

Abstract
Cervical cancers are the fourth most common and most deadly cancer in women worldwide. Despite being a tremendous public health burden, few novel approaches to improve care for these malignancies have been introduced. We discuss the potential for PCNA inhibition to address this need as well as the advantages and disadvantages for compounds that can therapeutically inhibit PCNA with a specific focus on cervical cancer.
Introduction
Most cervical cancer (CaCx) is caused by persistent high-risk human papillomavirus (HPV) infections. The HPV family is a large collection of over 400 viruses subdivided into 5 genera (alpha, beta, gamma, mu, and nu) based on similarities in their L1 capsid protein1. Not all these viruses are equally carcinogenic. Only the so-called high risk alpha HPVs (HR αHPVs) cause CaCx2–4. These HR α-HPVs are prevalent throughout the world with an estimated 11.5 million new infections occurring annually in the United States alone. In high income countries, regular screening and early intervention reduces the number of HR α-HPV infections that progress to CaCx. For example, the 11.5 million new HR α-HPV infections in the US result in approximately 13,000 CaCx diagnoses and about 4,000 deaths. The situation is more dire in low- and middle-income countries where 90% of CaCx cases and deaths occur. The death and disease caused by HR α-HPV worldwide is a significant public health problem. CaCx is the fourth most common cancer in women worldwide and kills someone about every 90 seconds5.
Overview of Current Chemotherapy and Radiotherapy Interventions for Cervical Cancer.
Care for CaCx, like most cancers, depends on how advanced the tumor is. Surgery alone is typically effective for early stage CaCx6. As tumors progress to regional or distant disease, surgery becomes less likely to be curative. At this point, radiotherapy and/or chemotherapy (most often a platinum-based drug) becomes the standard approach7. Both approaches work by causing largely indiscriminate DNA damage, achieving some degree of specificity since DNA damage is more lethal in replicating cells and tumor cells are more likely to be replicating than other cells.
Among platinum-based therapies, cisplatin and oxaliplatin are the most used. Both work similarly, by inducing replication stress in the form of bulky crosslinks in DNA that collapse replication forks into double strand breaks in DNA (DSBs)8. This represents a significant increase in the deleterious potential of the lesion as even a single unrepaired DSB can induce apoptosis. The chance that cisplatin induces a DSB is also more likely in CaCx cells in part because the HR-α HPV oncogenes (HR-α HPV E6 and E7) that drive cervical transformation make it harder for a cell to prevent replication stress-induced DSBs. HR-α HPV E7 directly causes replication stress, reducing the ability of the cell to tolerate more replication stress9,10. At the same time, HR-α HPV E6 inhibits the cellular pathway (translesion synthesis or TLS) that prevents replication stress from causing a DSB by preventing accumulation of polymerase eta (POLη), a critical factor for TLS11. Yet, cisplatin resistance is a persistent problem in CaCx care12. Analysis of the cancer genome atlas suggests that TLS is an important determinant of the platinum-based agent efficacy13. Twenty percent of cervical cancers have elevated expression of POLη or other TLS family polymerases (POLι, POLκ, REV1, or REV3L) and these changes are associated with significantly worse patient outcomes especially when those people are treated with cisplatin11.
Paclitaxel represents another class of chemotherapy used to treat CaCx. This drug is a naturally occurring plant alkaloid isolated from the bark of Pacific yew trees, representing one of the most well-known and most effective natural products. The mechanism of action for paclitaxel is well established, preferentially killing cells actively dividing cells by blocking the completion of mitosis14. Like platinum-based drugs, paclitaxel achieves a degree of specificity through the increased frequency that transformed cells undergo mitosis. However, instead of causing stalled replication forks, paclitaxel blocks cell cycle progression by preventing dissociation of microtubules.
Mechanistically, radiotherapy is a genotoxic agent like cisplatin or oxaliplatin. However, unlike therapeutic compounds, radiotherapy is administered by exposure to a radioactive source. Another difference between radiotherapy and platinum-based therapy is that radiation causes DSBs by directly, breaking the DNA backbone, or indirectly by splitting off free-radical species15. It is most commonly administered by a method known as external beam radiotherapy, where a high dose of radiation is delivered externally via high energy x-rays16. Brachytherapy, where a radiation source is placed inside or near the tumor, is an alternative delivery method for radiotherapy. Although less common, brachytherapy can be used with CaCx due to the accessible location of the primary tumor.
Fractionation of doses is a guiding principle of radiotherapy17. This means that if 20 gy of radiation is prescribed, that it will not be given in a single pulse. Instead, the radiation will be broken up into smaller units (e.g., 2 gy) and given as part of a series of doses. The so-called ‘5Rs’ of radiotherapy (repair, repopulation, redistribution, reoxygenation, and radiosensitivity) provide the rationale for this approach17. After any exposure to radiation, a fraction of the damage will be sublethal. This is true for normal and tumor cells, but the damage in a tumor cell is less likely to get “repaired”. Thus, the pause between treatments allows normal tissue to recover. The persistent but sublethal damage in cancer cells is then made lethal damage when compounded by the next wave of radiation. Pauses between rounds of radiation also help mitigate the rapid “repopulation” by tumor cells that is seen after a longer higher dose of radiation. “Redistribution” exploits the fact that radiation is more lethal to proliferating cells and that tumor cells are more likely to continue progression through the cell cycle (compared to normal cells). Thus, a pause between radiation doses makes it more likely that cancer cells will be “redistributed” into these more responsive portions of the cell cycle. Oxygen is a radiosensitizer. In response to an initial pulse of radiation, an increased flow of oxygen can occur in a previously hypoxic tumor. This resulting “reoxygenation” of the tumor makes the subsequent round of radiation more effective. The last “R” or “radio-resistance” is also related to the radiosensitizing properties of oxygen. Fully functioning mitochondria allow normal cells to increase their antioxidant supply by up regulating glycolysis. Decreased ability to derive energy from glycolysis is common in tumor cells, so they lack the ability to induce radio-resistance after an initial exposure.
Radiotherapy and chemotherapy are often used together as combinatorial therapies to treat CaCx. Among the possible combinations, the pairing of a platinum-based drug and radiation is probably the most common18. However, platinum-based drugs are also used in combination with paclitaxols19.
Outstanding Needs in the Treatment of Cervical Cancer.
While the approaches described in the preceding section have been studied and optimized extensively, there are limitations and room for further approval. One of the most widely known areas for improvement is the often-severe side effects associated with both radiotherapy and chemotherapy. Common severe adverse side effects associated with cisplatin, paclitaxel, oxaliplatin, and radiation are listed in Table 120. The consequences of these sequalae extend beyond extreme discomfort. These side effects can be so systemically damaging to the person receiving the care that cisplatin, oxailiplatin, radiation and paclitaxols cannot be given at the best dose to treat the tumor21–23. Furthermore, innate and acquired resistance to frontline therapies are also a barrier to their success. Resistance is a particularly common problem in advanced and recurrent CaCx24,25. Indeed, the five year survival rate for metastatic or otherwise advanced CaCx is below five percent when these malignancies are treated with front line therapeutics (e.g., radiation or cisplatin)26. Thus, there are two critical needs for CaCx care: novel agents that remain effective in CaCx that are resistant to mainline interventions and sensitizing agents that improve the therapeutic window of these interventions.
Table 1: Common Severe Side Effects During CaCx Treatment.
Sources: WebMD, American Cancer Society, Cancer Research UK, National Cancer Institute.
| Treatment | Side Effects | ||
|---|---|---|---|
| Cisplatin |
|
|
|
| Oxaliplatin |
|
|
|
| Paclitaxel |
|
|
|
| External beam radiation or brachytherapy |
|
|
|
There are several strategies being employed as novel therapeutics, including immune checkpoint (PD-1 and PD-L1) inhibitors27. These drugs work by reinvigorating the immune response against the tumor. At least 17 clinical trials have investigated this class of drugs in cervical cancer, focusing on pembrolizumab, nivolumab, sintilimab, cemiplimab, and atezolizumab with or without other drugs. These trials are ongoing and have had varying degrees of success. Because passive diffusion of oxygen and nutrients is inefficient beyond a short distance, tumor growth is dependent on generating new blood vessels. As a result, another approach is to inhibit the growth factors needed for angiogenesis. Specifically, inhibitors of vascular endothelial growth factors (VEGF) are an emerging therapeutic option for these difficult to treat CaCx. Most notably, bevacizumab has received FDA approval and can extend overall survival by at least 12 months24,28. While these approaches are promising and can be expected to improve with more refinement, the benefits are not likely to be universally enjoyed. Novel medications can be cost prohibitive, especially in low and middle income countries29. Given that the majority of CaCx occurs in these countries, this is particularly important to consider. There is also considerable interest in identifying sensitizing agents capable of reducing side effects by making lower doses of genotoxic agents effective. Such an approach could also be used to re-sensitize resistant tumors to standard interventions.
Targeting HPV Oncogene Induced Signaling Changes in CaCx.
Therapies targeted precisely to changes at the molecular level that uniquely occur in tumors, so called precision medicine, is an effective strategy for cancer care. However, diversity among tumor types often makes precision medicine both time and resource restrictive. This may not be the case for CaCx as most of these malignancies are caused by HPV infections. HPV+ CaCx have a notably high requirement for continued viral oncogene expression, as evidenced HeLa cells failing to acquire HPV oncogene independence after decades of passaging30. This commonality offers a potential therapeutic target in CaCx. For example, preventing the initial HPV infection would prevent HPV+ CaCx development. This is the rationale for several highly effective prophylactic vaccines that prevent HPV infection and have been available for over 15 years. As expected, there is mounting evidence that these vaccines are preventing CaCx development31. Perhaps more noteworthy, HPV vaccines appear to offer considerable and durable protection after a single dose32. However, the full benefit of these lifesaving vaccines has not been realized due to under-utilization, especially in low and middle income countries33,34. The reasons for this disappointing reality vary but include the difficulties of motivating people to take a preventative cure, religious concerns, and vaccine hesitance. Because the barriers to HPV vaccination hinder the prevention of CaCx, it is necessary to continue improving CaCx therapeutics.
Since HPV+ CaCx required continued HPV E6 and E7 expression, their manipulation of host cell signaling could result in synthetic lethalities35. To briefly review, HPV E6 degrades p53 by complexing with E6AP36,37 and activates telomerase38. HPV E7 degrades RB and RB family members39,40. Because the HPV oncogenes inactivate these tumor suppressors, HPV+ CaCx infrequently have mutations that inactivate p53 or RB13. This speaks to the power of HPV oncogenes to disrupt major tumor suppress pathways. p53 and RB inactivation are incredibly common across cancer types, but these changes are not readily targeted therapeutically. HPV oncogenes more broadly alter host cell signaling. As noted more briefly above, manipulation of cellular DNA repair responses is one of the principle ways that HPV oncogenes alter the host cell environment41–44. These changes are numerous (e.g., increased expression and activation of ATM, ATR, CHK1, and CHK2) and result in the impairment of major DNA repair pathways (e.g., homologous recombination). The homologous recombination pathway is impaired by mislocalization of repair factors away from sites of damage45. The ability of HPV oncogenes to relocalize DNA repair proteins has been independently twice. The Laimins’ lab demonstrated that the repair factors are recruited to sites of damage in the viral genome, while the Galloway lab demonstrated that HPV oncogenes hinder repair of DNA crosslink lesions by mislocalizing components of the Fanconi Anemia pathway46,47. Consistent with these findings, the Higginson group demonstrated that HPV oncogenes promote repair by microhomology-mediated end joining48, the repair mechanisms that I suppressed when the homologous recombination pathway is functional49. The inability to repair DSBs using the homologous recombination pathway makes cells sensitive to a category of drugs known as PARP inhibitors (e.g., Olaparib). This has been leveraged to better treat breast cancers that cannot complete the pathway due to mutations in the BRCA1 or BRCA250. Consistent with the ideas that HPV-induced changes in DNA repair can be targeted therapeutically and that HPV oncogenes impair the homologous recombination pathway, recurrent CaCx has been successful treated using Olaparib51.
The induction of replication stress by HPV E7 is another example of where the changes needed to facilitate the viral life cycle are important for the treatment of cancers caused by HPV9. Cisplatin kills cancer cells by crosslinking DNA, which causes lethal DSBs when these lesions result in replication fork collapse. Cells are more sensitive to cisplatin induced DSBs when either they have a reduced ability to activate the TLS pathway or are experiencing replication stress from other sources such as nucleotide deprivation. Figure 1 illustrates the latter point specifically for HeLa cells, an HPV+ CaCx cell line. In this data, the toxicity of cisplatin was measured with and without the addition of exogenous nucleosides, known to reduce endogenous replication stress. Hela cells were significantly less sensitive to a range of cisplatin doses when they were grown with supplemental nucleosides. These data suggest that the depletion of nucleoside reserves by HPV E7 induced replication stress contributes to the cisplatin sensitivity in HPV+ CaCx.
Figure 1:
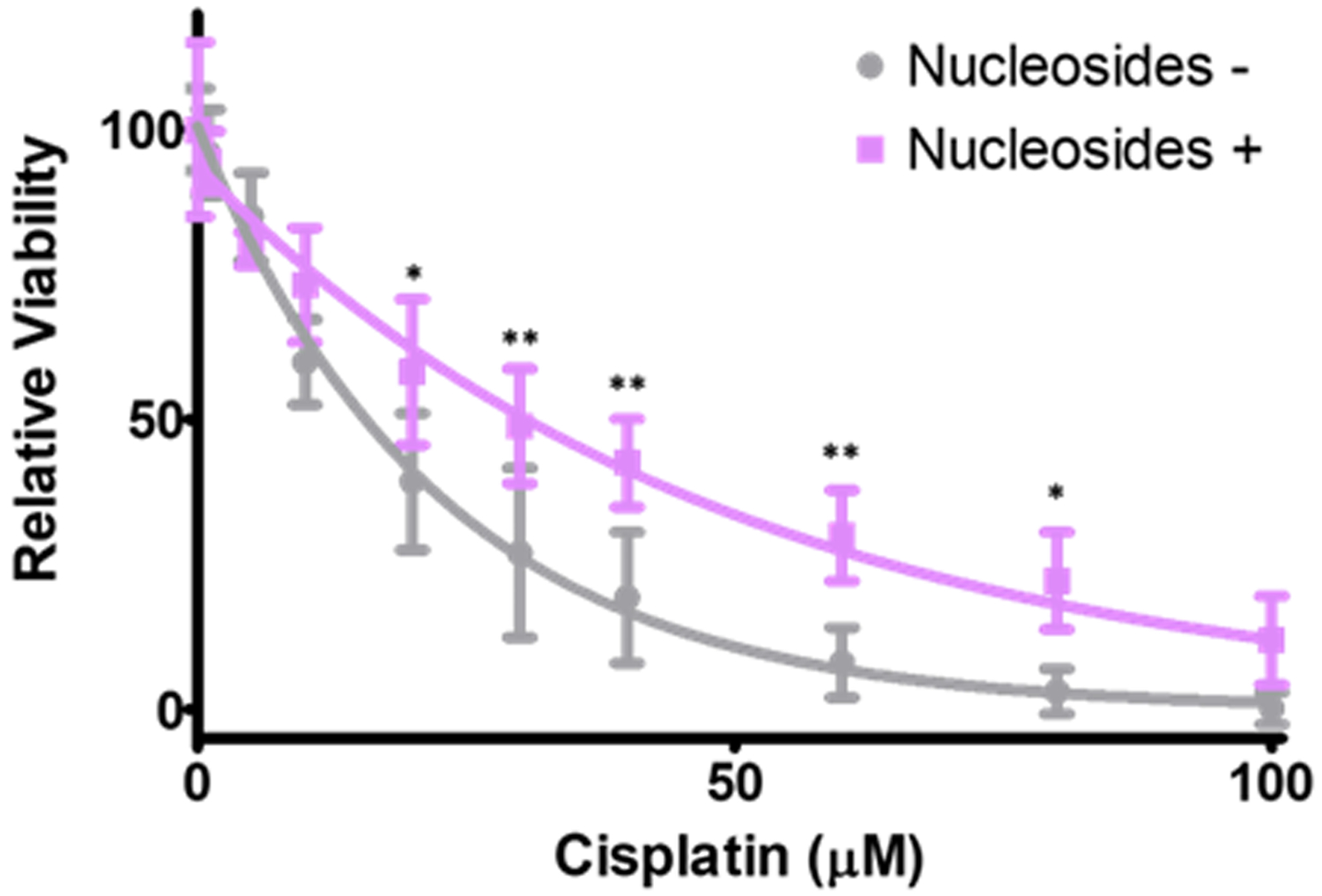
Nucleoside supplementation reduces cisplatin toxic in CaCx cells. HeLa cells were exposed to a gradient of cisplatin concentrations either with or without nucleoside supplementation to lower replication stress in these cells. Relative viability was determined by MTT assay. * and ** indicate statistically significant differences in viability at indicated points (p <0.05 and < 0.01, respectively).
We have demonstrated that the manipulation of host signaling by HPV E6 also contributes to the sensitivity of CaCx cells to cisplatin. Specifically, by destabilizing p53, HPV E6 prevents the induction of POLη in response to cisplatin-induced replication stress. This increases the likelihood for cisplatin induced DSBs and can be overcome by exogenous expression of POLη11,52. This could explain the observation that increased expression of TLS-specific polymerases results in worse patient outcomes11. People with increases TLS-specific polymerase expression had nearly a decade shorter median survival than people who did not. This suggests that HPV E6-mediated inhibition of TLS activation is notable contributor to the efficacy of cisplatin in treating CaCx. The efficacy of cisplatin was obviously determined long before these observations connecting the sensitivity of CaCx to the increased need for TLS (caused by HPV E7) and the decreased ability to complete TLS (caused by HPV E6). However, these observations offer retrospective support of the idea that it is possible to gain therapeutic benefit by exploiting HPV oncogene induced changes in cell signaling.
Proliferating Cell Nuclear Antigen, an Essential Repair and Replication Factor
Proliferating cell nuclear antigen or PCNA is the processivity factor of DNA polymerases and is involved in DNA repair, replication, the replication stress response53. It achieves these functions by forming a trimer that acts as a ring-shaped sliding clamp that encircles DNA with a 30 angstrom diameter54. PCNA also binds to DNA polymerases. The proximity of PCNA with DNA and the ability to bind polymerases, allows it to bring the two together most often at replication forks. By doing this, PCNA confers notably high processivity to DNA replication. Structurally, PCNA is made up of two independent domains connected by a short connector loop. The two domains are folded similarly and of approximately the same size, with the first containing 117 amino acids and the second 124 amino acids. The loop connecting these domains is much shorter and contains only 17 amino acids.
The ability to help catalyze DNA replication and the requirement of replication in DNA repair has earned PCNA the moniker of “master coordinator of DNA replication and repair”55. While PCNA consistently interacts with DNA, whether it is orchestrating DNA repair or replication, its interacting partners vary depending on the cellular process that it is facilitating. When PCNA is involved in standard replication of genomic DNA during the S-phase of the cell cycle, it interacts with polymerase delta and epsilon. In response to replication stress, PCNA interacts with TLS-specific polymerases (POLη, POLι, POLκ, or REV1)56. During homologous recombination, PCNA interacts primarily with polymerase delta and kappa, but also with other polymerases57. Interactions among PCNA and polymerases beta, delta, and epsilon facilitate the base excision repair pathway. Interactions with polymerase delta are also important for nucleotide excision repair. Finally, PCNA also interacts with cellular factors other than polymerases to facilitate alternative DNA repair pathways, including Fanconi Anemia repair (FANCM and FAN1), intrastrand crosslink repair (ZRANB3), nucleotide excision repair (XPA, SPV, and SPG), and mismatch repair (MutL alpha).
This great diversity of interactions makes PCNA an essential gene, defined by the embryonic lethality of knocking out PCNA in mice. The ability to play such an essential role in embryogenesis is dependent on the interactions facilitated by the PCNA interacting protein (PIP) binding motif or PIP box in target proteins with the short connector loop that separates two larger domains of PCNA58–60. Post-translational modifications in PCNA influence its interactions with cellular proteins. The most well studied among these post-translational modifications is the monoubiquitination of PCNA Lysine 164 that occurs in response to replication stress and is carried out by the RAD18/Rad6 complex61,62. This monoubiquitination shifts PCNA binding affinity from favoring replicative polymerases to favoring TLS-specific polymerases. This in turn facilitates the full activation of the TLS pathway. Once the lesion is bypassed, the deubiquination of PCNA facilitates the switch back to the higher fidelity replicative polymerases.
Another relevant post-translational modification of PCNA is a methyl-ester modification that occurs in amino acids 126 to 133 of the interdomain connector loop. This modification is found primarily in transformed tissues and was first identified in a study of breast cancers63. After ruling out the modification was the result of mutations or alternative splicing, this methyl-ester modified PCNA was dubbed cancer associated PCNA or caPCNA64,65. Since its discovery and initial characterization, caPCNA has lived up to its name being found in prostate cancer, hepatic carcinoma, high-grade prostatic intraepithelial neoplasia, and neuroblastoma, but always absent or in very low abundance in untransformed control tissue66–68. The specificity of caPCNA for transformed cells has several potential therapeutic benefits, such as a therapeutic target or biomarker. These benefits may extend to CaCx as the abundance of PCNA is increased in HPV+ cervical cancers and in primary cells expressing HPV E711,69,69,70. The increased abundance of PCNA in CaCx is not surprising given its role in DNA replication and repair. Further, our submitted work demonstrates that caPCNA is also found in CaCx (Wendel et al, pending). This suggests that it is worth considering the therapeutic value of inhibiting caPCNA for CaCx care.
Development of caPCNA Inhibition Strategies
The realization of the potential of caPCNA inhibition faced several barriers. In addition to the typical drug design, pharmacokinetic, and pharmacodynamic hurdles, the lack of intrinsic enzymatic activity represented a further challenge to targeting caPCNA. Because PCNA lacks a catalytic site, efforts to inhibit caPCNA have focused on blocking interactions with interacting proteins instead. Initially, these efforts included the generation of a peptide (R9-caPeptide) that inhibits the interaction of PCNA with its binding partners; presumably by acting as a “decoy” to the PIP box and AlkB homologue 2 PCNA-interacting motif (or APIM) proteins. R9- caPeptide was cytotoxic to breast cancer cell lines as well as neuroblastoma, pancreatic cancer and lymphoma cell lines71,72. The peptide was also able to transit past the plasma membrane and enter cells. While these characteristics are highly desirable, the use of peptides as therapeutic agents generally faces headwinds, most commonly because of poor stability73.
While there are a variety of delivery methods to improve peptide stability, the efforts to develop a clinically viable caPCNA inhibitor turned to small molecule inhibitors. This motivated further work to identify a first-in-class small molecule that could block PCNA interactions with other proteins and hinder PCNA function with notable specificity for caPCNA. Through the combination of computer modeling confirmed by cell viability assays, AOH39 was identified. This served as the backbone for further drug development and screening which ultimately identified AOH1160 as a powerful inhibitor of caPCNA. Like R9-caPeptide, AOH1160 was also cell membrane permeable and displayed broad toxicity for transformed cells while being minimally toxic to untransformed cells74. A subsequent round of screening AOH1160 analogs, identified AOH1996 as a caPCNA inhibitor with further improved therapeutic properties75. AOH1996 remains specifically lethal to malignant cells but has superior pharmacokinetic features including increased half-life and peak concentration in both mice and dogs. A notable feature of AOH1996 is its low toxicity both in vitro and in vivo75. This is likely due to its specificity fro caPCNA and in contrast to most other agents that target PCNA. See Table 2 for a list of compounds targeting PCNA or a post-translational modification of PCNA74,76–81. Our unpublished work determines the efficacy of AOH1996 in preventing the growth of in vitro and in vivo models of CaCx, providing evidence that the benefits of the drug likely extend to CaCx as well as mechanistic details on how AOH1996 kills CaCx cells.
Table 2: Existing PCNA inhibitors.
There are currently 3 peptide and 4 small molecular inhibitors of PCNA. They prevent the assembly of the PCNA trimer or PCNA:target protein interactions.
| Name | Target | Type | Reference |
|---|---|---|---|
| T2AA | PCNA-PIP Box | Small Molecule | Punchihewa et al 2012 |
| PCNA-I1 | PCNA Trimers | Small Molecule | Tan et al 2012 |
| AOH1160 | ca-PCNA | Small Molecule | Gu et al 2018 |
| AOH1996 | ca-PCNA | Small Molecule | Gu et al 2023 |
| p21C2 | p21-PCNA interaction | Peptide | Chen et al 1996 |
| p21PBP | p21-PCNA interaction | Peptide | Warbrick et al 1995 |
| Y211F CPPP | PCNA phosphorylation | Peptide | Zhao et al 2011 |
| Con1-Spop | PCNA | Peptide | Chang et al 2022 |
Relevant Outstanding Questions.
This review discussed caPCNA and caPCNA inhibition in the context of CaCx. With our closing remarks, we would like to propose four open questions that we believe will drive research to define the therapeutic potential of AOH1996 and other caPCNA inhibitors in CaCx. They are listed below:
How broadly effective will caPCNA inhibition be? Can it impair the growth of premalignant cervical lesions, other HPV driven cancers, or warts?
Will caPCNA inhibition be more effective in combination with other drugs? Will the most effective combinations differ between cancer types?
What are the possible mechanisms of resistance to caPCNA inhibition?
Which caPCNA-protein interactions are affected by AOH1996
Acknowledgements:
This research was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award numbers P20GM130448 (NAW) and P20 GM103418, the National Cancer Institute (NCI) award numbers R15CA242057 (NAW) and R21CA216789 (NSB), and the National Institute of Allergies and Infectious Disease (NIAID) R21AI173784 (NAW). Further support was provided by the Forever Pink Foundation (LM and NAW) and the Johnson Cancer Research Center (NAW).
Footnotes
Conflict of Interest Statement: The authors declare the following competing financial interest(s): City of Hope’s Office of Technology Licensing has been awarded a patent on AOH1996 and its analogs. L.H. Malkas, R.J. Hickey, D. Horne, and L. Gu are listed as inventors. Received: May 12, 2022; Revised: February 12, 2023; Accepted: July 10, 2023; Published: August 1, 2023.
Data Availability Statement:
One experiment is included in this review article. The raw data is available upon request.
Bibliography
- 1.Van Doorslaer K, Li Z, Xirasagar S, et al. The Papillomavirus Episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res. 2017;45(Database issue):D499–D506. doi: 10.1093/nar/gkw879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO | Human papillomavirus (HPV) and cervical cancer. WHO. Published November 18, 2016. Accessed November 18, 2016. http://www.who.int/mediacentre/factsheets/fs380/en/ [Google Scholar]
- 3.Carter JR, Ding Z, Rose BR. HPV infection and cervical disease: A review. Australian and New Zealand Journal of Obstetrics and Gynaecology. 2011;51(2):103–108. doi: 10.1111/j.1479-828X.2010.01269.x [DOI] [PubMed] [Google Scholar]
- 4.Berman TA, Schiller JT. Human papillomavirus in cervical cancer and oropharyngeal cancer: One cause, two diseases. Cancer. 2017;123(12):2219–2229. doi: 10.1002/cncr.30588 [DOI] [PubMed] [Google Scholar]
- 5.Cohen PA, Jhingran A, Oaknin A, Denny L. Cervical cancer. The Lancet. 2019;393(10167):169–182. doi: 10.1016/S0140-6736(18)32470-X [DOI] [PubMed] [Google Scholar]
- 6.Fader AN. Surgery in Cervical Cancer. N Engl J Med. 2018;379(20):1955–1957. doi: 10.1056/NEJMe1814034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pujade-Lauraine E, Tan DSP, Leary A, et al. Comparison of global treatment guidelines for locally advanced cervical cancer to optimize best care practices: A systematic and scoping review. Gynecologic Oncology. 2022;167(2):360–372. doi: 10.1016/j.ygyno.2022.08.013 [DOI] [PubMed] [Google Scholar]
- 8.Alexander JL, Orr-Weaver TL. Replication fork instability and the consequences of fork collisions from rereplication. Genes Dev. 2016;30(20):2241–2252. doi: 10.1101/gad.288142.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moody CA. Impact of Replication Stress in Human Papillomavirus Pathogenesis. Journal of Virology. 2019;93(2):e01012–17. doi: 10.1128/JVI.01012-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wendel SO, Stoltz A, Xu X, Snow JA, Wallace N. HPV 16 E7 alters translesion synthesis signaling. Virology Journal. 2022;19(1):165. doi: 10.1186/s12985-022-01899-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wendel SO, Snow JA, Bastian T, et al. High Risk α-HPV E6 Impairs Translesion Synthesis by Blocking POLη Induction. Cancers. 2021;13(1):28. doi: 10.3390/cancers13010028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo). 2018;73(Suppl 1). doi: 10.6061/clinics/2018/e478s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Integrated genomic and molecular characterization of cervical cancer | Nature. Accessed June 26, 2018. https://www.nature.com/articles/nature21386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25(18):2677–2681. doi: 10.1091/mbc.E14-04-0916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Riley PA. Free radicals in biology: oxidative stress and the effects of ionizing radiation. Int J Radiat Biol. 1994;65(1):27–33. doi: 10.1080/09553009414550041 [DOI] [PubMed] [Google Scholar]
- 16.O’Rourke SFC, McAneney H, Hillen T. Linear quadratic and tumour control probability modelling in external beam radiotherapy. Journal of Mathematical Biology. 2009;58(4–5):799–817. doi: 10.1007/s00285-008-0222-y [DOI] [PubMed] [Google Scholar]
- 17.Steel GG, McMillan TJ, Peacock JH. The 5Rs of Radiobiology. International Journal of Radiation Biology. 1989;56(6):1045–1048. doi: 10.1080/09553008914552491 [DOI] [PubMed] [Google Scholar]
- 18.Todo Y, Watari H. Concurrent chemoradiotherapy for cervical cancer: background including evidence-based data, pitfalls of the data, limitation of treatment in certain groups. Chin J Cancer Res. 2016;28(2):221–227. doi: 10.21147/j.issn.1000-9604.2016.02.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorusso D, Petrelli F, Coinu A, Raspagliesi F, Barni S. A systematic review comparing cisplatin and carboplatin plus paclitaxel-based chemotherapy for recurrent or metastatic cervical cancer. Gynecol Oncol. 2014;133(1):117–123. doi: 10.1016/j.ygyno.2014.01.042 [DOI] [PubMed] [Google Scholar]
- 20.Gold JM, Raja A. Cisplatin. In: StatPearls. StatPearls Publishing; 2023. Accessed May 17, 2023. http://www.ncbi.nlm.nih.gov/books/NBK547695/ [Google Scholar]
- 21.Oun R, Moussa YE, Wheate NJ. The side effects of platinum-based chemotherapy drugs: a review for chemists. Dalton Trans. 2018;47(19):6645–6653. doi: 10.1039/c8dt00838h [DOI] [PubMed] [Google Scholar]
- 22.Markman M Toxicities of the platinum antineoplastic agents. Expert Opinion on Drug Safety. 2003;2(6):597–607. doi: 10.1517/14740338.2.6.597 [DOI] [PubMed] [Google Scholar]
- 23.Stojanovska V, Sakkal S, Nurgali K. Platinum-based chemotherapy: gastrointestinal immunomodulation and enteric nervous system toxicity. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2015;308(4):G223–G232. doi: 10.1152/ajpgi.00212.2014 [DOI] [PubMed] [Google Scholar]
- 24.Tewari KS, Sill MW, Penson RT, et al. Bevacizumab for advanced cervical cancer: final overall survival and adverse event analysis of a randomised, controlled, open-label, phase 3 trial (Gynecologic Oncology Group 240). Lancet. 2017;390(10103):1654–1663. doi: 10.1016/S0140-6736(17)31607-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tewari KS, Sill MW, Long HJ, et al. Improved survival with bevacizumab in advanced cervical cancer. N Engl J Med. 2014;370(8):734–743. doi: 10.1056/NEJMoa1309748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pfaendler KS, Tewari KS. Changing Paradigms in the Systemic Treatment of Advanced Cervical Cancer. Am J Obstet Gynecol. 2016;214(1):22–30. doi: 10.1016/j.ajog.2015.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang W, Liu J, Xu K, Chen H, Bian C. PD-1/PD-L1 inhibitors for advanced or metastatic cervical cancer: From bench to bed. Front Oncol. 2022;12:849352. doi: 10.3389/fonc.2022.849352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skelton WP, Castagno J, Cardenas-Goicoechea J, Daily K, Yeung A, Markham MJ. Bevacizumab Eligibility in Patients with Metastatic and Recurrent Cervical Cancer: A Retrospective Review. Clin Med Insights Oncol. 2018;12:117955491877958. doi: 10.1177/1179554918779587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gyawali B, Iddawela M. Bevacizumab in Advanced Cervical Cancer: Issues and Challenges for Low- and Middle-Income Countries. J Glob Oncol. 2017;3(2):93–97. doi: 10.1200/JGO.2016.004895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singhania R, Khairuddin N, Clarke D, McMillan NA. RNA Interference for the Treatment of Papillomavirus Disease. Open Virol J. 2012;6:204–215. doi: 10.2174/1874357901206010204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng L, Wang Y, Du J. Human Papillomavirus Vaccines: An Updated Review. Vaccines (Basel). 2020;8(3):391. doi: 10.3390/vaccines8030391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baisley K, Kemp TJ, Kreimer AR, et al. Comparing one dose of HPV vaccine in girls aged 9–14 years in Tanzania (DoRIS) with one dose of HPV vaccine in historical cohorts: an immunobridging analysis of a randomised controlled trial. The Lancet Global Health. 2022;10(10):e1485–e1493. doi: 10.1016/S2214-109X(22)00306-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falcaro M, Castañon A, Ndlela B, et al. The effects of the national HPV vaccination programme in England, UK, on cervical cancer and grade 3 cervical intraepithelial neoplasia incidence: a register-based observational study. The Lancet. 2021;398(10316):2084–2092. doi: 10.1016/S0140-6736(21)02178-4 [DOI] [PubMed] [Google Scholar]
- 34.Bewley S HPV vaccination and cervical cancer screening. The Lancet. 2022;399(10339):1939. doi: 10.1016/S0140-6736(22)00110-6 [DOI] [PubMed] [Google Scholar]
- 35.Wallace NA. Catching HPV in the Homologous Recombination Cookie Jar. Trends Microbiol. 2020;28(3):191–201. doi: 10.1016/j.tim.2019.10.008 [DOI] [PubMed] [Google Scholar]
- 36.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75(3):495–505. [DOI] [PubMed] [Google Scholar]
- 37.Huibregtse JM, Scheffner M, Howley PM. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991;10(13):4129–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McMurray HR, McCance DJ. Degradation of p53, Not Telomerase Activation, by E6 Is Required for Bypass of Crisis and Immortalization by Human Papillomavirus Type 16 E6/E7. J Virol. 2004;78(11):5698–5706. doi: 10.1128/JVI.78.11.5698-5706.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Münger K, Scheffner M, Huibregtse JM, Howley PM. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products. Cancer Surv. 1992;12:197–217. [PubMed] [Google Scholar]
- 40.Zhang B, Chen W, Roman A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc Natl Acad Sci USA. 2006;103(2):437–442. doi: 10.1073/pnas.0510012103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace NA, Galloway DA. Manipulation of cellular DNA damage repair machinery facilitates propagation of human papillomaviruses. Semin Cancer Biol. 2014;26:30–42. doi: 10.1016/j.semcancer.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Studstill CJ, Moody CA. For Better or Worse: Modulation of the Host DNA Damage Response by Human Papillomavirus. Annu Rev Virol. Published online April 11, 2023. doi: 10.1146/annurev-virology-111821-103452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Banerjee NS, Moore D, Parker CJ, Broker TR, Chow LT. Targeting DNA Damage Response as a Strategy to Treat HPV Infections. Int J Mol Sci. 2019;20(21):5455. doi: 10.3390/ijms20215455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banerjee NS, Wang HK, Broker TR, Chow LT. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem. 2011;286(17):15473–15482. doi: 10.1074/jbc.M110.197574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallace NA, Khanal S, Robinson KL, Wendel SO, Messer JJ, Galloway DA. High Risk Alpha Papillomavirus Oncogenes Impair the Homologous Recombination Pathway. J Virol. Published online August 2, 2017:JVI.01084–17. doi: 10.1128/JVI.01084-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khanal S, Galloway DA. High-risk human papillomavirus oncogenes disrupt the Fanconi anemia DNA repair pathway by impairing localization and de-ubiquitination of FancD2. PLoS Pathog. 2019;15(2):e1007442. doi: 10.1371/journal.ppat.1007442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehta K, Laimins L. Human Papillomaviruses Preferentially Recruit DNA Repair Factors to Viral Genomes for Rapid Repair and Amplification. mBio. 2018;9(1):e00064–18. doi: 10.1128/mBio.00064-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leeman JE, Li Y, Bell A, et al. Human papillomavirus 16 promotes microhomology-mediated end-joining. PNAS. 2019;116(43):21573–21579. doi: 10.1073/pnas.1906120116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahrabi S, Sarkar S, Pfister SX, et al. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016;44(12):5743–5757. doi: 10.1093/nar/gkw326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ragupathi A, Singh M, Perez AM, Zhang D. Targeting the BRCA1/2 deficient cancer with PARP inhibitors: Clinical outcomes and mechanistic insights. Front Cell Dev Biol. 2023;11:1133472. doi: 10.3389/fcell.2023.1133472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gross M, Spencer RJ. Recurrent Cervical Cancer Treated Successfully with Single-Agent PARP-Inhibitor, Olaparib. Case Rep Obstet Gynecol. 2022;2022:6579715. doi: 10.1155/2022/6579715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wallace NA, Robinson K, Howie HL, Galloway DA. HPV 5 and 8 E6 Abrogate ATR Activity Resulting in Increased Persistence of UVB Induced DNA Damage. PLoS Pathog. 2012;8(7). doi: 10.1371/journal.ppat.1002807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.González-Magaña A, Blanco FJ. Human PCNA Structure, Function, and Interactions. Biomolecules. 2020;10(4):570. doi: 10.3390/biom10040570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dieckman LM, Freudenthal BD, Washington MT. PCNA structure and function: insights from structures of PCNA complexes and post-translationally modified PCNA. Subcell Biochem. 2012;62:281–299. doi: 10.1007/978-94-007-4572-8_15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Slade D Maneuvers on PCNA Rings during DNA Replication and Repair. Genes (Basel). 2018;9(8):416. doi: 10.3390/genes9080416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang W, Gao Y. Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annual Review of Biochemistry. 2018;87(1):239–261. doi: 10.1146/annurev-biochem-062917-012405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McVey M, Khodaverdian VY, Meyer D, Cerqueira PG, Heyer WD. Eukaryotic DNA Polymerases in Homologous Recombination. Annu Rev Genet. 2016;50:393–421. doi: 10.1146/annurev-genet-120215-035243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hingorani MM, O’Donnell M. Sliding clamps: A (tail)ored fit. Current Biology. 2000;10(1):R25–R29. doi: 10.1016/S0960-9822(99)00252-3 [DOI] [PubMed] [Google Scholar]
- 59.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116(Pt 15):3051–3060. doi: 10.1242/jcs.00653 [DOI] [PubMed] [Google Scholar]
- 60.Tsurimoto T PCNA binding proteins. Front Biosci. 1999;4:D849–858. doi: 10.2741/tsurimoto [DOI] [PubMed] [Google Scholar]
- 61.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419(6903):135–141. doi: 10.1038/nature00991 [DOI] [PubMed] [Google Scholar]
- 62.Hedglin M, Benkovic SJ. Regulation of Rad6/Rad18 Activity During DNA Damage Tolerance. Annu Rev Biophys. 2015;44:207–228. doi: 10.1146/annurev-biophys-060414-033841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Malkas LH, Herbert BS, Abdel-Aziz W, et al. A cancer-associated PCNA expressed in breast cancer has implications as a potential biomarker. Proc Natl Acad Sci U S A. 2006;103(51):19472–19477. doi: 10.1073/pnas.0604614103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoelz DJ, Arnold RJ, Dobrolecki LE, et al. The discovery of labile methyl esters on proliferating cell nuclear antigen by MS/MS. PROTEOMICS. 2006;6(17):4808–4816. doi: 10.1002/pmic.200600142 [DOI] [PubMed] [Google Scholar]
- 65.Bechtel PE, Hickey RJ, Schnaper L, et al. A unique form of proliferating cell nuclear antigen is present in malignant breast cells. Cancer Res. 1998;58(15):3264–3269. [PubMed] [Google Scholar]
- 66.Sandoval JA, Hickey RJ, Malkas LH. Isolation and characterization of a DNA synthesome from a neuroblastoma cell line. J Pediatr Surg. 2005;40(7):1070–1077. doi: 10.1016/j.jpedsurg.2005.03.054 [DOI] [PubMed] [Google Scholar]
- 67.Venturi A, Piaz FD, Giovannini C, Gramantieri L, Chieco P, Bolondi L. Human hepatocellular carcinoma expresses specific PCNA isoforms: an in vivo and in vitro evaluation. Lab Invest. 2008;88(9):995–1007. doi: 10.1038/labinvest.2008.50 [DOI] [PubMed] [Google Scholar]
- 68.Wang X, Hickey RJ, Malkas LH, et al. Elevated expression of cancer-associated proliferating cell nuclear antigen in high-grade prostatic intraepithelial neoplasia and prostate cancer. Prostate. 2011;71(7):748–754. doi: 10.1002/pros.21291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Noya F, Chien WM, Wu X, et al. The promoter of the human proliferating cell nuclear antigen gene is not sufficient for cell cycle-dependent regulation in organotypic cultures of keratinocytes. J Biol Chem. 2002;277(19):17271–17280. doi: 10.1074/jbc.M112441200 [DOI] [PubMed] [Google Scholar]
- 70.Chien WM, Parker JN, Schmidt-Grimminger DC, Broker TR, Chow LT. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S-phase entry. Cell Growth Differ. 2000;11(8):425–435. [PubMed] [Google Scholar]
- 71.Gu L, Smith S, Li C, et al. A PCNA-derived cell permeable peptide selectively inhibits neuroblastoma cell growth. PLoS One. 2014;9(4):e94773. doi: 10.1371/journal.pone.0094773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith SJ, Gu L, Phipps EA, et al. A Peptide Mimicking a Region in Proliferating Cell Nuclear Antigen Specific to Key Protein Interactions Is Cytotoxic to Breast Cancer. Mol Pharmacol. 2015;87(2):263–276. doi: 10.1124/mol.114.093211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li CM, Haratipour P, Lingeman RG, et al. Novel Peptide Therapeutic Approaches for Cancer Treatment. Cells. 2021;10(11):2908. doi: 10.3390/cells10112908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gu L, Lingeman RG, Yakushijin F, et al. The anti-cancer activity of a first-in-class small molecule targeting PCNA. Clin Cancer Res. Published online January 1, 2018:clincanres.0592.2018. doi: 10.1158/1078-0432.CCR-18-0592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gu L, Li M, Li CM, et al. Small molecule targeting of transcription-replication conflict for selective chemotherapy. Cell Chemical Biology. Published online August 2023:S2451945623002210. doi: 10.1016/j.chembiol.2023.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chang SC, Gopal P, Lim S, et al. Targeted degradation of PCNA outperforms stoichiometric inhibition to result in programed cell death. Cell Chem Biol. 2022;29(11):1601–1615.e7. doi: 10.1016/j.chembiol.2022.10.005 [DOI] [PubMed] [Google Scholar]
- 77.Punchihewa C, Inoue A, Hishiki A, et al. Identification of Small Molecule Proliferating Cell Nuclear Antigen (PCNA) Inhibitor That Disrupts Interactions with PIP-box Proteins and Inhibits DNA Replication*. Journal of Biological Chemistry. 2012;287(17):14289–14300. doi: 10.1074/jbc.M112.353201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tan Z, Wortman M, Dillehay KL, et al. Small-Molecule Targeting of Proliferating Cell Nuclear Antigen Chromatin Association Inhibits Tumor Cell Growth. Mol Pharmacol. 2012;81(6):811–819. doi: 10.1124/mol.112.077735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Warbrick E, Lane DP, Glover DM, Cox LS. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Current Biology. 1995;5(3):275–282. doi: 10.1016/S0960-9822(95)00058-3 [DOI] [PubMed] [Google Scholar]
- 80.Zhao H, Lo YH, Ma L, et al. Targeting Tyrosine Phosphorylation of PCNA Inhibits Prostate Cancer Growth. Molecular Cancer Therapeutics. 2011;10(1):29–36. doi: 10.1158/1535-7163.MCT-10-0778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen J, Peters R, Saha P, et al. A 39 Amino Acid Fragment of the Cell Cycle Regulator p21 Is Sufficient to Bind PCNA and Partially Inhibit DNA Replication in vivo. Nucleic Acids Research. 1996;24(9):1727–1733. doi: 10.1093/nar/24.9.1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
One experiment is included in this review article. The raw data is available upon request.