

Abstract
Catalytic borylations of sp3 C–H bonds occur with high selectivities for primary C–H bonds or secondary C–H bonds that are activated by nearby electron-withdrawing substituents. Catalytic borylation at tertiary C–H bonds has not been observed. Here we describe a broadly applicable method for the synthesis of boron-substituted bicyclo[1.1.1]pentanes and (hetero)bicyclo[2.1.1]hexanes by an iridium-catalysed borylation of the bridgehead tertiary C–H bond. This reaction is highly selective for the formation of bridgehead boronic esters and is compatible with a broad range of functional groups (>35 examples). The method is applicable to the late-stage modification of pharmaceuticals containing this substructure and the synthesis of novel bicyclic building blocks. Kinetic and computational studies suggest that C–H bond cleavage occurs with a modest barrier and that the turnover-limiting step of this reaction is an isomerization that occurs prior to reductive elimination that forms the C–B bond.
Recent developments in iridium-catalysed borylation have made the undirected borylation of alkyl C–H bonds an increasingly practical method to access alkyl boronic esters1–6. Generally, metal-catalysed borylation occurs at primary C–H bonds or, in the absence of accessible primary C–H bonds, at secondary C–H bonds activated by ring strain or electron-withdrawing substituents. Even recent radical-based borylations of C–H bonds, which could be expected to occur at tertiary C–H bonds, occurred at primary and secondary rather than tertiary C–H bonds7. Catalytic borylation of tertiary C–H bonds has not been reported, to the best of our knowledge (Fig. 1a). Our recent discovery of an iridium complex formed from 2-methylphenanthroline (2-mphen) that catalyses the borylation of primary and secondary alkyl C–H bonds with the alkane as the limiting reagent5 raised the question of whether the borylation of tertiary C–H bonds could be achieved. For this proposed reaction to occur and to be practical, the C–H functionalization must occur cleanly at a class of C–H bonds typically unreactive towards C–H activation-forming metal–carbon bonds, with minimal deleterious C–C bond cleavage and with the substrate as the limiting reagent.
Fig. 1 |. State of the borylation of alkyl C–H bonds, applications of BCPs and routes for their synthesis relevant to this study.
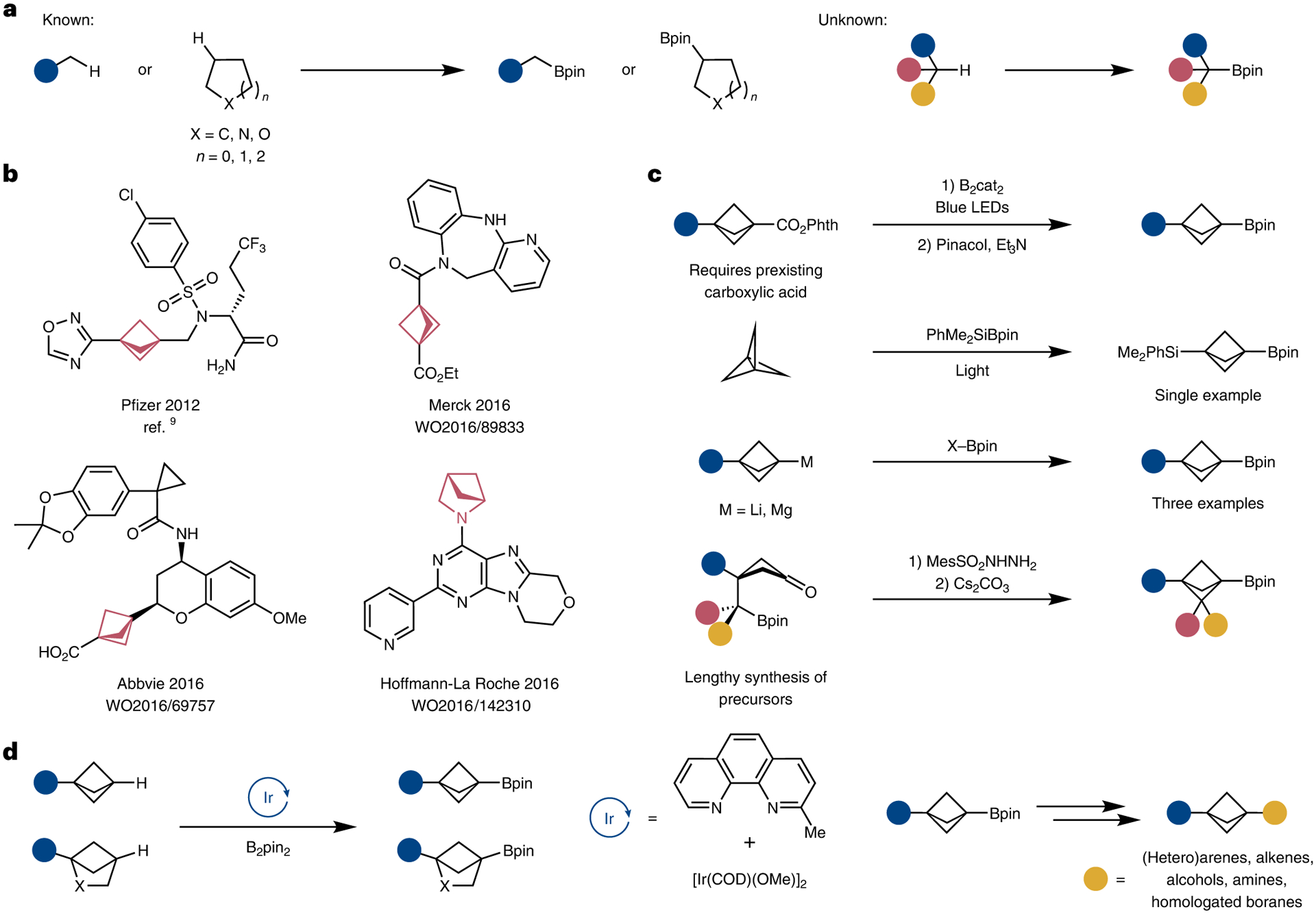
a, Borylation of primary and secondary C–H bonds is known, but borylation of tertiary C–H bonds is underexplored. b, Selected examples of drug candidates in which bioisosteric replacement of para-disubstituted benzenes and pyrrolidines with BCPs and (hetero)BCHs has been conducted. c, Existing approaches towards bridgehead-substituted boryl BCPs. Drawbacks include requirements for prefunctionalization, limitations in scope and lengthy syntheses. Bridgehead-substituted boryl BCHs are also underexplored. d, The direct bridgehead borylation presented in this work occurs with broad scope and provides a complementary route to access BCPs and XBCHs. B2cat2, bis(catecholato)diboron.
Since Lovering’s seminal paper8 introducing the concept of ‘escape from flatland’, the synthetic community has become increasingly interested in the incorporation of sp3-rich fragments into drug candidates. In particular, the highly strained bicyclo[1.1.1]pentane (BCP) motif is being applied as a potential bioisostere to mono- and para-disubstituted benzenes9–12, internal alkynes13 and tert-butyl groups14,15. Related bicyclo[2.1.1]hexanes (BCHs) and oxa- and aza-bicyclo[2.1.1]hexanes (XBCHs) have been proposed as possible bioisosteres to ortho- and meta-disubstituted benzenes16,17 and conformationally constrained equivalents of pyrrolidine, respectively18,19. In many cases, bioisosteric substitution of disubstituted benzenes and pyrrolidines for these saturated bicyclic scaffolds imparted greater rigidity, increased solubility and improved metabolic stability for drug candidates (Fig. 1b).
The increased demand for functionalized BCPs has led to a surge in the development of methods for the construction of substituted BCPs. Prevailing methods usually involve the manipulation of [1.1.1] propellane. Radical addition to the central bond of [1.1.1]propellane is well known and is part of radical chain and photoredox pathways to substituted BCPs (refs.20–30). The addition of organometallic compounds, such as Grignard reagents, to [1.1.1]propellane is also well known, and the resulting bicyclo[1.1.1]pentyl organometallic intermediates can be quenched by electrophiles or used as nucleophiles in cross-coupling reactions13,31–37. The insertion of donor–acceptor carbenes into the bridgehead C–H bond of BCPs also has been reported38. While these methods allow access to 1,3-disubstituted BCP derivatives, the intermediate bicyclopentyl species are not bench stable, and the limited scope of reaction partners that are compatible with the harsh reagents and with the conditions to install the second substituent makes rapid diversification of the scaffold difficult. Furthermore, most of these methods are limited to the synthesis of BCP derivatives and cannot be extended to BCHs and XBCHs.
An alternative and unifying strategy towards the construction of diverse BCP, BCH and XBCH building blocks is to enlist boronic esters, which are stable synthetic intermediates (Fig. 1c). The boronic ester motif could be derivatized into a variety of carbon- and heteroatom-based functional groups through well-established synthetic procedures. In this vein, Aggarwal developed a decarboxylative borylation of redox-active esters, including ester derivatives of BCPs (refs.39,40), Uchiyama reported the direct silaboration of propellane41 and Qin reported a Barluenga–Valdés-inspired cyclization that forges the BCP core from cyclobutanones42. The Anderson, Aggarwal and Walsh groups demonstrated the feasibility of trapping in situ-generated BCP organometallic species with boron electrophiles25,36,43,44. Many of these routes rely on the handling of [1.1.1]propellane or the lengthy synthesis of either a prefunctionalized BCP or cyclobutanone scaffold prior to installation of the boryl group, thereby limiting the general applicability of these groups to the preparation of the boronic esters of BCHs and XBCHs (Fig. 1c). A direct and straightforward strategy to construct these coveted boronic esters would be to repurpose existing, but underutilized, stable and commercially available 1-substituted strained building blocks via the direct functionalization of the bridgehead C–H bond (Fig. 1d).
In this Article, we report the iridium-catalysed borylation of the bridgehead tertiary C–H bonds of BCPs, BCHs and XBCHs. This reaction occurs selectively at this type of tertiary C–H bond rather than unactivated primary and secondary C–H bonds and tolerates a wide variety of functional groups. This C–H bond reacts with rates that are similar to those observed with cyclopropanes, leading to the broad scope. The boronic ester products of this method undergo a wide variety of reactions to form 1,3-disubstituted bicyclic building blocks.
Results and discussion
Reaction development of the borylation of bridgehead C–H bonds
We initiated our investigation of the borylation of the bridgehead tertiary C–H bonds of BCPs by testing reactions of the model substrate 4-tert-butylphenylbicyclopentane 1b in the presence of a series of catalysts reported for the borylation of alkyl C–H bonds (Supplementary Fig. 2)4,5,45. Studies on reaction times, stoichiometries and ligands for iridium showed that the iridium system formed from 2-mphen and (mesitylene)Ir(Bpin)3 (Bpin, 4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) catalysed the borylation of 1b to form 1a in high yield with the substrate as the limiting reagent and with selectivity exclusively for the bridgehead tertiary C–H bond. Reactions conducted with 2,9-dimethylphenanthroline (2,9-dmphen, L1) also occurred in a high yield, albeit measurably lower than those with 2-mphen. Reactions conducted with 3,4,7,8-tetramethylphenanthroline (tmphen) or 2,2′-((3-fluorophenyl)methylene)dipyridine (L3) (ref.4) occurred in moderate to low yields. Reactions conducted with ligands used primarily for the borylation of aromatic C–H bonds, such as 4,4′-di-tert-butylbipyridine (dtbpy, L2) (ref.46) or 5-methyl-2-(thiophen-3-yl)pyridine (L4) (ref.45), as ancillary ligands did not form product. Control experiments showed that both the ligand and the iridium precursor are necessary for the formation of the product. Reactions conducted with the commercially available precatalyst [Ir(COD)(OMe)]2 (COD, cycloocta-1,5-diene) were similar to those with (mesitylene)Ir(Bpin)3. The use of HBpin or alternative diboron reagents in place of bis(pinacolato)diboron (B2pin2) as the boron source resulted in lower yields. Although the desired boronic ester was obtained with L1, L3 or tmphen as the ligand, we used 2-mphen as the ligand for further studies on reaction scope because the reaction times could be shorter and these higher rates would be likely to reduce competing reactions with auxiliary functional groups and afford the products in superior yields (Supplementary Fig. 3).
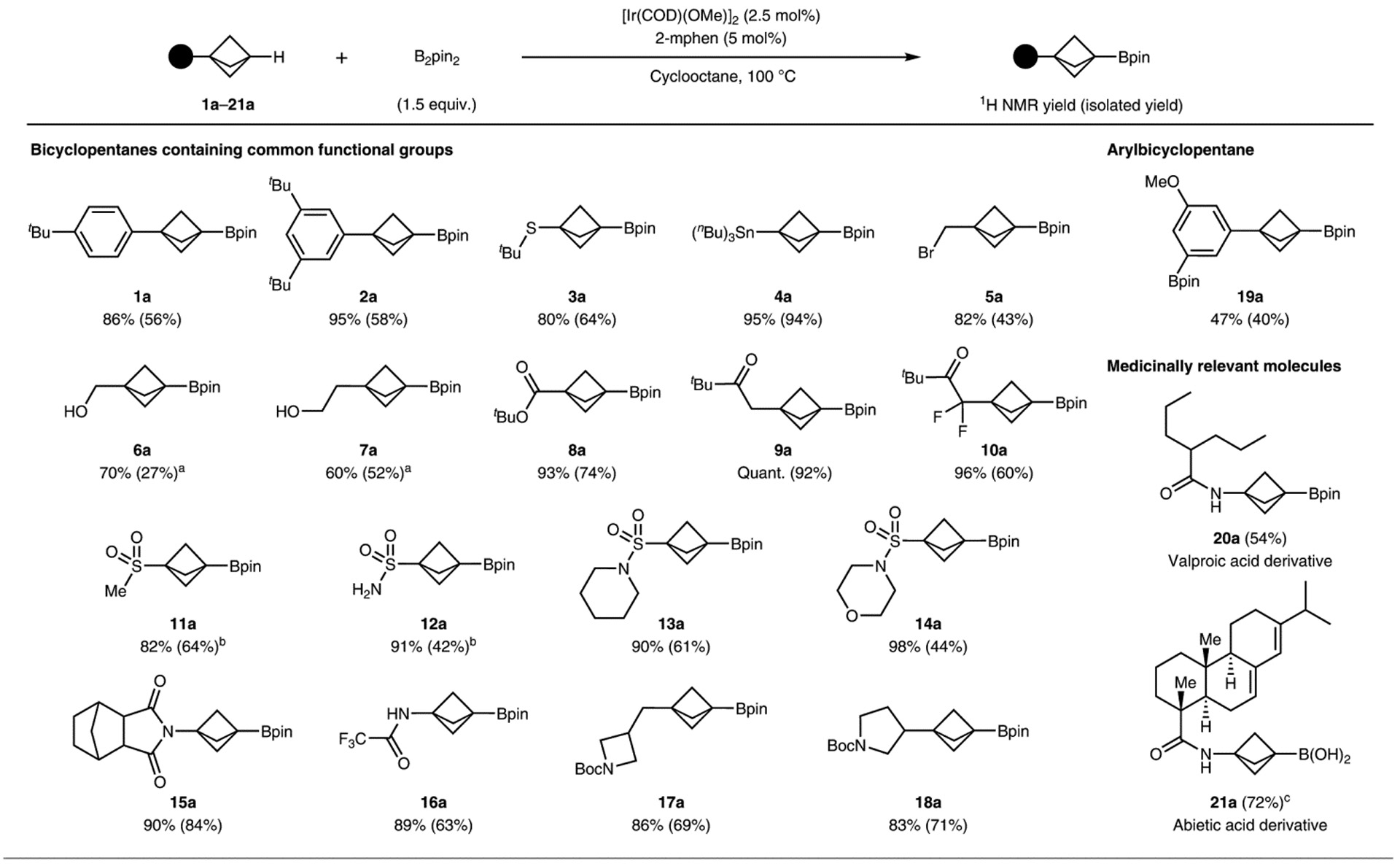
With conditions for the borylation of model substrate 1b in hand, we explored the scope of the undirected borylation of monosubstituted BCPs with B2pin2 catalysed by [Ir(COD)(OMe)]2 and 2-mphen. All substrates underwent borylation exclusively at the bridgehead C–H bond or concomitantly at the bridgehead and at an aryl or acidic primary alkyl C–H bond if those C–H bonds were not sufficiently hindered (Table 1). Competitive reactions at primary and secondary C–H bonds with fully substituted α-carbons were not observed, presumably due to steric hindrance. Arene, sulfide, stannane, bromide and ester functionalities did not interfere with borylation at the bridgehead tertiary C–H bond (1a–5a, 8a–10a). Alcohols were tolerated following an in situ protection procedure (6a, 7a)5. Reactions of BCPs containing sulfone and sulfonamide units gave products from borylation at the tertiary C–H bond (11a–14a); borylations at the methyl C–H bonds of a methyl sulfone and at the N–H bonds of the primary sulfonamide were observed, but the B–C and B–N bonds at those positions were labile and were selectively hydrolysed upon workup to afford the product containing a boronic ester solely at the bridgehead position (11a, 12a). Imides, amides and tert-butyloxycarbonyl(Boc)-protected cyclic amines also underwent borylation at the tertiary C–H bond (15a–18a). A substrate containing sterically accessible aryl C–H bonds underwent borylation of both the aryl and bridgehead C–H bonds (19a) (Table 1).
Table 1 |.
Examples of bicyclo-[1.1.1]-pentanes that undergo borylation of the bridgehead C–H bond
|
Compounds 1a–18a are examples of bicyclopentanes containing common functional groups that undergo borylation of the bridgehead C–H bond. Compound 19a is an example arylbicyclopentane that undergoes borylation of both the aryl and the bridgehead C–H bonds. Compounds 20a and 21a are examples of medicinally relevant molecules that contain the BCP substructure and undergo exclusive borylation of the bridgehead C–H bond. Reactions were conducted at 0.10–0.25 mmol scale. 1H NMR yields were determined relative to an internal standard of dibromomethane and the isolated yields are given in parenthesis. Standard conditions: substrate (0.10–0.25 mmol), B2pin2 (1.5 equiv.), [Ir(COD)(OMe)]2 (2.5 mol%), 2-mphen (5.0 mol%), cyclooctane (100 μl mmol−1), 100 °C. aSubstrate treated with HBpin (1.3 equiv.) prior to the reaction. bConducted with B2pin2 (3 equiv.). cSpontaneous hydrolysis to the corresponding boronic acid upon purification.
The broad functional group compatibility of this method prompted us to apply it to the borylation of medicinally relevant molecules containing the BCP fragment and to test the reaction on a larger scale. Exclusive borylation at the bridgehead C–H bond was observed for derivatives of abietic acid and valproic acid (20a, 21a) (Table 1). The method was easily amenable to syntheses on a gram scale. The borylation of 11b on a 4.0 mmol scale gave the corresponding boronic ester in 58% yield (631 mg), which was comparable to that of the reaction on a smaller scale (0.25 mmol, 64%). The borylation of 16b on a 4.0 mmol scale formed the functionalized product in 95% yield (1.89 g), which was also comparable to that of the reaction on a smaller scale (0.25 mmol, 94%).
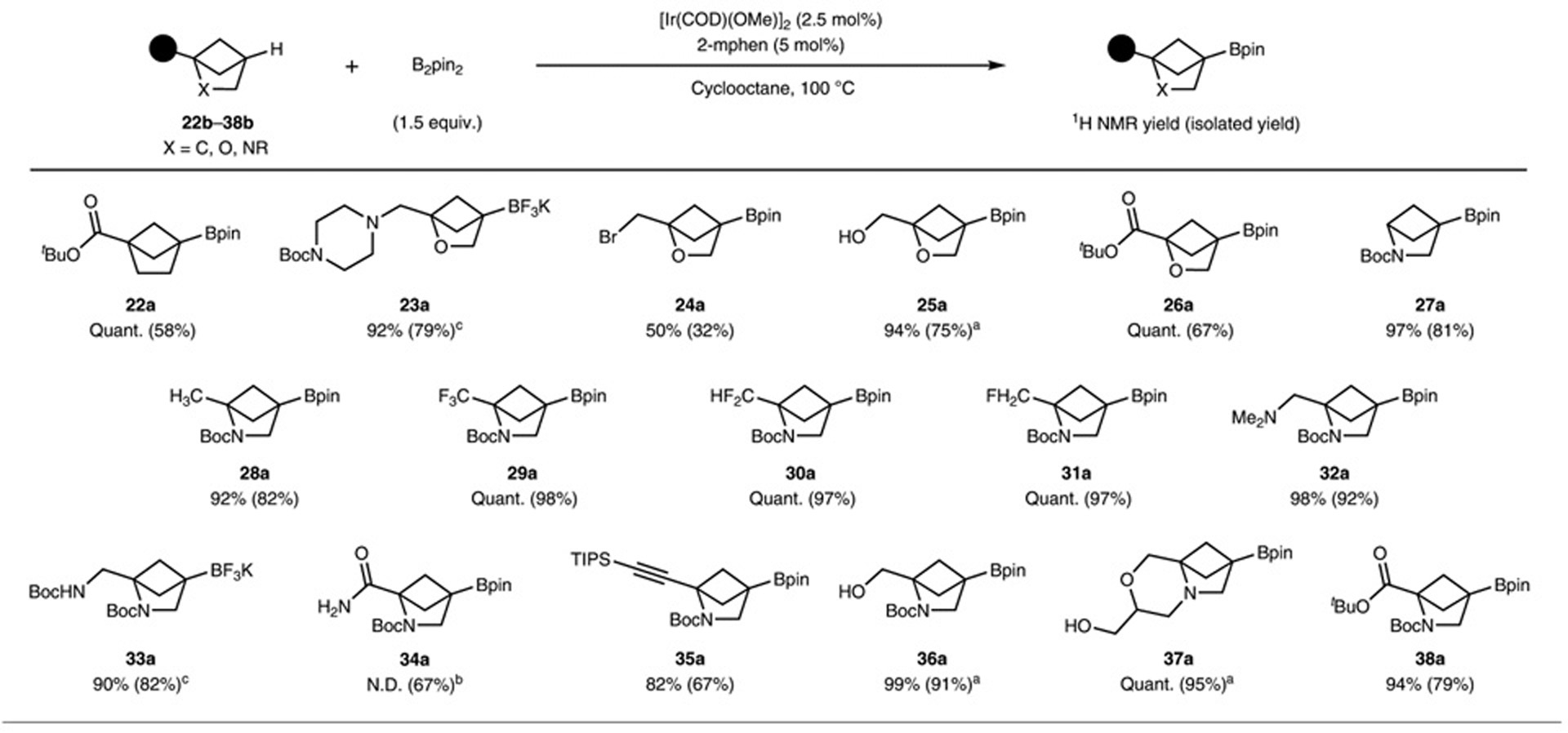
This reactivity extended to bridgehead tertiary C–H bonds in additional strained bi- or tri-cyclic systems. The all-carbon bicyclo-[2.1.1]-hexane 22a, the oxabicyclo-[2.1.1]-hexanes (23a–26a) and the azabicyclo-[2.1.1]-hexanes (27a–38a) all reacted cleanly at the tertiary C–H bonds to afford the corresponding boronic esters (Table 2). The methylene C–H bonds within the bicyclo-[2.1.1]-hexane cores were unreactive, which we attribute, again, to steric crowding of the methylene hydrogens by the quaternary centres vicinal to this position. Bicyclohexanes possessing esters, alcohols, bromides and amides underwent borylation at the tertiary C–H bond to afford the corresponding products (22a–31a, 33a, 36a–38a). The borylations of these bicyclic compounds were also compatible with a tertiary amine (32a), a primary amide (34a) and a triisopropylsilyl-protected alkyne (35a). Reactions conducted with bicyclo-[1.1.0]-butane and cubane structures gave complicated mixtures, while the less strained bicyclo-[2.2.1]-heptane (norbornane) did not react under our conditions (Supplementary Fig. 4).
Table 2 |.
Examples of (hetero)bicyclo-[2.1.1]-hexanes that undergo borylation of the bridgehead C–H bond
|
Reactions were conducted at 0.10–0.25 mmol scale. 1H NMR yields were determined relative to an internal standard of dibromomethane and the isolated yields are given in parenthesis. Standard conditions: substrate (0.10–0.25 mmol), B2pin2 (1.5 equiv.), [Ir(COD)(OMe)]2 (2.5 mol%), 2-mphen (5.0 mol%), cyclooctane (100 μl mmol−1), 100 °C. N.D., not determined. aSubstrate treated with HBpin (1.3 equiv.) prior to the reaction. bConducted with B2pin2 (3 equiv.). cCrude reaction mixture treated with KHF2 (5 equiv.) after completion of the borylation reaction.
Derivatization and applications of the bridgehead boronic esters
The bridgehead pinacol boronic ester fragments in the products of these borylation reactions are primed for conversion into a variety of functional groups (Fig. 2). The 3-boryl BCPs and 3-boryl XBCHs were readily converted into trifluoroboronates and boronic acids for use when more reactive boron derivatives were needed. Addition of bifluoride formed the corresponding trifluoroborate salts (1c, 11c, 16c) and addition of methyl boronic acid formed the corresponding boronic acids47 (1d, 11d, 28c). This set of 3-boryl BCPs underwent reactions at the B–C bond to form new C–C bonds. Cross-coupling with aryl electrophiles occurred under either metallaphotoredox conditions40 to form the 3-aryl BCP 1f from the trifluoroborate 1c or by activation with t-BuLi followed by Pd-catalysed coupling41 to form the 3-aryl BCP 1g from boronic ester 1a. An intermolecular Barluenga–Valdés coupling48 afforded 1e from boronic acid 1d and Zweifel olefination afforded the 3-vinyl BCP 11e from boronic ester 11a and the 3-vinyl XBCH 28c from boronic ester 28a. Heteroarylation afforded the 3-heteroaryl BCP 13c from boronic ester 13a and Matteson homologation formed the homologated boronic ester 1h from boronic ester 1a. The 3-boryl BCPs also underwent reactions to form heteroatom-substituted BCPs. For example, oxidation of boronic ester 16a occurred with urea hydrogen peroxide to afford the 3-hydroxy BCP 16d, while oxidation of boronic ester 27a afforded the 3-hydroxy XBCH 27c. Amination of the trifluoroborate 16c occurred under Matteson’s conditions49 to afford the benzyl amine 16e. C–C and C–N bond formation reactions from 3-boryl XBCHs are under investigation in our laboratories.
Fig. 2 |. Conversion of the boryl group of 3-boryl-bicyclo-[1.1.1]-pentanes and 3-boryl-bicyclo-[2.1.1]-hexanes into various functional groups.
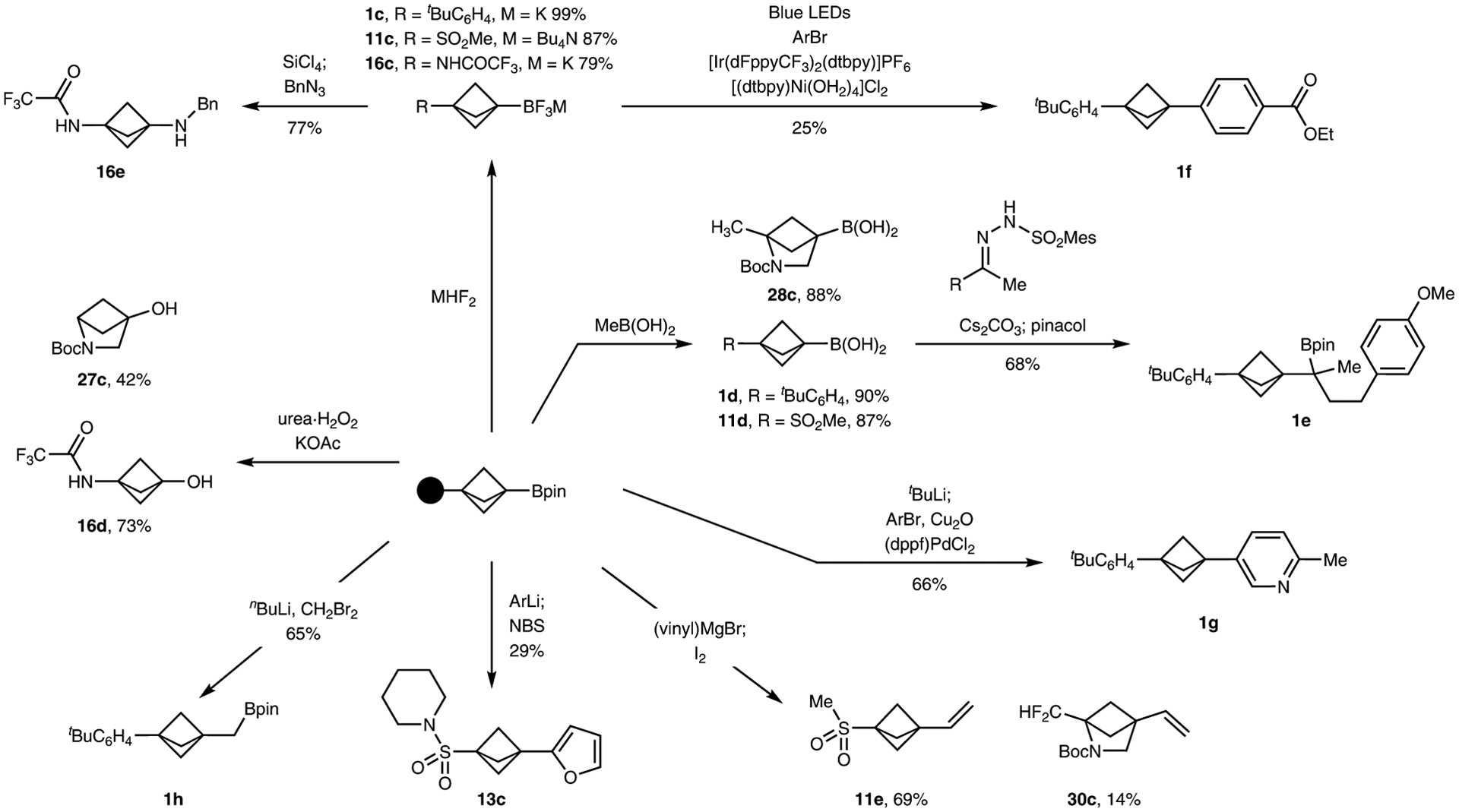
The boronic esters were converted into alternative boron species such as trifluoroborate salts or boronic acids. The boryl products reacted to form C–C bonds by 1,2-metallate rearrangements or cross coupling. The boryl products also reacted to form C–X bonds (X = O, N) through oxidation or aza-1,2-metallate rearrangements. The resultant BCPs and BCHs represent novel building blocks with substitution patterns that are difficult to access. See Supplementary Section 6 for detailed experimental conditions. (dFppy, 3,5-difluoro-2-(2-pyridinyl)phenyl; dppf, 1,1’-bis(diphenylphosphino)ferrocene; NBS, N-bromosuccinimide).
The examples in Fig. 2 also showcase the value of our method for medicinal chemistry. For example, 16d, 16e and 27c are analogues of electron-rich arenes such as aminophenol or aminoanilines. Compounds 11e, 13c and 30c represent examples of substitution patterns that would be difficult to access with existing methods.
Investigation of the mechanism of the tertiary C–H borylation
This borylation reaction presented an opportunity to compare the mechanism of the borylation at tertiary C–H bonds to that at primary C–H bonds and to assess the magnitude of the effect of strain on the borylation of C–H bonds. To obtain preliminary understanding of the functionalization of the tertiary C–H bond in BCPs under our conditions, we compared the relative rates for the borylation of the tertiary C–H bond in 4-tert-butylphenylbicyclopentane (1b) versus sp2 and other sp3 C–H bonds through a series of intermolecular competition studies (Fig. 3a). Because it is not necessary to conduct competition reactions under conditions without an induction period, these experiments were conducted with the most active catalyst, which was generated from [Ir(COD)(OMe)]2 and 2-mphen. The borylation of the bridgehead tertiary C–H bonds of BCP 1b occurred at a much lower rate than that of the aryl C–H bond of 1,3-dichlorobenzene and at roughly the same rate as that of the strained secondary C–H bonds of tert-butyl cyclopropanecarboxylate. This rate was much higher than that of the unactivated secondary C–H bonds of tetrahydrofuran and cyclohexane and much higher than that of the unactivated primary C–H bonds of tert-butyloctyl ether.
Fig. 3 |. Competition experiments and measurement of 1JC–H coupling constants.
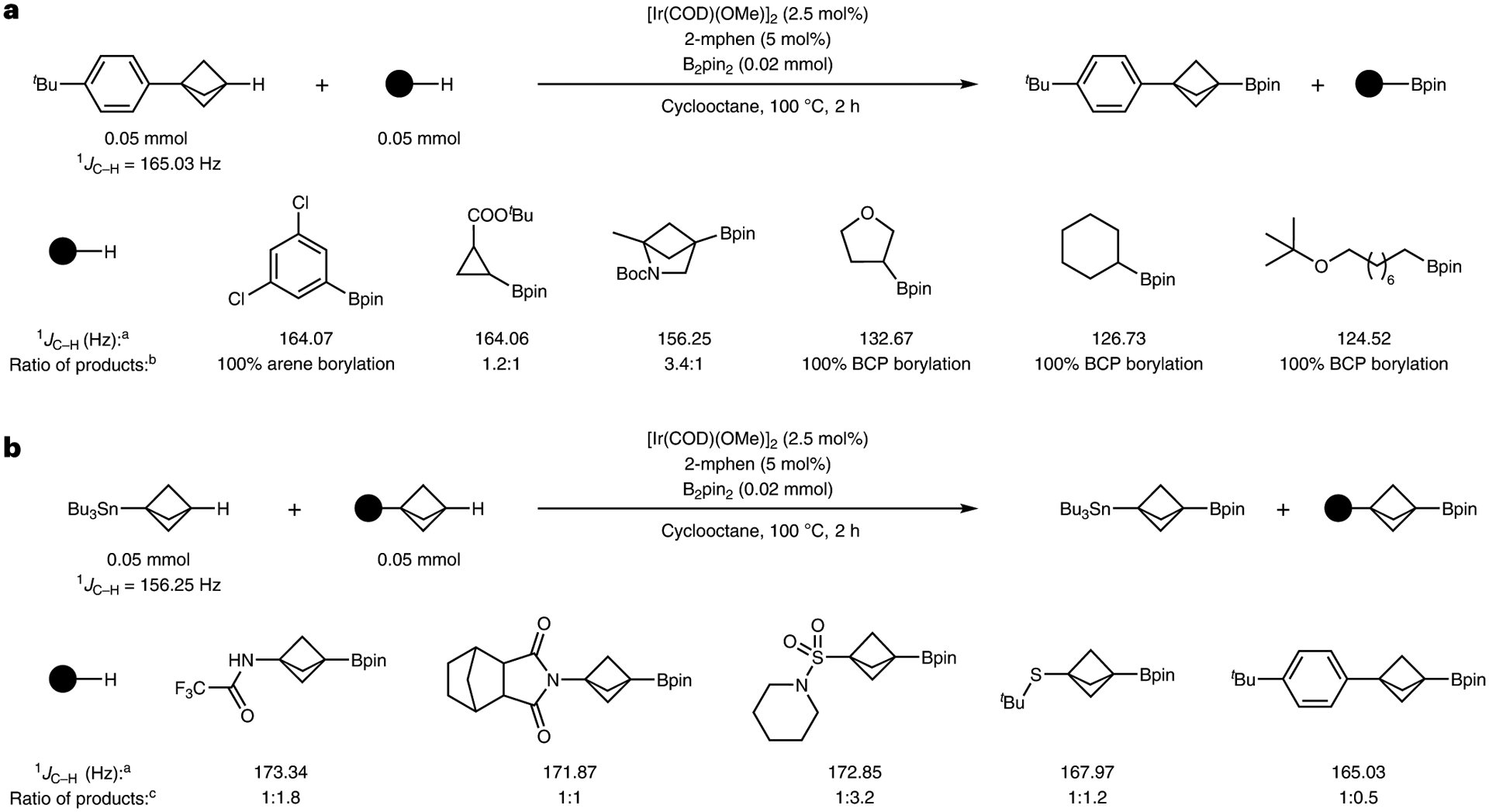
a, Competition experiments between BCPs and typical substrates for C–H borylation show that the borylations of BCPs and BCHs are comparable in rate to the borylation of cyclopropanes, much slower than the borylation of arenes and much faster than the borylation of unactivated alkyl C–H bonds. The relative reactivities correlate well to the 1JC–H coupling constants. b, Competition experiments between BCPs occur with no apparent correlation between the 1JC–H coupling constant or the electronegativity of the substituent and the rate of borylation. a 1JC–H coupling constant of the C–H bond that underwent borylation. b Ratio of products determined by gas chromatography analysis. c Ratio of products determined by NMR analysis.
We reasoned that the high activity of the BCPs towards borylation could be rationalized by the high degree of s-character in the C–H bond, which in turn has been shown to correlate with increased acidity50. To investigate the origin of the high levels of activity of the BCP substrates observed for the borylation reaction, relative to the low reactivity of typical tertiary C–H bonds, we measured the 1JC–H coupling constants of various BCPs and non-BCP substrates. We also measured the relative rates of reaction for various BCPs containing different substituents in the 1-position (Fig. 3b). Indeed, the measured 1JC–H coupling constants of BCPs and XBCHs were comparable to those of aryl and cyclopropyl C–H bonds. However, no clear correlation was observed between the relative rates of reaction of individual BCPs versus the variation of the 1JC–H coupling constants within the set of BCPs. Thus, the generally high reactivity of these C–H bonds towards undirected borylation can be explained by the s-character of the C–H bond, but the more precise reactivity of the individual BCPs requires a deeper mechanistic evaluation.
Kinetic isotope effect (KIE) data in previous reports on the borylation of aryl and alkyl C–H bonds suggested that cleavage of the C–H bond is likely to be irreversible and turnover limiting1,3,51. Yet, other studies have suggested that reductive elimination to form product or isomerization of an intermediate prior to reductive elimination is turnover limiting52–54. Because we observed no clear correlation between the relative rates of reaction and the electronic properties of the substituents on the BCPs and because the steric properties of the BCP substrates are similar, oxidative addition is unlikely to be turnover limiting.
To test further whether oxidative addition of the C–H bond was turnover limiting, we measured the KIE of the bridgehead C–H bond of the BCP. While direct measurements of the rates of reactions initiated with 2-mphen were hampered by a significant induction period, we obtained a KIE value from a pair of competition experiments. In separate flasks, a mixture of 3,5-di(tert-butyl)phenylbicyclopentane (2b) and either 4-tert-butylphenylbicyclopentane (1b) or 4-tert-butylphenylbicyclopentane-d1 (1b-d1) was allowed to react in the presence of B2pin2, [Ir(COD)OMe]2 and 2-mphen (Fig. 4a and see Supplementary Section 7.3 for details). These experiments yielded a KIE value of 1.8 ± 0.1, which is more consistent with an equilibrium isotope effect than a primary KIE.
Fig. 4 |. Mechanistic experiments.
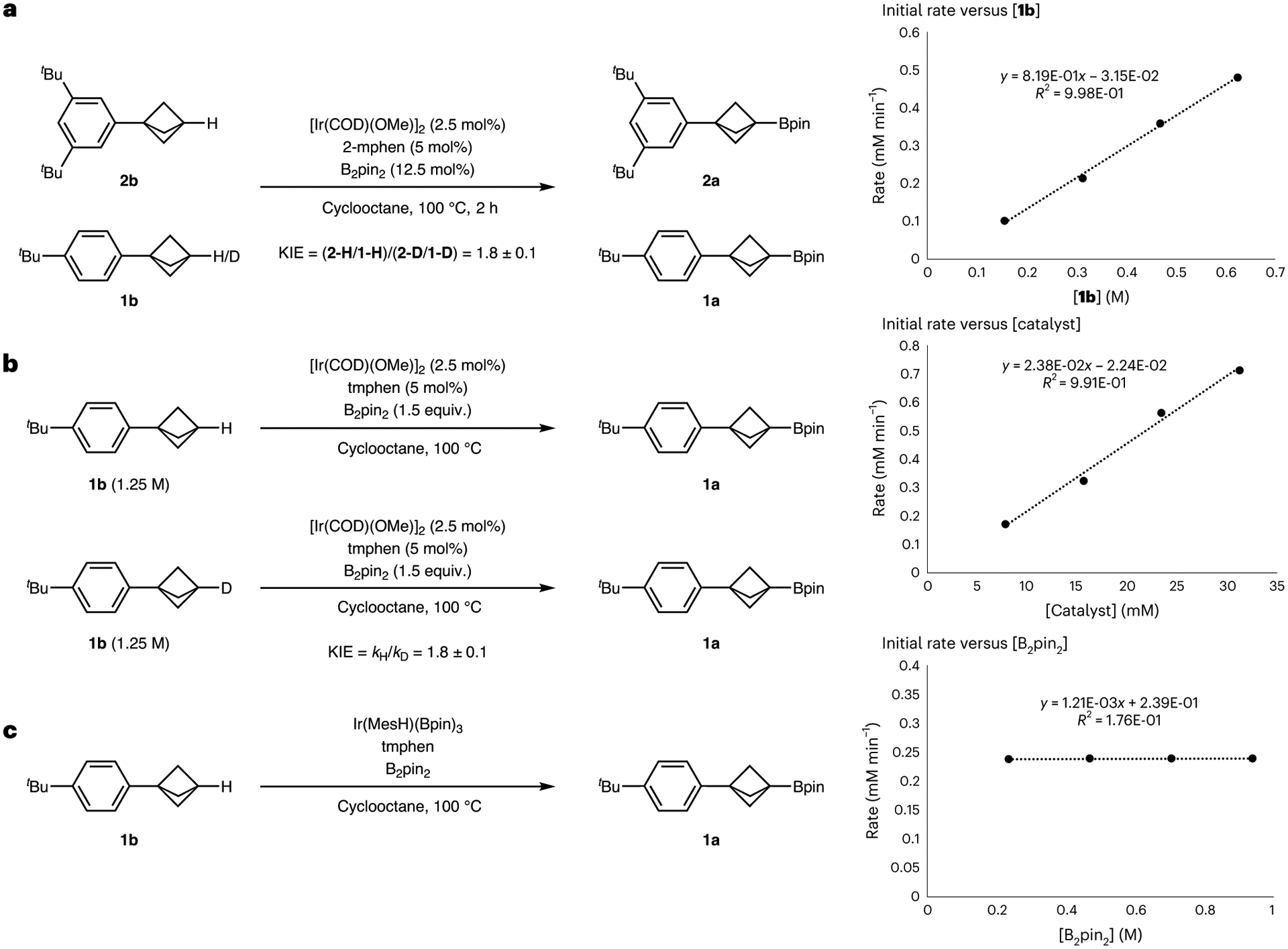
a, KIE obtained by competition experiments with 2-mphen as the ligand. The small magnitude of the KIE implies that cleavage of the C–H bond could be reversible. b, KIE obtained by measurement of rates in separate vessels with tmphen as the ligand. The small magnitude of the KIE implies that cleavage of the C–H bond may not be turnover limiting. c, Reaction orders were obtained by the method of initial rates. The borylation reaction is first order in the substrate, first order in the catalyst and zeroth order in the diboron reagent.
We conducted analogous rate measurements with tmphen as the ligand. The reaction with this ligand on iridium is slower than that with 2-mphen, but it occurred without a significant induction period, thereby enabling us to obtain clear kinetic data. The KIE value obtained from the initial rates of separate reactions initiated with tmphen as the ancillary ligand and either tert-butylphenylbicyclopentane (1b) or tert-butylphenylbicyclopentane-d1 (1b-d1) was, again, 1.8 ± 0.1 (Fig. 4b). The reaction with this catalyst was found to be first order in the BCP, first order in the catalyst and zeroth order in the diboron reagent (Fig. 4c). These results imply that the BCP and the iridium catalyst are involved in the turnover-limiting step, but the low KIE value and the lack of correlation between the 1JC–H coupling constants and rates of reaction of the BCPs suggest that oxidative addition is not likely to be turnover limiting and is likely to be reversible.
To understand the low KIE value and to determine the turnover-limiting step of the C–H borylation process, we conducted density functional theory (DFT) calculations on the borylation of unsubstituted bicyclopentane as a model substrate with tmphen as the ligand on iridium. The computed energy diagram for the reaction process is shown in Fig. 5. These calculations predict that oxidative addition of bicyclopentane (R–H) to the trisboryl species (tmphen)Ir(Bpin)3 ([Ir](Bpin)3) will be reversible with a barrier of 23.3 kcal mol−1 to form intermediate [Ir](Bpin)3(R)(H). This intermediate would then undergo an isomerization with a barrier of 17.9 kcal mol−1 that lies 30.1 kcal mol−1 above the starting complex and free substrate to form intermediate iso-[Ir](Bpin)3(R)(H). Reductive elimination from iso-[Ir](Bpin)3(R) (H) occurs with a barrier of just 3.6 kcal mol−1 to afford the product. Turnover with B2pin2 then regenerates [Ir](Bpin)3. The KIE value of the reaction by this path was computed to be 2.03 for bicyclopentane as the substrate, which is in good agreement with the experimental value of 1.8 for 4-tert-butylphenylbicyclopentane. A discussion of pathways involving Ir(I) species or alternative isomerization pathways is included in Supplementary Section 7. The higher computed barrier for isomerization following C–H bond cleavage is likely to result from the high s-character of the bridgehead position leading to more facile oxidative addition of the C–H bond to the iridium centre. As the isomerization step is less sensitive to the identity of the alkyl group, this step becomes turnover limiting. Similar conclusions have been drawn and overall barriers obtained from computational studies on the borylation of chlorosilanes and benzylic C–H bonds53,54.
Fig. 5 |. DFT studies.
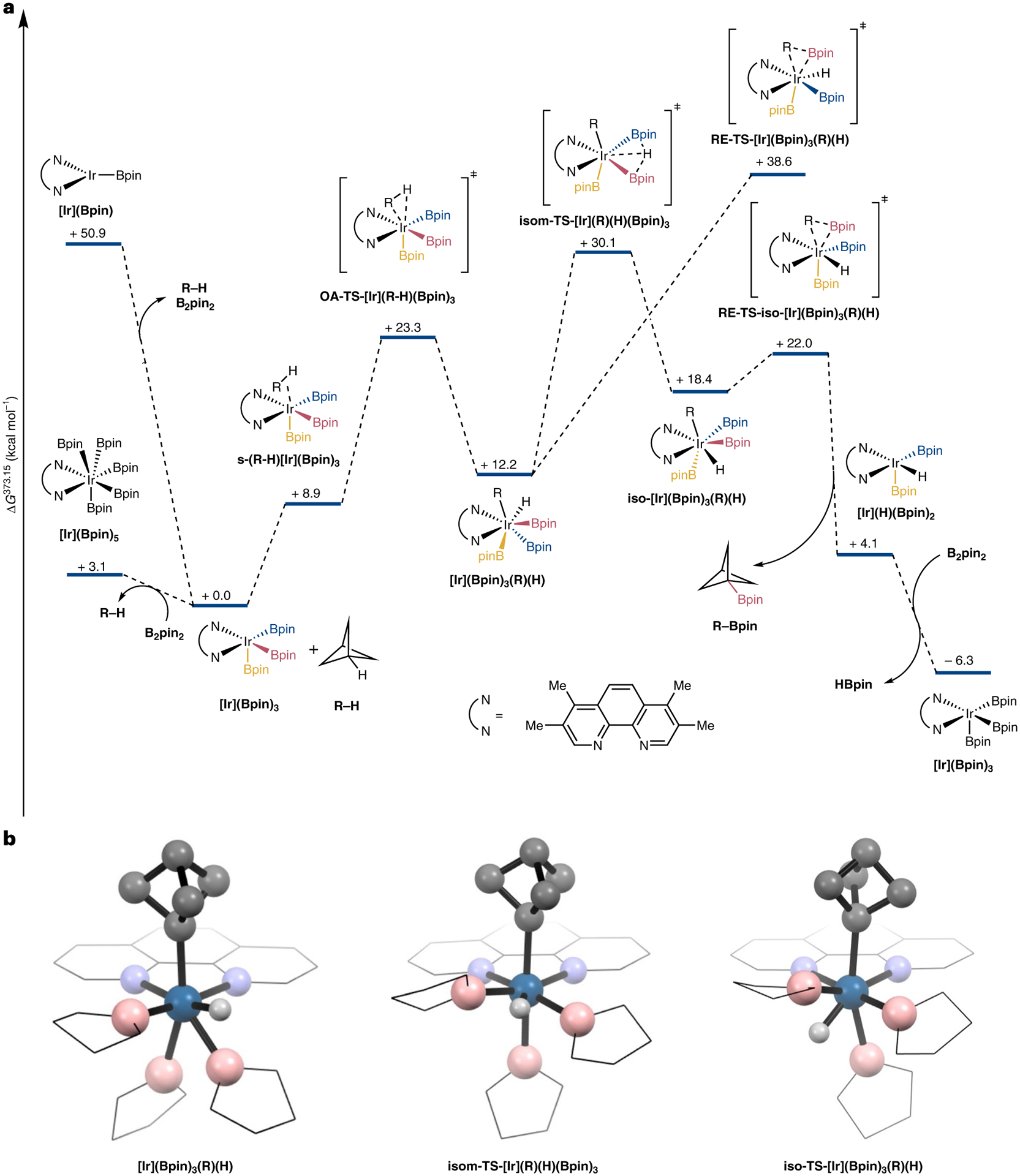
a, Energy diagram obtained by calculations using DFT. The resting state of the catalyst, [Ir](Bpin)3, engages the substrate and undergoes reversible oxidative addition of the tertiary C–H bond to generate [Ir](Bpin)3(R) (H). This initial product from oxidative addition undergoes rate-limiting isomerization to afford iso-[Ir](Bpin)3(R)(H). Facile reductive elimination occurs from iso-[Ir](Bpin)3(R)(H) to form the product. Energies are given in kcal mol−1 at 373.15 K. See Supplementary Section 8 for computational details and structures. Boryl groups have been colour coded for clarity. b, Ball-and-stick structures of the key isomerization process. For clarity, peripheral methyl groups and hydrogen atoms have been omitted and the phenanthroline ligand and boryl groups are represented as wireframes. The geometry of [Ir](Bpin)3(R) (H) is roughly pentagonal bipyramidal, in which one nitrogen of the tmphen ligand and the boryl ligand that later undergoes reductive elimination occupy axial positions. The geometry of iso-[Ir](Bpin)3(R)(H) is also roughly pentagonal bipyramidal, in which the other nitrogen of the tmphen ligand and a boryl ligand now occupy axial positions. The bicyclopenyl group and the boryl ligand, which are poised to undergo reductive elimination, lie in the equatorial plane. isom-TS refers to the transition state for isomerization.
Based on our experimental and computational data, we propose that the borylation of BCPs occurs by initial, reversible oxidative addition of the bridgehead C–H bond to (tmphen)Ir(Bpin)3 to generate the seven-coordinate Ir(V) species (tmphen)Ir(R)(H)(Bpin)3. Turnover-limiting isomerization of this intermediate leads to the seven-coordinate intermediate iso-(tmphen)Ir(R)(H)(Bpin)3, from which facile reductive elimination occurs to afford (tmphen)Ir(H) (Bpin)2 and the borylated product. The catalyst is then regenerated by reaction between (tmphen)Ir(H)(Bpin)2 and B2pin2 with HBpin as the by-product.
Conclusion
The undirected borylation of C–H bonds, which is known to occur at aryl, primary alkyl and some secondary alkyl C–H bonds, now has been shown to occur at a tertiary C–H bond. Although tertiary C–H bonds are typically inert under the conditions for the borylation of C–H bonds, the high strain in BCPs, BCHs and XBCHs and the resulting sp2 character of these C–H bonds lead to mild activation and functionalization, despite the steric hindrance of a tertiary position. This reactivity enables the synthesis of BCP, BCH and XBCH structures via the undirected borylation of the bridgehead C–H bonds and provides a direct route to these valuable building blocks from commercial or readily accessible reactants. As a result, this method tolerates a wide variety of functional groups of relevance to drug discovery and can be applied to the late-stage elaboration of complex structures containing these strained units. Mechanistic and computational studies show that the rate of reaction is relatively independent of the electronic properties of substituents on the BCP structures because the turnover-limiting step of the reaction is isomerization of the intermediate, seven-coordinate Ir complex, rather than C–H bond cleavage. Mechanistic insights gained in this study and the demonstration that a tertiary C–H bond can undergo undirected C–H bond functionalization by chemistry known to occur without radical intermediates should help guide future studies at hindered and typically unreactive alkyl C–H bonds.
Methods
In a nitrogen-filled glovebox, a 4 ml vial was sequentially charged with 2-mphen (2.43 mg, 12.5 μmol, 5.00 mol%), [Ir(COD)(OMe)]2 (6.25 μmol, 2.50 mol%), substrate (0.250 mmol, 1.00 equiv.) and B2pin2 (95.2 mg, 0.375 mmol, 1.50 equiv.); if the substrate was a liquid, it was added last. A magnetic stir bar and cyclooctane (0.2 ml) were added to the vial. The vial was then tightly sealed with a Teflon-lined cap. The vial was brought out of the glovebox and heated at 100 °C in a preheated aluminum heating block for the specified time. After cooling to ambient temperature, CDCl3 and CH2Br2 (internal standard) were added to the vial and a sample was taken and analysed by 1H NMR spectroscopy to quantify the conversion. The mixture was then co-evaporated 3 times with MeOH (5 ml) at 45 °C (Caution: vigorous gas evolution upon addition of MeOH!). The crude residue was purified by flash column chromatography (silica or C18 reverse phase) to give the borylated product.
Supplementary Material
Acknowledgements
This work was supported by the NIGMS of the NIH under R35GM130387. I.F.Y. and J.L.M. gratefully acknowledge the National Science Foundation Graduate Research Fellowship Program and the UC Berkeley Graduate Research Fellowship Program for support under DGE 1752814 and DGE 2146752. We thank H. Celik and A. Lund and UC Berkeley’s NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance, in particular for assistance and advice in acquiring J-resolved spectra. Instruments in the CoC-NMR are supported in part by NIH S10OD024998. We thank K. Durkin and D. Small and UC Berkeley’s Molecular Graphics and Computation Facility (MGCF) for assistance and resources for the computations. The MGCF is supported in part by NIH S10OD023532. We thank the QB3/Chemistry Mass Spectrometry Facility for assistance in obtaining high-resolution mass spectrometry data. We wish to thank T. W. Butcher and E. D. Kalkman for helpful discussions.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41557-023-01159-4.
Competing interests
A.E.P., P.K.M., S.V.R. and D.M.V. are employees of Enamine, which is a chemical supplier of reagents used in the studies in this paper. All other authors declare no competing interests.
Supplementary information
The online version contains supplementary material available at https://doi.org/10.1038/s41557-023-01159-4.
Data availability
Complete experimental procedures, computational details and compound characterization data are available in the Supplementary Information. Atomic coordinates of optimized structures are available as Supplementary Data 1.
References
- 1.Liskey CW & Hartwig JF Iridium-catalyzed borylation of secondary C–H Bonds in cyclic ethers. J. Am. Chem. Soc 134, 12422–12425 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Liskey CW & Hartwig JF Iridium-catalyzed C–H borylation of cyclopropanes. J. Am. Chem. Soc 135, 3375–3378 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Li Q, Liskey CW & Hartwig JF Regioselective borylation of the C–H bonds in alkylamines and alkyl ethers. Observation and origin of high reactivity of primary C–H bonds beta to nitrogen and oxygen. J. Am. Chem. Soc 136, 8755–8765 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Jones MR, Fast CD & Schley ND Iridium-catalyzed sp3 C–H borylation in hydrocarbon solvent enabled by 2,2′-dipyridylarylmethane ligands. J. Am. Chem. Soc 142, 6488–6492 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Oeschger R et al. Diverse functionalization of strong alkyl C–H bonds by undirected borylation. Science 368, 736 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kawazu R, Torigoe T & Kuninobu Y Iridium-catalyzed C(sp3)–H borylation using silyl-bipyridine pincer ligands. Angew. Chem. Int. Ed 10.1002/anie.202202327 [DOI] [PubMed] [Google Scholar]
- 7.Shu C, Noble A & Aggarwal VK Metal-free photoinduced C(sp3)–H borylation of alkanes. Nature 586, 714–719 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Lovering F, Bikker J & Humblet C Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem 52, 6752–6756 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Stepan AF et al. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem 55, 3414–3424 (2012). [DOI] [PubMed] [Google Scholar]
- 10.Measom ND et al. Investigation of a bicyclo[1.1.1]pentane as a phenyl replacement within an LpPLA2 inhibitor. ACS Med. Chem. Lett 8, 43–48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Auberson YP et al. Improving nonspecific binding and solubility: bicycloalkyl groups and cubanes as para-phenyl bioisosteres. ChemMedChem 12, 590–598 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Pu Q et al. Discovery of potent and orally available bicyclo[1.1.1] pentane-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors. ACS Med. Chem. Lett 11, 1548–1554 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makarov IS, Brocklehurst CE, Karaghiosoff K, Koch G & Knochel P Synthesis of bicyclo[1.1.1]pentane bioisosteres of internal alkynes and para-disubstituted benzenes from [1.1.1] propellane. Angew. Chem. Int. Ed 56, 12774–12777 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Barbachyn MR et al. U-87947E, a protein quinolone antibacterial agent incorporating a bicyclo[1.1.1]pent-1-yl (BCP) subunit. Bioorg. Med. Chem. Lett 3, 671–676 (1993). [Google Scholar]
- 15.Westphal MV, Wolfstädter BT, Plancher J-M, Gatfield J & Carreira EM Evaluation of tert-butyl isosteres: case studies of physicochemical and pharmacokinetic properties, efficacies, and activities. ChemMedChem 10, 461–469 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Denisenko A, Garbuz P, Shishkina SV, Voloshchuk NM & Mykhailiuk PK Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem. Int. Ed 59, 20515–20521 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Levterov VV, Panasyuk Y, Pivnytska VO & Mykhailiuk PK Water-soluble non-classical benzene mimetics. Angew. Chem. Int. Ed 59, 7161–7167 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Levterov VV et al. Photochemical in-flow synthesis of 2,4-methanopyrrolidines: pyrrolidine analogues with improved water solubility and reduced lipophilicity. J. Org. Chem 83, 14350–14361 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Homon AA et al. 4-(Di-/Trifluoromethyl)-2-heterabicyclo[2.1.1] hexanes: advanced fluorinated phenyl isosteres and proline analogues. Eur. J. Org. Chem 2021, 6580–6590 (2021). [Google Scholar]
- 20.Wiberg KB, Waddell ST & Laidig K [1.1.1]Propellane: Reaction with free radicals. Tetrahedron Lett 27, 1553–1556 (1986). [Google Scholar]
- 21.Kaszynski P & Michl J A practical photochemical synthesis of bicyclo [1.1.1] pentane-1, 3-dicarboxylic acid. J. Org. Chem 53, 4593–4594 (1988). [Google Scholar]
- 22.Wiberg KB & Waddell ST Reactions of [1.1.1] propellane. J. Am. Chem. Soc 112, 2194–2216 (1990). [Google Scholar]
- 23.Bunker KD, Sach NW, Huang Q & Richardson PF Scalable synthesis of 1-bicyclo[1.1.1]pentylamine via a hydrohydrazination reaction. Org. Lett 13, 4746–4748 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Kanazawa J, Maeda K & Uchiyama M Radical multicomponent carboamination of [1.1.1]propellane. J. Am. Chem. Soc 139, 17791–17794 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Caputo DFJ et al. Synthesis and applications of highly functionalized 1-halo-3-substituted bicyclo[1.1.1]pentanes. Chem. Sci 9, 5295–5300 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bär RM, Kirschner S, Nieger M & Bräse S Alkyl and aryl thiol addition to [1.1.1]propellane: scope and limitations of a fast conjugation reaction. Chem. Eur. J 24, 1373–1382 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Nugent J et al. A general route to bicyclo[1.1.1]pentanes through photoredox catalysis. ACS Catal. 9, 9568–9574 (2019). [Google Scholar]
- 28.Zhang X et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim JH, Ruffoni A, Al-Faiyz YSS, Sheikh NS & Leonori D Divergent strain-release amino-functionalization of [1.1.1] propellane with electrophilic nitrogen-radicals. Angew. Chem. Int. Ed 59, 8225–8231 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shin S, Lee S, Choi W, Kim N & Hong S Visible-light-induced 1,3-aminopyridylation of [1.1.1]propellane with N-aminopyridinium salts. Angew. Chem. Int. Ed 60, 7873–7879 (2021). [DOI] [PubMed] [Google Scholar]
- 31.Gianatassio R et al. Strain-release amination. Science 351, 241–246 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopchuk JM et al. Strain-release heteroatom functionalization: development, scope, and stereospecificity. J. Am. Chem. Soc 139, 3209–3226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shelp RA & Walsh PJ Synthesis of BCP benzylamines from 2-azaallyl anions and [1.1.1]propellane. Angew. Chem. Int. Ed 57, 15857–15861 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Hughes JME, Scarlata DA, Chen ACY, Burch JD & Gleason JL Aminoalkylation of [1.1.1]propellane enables direct access to high-value 3-alkylbicyclo[1.1.1]pentan-1-amines. Org. Lett 21, 6800–6804 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Trongsiriwat N et al. Reactions of 2-aryl-1,3-dithianes and [1.1.1] propellane. Angew. Chem. Int. Ed 58, 13416–13420 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu S, Jing C, Noble A & Aggarwal VK 1,3-Difunctionalizations of [1.1.1]propellane via 1,2-metallate rearrangements of boronate complexes. Angew. Chem. Int. Ed 59, 3917–3921 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Schwärzer K, Zipse H, Karaghiosoff K & Knochel P Highly regioselective addition of allylic zinc halides and various zinc enolates to [1.1.1]propellane. Angew. Chem. Int. Ed 59, 20235–20241 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garlets ZJ et al. Enantioselective C–H functionalization of bicyclo[1.1.1]pentanes. Nat. Catal 3, 351–357 (2020). [Google Scholar]
- 39.Fawcett A et al. Photoinduced decarboxylative borylation of carboxylic acids. Science 357, 283–286 (2017). [DOI] [PubMed] [Google Scholar]
- 40.VanHeyst MD et al. Continuous flow-enabled synthesis of bench-stable bicyclo[1.1.1]pentane trifluoroborate salts and their utilization in metallaphotoredox cross-couplings. Org. Lett 22, 1648–1654 (2020). [DOI] [PubMed] [Google Scholar]
- 41.Kondo M et al. Silaboration of [1.1.1]propellane: a storable feedstock for bicyclo[1.1.1]pentane derivatives. Angew. Chem. Int. Ed 59, 1970–1974 (2020). [DOI] [PubMed] [Google Scholar]
- 42.Yang Y et al. An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nat. Chem 13, 950–955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shelp RA et al. Strain-release 2-azaallyl anion addition/borylation of [1.1.1]propellane: synthesis and functionalization of benzylamine bicyclo[1.1.1]pentyl boronates. Chem. Sci 12, 7066–7072 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu S et al. Palladium-catalyzed stagewise strain-release-driven C–C activation of bicyclo[1.1.1]pentanyl alcohols. Angew. Chem. Int. Ed 10.1002/anie.202200052 [DOI] [PubMed] [Google Scholar]
- 45.Hoque ME, Hassan MMM & Chattopadhyay B Remarkably efficient iridium catalysts for directed C(sp2)–H and C(sp3)–H borylation of diverse classes of substrates. J. Am. Chem. Soc 143, 5022–5037 (2021). [DOI] [PubMed] [Google Scholar]
- 46.Ishiyama T et al. Mild iridium-catalyzed borylation of arenes. High turnover numbers, room temperature reactions, and isolation of a potential intermediate. J. Am. Chem. Soc 124, 390–391 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Hinkes SPA & Klein CDP Virtues of volatility: a facile transesterification approach to boronic acids. Org. Lett 21, 3048–3052 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Yang Y et al. Practical and modular construction of C(sp3)-rich alkyl boron compounds. J. Am. Chem. Soc 143, 471–480 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Matteson DS & Kim GY Asymmetric alkyldifluoroboranes and their use in secondary amine synthesis. Org. Lett 4, 2153–2155 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Tian Z & Kass SR Carbanions in the gas phase. Chem. Rev 113, 6986–7010 (2013). [DOI] [PubMed] [Google Scholar]
- 51.Boller TM et al. Mechanism of the mild functionalization of arenes by diboron reagents catalyzed by iridium complexes. Intermediacy and chemistry of bipyridine-ligated iridium trisboryl complexes. J. Am. Chem. Soc 127, 14263–14278 (2005). [DOI] [PubMed] [Google Scholar]
- 52.Zhong R-L & Sakaki S sp3 C–H borylation catalyzed by iridium(III) triboryl complex: comprehensive theoretical study of reactivity, regioselectivity, and prediction of excellent ligand. J. Am. Chem. Soc 141, 9854–9866 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Larsen MA, Wilson CV & Hartwig JF Iridium-catalyzed borylation of primary benzylic C–H bonds without a directing group: scope, mechanism, and origins of selectivity. J. Am. Chem. Soc 137, 8633–8643 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Huang G, Kalek M, Liao R-Z & Himo F Mechanism, reactivity, and selectivity of the iridium-catalyzed C(sp3)-H borylation of chlorosilanes. Chem. Sci 6, 1735–1746 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete experimental procedures, computational details and compound characterization data are available in the Supplementary Information. Atomic coordinates of optimized structures are available as Supplementary Data 1.