

Abstract
Wild-type reovirus serotype 3 Dearing (T3wt), a non-pathogenic intestinal virus, has shown promise as a cancer therapy in clinical trials, but it would benefit from an increased potency. Given that T3wt is naturally adapted to the intestinal environment (rather than tumors), we genetically modified reovirus to improve its infectivity in cancer cells. Various reovirus mutants were created, and their oncolytic potency was evaluated in vitro using plaque size as a measure of virus fitness in cancer cells. Notably, Super Virus 5 (SV5), carrying five oncolytic mutations, displayed the largest plaques in breast cancer cells among the mutants tested, indicating the potential for enhancing oncolytic potency through the combination of mutations. Furthermore, in a HER2+ murine breast cancer model, mice treated with SV5 exhibited superior tumor reduction and increased survival compared with those treated with PBS or T3wt. Intriguingly, SV5 did not replicate faster than T3wt in cultured cells but demonstrated a farther spread relative to T3wt, attributed to its reduced attachment to cancer cells. These findings highlight the significance of increased virus spread as a crucial mechanism for improving oncolytic virus activity. Thus, genetic modifications of reovirus hold the potential for augmenting its efficacy in cancer therapy.
Keywords: reovirus, oncolytic virus, breast cancer, immunotherapy, improved reovirus mutant, reovirus spread, virus spread, virus attachment, large-plaque-forming virus, improved oncolytic potency
Graphical abstract
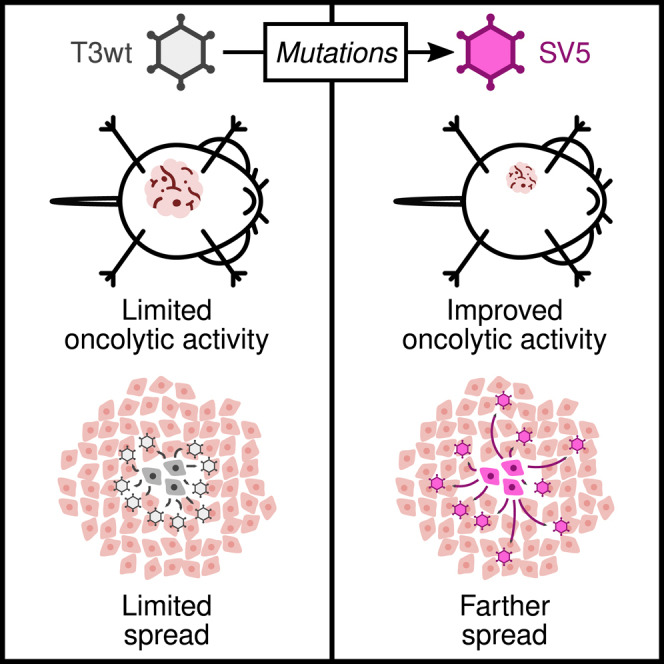
Shmulevitz and colleagues developed the mutant "Super Virus 5" (SV5) to enhance reovirus as an anti-cancer treatment. SV5 shrinks tumors and boosts survival in mice with breast cancer. Remarkably, SV5’s weaker binding to cancer cells enables its farther spread, potentially driving its enhanced cancer-fighting effects.
Introduction
Mammalian orthoreovirus (reovirus) is a non-pathogenic enteric virus that naturally infects intestinal cells but is rapidly cleared by innate and cellular immune responses. Reovirus has oncolytic potential since it selectively amplifies in tumor cells, but is rapidly subdued in untransformed or normal cells.1 Unmodified reovirus, specifically the PL laboratory strain of serotype 3 Dearing lineage (T3wt), is currently being evaluated in more than 30 clinical trials as a therapy for diverse types of cancer including metastatic breast cancer, prostate cancer, and colorectal cancer.2,3,4,5,6,7,8 In patients, T3wt has been well tolerated at maximal dose,9,10,11,12,13 but benefits of reovirus monotherapy have been brief and modest.11,14,15,16 For example, when reovirus was evaluated in combination with paclitaxel in patients with metastatic breast cancer, median overall survival was extended from 10.4 months to 17.4, but there was no increase in response rate (p = 0.87) or progression-free survival (p = 0.87).6 A recent summary of clinical trials of oncolytic viruses (OVs) as a whole, similarly highlights that only ∼20% of patients show some response to OVs.17 Similar to patient studies, T3wt fails to cure the majority of immunocompetent tumor-bearing animals in mouse cancer models.18,19,20,21 Altogether, while reovirus therapy seems promising, improvements are clearly necessary to achieve the potency required for reliable cancer treatment. To enhance T3wt therapy, clinical trials are focused on combining T3wt with chemotherapies and checkpoint inhibitors. As a complementary approach, we seek to re-engineer T3wt to enhance its oncolytic potency.
Given that replication in tumors was not the driving force for the evolution of T3wt, we reasoned that there might be considerable room for improvement of reovirus’ oncolytic activities by modifying the viral genome. Reovirus is a dsRNA virus with a genome consisting of 10 segments that encode 12 different proteins: 8 structural and 4 non-structural.22,23 Reovirus attaches to cells via junction adhesion molecule-A (JAM-A), sialic acid, and integrins, and enters by clathrin-mediated endocytosis.24,25,26 After entry, the virus outer capsid proteins σ3, μ1C, and σ1 are cleaved by cellular proteases, and the μ1C cleavage products mediate penetration through endosomal membranes to deliver transcriptionally active viral cores into the cytoplasm.27 Reovirus cores produce viral RNA and proteins within localized areas of virus amplification called factories, and new viral RNA and proteins ultimately assemble into thousands of new infectious virions in ∼24 h.28,29 Cell death accompanies the release of progeny viruses, which can infect more cells and amplify the oncolytic process. Previous studies discovered that single amino acid mutations in T3wt can improve oncolytic potency in cellulo and in vivo while retaining specificity toward transformed cells.30,31,32 Specifically, mutations in the cell attachment protein σ1 that decreased the number of σ1 on reovirions increased the probability that reovirions uncoated to cores in tumor cells,33 and doubled the survival time of animals in a melanoma syngeneic mouse model relative to T3wt.21 In addition, a mutation in the μ2 protein of reovirus that affects RNA synthesis and virus factory formation was advantageous at late stages of virus replication, leading to higher progeny production in tumor cells.34
With the objective of improving T3wt’s potency toward breast cancer without compromising safety, we initially assessed a library of reovirus mutations, either individually or in combination, for their potential to enhance reovirus oncolytic activity in tumor cells. We ultimately identified a combination of five mutations that improved oncolytic potency toward a panel of human and murine breast cancer cells and in the murine HER2+ TUBO cell breast cancer model. This reovirus mutant was named “Super Virus 5” or “SV5” for short. To reveal the mechanism of enhanced oncolysis by SV5, multiple steps of virus replication and cell-cell spread were evaluated, as were other possible mechanisms of virus oncolysis, such as cell signaling and anti-tumor immune responses. The mechanism revealed was unforeseen: mutations in SV5 reduced the probability of virus binding to tumor cells, which in turn resulted in viruses that travelled longer distances before infecting new tumor cells. In other words, thousands of viruses were produced by an SV5-infected cell but, rather than all new virions binding to the nearest cells to initiate reinfection, SV5 virus particles forwent binding to neighboring cells and diffused to infect tumor cells that were farther away. These results beget a new perspective on what features of a virus are required to promote efficient oncolysis; and suggest that a “cost,” such as reduced cell-binding efficiency, can produce a “benefit,” such as distal spread, that supersedes the cost in terms of ultimate outcome of infection.
Results
Multiple novel mutations can increase oncolytic potency of reovirus
We previously established an approach using the transformed murine tumorigenic L929 cell line to isolate mutations in reovirus that enhance infectivity.34 In short, T3wt was mutagenized randomly with 5-fluorouracil to create a quasispecies, and variants that produced larger plaques on L929 cells were plaque purified and amplified. Plaque size was used as a robust proxy for all stages of virus replication, cell death, and virus cell-cell spread, since enhancement of any of these stages would result in larger plaques relative to T3wt. Also, since a plaque allows to visualize areas of cell death, it provides a measure of in vitro oncolysis, i.e., tumor cell killing. Using this approach, two reovirus variants T3v1 and T3v2 were previously found to increase plaque size on melanoma B16-F10 in addition to L929 cell monolayers, and to significantly prolong survival in the B16-F10 mouse model of melanoma.21 In the current analysis, with the intent to expand the repertoire of potentially beneficial mutations, 10 additional large-plaque-forming reovirus variants isolated in L929 cells were selected for evaluation. These variants were named T3 variant “x” (T3v“x”), where “x” represents the chronological order of isolation. One of these variants, T3v2, was further mutagenized from which a larger-than T3v2 mutant named T3v12 was selected (Table S1). All 12 variants were amplified, gradient purified, and subsequently assessed for plaque size using a panel of tumor cell lines. This panel included murine B16-F10 melanoma, human H1299 lung cancer, human PanC pancreatic, and human T47D breast cancer cells, which were deliberately selected to represent diverse human and murine tumor types (Figure 1A). In general, all 12 variants produced larger plaques than T3wt on the majority of cancer cells. Some variants, such as T3v2, T3v4, T3v8, T3v11, and T3v12, consistently outperformed T3wt in the majority of cancer cells (Figure S1). Some variants exhibited enhanced plaque size in a cell-line-specific manner; for example, T3v16 plaques were notably larger on L929 and T47D cells but not in the remaining cell lines (Figure S1). Importantly, all variants were also evaluated on the non-transformed NIH3T3 mouse fibroblast cell line to ensure that they did not concurrently evolve the ability to propagate in non-transformed cells. Because T3wt poorly infects and spreads in non-transformed cell monolayers,35,36,37,38,39 T3wt does not produce a bona fide plaque on NIH3T3 cells but rather produces a focus of three to five infected cells visualized by immunohistochemical staining with reovirus-specific antibodies. Importantly, the 12 reovirus variants produced even smaller foci of infection than T3wt on NIH3T3 cell monolayers, indicating that restricted infection of non-transformed cells was maintained or even improved for the variants (Figure 1A).
Figure 1.

Isolated mutants selected by directed evolution harbor mutations that increase oncolytic potency evaluated in vitro by plaque size
(A) Plaque size relative to T3wt (set to a value of 1) of original isolated mutants in NIH3T3 (mouse non-transformed fibroblasts), L929 (mouse transformed fibroblasts), B16-F10 (mouse melanoma), H1299 (human lung carcinoma), PanC (human pancreatic carcinoma), and T47D (human breast cancer) cells. Each point represents an independent experiment (n = 2–6). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 relative to T3wt plaque size (p values determined with ANOVA with Tukey test). (B) Plaque size of the 24 reovirus variants harboring single mutations generated by site-directed mutagenesis and reverse genetics. Background color indicates the mutated protein in each virus (see legend). Each point represents an independent experiment performed in duplicate (n = 3–7). The green dashed line corresponds to the plaque size of T3wt and was set to 1. The red box shows mean ± standard error. ∗p < 0.05 relative to T3wt plaque size (p values determined with an ANOVA with Tukey test).
To determine which mutations in each variant contributed to the larger plaque phenotype, variants were first subjected to whole-genome sequencing. Each variant contained from two to four mutations (Table S1), with no variants sharing the same mutation(s), except for T3v12, which contained the S18I mutation in σ1 from the T3v2 parent from which it was derived. Given that multiple mutations were found in all variants, individual mutations were introduced separately into plasmids that were used to generate viruses with single mutations by reverse genetics. In total, 24 mutant reoviruses with single amino acid changes were generated. Each mutant was then assessed for plaque size on L929 cell monolayers relative to T3wt (Figure 1B). The mutations that significantly increased plaque size relative to T3wt were predominantly in two reovirus genes: the S1 gene that encodes the cell-binding protein σ1, and the L2 gene that encodes the pentameric protein λ2, which anchors σ1 at each reovirus vertex, but also contributes to the core structure and addition of 5′ 7-methylguanosine caps to virus mRNAs. Some mutations in the outer capsid σ3 protein encoded by the S4 gene (K64E and H230Q), and the inner capsid protein λ1 encoded by the L3 gene (A962S) also increased plaque size relative to T3wt, albeit not with statistical significance. Mutations that mapped to the L1-encoded λ3 viral polymerase did not increase plaque size (Figure 1B). Altogether, the new variants harbored novel mutations, previously unassociated with increased plaque size.
Oncolytic mutations can have additive effects on in vitro oncolysis
While single mutations can increase reovirus plaque size as a measure of in vitro oncolysis, it has yet to be tested whether mutations can be combined together in T3wt to provide additive enhancement of plaque size on tumorigenic cell monolayers. To establish if mutations could have additive effects, pairwise combinations of mutations were introduced into reovirus using reverse genetics rescue. First, the best mutation in L2-encoded λ2 (I1274T) was combined with the σ1 mutations S66I or N312R. The representative images (Figure 2A) and the quantification over several independent experiments (Figure 2B) show that the combination of λ2I1274T with σ1S66I or σ1N312R almost doubled plaque size relative to the individual mutations, and increased plaque size by 3.5-fold relative to T3wt. Although not statistically significant relative to the single mutated variants due to variation in plaque size among independent experiments, there is clearly a trend toward additive effects on plaque size. Not only did the plaque size increase when two plaque-enlarging mutations were combined into a single mutant virus, but when a mutation that increased plaque size such as σ1S18I was combined with a mutation that decreased plaque size such as λ1Y122H, the resulting double mutant produced smaller plaques relative to the σ1S18I single mutant (Figures 2C and 2D). These results show for the first time that combining mutations into reovirus can have additive effects on in vitro oncolysis. Whether the additive effects are negative or positive depends on whether the individual mutations reduce or enhance plaque size respectively.
Figure 2.

Oncolytic mutations can have additive effects on in vitro oncolysis in L929 cells
(A) Example of a positive additive effect on plaque size generated by the combination of the mutation I1274T in λ2 and the mutations S66I or N312R in σ1. (B) Graphical representation of (A). (C) Example of a negative additive effect on plaque size generated by the combination of mutations S18I in σ1 (which increases plaque size) and Y122H in λ1 (which decreases plaque size). (D). Graphical representation of (C). (E) Graph showing the relative plaque size of the combination of the best single mutation in σ1, S18I, with the mutation K64E in σ3 or the mutation N1148S in λ2. (F) Graph showing the relative plaque size of the combination of the best mutation in λ2, I1274T, with the mutations S18I, L28P, or R219Q in σ1. Gray dotted line corresponds to T3wt virus. Each point in the graphs represents an independent experiment performed in duplicate. Boxes in the graphs represent the average plaque size ± standard error of three or more independent experiments relative to T3wt (n = 3–8). The average plaque size is shown in gray for T3wt, red for the single mutated viruses, and purple for the combined viruses. ∗p < 0.05, ∗∗p < 0.01 relative to T3wt (p values determined with an ANOVA with Tukey test).
To establish if there are limits to plaque size enlargement, the best mutation σ1S18I was combined with other neutral or plaque-increasing mutations. When σ1S18I was combined with σ3K64E or λ2N1148, the plaque size produced by the combined viruses increased relative to T3wt but not relative to σ1S18I (Figure 2E). Similarly, combining mutations in σ1 (S18I, L28P, and R219Q) with the best mutation in λ2 (I1274T) does not increase plaque size relative to σ1S18I (Figure 2F). The results also suggest that, when a mutation such as σ1S18I strongly increases plaque size alone, there is minimal enhancement in plaque size by addition of other mutations. The lack of additive effects with σ1S18I could be due to mechanistic redundancies between mutations or due to a maximum threshold of plaque size achievable on L929 cell monolayers within the 5 days of feasible assay duration.
SV5 generates larger plaques than T3wt in breast cancer cell lines
Breast cancer continues to be a major cause of death in women, and since T3wt is underperforming as a therapeutic agent in breast cancer clinical trials, there is rationale for testing advanced oncolytic reovirus strategies in breast cancer models. Accordingly, the most promising reovirus mutations were evaluated individually and in combination in various breast cancer cells. The mutations σ1S18I, σ3K64E, and λ2I1274T were selected based on producing the largest plaques during screening with L929 cells (Figure 1B). The mutation μ2A612V was selected because it was previously found to increase viral protein synthesis.34 The mutation λ1A962S was chosen because it was the only mutation in T3v11, and this variant generated larger plaques in all human tumor cells lines evaluated (Figure 1A). SV5 was generated by reverse genetics to contain all five mutations. In vitro oncolysis was then evaluated on three breast cancer cell lines: MCF7 and T47D cells were used to evaluate sensitivity of human breast cancer cells, while the TUBO cell line, established from a BALB-NeuT mouse mammary carcinoma tumor, was used to permit transition of findings into in vivo immunocompetent mouse breast cancer models.
Traditional visualization of reovirus plaques by crystal violet staining of adherent cells surrounding regions of cell lysis is not feasible with all cell lines, since not all cell lines form contiguous cell monolayers and reovirus plaques are not always large enough to visualize on all cell lines. Accordingly, plaques of T3wt and mutant reoviruses on MCF7 and TUBO cell monolayers were visualized by immunohistochemical staining with reovirus-specific antibodies, while plaques on T47D cells were detected by crystal violet staining. SV5 generated larger plaques than T3wt on all breast cancer cell lines evaluated (Figure 3A). Relative to T3wt, SV5 enhanced plaque size by 3-, 5-, and 2-fold on MCF7, T47D, and TUBO cells, respectively (Figure 3B). When the contribution of single mutations to the plaque size was evaluated, cell-line-specific differences were remarkable. For example, when comparing only the single mutations, while the σ1S18I mutant produced the largest plaques on MCF7 and T47D cell monolayers, the σ3K64E mutant produced the largest plaques on TUBO cells (Figure 3B). Moreover, the λ2I1274T and σ3K64E mutations alone yielded larger plaques on T47D and TUBO monolayers relative to T3wt, but not on MCF7 monolayers (Figure 3B). These results suggest that, while the importance of any individual mutation for plaque size might fluctuate among breast cancer cell lines, the SV5 mutant that combines all five mutations may consistently produce larger plaques on all breast cancer cells. Importantly, the single mutated viruses and SV5 did not increase focus size on non-transformed foreskin BJ cells (Figure S2), indicating that SV5 retains specificity for cancer cells.
Figure 3.

Combined virus SV5 generates larger plaques than T3wt in breast cancer cell lines
(A) Representative images illustrating the plaque sizes generated by T3wt and SV5 in the breast cancer cell lines MCF7, T47D, and TUBO. (B) Graph showing plaque size of single mutated viruses and SV5 relative to T3wt in MCF7, T47D, and TUBO cells. Each point represents an independent experiment performed in duplicate. Boxes represent the average plaque size ± standard error of three or more independent experiments relative to T3wt (n = 3–8). Cells were infected with T3wt, single mutated viruses, and SV5. MCF7 and TUBO cells were fixed after 5 days of infection. T47Ds were fixed after 8 days. Immunocytochemistry with a polyclonal anti-reovirus antibody was performed in MCF7 and TUBO cells to detect plaques, while T47D cells were stained with crystal violet. ∗∗∗p < 0.001; ns, non-significant (ANOVA with Tukey test).
SV5 reduces tumor growth, prolongs survival, and induces an anti-tumor immune response in a mouse model of breast cancer
Having established that SV5 generated larger plaques than T3wt in all three breast cancer cell lines in vitro, the next key question was whether advantages of SV5 were conferred in vivo. To answer this, HER2/neu+ TUBO cells were implanted into the mammary fat pad of BALB/c mice and, when tumors were palpable, mice were separated into groups representing similar tumor size ranges. Mice were then treated intratumorally three times with either phosphate-buffered saline (PBS), T3wt, SV5, or the single mutated σS18I virus (Figure 4A). Tumor size was monitored regularly until endpoint of ∼1,500 mm3 tumor volume. On average, SV5 significantly reduced tumor growth relative to T3wt and the σS18I mutant (Figure 4B). When visualized as tumor growth for individual mice, there was a clear decrease in tumor growth in SV5-treated mice (Figure 4C). By 120 days, mice in all groups except the SV5 group had reached the endpoint, while two mice remained tumor free in the SV5-treated group (Figure 4C). Survival of mice in the SV5 group significantly exceeded that in the PBS group, while there were no differences in survival after treatment with T3wt or the σS18I mutant compared with PBS treatment (Figure 4D). These results suggest that the combination of mutations in SV5 improves oncolytic potency in vivo relative to T3wt and the single mutant σS18I. In addition, these findings are consistent with the formation of significantly larger plaques by SV5 compared with T3wt on TUBO cell monolayers, as opposed to the σS18I mutant, which did not exhibit a significant increase in plaque size relative to T3wt (Figure 3B).
Figure 4.

SV5 reduces tumor growth, prolongs survival, and induces an anti-tumor immune response
(A) Diagram of the animal experiment. TUBO cells were implanted into the mammary fat pad of BALB/c mice. When tumors were palpable, we performed three intratumoral injections (days 0, 2, and 4) with PBS or virus at 5 × 108 PFU/injection for T3wt and same infectivity levels (number of infected cells after 15 hpi) for the rest of the mutants checked by immunofluorescence. n = 5 per group. (B) Mean tumor growth of five mice per group until day 63 when all mice were alive. p values were calculated for each day with the non-parametric Wilcoxon test. PBS vs. T3wt: ∗on days 15 and 39, ∗∗day 12; PBS vs. SV5: ∗on days 15, 20, 25, 28, 31, 50, 57, and 63), ∗∗on days 10, 12, 35, 39, 43, 46, 53, and 60); T3wt vs. SV5: ∗on days 31, 35; virus σ1 S18I vs. SV5: ∗on days 31, 35, 46, 60, and 63. In the graph, statistics are displayed for T3wt relative to the PBS group (+p < 0.05, ++p < 0.001) and SV5 relative to PBS group (∗p < 0.05, ∗∗p < 0.001). (C) Tumor growth per individual mouse (each line represents an individual mouse) and shown by treatment until day 120 post first virus injections. (D) Survival. SV5 relative to PBS group: ∗p < 0.05 Significance calculated with Cox proportional hazards model test. This experiment finished with two cured mice from the SV5 group. (E) Diagram of TUBO re-challenge experiment. TUBO cells were re-implanted into the mammary fat pad opposite to the first cell injection site at day 210 after the first virus injection. Four mice that never received TUBO cells before were used as control mice. (F) Secondary, i.e., challenge, tumor growth (each line represents an individual mouse). The experiment finished at day 131 post TUBO cell re-implantation (day 341 post first virus injection), which was 6 weeks after the euthanasia of the last control mouse.
In many reports, when OVs kill tumor cells, they not only reduce tumor size, but they also promote anti-tumor immunity. To establish if SV5-treated mice developed a TUBO cell-directed immune response, TUBO cells were re-implanted into the mammary fat pad opposite to the original injection site of the surviving mice (Figure 4E). As a control to ensure that the re-implanted TUBO cells were capable of tumor formation, the same cells were also implanted into four control mice that had not previously received cells or viruses. As expected, tumors grew in the control mice. Conversely, tumors did not form in the SV5-treated mice, suggesting that SV5 treatment not only cleared the primary tumors, but established an anti-tumor immune response that prevented tumor reestablishment (Figure 4F).
T3wt and SV5 increase the proportion of cytotoxic T cells and decrease the proportion of myeloid-derived suppressor cells in the tumor microenvironment
The immunotherapeutic value of OVs is of growing interest, since establishing immunity toward the tumor cells could help clear cancer metastases and/or prevent recurrence caused by genotypically similar cancer stem cells. Ideally, OVs can reverse the immunosuppressed environment of tumors, reducing abundance of immunosuppressing immune cell populations and cytokines, while promoting recruitment of cytotoxic cells such as T cells. Given that SV5-treated mice established anti-tumor immune responses capable of preventing TUBO cell reestablishment, experiments were conducted to better understand which populations of immune cells were affected by SV5. The animal experiment described in Figure 4A was repeated with an endpoint at day 14 post first virus injection to characterize tumor and systemic changes in the immune system (Figure 5A). By 14 days, the trend of reduced tumor volume in SV5-treated mice relative to T3wt and PBS was again observed (Figure 5B). To study the immune response in the tumors, whole tumors were dissociated, immune cells purified on Percoll gradients and subjected to flow cytometric analysis for cell surface markers of specific immune cell populations.
Figure 5.

T3wt and SV5 increase cytotoxic T cells and decrease myeloid-derived suppressor cells in the tumor microenvironment
(A) Diagram of the animal experiment. TUBO cells were implanted into the mammary fat pad of BALB/c mice. When tumors were palpable, we performed three intratumoral injections (days 1, 3, and 5) with PBS or virus at 5 × 108 PFU/injection. Tumor size was measured until day 14 post first virus injection, when mice were euthanized, and tumors and spleens were collected for immune analysis by flow cytometry. Tumor samples were enriched for immune cells by Percoll gradient prior to flow cytometry. n = 5 per group. (B) Change of tumor volume per mouse and per group until 14 days post first virus injection. Each gray line represents an individual mouse and colored lines represent the average tumor growth. Tumor size was measured with calipers. (C) T cell (CD3+/CD45+) frequency from live immune-enriched cells in tumors. (D) T cell distribution in tumors. T cells (CD3+/CD45+) were divided between CD4+ T cells, regulatory T cells (CD4+ CD25+ cells) and CD8+ T cells. (E) Percentage of CD8+ Her2/neu rat p66 peptide+ T cells in tumors. These tumor-specific CD8+ T cells were detected by the tetramer H-2K(d) coupled with the Her2/neu rat p66 peptide TYVPANASL. (F) CD3–/CD45+/CD11b+ cell frequency from live immune-enriched cells in tumors. (G) Innate immune cell distribution in tumors. The total of CD3−/CD45+/CD11b+ cells from live cells were divided between dendritic cells (CD11c+), macrophages (F4/80+), NK cells (CD49b+), and myeloid-derived suppressor cells (MDSCs) (Ly-6G/Ly-6C+ (Gr-1)). (H) Innate immune cell distribution in spleens. The total numbers of CD3−/CD45+/CD11b+ cells from live cells were divided among dendritic cells (CD11c+), macrophages (F4/80+), NK cells (CD49b+), and MDSCs (Ly-6G/Ly-6C+ (Gr-1)). (I) T cell distribution in spleens. The total of T cells (CD3+/CD45+ cells from live cells) were divided among CD4+ T cells, regulatory T cells (CD4+ CD25+ cells), and CD8+ T cells. (J) Percentage of CD8+ Her2/neu rat p66 peptide+ T cells in spleens. These tumor-specific CD8+ T cells were detected by the tetramer H-2K(d) coupled with the Her2/neu rat p66 peptide TYVPANASL. p values were calculated with an ANOVA with Tukey test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Different T cell subsets can either promote or inhibit anti-tumor immunity. For example, tumor-infiltrating lymphocytes such as CD4+ and CD8+ T cells stimulate a pro-immunological environment,40,41 while T regulatory lymphocytes are immunosuppressive.40,41,42,43,44 In agreement with the literature on T3wt,45,46,47 we observed that both T3wt and SV5 increased the percentage of infiltrating CD3+/CD45+ T cells in the tumors relative to PBS treatment (Figure 5C). Of the CD3+/CD45+ T cells, the proportion of CD8+ cytotoxic T cells was increased 5-fold by T3wt and 6-fold by SV5, while CD4+ helper T cells were decreased relative to the PBS-treated group (Figure 5D). Although not statistically significant, there was a trend toward a reduction of the immunosuppressor regulatory T cell population (CD4+/CD25+) (Figure 5D). The differences in T cell subsets between treatments were not a consequence of attrition during sample processing, since the proportion of live versus dead immune-purified cells in the tumors was similar between the treatments (Figure S3A). When a tetramer was used to assess the frequency of CD8+ T cells directed against the immunodominant TUBO epitope p66 rat Her2/neu peptide, no differences were identified between treatments (Figure 5E). Likewise, there were no significant differences in B cell frequency between any group (Figure S4A). Therefore, while there was an overall increase in the tumor infiltration of pro-immunogenic CD8+ T cells, B cell infiltration and TUBO antigen-specific T cell amplification in the tumors were not yet achieved by any virus treatment at the 14 day post first virus injection endpoint.
Macrophages, dendritic cells, and NK cells also contribute to cancer immunosurveillance but in an antigen-independent manner. Conversely, myeloid-derived suppressor cells (MDSCs) contribute to immunosuppressive environments. To assess these cell populations within the entire tumor, while excluding T cell populations, we examined CD3-negative cells that were positive for CD45 and CD11b. Collectively, virus treatments led to a decrease in the levels of CD3−/CD45+/CD11b+ cells within the tumors (Figure 5F). Inclusion of additional surface markers to distinguish the specific cell populations revealed a decrease of approximately 17% in the proportion of MDSCs (Ly-6G/Ly-6C+ (Gr-1)) following treatment with either T3w or SV5 relative to PBS. (Figure 5G). The proportion of dendritic cells (CD11c+), macrophages (F4/80+), and NK cells (CD49b+) did not fluctuate by any treatment at the 14 day endpoint. These results alongside the increased infiltration of CD8+ T cells suggest a strong pro-immunogenic response generated by both T3wt and SV5.
To gain a deeper insight into the systemic immune response triggered by SV5 treatment, we conducted an analysis of immune cell frequencies in the spleens. The spleen processing yielded a high percentage of live cells, as shown in Figure S3B. Similar to their impact on tumors, both T3wt and SV5 treatments had no effect on the frequency of B cells (Figure S4B). However, these treatments did result in a decrease in the frequency of innate immune cells (Figure S3C), particularly macrophages, and interestingly there were no significant differences between the two virus treatment groups (Figure 5H). In the spleen, T cell frequency, mirroring the trend observed in tumors, was augmented in mice treated with both T3wt and SV5 (Figure S3D), with a significant increase in CD8+ T cells (Figure 5I). Although there was a trend toward an increase in TUBO-specific CD8+ T cells, particularly pronounced in the SV5 group, this change did not reach statistical significance (Figure 5J). Collectively, the data indicate the capacity of both T3wt and SV5 treatments to augment immune cell populations, including cytotoxic T cells, which have the potential to promote tumor immunity in both the tumor microenvironment and spleens. In addition, these treatments show promise in reducing immune suppressor cells such as MDSCs and regulatory T cells within the tumors.
As a complementary strategy to assess immunological changes in tumors between treatments, three tumors per group at 14 days post first virus injection were evaluated for changes in gene expression by RNA sequencing (RNA-seq). To relate gene expression to specific immune cell populations, gene expression levels were compared with publicly available databases using the murine Microenvironment Cell Populations-counter (mMCP-counter) method and the Immunedeconv R package (Table S5).48,49,50 Similar to flow cytometric analysis (Figure 5D), gene expression analysis suggested that CD8+ T cells were significantly increased in T3wt- and SV5-treated tumors (Figure S5). Also comparable with the flow cytometric analysis, gene expression signatures indicated a reduction, although not statistically significant, in the total monocyte population following T3wt and SV5 treatments (Figure S5). While changes in macrophage populations were minimal by flow cytometry, gene expression analysis suggested a significant increase in the macrophage population with virus treatments (Figure S5). This discrepancy may result from the well-known limitation of the flow cytometric surface marker F4/80 in detecting various subtypes of macrophage populations. The RNA-seq analysis also permitted evaluation of signatures for immune cells that were not evaluated by flow cytometry and revealed that SV5 and T3wt treatments also increased gene signatures representative of basophils (Figure S5). The role of basophils in tumor progression and anti-tumor immune response remains poorly understood.41,51 Future studies are needed to reveal the potential impact of changing basophil levels during OV treatment.
The transcriptomic data was also used to determine if any genes were upregulated differentially between T3wt and PBS treatments, or between T3wt and SV5 treatments. There were 230 upregulated and 22 downregulated genes in T3wt- versus PBS-treated mouse tumors, reflecting the large change in tumor transcriptome following oncolytic reovirus treatment (Figure S6A). In fact, a heatmap of differentially regulated genes clearly clusters PBS groups away from both T3wt- and SV5-treated groups (Figure S6B). When comparing T3wt- and SV5-treated tumors, only four genes were differentially expressed, suggesting minimal differences in host gene expression between these viruses (Figures S6C and S6D). Indeed, even when TUBO cells were infected in cell culture and assessed by qRT-PCR for the induction of IFN-dependent or NF-κB-dependent genes, there were no differences in gene induction between T3wt and SV5 (Figure S7). Important to note, however, is that only three independent tumors were used per condition for the RNA-seq experiment to enable probability value calculations; and so it is possible that SV5 versus T3wt might cluster apart from each other with considerably more samples. For example, among the three T3wt samples, sample “T3wt 2” clustered strongly with SV5 samples “SV5 2” and “SV5 3,” while “T3wt 1” and “T3wt 3” samples clustered with “SV5 1” (Figure S6B). Given that both T3wt and SV5 did have oncolytic activity in mice (Figure 4), albeit with better oncolytic activity by SV5 on average, the three transcriptomic samples from T3wt and SV5 might simply reflect the variation among mice in a single treatment group; and a greater sample number might be necessary to reveal transcriptomic differences between T3wt and SV5.
To better understand which cellular genes are modulated during reovirus oncolysis in vivo, the genes that were upregulated in the tumors by both T3wt and SV5 virus treatments relative to PBS were then divided into IFN receptor dependent/independent and RIG-I dependent/independent. Some of the genes from these categories were differentially expressed and can be found in Figure S8. These differentially expressed immune-regulated genes correlated with changes in tumor volume as a measure of tumor growth rate (Figure S9; Table S6). Remarkably, the increased expression of some genes highly correlated with a decrease in tumor progression. Some examples of these genes in each category are the IFNR-dependent RIG-dependent gene Irf5 (interferon regulatory factor 5), the IFNR-dependent RIG-I-independent gene Il2ra (interleukin-2 receptor subunit alpha), the IFNR-independent RIG-dependent gene Tbx1 (T-box transcription factor 1), and the IFNR-independent RIG-I-independent gene Hcst (hematopoietic cell signal transducer) (Figure S9). The data suggest that these correlative genes are strong predictors of suppression of tumor growth rate, regardless of the specific reovirus used for therapy.
In conclusion, changes induced by T3wt and SV5 in the immune cell frequency in tumors and spleens are also supported by gene expression analysis of tumors, which suggests increased cells and cytokines consistent with anti-tumor and anti-viral immunity. The immunotherapeutic activities of SV5 are evident in this study, particularly through the decreased tumor growth and prolonged animal survival. However, statistically significant differences between T3wt and SV5 treatments with respect to modulating immune cells and cytokines were not observed, suggesting that SV5 may impose additional immune-independent benefits to tumor clearance and survival.
SV5 binds less efficiently to tumor cells, but spreads farther compared with T3wt
SV5 treatment reduced tumor growth and improved animal survival relative to T3wt in vivo, but while both T3wt and SV5 promoted markers of immunotherapeutic value, differences in immune activation could not explain the superior oncolytic activity of SV5. Moreover, SV5 produced large plaques on breast cancer monolayers, and therefore clearly exhibits an evident advantage in the absence of immune cells. To better understand which characteristics of SV5 confer enhanced oncolytic activity, we next asked why SV5 produced larger plaques on breast cancer cells. As described in the introduction, reovirus infection involves a series of steps: cell attachment (binding), uncoating to cores, virus factory formation, RNA and protein synthesis, new virus assembly, cell lysis, and dissemination to neighboring cells. Increased plaque size could result from enhancement of any one or more of these steps. Two of the five mutations in SV5 were previously demonstrated to affect specific steps of reovirus replication. The σ1S18I mutation increased the probability of reovirus uncoating to cores,33 while the μ2A612V mutation increased formation of factories.34 But individual mutations also came with costs to other steps of virus replication; for example, μ2A612V decreased viral RNA synthesis. Since benefits of one mutation might be undone by costs of another mutation, it was pertinent to establish which steps of virus replication were enhanced in SV5; in other words, what were the ultimate efficiencies of each step of virus replication when all mutations were combined.
To assess the efficiency of virus attachment to tumor cells, L929 cells were exposed to input virus particles (“input”) and incubated at 4°C for 1 h thus permitting attachment without entry. Cells were then washed extensively to remove unbound virus particles before harvesting the lysate. Post-binding lysates and inputs were subjected to western blot analysis, and percentage of binding was calculated based on virus protein levels in lysates versus input. There were no significant differences in cell attachment among the single mutated reoviruses and T3wt (Figure 6A). Conversely, SV5 displayed a significant 40% binding reduction relative to T3wt. The requirement for combinations of mutations in SV5 to significantly reduced binding could be attributed to an additive effect of several mutations that each modestly reduce binding such as the σ1S18I and σ3K64E, or because combined mutations affect the virus in distinct ways relative to any individual mutation. The reduced binding of SV5 relative to T3wt was also consistent on TUBO cells (Figure S10).
Figure 6.

SV5 displays reduced binding but increased spread compared with T3wt in L929 cells
(A) Binding of T3wt, SV5, and single mutated viruses on L929 cells. Percentage of binding was calculated by western blot analysis comparing the amount of viral proteins bound to the cells at 0 hpi versus the input for each virus (n = 3). ∗p < 0.05; ns, not significant (ANOVA with Tukey test). (B) Representative agarose gel showing σ1 levels in intact virions. 1% agarose gels were loaded with the same number of virus particles. Band migration corresponds to σ1 trimer levels per virion (n = 3).52 (C) Graph showing the amount of virions presenting determined σ1 levels (n = 3). (D and F) Single-step growth curves showing cellular, total, and released titers (n = 2). L929 cells were infected at an MOI that infected more than 70% of the cells (checked by flow cytometry). Cellular lysates and supernatants (released titers) were collected at 0, 3, 6, 9 12, 15, 18, 24, 30, and 36 hpi and titered in L929 cells. Total titers were calculated as the sum of cellular and released titers. (G) Cell death evaluated by Zombie Aqua staining (by flow cytometry). Top: cell death evaluated at 24 hpi when L929 cells were infected at MOIs of 3 and 27 PFU/cell of T3wt or SV5. Bottom: cell death measured at different times of infection (15, 18, 24, 30, and 36 hpi) when L929 cells were infected at an MOI of 27 PFU/cell of T3wt or SV5. (H) Representative immunofluorescence images of T3wt and SV5 plaque formation at days 2, 3, and 4 post-infection (n = 3). In pink is anti-reovirus staining (rabbit polyclonal anti-reovirus), in green the σNS staining (non-structural reovirus protein, 2A9 mouse monoclonal antibody), in yellow the colocalization of both colors, and in blue the nuclei. Scale bars, 500 μm. (I) Quantification of the fluorescence from the edge of the circular plaque outward into the neighboring monolayer of at least 50 plaques per day post infection. Each separate line represents the average of the fluorescence presence from the edge of the plaque per plaque. Thick colored lines were calculated using a local polynomial regression fitting formula (LOESS).
Previous studies found that reovirus particles with fewer than three σ1 cell attachment proteins cannot attach efficiently to L929 cells.52 As a possible mechanistic basis for reduced binding by SV5, the levels of σ1 on the single mutated reovirus particles and SV5 were compared with T3wt. Purified virions were subjected to agarose gel electrophoresis and imperial protein staining; an approach that separates reovirus particles based on σ1 level per particle ranging from 0 to 12.52 While reduced σ1 levels were observed for virus particles of SV5 and single mutants σ1S18I and λ2I1274T (Figures 6B and 6C), there was no relationship between σ1 levels and reduced cell attachment. For example, σ1S18I (38% of which have ≥3 σ1 molecules per virion) and λ2I1274T (92% of which have ≥3 σ1 molecules per virion) mutants bound cells as efficiently as T3wt (95% of which have ≥3 σ1 molecules per virion). Therefore, the reduced attachment of SV5 to tumor cells is likely for reasons beyond simply having fewer σ1 molecules.
That SV5 bound less than T3wt to tumor cells was puzzling since we were expecting a step of virus replication to be enhanced rather than decreased in this more oncolytic mutant. We therefore continued to evaluate other steps of reovirus replication in search of enhancements. When evaluating post binding steps, the binding between T3wt and SV5 was equalized by adding higher amounts of SV5 virus particles to achieve equal numbers of cell-bound virus particles at onset. Virus uncoating was measured by the appearance of the δ fragment, the cleavage product of the outer capsid protein μ1C, at 2 and 4 h post infection (hpi) by western blot analysis. There were no differences between T3wt and any of the single mutated viruses or SV5 in uncoating (Figure S11A). Next, de novo viral protein synthesis was assessed by monitoring the levels of de novo μ1 protein at 6 hpi and was found to be similar between T3wt and the mutants (Figure S11B). To evaluate if SV5 produced more infectious virions than T3wt in a single replication cycle, L929 cells were infected at high multiplicities of infection (MOIs) and the kinetics of progeny virus production was quantified by plaque titration. One round of reovirus replication is ∼24 h, so titers at time points from 0 to 36 h were measured. Surprisingly, SV5 produced similar intracellular and total titers as T3wt, suggesting that there were no enhancements during virus production (Figures 6D and 6E). Finally, the efficiency of virus release and cell death was compared between T3wt and SV5. To measure virus release, extracellular virus titers were compared in a one-step growth curve and found to be similar (Figure 6F). To determine the percentage of dead cells, infected L929 cells were subjected to Zombie Aqua staining, and live/dead cells detected by flow cytometric analyses. At MOIs of both 3 and 27 plaque-forming units (PFU)/cell, and at various time points post infection, similar percentages of cell death were associated with SV5 and T3wt (Figure 6G). Altogether, SV5 bound less efficiently to cells, but otherwise performed equally to T3wt in subsequent steps of replication, cell death, and virus release.
SV5 bound less to cells, did not produce or release more infectious virions, yet still made larger plaques. To understand the mechanism of increased plaque size, the morphology of the plaques was visualized in a greater detail. Virus plaques were imaged at days 2, 3, and 4 post infection and reovirus-infected cells were detected by immunofluorescence using reovirus-specific antibodies. In representative images, SV5 plaques showed infected cells farther from the center-of-foci compared with T3wt as early as at 2 days post infection (dpi), which became further exaggerated at 3 and 4 dpi (Figure 6H). Moreover, it appeared that, regardless of the size of the zone of cell lysis at 4 dpi, there were infected cells at farther distance from the edge of the “zone of cell lysis” for SV5 (Figure 6H). To quantitatively assess the distance of infected cells, fluorescence presence relative to distance from the zone of cell lysis was measured, which showed that SV5 consistently infected cells more distally relative to T3wt (Figure 6I). Altogether, despite equivalent numbers of new viruses produced and released by infected cells, SV5 virions can infect cells that are farther away from the source of infection than T3wt can.
SV5 spreads farther than T3wt due to its reduced binding to cells
To our knowledge, this is the first time that increased oncolysis by reovirus corresponds with increased spread distance, and therefore we wanted to understand why SV5 infects cells at a farther distance relative to T3wt. In plaque assays, cells are overlaid with media stiffeners, such as agar, to reduce virus diffusion and contain infection to a localized focus. Similarly, the tumor microenvironment is composed of stiffeners such as the extracellular matrix. One possible explanation for the increased distance of spread was that SV5 might diffuse faster through the matrix, for example, by having a more condensed structure. To assess diffusion through agar in the absence of cells, equivalent numbers of SV5 and T3wt virus particles were added on top of a column of solidified agar. After 5 days of passive diffusion, the gel column was cut into sequential fractions of 100 μL, melted, and subjected to western blot analysis for reovirus proteins. While the majority of virus was maintained in the first fraction, similar levels of SV5 and T3wt diffused to the remaining fractions (Figure 7A).
Figure 7.

SV5 spreads farther than T3wt due to its reduced binding to cells
(A) Diffusion of T3wt and SV5 in 0.5% agar (same agar concentration that was used for plaque assays) in the absence of cells (n = 7). Each point represents an independent experiment. Colored lines were calculated using a local polynomial regression fitting formula (LOESS). (B) Consecutive-binding experiment showing the infectivity of unbound T3wt and SV5 virus particles to L929 cells over 10 consecutive binding opportunities (n = 4). Percentage of infected cells was evaluated by flow cytometry and the percentage of cells infected by T3wt was normalized to 1. p values were calculated with a Student’s t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant. (C) Plaque assays to evaluate plaque size of T3wt and SV5 in the presence of different concentrations of neuraminidase (0, 0.6, 1.2, 2.4, and 9.6 U/mL) to reduce virus binding (n = 3–4). T3wt and SV5 without neuraminidase were normalized to 1 in each of the plates. p values were calculated with a one-sample t test with multiple comparison adjustments. ∗p < 0.05, ∗∗p < 0.01; ns, not significant, relative to T3wt or SV5 without neuraminidase treatment.
One of the most remarkable features of SV5 was that it bound less to the cells (Figure 6A). Consequently, we wondered if the reduced binding to cells allowed the virus to move farther away from the initial infected cell, increasing the distance that the virus could spread. In other words, rather than 1,000 viruses released by the producer cells binding immediately to neighboring cells (note that the resulting high MOIs of neighboring cells would provide minimal advantage for the virus), perhaps reduced binding permits virus particles to diffuse to cells farther away from the site of production. To address this possibility, it was first important to evaluate if the unbound virus particles maintain their ability to infect new cells. A “consecutive-binding experiment” was developed, in which T3wt or SV5 were permitted to bind L929 cells for 15 min at 4°C to avoid virus internalization. The time for absorption was chosen to be 15 min based on optimization experiments demonstrating sufficient but sub-saturated binding at this duration (Figure S12A). Every 15 min, the unbound particles were transferred to fresh uninfected cell monolayers and provided an opportunity to bind for 15 min at 4°C again. This process was repeated for a total of 10 consecutive binding opportunities. After exposure to virus, the monolayers were washed twice and incubated in complete medium for 15 h to permit infection by bound virions. The percentage of infected cells was evaluated by flow cytometric analysis with reovirus-specific antibodies, and the ratio of infection was calculated comparing SV5 versus T3wt for each consecutive binding opportunity. Remarkably, binding and infection capacity of SV5 surpassed T3wt at the fourth binding opportunity and increased at each consecutive binding opportunity thereafter (Figure 7B). In addition, higher amounts of SV5 particles were maintained in the supernatants after each passage relative to T3wt (Figures S12B and S12C). These experimental results suggest that SV5 particles have a lower propensity to bind to the tumor cells and therefore remain unbound in the supernatant but retain the ability to bind tumor cells in future opportunities. In plaque assays and tumors, this phenomenon could lead to virus particles that travel a farther distance before re-infection, thereby expanding the zone of infection.
Finally, given the counter-intuitiveness of suggesting that reduced binding serves as a benefit to virus activity, a direct test was necessary to evaluate the relationship between binding and reovirus-induced plaque size independent of SV5 with its many mutations. It has been shown that reovirus binding to cells requires JAM-A and sialic acids.53 To reduce reovirus cell binding efficiency independent of mutations, the sialic acid-digesting enzyme neuraminidase was added to the plaque assays, and effects on plaque size were monitored. Neuraminidase was added at various concentrations to achieve conditions where sialic acids were reduced but not eliminated. If reduced binding favors larger distance of virus spread, then T3wt plaque size should increase in the presence of intermediate levels of neuraminidase. T3wt produced larger plaques in the presence of neuraminidase, reaching 1.78-fold larger at 0.6–1.2 U/mL of neuraminidase relative to the untreated control (Figure 7C). As neuraminidase concentrations were further increased beyond 1.2 U/mL, there was a dose-dependent reduction in the benefits to plaque size, suggesting that there is an optimal reduction in binding that leads to farther spread of reovirus. Interestingly, SV5 plaques also increased in size in the presence of neuraminidase, suggesting that SV5 might benefit from an even further reduction in cell binding than already achieved. Altogether, several approaches support the hypothesis that, for reovirus, reduced virus binding to the cells favors increased distance of virus spread.
Discussion
The Patrick Lee lab strain of reovirus serotype 3 Dearing (T3wt) has progressed to phase 2 clinical testing for various cancers including, breast cancers.8,14 Results from these trials so far indicate that T3wt is safe for human use and exhibits some positive effects on controlling the disease among certain patients, but that T3wt as a monotherapy would benefit from increased potency.11,15,16,54 Our study focused on identifying mutations in the reovirus that enhance its oncolytic potency both in vitro and in vivo. Our experimental results revealed that a reovirus variant called SV5, which contains five specific mutations, significantly enhanced reovirus oncolysis relative to T3wt. Paradoxically, the molecular basis for enhanced oncolysis of SV5 involves reduced efficiency in cell binding, allowing the virus to spread greater distances (Figure 8). In other words, SV5 virus particles can travel farther from the initial virus burst foci and infect cells at a greater distance. We also found SV5 to be a superior OV in vivo. In mice with TUBO breast tumors, both SV5 and T3wt exhibited the ability to recruit CD8+ T cells and reduce MDSCs, demonstrating their potential as immunotherapeutic agents. However, SV5 induced significantly greater tumor regression and prolongation of animal survival relative to T3wt.
Figure 8.

Final model
Compared with tumors treated with PBS, those injected with T3wt or SV5 viruses exhibited slower growth rates. However, SV5 was found to be more effective in reducing tumor growth than T3wt. In some mice, SV5 induced total tumor regression and significantly prolonged survival. Even though both viruses increased the CD8+/CD4+ T cell ratio in the tumors and decreased the MDSCs, SV5 demonstrated a greater oncolytic potential. In vitro results indicated that SV5 spreads farther than T3wt because its binding to cells is reduced. Since SV5 cannot bind efficiently to cells, it can move to cells farther away from the original infected cell, infecting these more distant cells, which could explain the enhanced oncolytic effect observed in vivo.
The most important new concept suggested by our study is that lower efficiencies of cell binding can be advantageous to virus fitness in the tumor niche. Reovirus, being a naturally occurring enteric virus, has evolved under the selective pressures associated with oral-fecal routes of infection. In the fast-flowing gut environment, it is believed that viruses depend on strong cell attachment to establish infection; this would explain the strong cell attachment capacity of the wild-type reovirus. Conversely, solid tumors exist under high pressure with less fluid movement,55,56 which is known to hamper therapeutic uptake but could similarly also restrict diffusion of therapeutic viruses. Unlike the fast-flowing enteric niche where reovirus binding must occur rapidly, the more-static tumor environment may favor dissemination for reovirus variants that have less-efficient cell binding.
Viruses with reduced binding to cells have been previously described in nature. For instance, reovirus variants with reduced binding to sialic acid have been isolated from both murine and human species.57,58 In addition, when infecting the apical or basolateral sides of polarized epithelial cells, a sialic-acid-binding-deficient reovirus variant exhibits higher infection than wild-type reovirus,59 suggesting a potential advantage of reduced binding during a natural infection. Similarly, rotavirus mutants that cannot bind to sialic acid exhibited slower replication and lower titers in MA104 cells, yet increased pathogenicity in mice.60 These findings demonstrate that the loss of binding to sialic acid by rotaviruses or reoviruses can confer benefits under specific conditions. The ability of a virus to spread farther in a monolayer of cells without increased virus replication has also been previously depicted for other viruses. With vaccinia virus, infected cells repel superinfecting virions, leading viruses to spread farther as we observed with SV5.61,62 With phage, decreased adsorption rate to host bacteria can also lead to an increase in plaque size.63 Furthermore, several virus variants of polyomavirus, parvovirus, and Sindbis virus with deficiencies in binding have been shown to generate larger plaques in vitro, and they have exhibited higher pathogenicity and greater spread in vivo.64,65,66,67
Our findings support that OVs that are better at dissemination in cancer cells also provide benefits to oncolytic potency in vivo. Many OVs have demonstrated safety and some efficacy in clinical trials, but insufficient oncolytic potency as monotherapies. Virus-based oncolytic therapy depends both on the direct activity of viruses on tumor cells themselves, in addition to stimulation of anti-tumor immunity. There is currently strong focus on enhancing the immunotherapeutic value of OVs through their combination with immune modulators such as checkpoint inhibitors.68,69,70 By demonstrating the beneficial nature of combining mutations in an oncolytic reovirus, our findings support a continued complementary investment into further advancing OVs to most effectively replicate and disseminate in the tumor environments. Oncolytic viruses capable of infecting and spreading farther within tumors would not only directly kill more tumor cells but also presumably release more tumor antigens, promote greater cytokine production, and facilitate enhanced immune cell recruitment. Although we did not observe significant differences in immune cell populations between T3wt and SV5 at 14 days post treatment, we did observe strong correlations between virus-induced tumor regression and the expression of various murine interferon- and Nfkb-specific host genes and cytokines. Precisely comparing the immunotherapeutic benefits between OVs at later time points poses several challenges, such as that when same endpoint dates are chosen, tumor sizes are drastically different due to differences in tumor regression. Conducting a large-scale time-course study with a substantial number of animals per group and multiple endpoints, while beyond the scope of this already comprehensive study, could significantly contribute to precisely associating virus oncolytic potency with specific changes in the immunological milieu. Similarly, investigating the distinctions between intravenous and intratumoral administration of T3wt and SV5 could provide valuable insights into virus distribution within the tumor and other organs, potentially positioning SV5 as a promising candidate for future reovirus therapy.
Finally, we turn to the specific inquiry of how mutations in SV5 contribute to the reduced efficiency of binding. As previously mentioned, no single mutation alone reduced the efficiency of cell attachment. It is crucial to understand that reovirus attachment to cells is a complex, multi-step process. In the case of T3wt, the tail domain of σ1 facilitates low-avidity attachment to sialic acid, while the head domain of σ1 enables high-avidity interactions with cellular JAM-A receptors.71 Conformational changes in σ1 subsequently facilitate interactions between λ2 turrets and β1 integrins.72,73 In addition, the outer capsid protein σ3 has been implicated in cellular binding through the neuronal Nogo receptor NgR1, although the specific domains of σ3 involved in this interaction have not been elucidated.74 Although the I1274T mutation in λ2 and the S18I mutation in σ1 do not directly reside in the binding domains for integrins or JAM-A/sialic acids, the combination of these mutations could potentially impact the required conformational changes. Similarly, mutations in the outer capsid protein σ3, inner capsid protein λ1, and even the interior-situated μ2 could influence the overall capsid structure and conformational flexibility, thereby affecting the availability of binding domains. Accordingly, to precisely determine how each mutation contributes to virus binding efficiency, it is necessary to test every combination of the five mutations to establish which mutations do or do not contribute to the reduction in binding efficiency.
In conclusion, this research has unveiled the significance of introducing multiple genetic modifications to augment the oncolytic activity of reovirus. Through the targeting of increased virus spread, we have successfully engineered an oncolytic reovirus that exhibits superior potency against breast cancer. Furthermore, this investigation has contributed significantly to our understanding of reovirus biology. Particularly noteworthy is the insight gained into the critical role of the outer capsid reovirus proteins in in vitro oncolysis. Moreover, we have demonstrated that a farther virus spread could be achieved through reduced binding to cells, a previously unreported advantage for reovirus or any virus in the context of oncolytic therapy.
Materials and methods
Animals
Animal experiments were performed according to the Canadian Council on Animal Care Guidelines and Policies with approval from the Animal Care and Use Committee: Health Sciences for the University of Alberta (Edmonton, Alberta, Canada). Female BALB/c mice between 6 and 8 weeks of age from Charles River were used for the tumor model experiments. They were housed in a biosafety level 2 containment suite in high-efficiency particulate air-filtered ventilated cages. They received LabDiet food and water ad libitum and had environmental enrichment in their cages.
Cell lines
All cell lines were cultured in a CO2 incubator at 37°C. L929, H1299, B16-F10, and BJ cells were generously provided by Dr. Patrick Lee at Dalhousie University. NIH3T3, PanC, T47D, and MCF7 cells were purchased from the American Type Culture Collection. BHK-T7 cells were a generous gift from Dr. Ursula Buchholz at NIH. TUBO mouse mammary carcinoma cells were provided by Dr. Landuzi from the Istituto Ortopedico Rizzoli. Prior to animal injections, TUBO cells were passed through a 21½G syringe to obtain single-cell suspensions. All cell lines were cultured in medium containing 1 mM sodium pyruvate (Sigma, no. S8636), 1% nonessential amino acids (Sigma, no. M7145), and fetal bovine serum (FBS) (Invitrogen, no. 102483-020). Specifically, L929 and BJ cells were cultured in minimal essential medium (MEM) (Sigma, no. M4655) supplemented with 10% FBS. H1299, T47D, and MCF7 cells were cultured in Roswell Park Memorial Institute (RPMI 1640) (Sigma, no. 8758) supplemented with 10% FBS. B16-F10, NIH3T3, PanC, and TUBO cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma, no. D5796) supplemented with 10% FBS. BHK-T7 cells were maintained in DMEM containing 5% FBS and supplemented with G418 (Sigma, no. A1720) every second passage.
Plaque assay
A confluent monolayer of the indicated cell line was infected with an amount of virus to allow the visualization of individual and distinct plaques. After 1 h of incubation at 37°C rocking gently every 5–7 min, a 0.5% overlay of agar was added to the cells. The overlay was prepared by mixing 2% agar (BD, no. 214010), 2× Joklik’s modified essential medium (JMEM) (Sigma, no. M0518), and 1× MEM in a 1:1:2 ratio. Once the agar solidified, the cells were returned to the 37°C incubator for 5–8 days, depending on the cell line. Subsequently, the cells were fixed with 4% paraformaldehyde (PFA). In MCF7 and TUBO cells, immunocytochemical staining was performed to visualize the plaques with a polyclonal anti-reovirus antibody as previously described.39 In the case of L929 and T47D cells, a 1% (wt/vol) crystal violet solution was used to stain the cellular monolayer. Plaque size analysis was conducted using the Fiji software with the particle analysis plugin. Most of the plaque size results are expressed as relative plaque size to T3wt, with T3wt normalized to a value of 1. In each independent experiment, a well containing T3wt was included, and each plate was normalized to T3wt. Plaque size analyses were performed with a minimum of three independent experiments in duplicate. In experiments involving neuraminidase (Sigma, no. N2876), the neuraminidase was diluted in MEM without supplements at concentrations of 0.6, 1.2, 2.4, and 9.6 U/mL. It was then added to the overlays of the plaque assay after virus absorption. Plaque assays were completed as described above.
Isolation of large-plaque forming viruses
Reovirus variants were isolated as described previously.34 In brief, L929 cells were infected with T3wt at an MOI of 1 in the presence of 200 μM of the mutagen 5-fluorouracil (Sigma, no. F6627-1G). Viruses from the cellular lysates were isolated in a 30% sucrose cushion by centrifugation at 100,000 × g for 1 h at 4°C. Plaque assays with these viruses were done and individual plaques were picked using sterile cotton-plugged glass Pasteur pipettes and rubber bulbs. Selected plaques were carefully extracted, dispensed into 1.5 mL centrifuge tubes with 200 μL of MEM (no additives), and incubated overnight at 4°C. Each of these viruses were sequenced to identify distinctive mutations relative to T3wt.34
Viruses
Except for the isolated viruses in Figure 1A, all the recombinant viruses utilized in this study were generated through site-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, no. 200524) and reverse genetics.75,76 Each T3D gene required for site-directed mutagenesis had been previously cloned into individual plasmids (pBacT7-S1T3D, Addgene, no. 33282) under the T7 promoter.76 The specific mutations were incorporated into the plasmids using the primers listed in Table S2. BHKT-7 cells were transfected with these plasmids, and lysates were collected after a 5-day incubation period. Cellular lysates obtained from BHK-T7 cells were then propagated in L929 cells and subsequently purified using ultracentrifugation on cesium chloride gradients, following established protocols.77
Evaluation of OVs in vivo: Tumor growth, survival, challenge experiment
A week post arrival to the animal facility, BALB/c mice were injected into the mammary fat pad with 1 × 106 of TUBO cells in 50% Matrigel (Corning, no. 354234). Once tumors reached a size larger than 50 mm3, which happened around 9–12 days post cell injection, mice were grouped based on similar tumor sizes. Virus solutions were prepared at 5 × 108 PFU (titers in L929 cells) in 50 μL for T3wt and the same infectivity levels (number of infected cells after 15 hpi) for the rest of the mutants checked by immunofluorescence. When tumors were palpable, intratumoral injections of viruses were administered three times, with each injection spaced every other day. Throughout the entire duration of the experiment, virus injections and tumor measurements were performed blindly. Mice were monitored twice daily, and tumor growth was measured with calipers twice per week. Tumor volume was calculated with the formula: tumor volume = (1/24) × π × tumor length × (tumor width + tumor height).2 Mice were euthanized when tumors reached 20 mm length in one dimension or 16 mm in two dimensions, and the endpoint was registered for each mouse. Survivor mice, at day 210 post first virus injection, were injected with fresh TUBO cells into the mammary fat pad opposite to the original injection site. Concurrently, four naive mice (6–8 weeks old) were injected with the same cells as controls. Tumor size was measured using calipers, and control mice were euthanized based on the same criteria described above. Since SV5-treated mice did not develop secondary tumors, they were maintained for an additional 6 weeks following the euthanasia of the last control mouse.
Processing of spleens and tumors
Spleens were collected at day 14 post first virus injection in isolation buffer (2 mM EDTA, 0.5% heat-inactivated FBS in PBS) on ice. They were weighed and cut into small pieces while maintaining the integrity of the tissue. Then they were mashed with a rubber plunger of a 3 mL syringe in a 70 μM pre-wet cell strainer. The strainer was washed with isolation buffer and all the filtered solution was centrifuged at 350 × g for 5 min at 4°C to recover the cells. The pellet was resuspended in red blood cell lysis buffer (BioLegend, no. 420301) and left to sit at room temperature for 5–10 min to lyse red blood cells. Cells were washed with isolation buffer and counted. Splenocytes (2 × 106) per sample per panel were stained for flow cytometric analyses.
Tumors were also collected at 14 days post first virus injection in cold HBSS buffer. After weighing them, tumors were cut into small pieces and added to GentleMACs C-tubes (Miltenyi Biotec, no. 130-093-237) containing medium with 0.5 mg/mL collagenase type 1A (Sigma-Aldrich, no. SCR103) and 10 μg/mL DNAse I (Roche, no. 03724751103). Tumors were dissociated using the GentleMACs dissociator (Miltenyi Biotec, no. 130-093-235) and the m-impTumor01.01 program, making sure that all the pieces were processed. Then the chunks of tumors were incubated in a shaker at 37°C and 250 rpm for 30 min to promote further dissociation. Tumor chunks were filtered through a pre-wet 70 μM cell strainer and the filtered solution was centrifuged at 350 × g for 5 min at 4°C to recover the cells. The pellet was resuspended in 40% Percoll (GE Healthcare, no. GE17-0891-01) and carefully overlaid in 80% Percoll. The gradient was centrifuged at 325 × g for 30 min at 4°C with a deceleration of zero and an ascending rate of five. Immune cells can be observed in a distinguished band between both Percoll solutions. To collect the band, the volume on top was carefully removed with a micropipette and then the band was carefully extracted and transferred to a clean separate tube. These cells were washed with isolation buffer and centrifuged at 500 × g for 10 min at 4°C. Tumor cells were resuspended in PBS and divided between the samples for flow cytometric analyses.
Immune cell frequency analysis by flow cytometry
Each sample derived from a tumor or a spleen was divided for two panels (innate immune cells and lymphocytes) and at least 10% of the sample was pooled together with the other samples for controls such as fluorescence minus one (FMO) and uninfected cells. Cells were washed with cell-staining buffer (BioLegend, no. 420201) and incubated with the Zombie Aqua viability dye (BioLegend, no. 423102) for 15 min at room temperature. After washing, cells were incubated for 5 min at room temperature with Fc block solution (TruStain FcX, anti-mouse CD16/32 antibody, BioLegend, no. 101320). Then, the conjugated primary antibodies (see Tables S3 and S4) were added to the samples and the FMO controls and incubated for 20 min at 4°C in dark. After two washes, cells were fixed with the fixation buffer (BioLegend, no. 420801). Finally, cells were analyzed in an AttuneNxT Acoustic Focusing Cytometer and data analyzed with the FlowJo software (BD).
Tumor processing for RNA-seq
Tumors were collected at day 14 post first virus injection and frozen at −80°C. After thawing on ice, tumors were minced and the pieces were added to 500 μL of sterile PBS in Lysing Matrix A tubes (MP Biomedicals, no. 6910050). These tubes containing ¼ inch ceramic beads were placed in Fastprep-24 Classic bead beating grinder and lysis system (MP Biomedicals, no. 6004500). To dissociate tumors, 2 to 3 cycles of 30 s at 6 m/s were done. To 200 μL of this lysate, 800 μL of TRIzol LS (Invitrogen, no. 10296010) was added. RNA was then extracted using a GenElute Mammalian Total RNA miniprep kit (Sigma, no. RTN350) following the manufacturer’s instructions. Poly(A) RNA-seq was performed by LC Sciences in Houston, TX.
Binding, uncoating, and de novo protein production
Confluent L929 cells were pre-chilled at 4°C for 1 h to prevent virus internalization by endocytosis. Virus inoculums (input) were added to the cells at an MOI in which similar binding resulted among viruses. Absorption of viruses was done for 1 h at 4°C mixing every 5–7 min. Unbound virus was removed, and cells were washed twice with MEM without supplements. For binding, cellular lysates were collected right after adding complete medium. After a couple of washes with PBS, radioimmunoprecipitation assay buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate) with protease inhibitors (Millipore Sigma, no. 11697498001) was added to the cells. After 5–10 min at 4°C, detached cells were collected and centrifuged at 800 × g for 10 min at 4°C to remove cellular DNA. After the centrifugation, the supernatant was mixed with protein sample buffer (50 mM Tris-HCl [pH 6.8], 1% sodium dodecyl sulfate [SDS], 9% glycerol, 1.8% β-mercaptoethanol, 0.01% bromophenol blue) and the mixture boiled at 100°C for 10 min. For the uncoating assay, cells were collected as described above at 2 and 4 hpi. For de novo protein production, lysates were collected at 6 hpi. To analyze binding, uncoating, and de novo protein production, the respective lysates were subjected to western blot analysis. Samples were loaded onto SDS-acrylamide gels, then separated proteins were transferred onto nitrocellulose membranes (Amersham/GE, no. 10600003) using the Trans-Blot transfer system (Bio-Rad). Membranes were blocked with 3% newborn calf serum (NCS) with 0.1% Tween 20 in Tris-buffered saline (TBS) for 1 h and then incubated overnight at 4°C with primary antibody. In all cases, a polyclonal anti-reovirus antibody was used to detect reovirus outer capsid proteins, such as σ3 and μ1, and a mouse β-actin (8H10D10) antibody (Cell Signaling, no. 3700) was used to normalize the samples. After the incubation with primary antibody, membranes were washed thrice with TBS 0.1% Tween 20 and incubated with an anti-rabbit horseradish peroxidase (HRP)-conjugated antibody (Jackson ImmunoResearch, no. 111-035-144) to detect reovirus bands and with an anti-mouse Alexa Fluor 647 antibody (Jackson ImmunoResearch, no. 115-605-146) to detect actin bands. ECL Plus Western Blotting Substrate (Thermo Fisher Scientific Pierce, no. 32132X3) was used as an HRP substrate. Membranes were visualized using an ImageQuant LAS4010 imager (GE Healthcare Life Sciences) and the intensity of the bands was calculated with ImageJ with the gel analyzer function. For binding, input samples (virus inoculums) were compared with 0 hpi samples. The difference between both resulted in the percentage of binding. For uncoating, lysates at 0, 2, and 4 hpi were compared and the appearance of δ fragment (characteristic of μ1C cleavage) at 2 and 4 hpi, was contrasted with the presence of μ1C at 0 hpi. For de novo protein production, 0 versus 6 hpi were compared. The difference between both time points indicated the increase in protein synthesis.
σ1 levels with agarose gels
Equivalent virus particles (calculated by absorbance at 260 nm) were diluted in water and mixed with a loading buffer containing 5% Ficoll and 0.05% bromophenol blue. Then, the virus particles were loaded in a 1% agarose gel and ran at 100 V for 2 h at room temperature. The resulting gel was stained with imperial protein stain for 2 h at room temperature. After an overnight distain with distilled water, bands characteristic of σ1 trimers were detected in the gel.52 Images of the gels were captured by an ImageQuant LAS4010 imager (GE Healthcare Life Sciences).
Virus growth
For the single-step growth curves, a 90%–100% confluent monolayer of L929 cells was infected with a sufficient MOI to infect more than 70% of the cells. At various time points (3, 6, 9, 12, 15, 18, and 24 hpi), supernatants were collected as well as the cellular lysate by scraping the monolayer of cells. Both fractions were subjected to three cycles of freeze-thawing and then titered on L929 cells. The supernatant fraction corresponded to the “released titers” fraction and the cell monolayer was the “cellular titers” fraction. The sum of both fractions represented the “total titers.”
Cell death assay
L929 cells were infected with T3wt or SV5 at an MOI of 3 or 27, for 15, 18, 24, 30, and 36 h. After the infection, the supernatants containing dead cells were collected and added to a 5 mL flow tube, then L929 infected cells were detached with 500 μL of CellStripper dissociation buffer (Corning, no. MT250566CI), and added to the same tube. Cells were washed twice with PBS and incubated with Zombie Aqua viability dye (BioLegend, no. 423102) for 20 min at room temperature. Afterward, cells were washed once with PBS and incubated with 4% PFA for 40 min at room temperature. PFA was removed and cells were washed twice with the flow staining buffer (PBS, 1 mM EDTA, 2% FBS) and once with PBS. Cells were analyzed in a Cell Analyzer Fortessa (BD LSRFortessa) in the channel V525. Data were analyzed with the FlowJo software (BD). At each time point, reovirus-infected cells were used as a control.
Virus spread: L929 cells were infected with T3wt or SV5 at a concentration to visualize between 50 and 100 separated plaques in a well from a 6-well plate. A plaque assay was conducted as previously described, but instead of stopping it at endpoint, it was stopped at days 2, 3, and 4 post infection. Cells were fixed with 4% PFA for 40 min at room temperature. Then the agar plugs were removed, and cells were washed twice with PBS. Cells were then incubated with blocking solution (3% NCF and 0.01% Triton X-100 in PBS) for 1 h at room temperature and left in primary antibody (rabbit polyclonal anti-reovirus antibody) at 4°C overnight. Cells were washed thrice with PBS 0.01% Triton X-100, and then incubated with DAPI and secondary antibody goat anti-rabbit Alexa Fluor 647 (Jackson ImmunoResearch, no. 111-605-144). After the incubation with secondary antibody, cells were washed thrice with PBS 0.01% Triton X-100. Plaques were imaged using an EVOS microscope (LifeTech, no. AMAFD1000). To study virus spread, images were analyzed with Fiji (ImageJ) using the Radial Profile plugin. First, a threshold was applied for the fluorescence corresponding to the anti-reovirus staining. Then a circular mask that covered all the plaque was added, and this was used to define the plaque area and its edge. A radial analysis was done for each plaque to measure the presence of fluorescence in the monolayer at specific distances relative to the center of the plaque (this measurement is called total distance of spread). Finally, to measure the distance of spread from the edge of the plaque into the intact monolayer, the radius of the plaque was subtracted from the total distance of spread (i.e., from the center of the plaque). The spread of virus into the intact cell monolayer is presented as average fluorescence presence per plaque at the indicated distances from the edge of the plaque. A minimum of 50 plaques per virus at 2, 3, and 4 dpi were quantified.
Virus diffusion assay
To evaluate virus diffusion in the absence of cells, the same overlay used for plaque assays (0.5% agar in MEM), was allowed to solidify in a 1 mL syringe barrel with the hub previously cut off (BD, no. 309659). After 30 min, equivalent virus particles of either T3wt or SV5 were added at the top of the syringe. After 5 days at room temperature, gel-containing syringes were placed at 4°C for 1 h. Gels were removed intact from the syringe using the plunger and cut with a razor into 100 μL pieces using the syringe barrel as volume reference. Each of the pieces was mixed with protein sample buffer (50 mM Tris-HCl [pH 6.8], 1% SDS, 9% glycerol, 1.8% β-mercaptoethanol, 0.01% bromophenol blue) and boiled for 10 min. Virus proteins in the samples were separated by SDS-PAGE and subjected to western blot analysis as described above. The presence of reovirus proteins in each fraction was identified with the rabbit polyclonal anti-reovirus antibody. Band intensities of reovirus outer capsid proteins σ3 and μ1C were quantified with Fiji ImageJ with the gel analyzer function.
Consecutive infection assay
T3wt or SV5 were added to chilled L929 cells at an MOI to infect a similar percentage of cells (∼80%). After absorption at 4°C for 15 min, the supernatant was transferred to another monolayer of cells. This procedure was repeated for 10 consecutive infections. After the 15 min of absorption, cells were washed twice with MEM with no supplements at 4°C. Then, complete MEM was added to the wells and cells were placed in the incubator at 37°C, 5% CO2 for 15 h. At 15 hpi, cells were washed twice with PBS, detached using CellStripper dissociation buffer (Corning, no. MT250566CI) and mixed immediately with 4% PFA. Cells were fixed for 40 min at room temperature. After fixing, cells were washed with flow staining buffer twice and incubated with blocking buffer (3% NCS and 0.01% Triton X-100 in PBS) for 1 h at room temperature. Then, cells were incubated overnight with primary polyclonal anti-reovirus antibody at 4°C. The next day, cells were washed thrice with flow staining buffer and incubated with secondary antibody goat rabbit 647 (Jackson ImmunoResearch, no. 111-605-144) for 1 h at room temperature. Cells were then washed thrice with flow staining buffer and left in PBS. Cells were analyzed in the Cell Analyzer Fortessa (BD LSR Fortessa) in channel R670. Data was analyzed with FlowJo software (BD).
Data analysis, representation, and statistics
Data were analyzed with Microsoft Excel version 16.88, FlowJo software (BD), Fiji ImageJ, and R 3.6 (with RStudio 1.2) using the tidyverse 1.3 package.78 A Student’s t test was used to compare the differences between two samples. To compare the differences among more than two samples, an ANOVA with Tukey as post-test or Student’s t test with the Holm adjustment method for multiple comparisons was used. Figures were made with ggplot 2.2. package (from tidyverse 1.3).78
Data and code availability
For access to the data supporting the findings of this study, please contact the corresponding author.
Acknowledgments
This publication is made possible through project grants awarded to M.S. and M.M.H. by several organizations, including the Li Ka Shing Institute of Virology (LKSIoV), the Canadian Cancer Society Research Institute (CCSRI), the Cancer Research Society (CRS), the Canadian Breast Cancer Foundation (CBCF), and the Canadian Institutes of Health Research (CIHR). M.S. also receives support from the Canada Research Chairs (CRC) through a salary award, and infrastructure support from the Canada Foundation for Innovation (CFI). Furthermore, F.C.’s funding for this work comes from a range of sources, including the John and Rose McAllister Graduate Scholarship award from the Faculty of Graduate Studies and Research at the University of Alberta, the Faculty of Medicine & Dentistry (FoMD) Dean’s Doctoral Award from the University of Alberta, the LKSIoV Doctoral Award, and the La Vie en Rose Scholarship for Breast Cancer Research from the Cancer Research Institute of Northern Alberta (CRINA). We express our appreciation to the Shmulevitz and Hitt laboratories for their invaluable insights and guidance in this project. Special thanks to Dr. Aja Rieger, the Flow Cytometry and High Content Analysis Core Manager at the Faculty of Medicine & Dentistry, for her instrumental role in designing the multi-color flow cytometry panels. We are also grateful to the NIH tetramer facility at Emory for providing the tetramer used in this study. Dr. Heather Eaton’s manuscript revisions and technical support are greatly appreciated. Finally, we extend our sincere thanks to Dr. Tomás Gutiérrez for his assistance in analyzing the RNA-seq data and for his guidance on the use of the R software.
Author contributions
F.C. was in charge of developing the methodology, conducting the experiments, collecting, and analyzing the data, researching the literature, writing the manuscript, and doing the editorial revisions. M.W. optimized the agarose gels to evaluate σ1 levels. Figure 6B was her contribution. N.N. characterized the plaque size of the original isolated mutants contributing with the data for Figure 1A. K.A. assisted with the animal experiments. M.M.H. and M.S. contributed with study supervision, technical and experimental guidance, and manuscript and editorial revisions.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omto.2023.100743.
Supplemental information
References
- 1.Hashiro G., Loh P.C., Yau J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch. Virol. 1977;54:307–315. doi: 10.1007/BF01314776. [DOI] [PubMed] [Google Scholar]
- 2.http://www.oncolyticsbiotech.com/reolysin/clinical-trials/
- 3.Cohn D.E., Sill M.W., Walker J.L., O'Malley D., Nagel C.I., Rutledge T.L., Bradley W., Richardson D.L., Moxley K.M., Aghajanian C. Randomized phase IIB evaluation of weekly paclitaxel versus weekly paclitaxel with oncolytic reovirus (Reolysin(R)) in recurrent ovarian, tubal, or peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017;146:477–483. doi: 10.1016/j.ygyno.2017.07.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villalona-Calero M.A., Lam E., Otterson G.A., Zhao W., Timmons M., Subramaniam D., Hade E.M., Gill G.M., Coffey M., Selvaggi G., et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer. 2016;122:875–883. doi: 10.1002/cncr.29856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eigl B.J., Chi K., Tu D., Hotte S.J., Winquist E., Booth C.M., Canil C., Potvin K., Gregg R., North S., et al. A randomized phase II study of pelareorep and docetaxel or docetaxel alone in men with metastatic castration resistant prostate cancer: CCTG study IND 209. Oncotarget. 2018;9:8155–8164. doi: 10.18632/oncotarget.24263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernstein V., Ellard S.L., Dent S.F., Tu D., Mates M., Dhesy-Thind S.K., Panasci L., Gelmon K.A., Salim M., Song X., et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res. Treat. 2018;167:485–493. doi: 10.1007/s10549-017-4538-4. [DOI] [PubMed] [Google Scholar]
- 7.Clements D., Helson E., Gujar S.A., Lee P.W. Reovirus in cancer therapy: an evidence-based review. Oncolytic Virother. 2014;3:69–82. doi: 10.2147/OV.S51321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Müller L., Berkeley R., Barr T., Ilett E., Errington-Mais F. Past, Present and Future of Oncolytic Reovirus. Cancers (Basel) 2020;12:3219. doi: 10.3390/cancers12113219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karapanagiotou E.M., Roulstone V., Twigger K., Ball M., Tanay M., Nutting C., Newbold K., Gore M.E., Larkin J., Syrigos K.N., et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012;18:2080–2089. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Black A.J., Morris D.G. Clinical trials involving the oncolytic virus, reovirus: ready for prime time? Expert Rev. Clin. Pharmacol. 2012;5:517–520. doi: 10.1586/ecp.12.53. [DOI] [PubMed] [Google Scholar]
- 11.Galanis E., Markovic S.N., Suman V.J., Nuovo G.J., Vile R.G., Kottke T.J., Nevala W.K., Thompson M.A., Lewis J.E., Rumilla K.M., et al. Phase II trial of intravenous administration of Reolysin((R)) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol. Ther. 2012;20:1998–2003. doi: 10.1038/mt.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lolkema M.P., Arkenau H.T., Harrington K., Roxburgh P., Morrison R., Roulstone V., Twigger K., Coffey M., Mettinger K., Gill G., et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011;17:581–588. doi: 10.1158/1078-0432.CCR-10-2159. [DOI] [PubMed] [Google Scholar]
- 13.Comins C., Spicer J., Protheroe A., Roulstone V., Twigger K., White C.M., Vile R., Melcher A., Coffey M.C., Mettinger K.L., et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. 2010;16:5564–5572. doi: 10.1158/1078-0432.CCR-10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chaurasiya S., Fong Y., Warner S.G. Oncolytic Virotherapy for Cancer: Clinical Experience. Biomedicines. 2021;9:419. doi: 10.3390/biomedicines9040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sborov D.W., Nuovo G.J., Stiff A., Mace T., Lesinski G.B., Benson D.M., Jr., Efebera Y.A., Rosko A.E., Pichiorri F., Grever M.R., Hofmeister C.C. A phase I trial of single-agent reolysin in patients with relapsed multiple myeloma. Clin. Cancer Res. 2014;20:5946–5955. doi: 10.1158/1078-0432.CCR-14-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samson A., Scott K.J., Taggart D., West E.J., Wilson E., Nuovo G.J., Thomson S., Corns R., Mathew R.K., Fuller M.J., et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018;10:eaam7577. doi: 10.1126/scitranslmed.aam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macedo N., Miller D.M., Haq R., Kaufman H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer. 2020;8:e001486. doi: 10.1136/jitc-2020-001486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiao J., Wang H., Kottke T., White C., Twigger K., Diaz R.M., Thompson J., Selby P., de Bono J., Melcher A., et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin. Cancer Res. 2008;14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pandha H.S., Heinemann L., Simpson G.R., Melcher A., Prestwich R., Errington F., Coffey M., Harrington K.J., Morgan R. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin. Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]
- 20.Yang W.Q., Lun X., Palmer C.A., Wilcox M.E., Muzik H., Shi Z.Q., Dyck R., Coffey M., Thompson B., Hamilton M., et al. Efficacy and safety evaluation of human reovirus type 3 in immunocompetent animals: racine and nonhuman primates. Clin. Cancer Res. 2004;10:8561–8576. doi: 10.1158/1078-0432.CCR-04-0940. [DOI] [PubMed] [Google Scholar]
- 21.Shmulevitz M., Gujar S.A., Ahn D.G., Mohamed A., Lee P.W.K. Reovirus variants with mutations in genome segments S1 and L2 exhibit enhanced virion infectivity and superior oncolysis. J. Virol. 2012;86:7403–7413. doi: 10.1128/JVI.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nibert M.L., Schiff L.A. In: Fields Virology. 4th Edition ed. Knipe D.M., editor. Lippincott Williams & Wilkins; 2001. Reoviruses and their replication; pp. 1679–1728. [Google Scholar]
- 23.Coombs K.M. Reovirus structure and morphogenesis. Curr. Top. Microbiol. Immunol. 2006;309:117–167. doi: 10.1007/3-540-30773-7_5. [DOI] [PubMed] [Google Scholar]
- 24.Guglielmi K.M., Kirchner E., Holm G.H., Stehle T., Dermody T.S. Reovirus binding determinants in junctional adhesion molecule-A. J. Biol. Chem. 2007;282:17930–17940. doi: 10.1074/jbc.M702180200. [DOI] [PubMed] [Google Scholar]
- 25.Danthi P., Holm G.H., Stehle T., Dermody T.S. Reovirus receptors, cell entry, and proapoptotic signaling. Adv. Exp. Med. Biol. 2013;790:42–71. doi: 10.1007/978-1-4614-7651-1_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chappell J.D., Gunn V.L., Wetzel J.D., Baer G.S., Dermody T.S. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein sigma1. J. Virol. 1997;71:1834–1841. doi: 10.1128/JVI.71.3.1834-1841.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agosto M.A., Ivanovic T., Nibert M.L. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc. Natl. Acad. Sci. USA. 2006;103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desmet E.A., Anguish L.J., Parker J.S.L. Virus-mediated compartmentalization of the host translational machinery. mBio. 2014;5:e01463-14. doi: 10.1128/mBio.01463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenorio R., Fernández de Castro I., Knowlton J.J., Zamora P.F., Sutherland D.M., Risco C., Dermody T.S. Function, Architecture, and Biogenesis of Reovirus Replication Neoorganelles. Viruses. 2019;11:288. doi: 10.3390/v11030288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohamed A., Konda P., Eaton H.E., Gujar S., Smiley J.R., Shmulevitz M. Closely related reovirus lab strains induce opposite expression of RIG-I/IFN-dependent versus -independent host genes, via mechanisms of slow replication versus polymorphisms in dsRNA binding sigma3 respectively. Plos Pathog. 2020;16:e1008803. doi: 10.1371/journal.ppat.1008803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohamed A., Smiley J.R., Shmulevitz M. Polymorphisms in the Most Oncolytic Reovirus Strain Confer Enhanced Cell Attachment, Transcription, and Single-Step Replication Kinetics. J. Virol. 2020;94:e01937-19–e01919. doi: 10.1128/JVI.01937-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohamed A., Johnston R.N., Shmulevitz M. Potential for Improving Potency and Specificity of Reovirus Oncolysis with Next-Generation Reovirus Variants. Viruses. 2015;7:6251–6278. doi: 10.3390/v7122936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohamed A., Teicher C., Haefliger S., Shmulevitz M. Reduction of virion-associated sigma1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J. Virol. 2015;89:4319–4334. doi: 10.1128/JVI.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yip, W. K. W.; Cristi, F.; Trifonov, G.; Narayan, N.; Kubanski, M.; Shmulevitz, M. The reovirus mu2 C-terminal loop inversely regulates NTPase and transcription functions versus binding to factory-forming muNS and promotes replication in tumorigenic cells. J. Virol. 2021, e02006-e02020. DOI: 10.1128/JVI.02006-20. [DOI] [PMC free article] [PubMed]
- 35.Marcato P., Shmulevitz M., Pan D., Stoltz D., Lee P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007;15:1522–1530. doi: 10.1038/sj.mt.6300179. [DOI] [PubMed] [Google Scholar]
- 36.Shmulevitz M., Lee P.W.K. Exploring host factors that impact reovirus replication, dissemination, and reovirus-induced cell death in cancer versus normal cells in culture. Methods Mol. Biol. 2012;797:163–176. doi: 10.1007/978-1-61779-340-0_12. [DOI] [PubMed] [Google Scholar]
- 37.Shmulevitz M., Marcato P., Lee P.W.K. Activated Ras signaling significantly enhances reovirus replication and spread. Cancer Gene Ther. 2010;17:69–70. doi: 10.1038/cgt.2009.46. [DOI] [PubMed] [Google Scholar]
- 38.Shmulevitz M., Marcato P., Lee P.W.K. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene. 2005;24:7720–7728. doi: 10.1038/sj.onc.1209041. [DOI] [PubMed] [Google Scholar]
- 39.Shmulevitz M., Pan L.Z., Garant K., Pan D., Lee P.W.K. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010;70:4912–4921. doi: 10.1158/0008-5472.CAN-09-4676. [DOI] [PubMed] [Google Scholar]
- 40.Tiwari A., Trivedi R., Lin S.Y. Tumor microenvironment: barrier or opportunity towards effective cancer therapy. J. Biomed. Sci. 2022;29:83. doi: 10.1186/s12929-022-00866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Labani-Motlagh A., Ashja-Mahdavi M., Loskog A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020;11:940. doi: 10.3389/fimmu.2020.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veglia F., Perego M., Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tesi R.J. MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharmacol. Sci. 2019;40:4–7. doi: 10.1016/j.tips.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Talmadge J.E., Gabrilovich D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer. 2013;13:739–752. doi: 10.1038/nrc3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groeneveldt C., Kinderman P., van den Wollenberg D.J.M., van den Oever R.L., Middelburg J., Mustafa D.A.M., Hoeben R.C., van der Burg S.H., van Hall T., van Montfoort N. Preconditioning of the tumor microenvironment with oncolytic reovirus converts CD3-bispecific antibody treatment into effective immunotherapy. J. Immunother. Cancer. 2020;8:e001191. doi: 10.1136/jitc-2020-001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thirukkumaran C.M., Nodwell M.J., Hirasawa K., Shi Z.Q., Diaz R., Luider J., Johnston R.N., Forsyth P.A., Magliocco A.M., Lee P., et al. Oncolytic viral therapy for prostate cancer: efficacy of reovirus as a biological therapeutic. Cancer Res. 2010;70:2435–2444. doi: 10.1158/0008-5472.CAN-09-2408. [DOI] [PubMed] [Google Scholar]
- 47.White C.L., Twigger K.R., Vidal L., De Bono J.S., Coffey M., Heinemann L., Morgan R., Merrick A., Errington F., Vile R.G., et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008;15:911–920. doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- 48.Sturm G., Finotello F., Petitprez F., Zhang J.D., Baumbach J., Fridman W.H., List M., Aneichyk T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics. 2019;35:i436–i445. doi: 10.1093/bioinformatics/btz363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sturm G., Finotello F., List M. Immunedeconv: An R Package for Unified Access to Computational Methods for Estimating Immune Cell Fractions from Bulk RNA-Sequencing Data. Methods Mol. Biol. 2020;2120:223–232. doi: 10.1007/978-1-0716-0327-7_16. [DOI] [PubMed] [Google Scholar]
- 50.Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., Selves J., Laurent-Puig P., Sautès-Fridman C., Fridman W.H., de Reyniès A. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marone G., Gambardella A.R., Mattei F., Mancini J., Schiavoni G., Varricchi G. Basophils in Tumor Microenvironment and Surroundings. Adv. Exp. Med. Biol. 2020;1224:21–34. doi: 10.1007/978-3-030-35723-8_2. [DOI] [PubMed] [Google Scholar]
- 52.Larson S.M., Antczak J.B., Joklik W.K. Reovirus exists in the form of 13 particle species that differ in their content of protein sigma 1. Virology. 1994;201:303–311. doi: 10.1006/viro.1994.1295. [DOI] [PubMed] [Google Scholar]
- 53.Barton E.S., Connolly J.L., Forrest J.C., Chappell J.D., Dermody T.S. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J. Biol. Chem. 2001;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 54.Mita A.C., Sankhala K., Sarantopoulos J., Carmona J., Okuno S., Goel S., Chugh R., Coffey M.C., Mettinger K., Mita M.M. A phase II study of intravenous (IV) wild-type reovirus (Reolysin) in the treatment of patients with bone and soft tissue sarcomas metastatic to the lung. J. Clin. Oncol. 2009;27:10524. doi: 10.1200/jco.2009.27.15_suppl.10524. [DOI] [Google Scholar]
- 55.Jain R.K. Barriers to drug delivery in solid tumors. Sci. Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 56.Wagner M., Wiig H. Tumor Interstitial Fluid Formation, Characterization, and Clinical Implications. Front. Oncol. 2015;5:115. doi: 10.3389/fonc.2015.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dermody T.S., Nibert M.L., Bassel-Duby R., Fields B.N. Sequence diversity in S1 genes and S1 translation products of 11 serotype 3 reovirus strains. J. Virol. 1990;64:4842–4850. doi: 10.1128/JVI.64.10.4842-4850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dermody T.S., Nibert M.L., Bassel-Duby R., Fields B.N. A sigma 1 region important for hemagglutination by serotype 3 reovirus strains. J. Virol. 1990;64:5173–5176. doi: 10.1128/JVI.64.10.5173-5176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Excoffon K.J.D.A., Guglielmi K.M., Wetzel J.D., Gansemer N.D., Campbell J.A., Dermody T.S., Zabner J. Reovirus preferentially infects the basolateral surface and is released from the apical surface of polarized human respiratory epithelial cells. J. Infect. Dis. 2008;197:1189–1197. doi: 10.1086/529515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yamasaki M., Kanai Y., Wakamura Y., Kotaki T., Minami S., Nouda R., Nurdin J.A., Kobayashi T. Characterization of Sialic Acid-Independent Simian Rotavirus Mutants in Viral Infection and Pathogenesis. J. Virol. 2023;97:e0139722. doi: 10.1128/jvi.01397-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doceul V., Hollinshead M., van der Linden L., Smith G.L. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science. 2010;327:873–876. doi: 10.1126/science.1183173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Condit R.C. Surf and turf: mechanism of enhanced virus spread during poxvirus infection. Viruses. 2010;2:1050–1054. doi: 10.3390/v2051050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gallet R., Kannoly S., Wang I.N. Effects of bacteriophage traits on plaque formation. BMC Microbiol. 2011;11:181. doi: 10.1186/1471-2180-11-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bauer P.H., Cui C., Liu W.R., Stehle T., Harrison S.C., DeCaprio J.A., Benjamin T.L. Discrimination between sialic acid-containing receptors and pseudoreceptors regulates polyomavirus spread in the mouse. J. Virol. 1999;73:5826–5832. doi: 10.1128/JVI.73.7.5826-5832.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rubio M.P., López-Bueno A., Almendral J.M. Virulent variants emerging in mice infected with the apathogenic prototype strain of the parvovirus minute virus of mice exhibit a capsid with low avidity for a primary receptor. J. Virol. 2005;79:11280–11290. doi: 10.1128/JVI.79.17.11280-11290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.López-Bueno A., Rubio M.P., Bryant N., McKenna R., Agbandje-McKenna M., Almendral J.M. Host-selected amino acid changes at the sialic acid binding pocket of the parvovirus capsid modulate cell binding affinity and determine virulence. J. Virol. 2006;80:1563–1573. doi: 10.1128/JVI.80.3.1563-1573.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byrnes A.P., Griffin D.E. Large-plaque mutants of Sindbis virus show reduced binding to heparan sulfate, heightened viremia, and slower clearance from the circulation. J. Virol. 2000;74:644–651. doi: 10.1128/jvi.74.2.644-651.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiu M., Armstrong E.J.L., Jennings V., Foo S., Crespo-Rodriguez E., Bozhanova G., Patin E.C., McLaughlin M., Mansfield D., Baker G., et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin. Biol. Ther. 2020;20:635–652. doi: 10.1080/14712598.2020.1729351. [DOI] [PubMed] [Google Scholar]
- 69.Malogolovkin A., Gasanov N., Egorov A., Weener M., Ivanov R., Karabelsky A. Combinatorial Approaches for Cancer Treatment Using Oncolytic Viruses: Projecting the Perspectives through Clinical Trials Outcomes. Viruses. 2021;13:1271. doi: 10.3390/v13071271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shi T., Song X., Wang Y., Liu F., Wei J. Combining Oncolytic Viruses With Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020;11:683. doi: 10.3389/fimmu.2020.00683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koehler M., Aravamudhan P., Guzman-Cardozo C., Dumitru A.C., Yang J., Gargiulo S., Soumillion P., Dermody T.S., Alsteens D. Glycan-mediated enhancement of reovirus receptor binding. Nat. Commun. 2019;10:4460. doi: 10.1038/s41467-019-12411-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Koehler M., Petitjean S.J.L., Yang J., Aravamudhan P., Somoulay X., Lo Giudice C., Poncin M.A., Dumitru A.C., Dermody T.S., Alsteens D. Reovirus directly engages integrin to recruit clathrin for entry into host cells. Nat. Commun. 2021;12:2149. doi: 10.1038/s41467-021-22380-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maginnis M.S., Forrest J.C., Kopecky-Bromberg S.A., Dickeson S.K., Santoro S.A., Zutter M.M., Nemerow G.R., Bergelson J.M., Dermody T.S. Beta1 integrin mediates internalization of mammalian reovirus. J. Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Konopka-Anstadt J.L., Mainou B.A., Sutherland D.M., Sekine Y., Strittmatter S.M., Dermody T.S. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe. 2014;15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eaton H.E., Kobayashi T., Dermody T.S., Johnston R.N., Jais P.H., Shmulevitz M. African Swine Fever Virus NP868R Capping Enzyme Promotes Reovirus Rescue during Reverse Genetics by Promoting Reovirus Protein Expression, Virion Assembly, and RNA Incorporation into Infectious Virions. J. Virol. 2017;91:e02416-16–e02416. doi: 10.1128/JVI.02416-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mohamed A., Clements D.R., Gujar S.A., Lee P.W., Smiley J.R., Shmulevitz M. Single Amino Acid Differences between Closely Related Reovirus T3D Lab Strains Alter Oncolytic Potency In Vitro and In Vivo. J. Virol. 2020;94:e01688-19–e01619. doi: 10.1128/JVI.01688-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mendez I.I., Hermann L.L., Hazelton P.R., Coombs K.M. A comparative analysis of Freon substitutes in the purification of reovirus and calicivirus. J. Virol. Methods. 2000;90:59–67. doi: 10.1016/s0166-0934(00)00217-2. [DOI] [PubMed] [Google Scholar]
- 78.Wickham H., Averick M., Bryan J., Chang W., McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J., et al. Welcome to the Tidyverse. J. Open Source Softw. 2019;4:1686. doi: 10.21105/joss.01686. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For access to the data supporting the findings of this study, please contact the corresponding author.