

Abstract
Most patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) do not benefit from current re-induction or approved targeted therapies. In the absence of targetable genetic mutations, there is minimal guidance on optimal treatment selection particularly in the R/R setting highlighting an unmet need for clinically useful functional biomarkers. Blood and bone marrow samples from patients treated on two clinical trials were used to test the combination of lenalidomide (LEN) and MEC (mitoxantrone, etoposide, and cytarabine) chemotherapy in R/R AML patients. The bone marrow samples were available to test the clinical utility of the mitochondrial apoptotic BH3 and dynamic BH3 profiling (DBP) assays in predicting response, as there was no clear genetic biomarker identifying responders. To test whether LEN-induced mitochondrial priming predicted clinical response to LEN-MEC therapy, we performed DBP on patient myeloblasts. We found that short-term ex vivo treatment with lenalidomide discriminated clinical responders from non-responders based on drug-induced change in priming (delta priming). Using paired patient samples collected before and after clinical LEN treatment (prior to MEC dosing), we confirmed LEN-induced increased apoptotic priming in vivo, suggesting LEN enhanced vulnerability of myeloblasts to cytotoxic MEC chemotherapy. This is the first study demonstrating the potential role of DBP in predicting clinical response to a combination regimen. Our findings demonstrate that functional properties of relapsed AML can identify active therapies.
1 |. INTRODUCTION
Beyond targeted therapies based on genetic mutations that drive disease, there are no clear strategies for drug selection for patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) who progress after standard therapies. Mitoxantrone, etoposide and cytarabine (MEC) is a common salvage chemotherapy regimen with modest response rates (18% to 25%) in R/R AML patients.1,2 We hypothesized that lenalidomide (LEN), an inducer of targeted degradation of several proteins including Ikaros (IKZF1)3 and casein kinase 1A1 (CK1α),4 may sensitize myeloblasts to cytarabine. A Stanford phase 1 trial adding LEN prior to MEC chemotherapy (LEN-MEC) led to complete remissions (CR) in 5 of 17 (29%) R/R AML patients.5 A separate phase 1 study of LEN-MEC with minor differences in dosing schema has previously reported that this combination was tolerable and yielded a CR rate of 41% (7/17) at the maximum tolerated dose (MTD).6
The mitochondrial pathway of apoptosis regulated by the BCL-2 family proteins is central to cellular response and resistance to chemotherapy. Cells commit to apoptosis when pro-apoptotic BH3-only proteins bind and inhibit pro-survival members or directly activate BAX and BAK, promote their homo-oligomerization, and cause mitochondrial outer membrane permeabilization (MOMP).7–10 The BH3 profiling measures baseline mitochondrial apoptotic priming (readiness to undergo apoptosis) via exposure of mitochondria to a panel of synthetic BH3 domain peptides.11,12 We have previously shown that the intrinsic priming status of myeloblasts as measured by BH3 profiling11 can identify clinical responders to conventional chemotherapy and the BCL-2 inhibitor (venetoclax) in AML.13,14 Additionally, the dynamic BH3 profiling (DBP) assay12 measures drug-induced increases in apoptotic priming following short-term ex vivo drug-treatment of cells.15 Results from DBP correlate with in vivo response to chemotherapy both in humans and in mice.12,16 We have previously shown that there is in vivo selection for decreased mitochondrial priming in paired cases of relapsed AML patients who had apoptotic priming measured prior to cytotoxic induction chemotherapy and upon disease relapse.13 Here, we hypothesized that select targeted therapies, such as LEN, could increase the priming of “low primed” myeloblasts in relapsed cases, and bring myeloblasts closer to the apoptotic threshold, resulting in increased chemosensitivity to subsequent chemotherapy, such as MEC. Hence, we asked if mitochondrial functional assays can identify R/R AML patients that are likely to achieve a CR to LEN-MEC therapy allowing potential future use to select patients for this regimen.
2 |. METHODS
2.1 |. Study cohort
Seventeen adults completed treatment on the Stanford IRB-approved clinical trial (NCT01904643; Table S1). Viably frozen (in 10% DMSO) mononuclear cells from blood (PBMCs) and bone marrow (BMMCs) that had >70% viability and contained >5% leukemia blasts from pre-treatment and post-LEN stages (collected prior to MEC chemotherapy) were studied from both Stanford5 (n = 15) and the Dana-Farber/Harvard Cancer Center (DF/HCC) (NCT01681537)6 (n = 8) phase 1 studies.
2.2 |. BH3 and DBP profiling analyses
The BMMCs/PBMCs were assessed by the Zombie Yellow fixable viability kit, and the leukemia blast population identified by SSClow and CD45mid/low (Figure S1) was subjected to BH3 profiling and DBP, as previously described (https://letailab.dana-farber.org/bh3-profiling.html).13,17 For BH3 profiling, BMMCs or PBMCs were exposed to synthetic BH3 peptides after plasma membrane permeabilization with digitonin,17 and sensitivity to BH3 peptides was measured by cytochrome c release by flow cytometry. The DMSO and alamethicin were used as negative and positive controls, respectively. For DBP, myeloblasts were exposed to ex vivo LEN treatment for 16 hours (0.5 μM and 1.0 μM), or DMSO (control) followed by BH3 profiling using BIM or PUMA peptides. The read-out for drug-induced change in priming is defined as “delta priming” (calculation: % delta [Δ] priming = % cytochrome c lossLEN - % cytochrome c lossDMSO).
2.3 |. Immuno-mass spectrometry
Frozen cell pellets were analyzed using immuno-multiple reaction monitoring mass spectrometry as previously described.18
2.4 |. Reagents and antibodies
Lenalidomide for ex vivo culturing was purchased at Selleck Chemicals. Other reagents purchased in this study were antibodies: BIM FITC (Cell Signaling), Cytochrome c (Alexa Fluor647, #558709, BD Biosciences), CD45-BV421 (563 879, BD Biosciences), and Zombie Yellow viability dye (423 102; BioLegend).
2.5 |. Definitions and statistical analysis
All biomarker studies were performed while the analyzer remained blinded to patient clinical response (defined as achievement of CR following ELN criteria19). For correlative studies, a comparison of responders and non-responders was assessed by a one-sided Wilcoxon rank-sum test. Optimal DBP threshold was determined via a recursive partitioning algorithm to distinguish responders from non-responders. Overall survival (OS) was estimated with Kaplan Meier analysis and the log-rank test was performed. A P value <.05 was considered to be statistically significant. Further details in Supplemental Methods.
3 |. RESULTS
Seventeen patients with R/R AML were enrolled and treated on the Stanford5 phase 1 clinical trial of lenalidomide followed by MEC chemotherapy, which terminated early due to poor patient accrual. The pretreatment characteristics of the enrolled patients are summarized in Figure 1A. The median age was 55 years (range, 22–72 years); six subjects were aged 60 and older (35%). Twelve subjects had relapsed AML (71%), including four subjects who were post-allogeneic hematopoietic stem cell transplantation. Five subjects had refractory AML (29%). Nearly 60% of the study population had received two or more prior therapies. Five of the 17 patients (29%) achieved a CR. Two additional patients achieved a partial remission (PR), of which one had only extramedullary involvement and the other progressed during the re-induction period. Bone marrow was evaluated by next-generation sequencing as part of routine clinical work up for the presence of mutations in TP53, DNMT3A, NRAS, IDH1, IDH2, CEBPa, NPM1, FLT3-TKD, and FLT3-ITD, and by allele-specific PCR for the presence of the D816V KIT mutation. Additional myeloid gene panels could not be subsequently performed due to sample availability. None of the patients with adverse risk cytogenetics, including six with complex karyotype, achieved a CR. The presence of common myeloid mutations did not inform response within this small heterogenous cohort. A separate but similar study investigated LEN at varying doses on days 1–10 and MEC on days 4–8, and an encouraging CR rate of 41% and a 1-year overall survival rate of 61% were observed at the MTD of 50 mg LEN (Figure 1B).6 No correlation between genetic profile and treatment response was noted, except perhaps a slightly worse chance of response in the presence of an NRAS mutation.
FIGURE 1.
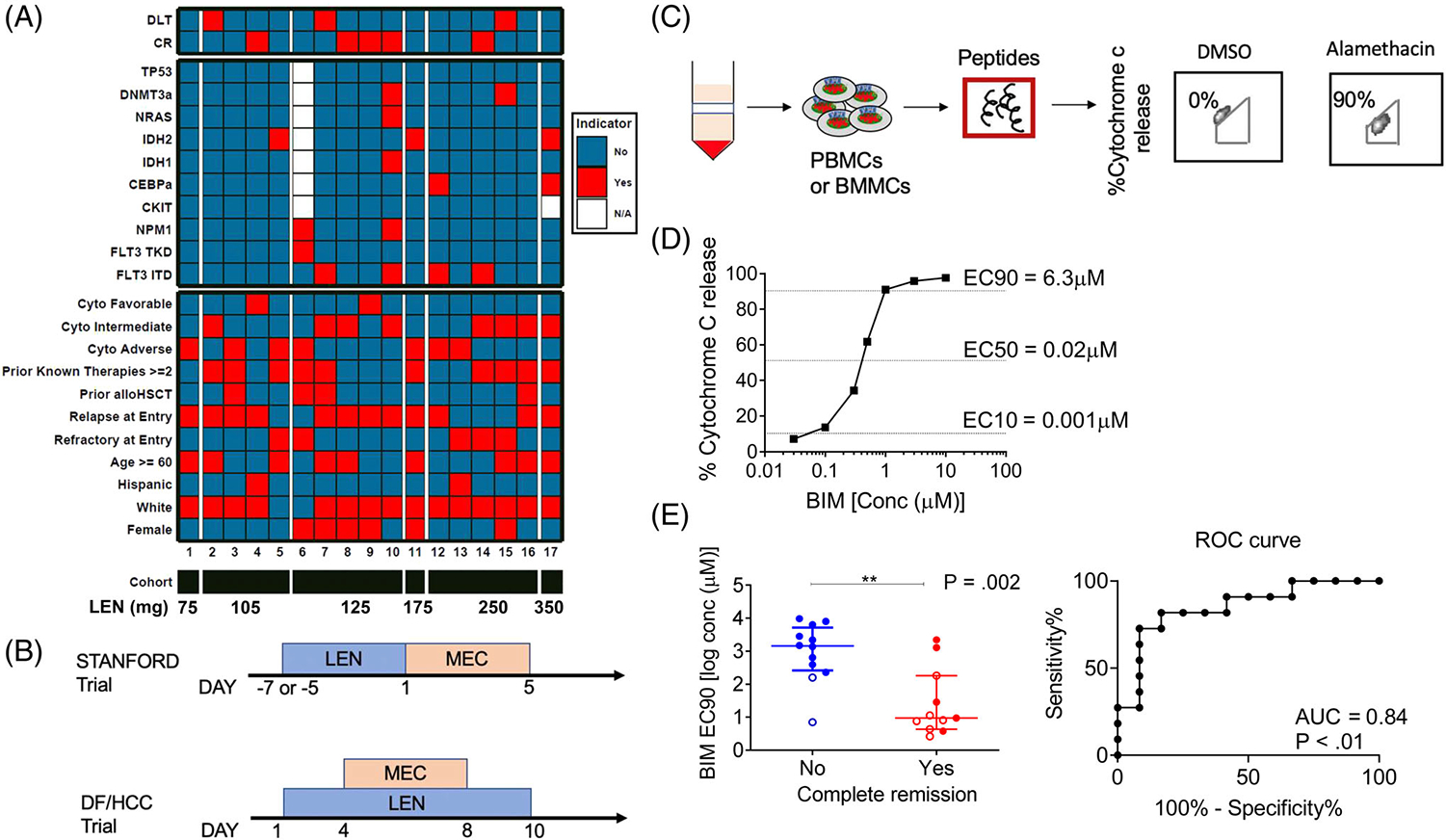
Patient cohorts and baseline BH3 profiling of pre-treatment myeloblasts. (A) Individual patient characteristics, mutation status, LEN dose assignment and clinical response from patients treated on the Stanford trial (NCT01904643). (B) Treatment schema of the Stanford and DF/HCC phase 1 LEN-MEC trials. In the Stanford trial, lenalidomide “priming” was administered for 5–7 days (per dose level, at doses ranging from 10–50 mg for 5–7 daily doses, see Supplemental Methods for details) prior to MEC chemotherapy (etoposide 100 mg/m2/day intravenous [IV] infusion over 1 hour on days 1–5, cytarabine 1000 mg/m2/day IV over 3 hours on days 1–5 and mitoxantrone 8 mg/m2/day IV over 15–30 minutes on days 1–5). Details of the DF/HCC phase 1 LEN-MEC trials including eligibility, patient demographics and disease characteristics have been previously described.6 (C) Schematic of BH3 assay performed on patient myeloblasts (see Figure S1 for myeloblast gating strategy). (D) BIM dose response curve demonstrating derivation of BIM EC10, EC50, and EC90 from individual patients. (E) Correlation of BIM EC90 derived from BH3 profiling and remission status of patients (left) with the corresponding receiver operating characteristic (ROC) curve (right). The horizontal line represents median with interquartile range and P values were calculated using one-sided Wilcoxon-rank sum test. Closed circles represent Stanford trial patients (n = 15) and open circles represent DF/HCC trial patients (n = 8)
Since the mutation spectrum and clinical characteristics of patients treated in the Stanford (Table S2) and DF/HCC6 LEN-MEC R/R AML trials did not predict a response, we pooled samples from these two separate studies (Figure 1B), and performed mitochondrial apoptotic pathway analyses to evaluate for a functional predictive biomarker. We first performed BH3 profiling on pre-treatment myeloblasts to measure baseline apoptotic priming status (Figure 1C). We have previously shown that the more sensitive mitochondria are to pro-apoptotic BH3 peptides such as BIM, the more primed they are for apoptosis-inducing treatments.11,13,14 We performed titrations of BIM peptide to determine the different effective concentrations required to induce cytochrome c release (Figure 1D). Previously, we found the least primed myeloblasts determined by the concentration of BIM peptide required to induce MOMP in all but 10% of the cells (or [BIM] EC90)within the bulk leukemia population, served as a better predictor of clinical response to cytotoxic therapy, compared to the median priming ([BIM] EC50) of the population.14 We observed that patients whose pretreatment myeloblasts had a lower [BIM] EC90 peptide ultimately achieved a CR in response to LEN-MEC therapy (P = .002, Figure 1E). This suggests that pre-treatment cases that had a relatively higher [BIM] EC90 contain a chemoresistant subpopulation and hence they require more BIM peptide to apoptotically prime myeloblasts compared to those with lower [BIM] EC90.
We next asked whether specific measurement of LEN-induced modulation in apoptotic signaling by DBP could more accurately predict a clinical response. We performed DBP by first exposing pretreatment myeloblasts to ex vivo LEN. At 16 hours post-treatment, we compared changes in mitochondrial signaling in response to BIM and PUMA, which are promiscuous BH3 peptides,10 set at concentrations identified to give the maximal delta priming percent. This was based on their respective dose-response curves, to capture the most dynamic range in mitochondrial depolarization across all samples (Figure 2A). An increase in mitochondrial priming caused by direct ex vivo exposure of myeloblasts to LEN, discriminated responders from non-responders based on response to the pro-apoptotic BIM (P < .001, Figures 2B and S2A) and PUMA (P < .001, Figures 2C and S2B). Corresponding ROC curve analyses indicated that the delta (Δ) priming was an excellent determinant for predicting achievement of CR, with AUC values approaching or equal to one for both BIM (P = .001) and PUMA (P < .001). These results suggest that LEN-induced apoptotic signaling is a good predictor of clinical response to LEN-MEC chemotherapy. Moreover, differential delta priming among patients showed that DBP may be able to identify “priming agents” that enhance myeloblasts response to subsequent chemotherapy - whether LEN or another therapeutic agent.
FIGURE 2.
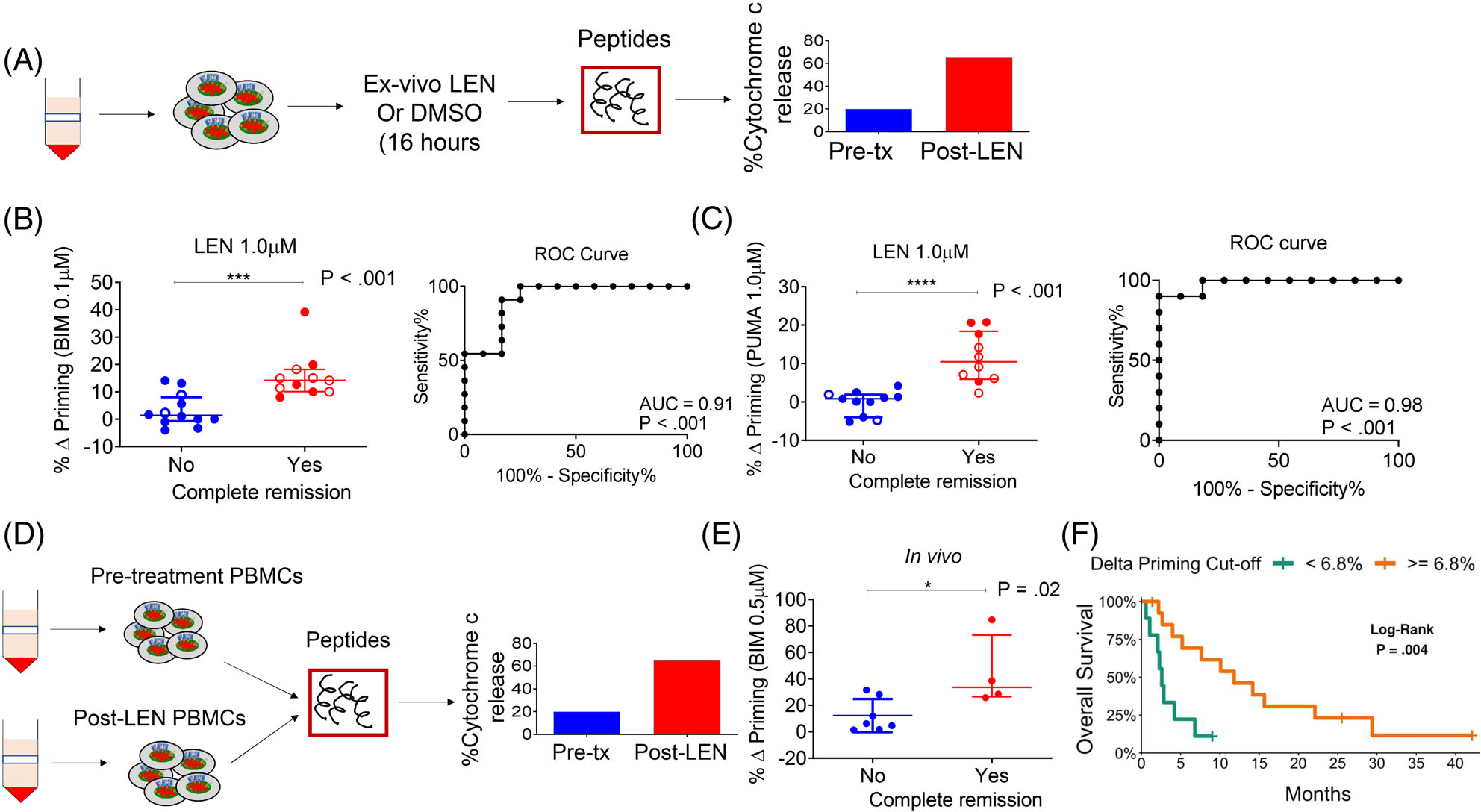
Lenalidomide-induced mitochondrial apoptosis priming correlates with clinical response. (A) Schematic of DBP assay performed on myeloblasts from patients treated on the two trials. (B and C) Correlation of % Δ priming and remission status of trial patients. The % Δ priming was derived from DBP analysis performed by first exposing pre-treatment myeloblasts to ex vivo LEN treatment, followed by measurement of priming response to BIM or PUMA peptides (left panels). The % Δ primingex vivo = % cytochrome c lossPost ex vivo LEN - % cytochrome c Post ex vivo DMSO. Corresponding ROC shows that DBP is a binary predictor of clinical response (right panels). Closed circles represent Stanford trial patients (n = 15) and open circles represent DF/HCC trial patients (n = 8). The DBP analysis using the PUMA peptide was restricted to 13 Stanford trial patients due to limited cell numbers. (D) Schematic of BH3 assays performed on myeloblasts collected from trial patients at pre-treatment stage and post-LEN alone (prior to MEC chemotherapy). (E) Correlation of in vivo % Δ priming (% Δ primingin vivo = % cytochrome c lossPost LEN - % cytochrome c losspre-treatment) with clinical response to LEN-MEC chemotherapy performed in paired samples collected from the Stanford trial (n = 11, restricted to those with leukemic blood at both pre-treatment and post-LEN/pre-MEC administration stages). (F) Using a recursive partitioning algorithm for categorizing response based on % Δ priming, a K-M curve was generated using the potential cut-point or threshold that distinguishes non-responders from responders. P-values were calculated using one-sided Wilcoxon-rank sum test. Horizontal line in B, C and E represents median with interquartile range
We next asked whether apoptotic signaling in patients’ peripheral myeloblasts changed after in vivo LEN administration, and if any changes correlated with response. We performed BH3 profiling on paired samples collected before and after completion of the LEN priming phase (Figure 2D). The LEN-induced delta priming of myeloblasts during treatment was significantly higher in responders compared to non-responders (P = .02, Figure 2E). This observation confirms that in vivo LEN treatment, which was administered as part of the clinical protocol, enhanced the mitochondrial apoptotic priming of myeloblasts and suggested that this increased priming was associated with enhanced sensitivity of myeloblasts to MEC chemotherapy. To determine if DBP could inform durable remission, we applied a recursive partitioning algorithm to the % Δ priming responses. We found 6.8% to be an acceptable % Δ priming threshold for distinguishing responders from non-responders (misclassification error 13%). Looking at OS as stratified by the 6.8% threshold, we found a positive association with increased delta priming (P = .004, Figures 2F and S2C).
Finally, to determine whether LEN’s effect was on target, we measured substrate protein levels before and after ex vivo and in vivo LEN-based therapy. Significant relative decreases in IKZF1 (ex vivo, P = .024; in vivo, P = .003) and CSNK1a1 protein levels (ex vivo, P = .003; in vivo, P = .007) were observed in all cases, including responders and non-responders, suggesting LEN-induced priming is mediated by substrate degradation in the malignant cells18 (Figure S3A,B).
4 |. DISCUSSION
In this study, initiation of apoptotic signaling by the targeted therapy LEN appears to be central to determining successful response to MEC re-induction chemotherapy. The selective increase in mitochondrial priming after ex vivo LEN treatment as measured by DBP, is an excellent predictor of in vivo clinical response. It is possible that LEN induced-substrate degradation may lead to activation of apoptosis. Although we observed on target degradation of substrates, further studies are needed to understand how this leads to increased apoptotic priming. Our observations from this study supports prospective evaluation of the DBP assay as an effective way to choose therapy in R/R AML, particularly in cases without available genetic targets. Note, DBP could identify “priming agents” that can enhance the apoptotic priming of myeloblasts from R/R AML patients, increasing the likelihood of chemoresistant cells to respond to additional therapies. Alternatively, DBP could identify R/R AML patients unlikely to respond to select therapies, thereby sparing them from ineffective and potentially toxic regimens particularly re-induction cytotoxic chemotherapies. The role of BH3 profiling and DBP as reliable biomarkers is being further explored in an on-going multicenter phase 2 study of LEN-MEC chemotherapy for R/R AML (NCT03118466).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank all the patients and the families that enrolled in these trials and the research staff at Stanford Cancer Center and DF/HCC. The conduct of the Stanford clinical trial was funded by Celgene. JSG is a recipient of a Career Development Award from Conquer Cancer Foundation and a Translational Research Project Award from the Leukemia and Lymphoma Society. SB is a recipient of a Career Development Award from the Leukemia and Lymphoma Society and Basic Cancer Research Fellowship award from AACR. AS was supported by a Ruth L. Kirschstein National Research Service Award and a Wong Family Foundation award. SC was supported by grants from the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium, NIH/NCI U24-CA210986 and NIH/NCI U01 CA214125. BE was supported by the Howard Hughes Medical Institute, the Edward P. Evans Foundation, the Leukemia and Lymphoma Society and the Adelson Medical Research Foundation. AL acknowledges support from P01 CA066996.
Footnotes
CONFLICT OF INTERESTS
SAC, BLE, and BCM have received research funding from Celgene. JSG has received research funding from Celgene, Abbvie, Pfizer and Genentech. JSG serves on the SAB for Abbvie. AL discloses that he has worked as a consultant for and his lab has received research support from AbbVie, Novartis, and Astra-Zeneca. He is an equity holder and cofounder of Flash Therapeutics and Vivid Bioscience. He is an equity holder and advisor for Dialectic Therapeutics. BLE has received research funding from Deerfield and consulting fees from GRAIL. RMS has received research funding from Novartis, Agios and Arog and consulting fees from Abbvie, Actinium Pharmaceuticals, Agios, Argenx, Arog Pharmaceuticals, Astellas, AstraZeneca Pharmaceuticals, Celgene, Cornerstone Biopharma, Fujifilm, and Jazz Pharmaceuticals. He is an advisor for Argenx, Celgene and Takeda Oncology. DJD has received research funding from Abbvie, Novartis, Glycomimetics and Blueprint and has served as a consultant for Amgen, Autolus, Celgene, Incyte, Takeda, Pfizer and Forty-Seven. All other authors have no conflicts of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg PL, Lee SJ, Advani R, et al. Mitoxantrone, etoposide, and cytarabine with or without valspodar in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome: a phase III trial (E2995). J Clin Oncol. 2004;22(6):1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kronke J, Fink EC, Hollenbach PW, et al. Lenalidomide induces ubiquitination and degradation of CK1alpha in del(5q) MDS. Nature. 2015;523(7559):183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia JS, Bhatt S, Fell G, et al. Dynamic BH3 profiling predicts for clinical response to Lenalidomide Plus chemotherapy in relapsed acute myeloid leukemia. Am Soc Hematology. 2018:4058. [Google Scholar]
- 6.DeAngelo DJ, Brunner AM, Werner L, et al. A phase I study of lenalidomide plus chemotherapy with mitoxantrone, etoposide, and cytarabine for the reinduction of patients with acute myeloid leukemia. Am J Hematol. 2018;93(2):254–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8(3):705–711. [DOI] [PubMed] [Google Scholar]
- 8.Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB. BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 2001;15 (12):1481–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9(5):351–365. [DOI] [PubMed] [Google Scholar]
- 11.Ni Chonghaile T, Sarosiek KA, Vo TT, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334(6059):1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montero J, Sarosiek KA, DeAngelo JD, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160(5):977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vo TT, Ryan J, Carrasco R, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151(2):344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhola PD, Mar BG, Lindsley RC, et al. Functionally identifiable apoptosis-insensitive subpopulations determine chemoresistance in acute myeloid leukemia. J Clin Invest. 2016;126(10):3827–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Etchin J, Montero J, Berezovskaya A, et al. Activity of a selective inhibitor of nuclear export, selinexor (KPT-330), against AML-initiating cells engrafted into immunosuppressed NSG mice. Leukemia. 2016;30(1):190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;30(1):183. [DOI] [PubMed] [Google Scholar]
- 17.Ryan J, Montero J, Rocco J, Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biol Chem. 2016;397(7):671–678. [DOI] [PubMed] [Google Scholar]
- 18.Sperling AS, Burgess M, Keshishian H, et al. Patterns of substrate affinity, competition and degradation kinetics underlie biological activity of thalidomide analogs. Blood. 2019;134(2):160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.