 392.2 ([M + H]+).
1-((9H-Fluoren-9-yl)methyl) 2-Cyclopropylmethyl (S)-5-Oxopyrrolidine-1,2-dicarboxylate (3d)
Starting material 2d (2.56 g). Product 3d was isolated as a light-yellow solid (3.98 g) in 70% yield. 1H NMR (500 MHz, CDCl3): δ 0.27 (d, J = 4.8, 2H), 0.55 (d, J = 8.0, 2H), 1.03–1.21 (m, 1H), 1.81–1.90 (m, 1H), 2.08–2.24 (m, 1H), 2.35–2.52 (m, 1H), 2.58 (ddd, J = 17.6, 9.3, 2.9, 1H), 2.73 (dt, J = 16.8, 10.0, 1H), 3.65–3.83 (m, 1H), 3.91–4.09 (m, 2H), 4.31 (t, J = 7.4, 1H), 4.44 (dd, J = 10.7, 7.4, 1H), 4.55 (dd, J = 10.5, 7.4, 1H), 4.70 (dd, J = 9.5, 2.5, 1H), 7.33 (t, J = 7.5, 2H), 7.41 (t, J = 7.5, 2H), 7.61–7.84 (m, 4H). ESI MS: 406.2 ([M + H]+).
1-((9H-Fluoren-9-yl)methyl) 2-tert-Butyl (S)-5-Oxopyrrolidine-1,2-dicarboxylate (3e)
Compound 3e was synthesized according to the published procedure,45 and the 1H NMR spectrum was in agreement with published data.
General Method for Synthesis of Compounds 4a–4e
Trimethylsilyl diazomethane (2 M solution in hexanes; 6.0 mL, 12.0 mmol, 1.2 equiv) was dissolved in anhydrous THF (75 mL), the reaction mixture was cooled to −98 °C, n-BuLi (2.5 M in hexanes; 4.9 mL, 12.3 mmol, 1.23 equiv) was added dropwise during 15 min, and the resulting yellow mixture was stirred for 30 min at the same temperature. This solution was transferred via cannula to a solution of compounds 3a–3e (10 mmol, 1 equiv) in anhydrous THF (100 mL) during 30 min under inert atmosphere at −98 °C. The resulting mixture was stirred for 30 min at the same temperature and then was allowed to heat to −78 °C and stirred for a further 2 h. The mixture was then quenched with saturated NH4Cl (50 mL) and water (50 mL). Phases were separated, the water phase was extracted with EtOAc (2 × 150 mL), and combined organic layers were washed with brine (100 mL) and dried over anhydrous MgSO4. Volatiles were removed under reduced pressure, and the solid residue was purified by LC on silica gel (cyclohexane/EtOAc, 2:1 to 1:1).
Methyl (S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-diazo-5-oxohexanoate (4a)
Starting material 3a (3.65 g). Product 4a was isolated as a light-yellow solid (2.65 g) in 65% yield. 1H NMR (500 MHz, CDCl3): δ 1.92–2.01 (m, 1H), 2.16–2.32 (m, 1H), 2.31–2.54 (m, 2H), 3.76 (s, 3H), 4.23 (t, J = 7.1, 1H), 4.34–4.45 (m, 3H), 5.26 (bs, 1H), 5.53 (d, J = 8.1, 1H), 7.28–7.37 (m, 2H), 7.41 (dd, J = 8.4, 6.6, 2H), 7.60 (t, J = 7.2, 2H), 7.77 (d, J = 7.5, 2H). ESI MS: 430.1 ([M + Na]+).
Ethyl (S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-diazo-5-oxohexanoate (4b)
Compound 4b was synthesized according to the published procedure,18 and the 1H NMR spectrum was in agreement with published data.
Cyclopropyl (S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-diazo-5-oxohexanoate (4c)
Starting material 3c (3.91 g). Product 4c was isolated as a light-yellow solid (2.05 g) in 47% yield. 1H NMR (500 MHz, CDCl3): δ 0.60–0.85 (m, 4H), 1.84–2.09 (m, 1H), 2.20 (s, 1H), 2.28–2.53 (m, 2H), 4.15–4.26 (m, 2H), 4.27–4.48 (m, 2H), 5.27 (bs, 1H), 5.51 (d, J = 8.2, 1H), 7.32 (t, J = 7.4, 2H), 7.41 (t, J = 7.5, 2H), 7.60 (t, J = 6.6, 2H), 7.77 (d, J = 7.6, 2H). ESI MS: 456.2 ([M + Na]+).
Cyclopropylmethyl (S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-diazo-5-oxohexanoate (4d)
Starting material 3d (4.05 g). Product 4d was isolated as a light-yellow solid (2.31 g) in 52% yield. 1H NMR (500 MHz, CDCl3): δ 0.29 (d, J = 5.2, 2H), 0.58 (d, J = 7.8, 2H), 1.98–2.11 (m, 1H), 2.18–2.32 (m, 1H), 2.33–2.55 (m, 2H), 3.99 (dd, J = 7.4, 4.0, 2H), 4.23 (t, J = 7.0, 1H), 4.34–4.46 (m, 3H), 5.27 (bs, 1H), 5.53 (d, J = 8.1, 1H), 7.32 (t, J = 7.4, 2H), 7.41 (t, J = 7.5, 2H), 7.60 (t, J = 7.1, 2H), 7.77 (d, J = 7.5, 2H). ESI MS: 470.2 ([M + Na]+).
tert-Butyl (S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-6-diazo-5-oxohexanoate (4e)
Starting material 3e (4.07 g). Product 4e was isolated as a light-yellow solid (3.03 g) in 67% yield. 1H NMR (401 MHz, CDCl3): δ 1.50 (s, 9H), 1.94–2.07 (m, 1H), 2.18–2.29 (m, 1H), 2.31–2.53 (m, 2H), 4.21–4.32 (m, 2H), 4.41 (d, J = 7.1, 2H), 5.29 (bs, 1H), 5.50 (d, J = 8.1, 1H), 7.34 (tt, J = 7.4, 1.4, 2H), 7.43 (t, J = 7.4, 2H), 7.63 (dd, J = 7.7, 4.0, 2H), 7.79 (d, J = 7.3, 2H). ESI MS: 472.2 ([M + Na]+).
General Method for Synthesis of Compounds 5a–5e
Compounds 4a–4e (4.00 mmol, 1 equiv) were dissolved in anhydrous DCM (18 mL), and piperidine (1.70 g, 1.98 mL, 20.0 mmol, 5 equiv) was added. The reaction mixture was stirred at rt under an inert atmosphere for 2–3.5 h. Volatiles were evaporated, and the residue was purified by LC on silica gel (DCM/MeOH, 30:1).
Methyl (S)-2-Amino-6-diazo-5-oxohexanoate (5a)
Starting material 4a (1.63 g); reaction time 2 h. Product 5a was isolated as a light-yellow oil (532 mg) in 72% yield. 1H NMR (401 MHz, CDCl3): δ 1.53 (bs, 2H), 1.76–1.87 (m, 1H), 2.03–2.15 (m, 1H), 2.42–2.55 (m, 2H), 3.44 (dd, J = 8.3, 5.1, 1H), 3.64 (s, 3H), 5.26 (bs, 1H). ESI MS: 186.1 ([M + H]+).
Ethyl (S)-2-Amino-6-diazo-5-oxohexanoate (5b)
Compound 5b was synthesized according to the published procedure,18 and the 1H NMR spectrum was in agreement with published data.
Cyclopropyl (S)-2-Amino-6-diazo-5-oxohexanoate (5c)
Starting material 4c (1.73 g); reaction time 3 h. Product 5c was isolated as a light-yellow oil (567 mg) in 67% yield. 1H NMR (401 MHz, CDCl3): δ 0.63–0.78 (m, 4H), 1.46–1.54 (m, 2H), 1.80 (dq, J = 14.5, 7.4, 1H), 2.02–2.14 (m, 1H), 2.41–2.53 (m, 2H), 3.39–3.42 (m, 1H), 4.14–4.18 (m, 1H), 5.27 (bs, 1H). ESI MS: 234.1 ([M + Na]+).
Cyclopropylmethyl (S)-2-Amino-6-diazo-5-oxohexanoate (5d)
Starting material 4d (1.79 g); reaction time 3 h. Product 5d was isolated as a light-yellow oil (443 mg) in a 49% yield. 1H NMR (401 MHz, CDCl3): δ 0.23–0.35 (m, 2H), 0.54–0.64 (m, 2H), 1.09–1.20 (m, 1H), 1. 52–1.61 (m, 2H), 2.10–2.21 (m, 1H), 2.30–2.40 (m, 1H), 2.55–2.77 (m, 2H), 3.95–4.08 (m, 3H), 5.54 (bs, 1H). ESI MS: 226.1 ([M + H]+).
tert-Butyl (S)-2-Amino-6-diazo-5-oxohexanoate (5e)
Starting material 4e (1.80 g); reaction time 3.5 h. Product 5e was isolated as a light-yellow oil (611 mg) in 67% yield. 1H NMR (401 MHz, CDCl3): δ 1.43 (s, 9H), 1.58 (bs, 2H), 1.72–1.81 (m, 1H), 1.99–2.09 (m, 1H), 2.38–2.50 (m, 2H), 3.30 (dd, J = 8.3, 5.0, 1H), 5.27 (bs, 1H). ESI MS: 228.1 ([M + H]+).
General Method for Synthesis of Compounds 6a–6e
Fmoc-l-Trp-OH (938 mg, 2.20 mmol, 1.1 equiv) and HATU (913 mg, 2.40 mmol, 1.2 equiv) were suspended in anhydrous DCM (20 mL) under inert atmosphere, and the reaction mixture was cooled to 0 °C. DIPEA (775 mg, 1.05 mL, 6.00 mmol, 3 equiv) was added, and the mixture was stirred for 5 min. Finally, a solution of amines 5a, 5c, and 5e (2.00 mmol, 1 equiv) in anhydrous DCM (10 mL) was slowly added over 5 min. The resulting mixture was stirred for 30 min at 0 °C and then for 2 h at rt. DCM (50 mL) was added, and the organic phase was washed with saturated NaHCO3 (50 mL), distilled H2O (50 mL), 10% KHSO4 (50 mL), distilled H2O (50 mL), and saturated NaCl (50 mL) and dried over anhydrous MgSO4. DCM was evaporated in vacuo, and the residue was purified by LC on silica gel (DCM/EtOAc, 3:1).
Methyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (6a)
Starting material 5a (370 mg). Product 6a was isolated as a light-yellow solid (1.01 g) with 85% yield. 1H NMR (401 MHz, CDCl3): δ 1.85–1.97 (m, 1H), 2.08–2.18 (m, 2H), 2.18–2.28 (m, 1H), 3.19 (dd, J = 14.5, 7.4, 1H), 3.41 (d, J = 8.6, 1H), 3.67 (s, 3H), 4.24 (t, J = 7.1, 1H), 4.34–4.51 (m, 3H), 4.52–4.60 (m, 1H), 5.08 (bs, 1H), 5.50 (d, J = 7.8, 1H), 6.57 (d, J = 7.2, 1H), 7.10 (bs, 1H), 7.16 (t, J = 7.4, 1H), 7.23 (ddd, J = 8.2, 7.0, 1.2, 1H), 7.33 (tdd, J = 7.5, 2.4, 1.1, 2H), 7.37–7.40 (m, 1H), 7.41–7.46 (m, 2H), 7.59 (dd, J = 7.5, 5.0, 2H), 7.70 (d, J = 7.8, 1H), 7.79 (d, J = 7.5, 2H), 8.19 (bs, 1H). ESI MS: 616.2 ([M + Na]+).
Ethyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (6b)
Compound 6b was synthesized according to the published procedure,18 and the 1H NMR spectrum was in agreement with published data.
Cyclopropyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (6c)
Starting material 5c (422 mg). Product 6c was isolated as a light-yellow solid (903 mg) in a 73% yield. 1H NMR (500 MHz, CDCl3): δ 0.58–0.76 (m, 4H), 1.75–1.95 (m, 1H), 1.96–2.27 (m, 3H), 3.17 (dd, J = 14.5, 7.3, 1H), 3.33–3.52 (m, 1H), 4.09 (s, 1H), 4.21 (t, J = 7.1, 1H), 4.29–4.63 (m, 4H), 5.05 (s, 1H), 5.46 (bs, 1H), 7.09 (bs, 1H), 7.15 (t, J = 7.5, 1H), 7.21 (t, J = 7.5, 1H), 7.28–7.34 (m, 2H), 7.35–7.46 (m, 3H), 7.57 (t, J = 7.0, 2H), 7.69 (d, J = 8.0, 1H), 7.77 (d, J = 7.6, 2H), 8.15 (bs, 1H). ESI MS: 642.2 ([M + Na]+).
Cyclopropylmethyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (6d)
Starting material 5d (451 mg). Product 6d was isolated as a light-yellow solid (1.20 g) in 95% yield. 1H NMR (500 MHz, CDCl3): δ 0.26 (s, 2H), 0.56 (d, J = 8.1, 2H), 1.08 (s, 1H), 1.83–2.02 (m, 1H), 2.02–2.37 (m, 3H), 3.17 (dd, J = 14.6, 7.3, 1H), 3.28–3.54 (m, 1H), 3.77–4.01 (m, 2H), 4.21 (t, J = 7.1, 1H), 4.27–4.42 (m, 1H), 4.41–4.50 (m, 2H), 4.50–4.61 (m, 1H), 5.07 (s, 1H), 5.46 (bs, 1H), 6.53 (bs, 1H), 7.08 (bs, 1H), 7.14 (t, J = 7.5, 1H), 7.20 (t, J = 7.5, 1H), 7.31 (td, J = 7.4, 3.2, 2H), 7.37 (d, J = 8.1, 1H), 7.40 (t, J = 7.4, 2H), 7.57 (t, J = 7.3, 2H), 7.68 (d, J = 7.9, 1H), 7.77 (d, J = 7.6, 2H), 8.15 (bs, 1H). ESI MS: 656.3 ([M + Na]+).
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (6e)
Starting material 5e (455 mg). Product 6e was isolated as a light-yellow solid (1.07 g) with 84% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.41 (s, 9H), 1.78–1.88 (m, 1H), 2.04–2.14 (m, 1H), 2.32–2.44 (m, 2H), 2.95 (dd, J = 14.7, 10.5, 1H), 3.12 (dd, J = 14.6, 4.0, 1H), 4.11–4.19 (m, 4H), 4.34 (ddd, J = 10.0, 8.3, 3.9, 1H), 6.02 (bs, 1H), 6.99 (t, J = 7.3, 1H), 7.07 (t, J = 7.2, 1H), 7.21 (d, J = 2.3, 1H), 7.25 (td, J = 7.5, 1.1, 1H), 7.30–7.36 (m, 2H), 7.36–7.45 (m, 2H), 7.53 (d, J = 8.5, 1H), 7.62 (d, J = 7.5, 1H), 7.66 (d, J = 7.4, 1H), 7.70 (d, J = 7.8, 1H), 7.88 (d, J = 7.5, 2H), 8.38 (d, J = 7.5, 1H), 10.83 (bs, 1H). ESI MS: 658.3 ([M + Na]+).
General Method for Synthesis of Compounds 7a–7e
Compounds 6a–6e (1.50 mmol, 1 equiv) were dissolved in anhydrous DCM (10 mL), diethylamine (1.10 g, 1.55 mL, 15.0 mmol, 10 equiv) was added, and the reaction mixture was stirred at rt under inert atmosphere for 3 h. Volatiles were evaporated, and the residue was purified by LC on silica gel (DCM/MeOH, 30:1 to 20:1).
Methyl (S)-2-((S)-2-Amino-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (7a)
Starting material 6a (890 mg). Product 7a was isolated as a light-yellow solid (514 mg) in 92% yield. 1H NMR (500 MHz, CDCl3): δ 1.91–2.01 (m, 1H), 2.05–2.29 (m, 1H), 2.55–2.67 (m, 2H), 3.06 (dd, J = 14.4, 8.1, 1H), 3.30 (dd, J = 14.4, 4.2, 1H), 3.73 (s, 3H), 3.74–3.78 (m, 1H), 4.57 (td, J = 8.2, 4.2, 1H), 5.11 (bs, 1H), 7.06–7.15 (m, 2H), 7.21 (t, J = 7.6, 1H), 7.37 (d, J = 8.1, 1H), 7.69 (d, J = 7.9, 1H), 7.86 (d, J = 8.3, 1H), 8.20 (bs, 1H). ESI MS: 394.2 ([M + Na]+).
Ethyl (S)-2-((S)-2-Amino-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (7b)
Compound 7b was synthesized according to the published procedure,18 and the 1H NMR spectrum was in agreement with published data.
Cyclopropyl (S)-2-((S)-2-Amino-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (7c)
Starting material 6c (930 mg). Product 7c was isolated as a light-yellow solid (536 mg) in a 90% yield. 1H NMR (500 MHz, CDCl3): δ 0.57–0.89 (m, 4H), 1.32–1.50 (m, 2H), 1.86–1.99 (m, 1H), 2.03–2.28 (m, 3H), 3.04 (dd, J = 14.4, 8.1, 1H), 3.30 (dd, J = 14.4, 4.2, 1H), 3.75 (dd, J = 8.1, 4.2, 1H), 4.11–4.19 (m, 1H), 4.51 (td, J = 8.4, 4.2, 1H), 5.10 (bs, 1H), 6.99–7.17 (m, 2H), 7.21 (ddd, J = 8.2, 7.1, 1.2, 1H), 7.37 (d, J = 8.2, 1H), 7.69 (d, J = 7.9, 1H), 7.87 (d, J = 8.3, 1H), 8.15 (bs, 1H). ESI MS: 420.2 ([M + Na]+).
Cyclopropylmethyl (S)-2-((S)-2-Amino-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (7d)
Starting material 6d (951 mg). Product 7d was isolated as a light-yellow solid (586 mg) in 95% yield. 1H NMR (401 MHz, CDCl3): δ 0.21–0.35 (m, 2H), 0.50–0.64 (m, 2H), 0.95–1.21 (m, 1H), 1.85–2.06 (m, 1H), 2.06–2.36 (m, 3H), 3.05 (dd, J = 14.4, 8.1, 1H), 3.30 (dd, J = 14.4, 4.2, 1H), 3.74 (dd, J = 8.1, 4.1, 1H), 3.95 (dd, J = 7.4, 1.5, 2H), 4.58 (td, J = 8.2, 3.7, 1H), 5.12 (bs, 1H), 7.06–7.15 (m, 2H), 7.19 (t, J = 7.5, 1H), 7.37 (d, J = 8.0, 1H), 7.68 (d, J = 7.9, 1H), 7.91 (d, J = 8.4, 1H), 8.42 (bs, 1H). ESI MS: 412.2 ([M + H]+).
tert-Butyl (S)-2-((S)-2-Amino-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (7e)
Starting material 6e (954 mg). Product 7e was isolated as a light-yellow solid (579 mg) in a 93% yield. 1H NMR (401 MHz, CDCl3): δ 1.48 (s, 9H), 1.88–2.00 (m, 1H), 2.01–2.28 (m, 5H), 3.07 (dd, J = 14.5, 8.1, 1H), 3.33 (dd, J = 14.5, 3.9, 1H), 3.79 (dd, J = 8.2, 4.1, 1H), 4.47 (td, J = 8.3, 4.2, 1H), 5.16 (bs, 1H), 7.09–7.16 (m, 2H), 7.21 (ddd, J = 8.1, 7.0, 1.2, 1H), 7.38 (dt, J = 8.1, 0.9, 1H), 7.69 (dd, J = 7.9, 1.0, 1H), 7.92 (d, J = 8.2, 1H), 8.51 (bs, 1H). ESI MS: 414.2 ([M + H]+).
General Method for Synthesis of Prodrugs P1–P5
To the solution of amines 7a–7e (0.300 mmol, 1 equiv) in anhydrous DMF (2 mL), pyridine (48 mg, 48 μL, 0.600 mmol, 2 equiv) was added followed by acetic anhydride (34 mg, 31 μL, 0.330 mmol, 1.1 equiv) at rt under inert atmosphere. The mixture was stirred at the same temperature for 2 h. Then, volatiles were removed under reduced pressure, and the residue was purified by LC on silica gel (DCM/MeOH, 30:1).
Methyl (S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (P1)
Starting material 7a (111 mg). Product P1 was isolated as a light-yellow solid (93 mg) in 75% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.76 (s, 3H), 1.77–1.91 (m, 1H), 1.92–2.07 (m, 1H), 2.37 (s, 2H), 2.81–2.95 (m, 1H), 3.00–3.15 (m, 1H), 3.61 (s, 3H), 4.19–4.34 (m, 1H), 4.47–4.62 (m, 1H), 6.03 (bs, 1H), 6.98 (t, J = 7.5, 1H), 7.06 (t, J = 7.5, 1H), 7.14 (s, 1H), 7.32 (d, J = 8.1, 1H), 7.62 (d, J = 7.9, 1H), 8.07 (d, J = 8.1, 1H), 8.46 (d, J = 7.6, 1H), 10.82 (bs, 1H). 13C NMR (101 MHz, d6-DMSO): δ 23.01, 26.24, 28.14, 51.71, 52.38, 53.59, 110.50, 111.72, 118.62, 118.94, 121.29, 124.09, 127.75, 136.48, 169.60, 172.56, 172.62, 194.63. ESI MS: 436.2 ([M + Na]+). HR ESI MS: calcd for C20H23N5NaO5 436.1592; found 436.1591.
Ethyl (S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (P2)
Starting material 7b (116 mg). Product P2 was isolated as a light-yellow solid (85 mg) in 66% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.17 (t, J = 7.1, 3H), 1.66–1.91 (m, 4H), 1.91–2.11 (m, 1H), 2.26–2.45 (m, 2H), 2.87 (dd, J = 14.7, 9.6, 1H), 3.08 (dd, J = 14.7, 4.5, 1H), 4.07 (td, J = 7.2, 6.2, 2H), 4.23 (ddd, J = 9.3, 7.4, 5.1, 1H), 4.46–4.65 (m, 1H), 6.04 (bs, 1H), 6.98 (ddd, J = 7.9, 7.0, 1.1, 1H), 7.06 (ddd, J = 8.1, 6.9, 1.2, 1H), 7.14 (d, J = 2.3, 1H), 7.31 (d, J = 7.8, 1H), 7.62 (d, J = 7.8, 1H), 8.06 (d, J = 8.1, 1H), 8.45 (d, J = 7.6, 1H), 10.82 (bs, 1H). 13C NMR (101 MHz, d6-DMSO): δ 14.49, 23.00, 26.26, 28.18, 51.84, 53.56, 61.01, 110.56, 111.73, 118.62, 118.92, 121.30, 124.07, 127.74, 136.49, 169.58, 172.05, 172.66. ESI MS: 450.2 ([M + Na]+). HR ESI MS: calcd for C21H25N5NaO5 450.1748; found 450.1750.
Cyclopropyl (S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (P3)
Starting material 7c (119 mg). Product P3 was isolated as a light-yellow solid (108 mg) with 82% yield. 1H NMR (401 MHz, d6-DMSO): δ 0.45–0.94 (m, 4H), 1.58–1.90 (m, 4H), 1.86–2.09 (m, 1H), 2.15–2.44 (m, 2H), 2.85 (dd, J = 14.6, 9.7, 1H), 2.95–3.17 (m, 1H), 3.95–4.14 (m, 1H), 4.14–4.28 (m, 1H), 4.53 (s, 1H), 6.04 (bs, 1H), 6.98 (t, J = 7.5, 1H), 7.06 (t, J = 7.5, 1H), 7.14 (bs, 1H), 7.32 (d, J = 8.0, 1H), 7.62 (d, J = 7.8, 1H), 8.05 (d, J = 7.9, 1H), 8.47 (d, J = 7.4, 1H), 10.82 (bs, 1H). 13C NMR (101 MHz, d6-DMSO): δ 4.59, 4.62, 22.36, 25.37, 27.55, 48.98, 51.16, 52.89, 111.13, 118.01, 118.28, 120.70, 123.45, 168.97, 172.08, 172.29, 193.98. ESI MS: 462.2 ([M + Na]+). HR ESI MS: calcd for C22H25N5NaO5 462.1748; found 462.1748.
Cyclopropylmethyl (S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (P4)
Starting material 7d (123 mg). Product P4 was isolated as a light-yellow solid (101 mg) in 74% yield. 1H NMR (500 MHz, CDCl3): δ 0.27 (s, 2H), 0.57 (d, J = 8.0, 2H), 1.04–1.16 (m, 1H), 1.88–1.98 (m, 1H), 2.00 (s, 3H), 2.06–2.22 (m, 2H), 2.28 (s, 1H), 3.16 (dd, J = 14.6, 7.5, 1H), 3.35 (dd, J = 14.5, 5.1, 1H), 3.83–3.95 (m, 2H), 4.44 (q, J = 7.6, 5.1, 1H), 4.74 (q, J = 7.0, 1H), 5.17 (bs, 1H), 6.16 (d, J = 7.6, 1H), 6.43–6.56 (m, 1H), 7.09–7.17 (m, 2H), 7.20 (t, J = 7.6, 1H), 7.36 (d, J = 8.1, 1H), 7.69 (d, J = 7.9, 1H), 8.15 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 3.45, 3.50, 9.76, 23.44, 27.06, 28.31, 36.26, 52.20, 54.06, 70.56, 110.41, 111.39, 118.84, 119.89, 122.33, 123.61, 127.72, 136.31, 170.24, 171.43, 171.50, 194.08. ESI MS: 476.2 ([M + Na]+). HR ESI MS: calcd for C23H27N5NaO5 476.1905; found 476.1902.
tert-Butyl (S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (P5)
Starting material 7e (124 mg). Product P5 was isolated as a light-yellow solid (126 mg) in 92% yield. 1H NMR (401 MHz, CDCl3): δ 1.42 (s, 9H), 1.81–1.92 (m, 1H), 1.94 (s, 3H), 2.01–2.17 (m, 2H), 2.18–2.30 (m, 1H), 3.19 (dd, J = 14.7, 6.6, 1H), 3.29 (dd, J = 14.7, 5.7, 1H), 4.31 (td, J = 7.7, 4.5, 1H), 4.77 (q, J = 6.4, 1H), 5.16 (bs, 1H), 6.35 (d, J = 7.7, 1H), 6.82 (d, J = 7.4, 1H), 7.04–7.12 (m, 2H), 7.16 (ddd, J = 8.2, 7.0, 1.3, 1H), 7.33 (dt, J = 8.1, 1.0, 1H), 7.63 (d, J = 7.9, 1H), 8.56 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 23.33, 27.11, 28.04 (3C), 28.16, 36.29, 52.70, 53.99, 54.92, 82.49, 110.30, 111.45, 118.75, 119.76, 122.25, 123.57, 127.72, 136.35, 170.34, 170.48, 171.51, 194.22. ESI MS: 478.2 ([M + Na]+). HR ESI MS: calcd for C23H29O5NaN5 478.20609; found 478.20567.
Synthesis of Prodrug P6
Starting material P2 (100 mg, 0.234 mmol, 1 equiv) was dissolved in a solution of 2 M methylamine in MeOH (6 mL), and the reaction mixture was heated to 60 °C for 20 h. Volatiles were evaporated, and the residue was purified by LC on silica gel (DCM/MeOH, 10:1 + 1% Et3N). Compound P6 was obtained as a light-yellow solid (63 mg) in 65% yield.
(S)-2-((S)-2-Acetamido-3-(1H-indol-3-yl)propanamido)-6-diazo-N-methyl-5-oxohexanamide (P6)
1H NMR (401 MHz, d6-DMSO): δ 1.66–1.77 (m, 1H), 1.81 (s, 3H), 1.88–2.01 (m, 1H), 2.20–2.31 (m, 2H), 2.53 (d, J = 4.6, 3H), 2.92 (dd, J = 14.6, 8.7, 1H), 3.12 (dd, J = 14.6, 5.2, 1H), 4.15 (td, J = 8.6, 5.2, 1H), 4.48–4.52 (m, 1H), 5.97 (bs, 1H), 6.98 (t, J = 7.3, 1H), 7.06 (t, J = 7.1, 1H), 7.16 (d, J = 2.1, 1H), 7.33 (d, J = 8.0, 1H), 7.45 (d, J = 4.5, 1H), 7.59 (d, J = 7.7, 1H), 8.03–8.09 (m, 1H), 8.22 (d, J = 7.0, 1H), 10.83 (d, J = 2.7, 1H). 13C NMR (101 MHz, d6-DMSO): δ 22.59, 25.59, 27.04, 27.46, 36.40, 51.98, 53.70, 54.44, 110.05, 111.30, 118.23, 118.52, 120.89, 123.66, 127.31, 136.05, 169.50, 171.24, 171.75, 194.34. ESI MS: 435.2 ([M + Na]+). HR ESI MS: calcd for C20H24O4NaN6 435.17512; found 435.17489.
General Method for Synthesis of Prodrugs P7–P9
Appropriate carboxylic acid (0.266 mmol, 1.1 equiv) and HATU (106 mg, 0.278 mmol, 1.15 equiv) were dissolved in anhydrous DMF (4 mL), and the reaction mixture was cooled to 0 °C. DIPEA (125 mg, 168 μL, 0.967 mmol, 4 equiv) was added, and the mixture was stirred for 5 min. Finally, a solution of compound 7e (100 mg, 0.242 mmol, 1 equiv) in anhydrous DMF (2 mL) was added over 5 min. The resulting mixture was stirred for 30 min at 0 °C and then at rt for 2 h. DMF was evaporated, EtOAc (100 mL) was added, and the organic phase was washed with saturated NaHCO3 (50 mL), distilled H2O (50 mL), and saturated NaCl (50 mL) and was dried over anhydrous MgSO4. The organic solvent was evaporated in vacuo. The residue was purified by LC on silica gel (various mobile phases) to afford the desired products.
tert-Butyl (S)-2-((S)-3-(1H-Indol-3-yl)-2-(2-morpholinoacetamido)propanamido)-6-diazo-5-oxohexanoate (P7)
Morpholinoacetic acid hydrochloride (48.3 mg); mobile phase: DCM/MeOH, 20:1. Product P7 was isolated as a light-yellow solid (107 mg) in 82% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.86–2.02 (m, 1H), 2.08–2.40 (m, 7H), 2.85 (d, J = 16.4, 1H), 2.98 (d, J = 16.4, 1H), 3.29 (t, J = 7.1, 2H), 3.38 (dtd, J = 14.0, 8.0, 6.6, 3.0, 4H), 4.38 (td, J = 7.5, 4.6, 1H), 4.77 (q, J = 6.8, 1H), 5.23 (bs, 1H), 6.84 (d, J = 7.4, 1H), 7.07–7.13 (m, 2H), 7.17 (ddd, J = 8.1, 7.0, 1.2, 1H), 7.34 (dt, J = 8.1, 1.0, 1H), 7.58 (d, J = 7.8, 1H), 7.63 (dd, J = 8.0, 1.1, 1H), 8.45 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 27.29, 27.65, 28.09 (3C), 36.33, 52.60, 53.44, 53.66 (2C), 54.90, 61.88, 66.80 (2C), 82.57, 110.35, 111.43, 118.76, 119.87, 122.41, 123.34, 127.71, 136.30, 170.50, 170.57, 171.39, 194.09. ESI MS: 541.3 ([M + H]+). HR ESI MS: calcd for C27H37O6N6 541.27691; found 541.27637.
tert-Butyl (S)-2-((S)-3-(1H-Indol-3-yl)-2-(quinuclidine-4-carboxamido) propanamido)-6-diazo-5-oxohexanoate (P8)
Quinuclidine-4-carboxylic acid hydrochloride (51.0 mg); mobile phase: DCM/MeOH, 5:1 + 1% Et3N. Product P8 was isolated as a light-yellow solid (119 mg) in 89% yield. 1H NMR (401 MHz, CDCl3): δ 1.42 (s, 9H), 1.55–1.67 (m, 6H), 1.82–1.94 (m, 1H), 2.00–2.17 (m, 2H), 2.17–2.32 (m, 1H), 2.82–2.89 (m, 6H), 3.17 (dd, J = 14.7, 6.8, 1H), 3.33 (dd, J = 14.7, 6.8, 1H), 4.29 (td, J = 7.5, 4.4, 1H), 4.74 (td, J = 7.0, 5.6, 1H), 5.17 (bs, 1H), 6.24 (d, J = 7.4, 1H), 6.72 (d, J = 7.2, 1H), 7.07–7.13 (m, 2H), 7.17 (ddd, J = 8.1, 6.9, 1.2, 1H), 7.35 (d, J = 8.1, 1H), 7.66 (d, J = 7.8, 1H), 8.69 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 27.03, 28.07 (3C), 28.12 (3C), 29.81, 36.30, 45.97, 47.29 (3C), 52.78, 53.64, 54.85, 82.49, 110.29, 111.47, 119.01, 119.75, 122.37, 123.61, 127.66, 136.40, 170.46, 171.43, 176.21, 194.13. ESI MS: 551.3 ([M + Na]+). HR ESI MS: calcd for C29H39O5N6 551.29764; found 551.29730.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-(1H-indol-3-yl)propanamido)-5-oxohexanoate (P9)
Dimethylglycine (27.4 mg); mobile phase: DCM/MeOH, 15:1. Product P9 was isolated as a light-yellow solid (106 mg) in 88% yield. 1H NMR (401 MHz, CDCl3): δ 1.40 (s, 9H), 1.84–1.94 (m, 1H), 2.02–2.12 (m, 1H), 2.11 (s, 6H), 2.17–2.30 (m, 2H), 2.87 (d, J = 16.1, 1H), 2.96 (d, J = 16.1, 1H), 3.26 (d, J = 6.9, 2H), 4.32 (q, J = 7.2, 1H), 4.76 (q, J = 6.9, 1H), 5.21 (bs, 1H), 7.01–7.17 (m, 4H), 7.32 (d, J = 8.1, 1H), 7.61 (d, J = 7.8, 1H), 7.85 (d, J = 8.0, 1H), 8.93 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 27.08, 27.98 (3C), 29.71, 36.32, 45.50 (2C), 52.64, 53.82, 54.83, 62.37, 82.31, 110.29, 111.43, 118.71, 119.43, 122.01, 123.45, 127.53, 136.34, 170.52, 171.53, 171.54, 194.26. ESI MS: 499.3 ([M + H]+). HR ESI MS: calcd for C25H35O5N6 499.26634; found 499.26585.
General Method for Synthesis of Compounds 8a–8l
Fmoc-AA-OH (4.84 mmol, 1.1 equiv) and HATU (1.92 g, 5.06 mmol, 1.15 equiv) were suspended in anhydrous DCM (30 mL) under inert atmosphere, and the reaction mixture was cooled to 0 °C. DIPEA (1.71 g, 2.30 mL, 13.2 mmol, 3 equiv) was added, and the mixture was stirred for 5 min. Finally, a solution of compound 5e (1.00 g, 4.40 mmol, 1 equiv) in anhydrous DCM (15 mL) was slowly added for 5 min. The resulting mixture was stirred for 30 min at 0 °C and then for 1–16.5 h at rt. DCM was evaporated, EtOAc (100 mL) was added, and the organic phase was washed with saturated NaHCO3 (50 mL), distilled H2O (50 mL), 10% KHSO4 (50 mL), distilled H2O (50 mL), and saturated NaCl (50 mL) and was dried over anhydrous MgSO4. EtOAc was evaporated, and the residue was purified by LC on silica gel (various mobile phases) to obtain desired products 8a–8l.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(1-methyl-1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (8a)
Starting material Fmoc-l-Trp(N-Me)-OH (2.13 g), reaction time 3 h, mobile phase: DCM/EtOAc, 3:1. Product 8a was isolated as a light-yellow solid (2.26 g) in 79% yield. 1H NMR (401 MHz, CDCl3): δ 1.43 (s, 9H), 1.81–1.93 (m, 1H), 2.05–2.25 (m, 3H), 3.16 (dd, J = 14.6, 7.1, 1H), 3.32–3.45 (m, 1H), 3.73 (s, 3H), 4.21 (t, J = 7.1, 1H), 4.33–4.40 (m, 2H), 4.44 (dd, J = 10.5, 7.3, 1H), 4.53 (d, J = 7.1, 1H), 5.04 (bs, 1H), 5.49 (d, J = 7.7, 1H), 6.55 (d, J = 7.6, 1H), 6.91 (bs, 1H), 7.13 (td, J = 7.4, 6.9, 1.2, 1H), 7.23 (ddd, J = 8.2, 6.8, 1.1, 1H), 7.27–7.33 (m, 3H), 7.40 (tdd, J = 7.6, 2.3, 1.3, 2H), 7.52–7.62 (m, 2H), 7.68 (d, J = 8.0, 1H), 7.74–7.79 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 27.36, 28.05 (3C), 28.38, 32.80, 36.33, 47.26, 52.57, 54.68, 55.61, 67.20, 82.50, 108.63, 109.43, 119.13, 119.48, 120.08, 120.09, 122.00, 125.25, 125.30, 127.22 (2C), 127.84 (2C), 128.03, 128.34, 137.17, 141.39 (2C), 143.89, 143.98, 156.09, 170.48, 171.30, 193.79. ESI MS: 672.3 ([M + Na]+). HR ESI MS: calcd for C37H39O6N5Na 672.27926; found 672.27867.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenylpropanamido)-6-diazo-5-oxohexanoate (8b)
Starting material Fmoc-l-Phe-OH (1.88 g); reaction time 16 h; mobile phase: DCM/EtOAc, 5:1. Product 8b was isolated as a light-yellow solid (2.00 g) in 76% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.40 (s, 9H), 1.76–1.87 (m, 1H), 1.93–2.06 (m, 1H), 2.35–2.43 (m, 2H), 2.79 (dd, J = 13.8, 10.9, 1H), 3.02 (dd, J = 13.8, 3.6, 1H), 4.05–4.21 (m, 4H), 4.29 (ddd, J = 10.9, 8.8, 3.6, 1H), 6.04 (bs, 1H), 7.15–7.22 (m, 1H), 7.22–7.44 (m, 8H), 7.63 (dd, J = 10.6, 7.5, 3H), 7.84–7.90 (m, 2H), 8.37 (d, J = 7.5, 1H). 13C NMR (101 MHz, d6-DMSO): δ 26.0, 27.6 (3C), 36.3, 37.4, 46.5, 52.1, 55.9, 56.6, 65.6, 80.7, 120.1 (2C), 125.3 (2C), 126.4, 127.0 (2C), 127.6 (2C), 128.0 (2C), 129.2 (2C), 138.1 (2C), 140.6, 143.7, 143.8, 155.8, 170.7, 171.8, 194.1. ESI MS: 619.3 ([M + Na]+). HR ESI MS: calcd for C34H36O6N4Na 619.25271; found 619.25162.
tert-Butyl (S)-2-((R)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-phenylpropanamido)-6-diazo-5-oxohexanoate (8c)
Starting material Fmoc-d-Phe-OH (1.88 g); reaction time 5 h; mobile phase: cyclohexane/EtOAc, 2:1. Product 8c was isolated as a light-yellow solid (2.52 g) in 87% yield. 1H NMR (401 MHz, d6-DMSO): 1.42 (s, 9H), 1.78–1.90 (m, 1H), 2.05–2.22 (m, 3H), 3.02–3.17 (m, 2H), 4.18 (t, J = 7.0, 1H), 4.24–4.34 (m, 1H), 4.39 (dd, J = 7.2, 10.5, 2H), 4.44–4.56 (m, 1H), 5.17 (bs, 1H), 5.51 (d, J = 7.8, 1H), 6.76 (bs, 1H), 7.23 (t, J = 7.2, 3H), 7.29 (t, J = 7.2, 4H), 7.39 (t, J = 7.4, 2H), 7.52 (t, J = 6.8, 2H), 7.75 (d, J = 7.5, 2H). 13C NMR (101 MHz, d6-DMSO): δ 27.2, 28.0, 36.2, 38.7, 47.1, 52.4, 53.5, 54.7, 56.3, 67.2, 82.6, 120.0, 125.1, 125.2, 127.1, 127.1, 127.8, 128.8, 129.4, 136.5, 141.3, 141.3, 143.8, 143.8, 156.0, 170.6, 170.8, 193.7. ESI MS: 619.2 ([M + Na]+). HR ESI MS: calcd for C34H36O6N4Na 619.25271; found 619.25300.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-2-phenylacetamido)-6-diazo-5-oxohexanoate (8d)
Starting material Fmoc-l-Phg-OH (1.81 g); reaction time 1.5 h; mobile phase: DCM/EtOAc, 10:1. Product 8d was isolated as a yellow solid (1.74 g) in 68% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.26 (s, 9H), 1.74–1.85 (m, 1H), 1.94 (dq, J = 14.2, 7.3, 1H), 2.30–2.42 (m, 2H), 4.08–4.16 (m, 1H), 4.23 (q, J = 5.7, 3H), 5.30 (d, J = 8.5, 1H), 6.02 (bs, 1H), 7.32 (ddd, J = 17.8, 8.0, 5.1, 5H), 7.38–7.49 (m, 4H), 7.77 (d, J = 7.5, 2H), 7.88 (d, J = 7.5, 2H), 8.07 (d, J = 8.5, 1H), 8.52 (d, J = 7.4, 1H). 13C NMR (101 MHz, CDCl3): δ 27.2, 27.9 (3C), 29.8, 36.5, 47.2, 53.1, 55.0, 59.0, 82.6, 120.0 (2C), 125.2, 125.2, 127.2 (2C), 127.4, 127.8 (2C), 128.7, 129.2 (2C), 129.3, 137.6, 141.4 (2C), 143.9, 144.0, 155.8, 169.8, 170.1, 194.0. ESI MS: 605.2 ([M + Na]+). HR ESI MS: calcd for C33H34O6N4Na 605.23706; found 605.23743.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-phenylbutanamido)-6-diazo-5-oxohexanoate (8e)
Starting material Fmoc-l-HomoPhe-OH (1.94 g); reaction time 1.5 h; mobile phase: DCM/EtOAc, 10:1. Product 8e was isolated as a yellow solid (2.31 g) in 86% yield. 1H NMR (401 MHz, CDCl3): δ 1.46 (s, 9H), 1.90–2.02 (m, 2H), 2.14–2.24 (m, 2H), 2.28–2.44 (m, 2H), 2.69 (d, J = 8.2, 2H), 4.21 (dt, J = 11.4, 6.8, 2H), 4.33–4.51 (m, 3H), 5.18 (bs, 1H), 5.37 (d, J = 8.1, 1H), 6.66 (d, J = 7.7, 1H), 7.16–7.23 (m, 3H), 7.27–7.35 (m, 4H), 7.40 (tdd, J = 7.5, 6.0, 2.6, 2H), 7.60 (d, J = 7.4, 2H), 7.73–7.79 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 27.1, 28.1 (3C), 31.7, 34.5, 36.5, 47.2, 52.6, 54.7, 59.8, 67.2, 82.6, 120.1, 120.1, 125.2, 125.2, 126.3, 127.3 (2C), 127.9 (2C), 128.5 (2C), 128.6 (2C), 140.9, 141.4 (2C), 143.9, 143.9, 156.2, 170.6, 171.6, 194.0. ESI MS: 633.3 ([M + Na]+). HR ESI MS: calcd for C35H38O6N4Na 633.26836; found 633.26825.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-fluorophenyl)propanamido)-6-diazo-5-oxohexanoate (8f)
Starting material Fmoc-l-Phe(4-F)-OH (1.96 g); reaction time 1.5 h; mobile phase: DCM/EtOAc, 5:1. Product 8f was isolated as a light-yellow solid (2.08 g) in 77% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.39 (s, 9H), 1.81 (dtd, J = 14.7, 9.0, 6.1, 1H), 1.92–2.05 (m, 1H), 2.30–2.44 (m, 2H), 2.77 (dd, J = 13.8, 10.9, 1H), 3.00 (dd, J = 13.7, 3.7, 1H), 4.05–4.21 (m, 4H), 4.27 (ddt, J = 10.8, 8.7, 3.7, 1H), 6.04 (bs, 1H), 7.08 (dd, J = 10.1, 7.7, 2H), 7.23–7.45 (m, 6H), 7.62 (dd, J = 8.2, 4.3, 3H), 7.88 (d, J = 7.5, 2H), 8.36 (d, J = 7.5, 1H). 13C NMR (101 MHz, CDCl3): δ 26.0, 27.6 (3C), 33.7, 36.6, 46.5, 52.1, 55.9, 59.0, 65.6, 80.7, 114.6, 114.8, 120.0 (2C), 125.2, 125.3, 127.0 (2C), 127.6 (2C), 131.0, 131.0, 134.2, 140.6, 143.7, 143.8, 155.8, 159.8, 162.2, 170.7, 171.7, 194.1. ESI MS: 637.3 ([M + Na]+). HR ESI MS: calcd for C34H35O6N4FNa 637.24328; found 637.24402.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(3-fluorophenyl)propanamido)-6-diazo-5-oxohexanoate (8g)
Starting material Fmoc-l-Phe(3-F)-OH (1.96 g); reaction time 16 h; mobile phase: DCM/MeOH, 40:1. Product 8g was isolated as a yellow solid (2.43 g) in 90% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.72–2.01 (m, 2H), 2.09–2.42 (m, 2H), 3.01–3.15 (m, 2H), 4.19 (t, J = 6.8, 1H), 4.26–4.53 (m, 4H), 5.18 (s, 1H), 5.45 (bs, 1H), 6.67–7.04 (m, 4H), 7.19–7.28 (m, 1H), 7.30 (t, J = 7.4, 2H), 7.40 (dd, J = 8.3, 6.9, 2H), 7.50–7.60 (m, 2H), 7.70–7.83 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 27.3, 28.0, 36.4, 38.3, 47.2, 52.6, 53.5, 54.9, 55.9, 67.2, 82.7, 114.0, 114.2, 116.4, 116.6, 120.1, 125.1, 125.2, 127.2, 127.9, 130.2, 130.2, 138.9, 141.4, 143.9, 155.9, 161.7, 164.2, 170.4, 170.5, 193.9. ESI MS: 637.4 ([M + Na]+). HR ESI MS: calcd for C34H35O6N4FNa 637.24328; found 637.24253.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-3-(4-(trifluoromethyl)phenyl)propanamido)-6-diazo-5-oxohexanoate (8h)
Starting material Fmoc-l-Phe(4-CF3)-OH (2.20 g); reaction time 3.5 h; without purification to the following step (low solubility). Product 8h was isolated as a light-yellow solid (2.92 g) in quantitative yield. 1H NMR (401 MHz, CDCl3): δ 1.40 (s, 9H), 1.77–1.89 (m, 1H), 1.93–2.06 (m, 1H), 2.36–2.45 (m, 2H), 2.84–2.94 (m, 2H), 4.10–4.22 (m, 4H), 4.30–4.39 (m, 1H), 6.03 (bs, 1H), 7.23–7.34 (m, 2H), 7.40 (dtd, J = 8.6, 4.6, 2.4, 3H), 7.56 (t, J = 8.7, 2H), 7.59–7.65 (m, 3H), 7.68 (d, J = 8.8, 1H), 7.85–7.91 (m, 2H), 8.40 (d, J = 7.5, 1H). ESI MS: 687.3 ([M + Na]+). HR ESI MS: calcd for C35H35O6N4F3Na 687.24009; found 687.23944.
tert-Butyl (S)-2-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)acetamido)-6-diazo-5-oxohexanoate (8i)
Starting material Fmoc-Gly-OH (1.44 g); reaction time 2 h; mobile phase: EtOAc. Product 8i was isolated as a light-yellow solid (2.05 g) in 92% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.97 (dt, J = 14.5, 7.5, 1H), 2.14–2.25 (m, 1H), 2.25–2.45 (m, 2H), 3.91 (d, J = 5.7, 2H), 4.21 (t, J = 7.2, 1H), 4.38 (d, J = 7.0, 2H), 4.48 (td, J = 8.1, 4.6, 1H), 5.27 (bs, 1H), 5.84 (t, J = 5.7, 1H), 7.06 (d, J = 7.8, 1H), 7.28 (t, J = 7.6, 2H), 7.37 (t, J = 7.4, 2H), 7.58 (d, J = 7.5, 2H), 7.73 (d, J = 7.6, 2H). 13C NMR (101 MHz, CDCl3): δ 27.32, 27.97 (3C), 36.44, 44.43, 47.09, 52.41, 54.87, 59.71, 67.30, 82.57, 120.00 (2C), 125.13, 125.15, 127.12 (2C), 127.76 (2C), 141.28, 141.28, 143.81, 143.83, 156.68, 169.16, 170.78, 193.89. ESI MS: 529.2 ([M + Na]+). HR ESI MS: calcd for C27H30O6N4Na 529.20576; found 529.20604.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)propanamido)-6-diazo-5-oxohexanoate (8j)
Starting material Fmoc-l-Ala-OH monohydrate (1.59 g); reaction time 4 h; mobile phase: DCM/EtOAc, 3:1. Product 8j was isolated as a light-yellow solid (2.24 g) in 98% yield. 1H NMR (401 MHz, CDCl3): δ 1.41 (d, J = 7.4, 3H), 1.44 (s, 9H), 1.97 (tt, J = 14.6, 7.2, 1H), 2.18 (ddd, J = 14.8, 7.1, 2.5, 1H), 2.25–2.48 (m, 2H), 4.21 (t, J = 7.1, 1H), 4.28 (t, J = 7.2, 1H), 4.37 (dd, J = 7.4, 3.1, 2H), 4.43 (td, J = 8.2, 4.6, 1H), 5.21 (bs, 1H), 5.59 (d, J = 7.5, 1H), 6.91 (d, J = 7.8, 1H), 7.30 (td, J = 7.5, 1.0, 2H), 7.39 (dd, J = 8.2, 6.7, 2H), 7.59 (d, J = 7.5, 2H), 7.75 (d, J = 7.5, 2H). 13C NMR (101 MHz, CDCl3): δ 18.93, 27.30, 28.04 (3C), 36.52, 47.18, 50.61, 52.56, 54.96, 67.19, 82.56, 120.07, 120.08, 125.19, 125.22, 127.19 (2C), 127.83 (2C), 141.36 (2C), 143.90 (2C), 155.99, 170.66, 172.37, 194.04. ESI MS: 543.2 ([M + Na]+). HR ESI MS: calcd for C28H32O6N4Na 543.22141; found 543.22096.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4-methylpentanamido)-6-diazo-5-oxohexanoate (8k)
Starting material Fmoc-l-Leu-OH (1.71 g); reaction time 2 h; mobile phase: cyclohexane/EtOAc, 1:1. Product 8k was isolated as a light-yellow solid (1.88 g) in 76% yield. 1H NMR (401 MHz, CDCl3): δ 0.82–1.00 (m, 7H), 1.45 (s, 9H), 1.51–1.74 (m, 2H), 1.96 (dq, J = 14.8, 7.7, 1H), 2.12–2.26 (m, 1H), 2.24–2.45 (m, 2H), 4.14–4.26 (m, 2H), 4.31–4.47 (m, 3H), 5.18 (bs, 1H), 5.27 (d, J = 8.3, 1H), 6.68 (d, J = 7.8, 1H), 7.31 (tt, J = 7.4, 1.0, 2H), 7.40 (tt, J = 7.5, 1.5, 2H), 7.59 (d, J = 7.5, 2H), 7.71–7.81 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 22.07, 23.11, 24.80, 27.46, 28.09 (3C), 36.51, 41.83, 47.27, 52.55, 53.71, 54.92, 67.20, 82.63, 120.11, 120.14, 125.18, 125.25, 127.24 (2C), 127.87, 127.88, 141.42 (2C), 143.90, 143.93, 156.27, 170.66, 172.24, 193.98. ESI MS: 585.2 ([M + Na]+). HR ESI MS: calcd for C31H38O6N4Na 585.26836; found 585.26795.
tert-Butyl (S)-2-((S)-2-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-4,4-dimethylpentanamido)-6-diazo-5-oxohexanoate (8l)
Starting material Fmoc-l-Ala(tBu)-OH (1.78 g); reaction time 16 h; mobile phase: DCM/MeOH, 50:1. Product 8l was isolated as a yellow solid (2.28 g) in 90% yield. 1H NMR (401 MHz, CDCl3): δ 0.97 (s, 9H), 1.44 (s, 9H), 1.78–2.01 (m, 2H), 2.09–2.44 (m, 2H), 4.15–4.36 (m, 2H), 4.36–4.50 (m, 2H), 5.16 (bs, 1H), 5.34 (d, J = 8.5, 1H), 6.78 (d, J = 7.9, 1H), 7.29 (td, J = 7.5, 1.2, 2H), 7.34–7.42 (m, 2H), 7.54–7.62 (m, 2H), 7.71–7.77 (m, 2H). 13C NMR (101 MHz, CDCl3): δ 28.0, 29.8, 30.6, 36.5, 46.0, 47.2, 52.5, 53.0, 53.5, 54.8, 67.2, 82.5, 120.1, 120.1, 125.1, 125.2, 127.2, 127.2, 127.8, 141.3, 143.8, 143.9, 156.0, 170.6, 172.7, 193.9. ESI MS: 599.5 ([M + Na]+). HR ESI MS: calcd for C32H41O6N4 577.30206; found 577.30234.
General Method for Synthesis of Compounds 9a–9l
Compounds 8a–8l (3.00 mmol, 1 equiv) were dissolved in anhydrous DCM (27 mL), and diethylamine (2.19 g, 3.10 mL, 30.0 mmol, 10 equiv) was added. The reaction mixture was stirred at rt for 1.5–7 h under an inert atmosphere. Volatiles were evaporated, and the residue was purified by LC on silica gel (various mobile phases) to afford products 9a–9l.
tert-Butyl (S)-2-((S)-2-Amino-3-(1-methyl-1H-indol-3-yl)propanamido)-6-diazo-5-oxohexanoate (9a)
Starting material 8a (1.95 g); reaction time 7 h; mobile phase: DCM/MeOH, 30:1. Product 9a was isolated as a yellow solid (1.15 g) in 90% yield. 1H NMR (401 MHz, CDCl3): δ 1.46 (s, 9H), 1.55 (bs, 2H), 1.86–2.00 (m, 1H), 2.07–2.32 (m, 3H), 2.98 (dd, J = 14.4, 8.5, 1H), 3.31 (dd, J = 14.0, 3.8, 1H), 3.72 (dd, J = 8.5, 4.1, 1H), 3.76 (s, 3H), 4.41–4.51 (m, 1H), 5.12 (bs, 1H), 6.94 (s, 1H), 7.12 (ddd, J = 8.0, 6.9, 1.1, 1H), 7.23 (ddd, J = 8.2, 6.9, 1.1, 1H), 7.29 (dt, J = 8.2, 1.0, 1H), 7.68 (dt, J = 8.0, 1.0, 1H), 7.87 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 27.95, 28.11 (3C), 30.76, 32.81, 36.74, 51.96, 54.66, 55.63, 82.40, 109.38, 110.07, 119.26, 119.41, 121.97, 128.08, 137.27 (2C), 171.15, 174.92, 193.85. ESI MS: 450.2 ([M + Na]+). HR ESI MS: calcd for C22H29O4N5Na 450.21118; found 450.21112.
tert-Butyl (S)-2-((S)-2-Amino-3-phenylpropanamido)-6-diazo-5-oxohexanoate (9b)
Starting material 8b (1.79 g); reaction time 2 h; mobile phase: DCM/MeOH, 30:1. Product 9b was isolated as a yellow solid (1.08 g) in 96% yield. 1H NMR (401 MHz, CDCl3): δ 1.40 (s, 9H), 1.71 (bs, 2H), 1.75–1.85 (m, 1H), 1.90–1.99 (m, 1H), 2.24–2.38 (m, 2H), 2.59 (dd, J = 13.4, 8.4, 1H), 2.95 (dd, J = 13.4, 4.5, 1H), 3.43 (dd, J = 8.4, 4.5, 1H), 4.07–4.17 (m, 1H), 6.05 (bs, 1H), 7.17–7.29 (m, 5H), 8.13 (d, J = 7.9, 1H). 13C NMR (101 MHz, CDCl3): δ 27.8, 28.1 (3C), 36.8, 41.1, 50.8, 54.8, 56.5, 82.5, 127.0, 128.8 (2C), 129.5 (2C), 137.8, 171.0, 174.4, 193.7. ESI MS: 397.2 ([M + Na]+). HR ESI MS: calcd for C19H26O4N4Na 397.18463; found 397.18427.
tert-Butyl (S)-2-((R)-2-Amino-3-phenylpropanamido)-6-diazo-5-oxohexanoate (9c)
Starting material 8c (1.79 g); reaction time 2.5 h; mobile phase: DCM/MeOH, 20:1. Product 9c was isolated as a yellow solid (1.10 g) in 98% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.88–2.00 (m, 1H), 2.10–2.21 (m, 1H), 2.22–2.41 (m, 2H), 2.64 (dd, J = 9.6, 13.8, 1H), 3.24 (dd, J = 4.3, 13.8, 1H), 3.57 (dd, J = 4.3, 9.5, 1H), 4.40–4.48 (m, 1H), 5.28 (bs, 1H), 7.16–7.24 (m, 3H), 7.24–7.31 (m, 2H), 7.74 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 27.8, 28.0, 36.8, 41.0, 52.0, 54.7, 56.7, 82.4, 126.8, 128.7, 129.3, 138.0, 170.9, 174.4, 193.7. ESI MS: 375.2 ([M + H]+). HR ESI MS: calcd for C19H27O4N4 375.20268; found 375.20286.
tert-Butyl (S)-2-((S)-2-Amino-2-phenylacetamido)-6-diazo-5-oxohexanoate (9d)
Starting material 8d (1.75 g); reaction time 3 h; mobile phase: DCM/MeOH, 30:1. Product 9d was isolated as a yellow solid (822 mg) in 76% yield. 1H NMR (401 MHz, CDCl3): δ 1.45 (s, 9H), 1.64 (bs, 2H), 1.93 (ddt, J = 11.7, 8.3, 3.6, 1H), 2.10–2.35 (m, 3H), 4.44 (td, J = 8.5, 4.0, 1H), 4.55 (s, 1H), 5.04 (bs, 1H), 7.27–7.45 (m, 5H), 7.80 (d, J = 8.3, 1H). 13C NMR (101 MHz, CDCl3): δ 27.9, 28.0 (3C), 36.5, 51.9, 54.6, 60.0, 82.4, 126.7 (2C), 128.0, 128.8 (2C), 140.9, 170.9, 173.2, 193.7. ESI MS: 361.2 ([M + H]+). HR ESI MS: calcd for C18H25O4N4 361.18703; found 361.18675.
tert-Butyl (S)-2-((S)-2-Amino-4-phenylbutanamido)-6-diazo-5-oxohexanoate (9e)
Starting material 8e (1.83 g); reaction time 3 h; mobile phase: DCM/MeOH, 30:1. Product 9e was isolated as a yellow solid (886 mg) in 76% yield. 1H NMR (401 MHz, CDCl3): δ 1.47 (s, 9H), 1.62 (bs, 2H), 1.79 (dtd, J = 14.3, 8.9, 6.0, 1H), 1.98 (dtd, J = 14.5, 8.5, 6.1, 1H), 2.15–2.25 (m, 2H), 2.36 (t, J = 21.3, 2H), 2.66–2.81 (m, 2H), 3.38 (dd, J = 8.4, 4.4, 1H), 4.46 (td, J = 8.4, 4.7, 1H), 5.28 (bs, 1H), 7.16–7.24 (m, 3H), 7.26–7.32 (m, 2H), 7.80 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 28.0, 28.1 (3C), 32.4, 37.0, 52.0, 53.5, 54.7, 55.0, 82.4, 126.2, 128.5 (2C), 128.6 (2C), 141.2, 171.1, 175.1, 193.7. ESI MS: 389.2 ([M + H]+). HR ESI MS: calcd for C20H29O4N4 389.21833; found 389.21798.
tert-Butyl (S)-2-((S)-2-Amino-3-(4-fluorophenyl)propanamido)-6-diazo-5-oxohexanoate (9f)
Starting material 8f (1.22 g); reaction time 3 h; mobile phase: DCM/MeOH, 30:1. Product 9f was isolated as a yellow solid (977 mg) in 83% yield. 1H NMR (401 MHz, CDCl3): δ 1.41 (bs, 2H), 1.46 (s, 9H), 1.97 (dtd, J = 14.3, 8.4, 5.6, 1H), 2.17 (td, J = 13.5, 5.7, 1H), 2.30 (d, J = 28.2, 2H), 2.75 (dd, J = 13.8, 8.9, 1H), 3.17 (dd, J = 13.8, 4.0, 1H), 3.61 (dd, J = 8.9, 4.1, 1H), 4.45 (td, J = 8.2, 4.6, 1H), 5.26 (bs, 1H), 6.96–7.05 (m, 2H), 7.15–7.22 (m, 2H), 7.80 (d, J = 8.2, 1H). 13C NMR (101 MHz, CDCl3): δ 27.8, 28.1 (3C), 36.7, 40.2, 52.0, 54.8, 56.5, 82.5, 115.5, 115.7, 130.9, 131.0, 133.4, 160.8, 171.0, 174.2, 193.7. ESI MS: 393.2 ([M + H]+). HR ESI MS: calcd for C19H26O4N4F 393.19326; found 393.19330.
tert-Butyl (S)-2-((S)-2-Amino-3-(3-fluorophenyl)propanamido)-6-diazo-5-oxohexanoate (9g)
Starting material 8g (1.22 g); reaction time 2 h; mobile phase: DCM/MeOH, 20:1. Product 9g was isolated as a yellow solid (1.04 mg) in 88% yield. 1H NMR (401 MHz, CDCl3): δ 1.42 (d, J = 1.3, 9H), 1.82–2.00 (m, 1H), 2.06–2.44 (m, 3H), 2.76 (dd, J = 13.7, 8.7, 1H), 3.15 (dd, J = 13.7, 4.0, 1H), 3.59 (ddd, J = 8.7, 4.1, 1.1, 1H), 4.41 (dtd, J = 8.3, 4.6, 2.3, 1H), 5.27 (bs, 1H), 6.84–7.04 (m, 3H), 7.18–7.32 (m, 1H), 7.82 (d, J = 8.3, 1H). 13C NMR (101 MHz, CDCl3): δ 28.0, 36.7, 40.7, 40.7, 52.0, 54.7, 56.2, 82.4, 113.7, 113.9, 116.1, 116.3, 125.1, 125.2, 130.1, 130.2, 140.2, 140.3, 161.7, 164.2, 170.9, 174.0, 193.7. ESI MS: 393.2 ([M + H]+). HR ESI MS: calcd for C19H26O4N4F 393.19326; found 393.19334.
tert-Butyl (S)-2-((S)-2-Amino-3-(4-(trifluoromethyl)phenyl)propanamido)-6-diazo-5-oxohexanoate (9h)
Starting material 8h (1.99 g); reaction time 1.5 h; mobile phase: DCM/MeOH, 30:1. Product 9h was isolated as a yellow solid (1.01 g) in 76% yield (over two steps). 1H NMR (401 MHz, CDCl3): δ 1.41 (bs, 2H), 1.44 (s, 9H), 1.88–2.05 (m, 1H), 2.11–2.42 (m, 3H), 2.82 (dd, J = 13.7, 8.9, 1H), 3.25 (dd, J = 13.7, 4.1, 1H), 3.64 (dd, J = 8.9, 4.1, 1H), 4.43 (td, J = 8.1, 4.4, 1H), 5.24 (bs, 1H), 7.34 (d, J = 7.9, 2H), 7.56 (d, J = 7.9, 2H), 7.82 (d, J = 8.2, 1H). 13C NMR (101 MHz, CDCl3): δ 27.55, 27.97 (3C), 36.57, 40.77, 52.03, 54.69, 56.20, 82.45, 125.58 (q, J = 3.7 Hz, 2C), 129.20 (q, J = 32.4 Hz), 129.74 (2C), 141.95 (2C), 170.84, 173.73, 193.50. ESI MS: 443.2 ([M + H]+). HR ESI MS: calcd for C20H26O4N4F3 443.19007; found 443.19016.
tert-Butyl (S)-2-(2-Aminoacetamido)-6-diazo-5-oxohexanoate (9i)
Starting material 8i (1.52 g); reaction time 3 h; mobile phase: DCM/MeOH, 10:1. Product 9i was isolated as a yellow-orange amorphous compound (768 mg) in 90% yield. 1H NMR (401 MHz, CDCl3): δ 1.43 (s, 9H), 1.73 (bs, 2H), 1.95 (dtd, J = 14.5, 8.6, 6.1, 1H), 2.17 (dddd, J = 13.4, 8.5, 6.7, 4.7, 1H), 2.26–2.48 (m, 2H), 3.34 (s, 2H), 4.47 (td, J = 8.4, 4.7, 1H), 5.31 (bs, 1H), 7.75 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 27.97, 28.04 (3C), 36.82, 44.75, 51.87, 54.82, 82.48, 171.04, 172.92, 193.85. ESI MS: 307.1 ([M + Na]+). HR ESI MS: calcd for C12H20O4N4Na 307.13768; found 307.13744.
tert-Butyl (S)-2-(2-Aminopropanamido)-6-diazo-5-oxohexanoate (9j)
Starting material 8j (1.56 g); reaction time 3 h; mobile phase: DCM/MeOH, 10:1 to 5:1. Product 9j was isolated as a yellow amorphous compound (841 mg) in 94% yield. 1H NMR (401 MHz, CDCl3): δ 1.36 (d, J = 7.0, 3H), 1.46 (s, 9H), 1.94–2.02 (m, 1H), 2.14–2.17 (m, 2H), 2.18–2.23 (m, 1H), 2.30–2.47 (m, 2H), 3.56 (q, J = 7.0, 1H), 4.40–4.47 (m, 1H), 5.34 (bs, 1H), 7.73–7.77 (m, 1H). 13C NMR (101 MHz, CDCl3): δ 21.08, 27.23, 27.68 (3C), 36.30, 50.27, 51.77, 54.70, 77.16, 81.98, 170.63, 175.25, 193.96. ESI MS: 299.2 ([M + H]+). HR ESI MS: calcd for C13H22O4N4Na 321.15387; found 321.15392.
tert-Butyl (S)-2-((S)-2-Amino-4-methylpentanamido)-6-diazo-5-oxohexanoate (9k)
Starting material 8k (1.69 g); reaction time 2 h; mobile phase: DCM/MeOH, 15:1. Product 9k was isolated as a yellow solid (950 mg) in 93% yield. 1H NMR (401 MHz, CDCl3): δ 0.91 (d, J = 6.3, 3H), 0.95 (d, J = 6.3, 3H), 1.27–1.36 (m, 1H), 1.44 (s, 9H), 1.47 (bs, 2H), 1.58–1.80 (m, 2H), 1.95 (dtd, J = 14.5, 8.6, 6.1, 1H), 2.10–2.24 (m, 1H), 2.25–2.47 (m, 2H), 3.37 (dd, J = 10.0, 3.8, 1H), 4.43 (td, J = 8.5, 4.7, 1H), 5.30 (bs, 1H), 7.82 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 21.36, 23.58, 24.99, 27.90, 28.09 (3C), 36.91, 44.31, 51.91, 53.64, 54.81, 82.39, 171.17, 175.92, 193.84. ESI MS: 341.2 ([M + H]+). HR ESI MS: calcd for C16H29O4N4 341.21833; found 341.21816.
tert-Butyl (S)-2-((S)-2-Amino-4,4-dimethylpentanamido)-6-diazo-5-oxohexanoate (9l)
Starting material 8l (1.73 g); reaction time 3 h; mobile phase: DCM/MeOH, 15:1. Product 9f was isolated as a yellow amorphous compound (1.01 g) in 95% yield. 1H NMR (401 MHz, CDCl3): δ 0.96 (s, 9H), 1.18 (dd, J = 14.3, 8.7, 1H), 1.43 (s, 9H), 1.87 (dd, J = 14.3, 2.5, 1H), 1.90–2.01 (m, 1H), 2.09–2.22 (m, 1H), 2.24–2.47 (m, 2H), 3.37 (dd, J = 8.6, 2.5, 1H), 4.40 (td, J = 8.5, 4.7, 1H), 5.30 (bs, 1H), 7.88 (d, J = 8.4, 1H). 13C NMR (101 MHz, CDCl3): δ 28.1, 30.1, 30.8, 36.9, 49.6, 52.0, 53.1, 54.8, 82.3, 171.1, 176.4, 193.8. ESI MS: 355.3 ([M + H]+). HR ESI MS: calcd for C17H31O4N4 355.23398; found 355.23361.
General Method A for Synthesis of Prodrugs P10, P11, P18, and P19
Dimethylglycine (113 mg, 1.10 mmol, 1.1 equiv) and HATU (456 mg, 1.20 mmol, 1.2 equiv) were dissolved in anhydrous DMF or DCM (15 mL) under inert atmosphere, the mixture was cooled to 0 °C, and DIPEA (388 mg, 523 μL, 3.00 mmol, 3 equiv) was added. After 5 min of stirring, a solution of amines 9a, 9b, 9i, and 9j (1.00 mmol, 1 equiv) in anhydrous DMF or DCM (10 mL) was added. The resulting mixture was stirred for 30 min at 0 °C and 1–2 h at rt. The solvent was evaporated, EtOAc (100 mL) was added, and the organic phase was washed with saturated NaHCO3 (70 mL), distilled H2O (70 mL), and saturated NaCl (50 mL) and was dried over anhydrous MgSO4, and solvent was evaporated. The crude product was purified by LC on silica gel (various mobile phases) to afford desired prodrugs P10, P11, P18, and P19.
General Method B for Synthesis of Prodrugs P12–17, P20, and P21
Compounds 9c–9h, 9k, 9l (1.00 mmol, 1 equiv) and 2,5-dioxopyrrolidin-1-yl dimethylglycinate (Dmg-ONSu)36 (220 mg, 1.10 mmol, 1.1 equiv) were dissolved in anhydrous DCM (5 mL) under inert atmosphere. The resulting mixture was stirred at rt for 2–20 h. DCM (50 mL) was added, and the organic phase was washed with saturated NaHCO3 (30 mL), distilled H2O (30 mL), and saturated NaCl (20 mL) and was dried over anhydrous MgSO4, and the solvent was evaporated. The crude product was purified by LC on silica gel (various mobile phases) to obtain prodrugs P12–P17, P20, and P21.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-(1-methyl-1H-indol-3-yl)propanamido)-5-oxohexanoate (P10)
General method A, starting material 9a (428 mg); solvent: DMF; reaction time 2 h; mobile phase: DCM/MeOH, 12:1. Prodrug P10 was isolated as a light-yellow solid (375 mg) in 73% yield. 1H NMR (401 MHz, CDCl3): δ 1.39 (s, 9H), 1.85 (dtd, J = 14.3, 8.4, 5.8, 1H), 2.01–2.30 (m, 3H), 2.09 (s, 6H), 2.80 (d, J = 16.2, 1H), 2.92 (d, J = 16.2, 1H), 3.21 (d, J = 6.8, 2H), 3.69 (s, 3H), 4.30 (td, J = 7.9, 4.7, 1H), 4.61–4.67 (m, 1H), 5.22 (bs, 1H), 6.92 (s, 1H), 7.00 (d, J = 7.6, 1H), 7.06 (ddd, J = 8.0, 6.9, 1.1, 1H), 7.16 (ddd, J = 8.2, 6.9, 1.2, 1H), 7.22 (dt, J = 8.3, 1.0, 1H), 7.60 (dt, J = 8.0, 1.0, 1H), 7.70 (d, J = 7.9, 1H). 13C NMR (101 MHz, CDCl3): δ 27.18, 27.66, 27.91 (3C), 32.61, 36.26, 45.71 (2C), 52.36, 53.51, 54.83, 62.78, 82.29, 108.83, 109.21, 118.87, 119.06, 121.72, 127.95, 136.97 (2C), 170.46, 171.26, 171.42, 194.28. ESI MS: 535.3 ([M + Na]+). HR ESI MS: calcd for C26H36O5N6Na 535.26394; found 535.26373.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-phenylpropanamido)-5-oxohexanoate (P11)
General method A, starting material 9b (374 mg); solvent: DCM; reaction time 2.5 h; mobile phase: DCM/MeOH, 15:1. Prodrug P11 was isolated as a yellow amorphous compound (307 mg) in 67% yield. 1H NMR (401 MHz, CDCl3): δ 1.40 (s, 9H), 1.82–1.97 (m, 1H), 2.06–2.17 (m, 1H), 2.10 (s, 6H), 2.17–2.39 (m, 2H), 2.76 (d, J = 16.3, 1H), 2.93 (d, J = 16.3, 1H), 2.97 (dd, J = 14.0, 8.5, 1H), 3.15 (dd, J = 14.0, 5.9, 1H), 4.32 (td, J = 7.9, 4.7, 1H), 4.66 (td, J = 8.3, 5.9, 1H), 5.30 (bs, 1H), 7.02 (d, J = 8.6, 1H), 7.13–7.19 (m, 3H), 7.19–7.25 (m, 2H), 7.55 (d, J = 8.1, 1H). 13C NMR (101 MHz, CDCl3): δ 27.22, 27.95 (3C), 36.34, 37.82, 45.83 (2C), 52.45, 53.93, 54.72, 62.86, 82.29, 126.92, 128.57 (2C), 129.21 (2C), 136.58, 170.39, 170.98, 171.10, 193.84. ESI MS: 460.3 ([M + H]+). HR ESI MS: calcd for C23H34O5N5 460.25545; found 460.25482.
tert-Butyl (S)-6-Diazo-2-((R)-2-(2-(dimethylamino)acetamido)-3-phenylpropanamido)-5-oxohexanoate (P12)
General method B, starting material 9c (374 mg); solvent: DCM; reaction time 16 h; mobile phase: DCM/MeOH, 20:1. Prodrug P12 was isolated as yellow solid (348 mg) in 76% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.81–1.94 (m, 1H), 2.04–2.33 (m, 9H), 2.78 (d, J = 16.3, 1H), 2.95–3.09 (m, 2H), 3.24 (dd, J = 6.6, 14.0, 1H), 4.38 (td, J = 4.5, 8.0, 1H), 4.62–4.71 (td, J = 6.6, 8.3, 1H), 5.24 (bs, 1H), 6.79 (d, J = 7.6, 1H), 7.16–7.34 (m, 6H), 7.57 (d, J = 7.9, 1H). 13C NMR (101 MHz, CDCl3): δ 27.1, 28.1, 36.4, 37.8, 46.0, 52.5, 54.2, 54.8, 63.0, 82.5, 127.1, 128.8, 129.3, 136.9, 170.6, 170.9, 171.5, 193.9. ESI MS: 460.3 ([M + H]+). HR ESI MS: calcd for C23H34O5N5 460.25545; found 460.25491.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-2-phenylacetamido)-5-oxohexanoate (P13)
General method B, starting material 9d (360 mg); reaction time 20 h; mobile phase: DCM/MeOH, 15:1. Prodrug P13 was isolated as a yellow amorphous compound (303 mg) in 68% yield. 1H NMR (401 MHz, CDCl3): δ 1.36 (s, 9H), 1.97 (dtd, J = 14.4, 8.0, 6.4, 1H), 2.11–2.25 (m, 1H), 2.30 (s, 6H), 2.32–2.51 (m, 2H), 2.92–3.04 (m, 2H), 4.39 (td, J = 7.9, 4.7, 1H), 5.31 (bs, 1H), 5.45 (d, J = 7.5, 1H), 6.52 (d, J = 7.5, 1H), 7.30–7.42 (m, 5H), 8.09 (d, J = 7.6, 1H). 13C NMR (101 MHz, CDCl3): δ 26.1, 27.1 (3C), 28.9, 45.3 (2C), 52.0, 55.2, 62.4, 69.8, 80.9, 126.7 (2C), 127.3, 128.0 (2C), 137.6, 169.3 (2C), 169.6, 193.4. ESI MS: 446.3 ([M + H]+). HR ESI MS: calcd for C22H32O5N5 446.23980; found 446.23917.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-4-phenylbutanamido)-5-oxohexanoate (P14)
General method B, starting material 9e (388 mg); reaction time 2 h; mobile phase: DCM/MeOH, 30:1 to 15:1. Prodrug P14 was isolated as a yellow amorphous compound (242 mg) in 51% yield. 1H NMR (401 MHz, CDCl3): δ 1.46 (s, 9H), 1.67–1.85 (m, 2H), 1.91–2.05 (m, 2H), 2.12–2.27 (m, 2H), 2.31 (s, 6H), 2.70 (t, J = 7.8, 2H), 2.98 (d, J = 1.2, 2H), 4.37–4.45 (m, 2H), 5.32 (bs, 1H), 6.76 (d, J = 7.7, 1H), 7.16–7.22 (m, 3H), 7.27–7.31 (m, 2H), 7.62 (d, J = 8.3, 1H). 13C NMR (101 MHz, CDCl3): δ 27.3, 28.1 (3C), 32.0, 34.0, 46.0 (2C), 52.6, 52.8, 55.1, 63.0, 70.7, 82.5, 126.3, 128.5 (2C), 128.6 (2C), 140.9, 170.6, 171.0, 171.4, 194.0. ESI MS: 474.4 ([M + H]+). HR ESI MS: calcd for C24H36O5N5 474.27110; found 474.27011.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-(4-fluorophenyl)propanamido)-5-oxohexanoate (P15)
General method B, starting material 9f (392 mg); reaction time 16 h; mobile phase: DCM/MeOH, 20:1. Prodrug P15 was isolated as a yellow solid (349 mg) in 73% yield. 1H NMR (401 MHz, CDCl3): δ 1.45 (s, 9H), 1.94 (dtd, J = 14.2, 8.0, 6.3, 1H), 2.09–2.16 (m, 1H), 2.18 (s, 6H), 2.23–2.43 (m, 2H), 2.80–2.90 (m, 1H), 2.94–3.06 (m, 2H), 3.14 (dd, J = 14.1, 6.6, 1H), 4.35 (td, J = 7.6, 4.8, 1H), 4.60 (td, J = 8.0, 6.6, 1H), 5.29 (bs, 1H), 6.73 (d, J = 7.4, 1H), 6.92–7.01 (m, 2H), 7.13–7.23 (m, 2H), 7.57 (d, J = 8.1, 1H). 13C NMR (101 MHz, CDCl3): δ 27.3, 28.1 (3C), 37.2, 46.0 (2C), 52.6, 54.2, 54.9, 63.0, 70.1, 82.6, 115.4, 115.6, 130.9, 130.9, 132.4, 163.2, 170.4, 170.7, 171.2, 193.9. ESI MS: 478.3 ([M + H]+). HR ESI MS: calcd for C23H33O5N5F 478.24602; found 478.24526.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-(3-fluorophenyl)propanamido)-5-oxohexanoate (P16)
General method B, starting material 9g (392 mg); reaction time 16 h; mobile phase: DCM/MeOH, 20:1. Prodrug P16 was isolated as a yellow amorphous compound (391 mg) in 82% yield. 1H NMR (401 MHz, CDCl3): δ 1.44 (s, 9H), 1.87–2.01 (m, 1H), 2.16 (s, 6H), 2.19–2.42 (m, 2H), 2.82 (d, J = 16.3, 1H), 2.98 (d, J = 16.3, 1H), 3.00–3.08 (m, 1H), 3.17 (dd, J = 14.0, 6.2, 1H), 4.32–4.39 (m, 1H), 4.65 (td, J = 8.1, 6.1, 1H), 5.28–5.33 (bs, 1H), 6.85–6.95 (m, 3H), 6.99 (d, J = 7.8, 1H), 7.19–7.26 (m, 1H), 7.59 (d, J = 8.2, 1H). 13C NMR (101 MHz, CDCl3): δ 27.3, 28.1, 36.4, 37.5, 37.6, 46.0, 52.6, 53.6, 53.9, 54.9, 63.0, 82.6, 113.9, 114.1, 116.2, 116.5, 125.0, 125.0, 130.1, 130.2, 139.2, 139.3, 161.7, 164.2, 170.4, 170.6, 171.2, 193.9. ESI MS: 478.3 ([M + H]+). HR ESI MS: calcd for C23H33O5N5F 478.24602; found 478.24527.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-3-(4-(trifluoromethyl)phenyl)propanamido)-5-oxohexanoate (P17)
General method B, starting material 9h (442 mg); reaction time 2 h; mobile phase: DCM/MeOH, 30:1. Prodrug P17 was isolated as a yellow solid (407 mg) in 77% yield. 1H NMR (401 MHz, CDCl3): δ 1.42 (s, 9H), 1.93 (dtd, J = 14.3, 8.0, 6.3, 1H), 2.14 (s, 6H), 2.06–2.18 (m, 1H), 2.20–2.42 (m, 2H), 2.81 (d, J = 16.3, 1H), 2.96 (d, J = 16.3, 1H), 3.07 (dd, J = 14.1, 8.0, 1H), 3.24 (dd, J = 14.1, 6.3, 1H), 4.34 (td, J = 7.7, 4.8, 1H), 4.72 (td, J = 8.1, 6.3, 1H), 5.28 (bs, 1H), 7.03 (d, J = 7.4, 1H), 7.32 (d, J = 8.4, 2H), 7.51 (d, J = 7.4, 2H), 7.59 (d, J = 8.3, 1H). 13C NMR (101 MHz, CDCl3): δ 27.05, 28.01 (3C), 36.35, 37.71, 45.93 (2C), 52.61, 53.61, 54.94, 62.96, 82.53, 125.51 (q, J = 3.7 Hz, 2C), 129.35 (q, J = 32.6 Hz), 129.78 (2C), 140.92, 140.93, 170.38, 170.52, 171.20, 193.89. ESI MS: 528.3 ([M + H]+). HR ESI MS: calcd for C24H33O5N5F3 528.24283; found 528.24252.
tert-Butyl (S)-6-Diazo-2-(2-(2-(dimethylamino)acetamido)acetamido)-5-oxohexanoate (P18)
General method A, starting material 9i (284 mg); solvent: DMF; reaction time 1.5 h; mobile phase: DCM/MeOH, 10:1 to 5:1. Prodrug P18 was isolated after lyophilization (MeCN/H2O, 4:1; 50 mL) as light-yellow solid (237 mg) in 64% yield. 1H NMR (401 MHz, CDCl3): δ 1.46 (s, 9H), 1.99 (dtd, J = 14.3, 8.0, 6.5, 1H), 2.19 (dtd, J = 15.7, 8.5, 7.9, 5.2, 1H), 2.33 (s, 6H), 2.34–2.46 (m, 2H), 2.99 (d, J = 16.3, 1H), 3.06 (d, J = 16.3, 1H), 3.93 (dd, J = 16.6, 5.9, 1H), 4.02 (dd, J = 16.6, 5.9, 1H), 4.44 (td, J = 8.0, 4.6, 1H), 5.31 (bs, 1H), 6.76 (d, J = 7.6, 1H), 7.72 (bs, 1H). 13C NMR (101 MHz, CDCl3): δ 27.25, 28.05 (3C), 36.54, 42.75, 46.15 (2C), 52.52, 54.98, 62.99, 82.57, 169.00, 170.74, 171.73, 194.01. ESI MS: 392.2 ([M + Na]+). HR ESI MS: calcd for C16H27O5N5Na 392.19044; found 392.19016.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)propanamido)-5-oxohexanoate (P19)
General method A, starting material 9j (298 mg); solvent: DMF; reaction time 2.5 h; mobile phase: DCM/MeOH, 10:1. Prodrug P19 was isolated as light-yellow amorphous compound (268 mg) in 70% yield. 1H NMR (401 MHz, d6-DMSO): δ 1.24 (d, J = 7.0, 3H), 1.39 (s, 9H), 1.78 (ddd, J = 14.3, 9.4, 6.0, 1H), 1.96 (dq, J = 14.0, 7.1, 1H), 2.21 (s, 6H), 2.33–2.42 (m, 2H), 2.79–2.93 (m, 2H), 4.09 (ddd, J = 9.1, 7.3, 5.1, 1H), 4.36 (p, J = 7.1, 1H), 6.06 (bs, 1H), 7.73 (d, J = 7.9, 1H), 8.26 (d, J = 7.6, 1H). 13C NMR (101 MHz, CDCl3): δ 18.2, 27.3, 28.1 (3C), 36.5, 46.1 (2C), 48.5, 52.6, 63.1, 70.6, 82.5, 170.7, 171.0, 172.2, 194.2. ESI MS: 384.2 ([M + H]+). HR ESI MS: calcd for C17H30O5N5 384.22415; found 384.22401.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-4-methylpentanamido)-5-oxohexanoate (P20)
General method B, starting material 9k (340 mg); reaction time 5 h; mobile phase: DCM/MeOH, 10:1. Prodrug P20 was isolated as a yellow amorphous compound (306 mg) in 72% yield. 1H NMR (401 MHz, CDCl3): δ 0.89 (d, J = 6.1, 3H), 0.92 (d, J = 6.1, 3H), 1.42 (s, 9H), 1.51–1.71 (m, 3H), 1.91 (dtd, J = 14.4, 8.4, 6.1, 1H), 2.08–2.21 (m, 1H), 2.28 (s, 6H), 2.20–2.45 (m, 2H), 2.96 (d, J = 3.1, 2H), 4.33–4.46 (m, 2H), 5.34 (bs, 1H), 6.90 (d, J = 7.7, 1H), 7.47 (d, J = 8.5, 1H). 13C NMR (101 MHz, CDCl3): δ 21.90, 23.08, 24.87, 27.38, 28.03 (3C), 29.74, 36.46, 41.00, 45.99, 51.39, 52.44, 54.85, 62.98, 82.36, 170.63, 170.93, 172.02, 194.00. ESI MS: 426.3 ([M + H]+). HR ESI MS: calcd for C20H36O5N5 426.27110; found 426.27057.
tert-Butyl (S)-6-Diazo-2-((S)-2-(2-(dimethylamino)acetamido)-4,4-dimethylpentanamido)-5-oxohexanoate (P21)
General method B, starting material 9l (354 mg); reaction time 2.5 h; mobile phase: DCM/MeOH, 20:1 to 15:1. Prodrug P21 was isolated as a yellow amorphous compound (404 mg) in 92% yield. 1H NMR (401 MHz, CDCl3): δ 0.90 (d, J = 1.7, 9H), 1.33–1.50 (m, 10H), 1.79–1.95 (m, 2H), 2.05–2.17 (m, 1H), 2.19–2.38 (m, 8H), 2.90 (s, 2H), 4.24–4.46 (m, 2H), 5.33 (bs, 1H), 6.83–6.94 (m, 1H), 7.43 (d, J = 8.3, 1H). 13C NMR (101 MHz, CDCl3): δ 28.0, 29.7, 30.5, 45.4, 46.1, 50.6, 52.4, 53.5, 63.0, 82.2, 82.3, 170.5, 170.8, 172.3, 193.9. ESI MS: 440.3 ([M + H]+). HR ESI MS: calcd for C21H38O5N5 440.28675; found 440.28632.
Metabolic Stability
Prodrugs were screened for metabolic stability in plasma and intestinal tissue homogenates in compliance with our previously reported methods.25,46 Briefly, intestinal tissue was homogenized over ice via probe sonication with 1:9 tissue to potassium phosphate buffer (0.1 M) conditions. Prodrugs were spiked in plasma or tissue homogenate at 10 μM final concentration with 0.2% DMSO v/v and incubated in triplicate for 0 and 60 min time points. At each time point, 100 μL of the sample was precipitated with 300 μL of methanol containing internal standard (IS; losartan, 0.5 μM). Precipitated samples were vortexed (30 s) and centrifuged at 10000g for 10 min at 4 °C. After centrifugation, 100 μL of supernatant was aliquoted and submitted for analysis. Prodrug disappearance over time was monitored by liquid chromatography–mass spectrometry (LC–MS) through peak area ratios between the analyte and internal standard.
Chromatographic analysis was performed on a Dionex ultra-high-performance LC system coupled with a Q Exactive Focus Orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham, MA). Separation was achieved using an Agilent Eclipse Plus column (100 × 2.1 mm i.d.; maintained at 35 °C) packed with a 1.8 μm C18 stationary phase. The mobile phase used was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution, starting with 2.5% organic phase (from 0 to 0.25 min) and linearly increasing to 99% (from 0.25 to 1.25 min) and re-equilibrating to 2.5% by 4 min. The total run time for each analyte was 5 min. Pumps were operated at a flow rate of 0.4 mL/min. The mass spectrometer controlled by Xcalibur software 4.0.27.13 (Thermo Scientific) was operated with an HESI ion source in positive ionization mode for all compounds. Compounds were identified in the full-scan mode with m/z ranging from 75 to 1125.
Pharmacokinetics of Prodrugs in C57BL/6/CES1–/– Mice
C57BL/6/CES1–/– mice were obtained as a gift from the United States Army Medical Research Institute of Chemical Defense, Maryland, USA; breeding was performed in the Johns Hopkins animal facility and was conducted according to protocols reviewed and approved by the Johns Hopkins Institutional Animal Care and Use Committee in compliance with the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC) and the Public Health Service Policy on the Humane Care and Use of Laboratory Animals (PHS Policy). Male and female C57BL/6 mice (weighing between 25 and 30 g) 6–8 weeks of age were used for the study. The animals were maintained on a 12 h light–dark cycle with ad libitum access to food (certified laboratory food: Teklad 18% Protein Extruded Rodent Diet) and water. EL4 mouse lymphoma cells were obtained as a gift from the laboratory of Dr. Jonathan Powell (Johns Hopkins University, Baltimore, MD) and maintained in RPMI 1640 medium 1X (Corning, cat. no. 10-040-CV) with 10% (v/v) fetal bovine serum (Corning, cat. no. 35-011-CV), 1% (v/v) antimycotic/antibiotic (Corning, cat. no. 30-004-CI), 2 mM l-glutamine (Corning, cat. no. 25-005-CI), and 10 mM HEPES (Corning, cat. no. 25-060-CI) in a 5% (v/v) CO2 and 95% (v/v) air incubator prior to subcutaneous (SC) injection (1 × 106 cells in 0.2 mL of phosphate-buffered saline) on the flank of each mouse.
Pharmacokinetic study was performed after tumors grew to a mean volume of around 400 mm3. Prior to dosing, the interscapular region was wiped with alcohol gauze. For single time point PK, prodrugs were dissolved immediately prior to dosing in ethanol:Tween 80:saline (5:10:85 v/v/v) and administered to mice as a single SC dose of 1 mg/kg DON equivalent. The mice were euthanized with carbon dioxide at 30 min post drug administration, blood samples (∼0.8 mL) were collected in heparinized microtubes by cardiac puncture, and jejunum as well as tumors were removed and flash frozen on dry ice. Blood samples were centrifuged at a temperature of 4 °C at 3000g for 10 min. All samples were maintained chilled throughout processing. Plasma samples (∼300 μL) were collected in polypropylene tubes and stored at −80 °C until bioanalysis. Flash-frozen jejunum and tumor samples were also stored at −80 °C until bioanalysis.
For complete pharmacokinetic evaluation of P11, similar methods (including vehicle) as described above were utilized except plasma, tumor, and GI sample collection for pharmacokinetics was conducted at 0–6 h post dose.
Bioanalysis of DON and Intact Prodrug P11
Plasma concentration levels of P11 were measured by precipitating 20 μL of plasma sample with 100 μL of methanol containing internal standard (losartan, 0.5 μM), followed by vortex mixing for 30 s and then centrifugation at 16000g for 5 min at 4 °C. Jejunum and tumor tissues were diluted 1:5 w/v with methanol containing losartan (0.5 μM) and homogenized, followed by vortex mixing and centrifugation at 16000g for 5 min at 4 °C.
Chromatographic analysis was performed on an Agilent ultra-high-performance LC system coupled with an Agilent 6520 QTOF mass spectrometer (Agilent, Santa Clara, CA). Separation was achieved using an Agilent Eclipse Plus column (100 × 2.1 mm i.d.; maintained at 35 °C) packed with a 1.8 μm C18 stationary phase. The mobile phase used was composed of 0.1% formic acid in acetonitrile and 0.1% formic acid in water with gradient elution, starting with 2.5% organic phase (from 0 to 0.5 min) and linearly increasing to 95% (from 0.5 to 5 min), maintaining for 1 min, and re-equilibrating to 2.5% by 7 min. Pumps were operated at a flow rate of 0.3 mL/min. Standards and QCs were prepared (0.01–100 nmol/mL) in naïve matched matrixes. P11 concentrations were determined by the AUC of high-resolution extracted chromatograms of P11 divided by internal standard (460.2554 m/z/423.1695 m/z).
Bioanalysis of DON in pharmacokinetic samples, plasma, and tissue homogenates was conducted as we have previously described.19,25 Briefly, standards and QCs were prepared in respective matrixes. Standards, QCs, and plasma samples (20 μL) were precipitated with 100 μL of methanol containing internal standard (5 μM glutamate-d5) in low-retention microcentrifuge tubes. Jejunum and tumor samples were processed by adding 5 μL of methanol containing internal standard per each milligram of the tissue sample and mechanically homogenized with three Spex 2150 stainless-steel beads operated in a Geno/Grinder for 3 min at 1500 rpm. For plasma, jejunum, and tumor, the mixture was vortex mixed and centrifuged at 16000g for 5 min at 4 °C. The supernatant (100 μL) was evaporated to dryness under vacuum at 45 °C for 1 h. Dried samples were derivatized using dabsyl chloride, as described above. Calibration curves were constructed over the range 0.03–100 nmol/mL for DON in plasma, jejunum, and tumor tissues.
Pharmacokinetic parameters of P11 and DON were calculated using noncompartmental analysis by PKanalix (PKanalix, Monolix Suite 2023R1, Lixoft, France). Parameters reported include maximum plasma and tissue concentration (Cmax), time to Cmax (Tmax), and area under the plasma and tissue concentration time curve (AUC). AUC was calculated to the last quantifiable sample (AUC0–last) by use of the log–linear trapezoidal rule.
Glutamine and FGAR Quantification
Glutamine and FGAR were extracted from the tumor by protein precipitation. Per milligram of tissue, 5 μL of methanol containing 10 μmol/L deuterated N-acetyl aspartic acid and deuterated glutamate (internal standards) was added. Tissue samples were homogenized as described above and centrifuged (16000g, 5 min). Standard concentration curves of glutamine and FGAR in untreated plasma and tumor tissues were prepared (separately). For FGAR quantification, supernatants (2 μL) were injected and separated on an UltiMate 3000 UHPLC coupled to a Q Exactive Focus Orbitrap mass spectrometer. Samples were separated on an Agilent Eclipse Plus C18 RRHD (1.8 μm) 2.1 × 100 mm column. The mobile phase consisted of 8 mmol/L dimethylhexylamine (DMHA) + 0.005% formic acid in water, pH 9 (A), and 8 mmol/L DMHA in acetonitrile (B). Separation was achieved at a flow rate of 0.4 mL/min using a gradient run. Quantification was performed in full MS negative mode. Data were acquired and quantified with Xcalibur software.
Glutamine analysis took place on an Agilent 1290 UPLC coupled to an Agilent 6520 quadrupole time-of-flight mass spectrometer. Samples (2 μL) were injected and separated on a Waters Acquity UPLC BEH Amide 1.7 μm 2.1 × 100 mm HILIC column with a flow rate of 0.3 mL/min. The mobile phases consisted of A (water +0.1% formic acid) and B (acetonitrile + 0.1% formic acid). The mass spectrometer was run in positive ion mode. Standard curves for FGAR and glutamine were fitted using a blank subtraction method47 to compensate for the presence of endogenous analyte levels in naïve matrixes.
Human Tumor Cell to Plasma Partitioning Assay
Human tumor cell to plasma partitioning assays were conducted as we have previously described.25 Briefly, P493B lymphoma cells were grown at 37 °C, in a humidified atmosphere with 5% CO2. Cell confluency was estimated, and cells were harvested after achieving >80% confluency and centrifuged at 200g for 5 min at 25 °C. The obtained cell pellet was resuspended in 20 mL of Dulbecco’s phosphate-buffered saline (DPBS; Gibco, USA, cat. no. 14-190-144) maintained at 37 °C, and cell count was determined using an automated cell counter (Bio-Rad, USA). Cell suspension in DPBS was further centrifuged at 200g for 5 min at 25 °C, and the cell pellet was resuspended in human plasma (Innovative Research, USA) for partitioning assessment. The final cell density after resuspending in plasma was 10 million cells/mL of plasma. Partitioning assessment was performed in triplicate. Preincubated (37 °C for 5 min) cell–plasma suspension was spiked with DRP-104 or P11 at a final concentration of 20 μM and incubated at 37 °C for 1 h. Following incubation, a 1 mL aliquot of cell–plasma suspension was centrifuged at 1000g for 5 min at 4 °C, and supernatant plasma as well as the cell pellet was collected and stored at −80 °C until bioanalysis. Both plasma and cell pellet fractions were analyzed for intact prodrug and DON levels.
For bioanalysis, cell pellets were resuspended in water and the total weight of cells was noted. The calibration curves (0.03–100 nmol/mL) were prepared in both human plasma and untreated P493B cells. A 50 μL volume of cell suspension/plasma was precipitated with 250 μL of methanol containing internal standards (glutamate-d5, 5 μM, and losartan, 0.5 μM). Samples were briefly vortexed for 30 s and centrifuged at 10000g for 10 min at 4 °C. DON, P11, and DRP-104 analyses were conducted as described above and previously.25
Cell Viability Assay
Cell proliferation assays were performed as we have previously described.19 Briefly, P493B lymphoma cells were plated in 96-well plates at a density of 20 000 cells/well in a final volume of 100 μL of growth media. P11, or DRP-104, was added to cells in half-log serial dilutions with a final concentration of 0.2% DMSO. Cells were allowed to proliferate for 72 h, and thereafter, 20 μL of CellTiter 96 AQueous (Promega no. 3580) was added per well and incubated for 2 h. Absorbance was measured at 490 nm. Relative cell viability was calculated from the difference between untreated cell wells (100%) and media well without cells (0%).
Solubility Assay
The aqueous solubility was determined as previously reported with minor modifications.48 Briefly, an excess amount of P11 or DRP-104 was added to PBS buffer (pH 7.4) and sonicated for 1 h at 37 °C to ensure saturation. After 1 h, the solution was filtered (0.45 μm PTFE), and the filtrate was diluted appropriately, and the concentration of each analyte was determined via LC–MS. Chemical stabilities of DRP-104 and P11 were done as previously described18 with modifications. For chemical stability, prodrugs were spiked (10 μM) in buffer at pH 7.4 and incubated at 37 °C for 24 h. Prodrug disappearance was monitored using the developed HRMS methods.
Glossary
Abbreviations Used
- AUC
area under the curve
- BLQ
below limit of quantification
- CES1
carboxylesterase 1
- CES1–/–
carboxylesterase 1 knockout
- DCC
N,N′-dicyclohexylcarbodiimide
- DIPEA
N,N-diisopropylethylamine
- DMAP
4-dimethylaminopyridine
- DON
6-diazo-5-oxo-l-norleucine
- FDA
Food and Drug Administration
- FGAR
formylglycinamide ribonucleotide
- Fmoc
9-fluorenylmethyloxycarbonyl
- GI
gastrointestinal
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HRMS
high resolution mass spectrometry
- LiHMDS
lithium bis(trimethylsilyl)amide
- MET ID
metabolite identification
- WT
wild type
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c01681.
Author Contributions
○ These authors have equally contributed to this work.
This research was supported by NIH Grants R01CA193895 (to B.S.S. and R.R.), R01CA229451 (to B.S.S. and R.R.), and R01NS103927 (to B.S.S. and R.R.). This work was also funded by institutional support from the Czech Academy of Sciences (RVO 61388963) and by Grant LTAUSA18166 from the Ministry of Education, Youth, and Sports of the Czech Republic (program INTER-EXCELLENCE) and by the project National Institute for Cancer Research (Programme EXCELES, ID Project No. LX22NPO5102).
The authors declare the following competing financial interest(s): K.N., L.T., J.A., P.M., B.S.S and R.R. are inventors on multiple Johns Hopkins University (JHU) patents covering novel glutamine antagonist prodrugs and their utility. These patents have been licensed to Dracen Pharmaceuticals Inc. P.M., B.S.S. and R.R. are founders of and hold equity in Dracen Pharmaceuticals Inc. P.M., B.S.S. and R.R. also serve/d as scientific consultants to Dracen. This arrangement has been reviewed and approved by the JHU in accordance with its conflict-of-interest policies. The authors declare no other competing interests.
Supplementary Material
References
- Cruzat V.; Macedo Rogero M.; Noel Keane K.; Curi R.; Newsholme P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10 (11), 1564. 10.3390/nu10111564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meynial-Denis D.Glutamine: Biochemistry, Physiology, and Clinical Applications; CRC Press: 2017. [Google Scholar]
- Wise D. R.; Thompson C. B. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35 (8), 427–33. 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnowski G. S.; Stock C. C. Effects of combinations of azaserine and of 6-diazo-5-oxo-L-norleucine with purine analogs and other antimetabolites on the growth of two mouse mammary carcinomas. Cancer research 1957, 17 (10), 1033–1039. [PubMed] [Google Scholar]
- Shelton L. M.; Huysentruyt L. C.; Seyfried T. N. Glutamine targeting inhibits systemic metastasis in the VM-M3 murine tumor model. International journal of cancer 2010, 127 (10), 2478–85. 10.1002/ijc.25431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovejera A. A.; Houchens D. P.; Catane R.; Sheridan M. A.; Muggia F. M. Efficacy of 6-diazo-5-oxo-L-norleucine and N-[N-gamma-glutamyl-6-diazo-5-oxo-norleucinyl]-6-diazo-5-oxo-norleucine against experimental tumors in conventional and nude mice. Cancer research 1979, 39 (8), 3220–3224. [PubMed] [Google Scholar]
- Catane R.; Von Hoff D. D.; Glaubiger D. L.; Muggia F. M. Azaserine, DON, and azotomycin: three diazo analogs of L-glutamine with clinical antitumor activity. Cancer treatment reports 1979, 63 (6), 1033–1038. [PubMed] [Google Scholar]
- Rahman A.; Smith F. P.; Luc P. T.; Woolley P. V. Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON). Investigational new drugs 1985, 3 (4), 369–74. 10.1007/BF00170760. [DOI] [PubMed] [Google Scholar]
- Kovach J. S.; Eagan R. T.; Powis G.; Rubin J.; Creagan E. T.; Moertel C. G. Phase I and pharmacokinetic studies of DON. Cancer treatment reports 1981, 65 (11–12), 1031–1036. [PubMed] [Google Scholar]
- Sullivan M. P.; Nelson J. A.; Feldman S.; Van Nguyen B. Pharmacokinetic and phase I study of intravenous DON (6-diazo-5-oxo-L-norleucine) in children. Cancer chemotherapy and pharmacology 1988, 21 (1), 78–84. 10.1007/BF00262746. [DOI] [PubMed] [Google Scholar]
- Sklaroff R. B.; Casper E. S.; Magill G. B.; Young C. W. Phase I study of 6-diazo-5-oxo-L-norleucine (DON). Cancer treatment reports 1980, 64 (12), 1247–1251. [PubMed] [Google Scholar]
- Lynch G.; Kemeny N.; Casper E. Phase II evaluation of DON (6-diazo-5-oxo-L-norleucine) in patients with advanced colorectal carcinoma. American journal of clinical oncology 1982, 5 (5), 541–3. 10.1097/00000421-198210000-00014. [DOI] [PubMed] [Google Scholar]
- Earhart R. H.; Koeller J. M.; Davis H. L. Phase I trial of 6-diazo-5-oxo-L-norleucine (DON) administered by 5-day courses. Cancer treatment reports 1982, 66 (5), 1215–1217. [PubMed] [Google Scholar]
- Earhart R. H.; Amato D. J.; Chang A. Y.-C.; Borden E. C.; Shiraki M.; Dowd M. E.; Comis R. L.; Davis T. E.; Smith T. J. Phase II trial of 6-diazo-5-oxo-L-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Investigational new drugs 1990, 8 (1), 113–119. 10.1007/BF00216936. [DOI] [PubMed] [Google Scholar]
- Eagan R. T.; Frytak S.; Nichols W. C.; Creagan E. T.; Ingle J. N. Phase II study on DON in patients with previously treated advanced lung cancer. Cancer treatment reports 1982, 66 (8), 1665–1666. [PubMed] [Google Scholar]
- Magill G. B.; Myers W. P.; Reilly H. C.; Putnam R. C.; Magill J. W.; Sykes M. P.; Escher G. C.; Karnofsky D. A.; Burchenal J. H. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-1-norleucine (DON) in human neoplastic disease. Cancer 1957, 10 (6), 1138–50. 10.1002/1097-0142(195711/12)10:6<1138::AID-CNCR2820100608>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Sullivan M. P.; Beatty E. C. Jr.; Hyman C. B.; Murphy M. L.; Pierce M. I.; Severo N. C. A comparison of the effectiveness of standard dose 6-mercaptopurine, combination 6-mercaptopurine and DON, and high-loading 6-mercaptopurine therapies in treatment of the acute leukemias of childhood: results of a coperative study. Cancer chemotherapy reports 1962, 18, 83–95. [PubMed] [Google Scholar]
- Rais R.; Jančařík A.; Tenora L.; Nedelcovych M.; Alt J.; Englert J.; Rojas C.; Le A.; Elgogary A.; Tan J.; Monincová L.; Pate K.; Adams R.; Ferraris D.; Powell J.; Majer P.; Slusher B. S. Discovery of 6-Diazo-5-oxo-l-norleucine (DON) Prodrugs with Enhanced CSF Delivery in Monkeys: A Potential Treatment for Glioblastoma. Journal of medicinal chemistry 2016, 59 (18), 8621–33. 10.1021/acs.jmedchem.6b01069. [DOI] [PubMed] [Google Scholar]
- Tenora L.; Alt J.; Dash R. P.; Gadiano A. J.; Novotná K.; Veeravalli V.; Lam J.; Kirkpatrick Q. R.; Lemberg K. M.; Majer P.; Rais R.; Slusher B. S. Tumor-Targeted Delivery of 6-Diazo-5-oxo-l-norleucine (DON) Using Substituted Acetylated Lysine Prodrugs. Journal of medicinal chemistry 2019, 62 (7), 3524–3538. 10.1021/acs.jmedchem.8b02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaford A. R.; Alt J.; Rais R.; Wang S. Z.; Kaur H.; Thorek D. L. J.; Eberhart C. G.; Slusher B. S.; Martin A. M.; Raabe E. H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Translational oncology 2019, 12 (10), 1314–1322. 10.1016/j.tranon.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M. H.; Sun I. H.; Zhao L.; Leone R. D.; Sun I. M.; Xu W.; Collins S. L.; Tam A. J.; Blosser R. L.; Patel C. H.; Englert J. M.; Arwood M. L.; Wen J.; Chan-Li Y.; Tenora L.; Majer P.; Rais R.; Slusher B. S.; Horton M. R.; Powell J. D. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Invest. 2020, 130 (7), 3865–3884. 10.1172/JCI131859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A. S.; da Costa Rosa M.; Stumpo V.; Rais R.; Slusher B. S.; Riggins G. J. The glutamine antagonist prodrug JHU-083 slows malignant glioma growth and disrupts mTOR signaling. Neuro-oncology advances 2021, 3 (1), vdaa149. 10.1093/noajnl/vdaa149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Wang R.; Liu Z.; Fan J.; Liu S.; Tan S.; Li X.; Li B.; Yang X. Unbalanced Glutamine Partitioning between CD8T Cells and Cancer Cells Accompanied by Immune Cell Dysfunction in Hepatocellular Carcinoma. Cells 2022, 11 (23), 3924. 10.3390/cells11233924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone R. D.; Zhao L.; Englert J. M.; Sun I. M.; Oh M. H.; Sun I. H.; Arwood M. L.; Bettencourt I. A.; Patel C. H.; Wen J.; Tam A.; Blosser R. L.; Prchalova E.; Alt J.; Rais R.; Slusher B. S.; Powell J. D. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (New York, N.Y.) 2019, 366 (6468), 1013–1021. 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais R.; Lemberg K. M.; Tenora L.; Arwood M. L.; Pal A.; Alt J.; Wu Y.; Lam J.; Aguilar J. M. H.; Zhao L.; Peters D. E.; Tallon C.; Pandey R.; Thomas A. G.; Dash R. P.; Seiwert T.; Majer P.; Leone R. D.; Powell J. D.; Slusher B. S. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Science advances 2022, 8 (46), eabq5925 10.1126/sciadv.abq5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y.; Estok T. M.; Wild R. Sirpiglenastat (DRP-104) Induces Antitumor Efficacy through Direct, Broad Antagonism of Glutamine Metabolism and Stimulation of the Innate and Adaptive Immune Systems. Molecular cancer therapeutics 2022, 21 (10), 1561–1572. 10.1158/1535-7163.MCT-22-0282. [DOI] [PubMed] [Google Scholar]
- Johnson M. L.; Doroshow D. B.; Seiwert T. Y.; Gibson M. K.; Velcheti V.; Lisberg A. E.; Patel S. A.; Scheffler M.; Lafleur F.; Dugan M. H.; Sharma S. Phase 1 and phase 2a, first-in-human (FIH) study, of DRP-104, a broad glutamine antagonist, in adult patients with advanced solid tumors. Journal of clinical oncology 2021, 39, TPS3149. 10.1200/JCO.2021.39.15_suppl.TPS3149. [DOI] [Google Scholar]
- Johnson M. L.; Doroshow D. B.; Seiwert T. Y.; Gibson M. K.; Velcheti V.; Lisberg A. E.; Patel S. A.; Scheffler M.; Lafleur F.; Dugan M. H.; Sharma S. Phase 1 and phase 2a, first-in-human (FIH) study, of DRP-104, a broad glutamine antagonist, in adult patients with advanced solid tumors. Journal of Clinical Oncology 2021, 39 (15_suppl), TPS3149. 10.1200/JCO.2021.39.15_suppl.TPS3149. [DOI] [Google Scholar]
- Zangaglia R.; Stocchi F.; Sciarretta M.; Antonini A.; Mancini F.; Guidi M.; Martignoni E.; Pacchetti C. Clinical experiences with levodopa methylester (melevodopa) in patients with Parkinson disease experiencing motor fluctuations: an open-label observational study. Clinical neuropharmacology 2010, 33 (2), 61–6. 10.1097/WNF.0b013e3181c5e60c. [DOI] [PubMed] [Google Scholar]
- Davies R. O.; Gomez H. J.; Irvin J. D.; Walker J. F. An overview of the clinical pharmacology of enalapril. British journal of clinical pharmacology 1984, 18 (S2), 215S–229S. 10.1111/j.1365-2125.1984.tb02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies R. O.; Gomez H. J.; Irvin J. D.; Walker J. F. An overview of the clinical pharmacology of enalapril. Br. J. Clin. Pharmacol. 1984, 18 (S2), 215S–229S. 10.1111/j.1365-2125.1984.tb02601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman R. L.; Ray A. S.; Hui H. C.; Zhang L.; Birkus G.; Boojamra C. G.; Desai M. C.; Douglas J. L.; Gao Y.; Grant D.; Laflamme G.; Lin K. Y.; Markevitch D. Y.; Mishra R.; McDermott M.; Pakdaman R.; Petrakovsky O. V.; Vela J. E.; Cihlar T. Discovery of GS-9131: Design, synthesis and optimization of amidate prodrugs of the novel nucleoside phosphonate HIV reverse transcriptase (RT) inhibitor GS-9148. Bioorganic & medicinal chemistry 2010, 18 (10), 3606–17. 10.1016/j.bmc.2010.03.041. [DOI] [PubMed] [Google Scholar]
- Davey M. S.; Malde R.; Mykura R. C.; Baker A. T.; Taher T. E.; Le Duff C. S.; Willcox B. E.; Mehellou Y. Synthesis and Biological Evaluation of (E)-4-Hydroxy-3-methylbut-2-enyl Phosphate (HMBP) Aryloxy Triester Phosphoramidate Prodrugs as Activators of Vγ9/Vδ2 T-Cell Immune Responses. Journal of medicinal chemistry 2018, 61 (5), 2111–2117. 10.1021/acs.jmedchem.7b01824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qandil A. M. Prodrugs of nonsteroidal anti-inflammatory drugs (NSAIDs), more than meets the eye: a critical review. International journal of molecular sciences 2012, 13 (12), 17244–74. 10.3390/ijms131217244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara S. J.; Meinig J. M.; Placzek A. T.; Banerji T.; McTigue P.; Hartley M. D.; Sanford-Crane H. S.; Banerji T.; Bourdette D.; Scanlan T. S. Ester-to-amide rearrangement of ethanolamine-derived prodrugs of sobetirome with increased blood-brain barrier penetration. Bioorganic & medicinal chemistry 2017, 25 (10), 2743–2753. 10.1016/j.bmc.2017.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisar J. S.; Grice C. A.; Jones T. K.; Wang D.-H.; Weber O.; Cravat B. F.; Niphakis M. J.; Cognetta A.; Chang J. W.. Preparation of carbamate compounds as modulators of MAGL and/or ABHD6. WO 2013103973 A1, 2013.
- Duysen E. G.; Koentgen F.; Williams G. R.; Timperley C. M.; Schopfer L. M.; Cerasoli D. M.; Lockridge O. Production of ES1 plasma carboxylesterase knockout mice for toxicity studies. Chemical research in toxicology 2011, 24 (11), 1891–8. 10.1021/tx200237a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Sedlacek M.; Manoharan I.; Boopathy R.; Duysen E. G.; Masson P.; Lockridge O. Butyrylcholinesterase, paraoxonase, and albumin esterase, but not carboxylesterase, are present in human plasma. Biochem. Pharmacol. 2005, 70 (11), 1673–1684. 10.1016/j.bcp.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Alt J.; Gori S. S.; Lemberg K. M.; Pal A.; Veeravalli V.; Wu Y.; Aguilar J. M. H.; Dash R. P.; Tenora L.; Majer P.; Sun Q.; Slusher B. S.; Rais R. Glutamine Antagonist GA-607 Causes a Dramatic Accumulation of FGAR which can be used to Monitor Target Engagement. Current drug metabolism 2021, 22 (9), 735–745. 10.2174/1389200222666210831125041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg K. M.; Zhao L.; Wu Y.; Veeravalli V.; Alt J.; Aguilar J. M. H.; Dash R. P.; Lam J.; Tenora L.; Rodriguez C.; Nedelcovych M. T.; Brayton C.; Majer P.; Blakeley J. O.; Rais R.; Slusher B. S. The Novel Glutamine Antagonist Prodrug JHU395 Has Antitumor Activity in Malignant Peripheral Nerve Sheath Tumor. Molecular cancer therapeutics 2020, 19 (2), 397–408. 10.1158/1535-7163.MCT-19-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenora L.; Alt J.; Dash R. P.; Gadiano A. J.; Novotna K.; Veeravalli V.; Lam J.; Kirkpatrick Q. R.; Lemberg K. M.; Majer P.; Rais R.; Slusher B. S. Tumor-Targeted Delivery of 6-Diazo-5-oxo-l-norleucine (DON) Using Substituted Acetylated Lysine Prodrugs. Journal of medicinal chemistry 2019, 62 (7), 3524–3538. 10.1021/acs.jmedchem.8b02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharpure S. J.; Nanda L. N.; Kumari D. Enantiospecific Total Synthesis of (+)-3-epi-Epohelmin A Using a Nitrogen-Substituted Donor-Acceptor Cyclopropane. Eur. J. Org. Chem. 2017, 2017 (27), 3917–3920. 10.1002/ejoc.201700498. [DOI] [Google Scholar]
- Zaminer J.; Brockmann C.; Huy P.; Opitz R.; Reuter C.; Beyermann M.; Freund C.; Müller M.; Oschkinat H.; Kühne R.; Schmalz H. G. Addressing protein-protein interactions with small molecules: a Pro-Pro dipeptide mimic with a PPII helix conformation as a module for the synthesis of PRD-binding ligands. Angewandte Chemie (International ed. in English) 2010, 49 (39), 7111–5. 10.1002/anie.201001739. [DOI] [PubMed] [Google Scholar]
- Chiha S.; Soicke A.; Barone M.; Müller M.; Bruns J.; Opitz R.; Neudörfl J.-M.; Kühne R.; Schmalz H.-G. Design and Synthesis of Building Blocks for PPII-Helix Secondary-Structure Mimetics: A Stereoselective Entry to 4-Substituted 5-Vinylprolines. Eur. J. Org. Chem. 2018, 2018 (4), 455–460. 10.1002/ejoc.201701584. [DOI] [Google Scholar]
- Calimsiz S.; Lipton M. A. Synthesis of N-Fmoc-(2S,3S,4R)-3,4-dimethylglutamine: An application of lanthanide-catalyzed transamidation. Journal of organic chemistry 2005, 70 (16), 6218–21. 10.1021/jo050518r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal A.; Gori S.; Yoo S. W.; Thomas A. G.; Wu Y.; Friedman J.; Tenora L.; Bhasin H.; Alt J.; Haughey N.; Slusher B. S.; Rais R. Discovery of Orally Bioavailable and Brain-Penetrable Prodrugs of the Potent nSMase2 Inhibitor DPTIP. Journal of medicinal chemistry 2022, 65 (16), 11111–11125. 10.1021/acs.jmedchem.2c00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakare R.; Chhonker Y. S.; Gautam N.; Alamoudi J. A.; Alnouti Y. Quantitative analysis of endogenous compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. 10.1016/j.jpba.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Zimmermann S. C.; Tichý T.; Vávra J.; Dash R. P.; Slusher C. E.; Gadiano A. J.; Wu Y.; Jančařík A.; Tenora L.; Monincová L.; Prchalová E.; Riggins G. J.; Majer P.; Slusher B. S.; Rais R. N-Substituted Prodrugs of Mebendazole Provide Improved Aqueous Solubility and Oral Bioavailability in Mice and Dogs. Journal of medicinal chemistry 2018, 61 (9), 3918–3929. 10.1021/acs.jmedchem.7b01792. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.