

Abstract
The present study focuses on the synthesis and characterization of lanthanide-containing paramagnetic ionic liquids (ILs), [CnC1Im]3[MCl3X3] (n = 4, 6, and 8; M = Gd, Dy, and Ho; X = Br and Cl), derived from 1-alkyl-3-methylimidazolium anions. These paramagnetic ILs exhibit low vapor pressure, high thermal stability, physiochemical stability, and tunability, along with significant magnetic susceptibility, making them of interest in advanced material applications that may take advantage of neat liquids with magnetic susceptibility. Structural and physical properties were determined using FTIR, 1H NMR, DSC, and TGA. The room temperature density and viscosity of the iron paramagnetic ILs were also reported. Accompanying this report of paramagnetic IL products, we reintroduce and highlight Evan’s NMR technique, an accessible magnetic susceptibility measurement technique that can utilize any available proton NMR to characterize the magnetic susceptibility of ILs. This work demonstrates the robustness of Evan’s technique by demonstrating the ability to account for the IL water content, a common issue for hygroscopic materials, during the measurement of magnetic susceptibility. A detailed comparison of the ILs is presented, with dysprosium- and holmium-containing paramagnetic ILs exhibiting the highest magnetic susceptibility reported for mononuclear ILs reported to date. These materials have been studied with an eye on applications for mass transfer, eventually seeking to optimize magnetic susceptibility and viscosity using magnetic field gradients to move paramagnetic ILs carrying solute or heat. The study of paramagnetic ILs is important not only for understanding the magnetic properties of these materials but also for potential applications in areas such as magnetic resonance imaging, biomedicine, environmental remediation, and mass transfer. These unique materials have the potential to bring about new advances and technologies in the fields of materials science and analytical chemistry.
Keywords: paramagnetic ionic liquids, magnetic ionic liquids, lanthanide anions, imidazolium-based ionic liquids, Evan’s NMR method, magnetic susceptibility
Introduction
The development of ionic liquids (ILs) has been an area of intense research in recent years due to their unique properties, such as negligible vapor pressure, high thermal and chemical stability, good solvation properties, and nonflammability.1 These properties make ILs promising candidates for a wide range of applications, including environmental remediation, liquid–liquid extractions, catalysis, cellulose dissolution, and carbon dioxide (CO2) absorption. Paramagnetic ILs (PILs) are a subclass of ILs that exhibit paramagnetic behavior in response to an external magnetic field. Imidazolium-based PILs have received particular attention in the literature, beginning with the work of Hayashi et al., iron(III) was incorporated into the anions to induce paramagnetism, and later work explored the development of a series of symmetric bis-alkylated imidazolium ILs with bromotrichloroferrate(III) as the anion.2,3
The introduction of paramagnetism into ILs is typically achieved by incorporating transition or lanthanide metals into the anion.4,5 Transition metal PILs containing iron, cobalt, nickel, and manganese anions are most common, but there has been an increase in reports of ILs containing rare earth metals.4−6 The ability of PILs to respond strongly to external magnetic fields makes them valuable candidates for advanced applications. PILs have been demonstrated to have potential in environmental remediation, liquid–liquid extractions, catalysis, cellulose dissolution, and CO2 absorption.7,8 The use of PILs for CO2 absorption is auspicious, given their high thermal and chemical stability, low vapor pressure, and ability to respond to magnetic fields.9 This makes PILs valuable avenues for continued research and development.
The development and utilization of PILs containing lanthanide complexes have garnered significant interest due to their unique combination of favorable optical and catalytic properties and high magnetic susceptibility compared to transition metal complexes. Nockemann et al. reported the first PILs containing lanthanide metals, incorporating imidazolium cations and lanthanide anions (La, Y, Pr, Nd, Sm, Eu, Gd, Tb, Ho, Er, and Yb) with thiocyanate ligands, which effectively reduced their melting points below 40 °C.5 The use of dysprosium anions in PILs with thiocyanate ligands was also reported shortly after.10 Del Sesto et al. demonstrated the synthesis of a tetraalkylphosphonium PIL with a gadolinium anion possessing a large magnetic dipole and a melting point below room temperature.4
Bwambok et al. demonstrated paramagnetic ILs to measure density via magnetic levitation.11 However, scarce detail is provided regarding the synthesis of lanthanide PILs containing holmium and dysprosium in their report. Moreover, Han et al. verify the [BMIm]3[LaCl6] general structure via crystallographic data and present a route for crystal growth of these materials.12
These works have inspired us to explore the synthesis of imidazolium-based lanthanide-containing PILs. We asked the basic question: could they be synthesized via the straightforward SN2 reaction followed by anion metathesis used to create typical imidazolium halide ILs? If successful, the result would be a simple route to PILs with a dramatically high magnetic susceptibility. To test this hypothesis, we synthesized imidazolium-based ILs with lanthanide metals containing high localized magnetic moments, specifically using dysprosium, holmium, and gadolinium.
In this study, we report the synthesis, characterization, and properties of a series of lanthanide-containing methylimidazolium PILs with the generic formula [CnC1Im]3[MCl3X3], (Im = imidazole; C1 = methyl; Cn = 4 (butyl), 6 (hexyl), and 8 (octyl); M = Dy, Ho, and Gd; X = Cl or Br). We also synthesized the previously reported iron imidazolium-based PILs, [CnC1Im][FeCl3X], for comparison.13 The thermal and physical properties of these compounds are characterized by thermogravimetric analysis (TGA), differential scanning calorimetry (DSC), Fourier-transform infrared (FTIR), nuclear magnetic resonance (NMR) spectroscopies, density measurements, viscosity measurements, and magnetic susceptibility properties. Moreover, we demonstrate Evan’s NMR method as a reliable, and arguably broadly accessible method for the measurement of magnetic susceptibility, while Gouy or similar susceptibility balances are simple, not all facilities have them.14,15 Given the relative scarcity and dramatically increasing cost of helium, Evan’s NMR method is considerably less costly, especially using newer benchtop NMR spectrometers, compared to using a superconducting quantum interference device (SQUID) magnetometer or a Gouy balance.14,15
The study of paramagnetic ILs is of significant importance due to their potential applications in advanced technologies, particularly in CO2 absorption.16 Our results provide further insights into the synthesis and characterization of lanthanide-containing PILs and demonstrate the feasibility of using Evan’s NMR method for magnetic susceptibility measurements. These findings serve as a valuable addition to the ongoing development and study of PILs and their potential applications.
Experimental Methods
General Considerations
All reagents were purchased through chemical manufacturers and were of high purity.
Spectroscopic Measurements
For [CnC1Im][X] series ILs, 1H NMR data was collected using a JEOL 500 MHz NMR, and d-chloroform was used as a reference solvent. For [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs, Evan’s NMR method data and COSY NMR were collected using Agilent 700 MHz NMR, and d4-methanol was used as a reference solvent. Moreover, PILs used in Evan’s NMR method were spiked with 30 μL of anhydrous methanol to elucidate the solvent shift between the sample and the pure reference contained in the coaxial insert and verify the solvent peaks in COSY NMR. All NMR spectra can be found in the Supporting Information.
FTIR-NIR spectra of all ILs were obtained using an Agilent 670 FTIR-ATR spectrometer. A sensitivity of 1 and a resolution of 8 cm–1 were used for all spectra. Each spectrum was generated after 32 scans for the background and sample measurements, respectively. A compilation of all spectra can be found in the Supporting Information (Figures S1–S30).
Density Measurement and Determination
Density was measured using two techniques. For samples of liquid at room temperature, namely, the [CnC1Im][FeCl3X] series, density was measured using precisely weighed Hamilton gastight syringes. The mass of each syringe was recorded using a Torbal AGZN200 analytical balance. The IL samples were drawn into the syringe, all air bubbles were expelled, and the syringe needle was removed. The sample was then equilibrated at 25 °C for 15 min. After equilibration, the syringe plunger was pushed to expel a small amount of liquid, which was wiped clean, and the volume was then recorded along with the added weight from the PIL. Density was calculated from the volume and added weight. The steps above were repeated three times for each sample.
For the viscous liquid PILs of the [CnC1Im]3[MCl3X3] series, a variation of the syringe method was used. Due to the high viscosity of this series, a larger gauge syringe needle was needed. Samples were heated to 90 °C before being drawn into the syringe. As before, the volume was recorded along with the added weight from the IL, and the density was calculated from the volume and added weight. The above steps were repeated three times for each sample.
For the higher melting (solid) PILs of the [CnC1Im]3[MCl3X3] series, the density was determined using a DV215CD OHAUS discovery balance with a density kit. Notably, this method requires measurement of mass while the sample is submerged in a well-characterized liquid. We found that acetone was a suitable liquid for these PILs, showing no PIL solubility. Solid PIL samples were extracted with tweezers, and the weight of the sample was recorded in the air. The sample was then carefully removed and placed on the sample holder, which was submerged in the acetone. The mass was allowed to stabilize, and the density value was calculated by the instrument. Each measurement was repeated with a fresh PIL sample three times to obtain an average density. The density of solid samples was determined using eq 1, where ρ is the density of the sample, A is the mass of the sample in air, B is the weight of the sample in the liquid, ρo is the density of the solvent, ρL is the density of air (0.0012 g cm–3), and α is a balance correction factor (0.99985)
| 1 |
Density Considerations for Water Containing ILs
The effect of the water content on the accurate measurement of the density of hygroscopic ILs is a significant concern. In this study, we report the synthesis of hygroscopic ILs and the measured densities of these wet ILs. Given the hygroscopic nature of these ILs and the use of hydrate containing precursors, several ILs contain a significant weight percent of water. To address the error associated with the density measurement and the water content, we employed TGA to measure the water content.
Based on the measured water content, we calculated a predicted density for the pure paramagnetic ILs. This calculation is based on the assumption of ideal mixtures, where the density of the mixture is a weighted average of the densities of the individual components. However, it is important to note the caveats and potential errors associated with this assumption. The assumption of ideal mixtures is a simplification, and real mixtures often deviate from ideal behavior due to interactions between the components. In the case of our paramagnetic ILs, the presence of ionic interactions and hydrogen bonding most certainly leads to nonideal behavior. This means that the actual density of the pure IL may deviate from the calculated value.
Despite these limitations, the calculated density provides a useful estimate and a starting point for understanding the properties of pure IL. Future work could involve more sophisticated models that take into account the specific interactions between the IL and water or the experimental determination of the density of the pure IL. The calculation and comparison of density values are provided in the Supporting Information and Table SI-81.
Added Water Density Measurement
In order to test the assumptions of the ideal mixtures, two samples of C4C1ImFeCl4, which are shown to have no residual water (see TGA data), were adulated with known quantities of water. Two 20 mL vials were placed onto a scale, and 4 g of dry C4C1ImFeCl4 was added, Sample A, 4.203 g, and Sample B, 4.123 g. Next, these vials on the scale were zeroed, and 10% by weight of water was added to sample A, 0.433 g, then 20% by weight of water was added to sample B, 0.830 g. In order to make the solutions homogeneous, a magnetic stir bar was placed into each individual vial, placed on a magnetic plate, and mixed for 15 min. During the 15 min, a glass syringe and plunger were placed and zeroed on a scale. Once the 15 min were over, about 1 mL of the solution was drawn up into the syringe and placed into an equilibrator for 15 min. After 15 min, the syringe was removed, the temperature was noted, along with the volume in the syringe and placed on the scale and noting the mass of the syringe. Next, 0.1–0.5 mL of the PIL was removed, and the process of equilibration was repeated. That was done for a total of three density calculations for each solution.
Water Content and Thermal Degradation
Thermal analysis of the [CnC1Im][X], [CnC1Im][FeCl3X], and [CnC1Im]3[MCl3X3] series of PILs was recorded on a TA Instruments Q500 from 25 to 500 °C with a ramp rate of 5 °C/min. [CnC1Im][X] series ILs and [CnC1Im][FeCl3X] series PILs showed no significant water content so the degradation was calculated at 5% mass loss from the initial thermal curve plateau. [CnC1Im]3[MCl3X3] series of PILs showed water loss as the IL was heated under ramping conditions. For these water-containing PILs, thermal degradation was determined for each paramagnetic IL depending on the thermal curve plateau after water mass loss. Water weight percent for each [CnC1Im]3[MCl3X3] series PIL was calculated from the difference of the starting mass from the plateau before degradation, and the degradation was calculated at 5% mass loss from that water loss plateau.
Viscosity Measurement
The dynamic viscosity of the [CnC1Im][FeCl3X] series of PILs was measured with a RheoSense m-VROC viscometer. The m-VROC is a microfluid small-volume viscometer ideally suited for expensive IL development. The device reports viscosity by measuring the pressure drop as microliter volumes of analyte flow through a rectangular microfluidic channel.
The IL was pushed at a constant flow rate of 100 L min–1 during the measurement. The viscosity of each IL was measured at room temperature (21–23 °C), and the shear rate range was within 400–2000 s–1. Each measurement was repeated in triplicate, and the uncertainty was calculated as the standard deviation.
Melting Point Measurement
DSC thermograms of the [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series were recorded on a TA Instruments Q200 over a temperature range of −85 to 175 °C. A heat/cool/heat/cool/heat cycle was used using a 5 °C ramp rate throughout the entire cycle, starting at room temperature (exo up). Tzero hermetic aluminum pans and lids were used for all samples. The mass weights of all samples ranged from 4 to 11 mg.
Synthesis
Reagents and solvents were used as received by the manufacturer. The manufacturer and purity of each compound are as follows: 1-methylimidazole (Sigma-Aldrich, 99%), 1-bromobutane (Sigma-Aldrich, 99%), 1-bromohexane (Alfa Aesar, 99%), 1-bromooctane (Acros Organics, 99%), 1-chlorobutane (Alfa Aesar, 99%), 1-chlorohexane (Acros Organics, 95%), 1-chlorooctane (Alfa Aesar, 99%), iron(III) chloride hexahydrate (Thermo Scientific, 97%), dysprosium(III) chloride hexahydrate (Alfa Aesar, 99.9%), holmium(III) chloride hexahydrate (Alfa Aesar, 99.9%), gadolinium(III) chloride hexahydrate (Alfa Aesar, 99.99%), anhydrous methanol (Sigma-Aldrich, ≥99.9%), anhydrous toluene (Alfa Aesar, ≥99.5%), anhydrous ethyl acetate (Sigma, 99.9%), and anhydrous acetonitrile (Sigma, 99.9%).
General Synthesis Procedure
The complete reaction pathway for the synthesis of 1-alkyl-3-methylimidazolium halide intermediates [CnC1Im][X], and the [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series of PILs is illustrated in Scheme 1.
Scheme 1. Chemical Synthesis Pathway for [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] PILs.

The [CnC1Im][X] series was synthesized by the reaction of equimolar amounts of 1-methylimidazole and either 1-chloroalkane or 1-bromoalkane.17 The reaction was performed in the absence of solvent under standard air atmospheric conditions at a temperature of 65–70 °C over 72–96 h. The intermediate ILs were purified by washing with ethyl acetate and then dried using rotary evaporation at 40 mbar and a temperature of 50 °C for 1.5 h to remove residual ethyl acetate. The [CnC1Im][X] series was characterized and stored in a desiccator under vacuum until further reactions were conducted.
The synthesis of the [CnC1Im][FeCl3X] series is achieved through the reaction of equimolar amounts of [CnC1Im][X] and iron(III) chloride hexahydrate. The reaction is conducted neatly under normal atmospheric conditions at 80 °C for 48 h.2,3 Using [CnC1Im][X] in excess results in the phase separation of the product and the floating of any unreacted material on top of the reaction. Synthesis of the [CnC1Im][FeCl3X] series has been previously demonstrated so we use these materials as a benchmark to compare our characterization techniques to known values, particularly magnetic susceptibility.2,3
The synthesis of [CnC1Im]3[MCl3X3] is performed through the reaction of 1 M equivalent of [CnC1Im][X] with 1 M equivalent of holmium(III) chloride hexahydrate, dysprosium(III) chloride hexahydrate, or gadolinium(III) chloride hexahydrate. The reaction mixture is refluxed in methanol (∼60 mL) at 90 °C for 72 h. After reflux, methanol is removed using rotary evaporation (40 mbar, 50 °C, 2 h). The IL containing the product is washed with diethyl ether to remove any residual unreacted imidazole; then, the entire product is dissolved in acetonitrile (∼100 mL). Running the reaction at a 1-to-1 molar ratio will result in excess lanthanide salt; however, when fully dissolved in an abundance of acetonitrile, the unreacted lanthanide(III) chloride precipitates readily from the solution during stirring (∼1 h). The solution of [CnC1Im]3[MCl3X3]/acetonitrile is then filtered through a packed column of glass wool/celite. Residual diethyl ether and acetonitrile are then removed via rotary evaporation (40 mbar, 50 °C, 3 h).
Starting Material Considerations
Both anhydrous and hydrated salts have been utilized. However, anhydrous lanthanide(III) chloride salts are significantly more expensive, and their anhydrous nature increases storage demands and handling difficulties in preventing water uptake. We found that the hydrated analogue is less hygroscopic and more cost-effective, and we did not observe significant differences in product quality or yield. Furthermore, although single-crystal X-ray diffraction (SCXRD) was not obtained, SCXRD of similar paramagnetic ILs synthesized in this report has been published by May et al.12 Alternatively, this reaction may be performed using a 3:1 stoichiometric ratio of the [CnC1Im][X] IL and MCl3·6H2O salt, respectively. However, the removal of excess [CnC1Im][X] series ILs is sedulous and less facile than the above method.
Synthesis of [C4C1Im][Cl]
A 250 mL two-neck round-bottom flask with a magnetic stir bar was placed in a heating mantle. A thermometer with a 24/40 thermometer adapter was placed in the side neck of the reaction flask and secured with a keck clamp. 1-Methylimidazole (8.2137 g, 0.100 mol) and 1-chlorobutane (9.2637 g, 0.100 mol) were weighed and analytically transferred to the reaction flask. A reflux condenser was connected to the top neck of the reaction flask and secured, and stirring was turned on. The solution was refluxed neat (96 h, 70 °C) under an atmosphere. After refluxing, the yellow liquid was transferred to a clean 250 mL single-neck round-bottom flask and washed with ethyl acetate (4 × 20 mL). The single-neck flask was connected to a rotary evaporator and dried (40 mbar, 50 °C, and 2 h). After rotary evaporation, the product was dried further via a Schlenk line (24 h, 0.05 Torr). After drying in vacuo, the light-yellow IL was stored in a vacuum desiccator for further characterization. The same procedure was adapted using 1-chlorohexane, 1-chlorooctane, 1-bromobutane, 1-bromohexane, and 1-bromooctane, respectively. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.4929 (s, 1H), 7.6142 (s, 1H), 7.4430 (s, 1H), 4.2127 (t, 2H), 4.0040 (s, 3H), 1.7791 (q, 2H), 1.2508 (s, 2H), 0.8278 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 87%.
Synthesis of [C6C1Im][Cl]
This was prepared similarly to [C4C1Im][Cl]. 1-Methylimidazole (8.2104 g, 0.100 mol) and 1-chlorohexane (12.0950 g, 0.100 mol) were used as the starting reagents. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.5561 (s, 1H), 7.6271 (s, 1H), 7.4129 (s, 1H), 4.2256 (t, 2H), 4.0398 (s, 3H), 1.8176 (q, 2H), 1.2179 (m, 6H), 0.7720 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 90%.
Synthesis of [C8C1Im][Cl]
This was prepared similarly to [C4C1Im][Cl]. 1-Methylimidazole (8.2212 g, 0.100 mol) and 1-chlorooctane (14.8763 g, 0.100 mol) were used as the starting reagents. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.5846 (s, 1H), 7.5906 (s, 1H), 7.3846 (s, 1H), 4.2505 (t, 2H), 4.0683 (s, 3H), 1.8433 (q, 2H), 1.2152 (m, 10H), 0.8001 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 94%.
Synthesis of [C4C1Im][Br]
This was prepared similarly to [C4C1Im][Cl]. 1-Methylimidazole (8.2227 g, 0.100 mol) and 1-bromobutane (13.7211 g, 0.100 mol) were used as the starting reagents. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.3033 (s, 1H), 7.5950 (s, 1H), 7.4677 (s, 1H), 4.2823 (t, 2H), 4.0699 (s, 3H), 1.8478 (q, 2H), 1.3222 (s, 2H), 0.8970 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 93%.
Synthesis of [C6C1Im][Br]
This was prepared similarly to [C4C1Im][Cl]. 1-Methylimidazole (8.2343 g, 0.100 mol) and 1-bromohexane (16.5135 g, 0.100 mol) were used as the starting reagents. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.2997 (s, 1H), 7.6125 (s, 1H), 7.4413 (s, 1H), 4.2577 (t, 2H), 4.0654 (s, 3H), 1.8478 (q, 2H), 1.2399 (m, 6H), 0.7958 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 97%.
Synthesis of [C8C1Im][Br]
This was prepared similarly to [C4C1Im][Cl]. 1-Methylimidazole (8.2211 g, 0.100 mol) and 1-bromooctane (19.3212 g, 0.100 mol) were used as the starting reagents. 1H NMR (500 MHz, CDCl3, 298.15 K): δ 10.2576 (s, 1H), 7.6134 (s, 1H), 7.2484 (s, 1H), 4.2403 (t, 2H), 4.0517 (s, 3H), 1.8304 (q, 2H), 1.1904 (m, 10H), 0.7756 (t, 3H). vmax/cm–1 3300–3400 (H2O), 3100 (C–H), 2840–3000 (C–H, aliphatic), 1610–1637 (C=C), 1565 (σ–π, ring vibration), 1462 (C–H), 1162 (C–N). Yield 96%.
Synthesis of [C4C1Im][FeCl4]
A 250 mL two-neck Schlenk flask with a magnetic stir bar was placed in a heating mantle. The inert air manifold was connected to the glass stopcock of the Schlenk flask using a rubber hose. Iron(III) chloride hexahydrate (8.1142 g, 0.0300 mol) and 1-butyl-3-methylimidazolium chloride (4.9216 g, 0.0282 mol) were weighed and analytically transferred to a Schlenk flask. An analytical grade thermometer with a 24/40 thermometer adapter was placed in the side neck of the Schlenk flask and secured with a keck clamp. A reflux condenser was connected to the top neck of the Schlenk flask and secured. A 24/40 flow control inlet adapter with a stopcock was attached to the top of the reflux condenser and secured with a keck clamp. The flow adapter was connected to the second port of the Schlenk line via a rubber hose. The reaction vessel was evacuated under a vacuum and refilled with nitrogen three times. With stirring, the solution was refluxed neat (48 h, 85 °C) under a N2 atmosphere. After refluxing, the dark brown solution was dried under a vacuum (8 h, 0.05 Torr). The solution was stored in a vacuum desiccator for future characterization. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 13.39 (s, 1H), 11.68 (s, 2H), 8.54 (s, 2H), 8.39 (s, 3H), 6.51 (s, 2H), 5.93 (s, 2H), 5.33 (s, 3H). Water content as determined by TGA: 0 wt %. Yield: dark red liquid, 9.3034 g, 74%.
Synthesis of [C6C1Im][FeCl4]
This was prepared similarly to [C4C1Im][FeCl4]. 1-Hexyl-3-methylimidazolium chloride (6.0862 g, 0.0300 mol) and iron(III) chloride hexahydrate (8.1118 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 12.96 (s, 1H), 11.26 (s, 2H), 8.13 (m, 2H), 7.95 (m, 3H), 6.07 (s, 2H), 5.47 (m, 2H), 5.36 (m, 2H), 5.26 (m, 2H), 4.69 (s, 3H). Water content as determined by TGA: 0 wt %. Yield: dark brown liquid, 10.0049 g, 70%.
Synthesis of [C8C1Im][FeCl4]
This was prepared similarly to [C4C1Im][FeCl4]. 1-Octyl-3-methylimidazolium chloride (6.9351 g, 0.0301 mol) and iron(III) chloride hexahydrate (8.1121 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 12.92 (s, 1H), 11.24 (s, 2H), 8.09 (s, 2H), 7.92 (s, 3H), 6.05 (s, 2H), 5.44 (m, 2H), 5.35 (m, 2H), 5.22 (m, 2H), 5.09 (m, 4H), 4.59 (s, 3H). Water content as determined by TGA: 0 wt %. Yield: dark brown liquid, 10.3471 g, 69%.
Synthesis of [C4C1Im][FeCl3Br]
This was prepared similarly to [C4C1Im][FeCl4]. 1-Butyl-3-methylimidazolium bromide (6.5988 g, 0.0301 mol) and iron(III) chloride hexahydrate (8.1183 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 11.45 (s, 1H), 9.92 (s, 2H), 6.64 (s, 2H), 6.42 (s, 3H), 4.44 (s, 2H), 3.91 (s, 2H), 3.42 (s, 3H). Water content as determined by TGA: 0 wt %. Yield: blood-red liquid, 9.2772 g, 63%.
Synthesis of [C6C1Im][FeCl3Br]
This was prepared similarly to [C4C1Im][FeCl4]. 1-Hexyl-3-methylimidazolium bromide (7.4119 g, 0.0300 mol) and iron(III) chloride hexahydrate (8.1100 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 11.90 (s, 1H), 10.32 (s, 2H), 7.06 (s, 2H), 6.86 (s, 3H), 4.92 (s, 2H), 4.35 (m, 2H), 4.28 (m, 2H), 4.22 (m, 2H), 3.70 (t, 3H). Water content as determined by TGA: 0 wt %. Yield: blood-red liquid, 10.9977 g, 71%.
Synthesis of [C8C1Im][FeCl3Br]
This was prepared similarly to [C4C1Im][FeCl4]. 1-Octyl-3-methylimidazolium bromide (8.2608 g, 0.0300 mol) and iron(III) chloride hexahydrate (8.1099 g, 0.300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 11.48 (s, 1H), 9.93 (s, 2H), 6.66 (s, 2H), 6.44 (s, 3H), 4.49 (s, 2H), 3.88 (m, 4H), 3.78 (m, 2H), 3.71 (m, 4H), 3.25 (s, 3H). Water content as determined by TGA: 0 wt %. Yield: blood-red liquid, 12.1188 g, 74%.
Synthesis of [C4C1Im]3[HoCl6]
A 250 mL 2-neck Schlenk flask with a magnetic stir bar was placed in a heating mantle. The inert air manifold was connected to the glass stopcock of the Schlenk flask using a rubber hose. 1-Butyl-3-methylimidazolium chloride (5.2466 g, 0.0300 mol) was heated on a hot plate at 80 °C and analytically transferred to the Schlenk flask. An analytical grade thermometer with a 24/40 thermometer adapter was inserted into the side neck of the Schlenk flask and secured. A reflux condenser was attached to the top neck of the Schlenk flask. A 24/40 flow control inlet adapter with a stopcock was attached to the top of the reflux condenser. Metal keck clamps were used to secure all of the joint attachments of the apparatus. The flow adapter was connected to the second port of the inert air manifold via a rubber hose. The reaction vessel was evacuated under vacuum and refilled with nitrogen three times.
Holmium(III) chloride hexahydrate (11.8605 g, 0.0313 mol) was dissolved in methanol. While the apparatus was under positive pressure (N2), the thermometer adapter was carefully removed from the side-neck of the Schlenk flask. To minimize atmospheric backdraft, the holmium(III) chloride hexahydrate solution and methanol (50 mL) were quickly added to the Schlenk flask via the side neck before the thermometer adapter. The solution was stirred and refluxed (4 days, 90 °C) under N2 atmosphere. After the mixture was refluxed, each N2 inlet stopcock was closed, and the apparatus was disassembled. The solution was transferred to a 250 mL single neck round-bottom flask and methanol was removed using rotary evaporation (6 h, 60 °C, 50 mbar). To remove excess metal salt, the product was washed with diethyl ether and dissolved in acetonitrile (∼100 mL). Unreacted metal salt precipitated out of solution, and the [C4C1Im]3[HoCl6]/acetonitrile solution was filtered through a packed column of glass wool and Celite. After filtration, the product was dried using rotary evaporation (40 mbar, 50 °C, 3 h) to remove acetonitrile/diethyl ether. After drying in vacuo, the IL was weighed, transferred into a scintillation vial, and stored in a vacuum desiccator for future characterization. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 13.58 (s, 1H) 12.27 (m, 1H) 12.23 (m, 1H) 8.85 (s, 2H) 8.56 (d, 3H) 6.50 (s, 2H) 6.01 (s, 2H) 5.62 (s, 3H). Water content as determined by TGA: 1.5 wt %. Yield: waxy, light-pink solid; 10.3752 g, approximately 60%.
Synthesis of [C6C1Im]3[HoCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium chloride (6.0603 g, 0.0299 mol) and holmium(III) chloride hexahydrate (11.9309 g, 0.0314 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 16.09 (s, 1H) 14.77 (s, 1H) 14.70 (s, 1H) 11.35 (s, 2H) 11.05 (s, 3H) 9.02 (s, 2H) 8.48 (s, 6H) 8.04 (s, 3H). Water content as determined by TGA:15.9 wt %. Yield: waxy, light-pink solid; 10.3752 g, approximately 70%.
Synthesis of [C8C1Im]3[HoCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium chloride (6.9065 g, 0.0299 mol) and holmium(III) chloride hexahydrate (11.5728 g, 0.0305 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 17.80 (s, 1H) 16.48 (d, 1H) 16.41 (d, 1H) 13.05 (s, 2H) 12.76 (s, 3H) 10.73 (s, 2H) 10.17 (s, 10H) 9.74 (s, 3H). Water content as determined by TGA:5.4 wt %. Yield: waxy, pink solid; 8.0924 g, approximately 40%.
Synthesis of [C4C1Im]3[HoCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Butyl-3-methylimidazolium bromide (6.5488 g, 0.0299 mol) and holmium(III) chloride hexahydrate (11.5474 g, 0.0304 mol) were used as the starting reagent. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 17.24 (s, 1H) 15.92 (d, 1H) 15.85 (d, 1H) 12.50 (s, 2H) 12.20 (s, 3H) 10.15 (s, 2H) 9.66 (s, 2H) 9.27 (s, 3H). Water content as determined by TGA: 9.4 wt %. Yield: waxy, pink solid; 7.0626 g, approximately 40%.
Synthesis of [C6C1Im]3[HoCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium bromide (7.3707 g, 0.0298 mol) and holmium(III) chloride hexahydrate (11.5945 g, 0.0306 mol) were used as the starting reagent. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 15.12 (s, 1H) 13.80 (s, 1H) 13.74 (s, 1H) 10.39 (s, 2H) 10.08 (s, 3H) 8.04 (s, 2H) 7.56 (s, 6H) 7.16 (s, 3H). Water content as determined by TGA: 10.4 wt %. Yield: waxy, pink solid; 9.4415 g, approximately 50%.
Synthesis of [C8C1Im]3[HoCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium bromide (8.3044 g, 0.0302 mol) and holmium(III) chloride hexahydrate (11.6289 g, 0.0307 mol) were used as the starting reagent. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 14.11 (s, 1H) 12.80 (d, 1H) 12.73 (d, 1H) 9.37 (s, 2H) 9.08 (s, 3H) 7.05 (s, 2H) 6.49 (s, 10H) 6.06 (s, 3H). Water content as determined by TGA: 14.2 wt %. Yield: waxy, pink solid; 8.3172 g, approximately 40%.
Synthesis of [C4C1Im]3[DyCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Butyl-3-methylimidazolium chloride (5.2252 g, 0.0299 mol) and dysprosium(III) chloride hexahydrate (11.3155 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 15.03 (s, 1H) 13.74 (s, 1H) 13.67 (s, 1H) 10.33 (s, 2H) 10.02 (s, 3H) 8.01 (s, 2H) 7.53 (s, 2H) 7.16 (s, 3H). Water content as determined by TGA: 13.3 wt %. Yield: waxy, golden solid; 9.7395 g, approximately 60%.
Synthesis of [C6C1Im]3[DyCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium chloride (6.1059 g, 0.0301 mol) and dysprosium(III) chloride hexahydrate (11.3173 g, 0.0300 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 17.80 (s, 1H) 16.53 (s, 1H) 16.44 (s, 1H) 13.11 (s, 2H) 12.80 (s, 3H) 10.83 (s, 2H) 10.35 (s, 6H) 9.94 (t, 3H). Water content as determined by TGA: 5.1 wt %. Yield: waxy, golden solid; 5.6108 g, approximately 30%.
Synthesis of [C8C1Im]3[DyCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium chloride (6.9823 g, 0.0303 mol) and dysprosium(III) chloride hexahydrate (11.3625 g, 0.0301 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 16.31 (s, 1H) 15.02 (s, 1H) 14.94 (s, 1H) 11.60 (s, 2H) 11.30 (s, 3H) 9.31 (s, 2H) 8.80 (s, 10H) 8.39 (s, 3H). Water content as determined by TGA: 3.8 wt %. Yield: waxy, golden solid; 8.6472 g, approximately 50%.
Synthesis of [C4C1Im]3[DyCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Butyl-3-methylimidazolium bromide (6.1635 g, 0.0302 mol) and dysprosium(III) chloride hexahydrate (11.3835 g, 0.0302 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 16.07 (s, 1H) 14.78 (s, 1H) 14.71 (s, 1H) 11.37 (s, 2H) 11.06 (s, 3H) 9.05 (d, 2H) 8.58 (s, 2H) 8.21 (s, 3H). Water content as determined by TGA: 6.1 wt %. Yield: waxy, cream-colored solid, 8.7815 g, approximately 50%.
Synthesis of [C6C1Im]3[DyCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium bromide (7.4272 g, 0.0300 mol) and dysprosium(III) chloride hexahydrate (11.4216 g, 0.0303 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 13.25 (s, 1H) 11.96 (s, 1H) 11.88 (s, 1H) 8.53 (s, 2H) 8.24 (s, 3H) 6.22 (s, 2H) 5.71 (s, 6H) 5.29 (t, 3H). Water content as determined by TGA: 16.2 wt %. Yield: waxy, cream-colored solid; 14.8309 g, approximately 80%.
Synthesis of [C8C1Im]3[DyCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium bromide (8.2565 g, 0.0300 mol) and dysprosium(III) chloride hexahydrate (11.3311 g, 0.0301 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 13.13 (s, 1H) 11.83 (s, 1H) 11.76 (s, 1H) 8.41 (s, 2H) 8.12 (s, 3H) 6.10 (s, 2H) 5.57 (s, 10H) 5.16 (s, 3H). Water content as determined by TGA: 10.8 wt %. Yield: waxy, cream-colored solid; 9.1622 g, approximately 50%.
Synthesis of [C4C1Im]3[GdCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Butyl-3-methylimidazolium chloride (4.9734 g, 0.0285 mol) and gadolinium(III) chloride hexahydrate (11.2582 g, 0.0303 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 11.68 (s, 1H) 10.36 (m, 1H) 10.30 (m, 1H) 6.94 (s, 2H) 6.65 (s, 3H) 4.59 (s, 2H) 4.10 (s, 2H) 3.71 (s, 3H). Water content as determined by TGA: 6.3 wt %. Yield: waxy, cream-colored solid; 9.7229 g, approximately 60%.
Synthesis of [C6C1Im]3[GdCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium chloride (6.0724 g, 0.0297 mol) and gadolinium(III) chloride hexahydrate (11.4661 g, 0.0309 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 11.73 (s, 1H) 10.42 (s, 1H) 10.35 (s, 1H) 6.99 (s, 2H) 6.70 (s, 3H) 4.66 (s, 2H) 4.13 (s, 6H) 3.69 (s, 3H). Water content as determined by TGA: 19.3 wt %. Yield: waxy, cream-colored solid; 10.0393 g, approximately 60%.
Synthesis of [C8C1Im]3[GdCl6]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium chloride (6.9268 g, 0.0300 mol) and gadolinium(III) chloride hexahydrate (11.5473 g, 0.0311 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 12.10 (s, 1H) 10.78 (s, 1H) 10.72 (s, 1H) 7.35 (s, 2H) 7.07 (s, 3H) 5.02 (s, 2H) 4.46 (s, 10H) 4.03 (s, 3H). Water content as determined by TGA: 14.0 wt %. Yield: waxy, cream-colored solid. No percent obtained.
Synthesis of [C4C1Im]3[GdCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Butyl-3-methylimidazolium bromide (6.5714 g, 0.0300 mol) and gadolinium(III) chloride hexahydrate (11.2241 g, 0.0302 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 12.12 (s, 1H) 10.80 (s, 1H) 10.73 (s, 1H) 7.38 (s, 2H) 7.09 (s, 3H) 5.03 (s, 2H) 4.53 (s, 2H) 4.14 (s, 3H). Water content as determined by TGA: 5.0 wt %. Yield: waxy, cream-colored solid; 14.8309 g, approximately 70%.
Synthesis of [C6C1Im]3[GdCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Hexyl-3-methylimidazolium bromide (7.4009 g, 0.0299 mol) and gadolinium(III) chloride hexahydrate (11.6137 g, 0.0312 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 12.49 (s, 1H) 11.17 (s, 1H) 11.11 (s, 1H) 7.75 (s, 2H) 7.46 (s, 3H) 5.40 (s, 2H) 4.90 (m, 6H) 4.51 (s, 3H). Water content as determined by TGA: 13.1 wt %. Yield: waxy, cream-colored solid; 7.4206 g, approximately 40%.
Synthesis of [C8C1Im]3[GdCl3Br3]
This was prepared similarly to [C4C1Im]3[HoCl6]. 1-Octyl-3-methylimidazolium bromide (8.2638 g, 0.0300 mol) and gadolinium(III) chloride hexahydrate (11.4155 g, 0.0307 mol) were used as the starting reagents. 1H NMR (700 MHz, CD3OD, 298.15 K): δ 13.50 (s, 1H) 12.17 (d, 1H) 12.11 (d, 1H) 8.74 (s, 2H) 8.46 (s, 3H) 6.41 (s, 2H) 5.84 (s, 10H) 5.42 (s, 3H). Water content as determined by TGA: 11.8 wt %. Yield: waxy, cream-colored solid; 3.8958 g, approximately 20%.
Results and Discussion
Physical Properties
The density, thermal degradation temperature, and molar magnetic susceptibility of the ILs are compiled in Table 1. Samples were dried using rotary evaporation (40 mbar, 50 °C, 2 h) to remove water and were stored in a desiccator under vacuum until characterization.
Table 1. Physical and Thermal Properties of [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] Series PILs.
| compound | measured density (g mL–1)a | thermal stability (°C)b | dynamic viscosity (mPa·s)c | χm (m3 mol–1) × 10–7 |
|---|---|---|---|---|
| [C4C1Im][FeCl4]d | 1.400 ± 0.032 | 31.1 ± 1.0 | 0.141 | |
| [C4C1Im][FeCl4] | 1.381 ± 0.011 | 337 | 45.8 ± 2.7 | 0.158 |
| [C6C1Im][FeCl4] | 1.338 ± 0.013 | 336 | 49.3 ± 2.1 | 0.157 |
| [C8C1Im][FeCl4] | 1.230 ± 0.038 | 320 | 60.9 ± 3.4 | 0.153 |
| [C4C1Im][FeCl3Br] | 1.505 ± 0.004 | 330 | 37.9 ± 2.6 | 0.143 |
| [C6C1Im][FeCl3Br] | 1.417 ± 0.004 | 318 | 51.9 ± 3.3 | 0.153 |
| [C8C1Im][FeCl3Br] | 1.375 ± 0.003 | 309 | 70.1 ± 5.9 | 0.131 |
| [C4C1Im]3[HoCl6] | 1.882 ± 0.059 | 306 | solid | 0.602 |
| [C6C1Im]3[HoCl6] | 1.672 ± 0.021 | 302 | solid | 0.881 |
| [C8C1Im]3[HoCl6] | 1.592 ± 0.057 | 358 | solid | 1.064 |
| [C4C1Im]3[HoCl3Br3] | 1.809 ± 0.005 | 355 | solid | 0.899 |
| [C6C1Im]3[HoCl3Br3] | 1.639 ± 0.021 | 346 | solid | 0.988 |
| [C8C1Im]3[HoCl3Br3] | 1.651 ± 0.031 | 341 | solid | 0.986 |
| [C4C1Im]3[DyCl6] | 1.859 ± 0.033 | 366 | solid | 0.881 |
| [C6C1Im]3[DyCl6] | 1.749 ± 0.074 | 371 | solid | 0.910 |
| [C8C1Im]3[DyCl6] | 1.582 ± 0.043 | 354 | solid | 0.865 |
| [C4C1Im]3[DyCl3Br3] | 1.770 ± 0.025 | 395 | solid | 0.863 |
| [C6C1Im]3[DyCl3Br3] | 1.531 ± 0.015 | 335 | solid | 0.820 |
| [C8C1Im]3[DyCl3Br3] | 1.515 ± 0.037 | 313 | solid | 0.682 |
| [C4C1Im]3[GdCl6] | 1.624 ± 0.019 | 358 | solid | 0.363 |
| [C6C1Im]3[GdCl6] | 1.484 ± 0.016 | 333 | solid | 0.492 |
| [C8C1Im]3[GdCl6] | 1.724 ± 0.035 | 344 | solid | 0.562 |
| [C4C1Im]3[GdCl3Br3] | 1.724 ± 0.021 | 366 | solid | 0.540 |
| [C6C1Im]3[GdCl3Br3] | 1.592 ± 0.017 | 371 | solid | 0.492 |
| [C8C1Im]3[GdCl3Br3] | 1.703 ± 0.026 | 354 | solid | 0.585 |
Density measured at 25 °C.
TGA temperature (N2, 5 °C/min) at which 5 wt % loss of compound is observed.
Dynamic viscosity (μ) at room temperature (25 °C).
As-received commercial reference material.
In general, the density of [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] were expected to decrease with an increase in alkyl chain length, which was observed. However, there were exceptions with [C8C1Im]3[HoCl3Br3] and [C8C1Im]3[GdCl3X3] PILs (Figure 1).3,18 The general observation in ILs is that the density decreases as the alkyl chain length increases if there are no significant interactions. This is typically attributed to the larger volume occupied by the longer alkyl chains, which reduces the overall number of ions per unit volume, thereby decreasing the density. However, in certain cases such as with [C8C1Im]3[HoCl3Br3] and [C8C1Im]3[GdCl3X3] ILs, this expected trend is not observed, indicating that other factors may be influencing the density.
Figure 1.

Density (g/mL) of the PILs as a function of the alkyl chain length for the [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs.
One possible explanation could be alterations in the structural arrangement of the IL as the alkyl chain length increases. Longer alkyl chains could potentially lead to more complex packing arrangements or even induce the formation of micelle-like or lamellar-like structures.3 These changes in structure could counteract the expected decrease in density due to the increased volume of the longer alkyl chains.
The role of the anion in these ILs should also be considered. While the molecular weight of the anion often influences the density of ILs, the specific nature of the anion, including its size, shape, and charge distribution, could impact the packing and interactions of the ions. For instance, in the case of [C8C1Im]3[GdCl3X3], the lower molecular weight of gadolinium compared to dysprosium or holmium might typically suggest a lower density. However, the specific characteristics of the gadolinium ion may affect the overall structure and interactions within the IL, leading to an unexpected density trend.
Furthermore, the interactions between the ions in the IL are crucial factors in determining its density. The presence of polarizable halides like Cl and Br, and the potential for complexation between the metal ions and the halides, could significantly alter the structure and dynamics of the IL. These changes could, in turn, affect the density.
When we measured the two solutions of C4C1ImFeCl4 with 10 and 20% water by weight, we observed a large discrepancy between the predicted weighted average density based on ideal mixtures and the experimentally measured density. The predicted density solutions of C4C1ImFeCl4 with 10 and 20% water were 1.34 g mL–1 and 1.30 g mL–1, respectively. The measured density values were 1.364 ± 0.12 g mL–1, and 1.358 ± 0.01 g mL–1. This is a striking difference and suggests that trace water content has a much smaller impact on the density of these PILs than we feared. More interestingly, the observation of very little density change between the 10 and 20 wt % samples suggests there is significant intermolecular void space within these PILs, that free water can occupy.
To gain a deeper understanding of these unexpected density trends, further studies could be beneficial. These might include experimental measurements such as X-ray diffraction or neutron scattering to investigate the structure of the ILs as well as theoretical calculations or simulations to explore the interactions and dynamics of the ions. Such studies could provide valuable insights into the factors influencing the density of these ILs.
The dynamic viscosities of the [CnC1Im][FeCl3X] series PILs (Figure 2) were measured at 25 °C and follow an expected trend of an increase in viscosity with an increase in the alkyl substituent chain length. The dynamic viscosity is not available for the ILs that are solid at room temperature. Generally, viscosity increases with the alkyl chain length, which is consistent with the expectation that larger molecules have higher viscosities due to increased intermolecular forces.
Figure 2.

Viscosity as a function of alkyl chain length for the [CnC1Im][FeCl3X] series PILs.
Thermal Degradation
Thermogravimetric analysis of the [CnC1Im][X] series ILs, [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs (Figure 3) were conducted in an N2 atmosphere, heated at a rate of 5 °C min–1, from RT up to 500 °C. The hygroscopic [CnC1Im]3[MCl3X3] PILs contain tightly bound water. The water content of the PILs does not show a discernible trend or pattern, and water mass loss ranged between 2 and 19% (Table 2).
Figure 3.

Thermal curves of all of the ILs. (A) TGA plot of [CnC1Im][X] series ILs; (B) TGA plots of [CnC1Im][FeCl3X] series PILs; (C) TGA plot of [CnC1Im]3[HoCl3X3] series PILs; (D) TGA plot of [CnC1Im]3[DyCl3X3] series PILs; (E) TGA plot of [CnC1Im]3[GdCl3X3] series PILs.
Table 2. Water Content of PILs Determined by TGA.
| compound | starting mass in TGA (mg) | mass after water loss (mg) | mass of water (mg) | percent water (%) |
|---|---|---|---|---|
| [C4C1Im][FeCl4] | 25.40 | N/a | N/a | N/a |
| [C6C1Im][FeCl4] | 22.20 | N/a | N/a | N/a |
| [C8C1Im][FeCl4] | 16.33 | N/a | N/a | N/a |
| [C4C1Im][FeCl3Br] | 21.81 | N/a | N/a | N/a |
| [C6C1Im][FeCl3Br] | 15.02 | N/a | N/a | N/a |
| [C8C1Im][FeCl3Br] | 15.77 | N/a | N/a | N/a |
| [C4C1Im]3[HoCl6] | 16.48 | 16.23 | 0.26 | 1.5 |
| [C6C1Im]3[HoCl6] | 18.92 | 15.92 | 3.00 | 15.9 |
| [C8C1Im]3[HoCl6] | 15.69 | 14.85 | 0.84 | 5.4 |
| [C4C1Im]3[HoCl3Br3] | 59.77 | 54.13 | 5.63 | 9.4 |
| [C6C1Im]3[HoCl3Br3] | 13.72 | 12.29 | 1.43 | 10.4 |
| [C8C1Im]3[HoCl3Br3] | 19.63 | 16.84 | 2.79 | 14.2 |
| [C4C1Im]3[DyCl6] | 24.08 | 20.88 | 3.20 | 13.3 |
| [C6C1Im]3[DyCl6] | 16.49 | 15.66 | 0.83 | 5.1 |
| [C8C1Im]3[DyCl6] | 17.57 | 16.90 | 0.67 | 3.8 |
| [C4C1Im]3[DyCl3Br3] | 45.71 | 42.93 | 2.78 | 6.1 |
| [C6C1Im]3[DyCl3Br3] | 21.45 | 17.98 | 3.47 | 16.2 |
| [C8C1Im]3[DyCl3Br3] | 29.96 | 26.73 | 3.22 | 10.8 |
| [C4C1Im]3[GdCl6] | 36.28 | 33.99 | 2.29 | 6.3 |
| [C6C1Im]3[GdCl6] | 20.92 | 16.89 | 4.03 | 19.3 |
| [C8C1Im]3[GdCl6] | 49.50 | 42.56 | 6.95 | 14.0 |
| [C4C1Im]3[GdCl3Br3] | 19.19 | 18.23 | 0.95 | 5.0 |
| [C6C1Im]3[GdCl3Br3] | 9.89 | 8.60 | 1.29 | 13.1 |
| [C8C1Im]3[GdCl3Br3] | 25.70 | 22.67 | 3.03 | 11.8 |
Most of the lanthanide metal containing PILs had water content between 5 and 13%. The increased water content is likely gained from hydrates present in the precursors during synthesis but could have also picked up water from the atmosphere. Due to cost, our synthesis was conducted using the hexahydrate form of the lanthanide(III) chloride instead of the anhydrous form.19 Thermal degradation of the [CnC1Im]3[MCl3X3] series of PILs was determined individually for each paramagnetic IL depending on the thermal curve plateau after water mass loss. There does not seem to be a correlation between thermal degradation temperature and alkyl chain length or halide coordination, albeit the PILs demonstrate dynamic thermal degradation behavior due to multiple plateaus observed during heating. All paramagnetic ILs displayed thermal stability above 300 °C.
DSC thermogram analysis of the [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs was conducted in an N2 atmosphere with a flow rate of 50 mL min–1. All samples were ramped at 5 °C min–1, from RT to 175 °C, from 175 to −85 °C, from −85 to 175 °C, from 175 to −85 °C, and from −85 to 175 °C to complete a heat/cool/heat/cool/heat cycle, respectively (Figure 4).
Figure 4.

DSC thermograms of the [CnC1Im][FeCl4] and [CnC1Im]3[MCl6] series PILs. (A) Iron PILs; (B) Holmium PILs; (C) Gadolinium PILs; (D) Dysprosium PILs. DSC thermograms were measured upon heating.
There were very few melting point transitions that appeared for the [CnC1Im]3[MCl3X3] series PILs; however, glass transition temperatures were observed. The glass transition temperature seems to increase with an increasing alkyl chain length for [CnC1Im]3[MCl3X3] series PILs. This is expected because there is good agreement that glass transition temperature increases with molecular weight.20 Unfortunately for the [CnC1Im][FeCl3X] series PILs, no glass transition or melting point peaks were observed except for the [C4C1Im][FeCl4] IL purchased from a manufacturer.
Magnetic Susceptibility
The magnetic susceptibility data was obtained by utilizing the Evans NMR method.14 This method offers a rapid, cost-effective, and straightforward alternative to determining the molar magnetic susceptibility of paramagnetic compounds compared to the Gouy balance and SQUID magnetometer. The Gouy balance requires the suspension of the sample between magnetic poles, making it impractical, while the SQUID magnetometer is expensive due to the necessity of helium gas. In contrast, NMR spectrometers, which are commonly found in academic chemistry departments, are widely available and make the Evans NMR method a more accessible option despite the cost associated with using liquid helium to maintain the NMR coil. Moreover, the advent of benchtop NMR spectrometers removes the cost associated with liquid helium.
Evan’s NMR method is used to determine the molar magnetic susceptibility by determining the difference in the chemical shift of the internal reference solvent and the solvent used to dissolve the paramagnetic sample (eq 2). Where χm is molar magnetic susceptibility,
| 2 |
Δf is the frequency difference in Hz between the paramagnetic solvent resonance and the pure solvent resonance (eq 3), F is the NMR radiofrequency in Hz, and c is the concentration of the paramagnetic sample (mol/mL).
| 3 |
For first row transition metals, the orbital angular momentum contributions can be negated and the effective magnetic moment is determined from spin momentum only (eq 4). However, for rare-earth metals spin–orbital coupling is strong, and the magnetic moment may be overestimated.
| 4 |
Paramagnetic compounds tend to have greater chemical shifts downfield compared to nonparamagnetic compounds from dipolar interactions that increase T1, or spin–lattice, relaxation times.21 In addition, unpaired electrons in metal ions have a greater polarizing affect in the local proximity than local nuclei due to a smaller mass and higher gyromagnetic ratio contribution. When performing Evan’s method, the elucidation of the chemical shift difference between the pure deuterated solvent and deuterated solvent-paramagnetic solute solution may be hard to identify or is nonobvious.
For paramagnetic species in general, there are two considerations of major importance when running NMR experiments: peak broadening and abnormal chemical resonance of nuclei. Peak broadening is an effect of perturbation of nuclei that are in the proximity of unpaired electrons of the metal ion. Therefore, resonance peak integration values in 1H NMR of paramagnetic compounds should be considered qualitative instead of quantitative as it is normally for nonparamagnetic compounds. However, peak broadening is unique for each metal ion coordination, and peak broadening may not be as significant for all metal-coordinated species. This is observed in the 1H NMR spectra of dysprosium and holmium PILs versus iron and gadolinium PILs, where peak broadening is less substantial for dysprosium and holmium PILs (see Supporting Information1H NMR spectra SI-31 to SI-78).
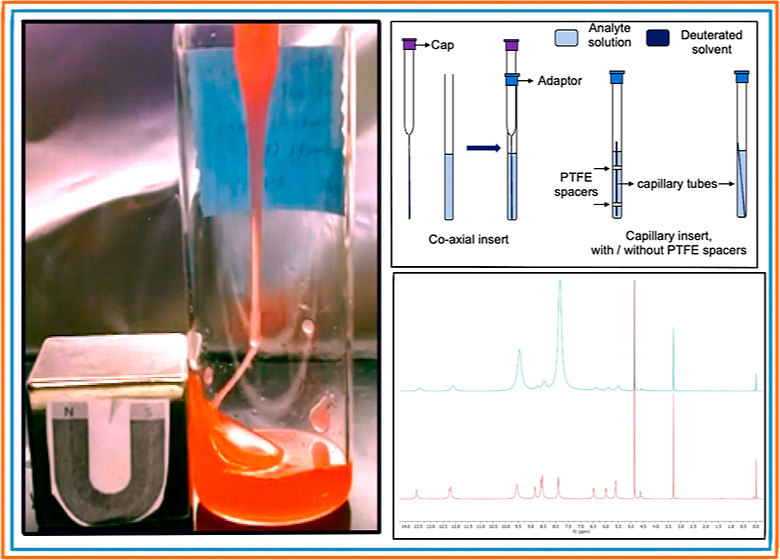
To elucidate the paramagnetic deuterated solvent peak, the addition of 10–30 μL of anhydrous solvent to the paramagnetic solution is sufficient (Figure 5). This approach verifies the chemical resonance of the deuterated solvent in the paramagnetic solution; however, one must calculate molar magnetic susceptibility from the original 1H NMR because of a change in concentration of the paramagnetic sample.
Figure 5.

Evan’s Method 1H NMR spectra of [C4C1Im]3[HoCl6]. Top: original NMR sample spiked with 30 μL of anhydrous methanol; bottom: original NMR sample.
Alternatively, instead of the addition of nondeuterated anhydrous solvent, COSY NMR can be an alternative method to verify the solvent reference peaks (Figure 6). Comparing the top spectra (Evan’s 1H NMR) to the bottom spectra (COSY NMR) of Figure 6, an absence of proton connectivity to the imidazolium cation is clearly observed for the two chemical resonances at 10.09 and 8.44 ppm, respectively. This confirms the shifted paramagnetic solution solvent shift that is observed in Figure 5 by the enlargement of the respective peaks. Integration of the Evan’s 1H NMR resonance peaks in Figure 6A matches up with the assignments given; however, these have been excluded from the spectra.
Figure 6.

Evan’s Method 1H NMR spectra of [C8C1Im]3[HoCl3Br3]. (A) 1H NMR spectra showing peak assignments and deuterated solvent shifts with dashed lines; (B) COSY NMR spectra with dashed lines to illustrate proton propinquity.
While the solvent reference analyte is generally acceptable and is typically used to determine magnetic susceptibility, we experienced a peak broadening challenge due to the water content of our samples. As described above, there are two resonance shifts associated with methanol (3.31 and 4.87 ppm). However, for our lanthinide samples with significant water content, there is a discrepancy in the two solvent peak shifts at 3.31 and 4.87 ppm. The variation observed is 0.10–0.16 ppm, 0.29–0.48 ppm, and 0.13–0.19 ppm for holmium, dysprosium, and gadolinium PILs, respectively. These slight variations in reference analyte shifts are most likely due to varying water content in the PILs as observed by TGA analysis. It has been shown previously, that the water–methanol interaction greatly contributes to peak broadening of the sample solution resonance downfield from the 4.87 ppm pure solvent resonance.22 In our situation, this requires us to average two different peak shifts to determine the susceptibility value. Due to the variation of these two shifts, error is introduced when taking the average to determine a molar magnetic susceptibility value.
Fortunately, we used tetramethyl silane (TMS) spiked solvents and were excited to find that the TMS peak also shifted with the interaction of paramagnetic solute. It is also easier to identify the shifted TMS peak among the analyte peaks, and because it is referenced as zero in the solvent, the paramagnetic shift calculation is as simple as a peak pick. So we happily report and recommend the use of the TMS resonance shifts to determine magnetic susceptibility.
As a result, the values presented in Table 3 are determined using TMS as the reference analyte, and the peak shifts are provided in Table SI-79. Molar magnetic susceptibility was determined using the Evan’s NMR method and calculated using eqs 2 and 3. It is highly advised to use TMS as the reference analyte for Evan’s NMR method when using deuterated solvents that have strong water–solvent interactions.
Table 3. Magnetic Susceptibility Data for of [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] Series PILs.
| compound | χm (m3 mol–1 × 10–7) | χg (m3 kg–1) × 10–7 | χ × 10–4 | μeff (μb) |
|---|---|---|---|---|
| [C4C1Im][FeCl4]* | 0.141 | 0.419 | 0.586 | 5.75 |
| [C4C1Im][FeCl4] | 0.158 | 0.469 | 0.648 | 6.09 |
| [C6C1Im][FeCl4] | 0.157 | 0.430 | 0.576 | 6.07 |
| [C8C1Im][FeCl4] | 0.153 | 0.389 | 0.479 | 5.99 |
| [C4C1Im][FeCl3Br] | 0.143 | 0.375 | 0.564 | 5.79 |
| [C6C1Im][FeCl3Br] | 0.153 | 0.374 | 0.530 | 5.99 |
| [C8C1Im][FeCl3Br] | 0.131 | 0.299 | 0.412 | 5.54 |
| [C4C1Im]3[HoCl6] | 0.602 | 0.758 | 1.426 | 11.88 |
| [C6C1Im]3[HoCl6] | 0.881 | 1.002 | 1.676 | 14.37 |
| [C8C1Im]3[HoCl6] | 1.064 | 1.105 | 1.759 | 15.80 |
| [C4C1Im]3[HoCl3Br3] | 0.899 | 0.968 | 1.752 | 14.52 |
| [C6C1Im]3[HoCl3Br3] | 0.988 | 0.976 | 1.599 | 15.22 |
| [C8C1Im]3[HoCl3Br3] | 0.986 | 0.899 | 1.484 | 15.21 |
| [C4C1Im]3[DyCl6] | 0.881 | 1.112 | 2.067 | 14.37 |
| [C6C1Im]3[DyCl6] | 0.910 | 1.038 | 1.816 | 14.61 |
| [C8C1Im]3[DyCl6] | 0.865 | 0.901 | 1.425 | 14.24 |
| [C4C1Im]3[DyCl3Br3] | 0.863 | 0.932 | 1.650 | 14.23 |
| [C6C1Im]3[DyCl3Br3] | 0.820 | 0.812 | 1.243 | 13.87 |
| [C8C1Im]3[DyCl3Br3] | 0.682 | 0.623 | 0.944 | 12.65 |
| [C4C1Im]3[GdCl6] | 0.363 | 0.461 | 0.749 | 9.23 |
| [C6C1Im]3[GdCl6] | 0.492 | 0.565 | 0.838 | 10.74 |
| [C8C1Im]3[GdCl6] | 0.562 | 0.588 | 1.014 | 11.48 |
| [C4C1Im]3[GdCl3Br3] | 0.540 | 0.587 | 1.011 | 11.25 |
| [C6C1Im]3[GdCl3Br3] | 0.492 | 0.490 | 0.780 | 10.74 |
| [C8C1Im]3[GdCl3Br3] | 0.585 | 0.537 | 0.915 | 11.71 |
The mass magnetic susceptibility and volume magnetic susceptibility were determined using the PILs molar masses and densities, respectively. The effective magnetic moment was calculated using eq 4. Overall, the magnetic susceptibility of the lanthanide paramagnetic ILs is significantly higher compared to the iron paramagnetic ILs for the [CnC1Im]3[MCl3X3] series PILs. The magnetic susceptibility of the iron paramagnetic ILs is close to reports in literature albeit slightly higher.8,23 The volume magnetic susceptibility of [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs are presented in Figure 7.
Figure 7.

Volume magnetic susceptibility of the [CnC1Im][FeCl3X] and [CnC1Im]3[MCl3X3] series PILs. (A) PILs with MCl4 and MCl6 anions; (B) PILs with MCl3Br and MCl3Br3 anions.
Verification of Evan’s Method Measurement by Accounting for Water Content
We noted previously that the water content in the lanthanide PILs presented challenges in calculating and interpreting the 1H NMR analysis presented here. However, if you are able to determine an accurate water weight percent as we have using the TGA analysis. The work presented here demonstrates the robustness of Evan’s method magnetic susceptibility measurements. One major advantage of TGA is that the water content of a sample can be verified. This allows us to correct the molarity of the PIL due to water weight in the sample, and if done while also measuring the paramagnetic shift with TMS, one can measure the paramagnetic susceptibility of wet hygroscopic PILs.
We verified this wet method by comparing the Evan’s method susceptibility measurements of two PILs, [C6C1Im]3[GdCl6] and [C6C1Im][HoCl6], that we were able to dry to completion in the TGA pan. We measured them as produced in the wet state and adjusted the molarity value to account for water and then also sampled the PIL from the TGA to measure the susceptibility of the dry PIL. To obtain dry samples, PIL was loaded into a TGA pan and held at an isotherm of 110 °C until the mass remained constant. The TGA experiment was stopped, and an NMR solution was prepared for the Evan’s NMR method using the dried PIL from the pan. A proton NMR spectrum was obtained from the dried sample, and the molar magnetic susceptibility was calculated from the TMS reference resonance. We demonstrate that the calculated magnetic susceptibility of a dry sample vs the calculated magnetic susceptibility of a wet sample (where the mass of water is subtracted from the starting mass in TGA) result in magnetic susceptibilities of similar value (see Supporting Information SI-80).
The data presented in Table 3 and Figure 7 provide a comprehensive overview of the magnetic properties of various ILs, including their molar magnetic susceptibility (χm), mass magnetic susceptibility (χg), magnetic susceptibility (χ), and effective magnetic moment (μeff). The effective magnetic moment of the iron paramagnetic ILs aligns with previous reports, suggesting the presence of five unpaired electrons.24 This is consistent with the electronic configuration of Fe3+ (3d5), which indeed has five unpaired electrons.
The lanthanide paramagnetic ILs exhibit higher molar magnetic susceptibilities compared to those of other ILs with different paramagnetic metal anions. This could be attributed to the larger number of unpaired electrons in the 4f orbitals of lanthanide ions, which contributes significantly to their magnetic moments. However, the effective magnetic moments of the [CnC1Im]3[MCl3X3] series ILs are slightly higher than those reported in the literature for Gd3+, Dy3+, and Ho3+ ions (7.937, 10.646, and 10.607, respectively). This discrepancy could be due to the fact that the calculation of the effective magnetic moment using eq 4 only considers the spin contribution and neglects the total orbital angular contribution. The orbital angular momentum can have a significant effect on the magnetic properties of lanthanide ions due to the partially filled 4f orbitals. The 4f orbitals are shielded by the outer 5s and 5p orbitals, which allow the 4f electrons to retain their orbital angular momentum and contribute to the overall magnetic moment.
The higher molar magnetic susceptibility of the [CnC1Im]3[DyCl3X3] series compared to dysprosium thiocyanate coordinate ILs synthesized by Mallick et al. could be due to differences in the coordination environment of the Dy3+ ion.10 The coordination environment can affect the splitting of the energy levels of the 4f orbitals and hence the magnetic properties. Similarly, the hydrate nature of the [CnC1Im]3[MCl3X3] series of ILs is still relatively unclear, and the determined magnetic susceptibility of these compounds may be slightly overestimated. The presence of water molecules could affect the coordination environment of the metal ions and alter their magnetic properties.
Further studies, such as magnetic measurements at different temperatures or under different magnetic fields, could provide more insight into the magnetic properties of these ILs. Computational studies could also be useful to understand the electronic structure of the metal ions and their interactions with the surrounding ions and molecules.
Conclusion
In the present study, we successfully synthesized a series of [CnC1Im][FeCl3X] paramagnetic ILs (PILs) and a substantial number of novel [CnC1Im]3[MCl3X3] lanthanide-containing PILs. Our findings reveal that dysprosium and holmium-containing PILs exhibit superior magnetic susceptibility compared to their gadolinium-containing counterparts. This observation is particularly noteworthy given that the magnetic susceptibility of these lanthanide PILs significantly surpasses the values reported in the existing literature.
Moreover, we demonstrated the practicality and cost-effectiveness of Evan’s NMR method for measuring magnetic susceptibility. With the increasing prevalence and accessibility of NMR spectrometers, including the advent of benchtop NMR, Evan’s method emerges as an attractive alternative to other techniques such as the Gouy balance. The latter, while precise, often suffers from limited availability and typically measures only a single parameter. Similarly, while SQUID magnetometers offer high accuracy, their maintenance can be prohibitively expensive, positioning Evan’s NMR method as a more practical option for many researchers.
However, our study also underscores the need for a more comprehensive understanding of the properties of PILs. While our focus has been primarily on the magnetic properties, it is crucial to recognize that the viscosity and gas solubility of PILs are equally important. These properties can significantly impact the performance of PILs in various applications, such as energy storage, magnetic cooling, and CO2 capture. For instance, the viscosity of PILs can influence the rate and efficiency of mass transfer during CO2 absorption and desorption, while the gas solubility can affect the capacity and selectivity of PILs for CO2 capture.
Our findings also highlight the need for more extensive research on the relationship between the magnetic properties, of PILs and other material properties, such as thermal and electrical conductivity. This could pave the way for the development of PILs with enhanced performance in various applications. Furthermore, the optimization of existing synthesis methods and the development of new ones could lead to the production of PILs with improved material properties and lower costs.
In the broader context, our study contributes to the growing body of knowledge in the field of ILs research. The synthesis of these paramagnetic ILs represents a significant advancement in the development of new technologies and has exciting implications for future research. However, it is also evident that there are still substantial gaps in both experimental work and theoretical and computational models. For instance, the unexpected density trends observed in our study underscore the need for more nuanced models that can accurately predict these properties. Similarly, the lack of comprehensive studies comparing the properties of similar PILs highlights the need for more systematic investigations in this area.
In conclusion, although our study has made significant strides in the synthesis and characterization of PILs, it also underscores the need for more comprehensive and systematic investigations in this field. By addressing these gaps, we can further enhance the performance of PILs in various applications and contribute to the development of more sustainable and efficient technologies.
Acknowledgments
This work was supported by an Early Career Faculty grant from NASA’s Space Technology Research Grants Program (80NSSC18K1513). We would also like to thank and acknowledge the Joint School of Nanoscience and Nanoengineering at the Universities of North Carolina A&T State University and the University of North Carolina at Greensboro for their abundance of support and contribution of instrument time and subject matter expertise. This work was performed in whole at the Joint School of Nanoscience and Nanoengineering, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (grant ECCS-2025462). We would like to acknowledge Jorge Abraham Aguilera for his contribution to the Table of Contents Abstract.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsaenm.3c00240.
FTIR and Proton NMR spectra, Evan’s NMR calculations, Magnetic susceptibility data of wet and dry samples, and comparison of measured density versus calculated pure density (PDF)
Author Contributions
† J.E.K. and J.R.A. contributed equally.
Author Contributions
The manuscript was written through contributions of all authors. James E. Knoop performed synthesis, designed the experiments, oversaw all characterization of the materials, and wrote the majority of the manuscript. Liberty Yoder conducted density measurements and 1H NMR analysis. Victor Hammed conducted density analysis experiments. Adesewa Maselugbo and Bolaji Sadiku conducted viscosity analysis and assisted in characterization of materials. Jeffrey R. Alston managed the research, secured funding, and planned and designed experiments. All authors have given approval to the final version of the manuscript.
This work was supported by an Early Career Faculty grant from NASA’s Space Technology Research Grants Program (80NSSC18K1513) and the National Science Foundation (Grant ECCS-2025462).
The authors declare no competing financial interest.
Supplementary Material
References
- Welton T. ILs: a brief history. Biophys. Rev. 2018, 10 (3), 691–706. 10.1007/s12551-018-0419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S.; Hamaguchi H.-o. Discovery of a Magnetic IL [bmim]FeCl4. Chem. Lett. 2004, 33 (12), 1590–1591. 10.1246/cl.2004.1590. [DOI] [Google Scholar]
- Greeson K. T.; Hall N. G.; Redeker N. D.; Marcischak J. C.; Gilmore L. V.; Boatz J. A.; Le T. C.; Alston J. R.; Guenthner A. J.; Ghiassi K. B. Synthesis and properties of symmetrical N,N′-bis(alkyl)imidazolium bromotrichloroferrate(III) paramagnetic, room temperature ILs with high short-term thermal stability. J. Mol. Liq. 2018, 265, 701–710. 10.1016/j.molliq.2018.06.016. [DOI] [Google Scholar]
- Del Sesto R. E.; McCleskey T. M.; Burrell A. K.; Baker G. A.; Thompson J. D.; Scott B. L.; Wilkes J. S.; Williams P. Structure and magnetic behavior of transition metal based ILs. Chem. Commun. 2008, (4), 447–449. 10.1039/B711189D. [DOI] [PubMed] [Google Scholar]
- Nockemann P.; Thijs B.; Postelmans N.; Van Hecke K.; Van Meervelt L.; Binnemans K. Anionic rare-earth thiocyanate complexes as building blocks for low-melting metal-containing ILs. J. Am. Chem. Soc. 2006, 128 (42), 13658–13659. 10.1021/ja0640391. [DOI] [PubMed] [Google Scholar]
- a Wu K.; Shen X. Designing a new type of magnetic IL: a strategy to improve the magnetic susceptibility. New J. Chem. 2019, 43 (40), 15857–15860. 10.1039/C9NJ03464A. [DOI] [Google Scholar]; b Abbasi N. M.; Zeger V. R.; Biswas A.; Anderson J. L. Synthesis and characterization of magnetic ILs containing multiple paramagnetic lanthanide and transition metal centers and functionalized diglycolamide ligands. J. Mol. Liq. 2022, 361, 119530. 10.1016/j.molliq.2022.119530. [DOI] [Google Scholar]; c McCourt E.; Esien K.; Zhenyu L.; Felton S.; Nockemann P. Designing Dimeric Lanthanide(III)-Containing ILs. Angew. Chem., Int. Ed. Engl. 2023, 62 (7), e201809334 10.1002/anie.201809334. [DOI] [PubMed] [Google Scholar]; d Wang N.; Li F.; Fan B.; Zhang S.; Bai L.; Zhang X. Rare-earth separation based on the differences of ionic magnetic moment via quasi-liquid strategy. Front. Chem. Sci. Eng. 2022, 16 (11), 1584–1594. 10.1007/s11705-022-2189-4. [DOI] [Google Scholar]; e Tang S.; Babai A.; Mudring A. V. Europium-based ILs as luminescent soft materials. Angew. Chem., Int. Ed. Engl. 2008, 47 (40), 7631–7634. 10.1002/anie.200801159. [DOI] [PubMed] [Google Scholar]; f Babai A.; Mudring A.-V. Anhydrous Praseodymium Salts in the IL [bmpyr] [Tf2N]: Structural and Optical Properties of [bmpyr]4[PrI6] [Tf2N] and [bmyr]2[Pr(Tf2N)5]. Chem. Mater. 2005, 17 (25), 6230–6238. 10.1021/cm051137x. [DOI] [Google Scholar]; g Pereira C. C. L.; Carretas J. M.; Monteiro B.; Leal J. P. Luminescent Ln-ILs beyond Europium. Molecules 2021, 26 (16), 4834. 10.3390/molecules26164834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Deng N.; Li M.; Zhao L.; Lu C.; de Rooy S. L.; Warner I. M. Highly efficient extraction of phenolic compounds by use of magnetic room temperature ILs for environmental remediation. J. Hazard. Mater. 2011, 192 (3), 1350–1357. 10.1016/j.jhazmat.2011.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lei Z.; Dai C.; Chen B. Gas solubility in ILs. Chem. Rev. 2014, 114 (2), 1289–1326. 10.1021/cr300497a. [DOI] [PubMed] [Google Scholar]; c Clark K. D.; Nacham O.; Yu H.; Li T.; Yamsek M. M.; Ronning D. R.; Anderson J. L. Extraction of DNA by magnetic ILs: tunable solvents for rapid and selective DNA analysis. Anal. Chem. 2015, 87 (3), 1552–1559. 10.1021/ac504260t. [DOI] [PubMed] [Google Scholar]; d Zhou C.; Yu X.; Ma H.; Huang X.; Zhang H.; Jin J. Properties and catalytic activity of magnetic and acidic ILs: Experimental and molecular simulation. Carbohydr. Polym. 2014, 105, 300–307. 10.1016/j.carbpol.2014.01.071. [DOI] [PubMed] [Google Scholar]; e Muraoka J.; Kamiya N.; Ito Y. Preparation and evaluation of cellulose-dissolving magnetic IL. J. Mol. Liq. 2013, 182, 76–78. 10.1016/j.molliq.2013.03.012. [DOI] [Google Scholar]; f Wang T.; Yu W.-h.; Li T.-x.; Wang Y.-t.; Tan J.-j.; Hu B.; Nie L.-h. Synthesis of novel magnetic ILs as high efficiency catalysts for extraction-catalytic oxidative desulfurization in fuel oil. New J. Chem. 2019, 43 (48), 19232–19241. 10.1039/C9NJ04015C. [DOI] [Google Scholar]; g Gholinejad M.; Zareh F.; Sheibani H.; Nájera C.; Yus M. Magnetic ILs as catalysts in organic reactions. J. Mol. Liq. 2022, 367, 120395. 10.1016/j.molliq.2022.120395. [DOI] [Google Scholar]
- Panja S. K.; Saha S. Recyclable, magnetic IL bmim[FeCl4]-catalyzed, multicomponent, solvent-free, green synthesis of quinazolines. RSC Adv. 2013, 3 (34), 14495. 10.1039/c3ra42039f. [DOI] [Google Scholar]
- Ma Y.; Gao J.; Wang Y.; Hu J.; Cui P. IL-based CO2 capture in power plants for low carbon emissions. Int. J. Greenhouse Gas Control 2018, 75, 134–139. 10.1016/j.ijggc.2018.05.025. [DOI] [Google Scholar]
- Mallick B.; Balke B.; Felser C.; Mudring A. V. Dysprosium room-temperature ILs with strong luminescence and response to magnetic fields. Angew. Chem., Int. Ed. Engl. 2008, 47 (40), 7635–7638. 10.1002/anie.200802390. [DOI] [PubMed] [Google Scholar]
- Bwambok D. K.; Thuo M. M.; Atkinson M. B. J.; Mirica K. A.; Shapiro N. D.; Whitesides G. M. Paramagnetic ILs for Measurements of Density Using Magnetic Levitation. Anal. Chem. 2013, 85 (17), 8442–8447. 10.1021/ac401899u. [DOI] [PubMed] [Google Scholar]
- Han Y.; Lin C.; Meng Q.; Dai F.; Sykes A. G.; Berry M. T.; May P. S. (BMI)3LnCl6 crystals as models for the coordination environment of LnCl3 (Ln = Sm, Eu, Dy, Er, Yb) in 1-butyl-3-methylimidazolium chloride ionic-liquid solution. Inorg. Chem. 2014, 53 (11), 5494–5501. 10.1021/ic500101x. [DOI] [PubMed] [Google Scholar]
- Sitze M. S.; Schreiter E. R.; Patterson E. V.; Freeman R. G. ILs Based on FeCl3 and FeCl2. Raman Scattering and ab Initio Calculations. Inorg. Chem. 2001, 40 (10), 2298–2304. 10.1021/ic001042r. [DOI] [PubMed] [Google Scholar]
- Evans D. F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc. 1959, 2003. 10.1039/jr9590002003. [DOI] [Google Scholar]
- Pierson S. A.; Nacham O.; Clark K. D.; Nan H.; Mudryk Y.; Anderson J. L. Synthesis and characterization of low viscosity hexafluoroacetylacetonate-based hydrophobic magnetic ILs. New J. Chem. 2017, 41 (13), 5498–5505. 10.1039/C7NJ00206H. [DOI] [Google Scholar]
- a Ferreira T. J.; Vera A. T.; de Moura B. A.; Esteves L. M.; Tariq M.; Esperanca J.; Esteves I. Paramagnetic IL/Metal Organic Framework Composites for CO(2)/CH(4) and CO(2)/N(2) Separations. Front. Chem. 2020, 8, 590191. 10.3389/fchem.2020.590191. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Santos E.; Albo J.; Rosatella A.; Afonso C. A. M.; Irabien A. ´. Synthesis and characterization of Magnetic ILs (MILs) for CO2 separation. J. Chem. Technol. Biotechnol. 2014, 89 (6), 866–871. 10.1002/jctb.4323. [DOI] [Google Scholar]
- a Bradley A. E.; Hardacre C.; Holbrey J. D.; Johnston S.; McMath S. E. J.; Nieuwenhuyzen M. Small-Angle X-ray Scattering Studies of Liquid Crystalline 1-Alkyl-3-methylimidazolium Salts. Chem. Mater. 2002, 14 (2), 629–635. 10.1021/cm010542v. [DOI] [Google Scholar]; b Downard A.; Earle M. J.; Hardacre C.; McMath S. E. J.; Nieuwenhuyzen M.; Teat S. J. Structural Studies of Crystalline 1-Alkyl-3-Methylimidazolium Chloride Salts. Chem. Mater. 2004, 16 (1), 43–48. 10.1021/cm034344a. [DOI] [Google Scholar]; c Dzyuba S. V.; Bartsch R. A. Efficient synthesis of 1-alkyl(aralkyl)-3-methyl(ethyl)imidazolium halides: Precursors for room-temperature ILs. J. Heterocycl. Chem. 2001, 38 (1), 265–268. 10.1002/jhet.5570380139. [DOI] [Google Scholar]
- Yoshida Y.; Saito G. Influence of structural variations in 1-alkyl-3-methylimidazolium cation and tetrahalogenoferrate(iii) anion on the physical properties of the paramagnetic ILs. J. Mater. Chem. 2006, 16 (13), 1254–1262. 10.1039/b515391c. [DOI] [Google Scholar]
- Cao Y.; Chen Y.; Wang X.; Mu T. Predicting the hygroscopicity of imidazolium-based ILs varying in anion by hydrogen-bonding basicity and acidity. RSC Adv. 2014, 4 (10), 5169. 10.1039/c3ra44464c. [DOI] [Google Scholar]
- Novikov V. N.; Rössler E. Correlation between glass transition temperature and molecular mass in non-polymeric and polymer glass formers. Polymer 2013, 54 (26), 6987–6991. 10.1016/j.polymer.2013.11.002. [DOI] [Google Scholar]
- Lauffer R. B. Paramagnetic Metal Complexes as Water Proton Relaxation Agents for NMR Imaging: Theory and Design. Chem. Rev. 1987, 87 (5), 901–927. 10.1021/cr00081a003. [DOI] [Google Scholar]
- a Corsaro C.; Maisano R.; Mallamace D.; Dugo G. 1H NMR study of water/methanol solutions as a function of temperature and concentration. Phys. A 2013, 392 (4), 596–601. 10.1016/j.physa.2012.11.008. [DOI] [Google Scholar]; b Gottlieb H. E.; Kotlyar V.; Nudelman A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62 (21), 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Hayashi S.; Saha S.; Hamaguchi H. A new class of magnetic fluids: bmim[FeCl/sub 4/] and nbmim[FeCl/sub 4/] ILs. IEEE Trans. Magn. 2006, 42 (1), 12–14. 10.1109/TMAG.2005.854875. [DOI] [Google Scholar]
- Burba C. M. C.; Chang H. C. The Nature of Cation-Anion Interactions in Magnetic ILs as Revealed Using High-Pressure Fourier Transform Infrared (FT-IR) Spectroscopy. Appl. Spectrosc. 2019, 73 (5), 511–519. 10.1177/0003702818805499. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.