

Abstract
In the future, zoonotic spillover events are expected to occur more frequently. Consequences of such events have clearly been demonstrated by recent outbreaks of monkeypox, Ebola virus, and the well-known severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Virus discovery has proven to be an important tool in the preparation against viral outbreaks, generating data concerning the diversity, quantity, and ecology of the vertebrate virome. Orthoparamyxoviruses, a subfamily within the Paramyxoviridae, are important biosurveillance targets, since they include several known animal, human, and zoonotic pathogens such as Nipah virus, measles virus, and Hendra virus. During this study, 127 bat samples, thirty-four rodent samples, and seventeen shrew samples originating from Belgium were screened for orthoparamyxovirus presence using nested reverse transcription–polymerase chain reaction assays and nanopore sequencing. We present here the complete genomes of six putative new viral species, belonging to the genera Jeilongvirus and Henipavirus. Characterization of these genomes revealed significant differences in gene composition and organization, both within viruses of the same genus and between viruses of different genera. Remarkably, a previously undetected gene coding for a protein of unknown function was identified in the genome of a putative new Henipavirus. Additionally, phylogenetic analysis of jeilongviruses and henipaviruses reveals a division of both genera into two clades, one consisting of bat-borne viruses and the other consisting of rodent- and shrew-borne viruses, elucidating the need for proper reclassification.
Keywords: virus discovery, nanopore sequencing, Henipavirus, Jeilongvirus
Introduction
Paramyxoviridae is a viral family belonging to the order Mononegavirales, consisting of four subfamilies, of which Orthoparamyxovirinae is the largest. Orthoparamyxoviruses include several important zoonotic, human, and animal pathogens such as Nipah virus (NiV), Measles virus (MeV), and Hendra virus (HeV) (Rima et al. 2019). Recently, a new zoonotic orthoparamyxovirus belonging to the genus Henipavirus was discovered in eastern China: Langya virus (LayV). Between April 2018 and August 2021, thirty-five infected patients were identified with symptoms ranging from cough, fatigue, and mild fever to severe pneumonia and loss of liver and kidney functions (Zhang et al. 2022). Although no human-to-human transmission has been reported yet, vigilance is recommended: multiple animal species such as shrews, goats, and dogs were identified as LayV hosts, highlighting the diverse opportunities for this virus to mutate and thus potentially adapt infection or transmission pathways over time (Taseen et al. 2023).
In the future, zoonotic spillover events are predicted to occur more frequently. Due to changes in human behavior, such as growing bushmeat markets, deforestation, and agricultural development, the frequency and intensity of contact with (potentially) infected animals will increase, and as a consequence, transmission to humans will be facilitated (Epstein and Anthony 2017). Performing more thorough virus discovery yields data concerning the diversity, quantity, and ecology of the vertebrate virome, therefore providing useful information to reduce the consequences of, or even fully prevent, future outbreaks (Epstein and Anthony 2017; Carroll et al. 2018). Orthoparamyxoviruses in particular are important targets for virus discovery: due to improvements in sequencing techniques, this subfamily has expanded greatly over the last few years, exposing a substantial lack of knowledge concerning these viruses. Furthermore, orthoparamyxoviruses can be found in a wide range of host species, e.g. shrews, rodents, and bats, and several zoonoses have already been identified within this subfamily (Zhu et al. 2022).
As members of a subfamily within the family Paramyxoviridae, orthoparamyxoviruses form pleomorphic, enveloped particles with single-stranded, non-segmented, negative-sense ribonucleic acid (RNA) genomes that can range in length from 14,296 nucleotides (Antarctic penguin virus B) to >21,523 nucleotides (Wenzhou pacific spadenose shark paramyxovirus) (Rima et al. 2019). For efficient replication, paramyxovirus genome lengths must be multiples of six (Calain and Roux 1993). Viruses belonging to this family generally share a similar genome organization that can be described as 3ʹ-N-P/V/C-M-F-RBP-L-5ʹ, with the different genes encoding six to ten different proteins (Rima et al. 2019). The fusion proteins (F) and receptor-binding proteins (RBPs) are two types of transmembrane glycoproteins responsible for host cell entry and receptor binding, respectively. The nucleocapsid protein (N), phosphoprotein (P), and large protein (L) form the nucleocapsid and are associated with the RNA genome inside the virus particle. The membrane-associated matrix protein (M) is responsible for assembling the virus particle by interacting with both the RNA-associated N protein and the transmembrane glycoproteins. Some paramyxoviruses can also encode for supplementary proteins such as a non-structural protein (C), a cysteine-rich Zn2+-binding protein (V), a transmembrane protein (TM), or a small integral membrane protein (SH) (Cox and Plemper 2017; Rima et al. 2019).
In this study, we screened 127 bats, thirty-four rodents, and seventeen shrews originating from Belgium for the presence of orthoparamyxoviruses by performing nested reverse transcription–polymerase chain reactions (RT-PCRs) and nanopore sequencing. Following detection of new viruses, attempts were made to completely assemble and characterize their genome sequences. In total, nine different orthoparamyxoviruses were detected during this study, of which six putatively represent new species belonging to the genera Henipavirus and Jeilongvirus. A complete genome sequence was determined for each of these six viruses.
Materials and methods
Ethical statement
Cadavers of bats, shrews, and rodents were used for the conducted experiments. Approval by ‘Agentschap voor Natuur en Bos’ was given to use the cadavers of the protected species (Approval No. ANB/BL/FF-V13-00172).
Sample collection and dissection
Two sample sets were investigated for the realization of this study. The first sample set consisted of 127 Belgian bats that were dead on arrival when brought to bird rescue centers or later euthanized due to severe injuries. Following dissection, kidneys were sent to the Laboratory of Clinical and Epidemiological Virology of the KU Leuven for further analysis. Exact species determination was carried out by amplifying and sequencing a part of the mitochondrial cytochrome b gene using primers developed by Schlegel et al. (2012). The second sample set consisted of thirty-four Belgian rodents and seventeen Belgian shrews that were killed by cats or vehicles. Organs of the cadavers were removed during dissection and treated with RNAlater (ThermoFisher Scientific, Waltham, MA, USA) for 24 h at 4°C, whereafter they were stored at −80°C. Determination of host species was carried out by assembling the nanopore sequencing data corresponding to the mitochondrial cytochrome b gene.
RNA extraction without viral enrichment
A total RNA extraction without viral enrichment was performed on kidneys of the 127 bat samples using the RNeasy Mini Kit (Qiagen, Hilden, Germany). Initially, ∼20 mg tissue was placed in a 2-ml tube containing five zirconium oxide beads (diameter 2.8 mm) (Bertin Technologies, Montigny-le-Bretonneux, France) and 100 µl phosphate-buffered saline was added. The sample was homogenized using the Bertin Minilys Homogenizer (Bertin Technologies) at 4,000 rotations per minute for 120 s. Six-hundred-microliter RNeasy Lysis Buffer RLT supplemented with 2 M dithiothreitol (DTT) was added to the homogenate, after which it was transferred to a new 1.5-ml tube and centrifuged at 17,000 × g for 3 min. Thereafter, the supernatant was transferred to a new 2-ml tube, 700 µl 70 per cent ethanol was added, and the complete volume was loaded onto an RNeasy Mini spin column. Further steps were performed as described in the RNeasy Mini Kit protocol. The extracted RNA was stored at −80°C.
Nested RT-PCR
A nested RT-PCR reaction was performed on the RNA extracts of bat, rodent, and shrew samples in order to determine the presence of orthoparamyxoviruses, using the OneStep RT-PCR Kit (Qiagen). Initially, 5 µl 5× OneStep RT-PCR Buffer, 10 nmol deoxynucleotide triphosphate mix, 30 pmol forward primer (5ʹ-ATGATGAARGGNCATGC-3ʹ) and reverse primer (5ʹ-GCYTTRTCYTTCATRTACAT-3ʹ), 1 µl OneStep RT-PCR Enzyme Mix, and 2 µl RNA extract and RNase-free water were added to a 0.2-ml tube to obtain a total volume of 25 µl. The following cycling conditions were used for the outer nested PCR: 30 min at 50°C, 15 min at 95°C, forty cycles of 30 s at 94°C, 30 s at 47°C, 1 min at 72°C, and finally 10 min at 72°C. Thereafter, the reaction was repeated using 2 µl outer nested RT-PCR product and inner forward primer (5ʹ-AARGGNCATGCHHTNTTCTG-3ʹ) and reverse primer (5ʹ-TTCATRTACATDGTNAGRTC-3ʹ) with identical cycling conditions minus the initial reverse transcription step of 30 min at 50°C. Results were visualized by agarose gel electrophoresis, whereafter positive samples were purified using ExoSAP-IT Express PCR Product Cleanup (ThermoFisher Scientific) and sent to Macrogen Europe (Amsterdam, the Netherlands) for Sanger sequencing. Retrieved sequences were trimmed using Chromas (v2.6.6) and joined with SeqMan (v7.0.0).
RNA extraction with viral enrichment and sequence-independent, single-primer amplification
In order to extract viral RNA, an RNA extraction with relative viral RNA enrichment was performed on kidney tissue of bat samples positive for paramyxoviruses and all rodent and shrew samples as described by Vanmechelen et al. (2022). Thereafter, with the purpose of converting viral RNA of all extracted samples to copy deoxyribonucleic acid (cDNA), sequence-independent, single-primer amplification (SISPA) was performed as described by Vanmechelen et al. (2022). The following primers were used: sol-primerA (40 pmol/µl, 5′-GTTTCCCACTGGAGGATA-N9-3′) and sol-primerB (100 pmol/µl, 5′-GTTTCCCACTGGAGGATA-3′) (Greninger et al. 2015).
Nanopore sequencing
Library preparation was performed using the Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies (ONT), Oxford, UK), as described in the manufacturer’s protocol. Multiplex sequencing was done using the Native Barcoding Expansion 1-12 Kit (EXP-NBD104, ONT), and clean-ups between steps were carried out using 0.4× AMPure XP beads (Beckman Coulter). Two hundred nanograms of cDNA per sample was used as initial input. On an FLO-MIN106 flow cell, approximately 120 fmol library was loaded and sequencing was carried out on a GridION X5 Mk1 device. Basecalling and demultiplexing were performed using the MinKNOW software (v22.03.02) on the GridION device. Thereafter, orthoparamyxovirus reads were detected by running a tBLASTx search (e-value = 1e-20) against a self-composed database containing all RefSeq orthoparamyxovirus sequences (retrieved from the National Center for Biotechnology Information (NCBI) Nucleotide database on 30 March 2022). Porechop (v0.2.4) (Wick et al. 2017) was used to remove sequencing adapters and sol-primer sequences added during SISPA.
Complete genome assembly of new viruses
Orthoparamyxovirus reads were assembled into contigs using Canu (v2.0) (minimum read length = 100 and minimum overlap length = 50) (Koren et al. 2017). Retrieved contigs were corrected and connected using CLC Genomics Workbench (v20.0.5) to obtain complete viral genomes. If genome endings were missing, rapid amplification of cDNA ends (RACE) was performed using the 5ʹ/3ʹ RACE Kit, 2nd Generation (Roche, Basel, Switzerland) according to the manufacturer’s instructions, with adapted oligo d(T)-anchor primers. A list of used primers can be found in Table 1. Results were visualized by agarose gel electrophoresis, whereafter samples were purified using ExoSAP-IT Express PCR Product Cleanup (ThermoFisher Scientific) and sent away to Macrogen Europe for Sanger sequencing. Retrieved sequences were trimmed using Chromas (v2.6.6), and genomes were completed using SeqMan (v7.0.0).
Table 1.
Primers used for the 5ʹ/3ʹ RACE.
| Virus | Primer | Sequence (5ʹ-…-3ʹ) |
|---|---|---|
| PipaV | SP1 | TACTGCATTTTTTGACCCGTAC |
| SP2 | GACATCAACAAGAAAAAGAAATGAGG | |
| SP3 | GGTCTCAAAGAGGTATCAACAATACC | |
| SP4 | GAGTGAAGAAGGCTGCAG | |
| SP5 | GGATCCAGGGGACGCTGATG | |
| NinYsV | SP1 | GGGTATGCTTTAAATCTGACTTGG |
| SP2 | CCAGGATTATCTTCTTAGGAGATT | |
| SP3 | ACCTGGTCATACGAATTGTCCAC | |
| SP4 | TTTTCTTGGGTCTTTCATTA | |
| SP5 | GCAATTGAGTTGATAACCCTCCC | |
| General | 5ʹ oligo d(G)-anchor | GACCACGCGTATCGATGTCGACGGGGGGGGGGGGGGACC |
| 3ʹ oligo d(T)-anchor | GACCACGCGTATCGATGTCGACTTTTTTTTTTTTACC | |
| PCR anchor | GACCACGCGTATCGATGTCGAC |
Phylogenetic analysis
Open reading frames (ORFs) of new viruses were determined using the NCBI Open Reading Frame Finder (https://www.ncbi.nlm.nih.gov/orffinder/). Representative sequences of all (putative) orthoparamyxovirus species were retrieved from the NCBI Nucleotide database for the composition of a Bayesian phylogenetic tree. Alignments of protein sequences (N, P, M, F, RBP, and L) of all selected viruses were created using MAFFT (auto settings) (v7.489) (Katoh, Rozewicki, and Yamada 2019) and trimmed using trimAl (‘gappyout’ mode) (v1.4.rev15) (Capella-Gutiérrez, Silla-Martínez, and Gabaldón 2009). Sequences were concatenated, an XML file was generated using BEAUti (v1.10.4) (Substitution Model = LG, Site Heterogeneity Model = Gamma + Invariant Sites), and phylogenetic trees were created using BEAST (v1.10.4) (Drummond and Rambaut 2007). Thereafter, this generated information was summarized into a single phylogenetic tree using TreeAnnotator (v1.10.4) with a burn-in of 10 per cent. Finally, the Bayesian phylogenetic tree was visualized using FigTree (v1.4.4). Comparison of paramyxovirus identities was carried out with the NIH Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Results
Host determination and paramyxovirus screening
Kidney tissue samples of 127 bats, thirty-four rodents, and seventeen shrews, all captured in Belgium, were screened during this study. Initially, bat host determination was performed by amplifying and sequencing a part of the mitochondrial cytochrome b gene, which resulted in the identification of six different species. Six rodent species and two shrew species were identified by retrieving the corresponding cytochrome b gene sequences from the nanopore sequencing data (Table 2). Paramyxovirus screening was performed on the bat and rodent/shrew samples, respectively, by nested RT-PCRs and nanopore sequencing. In total, thirty-five samples were positive for orthoparamyxovirus presence, resulting in a positivity rate of 19.10 per cent. A significant difference was observed between the positivity rates of bat samples and rodent/shrew samples: 10 out of 127 bat samples were positive (7.87 per cent) compared to twenty-five out of fifty-one positive rodent/shrew samples (49 per cent) (Table 2) (Supplementary Table S1). Spread over thirty-five positive samples, nine putative orthoparamyxovirus species were detected, including six previously unknown ones. Two bat-borne viruses, five rodent-borne viruses, and two shrew-borne viruses were distinguished, respectively, in two out of six bat species, four out of six rodent species, and two out of two shrew species. The bat-borne viruses, both previously unknown, were named piparella virus (PipaV) and plecomyxo virus (PlecoV), based on their virus family (Paramyxoviridae) and respective host species (Pipistrellus pipistrellus and Plecotus auritus). Three new rodent-borne viruses and one new shrew-borne virus were named ninapo virus (NinaV), denotus virus (DenoV), ninomys virus (NinYsV), and ninorex virus (NinExV), based on their location of origin (Ninove or Denderwindeke, Belgium) and respective host species (Apodemus sylvaticus, Microtus arvalis, Micromys minutus, and Sorex minutus). Additionally, two rodent-borne viruses (denalis virus and Ninove microtus virus) and one shrew-borne virus (denwin virus) that had already been identified during preceding research by Vanmechelen et al. (2022) were also detected but are not included in the following discussion. Viruses found more than once were consistently detected in the same host species. One sample, with M. arvalis as host, was positive for two viruses (denalis virus and DenoV). An overview of detected viruses, their abbreviations, host species, putative genera, and frequency of detection can be found in Table 3. Results of the nanopore sequencing of the rodent/shrew sample set were afterward validated by nested RT-PCR assays (Supplementary Table S2), which found no discrepancies.
Table 2.
Detected host species and Paramyxovirus positivity rates.
| Order | Host species | Common name | Paramyxovirus positivity rate |
|---|---|---|---|
| Chiroptera | Pipistrellus pipistrellus | Common pipistrelle | 9/98 |
| Pipistrellus nathusii | Nathusius’ pipistrelle | 0/18 | |
| Plecotus auritus | Brown long-eared bat | 1/4 | |
| Eptesicus serotinus | Serotine bat | 0/3 | |
| Myotis mystacinus | Whiskered bat | 0/3 | |
| Nyctalus leisleri | Lesser noctule | 0/1 | |
| Eulipotyphla | Crocidura russula | Greater white-toothed shrew | 11/15 |
| Sorex minutus | Eurasian pygmy shrew | 1/2 | |
| Rodentia | Apodemus sylvaticus | Long-tailed field mouse | 8/18 |
| Microtus arvalis | Common vole | 3/9 | |
| Rattus norvegicus | Brown rat | 0/4 | |
| Micromys minutus | Harvest mouse | 1/1 | |
| Microtus agrestis | Short-tailed field vole | 1/1 | |
| Sciurus vulgaris | Red squirrel | 0/1 |
Table 3.
An overview of detected viruses.
| Virus | Abbreviation | Host species | Frequency of detection | Putative genus |
|---|---|---|---|---|
| Piparella virus | PipaV | Pipistrellus pipistrellus | 8 | Jeilongvirus |
| Plecomyxo virus | PlecoV | Plecotus auritus | 2 | Jeilongvirus |
| Denwin virusa | DewV | Crocidura russula | 11 | Henipavirus |
| Denalis virusa | DenaV | Microtus arvalis | 1 | Narmovirus |
| Ninove microtus virusa | NiMiV | Microtus agrestis | 1 | Jeilongvirus |
| Ninapo virus | NinaV | Apodemus sylvaticus | 8 | Jeilongvirus |
| Denotus virus | DenoV | Microtus arvalis | 3 | Jeilongvirus |
| Ninorex virus | NinExV | Sorex minutus | 1 | Henipavirus |
| Ninomys virus | NinYsV | Micromys minutus | 1 | Jeilongvirus |
These are known viruses, previously identified by Vanmechelen et al. (2022).
Phylogenetic analysis
A Bayesian phylogenetic tree of the Orthoparamyxovirinae subfamily was composed based on the amino acid sequences of N, P, M, F, RBP, and L proteins (Fig. 1). DenoV, NinaV, and NinYsV seem to cluster within the genus Jeilongvirus and more specifically within the clade of other rodent- and shrew-borne jeilongviruses. DenoV is most closely related to Ninove microtus virus (OK623355.1) (percentage identity 76.10 per cent), which was also detected during this study. NinaV and NinYsV are most closely related to rodent paramyxovirus (KY370098.1) (percentage identity 77.63 per cent) and Longquan Niviventer fulvescens jeilongviruses 1 (MZ328279.1) and 2 (MZ328280.1) (percentage identity 76.97 and 75.47 per cent), respectively. Furthermore, two other new jeilongviruses were detected during this research: PlecoV and PipaV. Contrarily, these viruses cluster within the bat-borne clade of this genus. PlecoV is most closely related to Wenzhou Myotis laniger paramyxovirus 2 (OM030336.1) and Wenzhou Myotis davidii paramyxovirus 1 (OM030335.1) (percentage identity 72.59 and 70.47 per cent), while PipaV is most closely related to Wufeng Rhinolophus pearsonii paramyxovirus 1 (MZ328290.1) and Wufeng Murina leucogaster paramyxovirus 1 (OM030337.1) (percentage identity 71.70 and 70.17 per cent). In addition to these five newly discovered jeilongviruses, one virus that seems to cluster within the genus Henipavirus was identified: NinExV. Similar to the genus Jeilongvirus, a division between rodent- and shrew-borne viruses and bat-borne viruses can be observed within the henipaviruses. As expected from the determined host species, NinExV clusters within the rodent- and shrew-borne clade. Although this virus is most closely related to Henipavirus sp. strain CI17-32 (MZ574407.1) (percentage identity 67.84 per cent), Jingmen Crocidura shantungensis henipavirus 1 (OM030314.1) (percentage identity 67.86 per cent), and Wufeng Crocidura attenuata henipavirus 1 (OM030317.1) (percentage identity 67.95 per cent), it is only distantly related to all viruses within this clade, forming a separate sister lineage.
Figure 1.
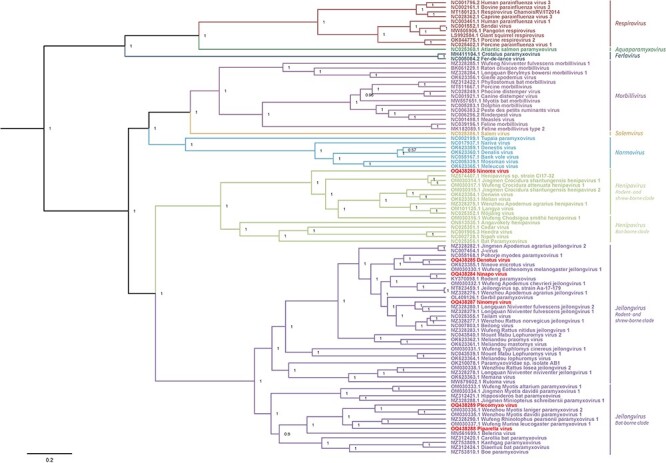
Bayesian phylogenetic tree of the Orthoparamyxovirinae subfamily. A Bayesian tree was composed based on inferred protein sequences (N, P, M, F, RBP, and L) of representative sequences of all (putative) orthoparamyxovirus species for which coding-complete genome sequences are available, retrieved from the NCBI Nucleotide database on 4 January 2023. Accession numbers of used viruses are shown in the tree. Genera are annotated next to the three according to the current classification by the International Committee on Taxonomy of Viruses (ICTV) (2019 release). Newly discovered viruses are annotated in bold . Numbers at node endings show posterior probabilities.
Genome assembly and characterization of five new (putative) jeilongviruses
According to the results of the phylogenetic analysis, five out of six newly determined viral species are proposed to be classified under the genus Jeilongvirus. Complete genome assembly of these new viruses was carried out through the processing of nanopore sequencing data and additional RACEs, after which ORFs were visualized using NCBI ORFfinder. The general paramyxovirus genome organization, 3ʹ-N-P/V/C-M-F-G-L-5ʹ, was recognized for all newly discovered jeilongviruses. However, some alterations were identified, with significant differences between the rodent- and bat-borne viruses. NinaV, DenoV, and NinYsV were, respectively, detected in host species A. sylvaticus, M. arvalis, and M. minutus and were therefore established as rodent-borne jeilongviruses. Their genomes were all characterized by an exceptionally large G ORF and additional ORFs for SH and TM proteins (Fig. 2). Due to these large G ORFs, genome lengths of these viruses are relatively long as well: the NinaV genome consists of 19,686 nucleotides, the DenoV genome of 20,502 nucleotides, and the NinYsV genome of 19,932 nucleotides. PipaV and PlecoV, on the other hand, were established as bat-borne jeilongviruses with respective host species P. pipistrellus and P. auritus. These viruses have significantly shorter genome lengths compared to the rodent-borne viruses (PipaV: 16,146 nucleotides and PlecoV: 16,038 nucleotides) due to the absence of exceptionally large G ORFs and SH ORFs. Additional ORFs for TM proteins, slightly larger than those in rodent-borne viruses, were observed (Fig. 2).
Figure 2.
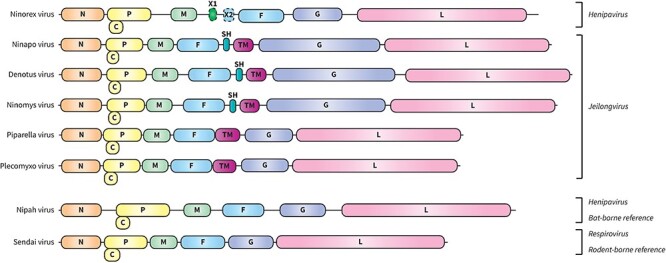
Genome organization of newly discovered orthoparamyxoviruses. All genomes are drawn to scale. Genome organizations of NiV and Sendai virus are also shown, as references of, respectively, bat- and rodent-borne orthoparamyxoviruses.
A comparison of the amino acid sequences of the TM proteins of all Jeilongvirus species used for our Bayesian phylogenetic tree was made (Supplementary Table S3). Between TM proteins of rodent- and shrew-borne viruses, a certain degree of conservation was observed: the highest percentage of similarity (97.5 per cent) was established between the TM proteins of Jeilongvirus sp. strain Aa-17-179 (MZ328282.1) and Wenzhou Apodemus agrarius jeilongvirus 2 (MZ328276.1), while the lowest percentage of similarity (4.4 per cent) was established between the TM proteins of Longquan Niviventer fulvescens jeilongvirus 2 (MZ328280.1) and ruloma virus (MW579602.1). Conservation between TM proteins of bat-borne viruses seems to be more limited, since similarity percentages are much lower in comparison with the rodent- and shrew-borne viruses: the highest percentage of similarity (69.2 per cent) was established between the TM proteins of Kanhgag paramyxovirus (MZ753809.1) and Diaemus bat paramyxovirus (MZ312424.1), while the lowest percentage of similarity (3.8 per cent) was established between the TM proteins of Hipposideros bat paramyxovirus (MZ312421.1) and Wenzhou Myotis davidii paramyxovirus 1 (OM030335.1).
For all five new jeilongviruses, the presence of gene start signals, intergenic regions, and gene end signals of all ORFs thought to represent functional genes was verified, confirming their actual coding capacity. Deviations from typically conserved motifs were observed in the intergenic regions between the F ORF and TM ORF of PipaV and between the M ORF and F ORF of PlecoV. An overview of these sequences can be found in Table 4. Furthermore, through verification of compliance with the rule of six (Calain and Roux 1993) and the presence of conserved sequences at 3ʹ- and 5ʹ-endings, the completeness of all genomes was confirmed.
Table 4.
Gene start signals, intergenic regions, and gene end signals established in newly discovered orthoparamyxoviruses. Individual sequences and general sequences are shown, conserved positions are annotated in bold. No independent gene start signal, intergenic region and gene end signal could be established for the hypothetical protein X2 of NinExV (indicated by ...).
| Gene end | Intergenic region | Gene start | |
|---|---|---|---|
| NinaV | |||
| N | AGGAGCAAGG | ||
| N/P | CTAAGAAAAA | CTT | AGGAGGAAGG |
| P/M | TTATAAAAAA | CTT | AGGAGGAAGG |
| M/F | ATAAGAAAAA | CTT | AGGGCGACAG |
| F/SH | TTAGAAAAAA | CTT | AGGACTAAGG |
| SH/TM | CTAGAAAAAA | CTT | AGGGCGAATG |
| TM/G | CTAAGAAAAA | CTT | AGGGCGAAAG |
| G/L | TTAAGAAAAA | CTT | AGGGGTAATG |
| L | TTAAGAAAAA | ||
| General sequence | HTADRAAAAA | CTT | AGGRSBAMDG |
| DenoV | |||
| N | AGGACTAAGG | ||
| N/P | TTAAGAAAAA | CTT | AGGATTAACG |
| P/M | TTAAGAAAAA | CTT | AGGAGTAAAG |
| M/F | ATATAAAAAA | CTT | AGGAGTCAAG |
| F/SH | TCATAAAAAA | CTT | AGGATGAACG |
| SH/TM | TTAGAAAAAA | CTT | AGGACAAATG |
| TM/G | CTAAGAAAAA | CTT | AGGACAAATG |
| G/L | TTAAGAAAAA | CTT | AGGAGCAATG |
| L | TTAAGAAAAA | ||
| General sequence | HYADRAAAAA | CTT | AGGABNMANG |
| NinYsV | |||
| N | AGGAGCAAAG | ||
| N/P | TTAAGAAAAA | CTT | AGGAACAAGG |
| P/M | TTATGAAAAA | CTT | AGGAATAAGG |
| M/F | CTAAGAAAAA | CTT | AGGTATAAGG |
| F/SH | ATACAAAAAA | CTT | AGGGTAAAAG |
| SH/TM | TTAAGAAAAA | CTT | AGGGCAAATG |
| TM/G | CTAAGAAAAA | CTT | AGGAGTAAAG |
| G/L | TTAAGAAAAA | CTT | AGGATCAAAG |
| L | TTAAGAAAAA | ||
| General sequence | HTAHRAAAAA | CTT | AGGDNHAADG |
| PipaV | |||
| N | AGGATGCAAG | ||
| N/P | TTAAGAAAAA | CTT | AGGATGCAAG |
| P/M | TTAAGAAAAA | CTT | AGGAGCAACG |
| M/F | GTAATAAAAA | CTT | AGGATCCAAG |
| F/TM | CTATAAAAAA | CAT | AGGATCCAAG |
| TM/G | TTAAGAAAAA | CTT | AGGAGACAAG |
| G/L | TTAAGAAAAA | CTT | AGGAGATAAC |
| L | TTAAGAAAAA | ||
| General sequence | BTAWDAAAAA | CWT | AGGAKVHAMS |
| PlecoV | |||
| N | AGGAATCAAG | ||
| N/P | TTAAGAAAAA | CTT | AGGATGAAAG |
| P/M | TTATAAAAAA | CTT | AGGGTTCAAG |
| M/F | TCAAGAAAAA | CAT | AGGGTTAATG |
| F/TM | TTAAGAAAAA | CTT | AGGATGAAAG |
| TM/G | TTAAGAAAAA | CTT | AGGATTCACG |
| G/L | TTAAGAAAAA | CTT | AGGATTCAAG |
| L | TTAAGAAAAA | ||
| General sequence | TYAWRAAAAA | CWT | AGGRWKMAHG |
| NinExV | |||
| N | AGGATCCAAG | ||
| N/P | TTAAGAAAAA | CTT | AGGATACAAG |
| P/M | TTAAGAAAAA | CTT | AGGATACAGG |
| M/X1 | TTAAGAAAAA | CTT | AGGATAAAAG |
| X1/X2 | TTAAAGAAAA | CTT | AGGATCCAGG |
| X2/F | … | … | … |
| F/G | TTAAGAAAAA | CTT | AGGATCCAAG |
| G/L | TTAAGAAAAA | CTT | AGGACACAAG |
| L | TTAAGAAAAA | ||
| General sequence | TTAARRAAAA | CTT | AGGAYMMARG |
NinExV: a new putative henipavirus with a formerly unobserved gene
One of the six newly discovered viruses proposedly belongs to the genus Henipavirus according to phylogenetic analysis: NinExV. The genome organization of this shrew-borne virus, identified from the determined host species S. minutus, can be described as 3ʹ-N-P-M-X1-(X2)-F-G-L-5ʹ (Fig. 2). Additional to the general paramyxovirus genome organization, two ORFs corresponding to hypothetical proteins X1 and X2 were observed. Both amino acid sequences were subjected to a blastp search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and a TMHMM search to predict the presence of transmembrane helices (https://services.healthtech.dtu.dk/service.php?TMHMM-2.0). Hypothetical protein X1 showed no similarity with other known sequences, and no transmembrane domain was revealed. However, gene start and end signals could be identified for the ORF encoding this protein, indicating that it represents an independent gene that is likely being expressed (Table 4). Conversely, no independent gene start or end signals could be established for ORF X2, although this ORF does fall within the boundaries of the F gene. However, the hypothetical protein encoded by this ORF, hypothetical protein X2, does show similarity with hypothetical proteins from other recently discovered putative henipaviruses: denwin virus (OK623354.1) (query cover 47 per cent, percentage identity 36.36 per cent), Wufeng Chodsigoa smithii henipavirus 1 (OM030316.1) (query cover 85 per cent, percentage identity 26.72 per cent), and Wenzhou Apodemus agrarius henipavirus 1 (MZ328275.1) (query cover 60 per cent, percentage identity 30.95 per cent). Comparable but less similar hypothetical proteins have been observed in other rodent- and shrew-borne henipaviruses as well. An alignment of these amino acid sequences was made using ClustalW (Thompson, Higgins, and Gibson 1994) and visualized by ESPript (v3.0) (Robert and Gouet 2014), showing some conserved positions (Fig. 3). Furthermore, as in the above-mentioned related hypothetical proteins, a transmembrane domain was detected within the amino acid sequence of hypothetical protein X2. For the six canonical genes identified in the NinExV genome, gene start signals, intergenic regions, and gene end signals were verified (Table 4). The completeness of the genome was confirmed through a genome length (19,158 nucleotides) divisible by six and the presence of conserved 3ʹ- and 5ʹ-endings.
Figure 3.

Alignment of the hypothetical X2 protein of NinExV with related hypothetical proteins in other henipaviruses. Completely conserved positions (indicated by *) and partially conserved positions (>70 per cent) are framed.
Discussion and conclusion
The aim of this project was to detect the presence of orthoparamyxoviruses in a bat and rodent/shrew sample set and to completely assemble genomes of newly discovered viruses. Two different approaches were used: for the bat samples, an initial nested RT-PCR with degenerate primers was carried out for orthoparamyxovirus screening, whereafter nanopore sequencing was done on positive samples. For the rodent and shrew samples, the initial screening step was skipped and all samples were subjected to nanopore sequencing. Afterward, a nested RT-PCR assay was carried out for these samples as well in order to validate nanopore sequencing results. Nested RT-PCR, eventually followed by sequencing, is to this day the standard for biosurveillance. However, it is more labor-intensive and time-consuming in comparison with solely using nanopore sequencing (Player et al. 2020). In this study, 10 out of 127 bat samples and twenty-five out of fifty-one rodent/shrew samples were positive for orthoparamyxovirus presence, corresponding to positivity rates of, respectively, 7.87 and 49 per cent. Significant differences between orthoparamyxovirus positivity rates in bat sample sets and rodent/shrew sample sets have been established in other research projects as well: Larsen et al. screened 358 North American bats and rodents for the presence of paramyxoviruses and hereby obtained positivity rates of, respectively, 9 and 36 per cent (Larsen et al. 2022). Furthermore, Kurth et al. detected three positive samples in a set of 120 German bats (positivity rate: 1.5 per cent) (Kurth et al. 2012), while Sasaki et al. identified ninety-six positives in a collection of 462 Zambian rodents and shrews (positivity rate: 20.8 per cent) (Sasaki et al. 2014). From these findings, it is suggested that direct nanopore sequencing without prior nested RT-PCR assays could be a fitting technique to screen rodent and shrew samples, since positivity rates in these small mammals have consistently been high in previously conducted studies. The validation with nested RT-PCR assays also demonstrated that no significant information was missed by directly using nanopore sequencing. The rather high cost of nanopore sequencing due to the inclusion of negative samples should, however, be considered. For bat samples, on the other hand, initial nested RT-PCR assays are still recommended in order to avoid unnecessary usage of sequencing reagents and materials.
In total, thirty-five analyzed samples were positive for the presence of orthoparamyxoviruses. Nine putative orthoparamyxovirus species were detected within these positive samples, including three species previously identified by Vanmechelen et al. (2022) and six formerly unknown species. Out of these six new species, five are thought to belong to the genus Jeilongvirus. This genus is relatively new, since it was only established in 2019, when it included six recognized member species (Amarasinghe et al. 2019). Currently, it is the most extensive genus within the Orthoparamyxovirinae subfamily, and more member species are regularly being discovered (Vanmechelen et al. 2022; Wells et al. 2022; Zhu et al. 2022). Due to this fast-paced expansion of the Jeilongvirus genus, problems concerning phylogenetical classification are arising. To accommodate the growing diversity of the genus Jeilongvirus, Su et al. (2023) proposed a new phylogenetical arrangement based on the analysis of complete L genes, biochemical criteria, and host ranges of included viral species. A division of the genus into four lineages is suggested: L1 and L4 consist of bat-borne viruses, L2 of rodent- and feline-borne viruses, and L3 of bat-, hedgehog-, and rodent-borne viruses. The phylogenetic analysis shown here, which is based on N, P, M, F, RBP, and L proteins of representative sequences of all (putative) orthoparamyxovirus species, is not in line with this suggestion but instead shows a clear and strongly supported division in just two clades. One clade includes rodent- and shrew-borne viruses, while the other clade includes bat-borne viruses. A notable exception is the presence of belerina virus (MN561699.1) within the bat-borne clade, since this virus was detected in a European hedgehog (Erinaceus europaeus) (Vanmechelen et al. 2020). Hedgehogs, as well as shrews, belong to the order Eulipotyphla. Hence, it would be expected that belerina virus clusters within the rodent- and shrew-borne clade. Similarly, according to a previous phylogenetic analysis by Vanmechelen et al. (2022), feline paramyxovirus (LC431581.1) clusters within the rodent- and shrew-borne clade between Paramyxoviridae sp. AB1 (OK210078.1), Meliandou lophuromys virus (OK623364.1), and Mount Mabu lophuromys virus 1 (NC043539.1). Feline paramyxovirus was not included in our Bayesian phylogenetic tree, since no coding-complete sequence was available for this virus. The classification of belerina virus and feline paramyxovirus is rather deviant, in our tree as well as in the phylogenetic tree established by Su et al. (2023). Unlike the proposition of Su et al. (2023), our analysis was conducted based on all major orthoparamyxovirus proteins. We suspect that by solely basing phylogenetic analysis on L genes, critical evolutionary information might be missed and that similarities and differences within other shared ORFs should be taken into account as well in order to perform a more robust phylogenetic analysis.
Three new (putative) rodent-borne jeilongviruses were observed in our analyzed sample set: NinaV, DenoV, and NinYsV. Their genomes, characterized by exceptionally large G ORFs and the presence of SH and TM ORFs, are similar to other rodent-borne jeilongviruses in line with their phylogenetical placement within our Bayesian tree. Additionally, two bat-borne jeilongviruses were detected as well: PipaV and PlecoV. Their genome organization clearly differs from that of the rodent-borne viruses, since they exhibit average-sized G ORFs and lack SH ORFs. The function of SH and TM proteins, distinctively present in jeilongviruses, has not been established to date. The only exception is the TM protein of J-virus (NC007454.1), which seems to promote cell-to-cell fusion according to the research by Li et al. (2015). Transmembrane domains are always observed within the amino acid sequences of SH and TM proteins, suggesting that these proteins might be involved in host cell entry or virus assembly and release (Ou, Dong, and Chou 2018). Contrary to the SH proteins, only found in some rodent- and shrew-borne jeilongviruses, TM proteins seem to be present in all Jeilongvirus species. When comparing amino acid similarities of the protein sequences of all TM proteins present in jeilongviruses, it appears that TM proteins of viruses clustering within the rodent- and shrew-borne clade are more similar than viruses clustering within the bat-borne clade. However, similarity percentages seem to correlate with branch lengths of the phylogenetic tree: the first subclade, including Jingmen Apodemus agrarius jeilongvirus 2 (MZ328282.1) to Meliandou mastomys virus (OK623361.1), exhibits shorter branch lengths than other subclades. Accordingly, similarity percentages of the TM proteins of viruses belonging to this subclade are significantly higher (Fig. 1, Supplementary Table S3). This branch length association seems to argue against host-specific evolutionary rates. TM mutation rates in general do seem to be higher than those of the six major canonical paramyxovirus proteins, as the similarity between TM proteins of different species is lower than what is typically observed for any of these six other proteins (Zhu et al. 2022).
In addition to the five jeilongviruses, one virus putatively belonging to the genus Henipavirus was also observed during this research project. Henipaviruses are important targets for virus discovery and biosurveillance, since this genus includes several zoonotic viruses such as HeV, NiV, Mòjiāng virus, and the recently discovered LayV (Wu et al. 2014; Centers for Disease Control and Prevention (CDC) 2022a,b; Mallapaty 2022). As is observed within the genus Jeilongvirus, phylogenetic analysis of henipaviruses demonstrates the division of this genus into two separate clades, one consisting of rodent- and shrew-borne viruses, among which are Mòjiāng virus and LayV, and the other consisting of bat-borne viruses, among which are HeV and NiV (Fig. 1). The novel virus described here, NinExV, clusters within the rodent- and shrew-borne clade of henipaviruses, which is in line with expectations according to its determined host species S. minutus (order Eulipotyphla). Interestingly, NinExV is only distantly related to other viruses within this clade and forms a separate sister lineage in our Bayesian tree, thus revealing the uniqueness of this putative viral species. Furthermore, the genome of NinExV is characterized by the presence of one additional gene and one additional ORF, respectively, corresponding to hypothetical proteins X1 and X2. The presence of hypothetical protein X1 is striking, since no comparable gene has been observed in any other henipavirus to date. Gene start signals, intergenic regions, and gene end signals were determined, implying that hypothetical protein X1 is being expressed. No transmembrane domain was established from the amino acid sequence, suggesting that the protein may function within the virion particle, together with proteins responsible for viral replication. Another possibility is that the protein may be excreted, hereby influencing immunological reactions or pathogenesis in its host species, thus hinting at a certain zoonotic potential of NinExV. However, the actual expression and function of the hypothetical protein X1 remain to be established. A second additional ORF, corresponding to hypothetical protein X2, was observed in the genome of NinExV as well. Contrary to hypothetical protein X1, comparable ORFs have been observed in other rodent- and shrew-borne henipaviruses (Vanmechelen et al. 2022). An alignment of these protein sequences was made (Fig. 3), showing some (semi-)conserved positions. Furthermore, hypothetical protein X2 does contain a transmembrane domain. The similarity between the function of these proteins in different henipaviruses, if functional at all, remains to be established. The possibility of resemblances between these hypothetical proteins in henipaviruses and TM or SH proteins in jeilongviruses should also be considered, since they all contain transmembrane domains. No gene start signals, intergenic regions, and gene end signals were established for hypothetical protein X2, implying that it falls within the boundaries of the F gene. Importantly, some significant similarities are observed between LayV and NinExV, highlighting the zoonotic potential of this newly discovered henipavirus. Both viruses are shrew-borne, and both viruses include the ORF corresponding to hypothetical protein X2. Since a transmembrane domain was established within the amino acid sequence of this protein, chances are that it concerns an additional membrane-associated protein playing a role in target cell entrance, which is crucial for the zoonotic potential of a virus.
In conclusion, the discovery of these novel viruses once again highlights that additional research concerning the function and expression of hypothetical proteins, TM proteins, and SH proteins in orthoparamyxoviruses is necessary, since these accessory proteins may contribute to host switch events or eventual zoonotic potential. Furthermore, the ongoing discovery of novel, diverse viruses highlights a clear need for a proper update and revision of Jeilongvirus and Henipavirus classification, and in extension of that of the whole Orthoparamyxovirinae subfamily.
Supplementary Material
Contributor Information
Jessica Van Bets, Department of Microbiology, Immunology and Transplantation, Laboratory of Clinical and Epidemiological Virology, Rega Institute for Medical Research, KU Leuven, Herestraat 49/Box 1040, Leuven BE3000, Belgium.
Tibe Joly Maes, Department of Microbiology, Immunology and Transplantation, Laboratory of Clinical and Epidemiological Virology, Rega Institute for Medical Research, KU Leuven, Herestraat 49/Box 1040, Leuven BE3000, Belgium.
Piet Maes, Department of Microbiology, Immunology and Transplantation, Laboratory of Clinical and Epidemiological Virology, Rega Institute for Medical Research, KU Leuven, Herestraat 49/Box 1040, Leuven BE3000, Belgium.
Bert Vanmechelen, Department of Microbiology, Immunology and Transplantation, Laboratory of Clinical and Epidemiological Virology, Rega Institute for Medical Research, KU Leuven, Herestraat 49/Box 1040, Leuven BE3000, Belgium.
Data availability
The viral genome sequences and host sequences obtained during this study have been submitted to GenBank (accession numbers OQ438284–OQ438289 and OQ939664–OQ939841).
Supplementary data
Supplementary data is available at Virus Evolution online.
Conflict of interest:
None declared.
References
- Amarasinghe G. K. et al. (2019) ‘Taxonomy of the Order Mononegavirales: Update 2019’, Archives of Virology, 164: 1967–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calain P., and Roux L. (1993) ‘The Rule of Six, a Basic Feature for Efficient Replication of Sendai Virus Defective Interfering RNA’, Journal of Virology, 67: 4822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutiérrez S., Silla-Martínez J. M., and Gabaldón T. (2009) ‘trimAl: A Tool for Automated Alignment Trimming in Large-scale Phylogenetic Analyses’, Bioinformatics, 25: 1972–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. et al. (2018) ‘The Global Virome Project’, Science, 359: 872–4. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) (2022a) Nipah Virus (NiV) <https://www.cdc.gov/vhf/nipah/index.html> accessed 22 Feb 2023.
- ——— (2022b) Hendra Virus Disease <https://www.cdc.gov/vhf/hendra/index.html> accessed 22 Feb 2023.
- Cox R. M., and Plemper R. K. (2017) ‘Structure and Organization of Paramyxovirus Particles’, Current Opinion in Virology, 24: 105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., and Rambaut A. (2007) ‘BEAST: Bayesian Evolutionary Analysis by Sampling Trees’, BMC Evolutionary Biology, 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J. H., and Anthony S. J. (2017) ‘Viral Discovery as a Tool for Pandemic Preparedness’, Revue Scientifique et Technique de l'OIE, 36: 499–512. [DOI] [PubMed] [Google Scholar]
- Greninger A. L. et al. (2015) ‘Rapid Metagenomic Identification of Viral Pathogens in Clinical Samples by Real-Time Nanopore Sequencing Analysis’, Genome Medicine, 7: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Rozewicki J., and Yamada K. D. (2019) ‘MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization’, Briefings in Bioinformatics, 20: 1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S. et al. (2017) ‘Canu: Scalable and Accurate Long-read Assembly via Adaptive K-mer Weighting and Repeat Separation’, Genome Research, 27: 722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth A. et al. (2012) ‘Novel Paramyxoviruses in Free-Ranging European Bats’, PLoS One, 7: e38688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen B. B. et al. (2022) ‘Evolution and Diversity of Bat and Rodent Paramyxoviruses from North America’, Journal of Virology, 96: e0109821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. et al. (2015) ‘Type II Integral Membrane Protein, TM of J Paramyxovirus Promotes Cell-to-Cell Fusion’, Proceedings of the National Academy of Sciences of the United States of America, 112: 12504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallapaty S. (2022) ‘New “Langya” Virus Identified in China: What Scientists Know So Far’, Nature, 608: 656–7. [DOI] [PubMed] [Google Scholar]
- Ou B. Y., Dong Y., and Chou J. J. (2018) ‘Structural and Functional Properties of Viral Membrane Proteins’, Advances in Membrane Proteins, 147–81. [Google Scholar]
- Player R. et al. (2020) ‘Comparison of the Performance of an Amplicon Sequencing Assay Based on Oxford Nanopore Technology to Real-Time PCR Assays for Detecting Bacterial Biodefense Pathogens’, BMC Genomics, 21: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rima B. et al. (2019) ‘ICTV Virus Taxonomy Profile: Paramyxoviridae’, Journal of General Virology, 100: 1593–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert X., and Gouet P. (2014) ‘Deciphering Key Features in Protein Structures with the New ENDscript Server’, Nucleic Acids Research, 42: 320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M. et al. (2014) ‘Molecular Epidemiology of Paramyxoviruses in Zambian Wild Rodents and Shrews’, Journal of General Virology, 95: 325–30. [DOI] [PubMed] [Google Scholar]
- Schlegel M. et al. (2012) ‘Molecular Identification of Small Mammal Species Using Novel Cytochrome B Gene-derived Degenerated Primers’, Biochemical Genetics, 50: 440–7. [DOI] [PubMed] [Google Scholar]
- Su H. et al. (2023) ‘Discovery and Characterization of Novel Paramyxoviruses from Bat Samples in China’, Virologica Sinica, 38: 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taseen S. et al. (2023) ‘Tip of the Iceberg: Emergence of Langya Virus in the Postpandemic World’, Journal of Medical Virology, 95: e28173. [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G., and Gibson T. J. (1994) ‘CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-specific Gap Penalties and Weight Matrix Choice’, Nucleic Acids Research, 22: 4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmechelen B. et al. (2020) ‘Common Occurrence of Belerina Virus, a Novel Paramyxovirus Found in Belgian Hedgehogs’, Scientific Reports, 10: 19341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmechelen B. et al. (2022) ‘The Characterization of Multiple Novel Paramyxoviruses Highlights the Diverse Nature of the Subfamily Orthoparamyxovirinae’, Virus Evolution, 8: veac061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells H. L. et al. (2022) ‘Classification of New Morbillivirus and Jeilongvirus Sequences from Bats Sampled in Brazil and Malaysia’, Archives of Virology, 167: 1977–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick R. R. et al. (2017) ‘Completing Bacterial Genome Assemblies with Multiplex MinION Sequencing’, Microbial Genomics, 3: e000132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z. et al. (2014) ‘Novel Henipa-like Virus, Mojiang Paramyxovirus, in Rats, China, 2012’, Emerging Infectious Diseases, 20: 1064–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-A. et al. (2022) ‘A Zoonotic Henipavirus in Febrile Patients in China’, New England Journal of Medicine, 387: 470–2. [DOI] [PubMed] [Google Scholar]
- Zhu W. et al. (2022) ‘Discovery and Evolutionary Analysis of a Novel Bat-Borne Paramyxovirus’, Viruses, 14: 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The viral genome sequences and host sequences obtained during this study have been submitted to GenBank (accession numbers OQ438284–OQ438289 and OQ939664–OQ939841).